

Abstract
Background
Thalassemia is an inherited hematological disorder categorized by a decrease or absence of one or more of the globin chains synthesis. Beta‐thalassemia is caused by one or more mutations in the beta‐globin gene. The absence or reduced amount of beta‐globin chains causes ineffective erythropoiesis which leads to anemia.
Methods
Beta‐thalassemia has been further divided into three main forms: thalassemia major, intermedia, and minor/silent carrier. A more severe form among these is thalassemia major in which individuals depend upon blood transfusion for survival. The high level of iron deposition occurs due to regular blood transfusion therapy.
Results
Overloaded iron raises the synthesis of reactive oxygen species (ROS) that are noxious and prompting the injury to the hepatic, endocrine, and vascular system. Thalassemia can be analyzed and diagnosed via prenatal testing (genetic testing of amniotic fluid), blood smear, complete blood count, and DNA analysis (genetic testing). Treatment of thalassemia intermediate is symptomatic; however; it can also be accomplished by folic supplementation and splenectomy.
Conclusion
Thalassemia major can be cured through regular transfusion of blood, transplantation of bone marrow, iron chelation management, hematopoietic stem cell transplantation, stimulation of fetal hemoglobin production, and gene therapy.
Keywords: blood transfusion, chelation therapy, gene therapy, hemoglobin, iron overload, reactive oxygen species, splenectomy, thalassemia
1. INTRODUCTION
Thalassemia is categorized by abnormal production or reduction in the rate of formation of normal α‐ or β‐globin subunits of hemoglobin (Hb) A. The genes responsible for making β‐globin are positioned on chromosome 11 while α‐globin genes are found on chromosome 16 (Adly et al., 2015). The Hb is a protein that is present in red blood cells which is accountable for carrying the oxygen from alveolus to tissues. Three forms of Hbs are found in normal adults such as HbA, HbA2, and HbF that consist of α2; β2, α2; δ2, and α2; γ2 subunits, respectively, as shown in Figure 1 (Thein, 2018).
FIGURE 1.
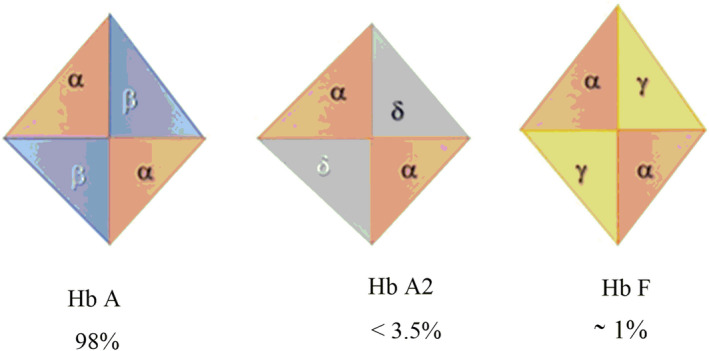
Types of hemoglobin in the normal adults
Thalassemia is categorized as β, α, δ γ, δβ, as well as γδβ, depending upon which globin chain is affected. The α‐ and β‐thalassemia are two major categories and their occurrence depends on four and two genes, respectively (Gibbs & Burdick, 2009; Sirachainan et al., 2016). It is produced by more than numerous hundred modifications in the consistent DNA segment. The unpaired globin chains are not stable. They are precipitous in cells which lead to immature destruction of precursors of RBCs and shortening of life span of mature RBCs in the blood. The Hb breaks down into iron and heme that catalyze chemical reactions in which free radicals or reactive oxygen species (ROS) are produce. These radicals and ROS cause the impairment of hepatocytes and functions of islets of Langerhans (Adly et al., 2015).
2. BETA‐THALASSEMIA
Beta‐thalassemia is described by the absence or reduction in the rate of production of the β‐globin chain (Galanello & Origa, 2010). It was the first time defined by Cooley and Lee in 1925 (Franco et al., 2014). The β‐thalassemia is a consequence of substitutions of bases on introns, exons as well as on the promoter regions of β‐globin genes while α‐thalassemia is a consequence of deletions that remove α gene (Stauder et al., 2018). It is further categorized according to decreased (β+) or absent (β0) globin chain production which might lead to microcytic and hypochromic anemia as well as a wide range of syndromicforms (Lei et al., 2019).
2.1. Types of beta‐thalassemia
2.1.1. Beta‐thalassemia major
It is the most severe type of thalassemia which is known as Cooley's anemia that occurs either when individuals are homozygous (B+/B0, B0/B0) or compounds heterozygous (B+/B+) for more severe mutations in β chain (Galanello & Origa, 2010; Tari et al., 2018). It usually induces between 6 months and 2 years. In major thalassemia, patients undergo severe anemia (heart failure, fatigue, and cachexia). The level of Hb might be <7 g/dl and Hb F <90%. The reduction in Hb resulted bone marrow expansion to compensate the loss of RBCs which led to bone abnormalities, enlargement of spleen and restriction of growth. The extreme hemolysis leads to pulmonary hypertension, lithiasis, and formation of the leg ulcer. Furthermore, hypercoagulability is also an impediment to this disorder. Regular management with transfusions of blood or blood products might be overload the iron in various organs which result in diabetes, hypopituitarism complications in the liver and endocrine glands such as hypothyroidism, hypopituitarism, hypoparathyroidism, dark metallic pigmentation of the skin, cirrhosis, cardiac arrhythmia, and myopathy which can lead to 71% death of patients who have thalassemia major (Leiet al., 2019). Other complications are HIV infection like prolonged hepatitis B and C, osteoporosis, and occlusion in blood. Furthermore, the patients with liver infection have a high risk of liver cancers (Borgna‐Pignatti et al., 2004).
2.1.2. Beta‐thalassemia intermedia
Thalassemia intermediate is a heterogeneous genetic mutation in which individuals have a little bit ability for the production of β chain of Hb (B+/B+, B+/B0). In some situations, both α and β mutations present simultaneously (Galanello & Origa, 2010). It occurs between 2 and 6 years of age. Thalassemia intermediate has milder anemia. In this case, level of Hb varies between 7 and 9–10 g/dl and transfusion of blood is not needed. The sufferer can survive without or only occasionally require blood transfusion as presented in Table 1 (Birgens & Ljung, 2007). When bone marrow expands with age, several complications like growth retardation, bone abnormalities, and infertility may develop in patients. On the other hand, the hemolysis raise the level of iron in different tissues (Taher et al., 2011).
TABLE 1.
