

Abstract
Objective
Although Follistatin-like protein 1 (FSTL1), as an “adipokine”, is highly expressed in preadipocytes, the detail role of FSTL1 in adipogenesis and obesity remains not fully understood.
Methods
In vitro differentiation of both Fstl1−/− murine embryonic fibroblasts (MEFs) and stromal vascular fraction (SVF) were measured to assess the specific role of FSTL1 in adipose differentiation. Fstl1 adipocyte-specific knockout mice were generated to evaluate its role in obesity development. Gene expression analysis and phosphorylation patterns were performed to check out the molecular mechanism of the biological function of FSTL1.
Results
FSTL1 deficiency inhibited preadipocytes differentiation in vitro and obesity development in vivo. Glycosylation at N142 site was pivotal for the biological effect of FSTL1 during adipogenesis; the conversion between PPARγ and p-PPARγ was the key factor for the function of FSTL1. Molecular mechanism studies showed that FSTL1 functions through the integrin/FAK/ERK signaling pathway.
Conclusions
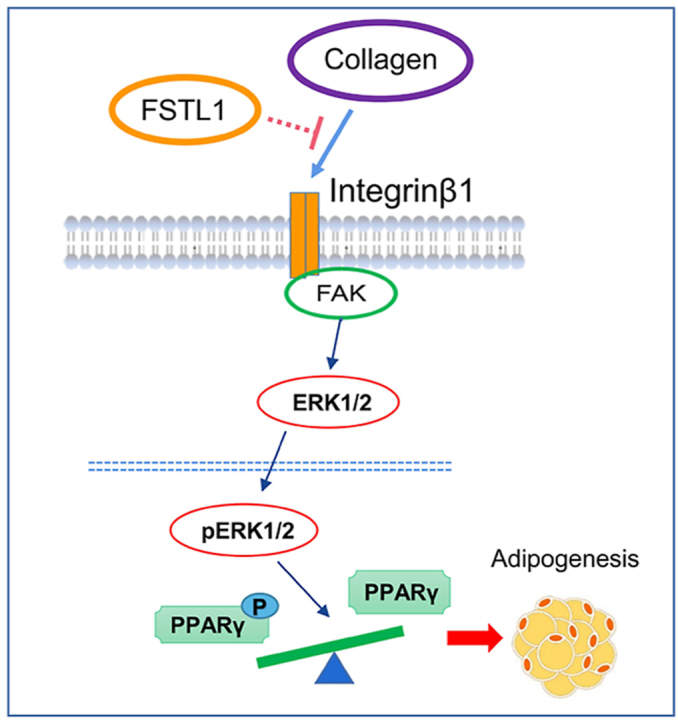
Our results suggest that FSTL1 promotes adipogenesis by inhibiting the conversion of PPARγ to p-PPARγ through the integrin/FAK/ERK signaling pathway. Glycosylated modification at N142 of FSTL1 is the key site to exert its biological effect.
Keywords: Obesity, Adipogenesis, FSTL1, Integrin/FAK/ERK, PPARγ/p-PPARγ
Graphical abstract
Highlights
-
•
FSTL1 is essential for adipogenic differentiation both in vitro and in vivo.
-
•
Glycosylated modification at Asp142 is sufficient for the biological function of FSTL1.
-
•
FSTL1 promotes adipogenesis by inhibiting the conversion of PPARγ to p-PPARγ.
-
•
FSTL1 inhibits PPARγ phosphorylation by disturbing the integrin-β1/FAK signaling pathway.
1. Introduction
Obesity has been recognized as one of the most serious public health problems, which is caused by the accumulation of white fat as a result of excessive energy intake. White adipose tissue (WAT) is the main “storage site” of excess energy, primarily in the form of triglycerides [1]. It has been reported that an increased adipocyte number (hyperplasia) and/or size (hypertrophy) contributes to development of obesity [[2], [3], [4]]. Study on exploring the mechanism of adipogenesis has great theoretical value and clinical significance for explaining the pathogenesis of obesity and combating obesity.
Adipogenesis is the process of mesenchymal stem cells (MSCs) undergo commitment to become preadipocytes and the program by which preadipocytes differentiate into adipocytes [5]. Members of the BMP and Wnt families are key mediators of stem cell commitment to produce preadipocytes [6,7]. During the maturing process, phosphorylation of the transcription factor CCATT enhancer binding protein β (C/EBPβ) triggers transcription of peroxisome proliferator–activated receptor-γ (PPARγ) and C/EBPα, which coordinately activate genes whose expression produces the adipocyte phenotype [8,9]. Among all transcription factors known to promote adipocyte development, none are as critical as PPARγ, which is considered the principal regulator of adipogenesis [[10], [11], [12]]. Phosphorylation of PPARγ was proved to inhibit PPARγ activity and that PPARγ-binding compounds that modulate serine phosphorylation within the PPARγ ligand-binding domain can be further developed as potential anti-obesity drugs [13,14].
Follistatin-like protein 1 (FSTL1), also known as Transforming growth factor-beta 1 (TGFβ)-induced clone 36 (TSC-36), is highly expressed during embryonic development, loss of FSTL1 can inhibit the development of respiratory system and tracheal chondrogenesis in fetal mice [[15], [16], [17]]. FSTL1 can also regulate the formation and differentiation of mesenchymal stem cells into cartilage, which is closely related to the occurrence and development of arthritis [18,19]. FSTL1 was found to express highly in adipose tissue and was considered as an “adipokine” [20,21]. During the differentiation of pre-adipocytes into adipocytes, Fstl1 showed a transient short high-level expression to become subsequently downregulated to the background level [22]. This suggests that FSTL1 may play a role in adipogenesis and obesity.
Glycosylated modification is a determinant of biological activity in cardiomyocytes, the glycosylated form promoting proliferation and the non-glycosylated working anti-apoptotic [23]. The glycosylation state shows differences between species and tissues, which might underlie the differences observed in in vitro studies. In the sequence of mouse FSTL1, three potential sites for N-glycosylation and two for O-glycosylation are present, and in vitro studies have shown that only the three aspartate residues Asp142, Asp173, and Asp178 are N-glycosylated [17]. A single N-Glycosylation site (N180) in human FSTL1 was shown to be sufficient to increase cardiomyocytes proliferation and cardiac regeneration [24].
Previous reports have shown that integrin-β1 regulates various biological functions, including cell migration and signal transduction, particularly in the context of cancer [25]. The activity of integrin-β1 is proposed to be impaired during adipogenesis. Adipocyte differentiation is regulated by the downstream signaling of integrin-β1 such as FAK and ERK [26,27]. FSTL1 was proposed as not only an antagonist of BMP4 but also an agonist of TGF-β1 signaling, which plays important roles during lung fibrosis. Deficiency of FSTL1 leads to reduced collagen accumulation, which is a key factor in lung injury [15,28]. Building on these reports, we proposed the hypothesis that FSTL1 may regulate adipogenesis through the integrin-β1/FAK signaling pathway.
In our previous study, we have shown that glycosylated FSTL1 secreted by brown adipocytes promotes the thermogenic program in BAT [29]; however, the expression pattern of FSTL1 is quite different in WAT and BAT. We planned to explore the role of FSTL1 in white fat, based on the current popular view that different shades of adipose tissue should be considered as different organs [30]. In this context, it is demonstrated that FSTL1 facilitates adipogenesis by inhibiting PPARγ phosphorylation which is regulated by the integrin-β1/FAK/ERK signaling pathway.
2. Materials and methods
2.1. Animal studies
Fstl1flox/+ mice of a mixed background (129Sv/C57BL/6J) were generated by Model Animal Research Center of Nanjing University; the details were shown in our previously study. Fstl1flox/+ mice were bred with EIIa-Cre and aP2(Fabp4)-Cre mice to generate Fstl1+/− mice and Fstl1ad KO mice. The primer set of Fstl1 is: forward, 5′-TCCCACCTTCGCCTCTAACT-3′; reverse, 5′-GAACTCTGCGGCTGCTCTG-3′. The primer set of aP2-Cre is: forward, 5′- ACCAGCCAGCTATCAACTCG-3’; reverse, 5′- TTACATTGGTCCAGCCACC-3’.
All animal experiments were performed in accordance with the Administration Regulations on Laboratory Animals of Beijing Municipality. All animals were maintained under 12 h/12 h light/dark cycle under constant conditions of room temperature (22 °C) and humidity with free access to food and water. For HFD feeding experiments, 6–8 weeks old male mice were fed with HFD (60 kcal%, Research Diets Inc. D12492) diets for up to 14 weeks. Mouse genotyping was performed using genomic DNA isolated from mouse tails.
2.2. Glucose tolerance test and insulin tolerance test
To determine glucose tolerance, 16-h-fasted mice were intraperitoneally administered with glucose (1 g/kg of body weight). To determine insulin sensitivity, 6-h-fasted mice were intraperitoneally administered with insulin (1 U/kg of body weight). Blood glucose level from tail vein blood was quantified by a NovaMax glucometer at the designated time after administration.
2.3. Cell cultures
For MEFs and SVF cell isolation, Fstl1+/− heterozygous mice were crossed to generate Fstl1−/− homozygous embryos. After the mice were pregnant for 13.5–14.5 days or 17.5–19.5 days, MEFs or SVF were isolated separately according to the instructions. MEFs were cultured in high glucose DMEM containing 10% FBS, 2 mM glutamine, 0.1 mM 2-mercaptoethanol, 50 U/mM penicillin, and 50 mg/mL streptomycin. Adipogenic differentiation was induced two days post confluence using DMEM/F12 Glutamax I, containing 10% fetal bovine serum (FBS) supplemented with 5 mg/mL insulin, 0.25 mmol/L dexamethasone, 0.5 mmol/L 3-isobutyl-1-methylxanthine (IBMX), and 10 mmol/L rosiglitazone for two days, after which the dexamethasone and IBMX were removed, and the cells were allowed to undergo full adipogenic differentiation.
