

Abstract
PURPOSE
Fanconi anemia (FA) and ataxia-telangiectasia (AT) are rare inherited syndromes characterized by abnormal DNA damage response and caused by pathogenic variants in key DNA repair proteins that are also relevant in the pathogenesis of breast cancer and other cancer types. The risk of cancer in children with these diseases is poorly understood and has never been assessed in a population-based cohort before.
METHODS
We identified 421 patients with FA and 160 patients with AT diagnosed between 1973 and 2020 through German DNA repair disorder reference laboratories. We linked patients' laboratory data with childhood cancer data from the German Childhood Cancer Registry.
RESULTS
Among 421 patients with FA, we observed 33 cases of childhood cancer (15 cases of myelodysplastic syndrome; seven cases of acute myeloid leukemia; two cases of lymphoma, carcinoma, medulloblastoma, and nephroblastoma, respectively; and one case of rhabdomyosarcoma, acute lymphoblastic leukemia, and glioma, respectively) versus 0.74 expected (on the basis of population-based incidence rates in Germany). This corresponds to a 39-fold increased risk (standardized incidence ratio [SIR] = 39; 95% CI, 26 to 56). For all FA subgroups combined, the cancer-specific SIR for myeloid neoplasms was 445 (95% CI, 272 to 687). Among the 160 patients with AT, we observed 19 cases of childhood cancer (15 cases of lymphoma, three cases of leukemia, and one case of medulloblastoma) versus 0.32 expected. This corresponds to a 56-fold increased risk (SIR = 56; 95% CI, 33 to 88). The cancer-specific SIR for Hodgkin lymphoma was 215 (95% CI, 58 to 549) and for non-Hodgkin lymphoma 470 (95% CI, 225 to 865).
CONCLUSION
Approximately 11% of patients with FA and 14% of patients with AT develop cancer by age 18 years.
INTRODUCTION
Fanconi anemia (FA; complementation groups A, B, C, D1, D2, E, F, G, I, J, L, N, O, P, Q, R, S, T, U, V, and W; Mendelian Inheritance in Man [MIM]: 227650, 300514, 227645, 605724, 227646, 600901, 603467, 614082, 609053, 609054, 614083, 610832, 613390, 613951, 615272, 617244, 617883, 616435, 617247, 617243, 617784) is a rare DNA-repair deficiency disorder characterized by congenital malformations, bone marrow failure, endocrine abnormalities, and an elevated cancer risk. The most common cancers are myelodysplastic syndrome (MDS), acute myeloid leukemia (AML), and squamous cell carcinoma.1 To date, germline pathogenic variants in 21 genes (FANCA-W) have been associated with FA, with complementation groups FA-A, FA-C, and FA-G representing the most common genetic subtypes.2 FA is usually a recessive condition with the exception of complementation group FA-B (X-linked) and FA-R (autosomal dominant).
CONTEXT
Key Objective
What is the cancer risk in children with Fanconi anemia (FA), a DNA-repair deficiency disorder caused by mainly biallelic germline variants in at least 21 different DNA repair genes, and what is the cancer risk in children with ataxia-telangiectasia (AT) caused by biallelic pathogenic variants in the ATM gene, also involved in DNA repair? We used a nationwide register-based cohort study enrolling 421 patients with FA and 160 patients with AT to address these questions.
Knowledge Generated
The risk of developing cancer before age 18 years is approximately 11% in children with FA and approximately 14% in children with AT. Compared with the general population, this amounts to a 39- and 56-fold increased risk, respectively.
Relevance
The study provides a robust and comprehensive data set for counseling and care of children with FA or AT.