Beta‐thalassemia genotypes, clinical features/laboratory features
| β Thalassemia | Globins Chain | β Gene | Clinical features | Laboratory features | References |
|---|---|---|---|---|---|
|
Thalassemia Major (Usually at 4–6 months or child younger than 2 years) |
α2 β2, α2 δ2, α2 γ2 α2 δ2, α2 γ2 |
β+/β+ β0/β0 |
Anemia Hepatosplenomegaly Growth retardation Require chronic Transfusion, iron overload |
Hb<7g/dl Hb F <90% HbA2 normal or high HbA absent |
Lei et al., (2019) |
|
Thalassemia Intermediate (presentation at later age) |
α 2 β2, α 2 δ2, α 2 γ2 |
β+/β0 β+/β+ |
Milder anemia hepatosplenomegaly |
Hb7‐10g/dl Hb F >10% HbA2 (4%–9%) HbA (5%–90%) |
Birgens and Ljung (2007) |
|
Thalassemia minor (Trait) |
α 2 β2, α2 δ2, α 2 γ2 |
β+/β β0/β |
Asymptomatic Normal to mild anemia No splenomegaly |
Hb > 10g/dl Hb F (2.5%–5%) HbA2 (4%–9%) HbA (>90%) |
Moi et al., (2004) |
2.1.3. Beta‐thalassemia minor
It is also termed as thalassemia carrier/trait that occurs when one copy of β globin gene is normal and one copy is defective (B0/B, B+/B; Galanello & Origa, 2010; Tariet al., 2018). Thalassemia minor mostly occurs during physiological stress or pregnancy and in childhood. It is an asymptomatic condition; sometimes has mild anemia due to abnormalities in the morphology of erythrocyte (Romanello et al., 2018). The level of Hb might be >10g/dl in β‐thalassemia minor or carrier patients. There is a 25% possibility of homozygous thalassemia at each gestation if both maternities are carriers (Moi et al., 2004).
3. HEREDITARY TRANSMISSION AND MUTATIONS OF BETA‐THALASSEMIA
Beta‐thalassemia is a congenital autosomal recessive condition. Children are obligate heterozygotes when parents are affected and bring mutation in a single copy of the β globin chromosome. In the beginning, every offspring of heterozygous parentage has a 25% possibility of being unaffected and not a carrier, 25% possibility of being affected, and 50% possibility of being an asymptomatic carrier (Canatan & Koç, 2004; Galanello & Origa, 2010).
The β form of thalassemia is heterogeneous on the molecular face and more than 200 mutations, mostly point mutations have been identified in functionally imperative regions of the β‐globin gene on chromosome 11 (Yatim et al., 2014). In β‐thalassemia, deletions are not common while it is caused by the absence or reduction in the rate of production of the β‐globin chain. According to ethnic distribution and severity, a list of mutations is reported in Table 2 (Al‐Akhras et al., 2016; Giardine et al., 2007).
TABLE 2.
List of mutations in beta‐globin gene
| Mutation in β‐gene | Severity | Population |
|---|---|---|
| −101 C→T | β++ | Mediterranean |
| −31 A→G | β++ | Japanese |
| −619 del | β0 | Indian |
| IVS2‐nt654 C→T | β+ | Chinese |
| −29 A→G | β++ | African |
| −28 A→C | β++ | Southeast Asian |
| AATAAA to AACAAA | β++ | African‐American |
3.1. Prevalence/incidence
The β‐thalassemia is most widespread among Mediterranean countries such as Central Asia, the Middle East, Southern China, India, South America, and countries alongside the north coast of Africa (Weatherall, 2010). In the world, Maldives has the highest incidence of thalassemia with an 18% carrier rate of the population. The estimated frequency of β‐thalassemia in Cyprus is up to 16%, Thailand 1%, Iran 5%–10%, and China 3%–8% (Weatherall, 2010). Migration and intermarriage of humans are the consequence of the establishment of β‐thalassemia in various regions of the world where thalassemia was formerly absent such as Northern Europe (Vichinsky, 2005). It has been assessed that annually ~60,000 symptomatic persons born and ~1.5% of people are carriers of β‐thalassemia from the global population (80–90 million people). It has been also estimated that the total annual incidence of symptomatic individuals is 1 in 10,000 individuals in the European Union and 1 in 100,000 throughout the world. Males and females both are equally affected by β‐thalassemia (Hossain et al., 2017).
4. PATHOPHYSIOLOGY
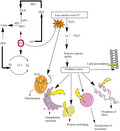
Ineffective erythropoiesis which leads to anemia is the consequence of deficiency (ß0) or declined (ß+) amount of β‐globin chain synthesis (Figure 2). Erythropoiesis is a result of excess unpaired α‐globin chains that form unbalanced and insoluble compounds that precipitate in erythroid precursors in the bone marrow and injured the plasma membrane of (RBCs) as well as leads to premature destruction of the erythrocytes. This progression mostly occurs in precursors of immature RBCs and in mature RBCs which are called ineffective erythropoiesis/hemolysis which cause anemia (Shariatiet al., 2016). Anemia encourages the production of erythropoietin which results in up to 25 to 30 times normal bone marrow expansion and the abnormalities occur in bones. The bone marrow compensates for the loss of RBCs with accelerated production of RBCs, while it is not sufficient to avoid severe anemia. Breakdown of erythrocytes causes a release of heme which results in increased iron absorption in the gastrointestinal tract (Sangkhae & Nemeth, 2017). High absorption of iron occurs due to inadequate repression of hepcidin (a protein that controls the duodenal intake of iron). Increased erythropoiesis and consistent blood exchange lead to overload the iron. Overloaded iron is extremely oxidative with lipid peroxidation of membranes and affects numerous organs, especially the heart by the formation of toxic reactive oxygen species (Hobanet al., 2015). The common inclusions are hemichromes which formed when oxidation of α‐chain subunits takes place which interacts with the proteins spectrin and ankyrin on the membrane. Due to these abnormalities, there are increases in cholesterol and phospholipids in membranes. The membranes become less stable and more rigid because α chain that is oxidized and interact with protein (Chakrabartiet al., 2013).
FIGURE 2.