For 3T3-L1 and SVF adipogenic differentiation, cells of 100% confluence were kept in growth medium for 2 days and induced with induction medium for 2 days, differentiation medium (without T3) for the following 6 days.
For PPARγ activation or P-ERK1/2 inhibition, rosiglitazone (5 μM) or U0126 (10 μM) was incubated with indicated durations as shown in figures. For dish coating, diluted Collagen Type I with 5 μg/cm2 was performed according to the instructions (Santa Cruz, sc-136157). Recombinant mouse FSTL1 protein was incubated with cells for 24 h (1738-FN, R&D Systems, 100 ng/ml).
2.4. Morphological study
For adipocytes Oil red O staining, cultured cells were washed with PBS and fixed with 10% formaldehyde for 15 min at room temperature. The cells were then stained using the Oil red O working solutions (5 g/L in isopropanol) and 4 ml ddH2O for 30 min. After staining, the cells were washed with 60% isopropanol and pictured. For adipose tissues, frozen sections were subjected with Oil red O solutions. The isolated adipose tissues were fixed in 4% formaldehyde/PBS and maintained at 4 °C until use. The fixed tissues were dehydrated and processed for paraffin embedding, and 4-mm sections were cut, followed by staining with hematoxylin-eosin.
2.5. RNA-sequencing, RNA interference and lentivirus production
To knock down FSTL1 in 3T3-L1 cells, lentiviral vector (hU6-MCS-Ubiquitin-EGFP-IRES-puromycin) containing the short-hairpin RNA (shRNA) specifically targeting FSTL1 or a negative control sequence was constructed (GeneChem, Shanghai, China). The sequences were as follows:
Fstl1-RNAi (55013-1) NM_008047 5′- AGAATGAAACAGCCATCAA -3′
Fstl1-RNAi (55014-1) NM_008047 5′- AGGTGAACACCAAAGAGAT -3′
Fstl1-RNAi (55015-1) NM_008047 5′- CTGCATTGAGCAATGCAAA -3′
Negative control RNAi: 5′- TTCTCCGAACGTGTCACGT -3′.
Primers for the glycosylation site mutation of N142, N173 and N178 were designed with Oligo7.0, asparagine (N) was mutated to glutamine (Q), lenti-virus were constructed by GeneChem corporation. The infection of virus was performed according to the manufacturer's instruction.
To overexpress FSTL1 in 3T3-L1 cells, GV141 vector (CMV-MCS-3FLAG-SV40-Neomycin) containing a FSTL1 coding sequence (NM_008047) was constructed (GeneChem, Shanghai, China). 3T3-L1 cells were incubated with 2 μg DNA and 6 μl Lipofectamine 3000 diluted in Opti-MEM for 48 h and then subjected to differentiation.
3T3-L1 cells with Fstl1 knockdown and control group were induced for differentiation with induction medium. After 2 days, total RNA was isolated using Trizol. RNA-sequencing and data sorting were performed by CapitalBio Technology.
2.6. Protein extraction and western blotting
Cells were lysed in the RIPA buffer with protease inhibitor and protein phosphatase inhibitor. Protein concentration was determined with BCA Protein assay kit (Thermo Scientific, Rockford, IL, USA), the amount of protein sample loading was 40–60 μg per lane. The primary antibodies were as follows: FSTL1 (1:2000, AF1738 R&D Systems), PPARγ (1:1000, #2443 Cell Signaling Technology), p-PPARγ (phospho-Ser112) (1:1000, LS-C416660 LifeSpan BioSciences), ERK1/2 (1:1000, #4695 Cell Signaling Technology), p-ERK1/2 (1:1000, #4370 Cell Signaling Technology), p-FAK (phospho Y397) (1:1000, ab81298), p-FAK (phospho Y576/577) (1:1000, #3281 Cell Signaling Technology), p-FAK (phospho Y925) (1:1000, #3284 Cell Signaling Technology), FAK (1:1000, #3285 Cell Signaling Technology), Flag (1:1000, #14793 Cell Signaling Technology), HA (1:1000, sc-7392 SANTA CRUZ), ITGB1 (1:1000, 26918-1-AP Proteintech), COLLAGEN1 (1:1000, ab270993), GAPDH (1:3000, #5174 Cell Signaling Technology).
2.7. Coimmunoprecipitation and GST pull-down assays
For coimmunoprecipitation of endogenous FSTL1, ITGB1, 293T cells were transfected with GV141 vector containing a Fstl1 coding sequence, GV417 vector containing a Itgb1 coding sequence (NM_010578). The whole experimental procedure was followed by instructions (26146; Thermo scientific). For immunoprecipitation, equal aliquots of tissue lysates were incubated with anti-FLAG antibody or anti-HA antibody overnight at 4 °C, normal rabbit IgG was used as control. Tissue lysates were then incubated with protein A/G-conjugated agarose beads at 4 °C for 4 h. Beads were washed with IP lysis/Wash Buffer three times, separately. Proteins were immunoblotted using antibodies as described above.
GST-fused constructs were expressed in BL21 Escherichia coli. In vitro transcription and translation experiments were done with rabbit reticulocyte lysate (TNT systems, Promega) according to the manufacturer's recommendation. For GST pull-down assays, about 5 mg of the appropriate GST fusion proteins with 30 ml of glutathione-Sepharose beads was incubated with 5–8 ml of in vitro transcribed/translated products in binding buffer (75 mM NaCl, 50 mM HEPES, pH 7.9) at 4 °C for 2 h in the presence of the protease inhibitor mixture. The beads were washed 5 times with binding buffer, resuspended in 30 ml of 2 × SDS-PAGE loading buffer, and detected by western blotting.
2.8. Real-time quantitative PCR analysis
Cellular mRNA was isolated using total RNA kit (DP419, TIANGEN). The concentration was measured with NanoDrop2000, cDNA synthesis with FastKing RT Kit (KR116, TIANGEN), PCR amplification was performed with SuperReal PreMix Plus (SYBR Green) Kit (FP205, TIANGEN). All the primer sequences are shown in Supplementary Table 1.
2.9. Reaction for protein deglycosylation
The reaction protocol for protein deglycosylation was followed according to the instructions of protein deglycosylation mix II kit (P6044; NEB). A total of 100 μg of glycoprotein was dissolved in 40 μl H2O. To the native glycoprotein, 5 μl 10x deglycosylation and mix buffer 1 were added. Next, 5 μl protein deglycosylation mix II was added, and the solution was mixed gently. The reaction mixture was incubated at 25 °C (room temperature) for 30 min. The reaction mixture was further incubated at 37 °C for 16 h. Proteins were immunoblotted using FSTL1 antibody to confirm the deglycosylation was effective. Brown adipocytes were incubated with supernatant, deglycosylation supernatant, or Deglycosylation Mix for 12 h.
2.10. Statistical analysis
All statistical analyses were performed using GraphPad Prism (v.5.0). Statistical significance was defined as ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.005, and determined by two-tailed Student's t tests (for comparison of two experimental conditions) or ANOVA (for comparison of three or more experimental conditions). Data are representative of at least three independent experiments.
3. Results
3.1. FSTL1 was identified as a potential adipogenic cytokine
To study the role of FSTL1 in adipogenic differentiation, the protein and mRNA levels of Fstl1 in different parts of adipose tissue of C57BL/6J mice were detected. FSTL1 protein was highly expressed and glycosylated in brown adipose tissue (BAT), and both glycosylated and non-glycosylated protein were detected in WAT, including inguinal white adipose tissue (iWAT), epididymal white adipose tissue (eWAT), and retroperitoneal white adipose tissue (rWAT) (Figure 1A). The mRNA level of Fstl1 was higher in WAT, but lower in BAT (Figure 1B). To further determine the effect of FSTL1 in white adipocyte formation, mouse embryo fibroblast (MEFs), stromal vascular fraction (SVF), and 3T3L1 were induced for differentiation. As the whole-body ablation Fstl1 homozygous mice died at birth, we obtained MEFs from Fstl1−/− embryos to study its function when FSTL1 is completely loss. Fstl1 protein and mRNA levels were initially upregulated after induction and then gradually reduced to basal level (Figure 1C–F). These results suggest that FSTL1 may play a role in adipocyte precursors differentiation.
Figure 1.

