

Abstract
PURPOSE
The double-blind, randomized, placebo-controlled phase III iNNOVATE study showed sustained efficacy of ibrutinib-rituximab in Waldenström's macroglobulinemia (WM). Here, we present the final analysis from iNNOVATE.
METHODS
Patients had confirmed symptomatic WM, either previously untreated or previously treated; patients with prior rituximab had at least a minor response to their last rituximab-based regimen. Patients were randomly assigned to once-daily ibrutinib 420 mg plus rituximab or placebo plus rituximab (n = 75 per arm). The primary end point was progression-free survival (PFS). Secondary end points included response rate, time to next treatment, hemoglobin improvement, overall survival, and safety.
RESULTS
With a median follow-up of 50 (range, 0.5-63) months, median (95% CI) PFS was not reached (57.7 months to not evaluable) with ibrutinib-rituximab versus 20.3 months (13.0 to 27.6) with placebo-rituximab (hazard ratio, 0.250; P < .0001). PFS benefit was regardless of prior treatment status, MYD88 and CXCR4 mutation status, or key patient characteristics. Higher response rates (partial response or better) were observed with ibrutinib-rituximab (76% v 31% with placebo-rituximab; P < .0001) and were sustained over time. Median time to next treatment was not reached with ibrutinib-rituximab versus 18 months with placebo-rituximab. More patients receiving ibrutinib-rituximab versus placebo-rituximab had sustained hemoglobin improvement (77% v 43%; P < .0001). Median overall survival was not reached in either arm. Ibrutinib-rituximab maintained a manageable safety profile; the prevalence of grade ≥ 3 adverse events of clinical interest generally decreased over time.
CONCLUSION
In the final analysis of iNNOVATE with a median follow-up of 50 months, ibrutinib-rituximab showed ongoing superiority across clinical outcomes in patients with WM regardless of MYD88 or CXCR4 mutation status, prior treatment, and key patient characteristics.
INTRODUCTION
Ibrutinib, a once-daily Bruton tyrosine kinase (BTK) inhibitor, is indicated as either a single-agent therapy or in combination with rituximab, a commonly used nonchemotherapy-based treatment, for patients with Waldenström's macroglobulinemia (WM) across all lines of therapy1 and is the only BTK inhibitor approved for the treatment of WM. Ibrutinib with or without rituximab has demonstrated high overall response rates (ORRs) in patients with WM,2-4 is an International Workshop for Waldenström's Macroglobulinemia (IWWM) preferred treatment option in the first line and for patients with relapsed or refractory disease, and is the only category-1–preferred regimen for primary therapy and previously treated WM recommended by the National Comprehensive Cancer Network (NCCN).5,6
CONTEXT
Key Objective
To present the final analysis of iNNOVATE, to our knowledge the first phase III study conducted in patients with Waldenström's macroglobulinemia (WM). This randomized study evaluated the efficacy and safety of ibrutinib plus rituximab versus placebo plus rituximab in patients with WM. The final analysis reports results after a median 50-month follow-up, building upon the 30-month median follow-up results from the primary analysis.
Knowledge Generated
Ibrutinib plus rituximab maintained a manageable safety profile, provided sustained efficacy, and significantly reduced the risk of disease progression or death compared with rituximab alone, regardless of patient genotype or prior treatment status. Additionally, time to response was shorter and time to next treatment was substantially longer with ibrutinib plus rituximab compared with rituximab alone.
Relevance (S. Lentzsch)
Long-term follow-up showed that ibrutinib plus rituximab had ongoing superiority across clinical outcomes compared to rituximab alone in patients with WM regardless of prior treatment status or MYD88/CXCR4 genotype, confirming its role as a standard of care for WM.*
*Relevance section written by JCO Associate Editor Suzanne Lentzsch, MD, PhD.
Mutations in MYD88 and CXCR4 are highly prevalent in patients with WM7 and drive constitutive activation of key BTK-dependent signaling pathways involved in clonal WM cell survival and proliferation,7,8 making the BTK pathway a favorable therapeutic target.9 A pivotal phase II study demonstrated high response rates in relapsed patients with WM who were treated with single-agent ibrutinib.10 In this study, lower response rates were observed in patients without MYD88 mutations and in those with concurrent MYD88 and CXCR4 mutations compared with patients with MYD88, but without CXCR4, mutations.
The phase III randomized iNNOVATE study (PCYC-1127; NCT02165397) evaluated the efficacy and safety of ibrutinib in combination with rituximab in both previously untreated and previously treated patients.2 At the primary analysis (median follow-up, 26.5 months), ibrutinib-rituximab demonstrated superior progression-free survival (PFS) and higher ORR versus single-agent rituximab, independent of patient genotype.2 Here, we present the final analysis from the randomized arms of the iNNOVATE study after a median follow-up of 50 (range, 0.5-63) months.
METHODS
Study Design
iNNOVATE (PCYC-1127) was a double-blind, randomized, placebo-controlled, multicenter, international, phase III study designed to assess the efficacy and safety of ibrutinib-rituximab versus placebo-rituximab in patients with WM. Study design details have been previously published.2 Patients age ≥ 18 years with centrally confirmed WM were eligible. Patients could have been previously untreated or previously treated and not refractory (ie, achieved at least a minor response [MR]) to the last rituximab-based therapy and had not received rituximab < 12 months before the study. Patients were randomly assigned 1:1 to ibrutinib-rituximab or placebo-rituximab (stratification factors: International Prognostic Scoring System for WM score, number of prior regimens, and Eastern Cooperative Oncology Group performance status). Patients received oral once-daily ibrutinib 420 mg or placebo until unacceptable toxicity or progressive disease (PD) and intravenous rituximab 375 mg/m2 on day 1 of weeks 1-4 and 17-20. Patients in the placebo-rituximab arm could cross over and receive single-agent ibrutinib after Independent Review Committee (IRC)–confirmed PD. The Protocol (online only) was approved by institutional review boards or independent ethics committees at participating institutions. The study was conducted according to the principles of the Declaration of Helsinki and the Good Clinical Practice guidelines from the International Conference on Harmonization. All patients provided written informed consent before screening.