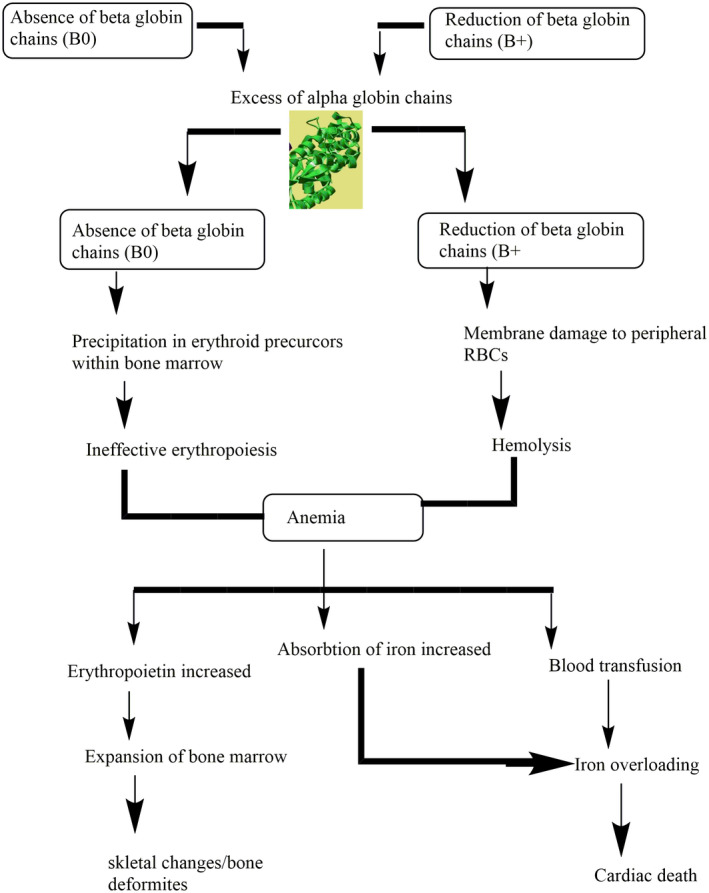
Pathophysiology of beta‐thalassemia
5. MEDICAL INDICES/COMPLICATIONS
5.1. Iron overload
Individuals who have more severe forms of β‐thalassemia major or intermediate deposit the iron within the reticuloendothelial system as a consequence of the exchange of blood. Over loaded iron appears in various endocrine tissues such as hepatic parenchyma, as well as more slowly in the tissues of the heart. Homeostasis of iron in normal humans is accomplished by controlling the concentration of iron (Ganz & Nemeth, 2011). Each day 1 mg of iron is forfeited from the physique, via detaching of the epithelium cells from the urinary system, colon, dermis, and other endodermal tissues, while 1 ml of transfused blood possess 1 mg of iron which results in 200 mg of iron is deposited in tissues. Patients with severe thalassemia who have a blood transfusion showed a significant capability of iron deposition due to lacking of iron excretory mechanisms from the body (Camaschella & Nai, 2016). During cellular apoptosis, overloaded iron discharges the RBCs which result in ineffective erythropoiesis, that prohibit the management of hepcidin through the hepatic system. Hepcidin is a peptide hormone consisting of 25 amino acid that prohibit the absorption of dietary iron and deleteriously normalizes the flow of iron into a serum. It also discharges the accumulation from hepatocytes and liberates the iron from Kupffer cells (Ganz & Nemeth, 2011; Kim & Nemeth, 2015). Ferroportin is a multipass transmembrane protein that acts as an iron exporter in the spleen and Kupffer cells of the liver. Primary human hepatocytes of thalassemia patients have suppressed the production of hepcidinin serum as well as the reduction of growth differentiation factor 15 (GDF15) prohibited the suppression of hepcidin (Sangkhae & Nemeth, 2017). Suppression of hepcidin manifestations allows iron release from macrophages and increases the absorption of iron from the intestine. Recently the pathological and normal regulation for the synthesis of hepcidin has been studied (Finianos et al., 2018). Synthesized miniature‐hepcidin that has a longer half‐life when orally given to mice showed a reduction in absorption of iron in mice and also has been recommended as prospective useful mediators in severe β‐thalassemia patient (Preza et al., 2011). After iron release from cells, it is conveyed to the bone marrow and other soft tissue, while up to 20_25 mg iron may circulate as transferrin‐bound iron in a 24‐hour round, and <1% of the total body iron remains in circulation at every time. Iron which is not bound with transferrin or non‐transferrin‐bound iron (NTBI) also develops numerous conformations in serum (Evanset al., 2008). Overloaded iron may cause damage to endocrine glands, blood vessels, diabetes, infertility, and cirrhosis in the liver (Figure 3 Hershko, 2010). Non‐transferrin‐bound iron enters into tissues through numerous cellular channels that are capable of destroying the cells rather than using the transferrin receptors (Wang et al., 2012). Thalassemia patients have the potential to diminish the non‐transferrin‐bound iron by administration of chelators (Evans et al., 2008; Fernandes et al., 2016). Administration of chelators through intravenously is more effective than subcutaneous administration at reducing NTBI. Various oral chelators such as desferrioxamine removed iron mainly by abolishing the liable plasma iron in serum whereas deferiprone and deferasirox were competent to efficiently admittance as well as diminish the intracellular labile iron segments (Jansová & Šimůnek, 2019). Various stratagems have formulated to perceive labile plasma iron (LPI), a portion of the non‐transferrin‐bound iron pool, activates metabolically by interrelating with components of membrane and affected the plasma membrane through invoking the production of free radicals or reactive oxygen species which stimulate the oxidative stress as well as lead to lipid peroxidation, oxidation of DNA and protein as shown in Figure 4 (Brissot et al., 2012; Hershko, 2010).
FIGURE 3.
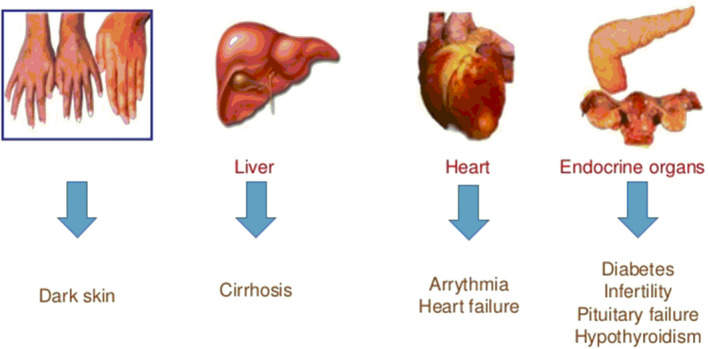
Features of iron overload
FIGURE 4.