The expression of FSTL1 in preadipocytes differentiation and adipose tissues of mice. (A) The protein levels of FSTL1 in inguinal white adipose tissue (iWAT), epididymal white adipose tissue (eWAT), retroperitoneal white adipose tissue (rWAT) and brown adipose tissue (BAT) of C57BL/6J mice. 50 kDa, glycosylated; 37 kDa, hypoglycosylated. (B) mRNA levels of Fstl1 in iWAT, eWAT, rWAT, and BAT were detected by RT-PCR. (C) Time course of FSTL1 protein levels during MEFs differentiation detected by western blot. (D) Time course of Fstl1, Pparγ, and C/ebpα gene expression during MEF differentiation by RT-PCR. (E, F) Time course of FSTL1 protein levels during SVF and 3T3L1 differentiation.
3.2. FSTL1 promoted adipogenesis by inhibiting the conversion of PPARγ to p-PPARγ
Adipocyte formation is a process enclosing six specific orchestrated steps but is broadly divided into two phases. The first phase is the differentiation of mesenchymal precursors, which can differentiate into preadipocytes, while the second phase consists of the differentiation of preadipocytes into mature adipocytes [31]. As the whole-body ablation Fstl1 homozygous mice died at birth, we obtained MEFs from E13.5 and E18.5 Fstl1−/− embryos to study the detail role of FSTL1 in whole period of adipogenic differentiation. The genotypes (Fstl1+/+ [WT], Fstl1−/− [KO]) were verified, and no FSTL1 protein was detected in KO MEFs (Figure s1A, B). The proliferation of MEFs, SVF was almost equal between WT and KO group (Figure s1C, D). MEFs and SVF were then induced for differentiation into adipocytes. Oil Red O staining for lipids revealed that there was almost no lipid accumulation in KO MEFs compared to WT group (Figure 2A,D). Histomorphological observation of mouse embryo sections by HE and Oil red O staining showed that the development of KO embryo adipose tissue was defective, and just a little bit of lipid accumulation was shown (Figure s1F, G). During adipogenic differentiation, the PPARγ/p-PPARγ ratio was significantly lower in the KO group than in the control group on the 4th and 6th days (Figure 2B,E). The expression of adipogenic genes (Fabp4, C/ebpα, Adiponectin) was much lower in KO cells, while Pparγ gene expression was almost equal (Figure 2C,F).
Figure 2.

Fstl1 deletion inhibited the differentiation of MEFs and SVF by promoting PPARγ phosphorylation. (A) MEFs extracted from E13.5 wild-type (WT) and Fstl1−/− (KO) mouse embryos were induced to differentiate into adipocytes for 10 days and stained with Oil Red O. The oil red O stain was extracted and quantitated by measuring absorbance at 492 nm (n = 3), Scale bar, 200 μm 100 μm, 50 μm. (B) Western blot analysis of protein levels of PPARγ and p-PPARγ during MEF differentiation. PPARγ and p-PPARγ band intensities were normalized relative to the GAPDH bands, and the PPARγ/p-PPARγ ratio was calculated and analyzed. (C) Relative mRNA levels of adipogenic genes (Pparγ, Fabp4, C/ebpα, Adipoq) of MEFs (as in A) detected by qPCR. Two-way ANOVA followed by Bonferroni post-tests, n = 3. (D) SVF extracted from iWAT of E18.5 wild-type (WT) and Fstl1−/− (KO) mouse embryos were induced to differentiate into adipocytes and stained with Oil Red O. The oil red O stain was extracted and quantitated by measuring absorbance at 492 nm (n = 3), Scale bar, 200 μm 100 μm, 50 μm. (E) Western blot analysis of protein levels of PPARγ and p-PPARγ during SVF differentiation. PPARγ and p-PPARγ band intensities were normalized relative to the GAPDH bands, and the PPARγ/p-PPARγ ratio was calculated and analyzed. (F) Relative mRNA levels of adipogenic genes (Pparγ, Fabp4, C/ebpα, Adipoq) of SVF (as in D) detected by qPCR. Two-way ANOVA followed by Bonferroni post-tests, n = 3. Data are expressed as mean ± SEM, ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.005.
We carried out a series of knockdown and overexpression experiments with 3T3L1 cell line to check the role of FSTL1 in the adipogenic process. Knockdown and overexpression of FSTL1 in 3T3L1 were verified by Western blot and RT-PCR (Figure s2A–D). The proliferation of 3T3L1 was not altered by Fstl1 knockdown ((Figure s1E). Consistent with the results of previous study, Fstl1 deficiency inhibited lipid accumulation and reduced the PPARγ/p-PPARγ ratio and adipogenic genes expression (Figure 3A–C). On the contrary, Fstl1 overexpression promoted lipid accumulation, PPARγ/p-PPARγ ratio, and adipogenic genes expression (Figure 3D–F). Consistent with previous study, Pparγ gene expression was not altered by Fstl1 reconstruction (Figure s2E, F). Taken together, these loss-of-function and gain-of-function data demonstrate that FSTL1 is essential for inducing the differentiation of adipocytes by inhibiting the conversion of PPARγ to p-PPARγ.
Figure 3.

