

Abstract
The transmittable spread of viral coronavirus (SARS-CoV-2) has resulted in a significant rise in global mortality. Due to lack of effective treatment, our aim is to generate a highly potent active molecule that can bind with the protein structure of SARS-CoV-2. Different machine learning and deep learning approaches have been proposed for molecule generation; however, most of these approaches represent the drug molecule and protein structure in 1D sequence, ignoring the fact that molecules are by nature in 3D structure, and because of this many critical properties are lost. In this work, a framework is proposed that takes account of both tertiary and sequential representations of molecules and proteins using Gated Graph Neural Network (GGNN), Knowledge graph, and Early Fusion approach. The generated molecules from GGNN are screened using Knowledge Graph to reduce the search space by discarding the non-binding molecules before being fed into the Early Fusion model. Further, the binding affinity score of the generated molecule is predicted using the early fusion approach. Experimental result shows that our framework generates valid and unique molecules with high accuracy while preserving the chemical properties. The use of a knowledge graph claims that the entire generated dataset of molecules was reduced by roughly 96% while retaining more than 85% of good binding desirable molecules and the rejection of more than 99% of fruitless molecules. Additionally, the framework was tested with two of the SARS-CoV-2 viral proteins: RNA-dependent-RNA polymerase (RdRp) and 3C-like protease (3CLpro).
Keywords: Molecule generation, Drug-target affinity prediction, Graph neural network, Deep learning
Introduction
Coronaviruses, which contain proactive RNA viruses that cause severe diseases in humans, belong to the coronaviridae group (Khan et al. 2020). Alpha, Beta, Delta, and Gamma coronavirus are among the four species in the group out of which SARS-CoV-2 belongs to the Beta category of viruses. According to current human genome data, SARS-CoV-2 has a positive-sense single-stranded RNA genome that comprises genes that encode 3CLpro, RdRp, spike protein, envelope proteins, and various other protein structures (Thiel et al. 2003). Furthermore, the epidemic was followed by increased fatalities, showing that efficient treatment at the outset is critical to preventing the progression of the virus (Khan et al. 2020).
Generating a novel molecule emerge as fundamental research activity that helps in the creation of new drug discovery by reducing the enormous expenses along with time, but it is a difficult process (Wouters et al. 2020). Recently machine learning (Janairo 2021) and deep generative approaches (Öztürk et al. 2018; Nguyen et al. 2021a) achieve remarkable performance in molecule generation tasks, they are mostly concentrated on training models to produce unique compounds and predicts the affinity score of drug-molecule-target-protein interactions (DTA) represented in simplified molecular input line entry system (SMILES) format. However, molecules are often represented as graphs with a certain number of nodes representing atoms and edges representing bonds, but the SMILES sequence does not capture all chemical properties (Nguyen et al. 2021a). Although sequence-based approaches are good but graph-based approaches are more useful in data formats for understanding compounds and have several benefits over strings, particularly when coupled using graph neural networks (GNNs) (Jiang et al. 2020). GNNs can acquire atom order sequence descriptions and can be trained on a GPU effectively and scalable to larger data (Jiang et al. 2020).
Similarly, for predicting the binding affinity of drug molecules (or generated molecules) with the target protein, there have been many computing prediction approaches presented (Öztürk et al. 2018). In current studies, the protein is simply composed of several amino acid residues denoted by sequences (Öztürk et al. 2018; Nguyen et al. 2021a). The disadvantage of using sequence data is that it does not reflect the protein’s three-dimensional structure. Obtaining a relative three-dimensional structure, on the other hand, is a difficult process. To illustrate tertiary protein structure, another reasonable option is to use two-dimensional residue contact maps (Senior et al. 2020). Now with a deep learning-based approach using GNNs, can effectively identify these contact maps with high accuracy. Furthermore, previously deep learning methods mostly employ a post concatenation method, in which the drugs and protein features are extracted independently and afterward concatenated at the end to predict the binding affinity score. This method although, neglects the notion that binding takes place in a pocket instead of throughout the entire protein. When a drug binds with a protein, it alters its functioning, resulting in the desired pharmacological actions. As a result, when the drug attaches to the protein, the post concatenation method is unable to capture these changes in protein structure. One way to tackle this challenge is to use the Graph Early Fusion Approach (Nguyen et al. 2021b), in which description features for a specific compound is identified first out of its molecule network structure. Before the actual proteins embedding phase, the molecule representation is incorporated into the protein network graph. The model can now consider changes in protein structure triggered by drug molecule’s bond formation thanks to its graph-in-graph algorithm architecture.
