

Abstract
The adaptor protein TNF receptor-associated factor 3 (TRAF3) is required for in vivo T cell effector functions, and for normal TCR/CD28 signaling. TRAF3-mediated enhancement of TCR function requires engagement of both CD3 and CD28, but the molecular mechanisms underlying how TRAF3 interacts with and impacts TCR/CD28-mediated complexes to enhance their signaling remains an important knowledge gap. We investigated how TRAF3 is recruited to, and regulates, CD28 as a TCR co-stimulator. Direct association with known signaling motifs in CD28 was dispensable for TRAF3 recruitment; rather, TRAF3 associated with the CD28-interacting protein Linker of activated T cells (LAT) in human and mouse T cells. TRAF3-LAT association required the TRAF3 TRAF-C domain and a newly identified TRAF2/3 binding motif in LAT. TRAF3 inhibited function of the LAT-associated negative regulatory protein Dok1, which is phosphorylated at an inhibitory tyrosine residue by the tyrosine kinase Breast tumor kinase (Brk/PTK6). TRAF3 regulated Brk activation in T cells, limiting association of Protein tyrosine phosphatase 1B (PTP1B) with the LAT complex. In TRAF3-deficient cells, LAT complex-associated PTP1B was associated with dephosphorylation of Brk at an activating tyrosine residue, potentially reducing its ability to inhibit Dok1. Consistent with these findings, inhibiting PTP1B activity in TRAF3-deficient T cells rescued basal and TCR/CD28-mediated activation of Src family kinases. These results reveal a new mechanism for promotion of TCR/CD28-mediated signaling through restraint of negative regulation of LAT by TRAF3, enhancing understanding of regulation of the TCR complex.
Introduction
TRAF3 is an intracellular adaptor protein with versatile cell type- and context-specific roles (1). While the roles of TRAF3 have been most extensively studied in B lymphocytes and myeloid cells, a growing body of literature supports its role as a key regulator in T cell biology (reviewed in 2). Mice with T cell-specific deletion of Traf3 from the CD4+CD8+ stage of development onward (T-Traf3−/−) have normal numbers of conventional CD4+ and CD8+ T cells. However, these T cells exhibit impaired TCR/CD28 signaling, associated with defective in vivo responses to antigen and pathogen challenge (3) and an altered balance of effector memory T cell populations (3, 4). In contrast to conventional T cells, development of invariant NK T (iNKT) cells is markedly reduced in the absence of TRAF3, as TCR-mediated upregulation of the key transcription factor Tbet is impaired (5). Strength of TCR-mediated signaling is important for the differentiation of helper T cell subsets (6-8), and can be an important predictor of successful T cell-mediated immune responses. While defective TCR/CD28 signaling contributes to the markedly defective T cell responses to immunization and infection displayed by the T-Traf3−/− mouse (3), TRAF3 also regulates the functions of additional T cell-expressed receptors. These include several types of cytokine receptors (5, 30) as well as T cell-expressed TNFR superfamily receptors (reviewed in 2), so assessment of complex in vivo immune responses mediated by TRAF3-deficient T cells cannot reveal precisely how TRAF3 regulates signaling by CD28. We thus turned to in vitro models to determine the molecular mechanisms underlying this important role of T cell TRAF3.
The earliest detectable defect in TCR signaling in TRAF3-deficient T cells is activation of the Src family kinase Lck. TRAF3-deficient T cells display increased localization of the inhibitory kinase Csk and the protein tyrosine phosphatase 22 (PTPN22) to the plasma membrane, where they negatively regulate Lck activation (9). T cell TRAF3 normally facilitates Lck activation via sequestration of both Csk and PTPN22 to the cytoplasm, resulting in enhanced TCR/CD28 signaling (9). Interestingly, while defects in TCR/CD3 signaling alone are seen in TRAF3-deficient T cells, the defects are much more evident in TCR + CD28-stimulated cells (3).
This initial observation of markedly defective TCR function in TRAF3-deficient T cells led to the unexpected finding that TRAF3 localizes to the TCR complex, with enhanced recruitment upon engagement of both CD3 and CD28; engagement of CD28 alone is not sufficient to effectively recruit TRAF3 to the complex (3). A key remaining knowledge gap has been identifying the molecular mechanisms by which TRAF3 interacts with the CD28 signaling complex, to both enhance TRAF3 recruitment to the TCR complex, and promote CD28 signaling. We hypothesized that TRAF3 is recruited directly via CD28, or by a CD28-associated adaptor protein, such as the important adaptor molecule LAT. LAT was of particular interest, because it balances TCR/CD28 signaling through interactions with both positive and negative regulatory molecules (10-13), although it has not been previously reported to interact with TRAF3.
Results presented here show that TRAF3 associated with LAT in both human and mouse T cells, and this association required a TRAF2/3 binding motif we newly identified in LAT. Association of TRAF3 with the TRAF2/3 binding motif in LAT, rather than directly with CD28, suggested that LAT, rather than CD28, mediated the recruitment of TRAF3 to the TCR/CD28 complex. We found that TRAF3 enhanced TCR/CD28 signaling concomitant with restraining negative regulators of LAT function. TRAF3 promoted the inactivation and degradation of the LAT-associated negative regulatory molecule Dok1 (11, 14-15). Further, TRAF3 regulated the tyrosine kinase Breast tumor kinase (Brk/PTK6) and the protein phosphatase PTP1B (16-18), in a manner that inhibited Dok1 activation, and enhanced LAT-mediated CD28 signaling. These findings reveal a new mechanism by which TCR/CD28 signaling is regulated by the multifaceted adapter protein TRAF3, and reveal TRAF3 as an important component of promoting LAT signaling in T cells.
Materials and Methods
Mice:
Traf3flox/flox mice, backcrossed extensively to C57Bl/6 mice (19), and bred to Cd4-Cre mice to generate T-Traf3−/− mice, were previously described (3-4, 9). CD28 AYAA and Y170F knock-in mice (20-21), were provided by Dr. Kelvin Lee (Roswell Park Comprehensive Cancer Center, Buffalo, NY). Adult mice (2-6 months; similar numbers of males and females) were used, and littermates were used as controls for all experiments. All mice were maintained under specific pathogen-free conditions at the University of Iowa and used in accordance with NIH guidelines under an animal protocol approved by the Animal Care and Use Committee at the University of Iowa.
Mouse primary T cell isolation:
Splenic T cells were purified from adult (2-6 months) mice as previously described (3, 9). Briefly, pan-T cells were isolated with a negative selection kit (STEMCELL Technologies, Vancouver, Canada) according to the manufacturer’s protocol. Isolated cells were washed with serum-free RPMI 1640 (Life Technologies, Grand Island, NY) prior to use.
Cell lines and culture:
HEK 293T cells were obtained from the ATCC (Manassas, VA) and maintained in DMEM supplemented with 10% heat-inactivated FBS (Life Technologies), 2 mM L-glutamine (Life Technologies), and 100 U/ml penicillin-streptomycin (Life Technologies).
HT28.11, a subclone of the human CD4 T cell line HuT78 transfected to stably express CD28 (22), was a gift from Dr. Arthur Weiss (University of California, San Francisco, CA). The TRAF3−/− subclone of HT28.11 cells, referred to here as crTRAF3−/− or crT3−/−, was described previously (9). The mouse T cell hybridoma line 2B4 was described previously (23). All T cell lines were maintained in RPMI 1640 (Life Technologies) supplemented with 10 μM 2-β-mercaptoethanol (VWR International, Radnor, PA), 10% heat-inactivated FBS (Life Technologies), 2 mM L-glutamine (Life Technologies), and 100 U/ml penicillin-streptomycin (Life Technologies).
Antibodies and reagents:
Antibodies (Abs) used for immunoblotting included the following. Rabbit anti-β-actin (13E5), rabbit anti-Brk (D4O2D), rat anti-CD3ε (CD3-12), rabbit anti-K48 Ub (D9D5), rabbit anti-LAT (E3U6J), rabbit anti-Lck (cat #2752), rabbit anti-phosphoSrc Y416 (D49G4, used to detect pFyn Y416 and pLck Y394), rabbit anti-PTP1B (cat #5311, used to detect PTP1B association with Brk in Fig 5H), and rabbit anti-TRAF3 (cat #4729) were purchased from Cell Signaling Technologies (Danvers, MA). Mouse anti-PTP1B (Ab38675) Ab, used to detect PTP1B expression in cell lysates, was purchased from Abcam (Cambridge, MA). Mouse Abs against FLAG (M2) and HA (HA-7) were purchased from Sigma-Aldrich (St. Louis, MO). Rabbit anti-phosphoDok1 Y362 (cat # PA5-37548) and rabbit anti-phosphoBrk Y447 (cat #PA5-38413) Abs were purchased from Invitrogen (Carlsbad, CA). Rabbit anti-cIAP Ab (cat #10022-1 AP) was purchased from Proteintech (Rosemont, IL). Mouse anti-Brk (G-6), goat anti-CD28 (M-20), mouse anti-Dok1 (A-3), mouse anti-GAPDH (6C5), and rabbit anti-TRAF3 (H-122 and M-20) Abs were purchased from Santa Cruz Biotechnology (Dallas, TX). Rabbit Abs against LAT (cat #06-807) and phosphoBrk Y342 (cat #09-144) were purchased from Millipore (Burlington, MA). Stimulatory monoclonal Abs (mAbs) against human and mouse CD3ε (OKT3 and 2C11, respectively) and CD28 (CD28.6 and 37.51, respectively) were purchased from eBioscience (San Diego, CA). The Brk inhibitor Cpd 4f was purchased from Millipore. The PTP1B inhibitor TCS-401 was purchased from Santa Cruz Biotechnology.
