

Abstract
We describe our experiences with organizing pro bono medical genetics and neurology outreach programs on several different resource-limited islands in the West Indies. Due to geographic isolation, small population sizes, and socioeconomic disparities, most Caribbean islands lack medical services for managing, diagnosing, and counseling individuals with genetic disorders. From 2015 to 2019, we organized 2–3 clinics per year on various islands in the Caribbean. We also organized a week-long clinic to provide evaluations for children suspected of having autism spectrum disorder. Consultations for over 100 different individuals with suspected genetic disorders were performed in clinics or during home visits following referral by locally registered physicians. When possible, follow-up visits were attempted. When available and appropriate, clinical samples were shipped to collaborating laboratories for molecular analysis. Laboratory tests included karyotyping, cytogenomic microarray analysis, exome sequencing, triplet repeat expansion testing, blood amino acid level determination, biochemical assaying, and metabolomic profiling. We believe that significant contributions to healthcare by genetics professionals can be made even if availability is limited. Visiting geneticists may help by providing continuing medical education seminars. Clinical teaching rounds help to inform local physicians regarding the management of genetic disorders with the aim of generating awareness of genetic conditions. Even when only periodically available, a visiting geneticist may benefit affected individuals, their families, their local physicians, and the community at large.
Keywords: Caribbean, clinical genetic testing, West Indies
1 ∣. INTRODUCTION
Disparities in access to healthcare are not uncommon. In countries with developed economies where specialty medical healthcare is generally available, socioeconomic status, location, and level of education affect the availability of genetic medicine and diagnostic services (Allford, Qureshi, Barwell, Lewis, & Kai, 2014; Hui, Barclay, Poulton, Hutchinson, & Halliday, 2018; Mikat-Stevens, Larson, & Tarini, 2015; Shea, Newschaffer, Xie, Myers, & Mandell, 2014). Small, resource-limited, and isolated islands have barriers to access due to their small population sizes, relatively low gross national product, and geographical isolation. Depending on how the region is defined, there are over 700 islands with 26 island nations in the West Indies or Caribbean region (Gibson, Fitzpatrick, Stone, Noel, & Macpherson, 2016). Most of these countries are geographically small, with small population sizes and a predominant dependence on the tourism industry (de Albuquerque & McElroy, 1992). They are described by the International Monetary Fund World Economic Outlook as emerging markets with a developing economy (https://www.imf.org, accessed Oct 01, 2020).
Most of the main islands, if their population is large enough, have a local hospital. Otherwise, healthcare is offered at community health centers or via privately practicing physicians. Many services that are taken for granted in resource-rich countries have either limited or periodic availability or are not available at all. Examples include computed tomography (CT) scanning, magnetic resonance imaging (MRI), retinal examination via optical coherence tomography (OCT), and electroencephalography (EEG). Because of low demand and/or cost, some standard medical testing is difficult to obtain. For example, tests for vitamin B12, lactate, serum ammonia, serum amino acid/organic acid, or alpha-fetoprotein levels might not be available. Imaging, such as MRI, when available, is usually at low resolution. Additionally, because of the lack of redundancy (if an island has only one MRI or CT service), technical issues may cause the service to be unavailable. Even when set up as an international collaboration, there are logistical challenges due to geographical distance and shipping resources, as some samples must be shipped frozen, or are difficult to batch because of rapid decay rates.
In general, genetic testing and specialist medical care for individuals with chronic neurodegenerative conditions or congenital anomalies are either difficult or almost impossible to obtain. In the general hospital, patients with neurologic disorders are typically referred to a generalist (internal medicine), and individuals with congenital disorders are managed by pediatricians. Alternatively, for those who have the resources, the patient and family must travel to a more developed country to seek additional care. Hospital medical care costs are often covered by nationalized government insurance plans or other arrangements. However, nonurgent testing, most prescription drugs, and imaging that is not part of a hospital admission are often covered out-of-pocket by the patient and family or by private insurance.
Genetic diagnosis is important at many levels, and outcomes are improved when a diagnosis is obtained (Malinowski et al., 2020). Ending the diagnostic odyssey, informing appropriate actions, avoiding costly (and sometimes dangerous) procedures and tests, and providing recurrence risk assessments are all benefits of a molecular diagnosis (Sawyer et al., 2016; Symonds & McTague, 2020). Here, we describe our efforts to provide genetic consultation and testing for individuals with rare disorders who live in the Caribbean region where these services are otherwise not available. We provide a summary of the laboratory testing we obtained from 2015 to 2019 and present five brief case reports to illustrate our activities.
2 ∣. METHODS
Clinical evaluations were performed at pro bono clinics under the auspices of locally registered consulting physicians. Due to the small population size of most Caribbean islands, our institutional review board (IRB) stipulated that we do not reveal the nationality or geographic locations of any affected individuals. All testing was initiated at the request of locally registered physicians. All individuals (if of consenting age), or their parents or legal guardians provided written informed consent for genetic testing and anonymous publication of results. The consenting process was approved by a local IRB to help ensure that the needs and values of the local population were respected. All individuals who provided samples for exome sequencing also underwent the consenting process of the collaborating laboratory. Biological samples, including whole blood, plasma, urine, purified DNA, saliva, and leukocytes were express-shipped to collaborating laboratories. Saliva was collected with DNA Genotek kits (Ontario, Canada). DNA was isolated from whole blood using Qiagen Puregene (Venlo, Netherlands).
Research exome sequencing and variant analyses were performed as previously described (Li et al., 2016). Identified variants with a minor allele frequency of >1% as established by public databases (dbSNP, 1,000 Genomes Project, NHLBI ESP6500SI, gnomAD, and Kaviar) and an in-house database of >10,000 exomes were filtered out. Variant prioritization based on deleterious prediction and biological relevance was done with reference to the HGMD and OMIM databases. Copy number variant (CNV) analyses were done using ExomeDepth (Plagnol et al., 2012).
The number of repeats in ATN1, ATXN1, ATXN2, ATXN3, CACNA1A, ATXN7, ATXN10, PPP2R2B, TBP, HTT, JPH3, AR, C9orf72, and FXN were determined using multiplex polymerase chain reaction (PCR), followed by capillary electrophoresis with internal standards. CEPH control samples were verified using Sanger sequencing for cross laboratory standardization (Ashizawa et al., 2013; Gan et al., 2017; Moscovich et al., 2015).
To test for fragile X syndrome, FMR1 CGG repeat quantification was obtained with a CGG repeat-primed PCR approach (Chen et al., 2010; Filipovic-Sadic et al., 2010). Global Metabolomics Assisted Pathway Screen (Global MAPS), an untargeted clinical metabolic analysis that included relative quantification of small molecules in plasma or urine was based on established protocols (Ford et al., 2020; Kennedy et al., 2018). Cytogenomic microarray analyses (CMA), karyotyping, Sanger sequencing, and dried blood spot amino acid (DBS AA) profiling were obtained using standard protocols.