The main goal of this work is to build a framework that can generate a highly potent active candidate molecule that can interact with the SARS-CoV-2 protein structure with a high binding score. The proposed framework is illustrated in Fig. 1 that contains three major modules. The first module deals with the molecule generation part using GGNN (Li et al. 2015). The second module deals with the screening part via the construction of a Knowledge Graph between drug–drug similarity, protein–protein similarity, and drug-target similarity. Finally, the third module employs a graph early fusion approach to estimate the binding affinity score of generated molecules with the target protein structure.
Fig. 1.

Workflow of the proposed framework carried out with three phases: (1) Molecule generation, (2) Molecule screening, and (3) Binding affinity prediction
Methodology
Molecule generation
For generating a molecule GGNN was used which is a type of GNNs that have recently gained popularity as effective tools for graph representation learning. In general, the GGNN receives the graph structure of the molecule, i.e. the adjacency matrix representing the connection of nodes within the graph, and the node features matrix for atom properties, as input and produces the processed node feature matrix, and the graph embedded feature matrix. The hidden node values (tensor) along with other propagation nodes within the graph structure are aggregated to produce the final output graph embedding. GGNN comprises three phases—Message Passing, Graph Readout, and Global Readout. Together these perform iterative actions on subgraphs of molecules to generate new molecules.
Message passing
The message-passing is processed through the different nodes within the graph network and mathematically represented as:
| 1 |
| 2 |
where represents the message coming from neighbor nodes and represents the present state of current node , shows the current neighbor of the node , and represents the feature for the edge linking two adjacent nodes i.e. and . Message passing and update operations are represented by and . Following the message passing step comes the graph readout phase.
Graph readout
| 3 |
where g denotes the output graph feature representation and R denotes the readout function captures the input and output of different node states, converts it, and creating a new graph representation.
Global readout
The global readout takes the hidden node and local graph property (g) to predict the global graph embedding. This embedding is used to compute the action probability distribution (APD) of every graph network, which is a value holding the potential actions for expanding a graph and instructs the GGNN how to construct a graph network. The 3 different actions that can be chosen are: firstly Inserting a node into the graph network followed by linking the graph’s latest added node to the existing node, and lastly bringing the graph building to a termination state. For a given graph network, GGNN must train to allocate null values to incorrect actions. The very first 2 multilayer perceptron (MLP) in the global readout phase output initial and , which could then be merged with graph readout output. This merged matrix is sent through the next MLPs, which then outputs the APD after being concatenated and normalized. It’s worth noting that is solely dependent on g.
| 4 |
| 5 |
| 6 |
| 7 |
| 8 |
| 9 |
Pre-processing
The training data set was preprocessed in such a manner that the algorithm can be trained on what to do to generate molecular graphs and how to rebuild graphs. To generate the data sets, the compounds in the training dataset are segmented one by one to determine the decoding pathway r for each compound. The graph network is split into and each time the associated for is determined; provides a mechanism to rebuild . Reversing the breadth first search technique is used to determine the order of node or edge deletion and it guarantees that unconnected segments throughout the graph are not produced after disconnecting the edge.
Dataset and evaluation metrics
The MOSES dataset, which was acquired out from the MOSES Git repository (Polykovskiy et al. 2020), was utilized to evaluate the GGNN model. The MOSES dataset is just a subset of the ZINC database and the description of dataset is mentioned in Table 1.
Table 1.
Dataset description of MOSES dataset
| Parameter | Values |
|---|---|
| Size of training set | 33M |
| (no. of graphs) | |
| Size of test set | 3.8M |
| (no. of graphs) | |
| Size of validation set | 210K |
| (no. of graphs) | |
| Types of atoms | (C, N, O, |
| F, S, Cl, Br) | |
| Maximum no. of | 27 |
| nodes in a graph | |
| Formal charges | [0] |
For evaluating the molecule generation module, following metrics were used.
Fraction of valid and unique compounds, shows the validity and uniqueness of the resulting molecule.
Novelty refers to the percentage of created compounds that were not in the train set.
Filters refer to the percentage of created compounds that travel through the filters used to create the dataset.
Fragment similarity (Frag) (Davis et al. 2011) is a measure that compares distributions in produced and source datasets. It is expressed like a cosine similarity. The limits of this metric are [0, 1].
Scaffold similarity (Scaff) (Blaschke et al. 2018) is a metric comparable to frag, as opposed to fragments it measures the patterns of all compound’s side chains and bridge fragments linking rings. The limits of this metric are [0, 1].
Similarity to the nearest neighbor (SNN) (Arús-Pous et al. 2019) shows the closeness among the fingerprints of a produced compound to its closest neighbor compound. The limits of this metric are [0, 1].