Figure 5. TRAF3-mediated regulation of Brk.
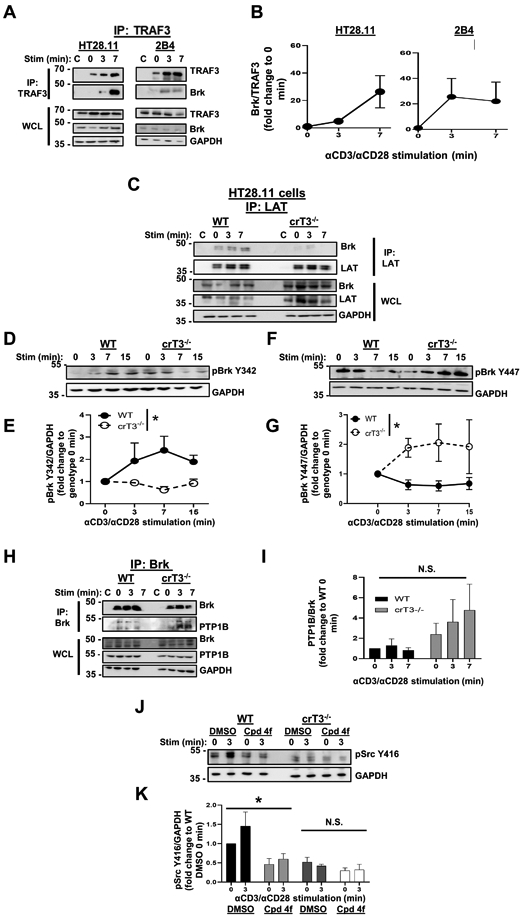
A) Human HT28.11 (left) and mouse 2B4 (right) T cells were stimulated as in Fig. 4A, and TRAF3 was immunoprecipitated. Association of Brk with TRAF3 was assessed by WB. Blots are representative of 3 independent experiments. B) Densitometric analysis of the ratio of Brk associated with TRAF3 at each timepoint shown in A, with each timepoint normalized to 0 min to determine the impact of CD3/CD28 stimulation on the kinetics of the TRAF3-Brk association. C) Human HT28.11 WT and crTRAF3−/− cells were stimulated and lysed as in Fig. 4C, and LAT was immunoprecipitated from lysates. Association of Brk with the LAT complex was assessed by WB. Blots are representative of 4 independent experiments. D) Human HT28.11 WT and crTRAF3−/− cells were stimulated as in Fig. 4A for the indicated times and lysed. Phosphorylation of Brk at Y342 was assessed by WB. Blots are representative of 6 independent experiments. E) Densitometric analysis of the ratio of the abundance of pBrk Y342 to that of GAPDH in each lane. Fold change was calculated by dividing the ratio of pBrkY342/GAPDH at each timepoint by the ratio of pBrk Y342/GAPDH at the 0 min timepoint for each cell line, to control for cell line-specific differences in abundance of pBrk Y342. The difference in pBrk Y342 abundance between WT and crTRAF3−/− cells was statistically significant (*p=0.0490). F) Human HT28.11 WT and crTRAF3−/− cells were stimulated as in Fig. 5D, and phosphorylation of Brk at Y447 was assessed by WB. Blots are representative of 7 independent experiments. G) Densitometric analysis of the ratio of the abundance of pBrk Y447 to that of GAPDH in each lane. Fold change was calculated by dividing the ratio of pBrkY447/GAPDH at each timepoint by the ratio of pBrk Y447/GAPDH at the 0 min timepoint for each cell line, to control for cell line-specific differences in abundance of pBrk Y447. The difference in pBrk Y447 abundance between WT and crTRAF3−/− cells was statistically significant (*p=0.0432). H) Human HT28.11 WT and crTRAF3−/− cells were stimulated and lysed as in Fig. 4A, and Brk was immunoprecipitated from lysates. Association of PTP1B with Brk was assessed by WB. Blots are representative of 4 independent experiments. I) Densitometric analysis of the ratio of PTP1B associated with Brk in each lane. Fold change was calculated by dividing the ratio of PTP1B/Brk at each timepoint by the ratio of PTP1B/Brk at WT 0 min. to determine the impact of genotype on the kinetics of the PTP1B-Dok1 association. The difference in PTP1B/Brk association between WT and crTRAF3−/− cells was not statistically significant. J) Human HT28.11 WT and crTRAF3−/− cells were treated with DMSO or the Brk inhibitor Cpd 4f for 2 hours, as described in Materials and Methods, then stimulated through CD3/CD28 for the times indicated and lysed. Phosphorylation of Fyn at Y416 and Lck at Y394 was detected by WB with an anti-pSrcY416 Ab. Blots are representative of 3 independent experiments. K) Densitometric analysis of the ratio of abundance of pSrc Y416 to that of GAPDH. Fold change was calculated by dividing the ratio of pSrc Y416/GAPDH in each lane to the ratio of pSrc Y416/GAPDH for DMSO-treated WT 0 min, to determine the impact of Cpd 4f treatment on Src kinase activation relative to a baseline value. The difference between DMSO-treated and Cpd 4f-treated WT cells was statistically significant (*p=0.0123). The difference between DMSO-treated and Cpd 4f-treated crTRAF3−/− cells was not statistically significant. ‘C’ indicates samples that only received stimulatory Abs, i.e. no IP Ab added after lysis (A, C, I). Graphs depict mean ± SEM. A 2-way ANOVA was performed to establish statistical significance in E, G, and I, and K. The reference group used to calculate fold change was not included in the analyses. *: p<0.05; ‘N.S.’: not significant.
Plasmid constructs:
pCMV3-FLAG-CD28 was purchased from Sino Biological (Chesterbrook, PA). pCDNA3-TRAF3-HA, pCDNA3-TRAF3ΔRing/Zn-HA, and pCDNA3-TRAF3ΔTRAFC-HA were described previously (4, 9, 24). pCDNA3-LATQ212A-FLAG was produced using subcloning by overlap extension PCR (25). PCR primers for generating the 5’ section of the construct were hLAT-F (ttataagcttgccgccaccatggaggaggccatcctgg) and Q212A-R (tctgcctccgcggaactcagggcggcaggctcag). Primers for the 3’ section were Q212A-F (gagttccgcggaggcagaggaagtggaggaagagggggctcc) and hLAT-FLAG-R (aaaatctagattcacttgtcatcgtcatccttgtagtcgttcagctcctgcagattctc). The 5’ and 3’ sections were joined by PCR using a mixture of the purified sections as template for amplification with hLAT-F and hLAT-FLAG-R. The final PCR product was purified, cut with HindIII and XbaI, and ligated into pCDNA3 cut with the same enzymes.
HEK 293T transfection:
1x106 HEK 293T cells per well of a 6 well plate were transiently transfected with the plasmids listed above as previously described (9), with the following modifications. 1 μg of plasmid DNA and 3 ul of Lipofectamine 3000 in Opti-MEM (both purchased from Thermo Fisher, Waltham, MA) were added per well, and cells were incubated for 48 hours. Cells were harvested and lysed for 30 minutes on ice with immunoprecipitation lysis buffer (0.5% Triton X-100, 100 mM NaCl, 40 mM Tris pH 7.5, 0.05 mg/ml DNase I, EDTA-free mini complete protease inhibitor, 1 mM CaCl2, and 1 mM MgSO4). Immunoprecipitations were performed by incubating lysates with anti-HA and anti-FLAG Abs (9).
Immunoprecipitation (IP):
TCR/CD28 IPs were performed as previously described (3). All other IPs were performed as follows. 15-30x106 T cells per timepoint were incubated with 10 μg/ml each of anti-CD3ε (OKT3 or 2C11) and anti-CD28 (CD28.6 or 37.51) mAbs for 30 minutes on ice prior to stimulation in a 37°C water bath for 3 or 7 minutes. Cells were lysed with 200 μl lysis buffer (0.5% Brij 97, 1% n-Octyl-β-d-glucopyranoside, 150 mM NaCl, 25 mM Tris pH 8, 0.05 mg/ml DNase I, EDTA-free mini complete protease inhibitor, 1 μM Na3VO4, 1 mM CaCl2, 1 mM MgSO4) on ice for 30 minutes. Precleared lysates were incubated with IP Abs at a concentration of 1:100 (in a total volume of 200 μl) on a rotator at 4°C overnight. Samples labeled ‘C’ received stimulatory Abs, but not IP Abs. The rest of the IP protocol was performed as previously described (9).