Evaluations for autism spectrum disorder (ASD) were conducted using the Autism Diagnostic Observation Schedule, second edition (Lord et al., 2012). All ASD assessments included parental interview to obtain developmental history and assessment of adaptive functioning as well as record review. Cognitive and/or behavioral functioning was determined either by standardized cognitive assessment or by requesting that parents complete appropriate questionnaires. Clinicians came to a consensus about ASD diagnosis in all patients using an evidenced-based team evaluation model (Gerdts et al., 2018).
3 ∣. RESULTS
3.1 ∣. Laboratory testing
To date, we have obtained exome sequencing for 31 affected individuals. Of these, we found a pathogenic, or likely pathogenic variant in 13 individuals, a variant of unknown significance (VUS) in 2, and uninformative results in 12 when classified by ACMG criteria (Richards et al., 2015). Four additional exomes await analysis. We believe the VUS found in exome 6 explains the pathology of the individual due to supportive biochemical evidence (manuscript submitted). Among the 13 exomes that contain a variant that explains the pathology of the affected individual, only two required trio sequencing, while a pathogenic or likely pathogenic variant was identified in the remaining 11 via proband-only exome sequencing (Table 1). In each case diagnosed via proband-only sequencing, we verified the genotypes of available parents by Sanger sequencing. Of the 12 exome sequences that were uninformative, six were of trios, one was a duo, and five were singletons.
TABLE 1.
Individuals affected with genetic disorders who were investigated with exome sequencing that yielded informative results
| Diagnosis confirmed by exome sequencing | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| # | Testing | Presenting features | Variant | Effect | Inherit. pattern |
OMIM | Note | ACMG classification | References |
| 1 | CMA, Trio exome | Irritable baby, doughy skin | COL5A1; c.944delC; p. (Thr315Argfs*242) | Stop gain | Autosomal dominant | 120,215 | Novel variant, apparent de novo | P (VS1 + PS2 + PM2) | 1 |
| 2 | Proband exome | Hypertonia, dystonia, encephalopathy | SYNJ1; c.242-2A > G | Splice site | Autosomal recessive | 604,297 | Novel homozygous variant | P (PVS1 + PS2 + PM2) | – |
| 3 | Proband exome | Autism, ID, dysmorphology | PHF8; c.1996delG; p. (Glu666Argfs*163) | Splice site; stop gain | X-linked | 300,560 | Novel variant; carrier mother | LP (PVS1 + PM2) | – |
| 4 | Proband exome, FRX | Autism, no dysmorphology | USP9X; c.1112G > T; p. (Arg371Leu) | Missense | X-linked | 300,072 | Novel variant; carrier mother, FRX neg | VUS (PM2 + PP3)a | – |
| 5 | Proband exome | Cornelia de Lange Syndrome | NIPBL; c.1885C > T; p. (Arg629*) | Stop gain | Autosomal dominant | 608,667 | Known variant | P (PVS1 + PS1 + PM2) | 2,3 |
| 6 | Proband exome, FRX | Autism with single regression episode | ALG13; c.2458-15_2486del | 15 aa deletion | X-linked | 300,776 | Novel variant, carrier mother; CDG, FRX neg | VUSb | – |
| 7 | Proband exome | Hypotonia, DD | NALCN; c.2203C > T; p. (Arg735*) | Stop gain | Autosomal recessive | 611,549 | Known variant | P (PVS1 + PM2 + PP1) | 4,5 |
| 8 | Proband exome | DD, ID, dysmorphology | KMT2A; c.6158 + 6 T > C | Splice site | Autosomal dominant | 159,555 | Known variant (ClinVar), affected mother | LP (PS1 + PM2 + PP1) | – |
| 9 | Proband exome, CMA | Craniosynostosis, normal intelligence | ~7.4 Mb duplication 6p22.3-6p22.1 | CNV | – | – | Detected by exome CNV analysis; CMA verified | – | – |
| 10 | Proband exome, FRX | Autism with single regression episode | ALG13; c.3013C > T; p. (Pro1005Ser) | Missense | X-linked | 300,776 | Novel variant; carrier mother and grandmother, CDG, FRX neg | LP (PS3 + PM2 + PP3) | – |
| 11 | Proband exome | Polycystic kidney disease | PKD1; c.11257C > T; p. (Arg3753Trp) | Missense | Autosomal dominant | 601,313 | Known variant, childhood onset | LP (PS1 + PM2) | 6,7 |
| 12 | Proband exome | Dysmorphology, DD, ID | SATB2; c.1165C > T; p. (Arg389Cys) | Missense | Autosomal dominant | 608,148 | Known variant | LP (PS1 + PM2 + PP3) | 8,9 |
| 13 | Proband exome, Global MAPS | Autism, DD, ID | PAH; c.1315 + 1G > A; c.740G > A; p.(Gly247Val) | Splice site, missense | Autosomal recessive | 612,349 | 2 known variants as compound heterozygote | P (PVS1 + PS1); LP (PS1 + PM3 + PP3) | 10,11 |
| 14 | Proband exome | Autism, ID, microcephaly | MECP2; c.455C > G; p. (Pro152Arg) | Missense | X-linked dominant | 300,005 | Known variant, apparent de novo | P (PS1 + PS2 + PM2) | 12 |
| 15 | Trio exome | Primary microcephaly | TUBG1; c.1022G > A; p. (Arg341Gln) | Missense | Autosomal dominant | 191,135 | Novel variant, apparent de novo | P (PS2 + PS3 + PS4 + PM2 + PP5) | 13 |
References: 1 (Wardeh et al., 2018); 2 (Miyake et al., 2005); 3 (Thompson et al., 2020); 4 (Helbig et al., 2016), 5 (Ope et al., 2020), 6 (Chang et al., 2013), 7 (Kim et al., 2000), 8 (Jiao et al., 2019), 9 (Zarate et al., 2018), 10 (Zurflüh et al., 2008), 11 (Razipour et al., 2017), 12 (Hoffbuhr et al., 2001), 13 (Yuen, Guella, Roland, Sargent, & Boelman, 2019).
In the absence of functional studies, we classified USP9X; c.1112G > T; p.(Arg371Leu) a VUS.
We consider the ALG13; c.2458-15_2486del (15 aa deletion) a pathogenic cause of the congenital disorder of glycosylation (CDG) in this individual because of biochemical evidence (manuscript submitted).
In total, we completed the exome analysis of 27 individuals and identified the genetic cause of their condition in 14 for a diagnosis rate of approximately 50%, similar to reported rates in well-resourced countries (Helbig et al., 2016; Ngo et al., 2020; Sawyer et al., 2016; Wright, FitzPatrick, & Firth, 2018). Currently, we have 11 additional exomes that await sequencing (Table 2). Among the cohort of 14 individuals whose diagnosis was informative via exome sequencing, 13 were children/adolescents and 1 was an adult. Of the 13 unsolved exomes, 6 were from children/adolescents, and 7 were from adults. Of the 15 that await sequence obtainment or analysis, 12 are from children/adolescents and 3 are from adults.