Internal diversity (IntDivp) (Sanchez-Lengeling et al. 2017) is a measure of chemical heterogeneity inside a group of molecules that have been generated. This measure captures the mode collapse of the model. The limits of this metric are [0, 1].
The Fréchet ChemNet Distance (FCD) (Prykhodko et al. 2019) is used to determine if produced compounds are diverse and also have comparable chemical and biological features to natural compounds.
The distribution of features is a vital component for viewing the molecular structures that have been produced. Four criteria were used to evaluate the distributions of produced and reference molecule:
Molecular weight is the total of a molecule’s atomic weight.
is the octanol/water partition coefficient.
Synthetic Accessibility Score (SA) (Degen et al. 2008) a qualitative assessment of how difficult (10) or simple (1) a particular compound is to synthesis.
Quantitative Estimation of Drug-likeness (QED) (Bemis and Murcko 1996) is the likelihood that a compound would be a suitable drug option. The limits of this metric are [0, 1].
Molecule screening
To optimize the results a custom-made Knowledge Graph was used to apply a screening process on the thousands of novel as well as FDA-approved drug molecules. Our knowledge graph is a heterogeneous graph generated from two homogeneous similarity matrices i.e. drug–drug and target-target similarity matrices and the DTI(Drug Target Interaction) data from the DAVIS dataset (Davis et al. 2011). A pictorial representation of our framed knowledge graph is shown in Fig. 2.
Fig. 2.
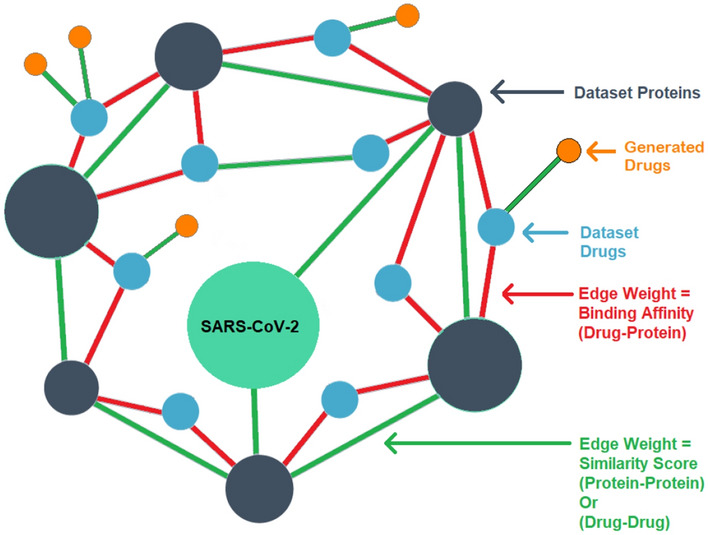
Knowledge graph build using two homogeneous similarity matrices and DTI data where the first-hop neighbor of the target protein includes the protein with the same structure from the dataset, the second-hop neighbor contains molecules from the dataset and the third hop contains the generated molecules
Drug–drug similarity matrix
Molecular similarity involves two major components:
Molecular Descriptors: Represent the structures of the molecules being compared.
Similarity coefficient: Metric used to compute a quantitative score for the degree of similarity based on the weighted values of structural descriptors.
The MACCS (Molecular ACCess System) keys (Durant et al. 2002) were used as molecular descriptors (2-D chemical fingerprints) as they are one of the most commonly used structural keys. The similarity coefficient between two drug molecules is calculated using Tanimoto Coefficient on their respective MACCS keys. Similarity scores for all pairs of drugs were calculated in the DAVIS dataset and generated the drug-drug similarity matrix.
Target–target similarity matrix
The standardized Smith–Waterman algorithm (Yamanishi et al. 2008) was used to calculate target–target similarity analysis of structural protein patterns. The Smith Waterman approach determines comparable regions between two strings of amino acid or structure of protein sequences by performing local sequencing.
Now, to construct the knowledge graph, it is required to define a threshold value for binding affinity as well as the similarity scores exceeding which would result in the creation of an edge between drug-target and target-target respectively. For binding affinity, we chose the value to be 7.0 ( value of 100 nM), since this value is a widely accepted threshold for the DAVIS dataset. To identify the threshold value of the target-target similarity score, the global clustering coefficient metric was used that was calculated from the knowledge graph.
Binding affinity prediction
The objective of the DTA concern is to predict the affinity score A of the generated drug-molecule D and target-protein P. This concern can mathematically be defined as a regression task:
| 10 |
where denotes the parameter values of the prediction function F.