Ataxia-telangiectasia (AT; MIM 208900) is an autosomal recessive disorder caused by biallelic pathogenic variants in ATM (ataxia-telangiectasia–mutated), a DNA damage–sensing kinase.3 Patients present with cerebellar ataxia, conjunctival telangiectasias, oculomotor apraxia, choreoathetosis, immunodeficiency, and an increased leukemia and lymphoma risk.4
Monoallelic pathogenic germline variants in ATM are associated with a two- to threefold increased breast cancer risk in women.5 Moreover, the pathogenic variants in rare FA genes such as FANCD1 (BRCA2), FANCN (PALB2), FANCS (BRCA1), and FANCO (RAD51C) are known to be associated with an increased breast cancer risk.6
The childhood cancer risk and the risk of individual cancer types in individuals with FA or AT have been studied in literature case series7,8 and volunteer cohorts.9-13 However, access to larger unbiased population data is limited. In this unique nationwide register study, we matched data from 581 patients with the German Childhood Cancer Registry (GCCR) in an encrypted approach. This allowed a population-based statistical analysis while minimizing the risk of selection bias. The diagnoses were genetically and/or functionally confirmed (FA: sensitivity to DNA crosslinking agents, AT: sensitivity to ionizing radiation) during a 47-year period by the major and nationwide German reference laboratories for DNA repair disorders for all patients enrolled in this study.
METHODS
We investigated the occurrence of childhood cancer in cohorts of 421 individuals with FA and 160 individuals with AT. In all cases, the suspected diagnosis FA was confirmed by cellular hypersensitivity toward mitomycin C or diepoxybutane (DEB) via chromosome breakage or cell cycle analysis,14,15 AT was confirmed by the presence of cellular radiosensitivity testing,16-18 and/or causative pathogenic variants have been identified. A majority of patient data have been published in various other studies on FA or AT dealing with mutation analysis, genotype-phenotype correlations, or new FA gene identification. The laboratory analyses used in the present study were performed or confirmed at the Department of Human Genetics, University of Würzburg, Biocenter, Germany, the major nationwide reference laboratory mainly for FA and also for AT and other DNA repair disorders, and/or at the Gynecology Research Unit of Hannover Medical School, Germany. Notably, in FA patients, the diagnosis can be confirmed by functional assays without knowledge of the precise FA subtype and without mutational analysis. In fact, a confirmed FA diagnosis often preceded the identification of the respective FA disease-causing gene variant.
Confirmed cases of FA and AT diagnosed between January 1, 1973, and September 22, 2020, were linked to the database of the GCCR (71,614 patients with childhood cancer on September 22, 2020), using a cryptographed stochastic record linkage approach on the basis of names and dates of births as described previously.19-21 Patients were considered under risk and contributed person-years to the analysis until death, a cancer diagnosis, or September 22, 2020, whichever came first. Vital status information at cutoff was available for 40% of the FA and AT cases. The GCCR is the nationwide population-based childhood cancer registry of Germany, monitoring incident cases of all malignancies and nonmalignant CNS tumors diagnosed in individuals age 0 to 17 years since 1980. Before 2009, the registry covered only diagnoses before age 15 years. On average, approximately 2,250 incident cases are observed annually on the basis of a population of about 13.5 million children annually below age 18 years. The study was approved by the ethical committee at the Hannover Medical School (no. 2850-2015).
All identifiable cases in the GCCR from 1980 until September 22, 2020, with German residence and below the ages 15 (until 2008) or 18 years (since 2009) at the time of diagnosis were included in the base cohort. The range of diagnoses is defined by the International Classification of Childhood Cancer.22 The GCCR follows patients with a first childhood cancer diagnosis and collects subsequent malignant neoplasms at any age as completely as possible from various sources, including the patients and their families (subsequent malignant neoplasm diagnoses are validated by treating physicians and histologically confirmed). Age-specific incidence rates for first neoplasms were calculated on the basis of this file (ie, excluding nonidentifiable patients) and corresponding population data from the Federal Statistical Office. This process ensures that the standardized incidence ratio (SIR) estimates are not biased by (1) the inclusion criterion for age changing in 2009 and (2) excluding nonidentifiable patients from the analyses. MDS was not registered systematically before 2000 as a result of international changes in coding, but as this applies to the cancer case ascertainment and the comparison incidence rates, this too does not lead to a biased SIR estimate. Cumulative incidences (risk until 18th birthday) were estimated as the sum of the age-specific incidence rates in the syndrome cohort. SIRs, comparing observed and expected numbers of cases, and their respective 95% CIs were calculated by the usual standard procedures. The process automatically excludes all person-years that occurred outside the respective age window and outside the time window 1980 to September 22, 2020. In genetic diseases, there is always the risk of cases going undetected, and, as a (suspected) cancer diagnosis can be the reason why a genetic assessment was performed, leading to an overestimation of the SIR. For 556 of 581 cases, the laboratory was able to provide the dates of the initial genetic reports and to assess whether a cancer diagnosis was the indication for genetic testing. The extent of this overestimation was estimated by running an analysis excluding every individual and their person-years for whom genetic testing was because of a (suspected) cancer diagnosis and/or the date of the syndrome diagnosis close to or after the cancer diagnosis as a sensitivity analysis. This analysis is unbiased, although the exclusion reduces power.