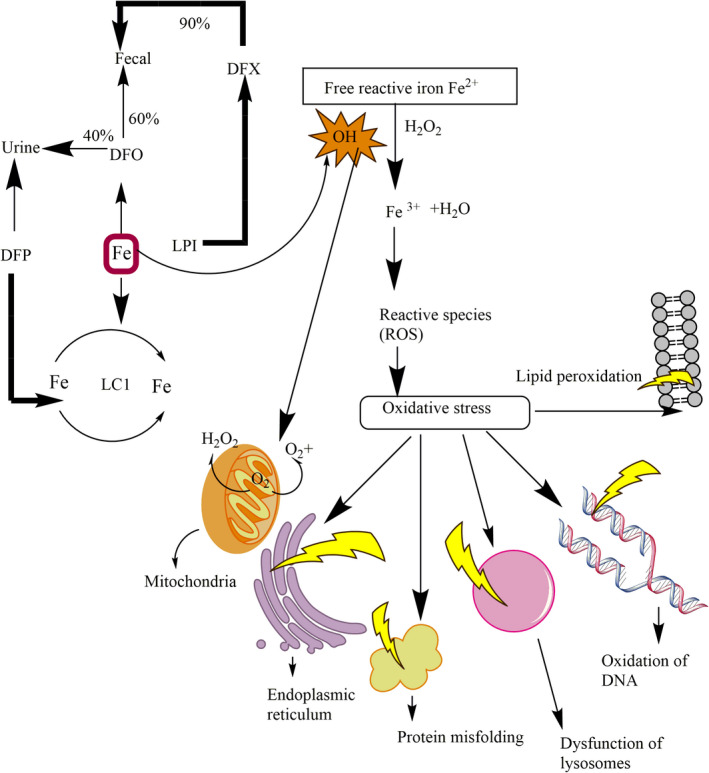
Amelioration of free iron species (labile cell iron (LCI) and labile plasma iron (LPI) by iron chelators. Labile plasma iron penetrates via plasma membrane which results in the deposition of LCI. Labile plasma iron and labile cell iron produce (ROS) such as free radicals (OH) which oxidize DNA, protein, and lipids. Deferiprone (DFP) chelates labile plasma iron and LCI alone or in combination with DFO. Deferasirox (DFX) mostly chelates labile plasma iron
5.2. Hepatitis
Patients of thalassemia have a high threat of virus‐related contagion. For example, pathological hepatitis due to prolong receiving blood and blood transfusion products. The incidence of hepatitis B for thalassemia patients and donors can greatly reduce due to the availability of vaccines but hepatitis C is greatly difficult among these patients due to lack of a vaccine (Mousaet al., 2016; Soliman et al., 2014).
5.3. Osteoporosis
Osteoporosis almost occurs in all thalassemia patients which results in multiple fissures such as pain in the bone. It also reflects the density of minerals in axial bone arises swiftly than fringe bone in old age. Endocrine deficiencies, expansion of bone marrow, the potential toxicity of chelators, and iron toxicity are also a result of osteoporotic in patients of β‐thalassemia (Terpos & Voskaridou, 2010). The accumulation of iron at a point of 7 mg/g (dry weight) in hepatocytes might be tolerated, but due to regular transfusion without iron chelation liver fibrosis can occur which result in iron overload and cannot eradicate from the liver which can increase the risk of liver carcinoma (Mancuso, 2010).
6. DIAGNOSIS
Thalassemia can be detected and diagnosed via several laboratory examinations such as DNA analysis (genetic testing), complete blood count (CBC), blood smear, prenatal testing (genetic testing of amniotic fluid), iron studies, and hemoglobinopathy (Alqahtani et al., 2018). DNA analysis test is used to help detect mutations in genes of β‐ and α‐globin chain. It can also help to determine the carrier status of thalassemia but it is not done routinely. Some mutations show no symptoms while some decline the formation of β‐globin chain in α‐thalassemia as well as some completely prevent the making of β‐globin (Sagar et al., 2015).
6.1. Hematologic diagnosis
Thalassemia intermediate is categorized by Hb concentration from 7 to 10 g/dl, mean corpuscular volume between 50 and 80 fl, as well as MCH between 16 and 24 pg. Thalassemia carrier/silent is categorized by increased Hb A2 level with a decline MCH and MCV. Thalassemia major is described by decline MCV >50 <70 fl, MCH >12 <20 pg, and (>10 g/dl) Hb level (Canatan & Koç, 2004; Galanello & Origa, 2010).
7. PREVENTION
Beta‐thalassemia might be prevented by the identification of carrier, prenatal diagnosis, and genetic counseling (Origa & Comitini, 2019). Genetic counseling offers the evidence for the risk of carriers of both parents and risk in offspring. Prenatal diagnosis can be carried out by a study of gene of fetus about 4–5 months of development or sampling at 11 weeks of gestation from chorionic villi. Examination of fetal DNA in the serum of mother and investigation of fetal cells in maternal blood might be useful to detect mutations in father (Mavrou et al., 2007). Lymphocytes, trophoblasts, and nucleated erythrocytes (NRBCs) are the three types of cells, which are utilized as sources of fetus DNA (Khan et al., 2019). The burden of thalassemia can be reduced by the prevention of the birth of homozygotes. Since the last two decades, numerous Mediterranean and Western countries have succeeded to make a substantial alteration in the population of homozygote. Some republics such as Turkey, Lebanon, Iran, Canada, Egypt, Malaysia, Pakistan, and China also have thalassemia control programs (Italia et al., 2019).
8. TREATMENT
No treatment is needed for people who have thalassemia traits. They might be deliberate genetic counseling because mutant gene can pass to their offsprings (Mutar et al., 2019). Individuals with β‐thalassemia intermedia will experience mild anemia in their life. They may be sentient like normal people, however, consistent monitoring will be required and blood transfusion can be occasionally needed. Iron supplementation is not given, while folic acid supplementation is often recommended (Kumar et al., 2012). Individuals with thalassemia intermedia will also experience hypersplenism which may cause growth retardation and worsening anemia and various other mechanical disturbances due to splenomegaly. Thalassemia intermediate is symptomatic, however; splenectomy is the treatment of thalassemia intermedia when the condition is acute (Origa, 2014).
8.1. Splenectomy
In thalassemia major and thalassemia intermedia, overactivity of the spleen occurs as a consequence of severe hemolysis. Splenomegaly in early age may be prevented after initiation of a regular blood transfusion. However, hypersplenism can develop in children among 5–10 years of age. Splenectomy protects the patients against poor health and growth retardation by decreasing the transfusion requirement, improving the level of Hb as well as decline the accumulation of iron (Pecorari et al., 2008). Removal of the spleen is suggested when a requirement of transfusion is greater than 200 to 220 ml RBCs/kg with 70% hematocrit as well as packed RBCs 250–275 ml/kg with 60% hematocrit per year. A meningococcal and pneumococcal vaccine before surgical removal of the spleen is recommended while after splenetic antimicrobial prophylaxis with penicillin is suggested for the reduction of irresistible infections (Ikeda et al., 2005).