End Points and Assessments
The primary end point was PFS per IRC. Secondary end points included ORR (MR or better) per IRC, hematologic improvement per change in serum hemoglobin (Hgb) levels, time to next treatment (TTNT), overall survival (OS), and safety.2 Patient-reported outcomes (PROs) were an exploratory end point. Major response rates (partial response or better) per IRC and changes in immunoglobulin M (IgM) levels were also assessed. Responses were assessed according to the modified consensus criteria from the Sixth International Workshop on WM.11
Genotype Analysis
MYD88 and CXCR4 mutational status was centrally assessed using the Personalis ACE Extended Cancer Panel (Menlo Park, CA) with > 500 × mean coverage depth. Calls of somatic variants for MYD88 and CXCR4 used the Personalis Cancer Panel DNA pipeline operating in the tumor-only mode with no matched normal samples and a 2% limit of detection.
PROs
PROs and disease-related symptoms were measured according to the Functional Assessment of Cancer Therapy-Anemia (FACT-An) and EuroQoL 5-Dimension 5-Level Questionnaire (EQ-5D-5L; copyright EuroQol Research Foundation. EQ-5D is a trademark of the EuroQol Research Foundation); additional methods are provided in the Data Supplement (online only).
Statistical Analysis
Efficacy was analyzed in the intent-to-treat population, which included all randomly assigned patients. Safety was assessed in the safety population, which included all patients who received ≥ 1 dose of any study drug.
This study was powered to evaluate the effect of treatment on the PFS end point, defined as the duration from date of random assignment to date of PD or death. Sample size and power calculations were based on a two-sided log-rank test for PFS, assuming a target hazard ratio (HR) of 0.5, minimum 80% power, and two-sided overall significance level of P = .05.
TTNT was measured as time from random assignment to start date of any subsequent WM treatment. Investigators could switch a patient to a second therapy if their disease was worsening or per investigator's decision after study unblinding per Data Monitoring Committee recommendation, even if the patient did not meet the criteria for PD. Sustained hematologic improvement was defined as improvement in Hgb of ≥ 2 g/dL (intent-to-treat population) or an increase to > 11 g/dL with an improvement of ≥ 0.5 g/dL (patients with baseline Hgb ≤ 11 g/dL) for ≥ 56 days.
Median PFS, OS, and TTNT were estimated using the Kaplan-Meier method. Treatment differences were analyzed using the stratified log-rank test. For patients who crossed over to single-agent ibrutinib, OS sensitivity analysis was performed using the two-stage Accelerated Failure Time model to adjust treatment effect as a result of crossover.12
Adverse events (AEs) were graded by the investigator according to National Cancer Institute Common Terminology Criteria for Adverse Events v4.03. SAS version 9.3 (SAS Institute Inc, Cary, NC) was used to perform all statistical analyses.
RESULTS
Patient Demographics and Characteristics
A total of 150 patients (n = 75 per randomized arm) were enrolled. Baseline demographics and disease characteristics were generally well-balanced between arms (Table 1 and Data Supplement). In patients treated with ibrutinib-rituximab, the median age was 70 years; 55% had received ≥ 1 prior systemic treatment for WM, with a median number of one (range, 0-5) prior treatment; the median baseline Hgb level was 10.5 g/dL (range, 6.9-15.5 g/dL); 59% of patients had a baseline Hgb level ≤ 11.0 g/dL. Mutational data were available for 91% of patients (n = 136) overall and for 92% (n = 69) in the ibrutinib-rituximab arm; of those in this arm, 46% (n = 32) had the MYD88L265P/CXCR4WT genotype, 38% (n = 26) the MYD88L265P/CXCR4WHIM genotype, and 16% (n = 11) the MYD88WT/CXCR4WT genotype.
TABLE 1.
Demographics and Clinical Characteristics of Patients at Baseline

Patient Disposition
The final analysis was performed upon study closure; the median follow-up was 50 (range, 0.5-63) months. The most common reasons for discontinuing ibrutinib or placebo were PD (ibrutinib-rituximab, n = 7 [9%]; placebo-rituximab, n = 34 [45%]) and investigator decision (ibrutinib-rituximab, n = 3 [4%]; placebo-rituximab, n = 29 [39%]) (Table 2; Data Supplement [Fig S1, Table S2]). The majority of patients (24 of 29 [83%]) in the placebo-rituximab arm who discontinued because of investigator decision did so because of study unblinding per Data Monitoring Committee recommendation. During the study, 35 patients (47%) in the placebo-rituximab arm crossed over to receive single-agent ibrutinib; in addition to those 35 patients, three additional patients from that arm received ibrutinib off-study, for a total of 38 patients (51%) who received ibrutinib as next-line therapy. Sixty-eight patients (45%) continued to receive ibrutinib in a commercial setting (n = 36) or through a treatment extension program (n = 32) after study closure.
TABLE 2.
Patient Disposition and Treatment Exposure

PFS
At the final analysis, median PFS was significantly longer in the ibrutinib-rituximab arm (median not reached; 95% CI, 58 to not evaluable [NE]) compared with the placebo-rituximab arm (median 20 months; 95% CI, 13 to 28) (Fig 1A), indicating a 75% reduction in risk of disease progression or death with ibrutinib-rituximab (HR, 0.25; 95% CI, 0.15 to 0.42; P < .0001). Survival benefits per standard Kaplan-Meier methodology were similar at timepoints of clinical significance relative to the median 50-month (range, 0.5-63 months) follow-up: the estimated 54-month PFS rate for ibrutinib-rituximab versus placebo-rituximab was 68% (95% CI, 55 to 78) versus 25% (95% CI, 15 to 37) compared with estimated 48-month PFS rates of 71% (95% CI, 58 to 80) versus 25% (95% CI, 15 to 37).
FIG 1.