Most existing methods for predicting binding affinity are using sequences of protein structure encoded in a feature vector, mostly with one-hot encoding. In our work, RaptorX tool (Wang et al. 2017) was used to extract 2D contact map data as the tertiary structure description in order to incorporate important structural features. Furthermore, instead of using one-hot encoding for the protein sequences, TAPE’s embedding model (Rao et al. 2019) was used, which was learned from a set of unlabeled amino acid sequence data. We also employ secondary structure data extracted using the RaptorX tool in the form of the likelihood of three different protein structure types i.e. alpha helix, beta-pleated sheet, and coil. The architecture of binding affinity prediction model is shown in Fig. 3.
Fig. 3.

Illustration of steps involved in Graph Early Fusion Model used for prediction of binding affinity includes: (1) Early fusion of drug and target to form drug-target graph, (2) Refinement of drug-target graph, and (3) Binding affinity prediction step
Graph early fusion approach
The main justification for utilizing Graph Early Fusion Approach(GEFA) (Nguyen et al. 2021b) is because it takes into account the changes made in target protein structure that occur as a result of drug interaction. The graph structure of drug and target are taken as an input and the binding affinity score ( where is Dissociation constant of the reaction) is given as output. The GEFA integrates the drug molecule graph into the protein graph using a self-attention mechanism to represent structural reforms within the binding site that occur throughout the interaction phase. To accomplish this, a two layer Graph Convolutional Network(GCN) with residual blocks was used to improve the vertex descriptions within the compound structure. Then, most of the drug representation were compressed into a single node in order to add it to the protein graph. This single node is now added to the protein structure, with the edges linking the compound node and residue nodes indicating their interaction. Each residue leads to the bond formation uniquely, and this is indicated in the drug-residue edges. A self-attention technique was utilized to learn this degree of compliance.
Binding affinity prediction
To improve the node descriptions of the combined drug-protein graph obtained from the early fusion process, a two-layer GCN with residual blocks was used once again. Now, before extracting the graph feature, the drug node was removed from the combined graph, therefore only the protein representation can be extracted. Next a max-pooling process afterward following a two-layer network was used to obtain the final structure of the protein graph network. Concatenating the drug features before and after the fusion procedure provides the final representation of the drug molecule. To estimate binding affinity scores, the final compound feature vector, as well as the protein drug feature vector were fused and sent to a three-layered fully connected deep network for predicting the binding affinity score.
Datasets and evaluation metrics
The DAVIS dataset (Davis et al. 2011), which is one of the most extensively used benchmark datasets for binding affinity prediction models was used to train the graph early fusion model. Reasons behind selecting this dataset for evaluating predictive models include: Data heterogeneity is not a problem when using the DAVIS dataset as it can be heterogeneous if we utilize data from other sources, which might lead to data inconsistency. In addition, the data in this dataset is of high quality that comprises interactions between 72 kinase inhibitors and 442 kinases, which cover more than 80% of the human active protein kinome. The dataset descriptions is shown in Table 2.
Table 2.
Dataset description of DAVIS dataset
| Parameter | Values |
|---|---|
| Total number of | 68 |
| Compounds | |
| The maximum length | 103 |
| of a compound SMILES | |
| Total number of Proteins | 442 |
| The maximum length | 2549 |
| Total number of Interactions of a protein sequence | 30056 |
| Formal Charges | [0] |
To analyze our model’s performance, four distinct metrics were employed, that are backed up by past research and are common for most models that work on binding affinity prediction. These metrics are:
Concordance Index (CI) to determine if the sequence of predicted binding affinity scores of pairs of drugs-target matches the higher sequence of actual values; the higher the CI score, the better the model.
Mean Squared Error (MSE) is the average value of the difference among the estimated and true output values.
R squared : represents the model’s global predictive accuracy.
Experiments and results
All the simulation of our framework comprising three modules are issued on a configuration of azure Standard NV12 with a vCPU size of 12 and RAM of 112 GiB.
Results
The GGNN model for molecule generation was evaluated on the MOSES dataset. Later on, the results were compared against different state-of-the-art methods including Variational Autoencoder (VAE) (Blaschke et al. 2018), Adversarial Autoencoder (AAE) (Blaschke et al. 2018), Character-level Recurrent Neural Networks (CharRNN) (Arús-Pous et al. 2019), Objective-Reinforced Generative Adversarial Network (ORGAN) (Sanchez-Lengeling et al. 2017), and LatentGAN (Prykhodko et al. 2019). The training was performed on mini-batches at a learning rate of 0.0001 and optimizer as adam with the default PyTorch parameters. For every epoch, 30,000 samples were simulated and the results were stored. The comparison of results obtained from different model is shown in Table 3.