TABLE 2.
Compilation of exome sequencing studies that were either uninformative, under analysis, or awaiting sequence obtainment
| # | Testing | Presenting features | Note |
|---|---|---|---|
| Uninformative exome | |||
| 16 | Trio exome, FRX | Autism in two full brothers | Awaiting exome of affected brother, FRX neg |
| 17 | Trio exome, Global MAPS | Microcephaly, ID, DD, seizure | Awaiting exome of three unaffected siblings |
| 18 | Trio exome, FRX | Autism, no dysmorphology | FRX neg |
| 19 | Trio exome, Global MAPS, | DD; two consanguineous sibs | Awaiting exome of affected sibling |
| 20 | Trio exome, HD/ataxia panel | Young onset ataxia | – |
| 21 | Trio exome karyotype, CMA, | Dysmorphology, ID, DD, | Normal karyotype, has family history |
| 22 | Proband exome | Hypertonia, ID, 45-year-old male | Singleton, cousin to the individual above |
| 23 | Duo exome, CMA | Joint contractures, arthrogryposis | VUS; chr19:56296992-56572894 (~0.3 Mb del) |
| 24 | Proband exome, HD/ataxia panel | Young onset Parkinson disease | Singleton, other family members unavailable |
| 25 | Proband exome, HD/ataxia panel | Ataxia, ocular photosensitivity | Family history of HD |
| 26 | Proband exome | Neurofibromas, café au lait spots | Singleton, other family members unavailable |
| 27 | Proband exome | Cerebral palsy | Singleton, other family members unavailable |
| Exome under analysis | |||
| 28 | Proband exome | Dysmorphology, DD, ID, club foot | – |
| 29 | Gene panel, duo exome | Adult with primary microcephaly | Research microcephaly panel uninformative |
| 30 | Proband exome, FRX | Autism, no dysmorphology | FRX negative |
| 31 | Proband exome, FRX | Autism, no dysmorphology | FRX negative |
| Awaiting exome sequencing | |||
| 32 | Proband exome | Suspected Mowat–Wilson syndrome | – |
| 33 | Proband exome | Suspected Noonan syndrome | – |
| 34 | Proband exome | Hydrocephalus, holoprosencephaly | – |
| 35 | Proband exome | Microcephaly, hypertelorism, ID | – |
| 36 | Proband exome | Dysmorphology, hydronephrosis | – |
| 37 | Proband exome | Dysmorphology, hypotonia, ID | – |
| 38 | Proband exome | Blindness, myopathy | Family history suggests autosomal dominant |
| 39 | Proband exome, FRX | Autism, no dysmorphology | FRX negative |
| 40 | Proband exome, FRX | Dysmorphology, ID, DD | FRX negative |
| 41 | Proband exome | ASD, no dysmorphology | Awaiting FRX test |
| 42 | Proband exome, FRX | Autism, behavioral problems | FRX negative |
We also obtained 24 karyotypes, 18 CMA tests, 15 FRX tests, and 12 other triplet repeat expansion tests (on a 14-gene panel). We have carried out three metabolic profiles (Global MAPS), with several additional Global MAPS analyses underway. Triplet repeat testing revealed two extended families with Huntington disease (HD), a large family with spinocerebellar ataxia 3 (SCA3) and one additional individual with SCA3. Karyotypes were used to confirm clinical diagnoses of Down syndrome (DS) and to rule out a familial translocation. Of 19 individuals suspected to have DS, 18 were confirmed. Trio exome for the one individual who did not have DS was uninformative. We did not identify any individuals who had DS caused by Robertsonian translocation, but we found one apparently unaffected female who had trisomy 21 mosaicism. All individuals who presented with suspected ASD in our clinics were tested for FRX, with none being positive.
Using karyotyping we identified an apparently de novo unbalanced translocation that resulted in deletion and duplication in one individual. The approximate breakpoints of the translocation were confirmed by CMA (Table 3). In two other unrelated individuals, a CNV was identified on the proband's exome sequence and confirmed with CMA. One of these CNVs was considered pathogenic as it involves a 7.4 megabase (Mb) duplication on chromosome 6. Karyotyping was not performed on this individual. The other CNV was a deletion which we classified as a VUS because it is approximately 0.3 Mb in size, and we could not predict if haploinsufficiency of any genes in the region would be pathogenic.
TABLE 3.
Compilation of individuals investigated with triplet repeat expansion testing, CMA, and karyotype
| Testing | Presenting |
Variant description | Effect | Diagnosis or note | References |
|---|---|---|---|---|---|
| Neurodegenerative disorders | |||||
| HD/Ataxia panel | Chorea | HTT; pathogenic CAG repeat expansion | Polyglutamine tract expansion | Confirmed HD in four individuals | 1 |
| HD/Ataxia panel | Ataxia | ATXN3; pathogenic CAG repeat expansion | Polyglutamine tract expansion | Confirmed SCA3 in four individuals | 2,3 |
| Fragile X testing (with no other testing) | |||||
| Fragile X test | Autism | Normal FMR1 (31 CAG repeats) | – | No further testing | – |
| Testing for suspected Down syndrome | |||||
| CMA | Down syndrome | – | Confirmed DS in 11 individuals | DS; CMA was the only test we could obtain at this time | 4 |
| Karyotype | Down syndrome | 47,XX + 21 and 47,XY + 21 | Trisomy 21 | Confirmed DS in 12 individuals | – |
| Karyotype | Unaffected mother | 46,XX/47,XX + 21 mosaic | Trisomy 21 mosaic | Explained recurrent DS in family | 5 |
| Karyotype | Unaffected mothers | 46,XX | Unaffected mothers | Confirmed normal karyotype in three unaffected mothers | – |
| Karyotype for disorder other than Down syndrome | |||||
| Karyotype followed by CMA | Dysmorphology, DD, ID | 46,XY,der(18)t(5;18) (p13.3;q22.3) | Translocation with deletion/duplication | – | – |
References: 1 (Charles et al., 2017), 2 (Yearwood et al., 2018), 3 (Mitchell et al., 2019), 4, (Kruszka et al., 2017), 5 (Sobering et al., 2017).
3.2 ∣. Case reports
3.2.1 ∣. Individual 1
Individual 1 was a 4-year-old female with developmental delay (DD). Pregnancy was complicated by gestational diabetes diagnosed at 6 months of pregnancy. She started walking at about 1.5 years and did not use expressive language. She was able to understand commands, request objects by pointing or reaching, but she did not verbalize. The mother reported that she was active while in daycare, and she slept well at night, usually 12 hr without waking. Her head circumference at age 4 was 49 cm (35th centile), inner canthal distance was 3 cm, and outer canthal distance 9 cm (IPD >95th centile). A formal evaluation by a medical geneticist is pending but based on analysis of photographs she had apparently deeply set and widely spaced eyes, a flat mid-face, and maloccluded large-appearing incisors (Figure 1a,b). Exome sequencing revealed a previously described c.1165C > T; p. (Arg389Cys) variant in SATB2 (Jiao et al., 2019; Zarate et al., 2018). This variant was not detected in her mother. Her father was not available for testing, but we were told that neither he nor other family members had similar features.