PFS (A) in the intention-to-treat population, (B) by genotype, and (C) by prior treatment status. For PFS by genotype and prior treatment status, Kaplan-Meier curves are shown for time points with ≥ 10 patients. HR, hazard ratio; NR, not reported; PFS, progression-free survival.
PFS benefit with ibrutinib-rituximab was observed regardless of genotype (Fig 1B). In patients with the MYD88WT/CXCR4WT genotype, the 54-month PFS rate was 70% with ibrutinib-rituximab versus 30% with placebo-rituximab. In patients with the MYD88 L265P/CXCR4WT genotype, the 54-month PFS rate was 72% with ibrutinib-rituximab versus 25% with placebo-rituximab. In patients with the MYD88 L265P/CXCR4WHIM genotype, the 54-month PFS rate was 63% with ibrutinib-rituximab versus 21% with placebo-rituximab. For all genotypes, estimated rates at 54 months were similar to those estimated at 48 months; in the ibrutinib-rituximab and placebo-rituximab arms, 48-month PFS rates were 79% and 25% in patients with the MYD88 L265P/CXCR4WT genotype, 63% and 21% in patients with the MYD88 L265P/CXCR4WHIM genotype, and 70% and 30% in patients with the MYD88WT/CXCR4WT genotype, respectively.
Likewise, PFS benefit with ibrutinib-rituximab was observed regardless of prior treatment status (Fig 1C). In previously untreated patients, the 54-month PFS rate was not evaluable in either the ibrutinib-rituximab or placebo-rituximab arm; the 48-month PFS rate in these subgroups was 70% and 32%, respectively. In previously treated patients, the 54-month PFS rate was 68% with ibrutinib-rituximab versus 20% with placebo-rituximab, with similar 48-month PFS rates of 71% and 20%, respectively. When analyzed by key patient characteristics, treatment with ibrutinib-rituximab significantly reduced risk of progression or death versus placebo-rituximab across all prespecified subgroups within the previously treated population, including by serum IgM level, Hgb, International Prognostic Scoring System for WM score, and MYD88 mutational status, and within most of these subgroups in the previously untreated population (Data Supplement [Fig S2]).
Response
At final analysis, ORR per IRC was significantly higher with ibrutinib-rituximab versus placebo-rituximab (92% v 44%) and time to overall response (MR or better) was faster (median 1 month [range, 1-21 months] v median 3 months [range, 1-22 months]). Major response rates (partial response or better) were also significantly higher with ibrutinib-rituximab versus placebo-rituximab (76% v 31%; P < .0001; Fig 2A). The depth of response to ibrutinib-rituximab increased over time, with major responses increasing from 72% at month 24 to 76% at month 60. One complete response occurred in each arm; both were achieved by month 18. The higher response rates and faster time to response with ibrutinib-rituximab versus placebo-rituximab were observed regardless of genotype and prior treatment status (Fig 2B). Among patients with the MYD88WT/CXCR4WT genotype, ORR was 82% with ibrutinib-rituximab versus 56% with placebo-rituximab. Similar results were observed among patients with the MYD88L265P/CXCR4WHIM and MYD88 L265P/CXCR4WTgenotypes, where ORRs with ibrutinib-rituximab were 100% and 94% versus 48% and 43% with placebo-rituximab, respectively.
FIG 2.

Overall response rates and major response rates in (A) the intention-to-treat population over time (n = 75 for each treatment arm at each timepoint) and (B) by genotypea and prior treatment status. aResponse rates exclude six patients in the ibrutinib-rituximab arm and eight patients in the placebo-rituximab arm who had unknown genotype. NOTE. Values over 100% are due to rounding. CR, complete response; MR, minor response; ORR, overall response rate; PR, partial response; VGPR, very good partial response.
Improvements in Hgb and IgM Levels
Significantly higher sustained Hgb levels were observed with ibrutinib-rituximab versus placebo-rituximab. At baseline, median Hgb was 10.5 g/dL with ibrutinib-rituximab and 10.0 g/dL with placebo-rituximab. At the final analysis, a greater proportion of patients receiving ibrutinib-rituximab versus placebo-rituximab had sustained Hgb improvement (77% [58 of 75] v 43% [32 of 75]; P < .0001), including in the subset of patients (n = 94) with Hgb ≤ 11.0 g/dL at baseline (95% [42 of 44] v 56% [28 of 50]; P < .0001).
Patients receiving ibrutinib-rituximab experienced rapid and sustained reductions in serum IgM levels. Baseline median serum IgM levels were similar between the treatment arms (ibrutinib-rituximab, 32.9 g/L; placebo-rituximab, 31.8 g/L). IgM decreased rapidly during the first year of treatment with ibrutinib-rituximab and reached a maximum median change of –33.5 g/L at 56 months. Initial decreases in IgM were not as pronounced in the placebo-rituximab arm and reached a maximum median change of –26.9 g/L at 57 months (Data Supplement [Fig S3]).
OS
Median OS was not reached in either treatment arm (HR, 0.81; 95% CI, 0.33 to 1.99; P = .6430). Estimated 54-month OS rates were similar with ibrutinib-rituximab and placebo-rituximab (86% [95% CI, 74 to 93] v 84% [95% CI, 71 to 92], respectively) (Data Supplement [Fig S4]) and consistent with the estimated 48-month OS rates of 90% [95% CI, 79 to 95] in the ibrutinib-rituximab arm and 88% [95% CI, 77 to 93] in the placebo-rituximab arm. At the final analysis, nine patients (12%) on ibrutinib-rituximab and 10 patients (13%) on placebo-rituximab had died. When adjusted for crossover using a two-stage Accelerated Failure Time model,12 OS remained consistent with the original analysis, with an improved HR of 0.64 (95% CI, 0.26 to 1.62) (Data Supplement [Fig S4]). In patients who crossed over to single-agent ibrutinib after PD on placebo-rituximab, median OS was not reached (95% CI, 47 months to NE) and the 54-month OS rate was not evaluable.