Table 3.
Comparison of results obtained from different models for molecule generation using MOSES dataset
| Metric | VAE | AAE | Char-RNN | ORGAN | LatentGAN | GGNN |
|---|---|---|---|---|---|---|
| Valid | 0.1535 | 0.6227 | 0.8161 | 0.756 | 0.6604 | 1 |
| unique@1K | 0.9999 | 0.9999 | 0.99990 | 0.9969 | 0.9969 | 1 |
| unique@10K | 0.9991 | 0.9995 | 0.9996 | 0.9942 | 0.9921 | 1 |
| FCD | 3.2761 | 1.3476 | 0.3249 | 73.595 | 3.8604 | 27.995 |
| SNN | 0.4788 | 0.5332 | 0.5442 | 0.3163 | 0.4412 | 0.2446 |
| Frag | 0.9883 | 0.9917 | 0.9992 | 0.7637 | 0.9832 | 0.5415 |
| Scaf | 0.7211 | 0.8555 | 0.8871 | 0 | 0.4500 | 0.0214 |
| IntDiv | 0.8551 | 0.8553 | 0.8538 | 0.4715 | 0.8525 | 0.8883 |
| IntDiv2 | 0.8483 | 0.8485 | 0.8479 | 0.4530 | 0.8461 | 0.8809 |
| Filters | 0.8503 | 0.9617 | 0.9766 | 0.9413 | 0.9374 | 0.3047 |
| logP | 0.2475 | 0.2606 | 0.0824 | 29.916 | 0.1985 | 0.9640 |
| SA | 0.2665 | 0.0674 | 0.0401 | 1.2772 | 0.4033 | 2.3227 |
| QED | 0.0237 | 0.0036 | 0.0043 | 0.7557 | 0.0379 | 0.1913 |
| Weight | 18.523 | 14.071 | 4.889 | 10.906 | 13.009 | 18.697 |
| Novelty | 0.9995 | 0.9986 | 0.9975 | 1 | 1 | 1 |
On the Davis dataset, the early fusion approach for predicting binding score prediction was compared with the traditional late fusion strategy like DeepDTA, GCNConvNet and DGraphDTA as shown in Table 4. The training was carried out on 128 mini-batches with a learning rate of 0.0005 and a learning decay of after 40 epochs, with no change in MSE in the validation data. The train, test and validation curve for GEFA is shown in Fig. 4. The model is trained till convergence using Adam as optimizer.
Table 4.
Comparison of results obtained from different models for binding affinity prediction using DAVIS dataset
Fig. 4.

a Shows the RMSE curve for train, test and validation curves of GEFA model, b shows the MSE curve for train, test and validation of GEFA model, and c shows the CI curve for train, test and validation of GEFA model
Usecase considering SARS-CoV-2 protein
Now, after completion of training our framework, two proteins of the coronavirus (SARS-CoV-2) namely, RdRp and 3CLpro were simulated with novel drug molecules generated using our optimized molecule generation model. The steps involved in this process are shown below.
For the simulation, firstly the SARS-CoV-2 proteins was added to the existing knowledge graph after calculating the similarity score with all other proteins. Then the new drug nodes were added to the graph iteratively by calculating drug-drug similarity with all other drugs in the graph and forming respective edges
Now, in the candidate selection phase, proteins that are direct neighbors of the coronavirus proteins were selected and ranked them as per their edge weight (normalized Smith-Waterman Score)
Then a list of drugs were generated that have a good binding affinity to these proteins. The drugs that have high similarity to the former were taken as candidate drugs for the SARS-CoV-2.
These candidate drugs were then fed into the Graph Early Fusion Model along with the SARS-CoV-2 proteins to predict their binding affinity scores. Those with binding affinity values higher than 7.0 were considered as potential drugs for coronavirus is shown in Table 5 and the corresponding molecular graph of generated compound with considerable binding affinity are shown in Fig. 5.
Table 5.