RESULTS
We identified 421 individuals with a confirmed diagnosis of FA and German residence between 1973 and 2020. In 301 of 421 cases, the molecular data were available: complementation group FA-A (n = 182), FA-G (n = 31), FA-C (n = 29), FA-D2 (n = 20), FA-D1 (n = 9), FA-I (n = 7), FA-P (n = 6), FA-B (n = 4), FA-N (n = 4), FA-E (n = 2), FA-J (n = 2), FA-L (n = 2), FA-F (n = 1), FA-Q (n = 1), and FA-T (n = 1). The 421 FA patients contributed a total of 5,245.1 person-years of observation time overlapping with the GCCR database. Patients were born between 1963 and 2017 and age at genetic testing ranged from 0 to 48 years (median: 7.2 years). The male-to-female ratio was 1.15. Cryptographed stochastic record linkage with the GCCR identified 33 patients who developed a primary cancer in childhood or adolescence of 419 patients overlapping with GCCR, of whom 10 patients additionally presented with a second neoplasm (Table 1).
TABLE 1.
Description of the 33 Individuals With Fanconi Anemia Who Developed Cancera

On the basis of all person-years and the age distribution of the studied FA population, 0.74 cases of childhood cancer would have been expected versus 33 observed, which corresponds to a 39-fold increased risk (SIR = 39; 95% CI, 26 to 56; Table 2). Age at cancer diagnosis ranged from 0.4 to 16.8 years (median: 11.1 years). The cancer SIRs for individual FA subgroups are given in Table 2. The highest cancer risks were observed in patients with biallelic mutations in FANCD1 (BRCA2; SIR, 324; 95% CI, 88 to 830) or FANCN (PALB2; SIR, 422; 95% CI, 51 to 1,526), whereas no cancer was observed among the 20 patients with biallelic mutations in FANCD2 (Table 2). The cumulative cancer risk before age 18 years was 10.6% in the entire FA cohort. Figure 1 shows cumulative incidences of AML, MDS, and other cancer types. SIRs of selected cancers in individuals with FA are given in Table 3. High SIRs were observed for MDS (FA, all subtypes combined: SIR = 1,104; 95% CI, 588 to 1,888) and AML (FA, all subtypes combined: SIR = 211; 95% CI, 85 to 435). To reduce selection bias, we conducted a sensitivity analysis after excluding nine FA cases in whom we cannot rule out a timely relationship between the diagnosis of FA and cancer. After exclusion of these cases, we observed a 29-fold increased cancer risk (SIR = 29; 95% CI, 16 to 48) for all FA patients combined.
TABLE 2.
Genotype-Dependent Categorization of Individuals With FA and ATa

FIG 1.

Cumulative cancer incidences in patients with (A) FA and (B) AT. AML, acute myeloid leukemia; AT, ataxia-telangiectasia; FA, Fanconi anemia; HL, Hodgkin lymphoma; MDS, myelodysplastic syndrome; NHL, non-Hodgkin lymphoma.
TABLE 3.