FIGURE 1.

Facial dysmorphology, clinical signs, and brain MRI of affected individuals. (a, b) A 4-year-old girl who has a known likely pathogenic SATB2; c.1165C > T; p.(Arg389Cys) variant. (c, d) A 5-year-old girl who has a known pathogenic MECP2; c.455C > G; p.(Pro152Arg) variant displaying the repetitive hand motions characteristic of Rett syndrome. (e) Transverse brain MRI showing white matter intensities (arrows) in a 4-year-old boy with a hemizygous novel USP9X; c.1112G > T; p.(Arg371Leu) VUS. (f–g) Sagittal brain MRI showing cerebellar atrophy in a 39-year-old woman with ataxia of unknown etiology
3.2.2 ∣. Individual 2
Individual 2 was a 6-year-old female who initially presented with DD and hyperactivity. She first began walking when she was 20 months old. Developmental regression was first noted when she was 2 years, 7 months. At this time, she also began to have seizures, up to nine per day. The seizures lasted approximately 1 min and were characterized by initially swaying from left to right and then falling to the ground with generalized hypertonicity with extended legs and plantar-flexed toes. During the seizures, she did not have urinary nor fecal incontinence. Her seizures have been managed with carbamazepine with relatively good control. EEGs performed initially and at age 4 years showed generalized and focal (parasagittal cerebral cortex) epileptogenic discharges with mild diffuse cerebral dysfunction. At age 5 years she displayed the repetitive hand movements characteristic of Rett syndrome (Banerjee, Miller, Li, Sur, & Kaufmann, 2019). (Figure 1c,d). At the age of 6 years, her head circumference was 48 cm (first centile). Currently, she does not speak and is not toilet trained. Exome sequencing showed a previously reported variant in MECP2; c.455C > G; p.(Pro152Arg) (Hoffbuhr et al., 2001). Sanger sequencing showed that neither parent had the variant suggesting an apparent de novo event.
3.2.3 ∣. Individual 3
Individual 3 was a 4-year-old male born to parents who were both approximately 40 years old at the time of his birth. He had an uneventful first year of life, achieving all milestones on time. By 13 months he could speak over 20 words and by 15 months he could say a few phrases. However, at approximately 18 months he showed regression of speech and at 25 months of age he had only a few garbled words. The loss of speech was associated with regression of social skills; for example, he began to prefer being alone with an electronic tablet device. His parents enrolled him in daycare but because of his lack of social skills he was removed. His hearing and vision appeared normal. He responded to electronic screens (iPad), video, and sound. In his fourth year of life he began eating a more restricted variety of foods, and to gain weight.
During an examination by a pediatrician at the age of 3 years, he appeared uncooperative and irritable. He did not use words, had poor social skills, and was hitting, biting, crying inappropriately, and was combative at times. At 3 years and 5 months, his head circumference was 51 cm (80th centile), inner canthal distance was 3 cm, and outer canthal distance was 10.5 cm (IPD 90th centile). Review of photographs by a medical geneticist suggested that he did not have dysmorphic features. A brain MRI demonstrated bilateral deep posterior parietal periventricular white matter hyperintensities in Figure 1e. He was formally diagnosed with autism at the age of 4 years. At 5 years of age, he can walk but he is completely nonverbal. Exome sequencing showed a novel hemizygous c.1112G > T; p. (Arg371Leu) variant in USP9X with the mother being a carrier. By the ACMG classification criteria, this variant is described as a VUS (Richards et al., 2015). No other variants that might explain the condition in this child were found. No other affected family members are known to be affected.
3.2.4 ∣. Individual 4
Individual 4 was a 5-year-old female who first came to our attention because of neonatal hypotonia. Her development has been delayed, she was nonverbal, and was not toilet trained. At the age of 4, she was diagnosed with ASD. Subsequently, proband-only exome sequencing showed that she had compound heterozygosity for known variants in PAH; c.1315 + 1G > A; c.740G > A; p. (Gly247Val) (Razipour et al., 2017; Zurflüh et al., 2008). Sanger sequencing showed that both parents are heterozygous. Phenylketonuria (PKU) was confirmed by DBS AA profiling with a blood phenylalanine level of 11.16 mg/dl (normal <2 mg/dl) and a blood tyrosine level of 0.58 mg/dl (normal <4.98 mg/dl). The phenylalanine/tyrosine molar ratio was 21.28 (normal <2.50). Two months before her fifth birthday her head circumference was 48.25 cm (20th centile). Currently, her diet is not controlled for reduced phenylalanine intake. After discussions about the diagnosis of PKU, her parents reported that she avoids red meat, chicken, and fish.
3.2.5 ∣. Individual 5
Individual 5 was a 43-year-old female with a 14-year history of progressively worsening ataxia. She had disconjugate eye movements with abnormal adduction of both eyes and extreme photosensitivity. Her gait was wide-based, and she was unable to walk unsupported. Brain MRI obtained approximately 10 years after symptom onset showed cerebellar atrophy but a normal cerebral cortex and basal ganglia (Figure 1f,g). She had a family history of HD with a maternal great-aunt and a maternal first cousin once removed both confirmed to be affected. Her mother, now deceased, had sickle cell disease, and her father was unavailable for evaluation. She had two maternal aunts who reportedly have some type of abnormal movements but attempts to locate them have been unsuccessful (Figure 2).
FIGURE 2.
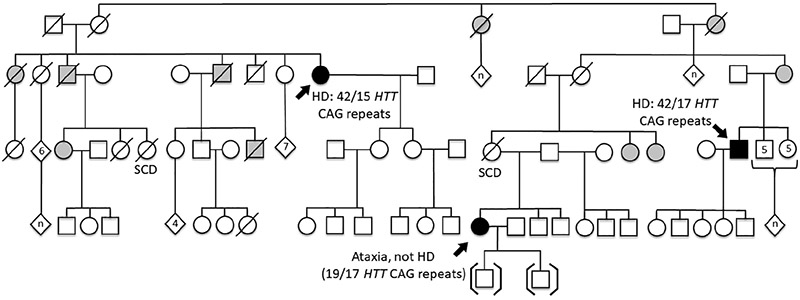
Pedigree of Individual 5. This family has two individuals affected with HD and one individual affected with ataxia and cerebellar degeneration of unknown etiology. Filled in symbols indicate individuals who were evaluated by a neurologist. Partially shaded symbols indicate individuals suggested to have a movement disorder by a family member but who have not been formally evaluated
Repeat expansion testing for the following chorea and ataxia neurodegenerative disorders was negative: dentatorubral-pallidoluysian atrophy, Friedreich ataxia, HD, HD-like 2, spinal and bulbar muscular atrophy, and SCA types 1, 2, 3, 6, 7, 10, 12, and 17. Exome sequencing for this woman was uninformative and no other immediate family members are available for genetic testing.