Binding affinity scores of two SARS-CoV-2 viral protein with generated compound
| ID | Compound-ISO-SMILES | Target-Name | Affinity Score |
|---|---|---|---|
| 8678 | CNC1CC2CCOC(C)(C2)C1O | YP_009725307.1 | 7.9265 |
| 1657 | CCC12CONC(CC1O)C2NC | YP_009725307.1 | 7.9091 |
| 8197 | CNC1CC(O)C2CCCC12O | YP_009725307.1 | 7.7064 |
| 3065 | CNC1C2C(C)=C3C(N)C(=O)N3C12 | YP_009725307.1 | 7.7062 |
| 306 | CNC1C2NC1(C)C(C)(O)C2=NO | YP_009725307.1 | 7.7059 |
| 11151 | CNC1C2NC3C(=O)C1C2(C)C30 | 6WQF-1 | 7.7042 |
| 3778 | CNC12CC3C1CNC3C1(C)OC21 | 6WQF-1 | 7.7002 |
| 4621 | CC1NNC(C2(C)CC2)CC=C1S | YP_009725307.1 | 7.6984 |
| 1458 | CNC1CNC2(C=CC2CO)C1C | YP_009725307.1 | 7.6983 |
| 308 | CCC1C(NC)C(C)(N)C1(O)CN | YP_009725307.1 | 7.6941 |
| 1924 | CNC1C2CC13CN(CC2O)C3C | YP_009725307.1 | 7.6900 |
| 2435 | CNN1C(C)C2=C(OC2)C2=CCC21 | YP_009725307.1 | 7.6883 |
| 4636 | CCOC1CSC(CN)=C1C1NN1 | YP_009725307.1 | 7.6882 |
| 4085 | CNC1C2NCC(=O)C1C2(C)CO | 6WQF-1 | 7.6877 |
| 8280 | CCC(CC)NC | YP_009725307.1 | 7.6848 |
| 4186 | CNC1CN2C(=O)C3CCC(N3)C12 | YP_009725307.1 | 7.6847 |
| 4715 | CNC(C)C(N)C1C[NH]C(=O)[NH]1 | YP_009725307.1 | 7.6803 |
| 2647 | C=C1SC2CC2OC(C)C1NNC | YP_009725307.1 | 7.6791 |
| 6890 | CNC(C)C1(C)COC1CC(C)=O | 6WQF-1 | 7.6762 |
| 7445 | CNC1C2NOC(C)C13CCCC23 | YP_009725307.1 | 7.6734 |
| 2330 | C=C1CC(NC)C(C)(O)C2C(O)C12 | YP_009725307.1 | 7.6700 |
| 9753 | CNC12CNC(CCO)C1(C)C2N | YP_009725307.1 | 7.6680 |
| 6826 | CNC(C)C(N)C1C[NH]C(=O)[NH]1 | 6WQF-1 | 7.6610 |
| 10485 | CCC(C)NC | YP_009725307.1 | 7.6607 |
| 1561 | CNC1CCN2NCCC1(C)C2C | 6WQF-1 | 7.6536 |
| 4801 | CNC1C2C(C)=C3C(N)C(=O)N3C1 | 6WQF-1 | 7.6518 |
| 7075 | C=C1SC2CC2OC(C)C1NNC | 6WQF-1 | 7.6513 |
Fig. 5.

A sample molecular graph representation of generated molecules with their IUPAC names that posses higher binding affinity score with the target SARS-CoV-2 viral proteins: RNA-dependent-RNA polymerase (RdRp) and 3C-like protease (3CLpro)
Out of 11164 novel generated molecules, only molecules were predicted to have binding affinity with the SARS-CoV-2 proteins. And after the screening process, 405 candidate molecules were selected of which, had binding affinity . Among 369 binding molecules, 316 were retained after screening and more than non-binding molecules were discarded in the screening process itself as demonstrated in Figure 6.
Fig. 6.
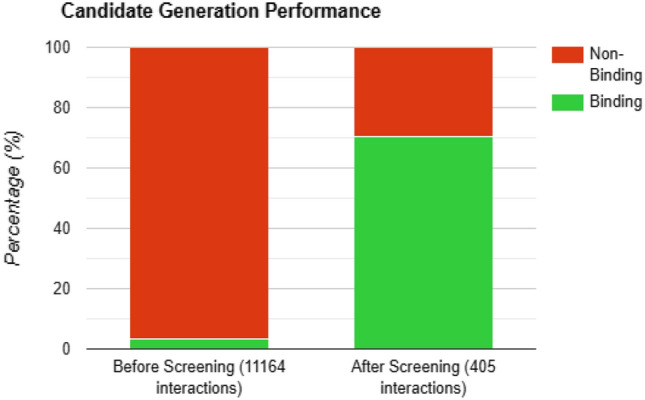
Performance of Candidate Selection process: before screening: 369 (3.31%) good binding molecules out of 11,164 novel molecules. After screening: 316 (70.37%) good binding molecules out of 405 candidate molecules
Discussion
Our study states that it’s not in the best interest to represent molecules in linear 1D representations while generative modeling, since it may lose out on many of their valuable properties which come from their tertiary structures. Now, it could therefore being said that GNNs are best suitable for most of the tasks surrounding chemical structures since they will preserve tertiary properties and will also allow us to use traditional graph algorithms surrounding graph data structures of computer science to perform various kinds of analysis. Experimental results support this claim as GGNN reproduced 100% valid and 100% unique novel molecules while preserving all chemical properties.