Dok1 degradation assay:
1x106 cells were plated in 500 μl serum-free RPMI in a 48 well plate. Cells were incubated at 37°C with 10 μg/ml each of anti-CD3ε (OKT3 or 2C11, eBioscience) and anti-CD28 (CD28.6 or 37.51, eBioscience) Abs for up to 6 hours. Cell pellets were resuspended in 2x SDS-PAGE sample buffer and sonicated to prepare whole cell lysates.
Inhibitor experiments:
HT28.11 cells were serum-starved for 24 hours, and 0.5x106 cells (HT28.11 cells) or 1x106 cells (primary mouse T cells) per well were plated in 210 μl serum-free RPM1 in a 48 well plate. Cells were treated with 10 μl of DMSO or inhibitor (for PTP1B inhibition, 5.3 μM TCS-401; for Brk inhibition, 20 nM Cpd 4f) and incubated at 37°C for 2 hours. Cells were washed three times with serum-free RPMI and stimulated with 10 μg/ml each of anti-CD3ε (OKT3 or 2C11, eBioscience) and anti-CD28 mAbs (CD28.6 or 37.51, eBioscience) for 3 minutes at 37°C. Cell pellets were resuspended in 2x SDS sample buffer and sonicated to prepare whole cell lysates.
Western blot analysis:
Proteins were separated by SDS-PAGE and transferred onto PVDF membranes for Western blotting as previously described (3-4, 9). Blots were imaged using Fujifilm LAS-4000, and densitometry was performed using Fujifilm Multi Gauge software (Fujifilm Life Sciences, Cambridge, MA).
Statistical analysis:
Results are presented as means ± SEM, and graphs and statistical analyses were prepared using GraphPad Prism software (San Diego, CA). Two-way ANOVA was used for statistical comparison of multiple groups, unless otherwise indicated. For analysis of data normalized to a reference group, the reference group used to calculate fold change was not included in the ANOVA. Statistical significance was set as p <0.05.
Results
TRAF3 recruitment to the TCR/CD28 complex
TRAF3 is recruited to the TCR/CD28 complex, with optimal recruitment seen upon engagement of both CD3 and CD28 (3). The cytoplasmic domain of CD28 contains multiple motifs previously identified as important for signal transduction, including the membrane-proximal YMNM motif and the membrane-distal PYAP motif (20-21, 26). The latter, which is particularly important for CD28-dependent proliferation and cytokine production (20-21, 26), resembles a putative TRAF2/3 binding motif (3). We thus tested the requirement for one or both signaling motifs for TRAF3 recruitment to the TCR/CD28 complex. Based upon our published studies of TRAF3 interactions with other proteins (9, 27-28), we predicted that the TRAF-C domain of TRAF3 would be required for TRAF3-CD28 interactions. To test these predictions, we transfected HEK 293T epithelial cells with plasmids encoding WT CD28, and either full length TRAF3, or mutant versions lacking the RING/Zn finger or TRAF-C domains (Fig. 1A). Neither full length nor mutant TRAF3 directly associated with CD28; this was confirmed by performing a reciprocal immunoprecipitation (IP) (Fig. 1B).
Figure 1. TRAF3 recruitment to the TCR complex does not require direct TRAF3-CD28 association.
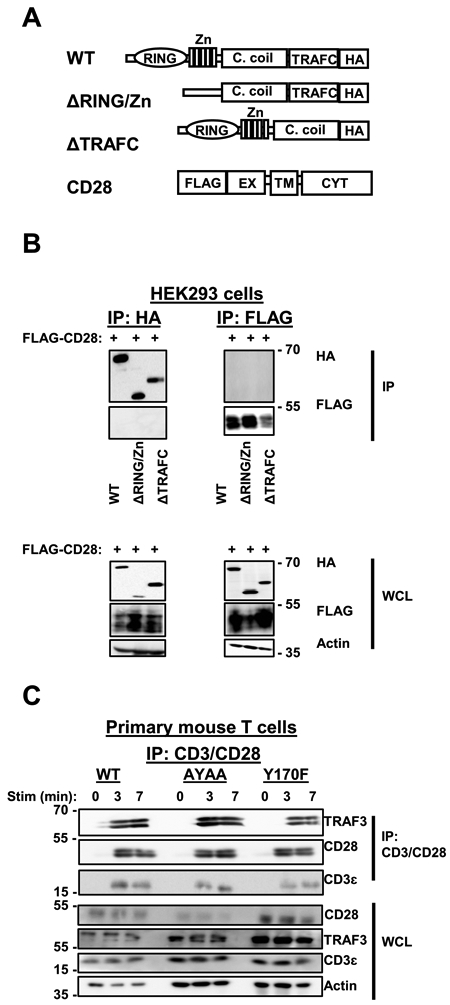
A) TRAF3 and CD28 constructs used in HEK 293T cell transfections. ‘C. coil’ denotes coiled coil domain, ‘EX’ denotes extracellular domain, ‘TM’ denotes transmembrane domain, and ‘CYT’ denotes cytoplasmic domain. B) HEK 293T cells were transfected with plasmids encoding HA-tagged TRAF3 and FLAG-tagged CD28 (constructs shown in Fig. 1A). IP with Abs against HA (left) or FLAG (right) was performed on cell lysates, followed by Western blot (WB) analysis to assess the association of TRAF3 constructs with CD28 (upper panels). Blots of whole cell lysates (WCL) appear in the lower panels. Blots are representative of 3 independent experiments. C) Primary mouse T cells were isolated from mice expressing wildtype CD28, CD28 AYAA, or CD28 Y170F as described in Materials and Methods. Cells were stimulated with anti-CD3/anti-CD28 Abs for the times indicated and lysed. The TCR/CD28 complex was immunoprecipitated with the stimulatory Abs, and samples were subjected to WB analysis to assess the association of TRAF3 with the TCR/CD28 complex. Blots are representative of 3 independent experiments.
We also tested the importance of the published CD28 signaling motifs in TRAF3 recruitment via a complementary approach, using primary mouse T cells expressing either wildtype CD28, CD28 with the PYAP motif mutated to AYAA, or CD28 with the YMNM motif mutated to FMNM (indicated as Y170F). There was no distinguishable difference in TRAF3 recruitment to the TCR/CD28 complex in T cells expressing wildtype vs. mutant CD28 (Fig. 1C). These results, together with those described above in HEK 293T cells, lead to the conclusion that direct association with CD28 itself is not critical for the recruitment of TRAF3 to the TCR/CD28 complex, and thus TRAF3 is likely recruited to this complex primarily through its interactions with other CD28-associated proteins.
Association of TRAF3 with LAT in T cells
We performed motif analysis of several CD28-associated proteins (Eukaryotic Linear Motif Database, 29), identifying a previously unreported putative TRAF2/3 binding motif in the cytoplasmic domain of LAT (SSQE, amino acids 211-213 in the human sequence). We thus tested if TRAF3 associated with LAT in intact T cells. This association was found in HT28.11 human T cells, 2B4 mouse T cells, and primary mouse T cells (Fig. 2A). The TRAF3-LAT association was observed in all T cell types examined (Fig. 2B), and in 2B4 and primary mouse T cells, there was a trend towards increased association with in vitro stimulation via CD3 and CD28. These results indicate that TRAF3 associated with the TCR/CD28 complex via its interaction with LAT. Given the finding that the association did not absolutely require TCR complex signaling, it is possible that TRAF3 associates constitutively with a fraction of LAT that is not TCR/CD28 associated in resting T cells. TRAF3 may then enhance LAT-promoted TCR/CD28 signaling when the complex is brought together in the plasma membrane by ligation.
Figure 2. TRAF3 association with LAT in T cells.