Differentiation of 3T3L1 cells was inhibited or promoted by Fstl1 knockdown or overexpression. (A) 3T3L1 fibroblasts expressing shFstl1 or vector control were differentiated into adipocytes and stained with Oil Red O at day 8. The oil red O stain was extracted and quantitated by measuring absorbance at 492 nm, Student's t test, n = 3. Scale bar, 100 μm. (B) The protein levels of PPARγ and p-PPARγ during differentiation of the 3T3L1-NC and 3T3L1-shFstl1 groups were detected by western blot. PPARγ and p-PPARγ band intensities were normalized relative to the GAPDH bands, and the PPARγ/p-PPARγ ratio was calculated and analyzed. (C) mRNA levels of adipogenic genes (Fabp4, C/ebpα, Adipoq) in 3T3L1 cells (as in A) were detected by qPCR. Two-way ANOVA followed by Bonferroni post-tests, n = 3. (D) 3T3L1 fibroblasts with Fstl1 overexpression or control were induced for differentiation using cocktail with no rosiglitazone, and stained with Oil Red O at day 8. The oil red O stain was extracted and quantitated by measuring absorbance at 492 nm, n = 3. Scale bar, 100 μm. (E) The protein levels of PPARγ and p-PPARγ during differentiation of 3T3L1 cells with Fstl1 OE and control groups were detected by western blot. The PPARγ/p-PPARγ ratio was analyzed as mentioned above. (F) mRNA levels of adipogenic genes in 3T3L1 cells (as in D) were detected by qPCR. Two-way ANOVA followed by Bonferroni post-tests, n = 3. Data are expressed as mean ± SEM, ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.005.
3.3. The biological activity of FSTL1 relied on N-glycosylated modification
It was reported that glycosylated or non-glycosylated FSTL1 showed different biological functions, suggesting that glycosylated modification is crucial for the FSTL1 protein [24]. To explore the role of glycosylation on the function of FSTL1 in the process of adipogenesis, we firstly isolated protein from 3T3L1 cells and supernatant. Western blot analysis showed that the molecular weight of FSTL1 in cells is 37kD, while the molecular weight of FSTL1 in supernatant is 50kD. When the medium was incubated with deglycosylation enzymes, the glycosylation on FSTL1 could be fully removed by the enzyme mix (Figure 4A). N-Glycosylation sites of FSTL1 were predicted by NetNGlyc 1.0 Server, and three sites, including N142, N173, N178, were verified. We constructed three mutants (N142Q, N173Q, N178Q) and transfected into 3T3L1 cells for adipogenic differentiation. The protein expression of FSTL1 was mostly unaltered, while the molecular weight of FSTL1 extracted from media of triple-mutant cells was obviously smaller than that of control (Figure 4B). When all the three sites were mutated totally, there was almost no lipid accumulation. A single site mutation of N178 resulted moderate decrease of lipid accumulation, and it is much less if N173 site was mutated, while N142 mutation resulted almost no lipid accumulation compared to WT control (Figure 4C). N142 mutation decreased adipogenic genes expression, and the PPARγ/p-PPARγ ratio compared to WT control (Figure 4D,E). These results suggest that N-glycosylation is necessary for the function of FSTL1 in adipogenesis, the mutation of a single site (N142Q) is sufficient.
Figure 4.

A single site mutation of N142 glycosylation was sufficient for the function of FSTL1. (A) Western blot analysis of FSTL1 protein extracted from 3T3L1 cells and supernatant (upper panel). Western blot analysis of FSTL1 protein extracted from supernatant or supernatant reacted with deglycosylation enzymes. (B) Western blot analysis of FSTL1 protein both in cell lysate and media of 3T3L1 with N-glycosylation mutants (N142, N173, N178). (C) Glycosylated modification sites (N142, N173, N178) were mutated separately or totally in 3T3L1 fibroblasts. Oil Red O staining was measured after inducing differentiation for 8 days. The oil red O stain was extracted and quantitated by measuring absorbance at 492 nm, n = 3. Scale bar, 100 μm. (D) mRNA levels of adipogenic genes (Fabp4, C/ebpα, Adipoq) of 3T3L1 cells with N142 site mutation after inducing differentiation for 8 days. Two-way ANOVA followed by Bonferroni post-tests, n = 3. (E) Relative protein levels of PPARγ and p-PPARγ during differentiation of 3T3L1 cells with N142 site mutation by western blot. PPARγ and p-PPARγ band intensities were normalized relative to the GAPDH bands, and the PPARγ/p-PPARγ ratio was calculated and analyzed. Data are expressed as mean ± SEM, ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.005.
3.4. FSTL1 attenuated the conversion of PPARγ to p-PPARγ by reducing ERK1/2 phosphorylation
It has been reported that the phosphorylation of PPARγ could be modified by p-ERK1/2 [32]. We tried to find out whether the effect of FSTL1 was also related to the phosphorylation of ERK1/2. The effectiveness of p-ERK1/2 inhibitor (U0126) was firstly confirmed at concentration of 10 μM (Figure 5A). Oil-red O staining showed that Fstl1 knockdown inhibited lipid accumulation of 3T3L1 cells as shown in previous studies, while it was rescued by p-ERK1/2 inhibition with U0126 (Figure 5B). Meanwhile, the down-regulated mRNA levels of adipogenic genes (Fabp4, C/ebpα, Adiponectin) and lipogenic genes (Acc, Fas) resulted by Fstl1 knockdown recovered by U0126 (Figure 5C). The conversion of PPARγ to p-PPARγ was also rescued by p-ERK1/2 inhibition (Figure 5D).
Figure 5.