Also a cutting-edge deep learning architecture was used that uses the Graph Early Fusion Approach and knowledge graphs to predict drug-target binding affinity, which is crucial for virtual drug repurposing and development. The Early Fusion Method is used to address the change in protein structure that occurs during the bond formation also with the compound molecule. Unlike previous deep learning methods that employed a late fusion approach, the early fusion technique incorporates drug molecule representation into the protein sequence training process, allowing the network to learn the potential binding sites that occur in the protein structure after the bond formation. As a result, the model is better to recognize since it reveals which residues and to what extent they contribute to the bond formation.
This claim is also supported by the fact that use of drug-drug and protein-protein similarity calculation to create and exploit drug target knowledge graphs for the screening purpose and optimization in efficiency for the discovery of highly potent drugs given a target protein. Without using screening process, all the generated novel molecules had to be tested against the target protein, even though among the 11,164 novel molecules generated, only 369 (3.31%) were predicted to be binding. While, after the screening process, only 405 molecules (out of 11164) were selected as candidate molecules out of which 316 (70.37%) of the binding molecules were retained. The screening process thus discarded 99% of the non-binding molecules before being fed into the Early Fusion model.
Conclusion
In this paper, a framework was introduced blending the three different techniques i.e. molecule generation using GGNN and novel candidate selection using knowledge graph and for further binding affinity calculations using early fusion approach for a molecular generation. This framework was tested with a simulated run on two of the SARS-CoV-2 viral proteins namely, RdRp and 3CLpro. The uniqueness of our framework consists in exploring the capability of GNN to process the structured data of molecular graphs. One major advantage of using GNNs includes dealing directly with the molecule’s graph representation, which the string representation lacks. GNNs can capture both the global and local context of the molecular graph whereas in few cases convolution operations performed on sequential data failed to capture the global context of the molecule. Finally, it could therefore being said that proposed framework will be the cornerstone for AI-based Drug Discovery.
Acknowledgements
We gratefully acknowledge support to MICROSOFT CORPORATION AND ITS AFFILIATES for providing us Microsoft Azure service under grant ID: 00011000011. The assistance provided by Mr. Hrithik Kumar and Mr. Archit Anand regarding the use of Azure services is highly appreciated.
Funding
This work is supported by Microsoft AI for Health COVID-19 project under grant ID: 00011000011.
Declarations
Conflict of interest
Amit Ranjan, Shivansh Shukla, Deepanjan Datta, and Rajiv Misra announce that they all have no conflict of interest.
Informed consent
Each individual involved in the research gave their informed consent.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Amit Ranjan, Email: amit_1921CS18@iitp.ac.in.
Shivansh Shukla, Email: shivansh.cs17@iitp.ac.in.
Deepanjan Datta, Email: deepanjan.cs17@iitp.ac.in.
Rajiv Misra, Email: rajivm@iitp.ac.in.
References
- Arús-Pous J, Johansson SV, Prykhodko O, Bjerrum EJ, Tyrchan C, Reymond J-L, Chen H, Engkvist O. Randomized smiles strings improve the quality of molecular generative models. J Cheminform. 2019;11(1):1–13. doi: 10.1186/s13321-019-0393-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bemis GW, Murcko MA. The properties of known drugs. 1. molecular frameworks. J Med Chem. 1996;39(15):2887–2893. doi: 10.1021/jm9602928. [DOI] [PubMed] [Google Scholar]
- Blaschke T, Olivecrona M, Engkvist O, Bajorath J, Chen H. Application of generative autoencoder in de novo molecular design. Mol Inf. 2018;37:1700123. doi: 10.1002/minf.201700123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, Hocker M, Treiber DK, Zarrinkar PP. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol. 2011;29:1046–1051. doi: 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- Degen J, Wegscheid-Gerlach C, Zaliani A, Rarey M. On the art of compiling and using ‘drug-like’ chemical fragment spaces. ChemMedChem. 2008;3(10):1503. doi: 10.1002/cmdc.200800178. [DOI] [PubMed] [Google Scholar]