TTNT
Nine patients (12%) in the ibrutinib-rituximab arm and 47 (63%) patients in the placebo-rituximab arm received subsequent treatment. Median TTNT was significantly longer with ibrutinib-rituximab (not reached [95% CI, NE to NE]) versus placebo-rituximab (18 months [95% CI, 11 to 33]; P < .0001) (Fig 3), with substantially more patients (87% v 29%) without subsequent treatment at the 54-month landmark timepoint.
FIG 3.
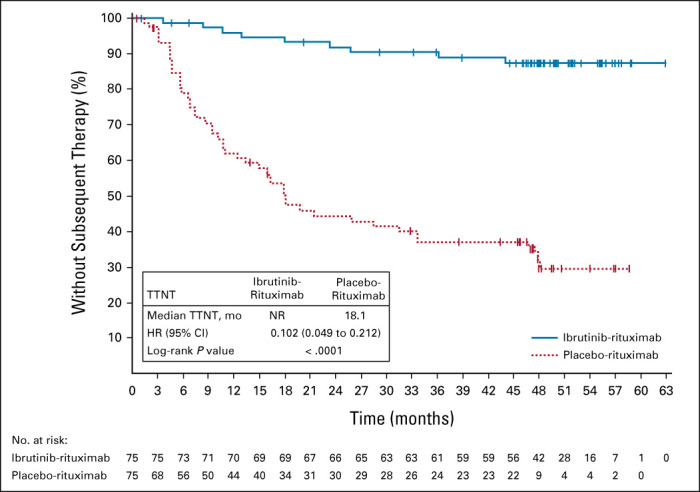
TTNT. HR, hazard ratio; NR, not reported; TTNT, time to next treatment.
Safety
At the final analysis, the median treatment duration for ibrutinib was 48 (range, 1-59) months. Patients had a mean relative ibrutinib dose intensity of 93%, with 59 patients (79%) having a relative dose intensity ≥ 90%. The median treatment duration for placebo-rituximab was 16 (range, 0.4-37) months; safety data for that arm were previously reported.2,13
Ibrutinib-rituximab was tolerable regardless of common concomitant medications, including acid-reducing agents (49%), antiplatelet agents (41%), anticoagulants (24%), and antihypertensives (40%) (Data Supplement [Table S3]). The most common grade ≥ 3 treatment-emergent adverse events of clinical interest that occurred with ibrutinib-rituximab were infections and infestations, anemia, atrial fibrillation, and hypertension; the prevalence of these grade ≥ 3 AEs generally decreased over time (Table 3). In total, 19 patients (25%) experienced any grade hypertension (grade 3 or 4, n = 11 [15%]) and 14 patients (19%) experienced any grade atrial fibrillation (grade 3 or 4, n = 12 [16%]). Grade ≥ 3 hypertension events (shown as prevalence) occurred primarily in the first 3 years of treatment, with only one event documented in year 5 (years 0-1, n = 5; years 1-2, n = 7; years 2-3, n = 6; years 3-4, n = 3; and years 4-5, n = 1). Similarly, the prevalence of atrial fibrillation events stabilized after the first 2 years of treatment (years 0-1, n = 6; years 1-2, n = 5) and remained infrequent (n ≤ 2) in years 3-5. Of 12 patients with grade 3 or 4 atrial fibrillation, nine (75%) remained on treatment. No major hemorrhage events were reported in the last 24 months of treatment. One death as a result of an AE of pneumonia occurred during year 4 and was deemed unrelated to study drug. Four patients discontinued treatment because of AEs (pneumonia, atrial fibrillation, small-cell lung cancer, and metastatic breast cancer) in the ibrutinib-rituximab arm during the last 24 months of treatment, for a total of eight patients overall who discontinued ibrutinib because of an AE (Table 3 and the Data Supplement [Table S2]). Overall, 17 patients had ≥ 1 AE leading to dose reduction (Data Supplement [Table S4]); 11 patients (15%) reduced their dose to 280 mg, and six (8%) reduced their dose to 140 mg. Following dose reduction, 92% of the AEs resolved.
TABLE 3.
Safety Summary

PROs
Patients in both arms reported clinically meaningful improvements in quality-of-life measures. EQ-5D-5L visual analog scale score, FACT-An score, and anemia subscale score improved from baseline. The proportion of patients reporting a clinically meaningful improvement in EQ-5D-5L visual analog scale score (51% in the ibrutinib-rituximab arm v 55% in the placebo-rituximab arm; P = .652) and utility score (47% v 36%; P = .168) was not significantly different between treatment arms. Overall, 56 patients (75%) in the ibrutinib-rituximab arm reported clinically meaningful improvement in total FACT-An score versus 44 (59%) in the placebo-rituximab arm (P = .039). A greater proportion of patients in the ibrutinib-rituximab arm versus the placebo-rituximab arm also experienced an improvement in anemia subscale score (67% v 48%; P = .025).
DISCUSSION
Consistent with the primary analysis of iNNOVATE,2 the results from the final analysis confirmed the durable efficacy of ibrutinib-rituximab in patients with WM, regardless of genotype or prior treatment status. With a median follow-up of 50 (range, 0.5-63) months, ibrutinib-rituximab showed ongoing superiority across clinical outcomes versus placebo-rituximab. Median PFS was not reached with ibrutinib-rituximab versus 20 months with placebo-rituximab; risk of disease progression or death was reduced by 75% with ibrutinib-rituximab. Major response rates were high (76% with ibrutinib-rituximab v 31% with placebo-rituximab), and because of increases in very good partial response rates (from 24% at month 24 to 29% at month 60), continued to deepen over time in the ibrutinib-rituximab arm. Furthermore, time to response was shorter and TTNT was substantially longer with ibrutinib-rituximab versus placebo-rituximab. Rapid and sustained Hgb improvements were observed with ibrutinib-rituximab, including in 95% of patients with anemia at baseline. As noted in the primary analysis, infusion-related reactions (43% v 59%) and IgM flare (8% v 47%) were less frequent with ibrutinib-rituximab versus placebo-rituximab2 and serum IgM levels remained low with extended follow-up.