8.2. Iron chelation therapy
In the case of regular transmission of blood, each RBC contains 200 mg of iron which results in 0.3–0.6 mg/kg per day iron accumulated (Sarker et al., 2014). Iron chelators are categorized into three classes: deferasirox (DFX), deferiprone (DFP), and deferoxamine (DFO). The removal of iron is one of the most important management for those individuals who have blood transfusion (Adewoyin & Oyewale, 2015). The DFO is a derivative from Streptomyces pilosus with a half‐life of 8–10 minutes and has a molecular weight of 657. It enters into parenchymal cells of the liver where it chelates the iron as the iron chelator deferoxamine in plasma and bile. The duration of dose differs from patient to patient and depends upon how much iron is overloaded after transfusion (Borgna‐Pignatti et al., 2004). The initial use of 1 week regularly in transfusion‐dependent patients of thalassemia, the commended dosage of deferoxamine is 30–40 mg/kg daily each week and 40–50 mg/kg and consequently to 60 mg/kg in teenagers and grownups, respectively. Chelation therapy inaugurates after 20–25 RBCs units are transferred between 2 to 4 years of age (Taher & Cappellini, 2018). In 1980s, DFP which is an artificial compound initially well known in New York. The half‐life of deferiprone is 1.5–4 hours in plasma and it is absorbed by the gastrointestinal tract. The dose is recommended daily 75 mg/kg daily, administration verbally in three allocated dosages with meal times and this dose might be augmented 100 mg/kg per day (Adel et al., 2019). Deferiprone infiltrates cell membranes more swiftly than deferoxamine and it is capable of chelating intracellular iron (Figure 4). It has efficacy for improving the function of cardiac by eradicating iron from cardiac as well as preventing cardiac diseases induced by overloaded iron (Buaboonnam & Charuvanij, 2017). Regular observation of complete blood count weekly is required as 1% of the patients treated with DFP in case of the prospective threat of agranulocytosis. Deferasirox DFX (Exjade), a hugely bioaccumulative chelator that is immersed in the gut and accepted in 2005 for use in transfusional overloaded individuals (Crichton et al., 2019). The half‐life of deferasirox is 12–18 hours and recommended orally once a day. The prescribed dosage is 20–30 mg/kg daily but some individuals may get assistance through increasing the dosage up to 40 mg/kg daily, it is effective both in grownups as well as offspring (Rivière et al., 2012).
8.3. Transplantation of bone marrow
The relocation of bone marrow remains the main conclusive treatment, reachable for patients of thalassemia (Majolino et al., 2017). The main efficacious transplantation of bone marrow was completed in the 1980s. The results in young patients are 3% mortality rate and 87% thalassemia free survival. But BMT has few disadvantages such as human leukocyte antigen matched compatible donor is required for this remedial process (Sabloff et al., 2011). The best results with very young individuals are: rejection rate is 23%, the mortality rate is 7%, and thalassemia free survival rate is 70%. Treatment for thalassemia through bone marrow transplantation is still not available for all Indian patients (Jeengar et al., 2017).
In low socio‐economic countries, the current management available for a majority of β‐thalassemia patients is effective chelating therapy and management of complications of overloaded iron as well as consistent transfusion of packed red cells (Grubovic et al., 2017).
8.4. Blood transfusion
The transfusion rehabilitation in β‐thalassemia major is to maintain the level of hemoglobin in plasma and to correct anemia which is the result of endogenous erythropoiesis (Cihanet al., 2017). Blood transfusion therapy should be started in case of severe anemia after confirmation of the diagnosis of thalassemia. Though, individuals, who have Hb > 7 g/dl, various aspects like growth retardation, increasing splenomegaly, facial changes as well as the expansion of bone, should be measured. Regular blood transmission would not be late till after the second‐third year, because multiple red cell antibodies might be developed and suitable blood donors are difficult to find. Numerous transfusion treatments have been projected over the years; however, the utmost extensively conventional goal at pre‐transfusion hemoglobin near to 9 to 10 g/dl as well as a post‐transfusion level of hemoglobin should be 13 to 14 g/dl. This inhibits impairment of organs, retardation of growth as well as malformations of bones which might lead to normal quality and activity of life (Shawkat et al., 2019). The frequency of blood transfusion depends on numerous causes such as the level of hematocrit and Hb as well as the weight of the patient (Taher & Cappellini, 2018). Blood transfusion therapy should not transfer RBCs more than 15 to 20 ml/kg daily to evade a profligate rise in the volume of blood. The efficiency of transfusion therapy should be examined via pre‐ and post‐transfusion levels of hemoglobin as well as hematocrit because these quantities can enable to monitor of iron intake and the requirement of RBCs (Galanello & Origa, 2010).
8.5. Gene therapy
In gene therapy, stem cells of patients are utilized for permanent cure of β‐thalassemia major. First, a hematopoietic stem and progenitor cells (HSPCs) of patients are harvested from umbilical cord blood, peripheral blood, or bone marrow. After that, the cells/tissues are susceptible to a treatment that leads to design variations in the DNA. The normal β or γ gene is transferred into the genome of a host cell via a lentiviral vector. Transfer of hemoglobin genome into pluripotent hematopoietic cells is also being deliberate in humans (Breda et al., 2010). The cells that contain desired genes are again inserted into patients where they multiply and proliferate in the bone marrow. Induced pluripotent stem cells (iPSCs) might be used to avoid the requirement for high‐variation proficiencies in future gene therapies (Figure 5). In this method, somatic cells consist of fibroblasts, first isolated from the patient and then remodeled into a pluripotent form through manifestations of numerous aspects such as Sox2, Klf4, Oct3/4, and C‐Myc (Yang et al., 2016). Then induced pluripotent stem cells are susceptible to accomplish the anticipated alteration in genes. Afterward adequate development, induced pluripotent stem cells are then distinguished into hematopoietic stem and progenitor cells. These HSPCs cells are then transferred back into the individual. However, this strategy will be overwhelmed but this therapy will control many of the extents in directly harvest action of HSPCs from the patient but HSPCs derived from iPSC is not yet feasible for HSCT because these cells have limitation for multiply in the bone marrow.
FIGURE 5.
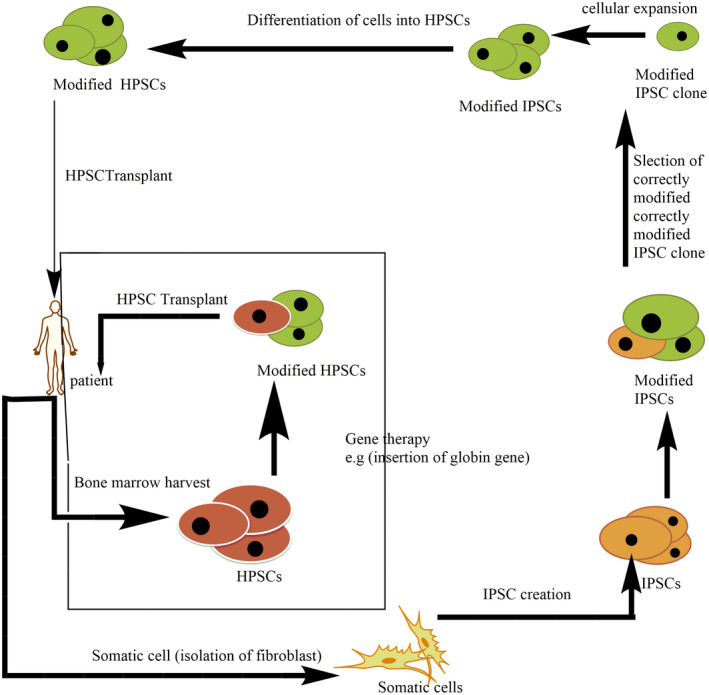
Gene therapy process. Induced pluripotent stem cells (iPSCs), Hematopoietic stem and progenitor cells (HSPCs)
8.6. Transplantation of hematopoietic stem cells
Thalassemia major might be treated by transplantation of stem cells (Angelucci et al., 2014). This scheme is used in the management of various ailments such as thalassemia. In this therapy, hematopoietic stem cells from the marrow squash of healthy individuals are sequestered and transmitted to patients of thalassemia. Nearly 80% of transplant recipients were successful by this treatment (Bernardo et al., 2012). Graft versus host disease (GVHD) is the most significant and hazardous problem in the transplantation of bone marrow which might lead to the death of transfer recipients (Cario, 2018).