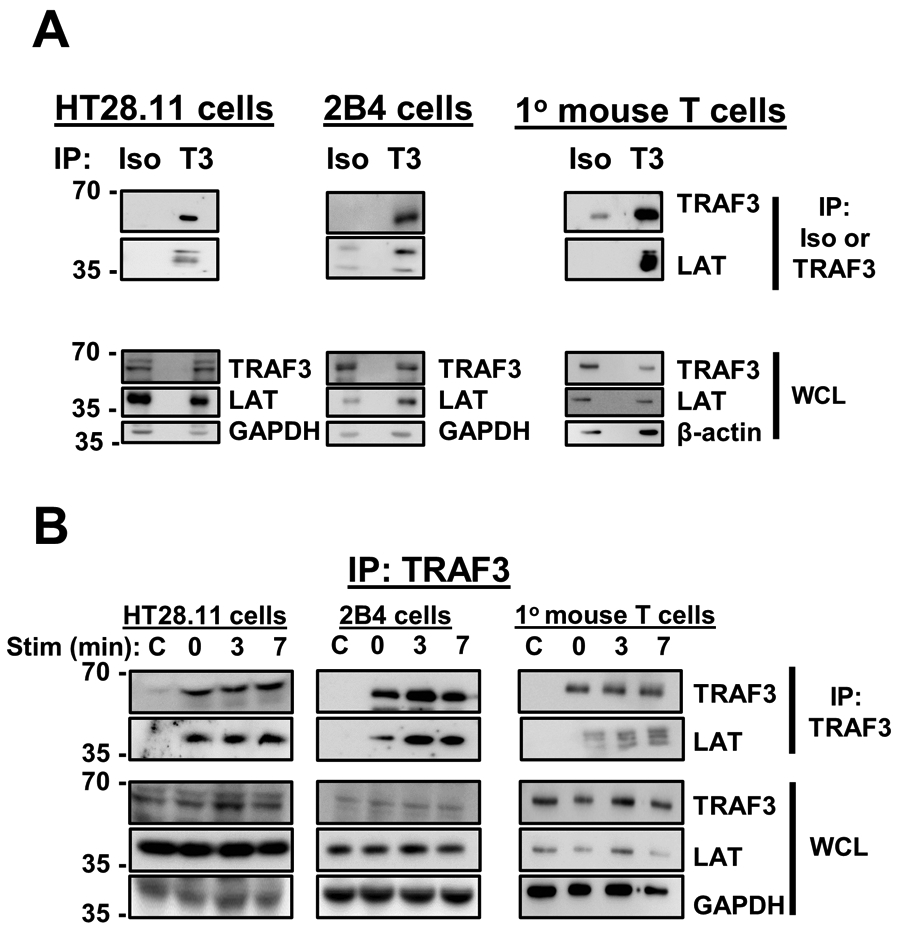
A) Unstimulated HT28.11 (human), 2B4 (mouse), and primary mouse T cells were lysed and subjected to IP with isotype control or anti-TRAF3 Abs. LAT association with TRAF3 was assessed by WB. Blots are representative of 4-6 independent experiments. B) T cells listed in A were stimulated with anti-CD3/anti-CD28 Abs for the times indicated and lysed. Lysates were subjected to IP with anti-TRAF3 Ab, followed by WB analysis to assess TRAF3 association with LAT. ‘C’ indicates samples that received stimulatory Abs only, i.e. no IP Ab added after lysis, to control for any association of LAT with the stimulatory Abs.
To investigate structural requirements for the association between TRAF3 and LAT, we transfected HEK 293T cells with constructs encoding full length and mutant TRAF3, or full length and mutant LAT. The LAT mutant has a single amino acid substitution in the putative TRAF2/3 binding motif that we identified, such that SSQE becomes SSAE, indicated as LATQ212A (Fig. 3A). The TRAF2/3 binding motif is conserved across species (Fig. 3B). The N-terminal RING/Zn finger domain of TRAF3 was dispensable for the TRAF3-LAT association, whereas the TRAF-C domain was required for the association with LAT (Fig. 3C). Mutation of a single amino acid in the TRAF2/3 binding motif of LAT ablated the interaction between TRAF3 and LAT (Fig. 3D). These results indicate that this newly identified TRAF2/3 binding motif in the cytoplasmic domain of LAT is necessary for the association between TRAF3 and LAT.
Figure 3. Association between TRAF3 and LAT requires the TRAF-C domain of TRAF3, and an intact TRAF2/3 binding motif in LAT.
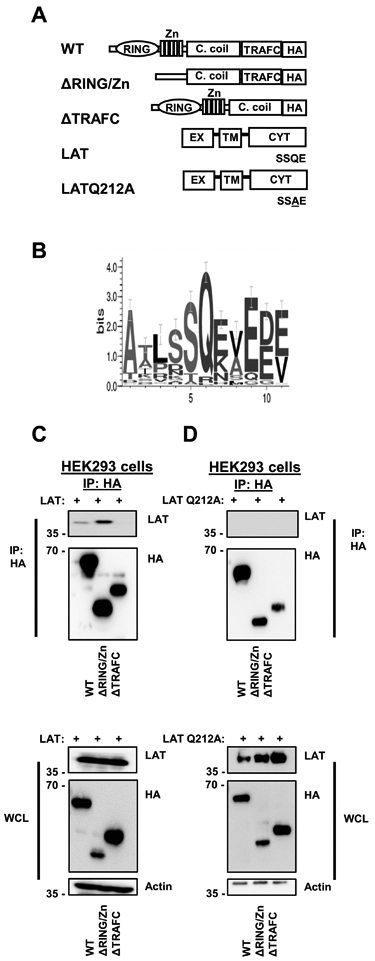
A) TRAF3 and LAT constructs used in HEK 293T cell transfections. Q212 refers to the amino acid sequence of human LAT. B) Sequence conservation across a wide range of species of the TRAF2/3 binding motif in LAT (generated with WebLogo). C) HEK 293T cells were transfected with the HA-TRAF3 and full-length LAT constructs depicted in A. HA was immunoprecipitated, and LAT association was assessed by WB (top). Protein expression in input whole cell lysates (WCL) is also shown (bottom). D) HEK 293T cells were transfected with HA-TRAF3 and LATQ212A constructs depicted in A, and HA was immunoprecipitated as in B. LATQ212A association with TRAF3 was assessed by WB (top), and protein expression in input WCL is also shown (bottom). Blots shown in B and C are from a single experiment and are representative of at least 3 independent experiments.
Effect of TRAF3 upon activity and stability of Dok1
The findings presented in Figs. 2-3 show TRAF3-LAT interactions as important for the localization of TRAF3 to the TCR/CD28 complex, and potentially necessary for TRAF3 to enhance the function of the complex. We next wished to determine the impact of TRAF3 upon LAT-mediated signaling. Based upon our recent findings that TRAF3 sequesters the Lck inhibitors PTPN22 and Csk in the cytoplasm to enhance TCR signaling (9), and additional evidence of restraint of inhibitory signaling pathways by TRAF3 in other circumstances (30), we focused our efforts upon TRAF3 interactions with the LAT complex-associated negative regulatory molecule Dok1 (11, 14-15). TRAF3 associated with Dok1 upon stimulation through CD3/CD28 in both human and mouse T cells (Fig. 4A, 4B). Phosphorylation of Dok1 at Y362 inhibits its activity and targets Dok1 for proteasomal degradation (17). We found less pDok1 Y362 (inactivated Dok1) associated with the LAT complex in TRAF3-deficient T cells compared to WT T cells (Fig. 4C). There was also less pDok1 Y362 relative to total Dok1 in TRAF3−/− T cells stimulated through CD3/CD28 (Fig 4C, 4D). Thus, TRAF3 in T cells may inhibit Dok1-mediated inactivation of LAT function.
Figure 4. Enhanced inactivation and degradation of Dok1 in the presence of TRAF3.
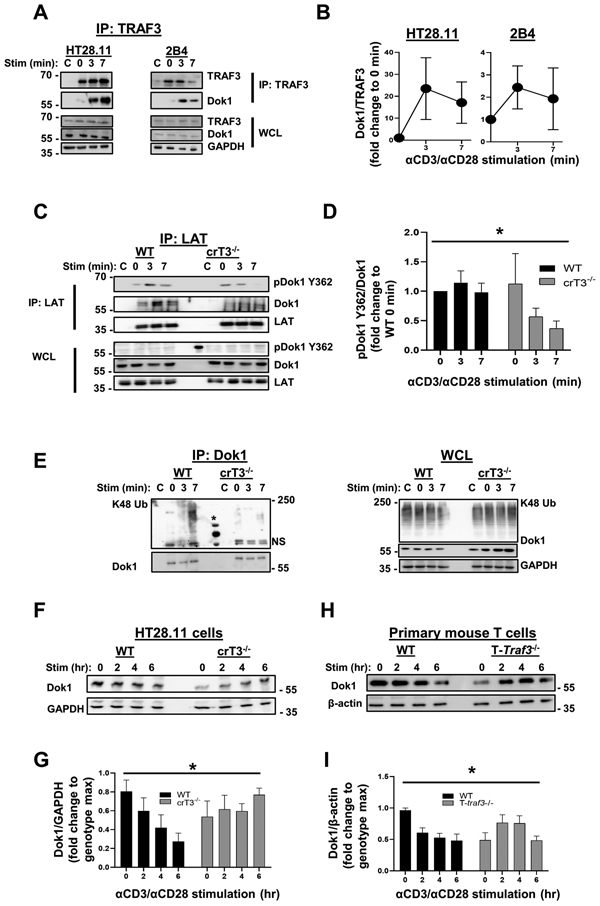
A) Human HT28.11 (left) and mouse 2B4 (right) T cell lines were stimulated with anti-CD3/anti-CD28 Abs for the times indicated, and TRAF3 was immunoprecipitated from lysates. Association of Dok1 with TRAF3 was assessed by WB. Blots are representative of 3-4 independent experiments. B) Densitometric analysis of the ratio of Dok1 associated with TRAF3 at each timepoint shown in A, with each timepoint normalized to 0 min, to determine the impact of CD3/CD28 stimulation on the kinetics of the TRAF3-Dok1 association. C) Human HT28.11 WT and crTRAF3−/− cells were stimulated with anti-CD3/anti-CD28 Abs for the times indicated and lysed, and LAT was immunoprecipitated from lysates. Association of pDok1 Y362 and total Dok1 with the LAT complex was assessed by WB. * in WCL indicates a band from the molecular weight (MW) ladder loaded in that lane. Blots are representative of 6 independent experiments. D) Densitometric analysis of the ratio of pDok1 Y362 associated with total Dok1, with each timepoint normalized to WT 0 min, to determine the impact of genotype on relative amount of pDok1 Y362 associated with the LAT complex. The difference in pDok1 Y362/Dok1 ratios between WT and crTRAF3−/− cells was statistically significant (*p=0.0174). E) Human HT28.11 WT and crTRAF3−/− cells were stimulated and lysed as in C, and Dok1 was immunoprecipitated from lysates. K48-linked polyubiquitin associated with Dok1 was assessed by WB. * indicates protein ladder. Blots are representative of 6 independent experiments. F) Human HT28.11 WT and crTRAF3−/− cells were stimulated with anti-CD3/anti-CD28 Abs for up to 6 hours. Levels of Dok1 in WCL were assessed by WB. G) Densitometric analysis of Dok1 abundance in WCL in F over time, presented as the ratio of Dok1 to GAPDH in each lane, normalized to the maximum ratio for each genotype, to control for cell line-specific differences in abundance of Dok1. The difference in Dok1 abundance over time between WT and crTRAF3−/− cells was statistically significant (*p=0.0159). Blots are representative of 6 independent experiments. H) Primary WT and T-Traf3−/− mouse T cells were stimulated as in F, and levels of Dok1 in WCL were assessed by WB. Blots are representative of 7 independent experiments. I) Densitometric analysis of Dok1 abundance in WCL in H over time, presented as the ratio of Dok1 to GAPDH in each lane, normalized to the maximum ratio for each genotype, to control for cell type-specific differences in abundance of Dok1. The difference in Dok1 abundance over time between WT and T-Traf3−/− cells was statistically significant (*p=0.0034).