No new safety signals emerged in this study with a median follow-up of 50 months. Consistent with observations from other studies, AEs were generally most common during the first year of treatment and decreased over time.14-17 Here, the prevalence of grade ≥ 3 hypertension was relatively stable from years 0-3, with no new events after year 3. The majority of patients with grade 3 or 4 atrial fibrillation were able to remain on treatment (9 of 12; 75%); no other ibrutinib discontinuations because of common grade 3 or 4 AEs occurred. As reported previously, 10 of 75 patients in the placebo-rituximab arm discontinued treatment because of an AE (70% from infusion-related reactions).2 In contrast, no patient receiving ibrutinib-rituximab discontinued because of an infusion-related reaction; a low rate of discontinuations because of AEs was maintained after a median of 48 months on treatment. Most AEs leading to an ibrutinib dose reduction resolved following dose reduction, suggesting that AEs can be managed effectively with dose modification, allowing patients to stay on therapy and maintain disease control.
MYD88 and CXCR4 mutational status can negatively affect treatment outcomes.18,19 The higher risk of transformation and death in patients expressing wild-type MYD88 may be due to activating mutations in the NFĸB pathway that overlap with those observed in diffuse large B-cell lymphoma and are further downstream of BTK. Patients with wild-type MYD88 exhibit a more aggressive disease course and have a lower probability of response to single-agent ibrutinib.18,20 Additionally, certain CXCR4 mutations, which occur in up to 40% of patients with WM, may affect response to ibrutinib.20 For example, a previous nonrandomized study in 63 patients treated with single-agent ibrutinib reported fewer major and very good partial responses and shorter PFS in patients with a CXCR4 mutation.10 Results from the open-label substudy of iNNOVATE demonstrate that single-agent ibrutinib is effective in patients with rituximab-refractory WM, with a high proportion of heavily pretreated patients achieving an overall response (87% [27 of 31]), including 86% (6 of 7) of patients with CXCR4 mutations.21 Notably, median PFS was shorter in patients with the MYD88 L265P/CXCR4WHIM genotype versus those with the MYD88 L265P/CXCR4WT genotype (18 months v not reached). In this final analysis from the iNNOVATE study, clinical benefit with ibrutinib-rituximab versus placebo-rituximab was independent of mutational status, with high overall (100%) and major response rates (77%) achieved in patients with CXCR4 mutations.
Although this study does not include a single-agent ibrutinib comparator arm, outcomes with single-agent ibrutinib suggest a potential benefit of adding rituximab to ibrutinib in those patients who are not refractory to rituximab.10 In contrast to outcomes reported with single-agent ibrutinib, response rates and PFS with ibrutinib-rituximab were independent of genotype; this is particularly important for patients with CXCR4 mutations and MYD88 wild-type patients who have a lower probability of response to single-agent ibrutinib10 and may also impact treatment decisions in the absence of genomic testing. Additionally, in the current study, there was a shorter time to major response with ibrutinib-rituximab (3 months) in patients with CXCR4 mutations than that reported in another study with single-agent ibrutinib in previously untreated (7.3 months)4 and previously treated (4.7 months)10 patients with WM and CXCR4 mutations. Although the results from the open-label substudy of iNNOVATE demonstrate that single-agent ibrutinib is effective in patients with rituximab-refractory disease,21 our results suggest a clinical benefit of combining ibrutinib and rituximab across all lines of therapy, particularly for patients with CXCR4 or wild-type genotypes.
Ibrutinib in combination with rituximab provided sustained efficacy and significantly reduced the risk of disease progression or death compared with rituximab alone, regardless of genotype and prior treatment. With a 24-month additional treatment follow-up since the previous analysis, ibrutinib-rituximab maintained a manageable safety profile. In conclusion, with a median follow-up of 50 (range, 0.5-63) months, ibrutinib-rituximab remains an efficacious and well-tolerated chemotherapy-free regimen for patients with WM regardless of prior treatment or MYD88 and CXCR4 mutational status.
ACKNOWLEDGMENT
We thank the patients who participated in the study and their supportive families; the investigators, study coordinators, study team, and nurses who cared for the patients; the iNNOVATE Study Group and the members of the European Consortium for Waldenström's Macroglobulinemia (ECWM) for contributing to this study; and Eva Briso de Montiano, PhD, for her contribution to the analyses.