The conversion of PPARγ to p-PPARγ was contributed by ERK1/2 phosphorylation when Fstl1 was deficient. (A) The protein levels of p-ERK1/2 and T-ERK1/2 in 3T3L1 cells treated with a p-ERK1/2 inhibitor (U0126) were determined by western blot. (B) Oil red O staining of the lipid droplets in three groups (Fstl1 knockdown, Fstl1 KD + U0126, control) of 3T3L1 cells after inducing differentiation for 8 days. Oil red O stain was quantified as mentioned above, One-way ANOVA followed by Dunnett's multiple comparison test. Scale bar, 100 μm. (C) mRNA levels of adipogenic (Fabp4, C/ebpα, Adipoq) and lipogenic genes (Acc, Fas) in 3T3L1 cells as in B. Two-way ANOVA followed by Bonferroni post-tests, n = 3. (D) Western blot analysis of protein levels of p-ERK1/2, T-ERK1/2, p-PPARγ, and PPARγ during differentiation of 3T3L1 cells as in B. p-ERK1/2, T-ERK1/2, p-PPARγ, and PPARγ band intensities were normalized relative to the GAPDH bands. p-ERK/T-ERK and PPARγ/p-PPARγ ratios were analyzed. Data are expressed as mean ± SEM, ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.005.
To further verify the molecular axis of FSTL1 by inhibiting ERK1/2 phosphorylation and downstream PPARγ phosphorylation. We attempted to investigate the effect of activated PPARγ on ERK phosphorylation; the effectiveness of PPARγ agonist rosiglitazone (RG) was confirmed at concentration of 5 μM, and p-ERK1/2 level was not altered by RG (Figure 6A). The inhibition of lipid droplet aggregation and expression of mRNA levels of adipogenic genes (Fabp4, C/ebpα, Adiponectin) and lipogenic genes (Acc, Fas) resulted by Fstl1 knockdown was rescued by RG (Figure 6B,C). The conversion of PPARγ to p-PPARγ was also rescued by PPARγ agonist, while the protein level of p-ERK1/2 was not disrupted (Figure 6D). These results suggest that FSTL1 maintains adipogenesis by inhibiting ERK1/2 phosphorylation and subsequently the PPARγ phosphorylation.
Figure 6.

Fstl1 deficiency promoted ERK1/2 phosphorylation and then the conversion of PPARγ to p-PPARγ. (A) Western blot analysis of protein levels of PPARγ, p-PPARγ, p-ERK1/2 and T-ERK1/2 in 3T3L1 cells treated with rosiglitazone at indicated concentrations. (B) Oil red O staining of lipid droplets in 3T3L1 cells of Fstl1 knockdown, Fstl1 KD + RG and control group after inducing differentiation for 8 days. The oil red O stain was extracted and quantitated by measuring absorbance at 492 nm, One-way ANOVA followed by Dunnett's multiple comparison test. Scale bar, 100 μm. (C) mRNA levels of adipogenic (Fabp4, C/ebpα, Adipoq) and lipogenic genes (Acc, Fas) in cells as mentioned in B. Two-way ANOVA followed by Bonferroni post-tests, n = 3. (D) Western blot analysis of protein levels of p-PPARγ, PPARγ, p-ERK1/2, and T-ERK1/2 during differentiation of 3T3L1 cells as in B. p-ERK1/2, T-ERK1/2, p-PPARγ, and PPARγ band intensities were normalized relative to the GAPDH bands, PPARγ/p-PPARγ and p-ERK/T-ERK ratios were analyzed. Data are expressed as mean ± SEM, ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.005.
3.5. FSTL1 inhibited ERK1/2 phosphorylation by disturbing the integrin-β1/FAK signaling pathway
As mentioned above, FSTL1 was glycosylated and secreted extracellular to promote adipogenesis. We attempted to determine the membrane receptor-mediated signaling pathway through which FSTL1 activates the intracellular response. We performed a gene expression analysis on 3T3L1-RNA of Fstl1 knockdown and control group after inducing differentiation (Figure s3 and s4). This analysis revealed more than 10 upregulated and 5 downregulated collagen-related genes in the differentiated 3T3L1 cells with Fstl1 knockdown compared with control (Figure 7A). A series of ECM-degradation genes, including matrix metalloproteinase and serine proteinase, were found to decrease in differentiated 3T3L1 cells with Fstl1 knockdown (Figure 7B). Gene ontology enrichment analysis revealed an overrepresentation of genes associated with integrin-ECM interaction, collagen formation, extracellular matrix organization, and focal adhesion (Figure 7C). However, no integrin-ECM associated pathway was enriched in the undifferentiated 3T3L1 cells with Fstl1 knockdown compared with control (Figure s5). On the basis of RNA-seq study, we hypothesized that FSTL1 might inhibit ERK phosphorylation by attenuating the integrin-β1 mediated signaling during preadipocytes differentiation. Western blot showed that the protein level of ITGB1 was increased in 3T3L1 cells with Fstl1 knockdown compared with control (Figure 7D). RNA-seq analysis indicated the potential effect of collagen for the function of FSTL1, and we coated the culturing dishes of 3T3L1 cells with Collagen Type I. Western blot showed that the protein levels of phosphorylated FAK (including s397, s576/577 and s925) and phosphorylated ERK1/2 were significantly increased in the Fstl1 knockdown group compared with the control group, while these proteins were decreased obviously in the Fstl1 overexpression group (Figure 7E,F). The relationship among FSTL1, COL1, and ITGB1 then needs to be clarified. To determine whether there is an interaction between FSTL1 and ITGB1, FSTL1 was coimmunoprecipitated with ITGB1 to form a complex by co-IP experiments (Figure 7G). Further, GST-pull down was performed to confirm that FSTL1 directly binds ITGB1 (Figure 7H) but not COL1 (Figure s2G). The binding of FSTL1 to ITGB1 diminished presentation of COL1 to the integrin-β1 receptor (Figure 7I). These data suggest that FSTL1 regulates the downstream FAK/ERK signaling pathway by attenuating the interaction between collagen and integrin-β1.
Figure 7.

FSTL1 exerted biological effects by inhibiting the integrin β1-mediated activation of the focal adhesion kinase pathway. (A, B) Heatmap of selected differential genes enrichment based on the RNA sequencing of the 3T3L1-NC and 3T3L1-shFstl1 groups after inducing for differentiation. (C) Gene Ontology analysis of differential genes in RNA-seq. (D) Western blot analysis of ITGB1 protein levels in the 3T3L1-NC and 3T3L1-shFstl1 groups after inducing for differentiation. GAPDH was used for loading control, ITGB1 band intensity was normalized relative to the GAPDH band. Student's t-test, ∗∗∗P < 0.005. (E, F) The culturing dishes were coated or not coated with Collagen Type I, and the protein levels of p-FAK (s397, s576/577, s925), FAK, p-ERK1/2 and T-ERK1/2 in 3T3L1 cells with Fstl1 knockdown (overexpression in F) and control group were analyzed by western blot. GAPDH was used for the loading control. (G) 293T cells were transfected with plasmids expressing Flag-tagged Fstl1 and HA-tagged Itgb1. Total cell lysate were subjected to IP with Flag or HA antibodies and immunoblotted for FSTL1 and ITGB1. Unspecific IgG antibodies were used as a control. (H) FSTL1 and ITGB1 protein complex was pulled down using glutathione-Sepharose beads and then subjected to western blot analyses with anti-FSTL1 antibody to confirm the presence of FSTL1 (upper). The presence of ITGB1 was detected with anti-ITGB1 antibody (lower). (I) 293T cells were transfected with HA-tagged Itgb1. The culturing dishes were coated or not coated with collagen Type I. ITGB1-HA was immunoprecipitated with HA-agarose and incubated with 100 ng FSTL1 protein. COL1 and ITGB1 were detected with anti-COL1 and anti-HA antibodies, respectively.
3.6. FSTL1 was required for HFD-induced obesity development
To evaluate the role of FSTL1 in obesity development, we generated adipocyte specific Fstl1 knockout mice using the aP2-Cre driver strain (Fstl1flox/flox × aP2-cre). Fstl1 gene expression was obviously lower in iWAT of f/f-aP2 mice than in the control group (Figure s6A). But the mRNA and protein levels of FSTL1 in SVF from iWAT were almost equal between the two groups (Figure s6B, C). Fstl1 deletion mice gained similar amounts of body weight and fat mass as the control group with equal amount of food intake under chow diet (CD) feeding (Figure s6D–F). The capability of glucose and insulin tolerance was also equal between the two groups (Figure s6G, H). During the switch from chow diet to high fat diet (HFD), Fstl1 deletion mice gained less amounts of body weight and fat mass (iWAT, eWAT) than control mice (Figure 8A–C). Intraperitoneal glucose tolerance test (IPGTT) showed that the glucose level of Fstl1 deletion mice was lower than that in the control group (Figure 8D). Smaller adipocytes were observed in iWAT and eWAT of Fstl1 deletion mice than that of control (Figure 8E,F). Western blot showed that the protein levels of phosphorylated FAK, ERK1/2 and PPARγ were increased in iWAT of Fstl1 deletion mice than control mice (Figure 8G). These data suggest that FSTL1 is necessary for obesity development in vivo.
Figure 8.