‘C’ indicates samples that received stimulatory Abs only, i.e. no IP Ab added after lysis. Graphs depict mean ± SEM. A 2-way ANOVA was performed to establish statistical significance in D, G, and I. The reference group used to calculate fold change was not included in the analysis in D. *: p<0.05; ‘N.S.’: not significant.
As mentioned above, phosphorylation of Dok1 at Y362 targets it for proteasomal degradation. We thus wished to determine the impact of TRAF3 on K48-linked polyubiquitination and degradation of Dok1. There was minimal difference in the association of the E3 ubiquitin ligase TRAF2, known to associate with TRAF3 to induce K48-mediated ubiquitination of target proteins, with the LAT complex in WT and TRAF3-deficient T cells (Supplemental Fig. 1A, 1B). Furthermore, we did not detect association of the TRAF2-binding E3 ubiquitin ligases cIAP1/2, with the LAT complex in either WT or TRAF3-deficient T cells (Supplemental Fig. 1C). This indicates that the TRAF2/cIAP complex is likely not responsible for enhanced K48-linked ubiquitination of Dok1 in the presence of TRAF3 (Fig. 4E).There was a decrease in total Dok1 protein in whole cell lysates from TRAF3-sufficient human (Fig. 4F, 4G) and mouse T cells (Fig. 4H, 4I) compared to TRAF3-deficient T cells. This is consistent with previous reports (17), and with our finding that there is K48-linked polyubiquitin associated with Dok1 in WT but not TRAF3-deficient T cells. These findings indicate that TRAF3 promotes the inactivation and degradation of Dok1, thus enhancing TCR/CD28 signaling.
TRAF3-mediated regulation of Brk
Phosphorylation of Dok1 at Y362 is mediated by the tyrosine kinase Brk (PTK6) (17). We thus asked if Brk was involved in TRAF3-mediated promotion of Dok1 inactivation. We found that unstimulated primary mouse T cells express Brk, and found similar Brk protein abundance in WT and TRAF3-deficient T cells (Supp. Fig. 2A, 2B). TRAF3 associated with Brk upon stimulation through CD3/CD28 in both human and mouse T cells (Fig. 5A, 5B). Brk also associated with the LAT complex in WT, but not TRAF3-deficient T cells (Fig. 5C). This suggests that TRAF3 promotes the recruitment of Brk to the TCR/CD28-associated LAT complex, allowing Brk better access to Dok1.
To determine the impact of TRAF3 on Brk activation, we examined the phosphorylation status of Brk in TRAF3-sufficient and deficient T cells. Phosphorylation of Brk at Y342 is activating, while phosphorylation at Y447 is inhibitory; overall Brk activity depends upon a balance between phosphorylation at these sites (18). We thus predicted higher Brk activation in the presence of TRAF3. Consistent with this prediction, we observed a stimulation-dependent increase in pBrk Y342 in WT T cells, while there was a decrease in pBrk Y342 upon stimulation in TRAF3−/− T cells (Fig. 5D, 5E). The pattern of phosphorylation was the opposite for pBrk Y447, with a decrease in pBrk Y447 upon stimulation through CD3/CD28 in WT T cells, and an increase in pBrk Y447 in TRAF3−/− T cells (Fig. 5F, 5G). These results suggest that TRAF3 restrains the inhibitory activity of Dok1 by promoting the activity of Brk. This in turn leads to enhanced LAT function and TCR/CD28 signaling.
Dephosphorylation of Brk at Y342 is mediated by the protein phosphatase PTP1B (31-32), so we examined the impact of T cell TRAF3 expression on the association between Brk and PTP1B. We found that this association was enhanced in TRAF3−/− T cells, compared to their WT counterparts (Fig. 5H, 5I), consistent with the decreased levels of pBrk Y342 in T cells lacking TRAF3. There is less Brk associated with the LAT complex in TRAF3−/− T cells; it is possible that dephosphorylation of Brk at Y342 by PTP1B diminishes Brk association with the LAT complex.
To determine the role of Brk in TCR/CD28 signaling, we treated TRAF3-sufficient and -deficient T cells with the selective Brk inhibitor Cpd 4f (32). Based upon our recent findings that TRAF3 is required for optimal activation of the Src family kinases, one of the earliest events following TCR engagement (9), we investigated the impact of Brk inhibition upon phosphorylation of Fyn and Lck. Cpd 4f-treated WT human (Fig. 5J, K) and primary mouse T cells (Supp. Fig. 2C, 2D) exhibited decreased Src kinase activation, compared to their DMSO-treated counterparts. In contrast, there was no detectable difference in Src kinase activation between Cpd 4f-treated TRAF3-deficient T cells and their DMSO-treated counterparts. These results are consistent with the conclusion that TRAF3-Brk interactions may contribute to the role played by TRAF3 in promoting TCR/CD28 signaling.
We next examined the impact of Brk inhibition on the activity of Dok1 associated with the LAT complex, to confirm that Dok1 Y362 is a substrate of Brk (Supp. Fig 3A, 3B). Consistent with the impaired TCR complex-mediated Src kinase activation observed in WT T cells treated with the Brk inhibitor Cpd 4f (Fig. 5J, 5K, and Supp. Fig. 2B, 2C), there is less pDok1 Y362 associated with the LAT complex in Cpd 4f-treated WT T cells. Thus, Brk inhibition is associated with greater activation of LAT-associated Dok1. This in turn can facilitate Dok1-mediated restraint of LAT function and TCR/CD28 signaling.
Interaction between TRAF3 and PTP1B
TRAF3 constitutively associated with the phosphatase PTP1B in T cells (Fig. 6A, 6B). To understand how this interaction contributes to the TRAF3-mediated enhancement of TCR/CD28 signaling, we examined PTP1B recruitment in WT and TRAF3−/− T cells, finding enhanced recruitment of PTP1B to the LAT complex in TRAF3−/− T cells. (Fig. 6C). This suggests that in the absence of TRAF3, PTP1B preferentially localizes to the LAT complex, where it can dephosphorylate Brk at Y342 and facilitate the restraint of TCR/CD28 signaling.
Figure 6. Interaction between TRAF3 and PTP1B.