Standardized Incidence Ratio for Specific Cancers in Patients With FA and ATa

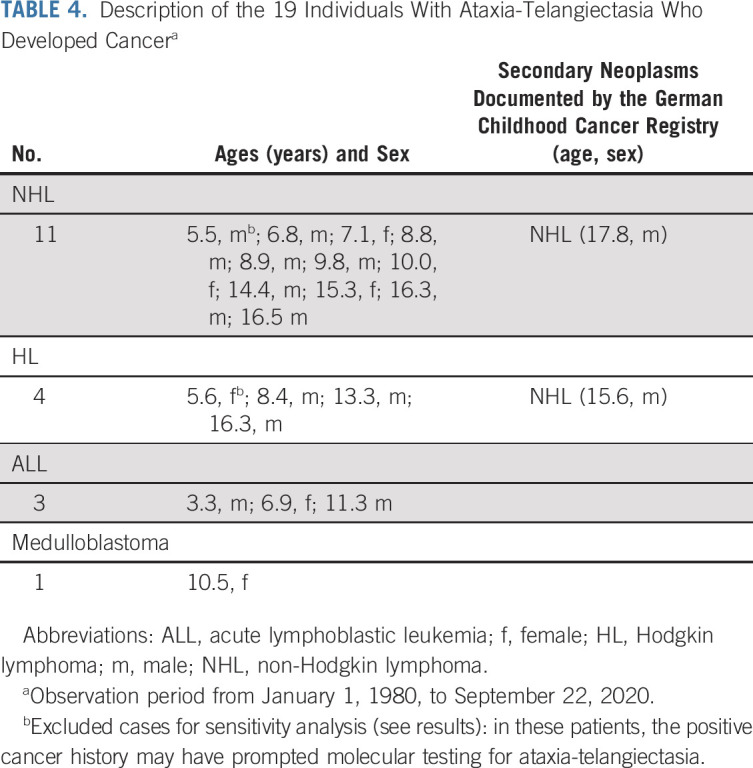
We identified 157 of 160 AT patients whose childhood period overlapped with the GCCR database. These patients contributed 2,267.9 person-years of observation time. Patients were born between 1968 and 2015 and the age at genetic and/or sensitivity to ionizing radiation testing ranged from 0 to 35 years (median age: 7.2 years). The male-to-female ratio was 1.44. Nineteen patients developed childhood cancer (Table 4). On the basis of all person-years and the age distribution of the studied AT population, 0.32 cases of childhood cancer would be expected versus 19 observed, a 56-fold increase (SIR = 56; 95% CI, 33 to 88; Table 2). Age at cancer diagnosis ranged from 3 to 17 years (median age: 9.8 years). The risk of cancer diagnosed before age 18 years was 14.3%. Cancer-specific SIRs are provided in Table 3. High SIRs were found for Hodgkin lymphoma (SIR = 215; 95% CI, 58 to 549) and non-Hodgkin lymphoma (SIR = 470; 95% CI, 225 to 865). The corresponding cumulative incidences are depicted in Figure 1. When excluding two cases in whom the cancer diagnosis was known before the AT diagnosis, we calculated a 34-fold increased cancer risk (SIR = 34; 95% CI, 9 to 88).
TABLE 4.
Description of the 19 Individuals With Ataxia-Telangiectasia Who Developed Cancera
DISCUSSION
We conducted a nationwide population-based cohort study to quantify the cancer risk in children with FA and AT and observed a significant excess risk for all childhood cancers combined compared with the general population. The elevated cancer risk was primarily because of significant excesses of MDS and AML in children with FA and lymphoma in children with AT. The highest cancer risks were observed in children belonging to subgroups FA-D1 (BRCA2) and FA-N (PALB2). In addition to AML and MDS, latter patients developed a range of other neoplasms such as rhabdomyosarcoma, medulloblastoma, nephroblastoma, and lymphoma, which is consistent with previous reports.8 Notably, some patients developed multiple cancers within the first years of life.
Our cohort is unique because of the unbiased case ascertainment over a long interval of almost five decades. Especially the FA cohort is substantially larger than any other cohort that has been analyzed in the past.9-12 A linkage of patient data with the GCCR allowed us to identify cases of cancer in an unbiased manner and to calculate SIRs. Therefore, our results provide robust cancer risk estimates for the childhood period of patients with both conditions. Of note, on the basis of these results and lifetime estimates, we appreciate that a considerable proportion of cancer burden in FA and AT patients is beyond childhood.9-12 The fact that the sensitivity analyses resulted in only slightly smaller SIRs shows that the estimates are only minimally biased.