FSTL1 was required for obesity development in adipose knockout mice. (A) Adipocyte-specific Fstl1 mutant (f/f-aP2) and control (f/f) mice were fed high fat diet (HFD) for 14 weeks. Total body weight was determined weekly. Student's t-test, n = 9 for controls and n = 11 for KOs. (B) Weights of individual fat pads from f/f-aP2 and control mice following 14 weeks on HFD. n = 10 per group. (C) Representative images of wet fat pads of iWAT and eWAT from f/f-aP2 and f/f mice following 14 weeks on HFD. (D) Intraperitoneal glucose tolerance test (1.5 mg/kg) in male, HFD feeding f/f-aP2 and f/f mice. Two-way ANOVA followed by Bonferroni post-tests, n = 6 per group. (E, F) Cell size of iWAT and eWAT from male f/f-aP2 and f/f mice following 14 weeks on HFD. Representative images from hematoxylin and eosin (H&E) stained sections. Scale bar, 100 μm. Mean adipocyte area of the representative section from iWAT and eWAT was analyzed. Two-way ANOVA followed by Bonferroni post-tests, n = 3 per group. (G) The protein levels of FSTL1, p-PPARγ, PPARγ, p-ERK1/2, T-ERK1/2, p-FAK (s397), and FAK in iWAT of mice were analyzed by western blot. GAPDH was used for loading control. Data are expressed as mean ± SEM, ∗P < 0.05, ∗∗P < 0.01.
4. Discussion
Obesity has become a global pandemic, and it is closely associated with other metabolic diseases [33]. Because obesity is characterized by excessive accumulation of white adipose tissue, targeting adipogenesis represents a promising strategy to manage obesity [34]. Serum levels of FSTL1 were found to be higher in overweight and obese subjects than in controls, which indicates a potential pro-obesity effect [35,36]. The specific role and molecular mechanism of FSTL1 in adipogenesis have not been clarified. In this study, we carried out a series of Fstl1 knockdown and overexpression experiments in vitro and verified its function in promoting adipogenesis. In vivo studies, our adipose tissue-specific Fstl1 deletion mice indeed have defects in gaining body weight under HFD feeding. We comprehensively elucidated the role of FSTL1 in adipogenesis both in vitro and in vivo. During differentiation of pre-adipocytes into adipocytes, Fstl1 showed a transient short high-level expression to become subsequently downregulated to background levels at both the mRNA and protein level (Figure 1). We desired a Cre strain that expresses during the adipogenic process in animal studies. Adiponectin-cre lines are the best Cre strains used for targeting mature adipocytes, while the Pdgfrα-Cre lines are preferable for targeting adipocyte progenitors in vivo. Ap2-Cre mediated recombination occurs not only in mature adipocytes, but also in adipocytes progenitors [37]. We chose Ap2-cre in our animal model, although it was studied to be expressed not only in adipose tissue but some other cell types. However, we found that FSTL1 was not efficiently deleted in adipocyte precursor cells of KO mice. The deficiency of FSTL1 in the KO mice was to inhibit the size of adipocytes (hypertrophy) rather than the number (hyperplasia) of precursor cells. Compared to the control group, the KO mice show an improved metabolic profile when fed a high fat diet. For further studies, Adiponectin-cre and Pdgfra-Cre should be considered for using in our animal model.
It was reported that non-glycosylated FSTL1 induced cardiomyocytes proliferation, whereas the glycosylated protein working anti-apoptotic [23]. This indicates that glycosylated modification is a determination for the function of FSTL1. In our previous studies, we identified that glycosylated modification was essential for FSTL1 in promoting BAT thermogenesis during cold exposure or β3-adrenergic signaling activation [29]. In this study, we confirmed that N-glycosylation is necessary for FSTL1 in promoting white adipogenic differentiation. We performed N-glycosylation site mutation and found that Asp142 modification was of great significance for the function of FSTL1. Our previous reports and the current study together reveal that N-glycosylation is necessary for the biological effect of FSTL1 in both BAT and WAT. It is well known that WAT acts to store energy in the form of triglycerides, whereas BAT dissipates heat against cold. FSTL1 is essential for both WAT and BAT formation through our studies. Upon cold exposure, BAT secretes more FSTL1 to promote thermogenesis of itself. During HFD-induced obesity development, FSTL1 is necessary for WAT hypertrophy. It is complicated in vivo studies, and FSTL1 is secreted by many tissues and organs. More types of conditional knockout mice should be measured to further illustrate the role of FSTL1 in obesity development.
Peroxisome proliferator-activated receptor γ (PPARγ) is a key regulatory factor for adipogenesis and obesity [9]. Phosphorylated modification was proved to inhibit the activity of PPARγ, and thus the process of adipogenesis. PPARγ phosphorylation was found to increase in the obese state [13]. Treatment of mice with classical and non-classical thiazolidinediones (TZDs), such as rosiglitazone, prevents PPARγ phosphorylation and inducing expression of a constituent lipid gene [38]. The efficacy of synthetic ligands of PPARγ in controlling blood glucose has been widely recognized. However, their clinical usage is limited due to side effects, including weight gain, fluid retention, bone fracture, cardiovascular disease, and bladder cancer [39]. Many recent studies have focused on the post-translational modifications of PPARγ, such as phosphorylation, acetylation, ubiquitination et,al. In our study, we find that FSTL1 inhibits the conversion of PPARγ to p-PPARγ, which provides a potential therapeutic target in combating obesity.
FSTL1 was involved in multiple signaling pathways; binding to membrane receptors has been shown, but other molecular partners probably exist [[40], [41], [42], [43]]. In our study, we found that FSTL1 inhibited PPARγ phosphorylation to promote adipogenesis. It was reported that the phosphorylation of PPARγ could be modified by CDK5, p-ERK1/2 and other cytokines [26,32]. We proved that FSTL1 inhibited the conversion of PPARγ to p-PPARγ by reducing ERK1/2 phosphorylation. We were then curious about the signaling pathway through which FSTL1 regulates ERK1/2 phosphorylation. Our gene expression analysis showed that the organization of extracellular matrix (collagen, tenascin, MMP et al.) was disrupted when FSTL1 was lost, and the signaling pathways associated with ECM-receptor interaction were enriched. It was reported that the downstream signaling of integrin-β1 such as FAK and ERK is involved in adipocyte differentiation [44]. We finally concluded that the role of FSTL1 in the late stage of adipogenic differentiation was related with FAK/ERK signaling pathway; however, its role in the phase of mitotic clonal expansion prior to differentiation and the phase of determination was possibly otherwise related. Here, our study demonstrates the conversion between PPARγ and p-PPARγ as a potential downstream target of FSTL1, and we propose a new mechanism of FSTL1 as a component of extracellular matrix. We have proven that FSTL1 directly binds with ITGB1, but how FSTL1 inhibits the binding between collagen and integrin-β1 is still unclear in this research. Previous reports have shown that integrin-β1 regulates various biological functions, including cell migration and signal transduction, particularly in the context of cancer [[45], [46], [47]]. It will also be interesting to determine whether morphofunctional modifications of adipocytes are involved in the action of FSTL1 in promoting adipogenesis.
Together, our studies reveal that FSTL1 is an important adipokine in promoting adipogenesis and obesity development. N-glycosylation is necessary for the function of FSTL1 in adipogenesis, and modification at Asp142 is of great significance. FSTL1 promotes adipogenesis by disrupting the interaction between collagen and integrin-β1, which caused the inhibitory effect of FAK and ERK1/2 phosphorylation, and then the PPARγ phosphorylation. Thus, PPARγ phosphorylation regulated by FSTL1 might prove to be a novel target in adipogenesis and obesity.
Author contributions
Y.G. and D.L.F. conceived the project and designed research. D.L.F. and Y.X.S performed the experiments. X.W.J., Ch.Y., and T.L. provided help in tissue processing and assistance. L.L.W., L.L., and B.P.D. helped with cell culture studies. L.S. helped with RNA-seq and data analysis. D.L.F. and Y.G. analyzed the data and wrote the original draft.
Acknowledgments
We thank Dr. H.B.R. from University of Minnesota for providing counseling on this study and final editing of the manuscript. The present study was supported by the Beijing Natural Science Foundation of China (grant no. 5214022; 5202004).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.molmet.2021.101400.
Conflict of interest
The authors declare no conflicts of interest.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
multimedia component 2
multimedia component 3
multimedia component 4
multimedia component 5
multimedia component 6
multimedia component 7
Figure s1.