8.7. Induction of fetal hemoglobin production
Individuals with long‐term manifestation of thalassemia induction fetal hemoglobin enhance the lifetime of RBCs. Various drugs are used for stimulation of the production of fetal hemoglobin such as hydroxyl urea. Hydroxyurea might be used for the cure of both sickle cell disease and thalassemia. Hydroxyurea enhances the production of γ‐globin and also improves hematological and quantifiable signs in patients of thalassemia intermedia (Tari et al., 2018; Wilber et al., 2011). Hydroxyurea acts as a cytotoxic compound for the synthesis phase of the cell cycle and also a ribonucleotide reductase inhibitor (Finotti et al., 2015). It regulates and increases expressions of fetal hemoglobin gene GATA‐2 related to apoptosis and cell cycle as well as suppresses the countenance of the GATA‐1 gene. It can also induce the proliferation of progenitor cells and enhance the amount of erythropoietin (Pace et al., 2015).
9. CONCLUSIONS
Beta‐thalassemia is induced by modifications in the β‐globin gene which results in complete absent or reduction in the rate of synthesis of normal β‐globin chain. The absence of β‐globin chain and excessiveness of the unmatched α‐globin chains cause oxidative strain and premature destruction of RBCs which result in severe anemia. Blood transfusion therapy recovers the anemia but aggravates the iron overload. It can be prevented by premarital screening, prenatal diagnosis, genetic counseling, and carrier detection. Gene therapy and transplantation of bone marrow is remained the only absolute therapy accessible for thalassemia patients.
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
ACKNOWLEDGMENT
The authors wish to heartfelt thanks to all the authors for their assistance in this study.
Ali, S. , Mumtaz, S. , Shakir, H. A. , Khan, M. , Tahir, H. M. , Mumtaz, S. , Mughal, T. A. , Hassan, A. , Kazmi, S. A. R. , Saida, Irfan, M. , & Khan, M. A. (2021). Current status of beta‐thalassemia and its treatment strategies. Molecular Genetics & Genomic Medicine, 9, e1788. 10.1002/mgg3.1788
REFERENCES
- Adel, N. , Mantawy, E. M. , El‐Sherbiny, D. A. , & El‐Demerdash, E. (2019). Iron chelation by deferasirox confers protection against concanavalin A‐induced liver fibrosis: A mechanistic approach. Toxicology and Applied Pharmacology, 382, 114–148. 10.1016/j.taap.2019.114748 [DOI] [PubMed] [Google Scholar]
- Adewoyin, A. S. , & Oyewale, O. A. (2015). Complications of allogeneic blood transfusion: Current approach to diagnosis and management. International Blood Research & Reviews, 4, 135–151. 10.9734/IBRR/2015/17874 [DOI] [Google Scholar]
- Adly, A. A. M. , El‐Sherif, N. H. , Ismail, E. A. R. , El‐Zaher, Y. A. , Farouk, A. , El‐Refaey, A. M. , & Wahba, M. S. (2015). Vascular dysfunction in patients with young β‐thalassemia: Relation to cardiovascular complications and subclinical atherosclerosis. Clinical and Applied Thrombosis/Hemostasis, 21(8), 733–744. 10.1177/1076029614541515 [DOI] [PubMed] [Google Scholar]
- Al‐akhras, A. , Badr, M. , El‐safy, U. , Kohne, E. , Hassan, T. , Abdelrahman, H. , Mourad, M. , Brintrup, J. , & Zakaria, M. (2016). Impact of genotype on endocrinal complications in β‐thalassemia patients. Biomedical Reports, 4(6), 728–736. 10.3892/br.2016.646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alqahtani, R. S. , Bedaiwi, A. A. , Alburkani, A. M. , AlFahed, M. M. , Alhoraibi, R. A. , & Tarawah, A. M. (2018). Knowledge and response of the community to premarital screening program (Sickle Cell Anemia\Thalassemia); AlMadinah, Saudi Arabia. Journal of Applied Hematology, 9(2), 59. 10.4103/joah.joah_1_18 [DOI] [Google Scholar]
- Angelucci, E. , Matthes‐Martin, S. , Baronciani, D. , Bernaudin, F. , Bonanomi, S. , Cappellini, M. D. , Dalle, J.‐H. , Di Bartolomeo, P. , de Heredia, C. D. , Dickerhoff, R. , Giardini, C. , Gluckman, E. , Hussein, A. A. , Kamani, N. , Minkov, M. , Locatelli, F. , Rocha, V. , Sedlacek, P. , Smiers, F. , … Peters, C. (2014). Hematopoietic stem cell transplantation in thalassemia major and sickle cell disease: Indications and management recommendations from an international expert panel. Haematologica, 99(5), 811–820. 10.3324/haematol.2013.099747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardo, M. E. , Piras, E. , Vacca, A. , Giorgiani, G. , Zecca, M. , Bertaina, A. , Pagliara, D. , Contoli, B. , Pinto, R. M. , Caocci, G. , Mastronuzzi, A. , La Nasa, G. , & Locatelli, F. (2012). Allogeneic hematopoietic stem cell transplantation in thalassemia major: Results of a reduced‐toxicity conditioning regimen based on the use of treosulfan. Blood, 120(2), 473–476. 10.1182/blood-2012-04-423822 [DOI] [PubMed] [Google Scholar]
- Birgens, H. , & Ljung, R. (2007). The thalassaemia syndromes. Scandinavian Journal of Clinical and Laboratory Investigation, 67(1), 11–26. 10.1080/00365510601046417 [DOI] [PubMed] [Google Scholar]
- Borgna‐Pignatti, C. A. T. E. R. I. N. A. , Rugolotto, S. I. M. O. N. E. , De Stefano, P. , Zhao, H. U. A. Q. I. N. G. , Cappellini, M. D. , Del Vecchio, G. C. , & Piga, A. (2004). Survival and complications in patients with thalassemia major treated with transfusion and deferoxamine. Haematologica, 89(10), 1187–1193. [PubMed] [Google Scholar]
- Breda, L. , Kleinert, D. A. , Casu, C. , Casula, L. , Cartegni, L. , Fibach, E. , Mancini, I. , Giardina, P. J. , Gambari, R. , & Rivella, S. (2010). A preclinical approach for gene therapy of β‐thalassemia. Annals of the New York Academy of Sciences, 1202, 134. 10.1111/j.1749-6632.2010.05594.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brissot, P. , Ropert, M. , Le Lan, C. , & Loréal, O. (2012). Non‐transferrin bound iron: A key role in iron overload and iron toxicity. Biochimica Et BiophysicaActa (BBA)‐General Subjects, 1820(3), 403–410. 10.1016/j.bbagen.2011.07.014 [DOI] [PubMed] [Google Scholar]