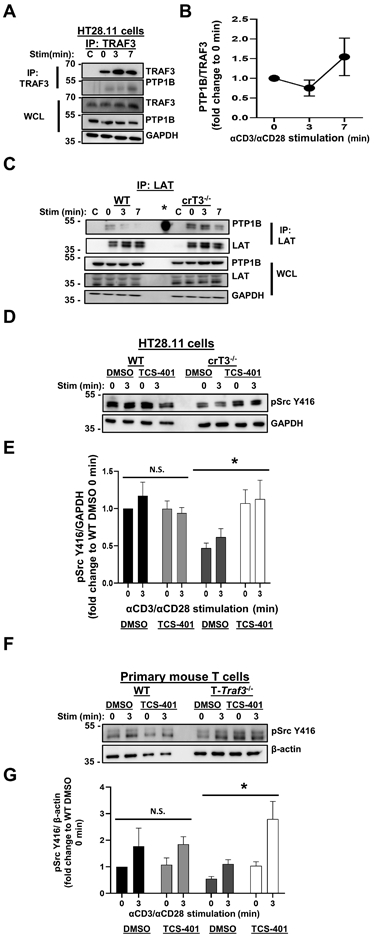
A) Human HT28.11 T cells were stimulated through CD3/CD28 for the times indicated and lysed, and TRAF3 was immunoprecipitated. Association of PTP1B with TRAF3 was assessed by WB. Blots are representative of 4 independent experiments. B) Densitometric analysis of the ratio of PTP1B associated with TRAF3 at each timepoint shown in A, with each timepoint normalized to 0 min, to determine the impact of CD3/CD28 stimulation on the kinetics of the TRAF3-Brk association. C) Human HT28.11 WT and crTRAF3−/− cells were stimulated as in A, and LAT was immunoprecipitated. Association of PTP1B with the LAT complex was assessed by WB. * in IP indicates a band from the molecular weight (MW) ladder loaded in that lane. Blots are representative of 4 independent experiments. D) Human HT28.11 WT and crTRAF3−/− cells were treated with DMSO or the PTP1B inhibitor TCS-401 for 2 hours, as described in Materials and Methods, then stimulated through CD3/CD28 for the times indicated and lysed. Phosphorylation of Fyn at Y416 and Lck at Y394 was detected by WB with an anti-pSrcY416 Ab. Blots are representative of 5 independent experiments. E) Densitometric analysis of the ratio of abundance of pSrc Y416 to that of GAPDH. Fold change was calculated by dividing the ratio of pSrc Y416/GAPDH in each lane to the ratio of pSrc Y416/GAPDH for DMSO-treated WT 0 min, to determine the impact of TCS-401 treatment on Src kinase activation relative to a baseline value. The difference between DMSO-treated crTRAF3−/− cells and TCS-401-treated crTRAF3−/− cells was statistically significant (*p=0.0173). F) Primary WT and T-Traf3−/− mouse T cells were treated as in D. Phosphorylation of Fyn at Y416 and Lck at Y394 was detected by WB with an anti-pSrc Y416 Ab. Blots are representative of 4 independent experiments. G) Densitometric analysis of the ratio of abundance of pSrc Y416 to that of β-actin. Fold change was calculated by dividing the ratio of pSrc Y416/β-actin in each lane to the ratio of pSrc Y416/ β-actin for DMSO-treated WT 0 min, to determine the impact of TCS-401 treatment on Src kinase activation relative to a baseline value. The difference between DMSO-treated T-Traf3−/− cells and TCS-401-treated T-Traf3−/− cells was statistically significant (*p=0.0117). ‘C’ indicates samples that only received stimulatory Abs, i.e. no IP Ab added after lysis (A, C). Graphs depict mean ± SEM. An unpaired two-tailed t test was performed to establish statistical significance at each timepoint in E. A 2-way ANOVA was performed to establish statistical significance in E and G. The reference group used to calculate fold change was not included in the analyses in E and G. *: p<0.05; ‘N.S’.: not significant.
To further address the role of PTP1B in restraint of TCR/CD28 signaling, we treated TRAF3-sufficient and deficient T cells with TCS-401, a selective competitive inhibitor of PTP1B (34). We first investigated the impact of PTP1B inhibition upon activation of the Src family kinases Fyn and Lck, the earliest observed defect in CD3/CD28 signaling in TRAF3-deficient T cells (9). TCS-401-treated TRAF3-deficient human (Fig. 6D, 6E) and primary mouse T cells (Fig. 6F, 6G) exhibited increased Src kinase activation, compared to their DMSO-treated counterparts. TCS-401-treated WT human T cells exhibited increased phosphorylation of Brk at Y342, confirming the role of PTP1B in regulation of Brk activity via dephosphorylation at Y342 (Supp. Fig. 4A, 4B). There was minimal difference in phosphorylation of Brk at Y447 between DMSO-treated and TCS-401-treated WT cells. Interestingly, in contrast to their WT counterparts, TCS-401-treated TRAF3-deficient T cells exhibited increased phosphorylation of Brk at Y447 (Supp. Fig. 4C, 4D).This may be due to altered autophosphorylation of Brk, or to the unhindered activity of the tyrosine kinase SRMS, which also phosphorylates Brk at Y447, in the absence of functional PTP1B (32). These results indicate that lack of normal restraint of PTP1B may contribute to the impairment in LAT-mediated CD3/CD28 signaling observed in TRAF3-deficient T cells. Thus, T cell TRAF3 plays key mechanistic roles in ‘inhibiting the inhibitors’ of normal LAT-enhanced TCR complex signaling, to ultimately promote TCR/CD28-mediated functions.
Discussion
It is becoming increasingly clear that TRAF3 plays multiple important roles in T cell biology (reviewed in 2). Our group’s initial characterization of the T-Traf3−/− mouse, which lacks TRAF3 in all mature T cells, revealed that TRAF3 is important for TCR/CD28-mediated effector function and cytokine production (3). We continue to expand our understanding of the role of TRAF3 in various aspects of T cell biology (4-5, 9, 35).
The initially unexpected finding that TRAF3 is recruited to the TCR/CD28 complex in normal T cells (3) led us to question how TRAF3 is recruited to this complex, and how it enhances CD28 signaling downstream of Lck activation, one of the earliest events in the signaling cascade. As TRAF3 regulates various types of receptors in T cells, alterations in multiple signaling pathways undoubtedly contribute to the documented defective in vivo T cell responses in T-Traf3−/− mice. Thus, analysis of T cell responses in vivo cannot reveal precisely how TRAF3 regulates signaling by a specific receptor complex. We thus turned to in vitro models to identify the molecular mechanisms by which TRAF3 is recruited to, and enhances the function of, the TCR/CD28 complex. The data presented here uncover an important interaction between TRAF3, via the TRAF-C domain, and LAT, via a highly conserved TRAF2/3 binding motif, in recruitment of TRAF3 to the TCR/CD28 complex. To our knowledge, this is the first time that LAT has been shown to associate with a signaling protein independent of phosphorylated tyrosine residues in the LAT intracellular domain.
LAT can be recruited to the TCR complex upon engagement of CD3 alone, which raises the question of why TRAF3 recruitment is difficult to detect upon engagement of only CD3 (3), if TRAF3 can constitutively associate with LAT. It should be noted that T cell TRAF3 deficiency does in fact modestly impact activation of TCR-associated signaling molecules following engagement of CD3 alone (3), so there may be a small amount of recruitment in the absence of CD28 engagement, not easily detected by co-IP. However, CD28 stimulation is likely to enhance the recruitment of LAT specifically to the TCR complex, and/or promote clustering of key signaling molecules at the plasma membrane via modulation of cytoskeletal dynamics and vesicular trafficking, as suggested by a recent report (36). Additionally, there are multiple pools of LAT in the cell, including plasma membrane-associated and vesicular LAT, as well as multiple pools of TRAF3 (37). CD28 signaling may enhance the trafficking of vesicles containing LAT associated with TRAF3 to the plasma membrane, facilitating the recruitment of TRAF3 to the TCR/CD28 complex.
Mutation of a single amino acid in the TRAF2/3 binding motif in LAT prevented the interaction between TRAF3 and LAT. This suggests the possibility that the Q212A LAT mutation could phenocopy some of the features of TRAF3 deficiency in T cells. Unfortunately, direct experimental testing of this possibility faces technical challenges. Cells lacking normal LAT would be the optimal model in which to test functions of introduced WT vs. mutant forms of LAT. However, LAT deficiency in vivo results in a complete absence of T cells, and inhibition of LAT expression in the HuT78 T cell line interferes with TCR surface expression and signaling (JCDH, unpublished data), which prevents interpretation of such experiments. Thus, we have not created a LAT Q212A-expressing T cell line. LAT-deficient Jurkat T cell lines with functional TCRs exist (38), but data interpretation is complicated because Jurkat T cells lack the negative regulatory phosphatase PTEN (39). Thus, we have not used Jurkat as a model for TRAF3 signaling, as TRAF3 impacts receptor-associated phosphatase recruitment and activity via several distinct receptors (30, 35), including PTP1B in the present study. The impact of the LAT Q212A mutation on TRAF3 recruitment to, and function of, the TCR/CD28 complex is an area for future investigation.
The present study shows that TRAF3 regulated the LAT-associated negative regulatory molecule Dok1, by promoting its phosphorylation at Y362 and subsequent inactivation and proteasomal degradation. To date, studies of Dok1 have focused upon its role as a negative regulator of CD3 and the CD3-associated signaling molecule Zap70 (14, 40). Our results show that Dok1 plays a previously unrecognized role as a negative regulator of signaling more proximal to the LAT complex. TRAF3 often promotes K48-mediated ubiquitination leading to protein degradation by recruiting the TRAF2/cIAP complex to TRAF3-associated proteins (1, 41). However, we showed here that there was minimal difference in recruitment of TRAF2, and no recruitment of cIAP, to the LAT complex in TRAF3-sufficient versus -deficient T cells, indicating that the TRAF2/cIAP complex is highly unlikely to mediate the ubiquitination of Dok1 we observed.