Figure s2.

Figure s3.

Figure s4.

Figure s5.

Figure s6.

References
- 1.Blüher M. Obesity: global epidemiology and pathogenesis. Nature Reviews Endocrinology. 2019;15(5):288–298. doi: 10.1038/s41574-019-0176-8. [DOI] [PubMed] [Google Scholar]
- 2.Cinti S. Adipose organ development and remodeling. Comprehensive Physiology. 2018;8(4):1357. doi: 10.1002/cphy.c170042. [DOI] [PubMed] [Google Scholar]
- 3.Petrus P., Mejhert N., Corrales P., Lecoutre S., Li Q., Maldonado E., et al. Transforming growth factor-β3 regulates adipocyte number in subcutaneous white adipose tissue. Cell Reports. 2018;25(3):551–560. doi: 10.1016/j.celrep.2018.09.069. e5. [DOI] [PubMed] [Google Scholar]
- 4.Hepler C., Gupta R.K. The expanding problem of adipose depot remodeling and postnatal adipocyte progenitor recruitment. Molecular and Cellular Endocrinology. 2017;445:95–108. doi: 10.1016/j.mce.2016.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sanchez-Gurmaches J., Guertin D.A. Adipocytes arise from multiple lineages that are heterogeneously and dynamically distributed. Nature Communications. 2014;5(1) doi: 10.1038/ncomms5099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Modica S., Straub L.G., Balaz M., Sun W., Varga L., Stefanicka P., et al. Bmp4 promotes a Brown to white-like adipocyte shift. Cell Reports. 2016;16(8):2243–2258. doi: 10.1016/j.celrep.2016.07.048. [DOI] [PubMed] [Google Scholar]
- 7.Chen N., Wang J. Wnt/β-Catenin signaling and obesity. Frontiers in Physiology. 2018;9 doi: 10.3389/fphys.2018.00792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen Z., Torrens J.I., Anand A., Spiegelman B.M., Friedman J.M. Krox20 stimulates adipogenesis via C/EBPβ-dependent and -independent mechanisms. Cell Metabolism. 2005;1(2):93–106. doi: 10.1016/j.cmet.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 9.Lee J., Schmidt H., Lai B., Ge K. Transcriptional and epigenomic regulation of adipogenesis. Molecular and Cellular Biology. 2019;39(11) doi: 10.1128/MCB.00601-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wagner R., Hieronimus A., Lamprinou A., Heni M., Hatziagelaki E., Ullrich S., et al. Peroxisome proliferator-activated receptor gamma (PPARG) modulates free fatty acid receptor 1 (FFAR1) dependent insulin secretion in humans. Molecular Metabolism. 2014;3(6):676–680. doi: 10.1016/j.molmet.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mayoral R., Osborn O., McNelis J., Johnson A.M., Oh D.Y., Izquierdo C.L., et al. Adipocyte SIRT1 knockout promotes PPARγ activity, adipogenesis and insulin sensitivity in chronic-HFD and obesity. Molecular Metabolism. 2015;4(5):378–391. doi: 10.1016/j.molmet.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lasar D., Rosenwald M., Kiehlmann E., Balaz M., Tall B., Opitz L., et al. Peroxisome proliferator activated receptor gamma controls mature Brown adipocyte inducibility through glycerol kinase. Cell Reports. 2018;22(3):760–773. doi: 10.1016/j.celrep.2017.12.067. [DOI] [PubMed] [Google Scholar]
- 13.Choi J.H., Banks A.S., Estall J.L., Kajimura S., Boström P., Laznik D., et al. Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARγ by Cdk5. Nature. 2010;466(7305):451–456. doi: 10.1038/nature09291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paula Mota De Sa A.J.R.H. Transcriptional regulation of adipogenesis. Comprehensive Physiology. 2017;7:635–674. doi: 10.1002/cphy.c160022. [DOI] [PubMed] [Google Scholar]
- 15.Geng Y., Dong Y., Yu M., Zhang L., Yan X., Sun J., et al. Follistatin-like 1 (Fstl1) is a bone morphogenetic protein (BMP) 4 signaling antagonist in controlling mouse lung development. Proceedings of the National Academy of Sciences. 2011;108(17):7058–7063. doi: 10.1073/pnas.1007293108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu X., Liu Y., Li X., Zhao J., Geng Y., Ning W. Follistatin like-1 (Fstl1) is required for the normal formation of lung airway and vascular smooth muscle at birth. PLoS One. 2017;12(6) doi: 10.1371/journal.pone.0177899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hambrock H.O., Kaufmann B., Müller S., Hanisch F., Nose K., Paulsson M., et al. Structural characterization of TSC-36/flik. Journal of Biological Chemistry. 2004;279(12):11727–11735. doi: 10.1074/jbc.M309318200. [DOI] [PubMed] [Google Scholar]
- 18.Li D., Wang Y., Xu N., Wei Q., Wu M., Li X., et al. Follistatin-like protein 1 is elevated in systemic autoimmune diseases and correlated with disease activity in patients with rheumatoid arthritis. Arthritis Research & Therapy. 2011;13(1):R17. doi: 10.1186/ar3241. R17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Y., Li D., Xu N., Tao W., Zhu R., Sun R., et al. Follistatin-like protein 1: a serum biochemical marker reflecting the severity of joint damage in patients with osteoarthritis. Arthritis Research & Therapy. 2011;13(6):R193. doi: 10.1186/ar3522. R193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Raschke S., Eckel J. Adipo-Myokines: two sides of the same coin—mediators of inflammation and mediators of exercise. Mediators of Inflammation. 2013;2013:1–16. doi: 10.1155/2013/320724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rodríguez A., Becerril S., Ezquerro S., Méndez-Giménez L., Frühbeck G. Crosstalk between adipokines and myokines in fat browning. Acta Physiologica. 2017;219(2):362–381. doi: 10.1111/apha.12686. [DOI] [PubMed] [Google Scholar]