4 ∣. DISCUSSION
In this report we present five brief case reports representative of our clinical encounters and summarize the testing that we have offered to date. Individuals 1 and 2 had dysmorphic features, ID, and DD. By exome sequencing we were able to determine that the cause of their disorder was due to known variants. The features of Individual 1 were explained by a previously described likely pathogenic variant in SATB2 (Bengani et al., 2017; Zarate et al., 2018). Individual 2 had Rett syndrome (Banerjee et al., 2019) due to a previously described apparent de novo pathogenic variant in MECP2 (Hoffbuhr et al., 2001). Obtaining the diagnosis for these individuals has given the parents an explanation for the disorder in their children. Following the diagnoses, we were able to suggest to the parents that they contact appropriate family support and patient advocacy groups. Individual 3 had DD and ID. Exome analysis revealed a VUS in USP9X with the mother being a carrier.
The X-linked USP9X gene encodes a substrate-specific deubiquitylating enzyme (Homan et al., 2014); complete loss of function of this gene is lethal in the mouse (Stegeman et al., 2013). Pathogenic de novo variants in USP9X cause congenital malformation in females, DD, ID, and brain abnormalities on MRI (Reijnders et al., 2016). Males with likely pathogenic USP9X variants have similar features including brain malformations and a more variable facial dysmorphology. Additionally, several males with USP9X VUSs were described who have a similar, but more variable phenotype, with observable features found at a reduced frequency (Johnson et al., 2020). Because of the absence of functional data for the USP9X c.1112G > T; p.(Arg371Leu) variant, and its classification as a VUS, we have not discussed this variant with the family.
We formally diagnosed Individual 4 with ASD during a pro bono clinic. Subsequent exome analysis showed her to have compound heterozygosity for known pathogenic variants in PAH (Razipour et al., 2017; Zurflüh et al., 2008). Follow-up biochemical testing with DBS AA analysis showed elevated serum phenylalanine, with depressed tyrosine confirming PKU. Unfortunately, the absence of a newborn screening program for inborn errors of metabolism allowed the PKU in this individual to remain undetected until exome sequencing indicated that she had the disorder.
Having the molecular diagnosis with blood amino acid measurement confirmation has allowed us to begin an effort to obtain PKU formula and train a medical nutritionist to oversee a low-protein, phenylalanine restricted diet. Difficulties in implementing this treatment include obtaining legal affidavits from the family absolving anybody who donates PKU formula from liability due to the possibility of secondary nutritional deficiencies or malnourishment (van Wegberg et al., 2017). Coordinating appropriate training of the local medical nutritionist to help oversee the dietary restriction therapy is also a challenge. However, our biggest obstacle has been organizing donations of the PKU formula which is needed by this child to slow her neurological decline.
We also learned that this individual has a pathogenic variant that was associated with benefit from administration of sapropterin dihydrochloride (Zurflüh et al., 2008), and we are currently looking into the possibility of obtaining the drug. Another possible positive impact, albeit in the future, is that the diagnosis will allow us to provide education to the parents and the local physician about the potential for maternal PKU syndrome (Hoeks, den Heijer, & Janssen, 2009).
Individual 5 presented with ataxia, cerebellar degeneration, unusual eye movements, and photosensitivity. She had two relatives confirmed to have HD, however, this diagnosis was excluded, as was SCA3 which we previously had diagnosed in a different family in this small population (Yearwood, Rethi, Figueroa, Walker, & Sobering, 2018). Other spinocerebellar ataxias (types, 2, 3, 6, 7, 10, 12, and 17) were also excluded. Proband exome sequencing was also uninformative, and she remains undiagnosed. Difficulties in obtaining care were experienced with Individual 5. After coordination with a visiting eye clinic, we were unable to obtain OCT because of outdated equipment, and so she was left with dilated eyes and no retinal scan, which we needed to help us understand her unusual symptoms.
In addition to the five individuals described above, we were able to have an impact on several other patients. Among the individuals whose exomes are currently being analyzed is an adult male with primary microcephaly. His mother expressed her appreciation to us because we helped her to consider genetic reasons for her son's condition. As her child was growing up, she had to endure years of others telling her the reason that she had a child with microcephaly was that she “looked at a turtle” when she was pregnant. Sequence-based testing with a research panel for approximately 70 genes known to be involved with microcephaly was negative, but with exome sequencing we hope to give this mother a definitive explanation for her son's condition.
Another example is that of a patient in whom the physician initially suspected a metabolic disorder but had concerns about the inability to obtain testing for blood ammonia levels. With the aid of a medical geneticist, we were able to quickly learn that the disorder in the child was neurological and not a urea cycle defect. Follow-up testing helped to direct management for this individual and her affected sibling (Ope et al., 2020). In yet another individual, we helped a parent understand that her child's ataxia was due to SCA3 (Mitchell, LaTouche, Nelson, Figueroa, & Walker, 2019) and have been able to coordinate continuing community support to this family.
Neurological consultations may also help to redirect the focus of disorder causation away from a neurological, or genetics etiology, and refine the differential diagnosis. As an example, we saw a young boy who had ataxia which at first appeared to be of neurological cause. By quickly obtaining a brain MRI, and an evaluation by a neurologist, we were able to determine that a neurological cause for the ataxia was less likely. It transpired that the child had pseudo-ataxia caused by an osteoid osteoma which was cured with surgery (McKenzie, Oettel-Flaherty, Noel, Walker, & Sobering, 2019).
We have also received evidence of intangible benefits to our patients and their physicians. We have had unanimous positive feedback from the physicians with whom we interacted. Although non-systematically obtained, comments included appreciation for our efforts which have helped them to understand some of their most challenging patients. Without our efforts, they would be left without answers. From casual discussion with parents we also find a general sense of appreciation and gratitude. One mother told us how she now has additional peace of mind knowing that her child has a specific disorder and not some unknown disease entity. In the words of one parent after we diagnosed an ID syndrome in her child, “Thank you, now I know what I am dealing with.” She expressed relief to have the diagnosis and that her child's condition was understood, and she no longer felt guilt that she did something to cause the disorder. This is an example of how we helped to end the diagnostic odyssey for this family (Sawyer et al., 2016). In the future we hope to formally study the impact of our efforts in this community.
Visiting geneticists, neurologists, and psychologists who have expertise in ASD and various other specialties can offer additional benefits to medical practitioners by helping with continuing medical education (CME). Over the past 7 years, we have facilitated annual CME seminars to help physicians understand, diagnose, and manage their patients who have genetic and neurodegenerative disorders. Additionally, we have offered public seminars to help parents understand ASD. One of these events was attended by over 40 interested parents showing that there is a need for more educational outreach to the community. We believe that these educational seminars are a small step toward the acceptance of individuals with genetic disorders into the general community.