- Durant JL, Leland BA, Henry DR, Nourse JG. Reoptimization of mdl keys for use in drug discovery. J Chem Inf Comput Sci. 2002;42:1273–1280. doi: 10.1021/ci010132r. [DOI] [PubMed] [Google Scholar]
- Janairo GIB, Yu DEC, Janairo JIB. A machine learning regression model for the screening and design of potential sars-cov-2 protease inhibitors. Netw Model Anal Health Inf Bioinform. 2021;10:1–8. doi: 10.1007/s13721-021-00326-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, Li Z, Zhang S, Wang S, Wang X, Yuan Q, Wei Z. Drug-target affinity prediction using graph neural network and contact maps. RSC Adv. 2020;10:20701–20712. doi: 10.1039/D0RA02297G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M, Adil SF, Alkhathlan HZ, Tahir MN, Saif S, Khan M, Khan ST. Covid-19: a global challenge with old history, epidemiology and progress so far. Molecules. 2020;26:39. doi: 10.3390/molecules26010039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Tarlow D, Brockschmidt M, Zemel R (2015) Gated graph sequence neural networks. arXiv preprint arXiv:1511.05493
- Nguyen T, Le H, Quinn TP, Nguyen T, Le TD, Venkatesh S. Graphdta: predicting drug-target binding affinity with graph neural networks. Bioinformatics. 2021;37:1140–1147. doi: 10.1093/bioinformatics/btaa921. [DOI] [PubMed] [Google Scholar]
- Nguyen TM, Nguyen T, Le TM, Tran T (2021b) GEFA: Early fusion approach in drug-target affinity prediction. In: IEEE/ACM transactions on computational biology and bioinformatics 1. 10.1109/tcbb.2021.3094217 [DOI] [PubMed]
- Öztürk H, Özgür A, Ozkirimli E (2018) Deepdta: deep drug–target binding affinity prediction. Bioinformatics 34 [DOI] [PMC free article] [PubMed]
- Polykovskiy D, Zhebrak A, Sanchez-Lengeling B, Golovanov S, Tatanov O, Belyaev S, Kurbanov R, Artamonov A, Aladinskiy V, Veselov M, Kadurin A, Johansson S, Chen H, Nikolenko S, Aspuru-Guzik A, Zhavoronkov A. Molecular sets (moses): a benchmarking platform for molecular generation models. Front Pharmacol. 2020;11:1931. doi: 10.3389/fphar.2020.565644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prykhodko O, Johansson SV, Kotsias P-C, Arús-Pous J, Bjerrum EJ, Engkvist O, Chen H. A de novo molecular generation method using latent vector based generative adversarial network. J Cheminform. 2019;11:1–13. doi: 10.1186/s13321-019-0397-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao R, Bhattacharya N, Thomas N, Duan Y, Chen X, Canny J, Abbeel P, Song YS. Evaluating protein transfer learning with tape. Adv Neural Inf Process Syst. 2019;32:9689. [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Lengeling B, Outeiral C, Guimaraes GL, Aspuru-Guzik A (2017) Optimizing distributions over molecular space. An Objective-Reinforced Generative Adversarial Network for Inverse-design Chemistry (ORGANIC). 10.26434/chemrxiv.5309668.v2
- Senior AW, Evans R, Jumper J, Kirkpatrick J, Sifre L, Green T, Qin C, Žídek A, Nelson AWR, Bridgland A, Penedones H, Petersen S, Simonyan K, Crossan S, Kohli P, Jones DT, Silver D, Kavukcuoglu K, Hassabis D. Improved protein structure prediction using potentials from deep learning. Nature. 2020;577:706–710. doi: 10.1038/s41586-019-1923-7. [DOI] [PubMed] [Google Scholar]
- Thiel V, Ivanov KA, Putics Á, Hertzig T, Schelle B, Bayer S, Weißbrich B, Snijder EJ, Rabenau H, Doerr HW, Gorbalenya AE, Ziebuhr J. Mechanisms and enzymes involved in sars coronavirus genome expression. J Gen Virol. 2003;84:2305–2315. doi: 10.1099/vir.0.19424-0. [DOI] [PubMed] [Google Scholar]
- Wang S, Sun S, Li Z, Zhang R, Xu J. Accurate de novo prediction of protein contact map by ultra-deep learning model. PLoS Comput Biol. 2017;13:e1005324. doi: 10.1371/journal.pcbi.1005324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wouters OJ, McKee M, Luyten J. Estimated research and development investment needed to bring a new medicine to market, 2009–2018. JAMA. 2020;323:844–853. doi: 10.1001/jama.2020.1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanishi Y, Araki M, Gutteridge A, Honda W, Kanehisa M. Prediction of drug-target interaction networks from the integration of chemical and genomic spaces. Bioinformatics. 2008;24(13):i232–i240. doi: 10.1093/bioinformatics/btn162. [DOI] [PMC free article] [PubMed] [Google Scholar]