In addition to the different strategies of how patients with FA and AT as well as cases of cancer were ascertained in previous studies compared with ours, there are further methodical differences that challenge a direct comparison of our study with previous risk estimates. Alter et al11,12 used competing risk analyses and excluded the cases of MDS from cancer incidence estimates. Indeed, MDS is not always a malignant condition and children with refractory cytopenia of childhood can remain stable without transformation. We were able to include MDS in our cancer risk analysis because all MDS cases were confirmed by the European Working Group on MDS in Childhood and the included patients displayed definite signs of transformation, such as elevated blast percentage or monosomy 7. Despite these different approaches, the cancer risks that result from our analysis are consistent with results from previous reports.7-13,23,24 Notably, the previous studies on FA provide valuable information on the high cancer risks (mainly squamous cell carcinoma) in adults with FA.7-12
Our study has several limitations: (1) We were unable to ascertain cancer diagnoses in patients being older than 17 years for statistical analysis, as the case-identifying resource in this study was the GCCR. (2) We assume that the FA and AT diagnoses were prompted by the cancer diagnosis in some cases (eg, in all children with MDS, FA is routinely ruled out in Germany) potentially leading to a selection bias. If we exclude cases in whom the cancer diagnosis occurred in timely relationship with the syndrome diagnosis, the cancer risks remain significantly elevated. Conversely, patients with FA and AT are more likely to undergo cancer surveillance and this may potentially lead to an overestimation of the cancer risk, although such surveillance effects are not known for AML, MDS, or lymphoma. (3) Patients in whom the diagnosis of FA or AT was established as adults are biased toward those that did not develop childhood cancer leading to an underestimation of the childhood cancer risk. (4) Vital status information of the FA and AT cases was incomplete; individuals without a specified status were assumed to be alive at the cutoff date by default potentially leading to an underestimation of the cancer risk. (5) Although we calculated FA gene–dependent differences in cancer risk (Table 2), our sample is too small to draw a definitive conclusion and the SIR CIs overlap. Especially estimating the difference in cancer risk for subtypes with only four patients (eg, FA-N) is unlikely to be clinically meaningful. (6) Factors such as mosaicism in the hematopoietic system or hematopoietic stem-cell transplantation (HSCT) may influence (reduce) the risk for MDS and AML. Mosaicism is observed at different rates in different FA subtypes. The relatively frequent occurrence in FA-A may perhaps explain their relatively lower group-specific and MDS/AML-specific SIR rates compared with FA-C and FA-G patients. Moreover, HSCT at a young age, as is often performed for early bone marrow failure in FA-D2 patients,25 may contribute to the zero incidence rates for MDS and AML in the FA-D2 patients in our study. Notably, we observed no myeloid neoplasms in children following HSCT for bone marrow failure (data not shown). (7) We were unable to determine specific lymphoma subentities associated with AT as the data source was the GCCR and not the original pathology report. (8) We were unable to include patients with cancer who were not registered in the GCCR leading to a potential underestimation of the cancer risk. (9) We were unable to analyze details on treatment response and toxicities as these data are not part of the GCCR data set. (10) We cannot exclude that ancestry influences cancer risks in FA and AT patients.
Despite these limitations, our risk estimates are based on a unique and large cohort of patients and provide clinically valuable and robust information on the cancer risks and spectra in children with FA and AT. The cancer risk in children with FA and AT is high but not as high as in children with Li-Fraumeni syndrome (approximately 40% of patients develop cancer by age 18 years) or constitutional mismatch repair deficiency (patients generally develop cancer by age 18 years). For patients with either of these conditions, surveillance has been documented to improve survival.26-29 FA- and AT-specific recommendations for childhood cancer screening and surveillance already exist for these children30 and are supported by our results. Notably, among patients with FA-A, FA-C, and FA-G, MDS and AML did not occur before age 4 years. Therefore, our data suggest that it may be safe to start bone marrow surveillance in FA patients of these subgroups following the third year of life.
ACKNOWLEDGMENT
We thank Traute M. Schroeder-Kurth for her facilitation of including some FA patients diagnosed by her Cytogenetics Section in the former Institute for Anthropology and Human Genetics at the University of Heidelberg, Germany. We thank the Deutsche Fanconi-Anämie-Hilfe for procuring contacts and exchange between FA families and the support group on the one hand and physicians and scientists on the other hand. We are grateful to Richard Friedl and Christin Berger, University of Würzburg, Würzburg, Germany, and Peter Schürmann, Hannover Medical School, Hannover, Germany, for their excellent technical assistance. Studies by the Würzburg Fanconi anemia group have been supported over time by several grants from the Schroeder-Kurth Fund. We thank Charlotte Niemeyer, University Medical Center Freiburg, Freiburg, Germany, for the insightful discussion.