We view each patient encounter as an opportunity to educate the physicians with whom we are working in addition to ourselves. We learn more about how peoples in countries outside of our own manage genetic disorders, and we may be able to advise local physicians on current best practices for disease management. We ensure that a locally registered physician is present at any patient encounter where clinical information is discussed. This is important to maintain trust and ensure that the encounter is appropriately recorded in the patient's medical record. Having a local physician is also important to ensure that licensure and liability are not an issue. The role of the visiting medical geneticist or neurologist then becomes defined as an advisor. Local clinicians also help with communication because the accent and idioms that are sometimes used may lead to difficulties during discussions.
Since inception of this project, we have organized one formal video consultation clinic, and many impromptu brief video calls to answer emergent questions. These experiences have raised our awareness of telemedicine (Duis et al., 2019; Otten, Birnie, Lucassen, Ranchor, & Van Langen, 2016; Srinivasan et al., 2020). We hope to increase the use of video consultations specifically to support our genetics outreach program (Hilgart, Hayward, Coles, & Iredale, 2012). Telemedicine is becoming even more important in the face of the interruptions to our program during the global response to the COVID-19 pandemic (Mann, Chen, Chunara, Testa, & Nov, 2020).
Because of high shipping costs, samples are typically collected and batched together to save money. Having a local laboratory to purify DNA is, therefore, indispensable for this project, since not all individuals are able to give a saliva sample. Additionally, in cases where DNA must be sent to more than one lab, a blood draw is important. Delays imposed by periodic shipping, in combination with the typically longer turn-around times associated with research-based testing, leads to excessive wait times. However, even when results are delayed, all who are involved appreciate our efforts. The lack of an organized system for the entire project means that the coordinating individuals often must put in much of their effort during their spare time, and that they are unremunerated. Difficulties associated with this type of project are compounded by the need for air travel. Unexpected interruptions occurred in early 2020 and unfortunately, planned adult neurology, pediatric genetics, and ASD evaluation clinics were canceled due to the COVID-19 pandemic.
Individuals from diverse populations tend to be underrepresented in the medical literature, leading to difficulties in identifying human malformation syndromes (Muenke, Adeyemo, & Kruszka, 2016). Increased representation of individuals from diverse ancestry is therefore important. The genetic makeup of the Caribbean population is particularly interesting in that it is composed of a broad assortment of ancestral genetic contributions, from West African, European, Asian, and Amerindian populations (Murray et al., 2010).
5 ∣. CONCLUSION
This project was enabled by several different successful collaborations between physicians and researchers in a country of low socioeconomic status and several USA-based research laboratories. Without our efforts, the individuals we encountered who are affected with genetic disorders would have remained without a molecular diagnosis. Even when only available for a few days per year, visiting medical geneticists and neurologists may offer benefits to under-served communities. Some people might consider genetic testing a low priority with so many other pressing issues in global medicine, especially in the infectious disease realm, but when appropriately implemented, this resource can have positive long-reaching effects for individuals, families, and the health care system.
ACKNOWLEDGMENTS
We thank the patients, their families, and the physicians who worked with us for participating in this study. We also thank Dr. Taraka Donti and Perkin-Elmer, Inc. for newborn screening tests and Theresa Wilson for metabolic sample management.
Funding information
Brotman Baty Institute; Jordan's Guardian Angels; National Institute of Neurological Disorders and Stroke, Grant/Award Number: K08NS092898; St. George's University Small Research Grant Initiative
Footnotes
CONFLICT OF INTEREST
F.T. has received funding from the Azrieli Foundation, Zynerba Inc., and Asuragen Inc. for studies of Fragile X syndrome. R.H.W. has received honoraria from the International Parkinson Disease and Movement Disorders Society, the International Association of Parkinsonism and Related Disorders, and consulting fees from Neurocrine Biosciences, Inc., and Teladoc Health, Inc. All other authors have no conflict of interest to declare.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
REFERENCES
- Allford A, Qureshi N, Barwell J, Lewis C, & Kai J (2014). What hinders minority ethnic access to cancer genetics services and what may help. European Journal of Human Genetics, 22(7), 866–874. 10.1038/ejhg.2013.257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashizawa T, Figueroa KP, Perlman SL, Gomez CM, Wilmot GR, Schmahmann JD, … Subramony S (2013). Clinical Characteristics of Spinocerebellar Ataxias 1, 2, 3 and 6 in the US; a prospective observational study. Orphanet Journal of Rare Diseases, 8(177), 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee A, Miller MT, Li K, Sur M, & Kaufmann WE (2019). Towards a better diagnosis and treatment of Rett syndrome: A model synaptic disorder. Brain, 142(2), 239–248. 10.1093/brain/awy323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengani H, Handley M, Alvi M, Ibitoye R, Lees M, Lynch SA, … FitzPatrick DR (2017). Clinical and molecular consequences of disease-associated de novo mutations in SATB2. Genetics in Medicine, 19(8), 900–908. 10.1038/gim.2016.211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang M-Y, Chen H-M, Jenq C-C, Lee S-Y, Chen Y-M, Tian Y-C, … Wu-Chou Y-H (2013). Novel PKD1 and PKD2 mutations in Taiwanese patients with autosomal dominant polycystic kidney disease. Journal of Human Genetics, 58(11), 720–727. 10.1038/jhg.2013.91. [DOI] [PubMed] [Google Scholar]
- Charles J, Lessey L, Rooney J, Prokop I, Yearwood K, Da Breo H, … Sobering AK (2017). Presentation and care of a family with Huntington disease in a resource-limited community. Journal of Clinical Movement Disorders, 4(4), 1–8. 10.1186/s40734-017-0050-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Hadd A, Sah S, Filipovic-Sadic S, Krosting J, Sekinger E, … Latham GJ (2010). An information-rich CGG repeat primed PCR that detects the full range of fragile X expanded alleles and minimizes the need for southern blot analysis. Journal of Molecular Diagnostics, 12(5), 589–600. 10.2353/jmoldx.2010.090227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Albuquerque K, & McElroy JL (1992). Caribbean small-island tourism styles and sustainable strategies. Environmental Management, 16(5), 619–632. 10.1007/BF02589017 [DOI] [Google Scholar]