- Buaboonnam, J. , & Charuvanij, S. (2017). Severe deferiprone‐induced arthropathy in young adolescent successfully treated with intraarticular triamcinolone acetonide injection: A case report. Journal of the Medical Association of Thailand, 100(7), 815. [Google Scholar]
- Camaschella, C. , & Nai, A. (2016). Ineffective erythropoiesis and regulation of iron status in iron loading anaemias. British Journal of Haematology, 172(4), 512–523. 10.1111/bjh.13820 [DOI] [PubMed] [Google Scholar]
- Canatan, D. , & Koç, N. (2004). The effect of transfusion on pulmonary function tests in patients with thalassemia. Turkish Journal of Haematolgy, 21(3), 137–139. [PubMed] [Google Scholar]
- Cario, H. (2018). Hemoglobinopathies: Genetically diverse, clinically complex, and globally relevant. European Medical Oncology, 11(3), 235–240. 10.1007/s12254-018-0402-4 [DOI] [Google Scholar]
- Chakrabarti, A. , Bhattacharya, D. , Deb, S. , & Chakraborty, M. (2013). Differential thermal stability and oxidative vulnerability of the hemoglobin variants, HbA2 and HbE. PLoS One, 8(11), 81–820. 10.1371/journal.pone.0081820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cihan, M. K. , Belen, B. , Bolat, F. , Bülbül, Ö. G. , Korgalı, E. Ü. , & Koçak, Ü. (2017). The impact of transfusion and chelation on oxidative stress in immigrant Syrian children with β‐thalassemia. Indian Journal of Hematology and Blood Transfusion, 33(4), 552–558. 10.1007/s12288-017-0791-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crichton, R. R. , Ward, R. J. , & Hider, R. C. (2019). The efficacy of iron chelators for removing iron from specific brain regions and the pituitary—ironing out the brain. Pharmaceuticals, 12(3), 138. 10.3390/ph12030138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans, R. W. , Rafique, R. , Zarea, A. , Rapisarda, C. , Cammack, R. , Evans, P. J. , Porter, J. B. , & Hider, R. C. (2008). Nature of non‐transferrin‐bound iron: Studies on iron citrate complexes and thalassemic sera. JBIC Journal of Biological Inorganic Chemistry, 13(1), 57–74. 10.1007/s00775-007-0297-8 [DOI] [PubMed] [Google Scholar]
- Fernandes, J. L. , Loggetto, S. R. , Veríssimo, M. P. A. , Fertrin, K. Y. , Baldanzi, G. R. , Fioravante, L. A. B. , Tan, D. M. , Higa, T. , Mashima, D. A. , Piga, A. , Coelho, O. R. , Costa, F. F. , & Saad, S. T. (2016). A randomized trial of amlodipine in addition to standard chelation therapy in patients with thalassemia major. Blood, 128(12), 1555–1561. 10.1182/blood-2016-06-721183 [DOI] [PubMed] [Google Scholar]
- Finianos, A. , Matar, C. F. , & Taher, A. (2018). Hepatocellular carcinoma in β‐thalassemia patients: Review of the literature with molecular insight into liver carcinogenesis. International Journal of Molecular Sciences, 19(12), 4070. 10.3390/ijms19124070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finotti, A. , Breda, L. , Lederer, C. W. , Bianchi, N. , Zuccato, C. , Kleanthous, M. , & Gambari, R. (2015). Recent trends in the gene therapy of β‐thalassemia. Journal of Blood Medicine, 6, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco, S. S. , De Falco, L. , Ghaffari, S. , Brugnara, C. , Sinclair, D. A. , Matte’, A. , Iolascon, A. , Mohandas, N. , Bertoldi, M. , An, X. , Siciliano, A. , Rimmele, P. , Cappellini, M. D. , Michan, S. , Zoratti, E. , Anne, J. , & De Franceschi, L. (2014). Resveratrol accelerates erythroid maturation by activation of FoxO3 and ameliorates anemia in beta‐thalassemic mice. Haematologica, 99(2), 267–275. 10.3324/haematol.2013.090076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galanello, R. , & Origa, R. (2010). Beta‐thalassemia. Orphanet Journal of Rare Diseases, 5(1), 11. 10.1186/1750-1172-5-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganz, T. , & Nemeth, E. (2011). Hepcidin and disorders of iron metabolism. Annual Review of Medicine, 62, 347–360. 10.1146/annurev-med-050109-142444 [DOI] [PubMed] [Google Scholar]
- Giardine, B. , van Baal, S. , Kaimakis, P. , Riemer, C. , Miller, W. , Samara, M. , & Hardison, R. C. (2007). HbVar database of human hemoglobin variants and thalassemia mutations. Human Mutation, 28(2), 206. [DOI] [PubMed] [Google Scholar]
- Gibbs, G. , & Burdick, C. O. (2009). Separating thalassemia trait and iron deficiency by even simpler inspection. American Journal of Clinical Pathology, 132(4), 643–644. 10.1309/AJCPORRCP16WILSB [DOI] [PubMed] [Google Scholar]
- Grubovic, R. M. , Georgievski, B. , Cevreska, L. , Genadieva‐Stavric, S. , & Grubovic, M. R. (2017). Analysis of factors that influence hematopoietic recovery in autologous transplanted patients with hematopoietic stem cells from peripheral blood. Open Access Macedonian Journal of Medical Sciences, 5(3), 324. 10.3889/oamjms.2017.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershko, C. (2010). Pathogenesis and management of iron toxicity in thalassemia. Annals of the New York Academy of Sciences, 1202(1), 1–9. 10.1111/j.1749-6632.2010.05544.x [DOI] [PubMed] [Google Scholar]
- Hoban, M. D. , Mendel, M. C. , Romero, Z. , Kaufman, M. L. , Joglekar, A. V. , Ho, M. , & Urbinati, F. (2015). Correction of the sickle‐cell disease Mutation in human hematopoietic stem/Progenitor cells. Molecular Therapy, 125(17), 2597–2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossain, M. S. , Raheem, E. , Sultana, T. A. , Ferdous, S. , Nahar, N. , Islam, S. , Arifuzzaman, M. , Razzaque, M. A. , Alam, R. , Aziz, S. , Khatun, H. , Rahim, A. , & Morshed, M. (2017). Thalassemias in South Asia: Clinical lessons learnt from Bangladesh. Orphanet Journal of Rare Diseases, 12(1), 93. 10.1186/s13023-017-0643-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda, M. , Sekimoto, M. , Takiguchi, S. , Kubota, M. , Ikenaga, M. , Yamamoto, H. , Fujiwara, Y. , Ohue, M. , Yasuda, T. , Imamura, H. , Tatsuta, M. , Yano, M. , Furukawa, H. , & Monden, M. (2005). High incidence of thrombosis of the portal venous system after laparoscopic splenectomy: A prospective study with contrast‐enhanced CT scan. Annals of Surgery, 241(2), 208. 10.1097/01.sla.0000151794.28392.a6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Italia, K. , Chandrakala, S. , Nadkarni, A. , Ghosh, K. , & Colah, R. B. (2019). The genetic determinants for long‐term response to hydroxyurea therapy in indian β‐thalassemia patients. Clinical Research in Hematology, 2(1), 1–8. [Google Scholar]