Christian Buske
Honoraria: Roche/Genentech, Janssen, BeiGene, Novartis, Pfizer, Incyte, AbbVie, Gilead Sciences, Celltrion, MorphoSys, Regeneron
Consulting or Advisory Role: Gilead Sciences, Janssen, Roche, Pfizer, BeiGene, Celltrion, AbbVie, Incyte, Regeneron, MorphoSys, Novartis
Speakers' Bureau: Roche, Janssen, BeiGene, Celltrion, AbbVie, Pfizer, Gilead Sciences
Research Funding: Roche/Genentech, Janssen, Celltrion, MSD, Pfizer, Amgen,
Alessandra Tedeschi
Consulting or Advisory Role: Janssen, BeiGene, AstraZeneca, AbbVie
Speakers' Bureau: AbbVie, AstraZeneca, Janssen, BeiGene
Judith Trotman
Research Funding: BeiGene, Roche/Genentech, Pharmacyclics, Janssen-Cilag, Takeda, Celgene
Travel, Accommodations, Expenses: Roche/Genentech
Ramón García-Sanz
Honoraria: Janssen, Takeda, Amgen, BeiGene, Novartis
Consulting or Advisory Role: Janssen
Research Funding: Gilead Sciences, Incyte
Patents, Royalties, Other Intellectual Property: BIOMED-2 primers
Travel, Accommodations, Expenses: Janssen, Takeda (I)
Other Relationship: Spanish Society of Hematology (SEHH)
David MacDonald
Research Funding: Celgene, Servier
Veronique Leblond
Honoraria: AstraZeneca, Roche Pharma AG, BeiGene, Amgen, Janssen Oncology, AbbVie, MSD Oncology, Lilly
Consulting or Advisory Role: BeiGene, Janssen, AstraZeneca, Lilly, AbbVie
Speakers' Bureau: BeiGene, AstraZeneca, AbbVie
Travel, Accommodations, Expenses: AbbVie
Charles Herbaux
Honoraria: Roche, Janssen-Cilag, AbbVie
Research Funding: Takeda
Travel, Accommodations, Expenses: Janssen-Cilag, AbbVie, Roche
Jeffrey V. Matous
Consulting or Advisory Role: Pharmacyclics, BeiGene
Constantine S. Tam
Honoraria: Janssen-Cilag, AbbVie, Novartis, BeiGene, Pharmacyclics
Consulting or Advisory Role: Janssen, Loxo, Roche, AbbVie
Research Funding: Janssen-Cilag, AbbVie
Leonard T. Heffner
Speakers' Bureau: Kite, a Gilead company
Research Funding: Pharmacyclics, Genentech, Kite, a Gilead Company, ADC Therapeutics, Astex Pharmaceuticals, Loxo, Cellectar
Marzia Varettoni
Consulting or Advisory Role: Janssen-Cilag, Roche, Janssen, AstraZeneca
Travel, Accommodations, Expenses: Gilead Sciences, Janssen-Cilag, AbbVie, Janssen
Lia Palomba
Stock and Other Ownership Interests: Seres Therapeutics (I)
Honoraria: Flagship Biosciences (I), Evelo Therapeutics (I), Jazz Pharmaceuticals (I), Therakos (I), Amgen (I), Merck (I), Seres Therapeutics (I)
Consulting or Advisory Role: Flagship Biosciences (I), Novartis (I), Evelo Therapeutics (I), Jazz Pharmaceuticals (I), Therakos (I), Amgen (I), Merck (I), Seres Therapeutics (I), Kite, a Gilead Company, BeiGene
Research Funding: Seres Therapeutics (I)
Patents, Royalties, Other Intellectual Property: Intellectual Property Rights (I), Juno Intellectual Property Rights
Chaim Shustik
Expert Testimony: Janssen Oncology
Efstathios Kastritis
Honoraria: Amgen, Genesis Pharma, Janssen Oncology, Takeda, Prothena, Pfizer
Consulting or Advisory Role: Amgen, Janssen Oncology, Takeda, Genesis Pharma, Prothena, Pfizer
Research Funding: Janssen Oncology, Amgen
Travel, Accommodations, Expenses: Janssen Oncology, Genesis Pharma, Takeda, Pfizer
Steven P. Treon
Consulting or Advisory Role: Janssen, Pharmacyclics, BeiGene, X4 Pharmaceuticals, Bristol Myers Squibb
Research Funding: Pharmacyclics, Bristol Myers Squibb, X4 Pharmaceuticals, Lilly, BeiGene, AbbVie
Patents, Royalties, Other Intellectual Property: My institution holds patents related to the use of MYD88 and CXCR4 testing for which a predetermined financial distribution to the laboratory and individuals is provided. I have not received any income to this date related to these patents.
Travel, Accommodations, Expenses: Janssen Oncology
Other Relationship: Janssen, Pharmacyclics, BeiGene
Jerry Ping
Employment: AbbVie
Stock and Other Ownership Interests: AbbVie
Travel, Accommodations, Expenses: AbbVie
Bernhard Hauns
Employment: AbbVie/Pharmacyclics
Stock and Other Ownership Interests: AbbVie
Travel, Accommodations, Expenses: AbbVie
Israel Arango-Hisijara
Employment: Janssen Oncology, Abbvie/Pharmacyclics
Stock and Other Ownership Interests: Bristol Myers Squibb/Celgene, Abbvie
Honoraria: Janssen Oncology, Abbvie/Pharmacyclics
Meletios A. Dimopoulos
Honoraria: Amgen, Takeda, Janssen-Cilag, Bristol Myers Squibb, BeiGene
Consulting or Advisory Role: Amgen, Janssen-Cilag, Takeda, Bristol Myers Squibb, BeiGene
No other potential conflicts of interest were reported.
PRIOR PRESENTATION
Presented in part at the 62nd ASH Annual Meeting and Exposition (virtual), December 5-8, 2020.
SUPPORT
Supported by Pharmacyclics LLC, an AbbVie Company. Pharmacyclics LLC sponsored and designed the study. Study investigators and their research teams collected the data. The sponsor confirmed data accuracy and performed analysis of the data. Medical writing support was funded by the sponsor. Editorial support was provided by Cindi Hoover, PhD, and funded by Pharmacyclics LLC, an AbbVie Company.
CLINICAL TRIAL INFORMATION
DATA SHARING STATEMENT
Requests for access to individual participant data from clinical studies conducted by Pharmacyclics LLC, an AbbVie Company, can be submitted through Yale Open Data Access (YODA) Project site at http://yoda.yale.edu.