- 22.Wu Y., Zhou S., Smas C.M. Downregulated expression of the secreted glycoprotein follistatin-like 1 (Fstl1) is a robust hallmark of preadipocyte to adipocyte conversion. Mechanisms of Development. 2010;127(3–4):183–202. doi: 10.1016/j.mod.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wei K., Serpooshan V., Hurtado C., Diez-Cuñado M., Zhao M., Maruyama S., et al. Epicardial FSTL1 reconstitution regenerates the adult mammalian heart. Nature. 2015;525(7570):479–485. doi: 10.1038/nature15372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Magadum A., Singh N., Kurian A.A., Sharkar M.T.K., Chepurko E., Zangi L. Ablation of a single N-glycosylation site in human FSTL 1 induces cardiomyocyte proliferation and cardiac regeneration. Molecular Therapy - Nucleic Acids. 2018;13:133–143. doi: 10.1016/j.omtn.2018.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Howe E.N., Burnette M.D., Justice M.E., Schnepp P.M., Hedrick V., Clancy J.W., et al. Rab11b-mediated integrin recycling promotes brain metastatic adaptation and outgrowth. Nature Communications. 2020;11(1) doi: 10.1038/s41467-020-16832-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fuentes P., Acuña M.J., Cifuentes M., Rojas C.V. The anti-adipogenic effect of angiotensin II on human preadipose cells involves ERK1,2 activation and PPARG phosphorylation. Journal of Endocrinology. 2010;206(1):75–83. doi: 10.1677/JOE-10-0049. [DOI] [PubMed] [Google Scholar]
- 27.Ruiz-Ojeda F.J., Wang J., Bäcker T., Krueger M., Zamani S., Rosowski S., et al. Active integrins regulate white adipose tissue insulin sensitivity and brown fat thermogenesis. Molecular Metabolism. 2021;45:101147. doi: 10.1016/j.molmet.2020.101147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dong Y., Geng Y., Li L., Li X., Yan X., Fang Y., et al. Blocking follistatin-like 1 attenuates bleomycin-induced pulmonary fibrosis in mice. Journal of Experimental Medicine. 2015;212(2):235–252. doi: 10.1084/jem.20121878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fang D., Shi X., Lu T., Ruan H., Gao Y. The glycoprotein follistatin-like 1 promotes brown adipose thermogenesis. Metabolism. 2019;98:16–26. doi: 10.1016/j.metabol.2019.05.008. [DOI] [PubMed] [Google Scholar]
- 30.Peirce V., Carobbio S., Vidal-Puig A. The different shades of fat. Nature. 2014;510(7503):76–83. doi: 10.1038/nature13477. [DOI] [PubMed] [Google Scholar]
- 31.Rosen E.D., MacDougald O.A. Adipocyte differentiation from the inside out. Nature Reviews Molecular Cell Biology. 2006;7(12):885–896. doi: 10.1038/nrm2066. [DOI] [PubMed] [Google Scholar]
- 32.Banks A.S., McAllister F.E., Camporez J.P.G., Zushin P.H., Jurczak M.J., Laznik-Bogoslavski D., et al. An ERK/Cdk5 axis controls the diabetogenic actions of PPARγ. Nature. 2015;517(7534):391–395. doi: 10.1038/nature13887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sung H., Siegel R.L., Torre L.A., Pearson Stuttard J., Islami F., Fedewa S.A., et al. Global patterns in excess body weight and the associated cancer burden. CA: A Cancer Journal for Clinicians. 2018;69:88–112. doi: 10.3322/caac.21499. [DOI] [PubMed] [Google Scholar]
- 34.Ghaben A.L., Scherer P.E. Adipogenesis and metabolic health. Nature Reviews. Molecular Cell Biology. 2019;20(4):242–258. doi: 10.1038/s41580-018-0093-z. [DOI] [PubMed] [Google Scholar]
- 35.Fan N., Sun H., Wang Y., Wang Y., Zhang L., Xia Z., et al. Follistatin-like 1: a potential mediator of inflammation in obesity. Mediators of Inflammation. 2013;2013:1–12. doi: 10.1155/2013/752519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee S.Y., Kim D.Y., Kyung K.M., Hee A.S., Kim H., Kim B.J., et al. High circulating follistatin-like protein 1 as a biomarker of a metabolically unhealthy state. Endocrine Journal. 2019;66(3):241–251. doi: 10.1507/endocrj.EJ18-0352. [DOI] [PubMed] [Google Scholar]
- 37.Liu J., Xu Z., Wu W., Wang Y., Shan T. Cre recombinase strains used for the study of adipose tissues and adipocyte progenitors. Journal of Cellular Physiology. 2017;232(10):2698–2703. doi: 10.1002/jcp.25675. [DOI] [PubMed] [Google Scholar]
- 38.Choi J.H., Choi S., Kim E.S., Jedrychowski M.P., Yang Y.R., Jang H., et al. Thrap3 docks on phosphoserine 273 of PPARγ and controls diabetic gene programming. Genes & Development. 2014;28(21):2361–2369. doi: 10.1101/gad.249367.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ahmadian M., Suh J.M., Hah N., Liddle C., Atkins A.R., Downes M., et al. PPARγ signaling and metabolism: the good, the bad and the future. Nature Medicine. 2013;19(5):557–566. doi: 10.1038/nm.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Benjamin E.J., Blaha M.J., Chiuve S.E., Cushman M., Das S.R., Deo R., et al. Heart disease and stroke statistics—2017 update: a report from the American heart association. Circulation. 2017;135(10) doi: 10.1161/CIR.0000000000000485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maruyama S., Nakamura K., Papanicolaou K.N., Sano S., Shimizu I., Asaumi Y., et al. Follistatin-like 1 promotes cardiac fibroblast activation and protects the heart from rupture. EMBO Molecular Medicine. 2016;8(8):949–966. doi: 10.15252/emmm.201506151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2019. CA. A Cancer Journal for Clinicians. 2019;69(1):7–34. doi: 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- 43.Sundaram G.M., Ismail H.M., Bashir M., Muhuri M., Vaz C., Nama S., et al. EGF hijacks miR-198/FSTL1 wound-healing switch and steers a two-pronged pathway toward metastasis. Journal of Experimental Medicine. 2017;214(10):2889–2900. doi: 10.1084/jem.20170354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kaburagi T., Kizuka Y., Kitazume S., Taniguchi N. The inhibitory role of α2,6-sialylation in adipogenesis. Journal of Biological Chemistry. 2017;292(6):2278–2286. doi: 10.1074/jbc.M116.747667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sulzmaier F.J., Jean C., Schlaepfer D.D. FAK in cancer: mechanistic findings and clinical applications. Nature Reviews Cancer. 2014;14(9):598–610. doi: 10.1038/nrc3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cooper J., Giancotti F.G. Integrin signaling in cancer: mechanotransduction, stemness, epithelial plasticity, and therapeutic resistance. Cancer Cell. 2019;35(3):347–367. doi: 10.1016/j.ccell.2019.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhou J., Yi Q., Tang L. The roles of nuclear focal adhesion kinase (FAK) on Cancer: a focused review. Journal of Experimental & Clinical Cancer Research. 2019;38(1) doi: 10.1186/s13046-019-1265-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
multimedia component 2
multimedia component 3
multimedia component 4
multimedia component 5
multimedia component 6
multimedia component 7