TRAF3 promoted the inhibition of Dok1 by enhancing its interactions with the inhibitory kinase Brk (Fig. 5). Prior to our study, Brk was not well studied in lymphocytes; we show here for the first time that Brk associates with both TRAF3 and the LAT complex in T cells. Interestingly, Brk contains a predicted TRAF2/3 binding motif, as does PTP1B, which dephosphorylates Brk at its activating tyrosine (Y342) in the absence of TRAF3. Our data show that the association between Brk and PTP1B was enhanced, as was localization of PTP1B to the LAT complex, in TRAF3-deficient T cells. This raises the possibility that TRAF3 acts as a barrier between Brk and PTP1B, preventing Brk from being inactivated, by sequestering PTP1B from the LAT complex. It is also possible that interactions between TRAF3 and Brk alter intramolecular interactions within Brk (42-43), and promote phosphorylation of Y342, whereas Brk may be more likely to exist in an autoinhibited conformation in the absence of TRAF3, and/or is more easily targeted by the tyrosine kinase SRMS (32).Such molecular interactions may partially explain the increased phosphorylation at Y447 observed in TCS-401-treated TRAF3−/− T cells (Supp. Fig. 4C-D). It would be of interest in future work to determine if TRAF3 prevents Brk and PTP1B from physically interacting with each other, and/or if TRAF3 affects the phosphorylation status of PTP1B. Recent studies indicate that the stability of PTPN22, which TRAF3 also regulates to promote TCR/CD28 signaling, is enhanced upon Ser751 phosphorylation (44).
We previously demonstrated that TRAF3 does not detectibly alter the differentiation of CD4+ T cells into TH1 and TH2 subsets in vitro (3). However, TRAF3 does play a role in maintenance of memory CD8+ T cell populations (4). TRAF3 is also required for TCR signaling-mediated upregulation of Tbet, for normal IL-15 receptor function in developing iNKT cells, and for restraint of IL-2R signaling, which has implications for Treg differentiation (3-5, 35). TRAF3 impacts signaling through the TCR, cytokine receptors, and members of the TNFR superfamily, all of which play a role in T cell subset differentiation; the results of studies interrogating the impact of TRAF3 on T cell differentiation are thus complex to interpret, as discussed above (3, 5, 8). The role of TRAF3 in the differentiation of other T cell subsets, such as TFH, TH17, and TRM cells, is currently unknown and is an area of future interest.
Our study provides a mechanistic basis to explain how TRAF3 associates with and regulates T cell CD28 function, to ultimately promote early TCR complex signaling. Our results also introduce Brk and PTP1B as two new participants in the regulation of TCR/CD28 signaling via LAT. Fig. 7 presents a model depicting how we propose TRAF3 regulates the activity status of Dok1, and subsequently impacts TCR/CD28 signaling (Fig. 7). These results add new mechanisms of regulation, and additional signaling molecules, to our understanding of TCR/CD28-mediated activation, potentially identifying additional targets for manipulation of T cell functions.
Figure 7. Impact of T cell TRAF3 upon regulation of Dok1 activity and TCR/CD28 signaling via LAT.
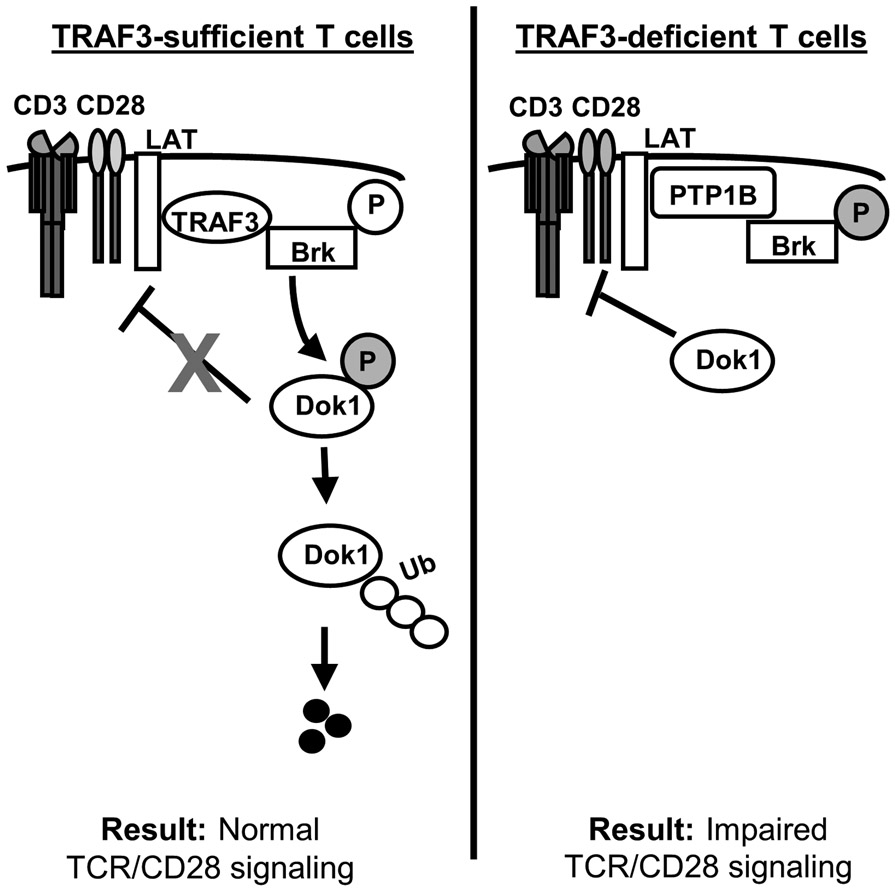
In TRAF3-sufficient T cells (left), TRAF3 associates with LAT and recruits Brk to the LAT complex. Brk activation via phosphorylation at Y342 is enhanced in the presence of TRAF3, allowing Brk to phosphorylate and inactivate Dok1. Phosphorylated Dok1 is ubiquitinated and targeted to the proteasome for degradation, and TCR/CD28 signaling proceeds normally.
In TRAF3-deficient T cells (right), the association between PTP1B and Brk is enhanced as a result of increased PTP1B localization to the plasma membrane, and Brk is inactivated via PTP1B-mediated dephosphorylation at Y342. In the absence of Brk-mediated inhibitory phosphorylation, active Dok1 facilitates the inhibition of LAT-mediated TCR/CD28 signaling. ‘P’ in open circles represents activating phosphorylation; ‘P’ in shaded circles represents inhibitory phosphorylation.
Supplementary Material
Key points:
TRAF3 is recruited to the TCR/CD28 complex and interacts with LAT.
TRAF3 enhances TCR/CD28 signaling and restrains the negative LAT regulator Dok1.
TCR-induced kinase activation in TRAF3−/− T cells is enhanced by inhibiting PTP1B.
Acknowledgements:
The authors acknowledge the statistical support of Sarah Bell at the Holden Comprehensive Cancer Center Biostatistics Core.
This study was supported by NIH R01 AI123107 to G.A.B. T.A. received support from NIH T32 AI007485 and T32 GM007337. This work was also supported by the Holden Comprehensive Cancer Center at The University of Iowa and its National Cancer Institute Award P30CA086862. This material is based upon work supported in part by facilities and equipment provided by the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development.
Abbreviations used in this article:
- Abs
Antibodies
- Brk (PTK6)
Breast tumor kinase
- IP
Immunoprecipitation
- iNKT cells
invariant NK T cells
- LAT
Linker of Activated T cells
- TCR
T cell receptor
- PTP1B
Protein tyrosine phosphatase 1B
- PTPN22
Protein tyrosine phosphatase 22
- SEM
Standard error of the mean
- TRAF3
TNF receptor-associated factor 3
- WB
Western blot
- WCL
Whole cell lysate
- WT
Wildtype
Footnotes
Competing interests: The authors declare that they have no competing interests.
The online version of this article contains supplemental material.