See accompanying editorial on page 5
PRIOR PRESENTATION
Presented in part at the 2nd annual meeting of the European Society of Pediatric Oncology, April 28-30, 2021 (virtual meeting).
SUPPORT
C.P.K. has been supported by the BMBF ADDRess (01GM1909A) and by the Deutsche Kinderkrebsstiftung (DKS2019.13) and D.S. and R.K. by the BMBF ADDRess (01GM1909B). F.E. and C.D. have received support from the Gerdes Foundation. T.D. has been supported by the German Research Foundation (Do761/2-1). D.S. has been supported by the Schroeder-Kurth Fund.
D.S., R.K., and C.P.K. contributed equally to this work.
AUTHOR CONTRIBUTIONS
Conception and design: Christina M. Dutzmann, Claudia Spix, Friederike Erdmann, Detlev Schindler, Reinhard Kalb, Christian P. Kratz
Collection and assembly of data: Christina M. Dutzmann, Claudia Spix, Isabell Popp, Melanie Kaiser, Miriam Erlacher, Thilo Dörk, Detlev Schindler, Reinhard Kalb, Christian P. Kratz
Data analysis and interpretation: All authors
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Cancer in Children With Fanconi Anemia and Ataxia-Telangiectasia—A Nationwide Register-Based Cohort Study in Germany
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/authors/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
No potential conflicts of interest were reported.
REFERENCES
- 1.Savage SA, Walsh MF: Myelodysplastic syndrome, acute myeloid leukemia, and cancer surveillance in Fanconi anemia. Hematol Oncol Clin North Am 32:657-668, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Knies K, Inano S, Ramirez MJ, et al. : Biallelic mutations in the ubiquitin ligase RFWD3 cause Fanconi anemia. J Clin Invest 127:3013-3027, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Savitsky K, Bar-Shira A, Gilad S, et al. : A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 268:1749-1753, 1995 [DOI] [PubMed] [Google Scholar]
- 4.Amirifar P, Ranjouri MR, Lavin M, et al. : Ataxia-telangiectasia: Epidemiology, pathogenesis, clinical phenotype, diagnosis, prognosis and management. Expert Rev Clin Immunol 16:859-871, 2020 [DOI] [PubMed] [Google Scholar]
- 5.Marabelli M, Cheng SC, Parmigiani G: Penetrance of ATM gene mutations in breast cancer: A meta-analysis of different measures of risk. Genet Epidemiol 40:425-431, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hu C, Hart SN, Gnanaolivu R, et al. : A population-based study of genes previously implicated in breast cancer. N Engl J Med 384:440-451, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alter BP: Cancer in Fanconi anemia, 1927-2001. Cancer 97:425-440, 2003 [DOI] [PubMed] [Google Scholar]
- 8.Alter BP, Rosenberg PS, Brody LC: Clinical and molecular features associated with biallelic mutations in FANCD1/BRCA2. J Med Genet 44:1-9, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosenberg PS, Alter BP, Ebell W: Cancer risks in Fanconi anemia: Findings from the German Fanconi Anemia Registry. Haematologica 93:511-517, 2008 [DOI] [PubMed] [Google Scholar]
- 10.Tamary H, Nishri D, Yacobovich J, et al. : Frequency and natural history of inherited bone marrow failure syndromes: The Israeli Inherited Bone Marrow Failure Registry. Haematologica 95:1300-1307, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alter BP, Giri N, Savage SA, et al. : Malignancies and survival patterns in the National Cancer Institute inherited bone marrow failure syndromes cohort study. Br J Haematol 150:179-188, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alter BP, Giri N, Savage SA, et al. : Cancer in the National Cancer Institute inherited bone marrow failure syndrome cohort after fifteen years of follow-up. Haematologica 103:30-39, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suarez F, Mahlaoui N, Canioni D, et al. : Incidence, presentation, and prognosis of malignancies in ataxia-telangiectasia: A report from the French national registry of primary immune deficiencies. J Clin Oncol 33:202-208, 2015 [DOI] [PubMed] [Google Scholar]