- Duis J, van Wattum PJ, Scheimann A, Salehi P, Brokamp E, Fairbrother L, … Miller JL (2019). A multidisciplinary approach to the clinical management of Prader–Willi syndrome. Molecular Genetics and Genomic Medicine, 7(3), 1–21. 10.1002/mgg3.514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipovic-Sadic S, Sah S, Chen L, Krosting J, Sekinger E, Zhang W, … Tassone F (2010). A novel FMR1 PCR method for the routine detection of low abundance expanded alleles and full mutations in fragile X syndrome. Clinical Chemistry, 56(3), 399–408. 10.1373/clinchem.2009.136101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford L, Kennedy AD, Goodman KD, Pappan KL, Evans AM, Miller LAD, … Toal DR (2020). Precision of a clinical metabolomics profiling platform for use in the identification of inborn errors of metabolism. The Journal of Applied Laboratory Medicine, 05, 342–356. 10.1093/jalm/jfz026 [DOI] [PubMed] [Google Scholar]
- Gan SR, Wang J, Figueroa KP, Pulst SM, Tomishon D, Lee D, … Kuo SH (2017). Postural tremor and ataxia progression in spinocerebellar ataxias. Tremor and Other Hyperkinetic Movements, 7, 1–7. 10.7916/D8GM8KRH [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdts J, Mancini J, Fox E, Rhoads C, Ward T, Easley E, & Bernier RA (2018). Interdisciplinary team evaluation: An effective method for the diagnostic assessment of autism spectrum disorder. Journal of Developmental and Behavioral Pediatrics, 39(4), 271–281. 10.1097/DBP.0000000000000549 [DOI] [PubMed] [Google Scholar]
- Gibson KE, Fitzpatrick DM, Stone D, Noel TP, & Macpherson CNL (2016). Vector-borne diseases in the Caribbean: History and current status. CAB Reviews, 11(022), 1–28. 10.1079/PAVSNNR201611022 [DOI] [Google Scholar]
- Helbig KL, Farwell Hagman KD, Shinde DN, Mroske C, Powis Z, Li S, … Helbig I (2016). Diagnostic exome sequencing provides a molecular diagnosis for a significant proportion of patients with epilepsy. Genetics in Medicine, 18, 898–905. 10.1038/gim.2015.186 [DOI] [PubMed] [Google Scholar]
- Hilgart JS, Hayward JA, Coles B, & Iredale R (2012). Telegenetics: A systematic review of telemedicine in genetics services. Genetics in Medicine, 14(9), 765–776. 10.1038/gim.2012.40 [DOI] [PubMed] [Google Scholar]
- Hoeks MPA, den Heijer M, & Janssen MCH (2009). Adult issues in phenylketonuria. Netherlands Journal of Medicine, 67(1), 2–7. [PubMed] [Google Scholar]
- Hoffbuhr K, Devaney JM, LaFleur B, Sirianni N, Scacheri C, Giron J, … Naidu S (2001). MeCP2 mutations in children with and without the phenotype of Rett syndrome. Neurology, 56(11), 1486–1495. 10.1212/WNL.56.11.1486 [DOI] [PubMed] [Google Scholar]
- Homan CC, Kumar R, Nguyen LS, Haan E, Raymond FL, Abidi F, … Jolly LA (2014). Mutations in USP9X are associated with x-linked intellectual disability and disrupt neuronal cell migration and growth. American Journal of Human Genetics, 94(3), 470–478. 10.1016/j.ajhg.2014.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui L, Barclay J, Poulton A, Hutchinson B, & Halliday JL (2018). Prenatal diagnosis and socioeconomic status in the non-invasive prenatal testing era: A population-based study. The Australian & New Zealand Journal of Obstetrics & Gynaecology, 58(4), 404–410. 10.1111/ajo.12778 [DOI] [PubMed] [Google Scholar]
- Jiao Q, Sun H, Zhang H, Wang R, Li S, Sun D, … Jin Y (2019). The combination of whole-exome sequencing and copy number variation sequencing enables the diagnosis of rare neurological disorders. Clinical Genetics, 96(2), 140–150. 10.1111/cge.13548 [DOI] [PubMed] [Google Scholar]
- Johnson BV, Kumar R, Oishi S, Alexander S, Kasherman M, Vega MS, … Jolly LA (2020). Partial loss of USP9X function leads to a male neurodevelopmental and behavioral disorder converging on transforming growth factor β signaling. Biological Psychiatry, 87, 100–112. 10.1016/j.biopsych.2019.05.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy AD, Wittmann BM, Evans AM, Miller LAD, Toal DR, Lonergan S, … Pappan KL (2018). Metabolomics in the clinic: A review of the shared and unique features of untargeted metabolomics for clinical research and clinical testing. Journal of Mass Spectrometry, 53, 1143–1154. 10.1002/jms.4292 [DOI] [PubMed] [Google Scholar]
- Kim UK, Jin DK, Ahn C, Shin JH, Lee KB, Kim SH, …Lee CC (2000). Novel mutations of the PKD1 gene in Korean patients with autosomal dominant polycystic kidney disease. Mutation Research/Mutation Research Genomics, 432(1-2), 39–45. 10.1016/s1383-5726(99)00013-8. [DOI] [PubMed] [Google Scholar]
- Kruszka P, Porras AR, Sobering AK, Ikolo FA, La Qua S, Shotelersuk V, … Muenke M (2017). Down syndrome in diverse populations. American Journal of Medical Genetics, Part A, 173A, 42–53. 10.1002/ajmg.a.38043 [DOI] [PubMed] [Google Scholar]
- Li D, Yuan H, Ortiz-Gonzalez XR, Marsh ED, Tian L, McCormick EM, … Falk MJ (2016). GRIN2D recurrent de novo dominant mutation causes a severe epileptic encephalopathy treatable with NMDA receptor channel blockers. American Journal of Human Genetics, 99(4), 802–816. 10.1016/j.ajhg.2016.07.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord C, Rutter M, DiLavore P, Risi S, Gotham K, & Bishop S, (2012). Autism diagnostic observation schedule (et al. ed.). Torrance, CA: Western Psychological Services. [Google Scholar]
- Malinowski J, Miller DT, Demmer L, Gannon J, Pereira EM, Schroeder MC, … Shen J (2020). Systematic evidence-based review: Outcomes from exome and genome sequencing for pediatric patients with congenital anomalies or intellectual disability. Genetics in Medicine, 22(6), 986–1004. 10.1038/s41436-020-0771-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann DM, Chen J, Chunara R, Testa PA, & Nov O (2020). COVID-19 transforms health care through telemedicine: Evidence from the field. Journal of the American Medical Informatics Association, 27(7), 1132–1135. 10.1093/jamia/ocaa072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie J, Oettel-Flaherty C, Noel D, Walker RH, & Sobering AK (2019). Pseudo-ataxia due to osteoid osteoma. Tremor and Other Hyperkinetic Movements, 9, 1–4. 10.7916/vt1n-ga19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikat-Stevens NA, Larson IA, & Tarini BA (2015). Primary-care providers' perceived barriers to integration of genetics services: A systematic review of the literature. Genetics in Medicine, 17(3), 169–176. 10.1038/gim.2014.101 [DOI] [PubMed] [Google Scholar]