References:
- 1.Bishop GA 2016. TRAF3 as a powerful and multitalented regulator of lymphocyte functions. J Leukoc Biol. 100:919–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arkee T and Bishop GA. 2020. TRAF family molecules in T cells: Multiple receptors and functions. J Leukoc Biol. 107:907–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xie P, Kraus ZJ, Stunz LL., Liu Y, and Bishop GA. 2011. TNF receptor-associated factor 3 is required for T cell-mediated immunity and TCR/CD28 signaling. J Immunol. 186:143–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yi Z, Stunz LL, Lin WW, and Bishop GA. 2014. TRAF3 regulates homeostasis of CD8+ central memory T cells. PLoS One. 9:e102120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yi Z, Stunz LL, and Bishop GA. 2013. TRAF3 plays a key role in development and function of invariant natural killer T cells. J Exp Med. 210:1079–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morel PA Differential T-cell receptor signals for T helper cell programming. 2018. Immunology. 155:63–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bhattacharyya ND and Feng CG. 2020. Regulation of T helper cell fate by TCR signal strength. Front Immunol. 11:624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hwang JR, Byeon Y, Kim D, and Park SG. 2020. Recent insights of T cell receptor-mediated signaling pathways for T cell activation and development. Exp Mol Med. 52:750–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wallis AM, Wallace EC, Hostager BS, Yi Z, Houtman JCD, and Bishop GA. 2017. TRAF3 enhances TCR signaling by regulating the inhibitors Csk and PTPN22. Sci Rep. 7:2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Malbec O, Malissen M, Isnardi I, Lesourne R, Mura A, Fridman WH, Malissen B, and Daëron M. 2004. LAT integrates positive and negative signaling in mast cells. J Immunol. 173:5086–5094. [DOI] [PubMed] [Google Scholar]
- 11.Acuto O, Di Bartolo V, and Michel F. 2008. Tailoring T-cell receptor signals by proximal negative feedback mechanisms. Nat Rev Immunol. 8:699–712. [DOI] [PubMed] [Google Scholar]
- 12.Fuller DM and Zhang W. 2009. Regulation of lymphocyte development and activation by the LAT family of adapter proteins. Immunol Rev. 232:72–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bartelt RR and Houtman JC. 2013. The adaptor protein LAT serves as an integration node for signaling pathways that drive T cell activation. Wiley Interdiscip Rev Syst Biol Med. 5:101–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dong S, Corre B, Foulon E, Dufour E, Veillette A, Acuto O, and Michel F. 2006. TCR induces LAT-dependent activation of a negative signaling complex involving Dok-2, SHIP-1, and Grb-2. J Exp Med. 203:2509–2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yasuda T, Bundo K, Hino A., Honda K, Inoue A, Shirakata M, Osawa M, Tamura T, Nariuchi H, Oda H, Yamamoto T, and Yamanashi Y. 2007. Dok-1 and Dok-2 are negative regulators of TCR signaling. Int Immunol. 19:487–495. [DOI] [PubMed] [Google Scholar]
- 16.Kasprzycka M, Majewski M, Wang ZJ, Ptasznik A, Wysocka M, Zhang Q, Marzec M, Gimotty P, Crompton MR, and Wasik MA. 2006. Expression and oncogenic role of Brk (PTK6/Sik) protein tyrosine kinase in lymphocytes. Am J Pathol. 168:1631–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miah S, Goel RK, Dai C, Kalra N, Beaton-Brown E, Bagu ET, Bonham E, and Lukong KE. 2014. BRK targets Dok1 for ubiquitin-mediated proteasomal degradation to promote cell proliferation and migration. PLoS One. 9:e87684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wozniak DJ, Kajdacsy-Balla A, Macias V, Ball-Kell S, Zenner ML, Bie W, and Tyner AL. 2017. PTEN is a protein phosphatase that targets active PTK6 and inhibits PTK6 oncogenic signaling in prostate cancer. Nat Commun. 8:1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xie P, Stunz LL, Larison KD, Yang B, and Bishop GA. 2007. TRAF3 is a critical regulator of B cell homeostasis in secondary lymphoid organs. Immunity. 27:253–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Friend LD, Shah DD, Deppong C, Lin J, Bricker TL, Juehne TI, Rose CM, and Green JM. 2006. A dose-dependent requirement for the proline motif of CD28 in cellular and humoral immunity revealed by a targeted knockin mutant. J Exp Med. 203:2121–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dodson LF, Boomer JS, Deppong CM, Shah DD, Sim J, Bricker TL, Russell JH, and Green JM. 2009. Targeted knock-in mice expressing mutations of CD28 reveal an essential pathway for costimulation. Mol Cell Biol. 29:3710–3721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stein PH, Fraser JD, and Weiss A. 1994. The cytoplasmic domain of CD28 is both necessary and sufficient for costimulation of IL-2 secretion and association with phosphatidylinositol 3'-kinase. Mol Cell Biol. 14:3392–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ashwell JD, Longo DL, and Bridges SH. 1987. T-cell tumor elimination as a result of TCR-mediated activation. Science. 237:61–64. [DOI] [PubMed] [Google Scholar]
- 24.Hostager BS and Bishop GA. 1999. Cutting edge: contrasting roles of TRAF2 and TRAF3 in CD40-activated B lymphocyte differentiation. J Immunol. 162:6307–6311. [PubMed] [Google Scholar]
- 25.Ho SN, Hunt HD, Horton RM, Pullen JK, and Pease LR. 1989. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 77:51–59. [DOI] [PubMed] [Google Scholar]
- 26.Boomer JS and Green JM. 2010. An enigmatic tail of CD28 signaling. Cold Spring Harb Perspect Biol. 2:a002436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mambetsariev N, Lin WW, Stunz LL, Hanson BM, Hildebrand JM, and Bishop GA. 2016. Nuclear TRAF3 is a negative regulator of CREB in B cells. Proc Natl Acad Sci U S A. 113:1032–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hildebrand JM, Yi Z, Buchta CM, Poovassery J, Stunz LL, and Bishop GA. 2011. Roles of TRAF3 and TRAF5 in immune cell functions. Immunol Rev. 244:55–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kumar M, Gouw M, Michael S, Samano-Sanchez H, Pancsa R, Glavina J, Diakogianni A. Alvarado Valverde J, Bukirova D, Calyseva J, Palopoli N, Davey NE, Chemes LB, and Gibson TJ. 2020. ELM – the eukaryotic linear motif resource in 2020. Nucleic Acids Res. 48: D296–D306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wallis AM and Bishop GA. 2018. TRAF3 regulation of inhibitory signaling pathways in B and T lymphocytes by kinase and phosphatase localization. J Leukoc Biol. 103:1089–1098. [DOI] [PubMed] [Google Scholar]
- 31.Fan G, Lin G, Lucito R, and Tonks NK. 2013. PTP1B antagonized signaling by insulin-like growth factor-1 receptor and kinase BRK/PTK6 in ovarian cancer cells. J Biol Chem. 288:24923–24934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fan G, Aleem S, Yang M, Miller WT, and Tonks NK. 2015. Protein-tyrosine phosphatase and kinase specificity in regulation of SRC and Brk. J Biol Chem. 290:15934–15947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mahmoud KA, Krug M, Wersig T, Slynko I, Schachtele C, Totzke F, Sippl W, and Hilgeroth A. 2014. Discovery of 4-anilo α-carbolines as novel Brk inhibitors. Bioorg Med Chem Lett. 24:1948–1951. [DOI] [PubMed] [Google Scholar]
- 34.Du ZD, Hu LT, Zhao GQ, Li Y, and Ma ZZ. 2015. PTP1B regulates the activity of retinal pigment epithelial cells. Mol Vis. 21:523–531. [PMC free article] [PubMed] [Google Scholar]
- 35.Yi Z, Lin WW, Stunz LL, and Bishop GA. 2014. The adaptor TRAF3 restrains the lineage determination of thymic regulatory T cells by modulating signaling via the receptor for IL-2. Nat Immunol. 15(9):866–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vivar OI, Masi G, Carpier JM, Magalhaes JG, Galgano D, Pazour GJ, Amigorena S, Hivroz C, Baldari CT. 2016. IFT20 controls LAT recruitment to the immune synapse and T-cell activation in vivo. Proc Natl Acad Sci U S A. 113:386–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Balagopalan L, Yi J, Nguyen T, McIntire KM, Harned AS, Narayan K, and Samelson LE. 2018. Plasma membrane LAT activation precedes vesicular recruitment defining two phases of early T-cell activation. Nat Commun. 9:2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tremblay MM, Ollinger T, and Houtman JCD. 2020. The membrane proximal proline-rich region and correct order of C-terminal tyrosines on the adaptor protein LAT are required for TCR-mediated signaling and downstream functions. Cell Signal. 76:109790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shan X, Czar MJ, Bunnell SC, Liu P, Liu Y, Schwartzberg PL, and Wange RL. 2000. Deficiency of PTEN in Jurkat T cells causes constitutive localization of Itk to the plasma membrane and hyperresponsiveness to CD3 stimulation. Mol Cell Biol. 20:6945–6957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Davidson D, Zhong MC, Pandolfi PP, Bolland S, Xavier RJ, Seed B, Li X, Gu H, and Veillette A. 2016.The Csk-associated adaptor PAG inhibits effector T cell activation in cooperation with phosphatase PTPN22 and Dok adaptors. Cell Rep. 217:2776–2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shi JH and Sun SC. 2018. TRAF Regulation of NF-κB and MAPK pathways. Front Immunol. 9:1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qiu H and Miller WT. 2002. Regulation of the nonreceptor tyrosine kinase Brk by autophosphorylation and autoinhibition. J Biol Chem. 277:34634–34641. [DOI] [PubMed] [Google Scholar]
- 43.Qiu H and Miller WT. 2004. Role of the Brk SH3 domain in substrate recognition. Oncogene. 23:2216–2223. [DOI] [PubMed] [Google Scholar]
- 44.Yang S, Svensson MND, Harder NHO, Hsieh WC, Santelli E, Kiosses WB, Moresco JJ, Yates JR, King CC, Liu L, Stanford SM, and Bottini N. 2020. PTPN22 phosphorylation acts as a molecular rheostat for the inhibition of TCR signaling. Sci Signal. 13:eaaw8130. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.