- 14.Schindler D, Kubbies M, Hoehn H, et al. : Presymptomatic diagnosis of Fanconi's anaemia. Lancet 1:937, 1985 [DOI] [PubMed] [Google Scholar]
- 15.Schindler D, Kubbies M, Hoehn H, et al. : Confirmation of Fanconi's anemia and detection of a chromosomal aberration (1Q12-32 triplication) via BrdU/Hoechst flow cytometry. Am J Pediatr Hematol Oncol 9:172-177, 1987 [DOI] [PubMed] [Google Scholar]
- 16.Schindler D, Seyschab H, Poot M, et al. : Screening test for ataxia telangiectasia. Lancet 2:1398-1399, 1987 [DOI] [PubMed] [Google Scholar]
- 17.Seyschab H, Schindler D, Friedl R, et al. : Simultaneous measurement, using flow cytometry, of radiosensitivity and defective mitogen response in ataxia telangiectasia and related syndromes. Eur J Pediatr 151:756-760, 1992 [DOI] [PubMed] [Google Scholar]
- 18.Heinrich T, Prowald C, Friedl R, et al. : Exclusion/confirmation of ataxia-telangiectasia via cell-cycle testing. Eur J Pediatr 165:250-257, 2006 [DOI] [PubMed] [Google Scholar]
- 19.Hammer GP, Seidenbusch MC, Schneider K, et al. : A cohort study of childhood cancer incidence after postnatal diagnostic X-ray exposure. Radiat Res 171:504-512, 2009 [DOI] [PubMed] [Google Scholar]
- 20.Kratz CP, Franke L, Peters H, et al. : Cancer spectrum and frequency among children with Noonan, Costello, and cardio-facio-cutaneous syndromes. Br J Cancer 112:1392-1397, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coktu S, Spix C, Kaiser M, et al. : Cancer incidence and spectrum among children with genetically confirmed Beckwith-Wiedemann spectrum in Germany: A retrospective cohort study. Br J Cancer 123:619-623, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Steliarova-Foucher E, Stiller C, Lacour B, et al. : International classification of childhood cancer, third edition. Cancer 103:1457-1467, 2005 [DOI] [PubMed] [Google Scholar]
- 23.Morrell D, Cromartie E, Swift M: Mortality and cancer incidence in 263 patients with ataxia-telangiectasia. J Natl Cancer Inst 77:89-92, 1986 [PubMed] [Google Scholar]
- 24.Olsen JH, Hahnemann JM, Borresen-Dale AL, et al. : Cancer in patients with ataxia-telangiectasia and in their relatives in the nordic countries. J Natl Cancer Inst 93:121-127, 2001 [DOI] [PubMed] [Google Scholar]
- 25.Cagnan I, Gunel-Ozcan A, Aerts-Kaya F, et al. : Bone marrow mesenchymal stem cells carrying FANCD2 mutation differ from the other Fanconi anemia complementation groups in terms of TGF-beta1 production. Stem Cell Rev Rep 14:425-437, 2018 [DOI] [PubMed] [Google Scholar]
- 26.Villani A, Shore A, Wasserman JD, et al. : Biochemical and imaging surveillance in germline TP53 mutation carriers with Li-Fraumeni syndrome: 11 year follow-up of a prospective observational study. Lancet Oncol 17:1295-1305, 2016 [DOI] [PubMed] [Google Scholar]
- 27.Bougeard G, Renaux-Petel M, Flaman JM, et al. : Revisiting Li-Fraumeni syndrome from TP53 mutation carriers. J Clin Oncol 33:2345-2352, 2015 [DOI] [PubMed] [Google Scholar]
- 28.Wimmer K, Kratz CP: Constitutional mismatch repair-deficiency syndrome. Haematologica 95:699-701, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Durno C, Ercan AB, Bianchi V, et al. : Survival benefit for individuals with constitutional mismatch repair deficiency undergoing surveillance. J Clin Oncol 39:2779-2790, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Walsh MF, Chang VY, Kohlmann WK, et al. : Recommendations for childhood cancer screening and surveillance in DNA repair disorders. Clin Cancer Res 23:e23-e31, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]