AUTHOR CONTRIBUTIONS
Conception and design: Christian Buske, Bernhard Hauns, Israel Arango-Hisijara
Provision of study materials or patients: Alessandra Tedeschi, Judith Trotman, Ramón García-Sanz, David MacDonald, Veronique Leblond, Beatrice Mahe, Charles Herbaux, Jeffrey V. Matous, Constantine S. Tam, Leonard T. Heffner, Marzia Varettoni, M. Lia Palomba, Chaim Shustik, Efstathios Kastritis, Steven P. Treon, Meletios A. Dimopoulos
Collection and assembly of data: Christian Buske, Alessandra Tedeschi, Judith Trotman, Ramón García-Sanz, David MacDonald, Veronique Leblond, Beatrice Mahe, Charles Herbaux, Jeffrey V. Matous, Constantine S. Tam, Leonard T. Heffner, Marzia Varettoni, M. Lia Palomba, Chaim Shustik, Efstathios Kastritis, Steven P. Treon, Jerry Ping, Israel Arango-Hisijara, Bernhard Hauns, Meletios A. Dimopoulos
Data analysis and interpretation: All authors
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Ibrutinib Plus Rituximab Versus Placebo Plus Rituximab for Waldenström's Macroglobulinemia: Final Analysis From the Randomized Phase III iNNOVATE Study
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/authors/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Christian Buske
Honoraria: Roche/Genentech, Janssen, BeiGene, Novartis, Pfizer, Incyte, AbbVie, Gilead Sciences, Celltrion, MorphoSys, Regeneron
Consulting or Advisory Role: Gilead Sciences, Janssen, Roche, Pfizer, BeiGene, Celltrion, AbbVie, Incyte, Regeneron, MorphoSys, Novartis
Speakers' Bureau: Roche, Janssen, BeiGene, Celltrion, AbbVie, Pfizer, Gilead Sciences
Research Funding: Roche/Genentech, Janssen, Celltrion, MSD, Pfizer, Amgen,
Alessandra Tedeschi
Consulting or Advisory Role: Janssen, BeiGene, AstraZeneca, AbbVie
Speakers' Bureau: AbbVie, AstraZeneca, Janssen, BeiGene
Judith Trotman
Research Funding: BeiGene, Roche/Genentech, Pharmacyclics, Janssen-Cilag, Takeda, Celgene
Travel, Accommodations, Expenses: Roche/Genentech
Ramón García-Sanz
Honoraria: Janssen, Takeda, Amgen, BeiGene, Novartis
Consulting or Advisory Role: Janssen
Research Funding: Gilead Sciences, Incyte
Patents, Royalties, Other Intellectual Property: BIOMED-2 primers
Travel, Accommodations, Expenses: Janssen, Takeda (I)
Other Relationship: Spanish Society of Hematology (SEHH)
David MacDonald
Research Funding: Celgene, Servier
Veronique Leblond
Honoraria: AstraZeneca, Roche Pharma AG, BeiGene, Amgen, Janssen Oncology, AbbVie, MSD Oncology, Lilly
Consulting or Advisory Role: BeiGene, Janssen, AstraZeneca, Lilly, AbbVie
Speakers' Bureau: BeiGene, AstraZeneca, AbbVie
Travel, Accommodations, Expenses: AbbVie
Charles Herbaux
Honoraria: Roche, Janssen-Cilag, AbbVie
Research Funding: Takeda
Travel, Accommodations, Expenses: Janssen-Cilag, AbbVie, Roche
Jeffrey V. Matous
Consulting or Advisory Role: Pharmacyclics, BeiGene
Constantine S. Tam
Honoraria: Janssen-Cilag, AbbVie, Novartis, BeiGene, Pharmacyclics
Consulting or Advisory Role: Janssen, Loxo, Roche, AbbVie
Research Funding: Janssen-Cilag, AbbVie
Leonard T. Heffner
Speakers' Bureau: Kite, a Gilead company
Research Funding: Pharmacyclics, Genentech, Kite, a Gilead Company, ADC Therapeutics, Astex Pharmaceuticals, Loxo, Cellectar
Marzia Varettoni
Consulting or Advisory Role: Janssen-Cilag, Roche, Janssen, AstraZeneca
Travel, Accommodations, Expenses: Gilead Sciences, Janssen-Cilag, AbbVie, Janssen
Lia Palomba
Stock and Other Ownership Interests: Seres Therapeutics (I)
Honoraria: Flagship Biosciences (I), Evelo Therapeutics (I), Jazz Pharmaceuticals (I), Therakos (I), Amgen (I), Merck (I), Seres Therapeutics (I)
Consulting or Advisory Role: Flagship Biosciences (I), Novartis (I), Evelo Therapeutics (I), Jazz Pharmaceuticals (I), Therakos (I), Amgen (I), Merck (I), Seres Therapeutics (I), Kite, a Gilead Company, BeiGene
Research Funding: Seres Therapeutics (I)
Patents, Royalties, Other Intellectual Property: Intellectual Property Rights (I), Juno Intellectual Property Rights
Chaim Shustik
Expert Testimony: Janssen Oncology
Efstathios Kastritis
Honoraria: Amgen, Genesis Pharma, Janssen Oncology, Takeda, Prothena, Pfizer
Consulting or Advisory Role: Amgen, Janssen Oncology, Takeda, Genesis Pharma, Prothena, Pfizer
Research Funding: Janssen Oncology, Amgen
Travel, Accommodations, Expenses: Janssen Oncology, Genesis Pharma, Takeda, Pfizer
Steven P. Treon
Consulting or Advisory Role: Janssen, Pharmacyclics, BeiGene, X4 Pharmaceuticals, Bristol Myers Squibb
Research Funding: Pharmacyclics, Bristol Myers Squibb, X4 Pharmaceuticals, Lilly, BeiGene, AbbVie
Patents, Royalties, Other Intellectual Property: My institution holds patents related to the use of MYD88 and CXCR4 testing for which a predetermined financial distribution to the laboratory and individuals is provided. I have not received any income to this date related to these patents.
Travel, Accommodations, Expenses: Janssen Oncology
Other Relationship: Janssen, Pharmacyclics, BeiGene
Jerry Ping
Employment: AbbVie
Stock and Other Ownership Interests: AbbVie
Travel, Accommodations, Expenses: AbbVie
Bernhard Hauns
Employment: AbbVie/Pharmacyclics
Stock and Other Ownership Interests: AbbVie
Travel, Accommodations, Expenses: AbbVie
Israel Arango-Hisijara
Employment: Janssen Oncology, Abbvie/Pharmacyclics
Stock and Other Ownership Interests: Bristol Myers Squibb/Celgene, Abbvie
Honoraria: Janssen Oncology, Abbvie/Pharmacyclics
Meletios A. Dimopoulos
Honoraria: Amgen, Takeda, Janssen-Cilag, Bristol Myers Squibb, BeiGene
Consulting or Advisory Role: Amgen, Janssen-Cilag, Takeda, Bristol Myers Squibb, BeiGene
No other potential conflicts of interest were reported.