- Mitchell N, LaTouche GA, Nelson B, Figueroa KP, Walker RH, & Sobering AK (2019). Childhood-onset spinocerebellar ataxia 3: Tongue dystonia as an early manifestation. Tremor and Other Hyperkinetic Movements, 9, 1–4. 10.7916/tohm.v0.704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake N, Visser R, Kinoshita A, Yoshiura K, Niikawa N, Kondoh T, … Kurosawa K (2005). Four novelNIPBL mutations in Japanese patients with Cornelia de Lange syndrome. American Journal of Medical Genetics Part A, 135A(1), 103–105. 10.1002/ajmg.a.30637. [DOI] [PubMed] [Google Scholar]
- Moscovich M, Okun MS, Favilla C, Figueroa KP, Pulst SM, Perlman S, … Subramony SH (2015). Clinical evaluation of eye movements in spinocerebellar ataxias: A prospective multicenter study. Journal of Neuro-Ophthalmology, 35(1), 1–11. 10.1097/WNO.0000000000000167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muenke M, Adeyemo A, & Kruszka P (2016). An electronic atlas of human malformation syndromes in diverse populations. Genetics in Medicine, 18(11), 1085–1087. 10.1038/gim.2016.3 [DOI] [PubMed] [Google Scholar]
- Murray T, Beaty TH, Mathias RA, Rafaels N, Grant AV, Faruque MU, … Barnes KC (2010). African and non-African admixture components in African Americans and an African Caribbean population. Genetic Epidemiology, 34, 561–568. 10.1002/gepi.20512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo KJ, Rexach JE, Lee H, Petty LE, Perlman S, Valera JM, … Fogel BL (2020). A diagnostic ceiling for exome sequencing in cerebellar ataxia and related neurological disorders. Human Mutation, 41, 487–501. 10.1002/humu.23946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ope O, Bhoj EJ, Nelson B, Li D, Hakonarson H, & Sobering AK (2020). A homozygous truncating NALCN variant in two Afro-Caribbean siblings with hypotonia and dolichocephaly. American Journal of Medical Genetics, Part A, 182(8), 1877–1880. 10.1002/ajmg.a.61744 [DOI] [PubMed] [Google Scholar]
- Otten E, Birnie E, Lucassen AM, Ranchor AV, & Van Langen IM … (2016). Telemedicine uptake among genetics professionals in Europe: Room for expansion. European Journal of Human Genetics, 24(2), 157–163. 10.1038/ejhg.2015.83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plagnol V, Curtis J, Epstein M, Mok KY, Stebbings E, Grigoriadou S, … Nejentsev S (2012). A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics, 28(21), 2747–2754. 10.1093/bioinformatics/bts526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razipour M, Alavinejad E, Sajedi SZ, Talebi S, Entezam M, Mohajer N, … Keramatipour M (2017). Genetic study of the PAH locus in the Iranian population: familial gene mutations and minihaplotypes. Metabolic Brain Disease, 32(5), 1685–1691. 10.1007/s11011-017-0048-7 [DOI] [PubMed] [Google Scholar]
- Reijnders MRF, Zachariadis V, Latour B, Jolly L, Mancini GM, Pfundt R, … Kleefstra T (2016). De novo loss-of-function mutations in USP9X cause a female-specific recognizable syndrome with developmental delay and congenital malformations. American Journal of Human Genetics, 98(2), 373–381. 10.1016/j.ajhg.2015.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, … Rehm HL (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer SL, Hartley T, Dyment DA, Beaulieu CL, Schwartzentruber J, Smith A, … Boycott KM (2016). Utility of whole-exome sequencing for those near the end of the diagnostic odyssey: Time to address gaps in care. Clinical Genetics, 89(3), 275–284. 10.1111/cge.12654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shea L, Newschaffer CJ, Xie M, Myers SM, & Mandell DS (2014). Genetic testing and genetic counseling among medicaid-enrolled children with autism spectrum disorder in 2001 and 2007. Human Genetics, 133(1), 111–116. 10.1007/s00439-013-1362-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobering AK, Stevens JB, Smith JL, Nelson B, Donald T, & Elsea SH (2017). Genetic diagnosis of Down syndrome in an under-served community. American Journal of Medical Genetics, Part A, 176, 1–4. 10.1002/ajmg.a.38573 [DOI] [PubMed] [Google Scholar]
- Srinivasan R, Ben-Pazi H, Dekker M, Cubo E, Bloem B, Moukheiber E, … Guttman M (2020). Telemedicine for hyperkinetic movement disorders. Tremor and Other Hyperkinetic Movements, 10, 1–8. 10.7916/tohm.v0.698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegeman S, Jolly LA, Premarathne S, Gecz J, Richards LJ, Mackay-Sim A, & Wood SA (2013). Loss of Usp9x disrupts cortical architecture, hippocampal development and TGFβ-mediated axonogenesis. PLoS One, 8(7), 1–12. 10.1371/journal.pone.0068287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symonds JD, & McTague A (2020). Epilepsy and developmental disorders: Next generation sequencing in the clinic. European Journal of Paediatric Neurology, 24, 15–23. 10.1016/j.ejpn.2019.12.008 [DOI] [PubMed] [Google Scholar]
- Thompson W, Carey PZ, Donald T, Nelson B, Bhoj EJ, Li D, … Sobering AK (2020). Application of exome sequencing to diagnose a novel presentation of the Cornelia de Lange syndrome in an Afro-Caribbean family. Molecular Genetics & Genomic Medicine, 8(8), 1–7. 10.1002/mgg3.1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Wegberg AMJ, MacDonald A, Ahring K, Bélanger-Quintana A, Blau N, Bosch AM, … Spronsen van FJ (2017). The complete European guidelines on phenylketonuria: Diagnosis and treatment. Orphanet Journal of Rare Diseases, 12(162), 1–56. 10.1186/s13023-017-0685-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wardeh A, Jackson T, Nelson B, Ernst C, Théroux J-F, Al-Hertani W, … Maj MC (2018). Identification of a de novo case of COL5A1 -related Ehlers-Danlos syndrome in an infant in the West Indies leading to improved targeted clinical care. Clinical Case Reports, 6(11), 2256–2261. 10.1002/ccr3.1873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright CF, FitzPatrick DR, & Firth HV (2018). Paediatric genomics: Diagnosing rare disease in children. Nature Reviews Genetics, 19(5), 253–268. 10.1038/nrg.2017.116 [DOI] [PubMed] [Google Scholar]
- Yearwood AK, Rethi S, Figueroa KP, Walker RH, & Sobering AK (2018). Diagnosis of spinocerebellar ataxia in the West Indies. Tremor and Other Hyperkinetic Movements, 8, 567. 10.7916/D8DV329C [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen YTK, Guella I, Roland E, Sargent M, & Boelman C (2019). Case reports: novel TUBG1 mutations with milder neurodevelopmental presentations. BMC Medical Genetics, 20(95), 1–7. 10.1186/s12881-019-0827-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarate YA, Smith-Hicks CL, Greene C, Abbott MA, Siu VM, Calhoun ARUL, … Chung WK (2018). Natural history and genotype-phenotype correlations in 72 individuals with SATB2-associated syndrome. American Journal of Medical Genetics, Part A, 176(4), 925–935. 10.1002/ajmg.a.38630 [DOI] [PubMed] [Google Scholar]
- Zurflüh MR, Zschocke J, Lindner M, Feillet F, Chery C, Burlina A, … Blau N (2008). Molecular genetics of tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency. Human Mutation, 29(1), 167–175. 10.1002/humu.20637 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.