REFERENCES
- 1.Imbruvica (ibrutinib) [prescribing information]. Sunnyvale, CA: Pharmacyclics LLC, an AbbVie Company, 2020 [Google Scholar]
- 2.Dimopoulos MA, Tedeschi A, Trotman J, et al. : Phase 3 trial of ibrutinib plus rituximab in Waldenström's macroglobulinemia. N Engl J Med 378:2399-2410, 2018 [DOI] [PubMed] [Google Scholar]
- 3.Dimopoulos MA, Trotman J, Tedeschi A, et al. : Ibrutinib for patients with rituximab-refractory Waldenström's macroglobulinaemia (iNNOVATE): An open-label substudy of an international, multicentre, phase 3 trial. Lancet Oncol 18:241-250, 2017 [DOI] [PubMed] [Google Scholar]
- 4.Treon SP, Gustine J, Meid K, et al. : Ibrutinib monotherapy in symptomatic, treatment-naive patients with Waldenström macroglobulinemia. J Clin Oncol 36:2755-2761, 2018 [DOI] [PubMed] [Google Scholar]
- 5.National Comprehensive Cancer Network : NCCN Clinical Practice Guidelines in Oncology. Waldenstrom Macroglobulinia Lymphoplasmatic Lymphoma. Version 1.2021. Plymouth Meeting, PA, National Comprehensive Cancer Network, 2020 [Google Scholar]
- 6.Castillo JJ, Advani RH, Branagan AR, et al. : Consensus treatment recommendations from the tenth International Workshop for Waldenström Macroglobulinaemia. Lancet Haematol 7:e827-e837, 2020 [DOI] [PubMed] [Google Scholar]
- 7.Hunter ZR, Xu L, Yang G, et al. : The genomic landscape of Waldenström macroglobulinemia is characterized by highly recurring MYD88 and WHIM-like CXCR4 mutations, and small somatic deletions associated with B-cell lymphomagenesis. Blood 123:1637-1646, 2014 [DOI] [PubMed] [Google Scholar]
- 8.Treon SP, Cao Y, Xu L, et al. : Somatic mutations in MYD88 and CXCR4 are determinants of clinical presentation and overall survival in Waldenström macroglobulinemia. Blood 123:2791-2796, 2014 [DOI] [PubMed] [Google Scholar]
- 9.Yang G, Zhou Y, Liu X, et al. : A mutation in MYD88 (L265P) supports the survival of lymphoplasmacytic cells by activation of Bruton tyrosine kinase in Waldenström macroglobulinemia. Blood 122:1222-1232, 2013 [DOI] [PubMed] [Google Scholar]
- 10.Treon SP, Meid K, Gustine J, et al. : Long-term follow-up of ibrutinib monotherapy in symptomatic, previously treated patients with Waldenström macroglobulinemia. J Clin Oncol 39:565-575, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Owen RG, Kyle RA, Stone MJ, et al. : Response assessment in Waldenström macroglobulinaemia: Update from the VIth International Workshop. Br J Haematol 160:171-176, 2013 [DOI] [PubMed] [Google Scholar]
- 12.Latimer NR, Abrams KR, Lambert PC, et al. : Adjusting survival time estimates to account for treatment switching in randomized controlled trials – an economic evaluation context: Methods, limitations, and recommendations. Med Decis Making 34:387-402, 2014 [DOI] [PubMed] [Google Scholar]
- 13.Buske C, Tedeschi A, Trotman J, et al. : Ibrutinib treatment in Waldenström's macroglobulinemia: Follow-up efficacy and safety from the iNNOVATE study. Poster presented at the 60th American Society of Hematology Annual Meeting and Exposition, San Diego, CA, December 1-4, 2018 [Google Scholar]
- 14.Burger JA, Barr PM, Robak T, et al. : Long-term efficacy and safety of first-line ibrutinib treatment for patients with CLL/SLL: 5 years of follow-up from the phase 3 RESONATE-2 study. Leukemia 34:787-798, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coutre SE, Byrd JC, Hillmen P, et al. : Long-term safety of single-agent ibrutinib in patients with chronic lymphocytic leukemia in 3 pivotal studies. Blood Adv 3:1799-1807, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O'Brien S, Furman RR, Coutre S, et al. : Single-agent ibrutinib in treatment-naive and relapsed/refractory chronic lymphocytic leukemia: A 5-year experience. Blood 131:1910-1919, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gustine JN, Meid K, Dubeau T, et al. : Ibrutinib discontinuation in Waldenström macroglobulinemia: Etiologies, outcomes, and IgM rebound. Am J Hematol 93:511-517, 2018 [DOI] [PubMed] [Google Scholar]
- 18.Treon SP, Gustine J, Xu L, et al. : MYD88 wild-type Waldenström macroglobulinaemia: Differential diagnosis, risk of histological transformation, and overall survival. Br J Haematol 180:374-380, 2018 [DOI] [PubMed] [Google Scholar]
- 19.Tam CS, Opat S, D'Sa S, et al. : A randomized phase 3 trial of zanubrutinib vs ibrutinib in symptomatic Waldenström macroglobulinemia: The ASPEN study. Blood 136:2038-2050, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Treon SP, Xu L, Guerrera ML, et al. : Genomic landscape of Waldenström macroglobulinemia and its impact on treatment strategies. J Clin Oncol 38:1198-1208, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trotman J, Buske C, Tedeschi A, et al. : Long-term follow-up of ibrutinib treatment for rituximab-refractory Waldenström's macroglobulinemia: Final analysis of the open-label substudy of the phase 3 iNNOVATE™ trial. Blood 136:38-39, 2020 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Requests for access to individual participant data from clinical studies conducted by Pharmacyclics LLC, an AbbVie Company, can be submitted through Yale Open Data Access (YODA) Project site at http://yoda.yale.edu.