- Jansová, H. , & Šimůnek, T. (2019). Cardioprotective potential of iron chelators and prochelators. Current Medicinal Chemistry, 26(2), 288–301. 10.2174/0929867324666170920155439 [DOI] [PubMed] [Google Scholar]
- Jeengar, R. K. , Upadhyaya, A. , Agarwal, N. , & Mehta, A. (2017). Red cell alloimmunization in repeatedly transfused children with beta thalassemia major. International Journal of Contemporary Pediatr, 4(3), 775–779. 10.18203/2349-3291.ijcp20171486 [DOI] [Google Scholar]
- Khan, M. A. , Khan, M. A. , Seedat, A. M. , Khan, M. , Khuwaja, S. F. , Kumar, R. , Usama, S. M. , & Fareed, S. (2019). Sensorineural hearing loss and its relationship with duration of chelation among major β‐Thalassemia Patients. Cureus,11(8). 10.7759/cureus.5465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, A. , & Nemeth, E. (2015). New insights into iron regulation and erythropoiesis. Current Opinion in Hematology, 22(3), 199. 10.1097/MOH.0000000000000132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, R. , Sharma, D. C. , & Kishor, P. (2012). Hb E/β‐Thalassemia: The second most common cause of transfusion‐dependent thalassemia in the Gwalior‐Chambal region of central India. Hemoglobin, 36(5), 485–490. 10.3109/03630269.2012.699489 [DOI] [PubMed] [Google Scholar]
- Lei, M. , Sun, L. , Luo, X. S. , Yang, X. , Yu, F. , Chen, X. , & Wang, Z. (2019). Distinguishing iron deficiency anemia from thalassemia by the red blood cell lifespan with a simple CO breath test: A pilot study. Journal of Breath Research, 2, 4–6. 10.1088/1752-7163/aafe4f [DOI] [PubMed] [Google Scholar]
- Majolino, I. , Othman, D. , Rovelli, A. , Hassan, D. , Rasool, L. , Vacca, M. , & Ali, K. (2017). The start‐up of the first hematopoietic stem cell transplantation center in the Iraqi Kurdistan: A capacity‐building cooperative project by the Hiwa Cancer Hospital, Sulaymaniyah, and the Italian Agency for Development Cooperation: An innovative approach. Mediterranean Journal of Hematology and Infectious Diseases, 9(1), 45–54. 10.4084/mjhid.2017.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancuso, A. (2010). Hepatocellular carcinoma in thalassemia: A critical review. World Journal of Hepatology, 2(5), 171. 10.4254/wjh.v2.i5.171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavrou, A. , Kouvidi, E. , Antsaklis, A. , Souka, A. , Kitsiou Tzeli, S. , & Kolialexi, A. (2007). Identification of nucleated red blood cells in maternal circulation: A second step in screening for fetal aneuploidies and pregnancy complications. Prenatal Diagnosis, 27(2), 150–153. 10.1002/pd.1640 [DOI] [PubMed] [Google Scholar]
- Moi, P. , Faà, V. , Marini, M. G. , Asunis, I. , Ibba, G. , Cao, A. , & Rosatelli, M. C. (2004). A novel silent β‐thalassemia mutation in the distal CACCC box affects the binding and responsiveness to EKLF. British Journal of Haematology, 126(6), 881–884. 10.1111/j.1365-2141.2004.05146.x [DOI] [PubMed] [Google Scholar]
- Mousa, S. M. O. , Afifi, M. F. , Saedii, A. A. , & El‐Setohy, A. A. (2016). Ischemia modified albumin in children with transfusion‐dependent β‐thalassemia: A new marker for an old problem. The Egyptian Journal of Haematology, 41(2), 45. 10.4103/1110-1067.186397 [DOI] [Google Scholar]
- Mutar, M. T. , Majid, M. , Jaleel, A. , Saad, A. , Abdulmortafea, A. , & Talib, H. (2019). Awareness among parents of beta thalassemia major and intermedia patients in three centers in Baghdad and Al‐Nasiriyah, Iraq in 2017. International Journal of Medical Sciences, 7(1), 6–10. 10.5195/ijms.2019.315 [DOI] [Google Scholar]
- Origa, R. (2014). Combination therapies in iron chelation. Thalassemia Reports, 4(3), 452. [Google Scholar]
- Origa, R. , & Comitini, F. (2019). Pregnancy in thalassemia. Mediterranean Journal of Hematology and Infectious Diseases, 11(1), 120. 10.4084/mjhid.2019.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace, B. S. , Liu, L. , Li, B. , & Makala, L. H. (2015). Cell signaling pathways involved in drug‐mediated fetal hemoglobin induction: Strategies to treat sickle cell disease. Experimental Biology and Medicine, 240(8), 1050–1064. 10.1177/1535370215596859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pecorari, L. , Savelli, A. , Cuna, C. D. , Fracchia, S. , & Borgna‐Pignatti, C. (2008). The role of splenectomy in thalassemia major. An update. Acta Pediatrics Mediterranea, 24, 57–60. [Google Scholar]
- Preza, G. C. , Ruchala, P. , Pinon, R. , Ramos, E. , Qiao, B. O. , Peralta, M. A. , Sharma, S. , Waring, A. , Ganz, T. , & Nemeth, E. (2011). Minihepcidins are rationally designed small peptides that mimic hepcidin activity in mice and may be useful for the treatment of iron overload. The Journal of Clinical Investigation, 121(12), 4880–4888. 10.1172/JCI57693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivière, I. , Dunbar, C. E. , & Sadelain, M. (2012). Hematopoietic stem cell engineering at a crossroads. Journal of Ameican Society and Hematology, 119(5), 1107–1116. 10.1182/blood-2011-09-349993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanello, K. S. , Teixeira, K. K. L. , Silva, J. P. M. O. , Nagamatsu, S. T. , Bezerra, M. A. C. , Domingos, I. F. , Martins, D. A. P. , Araujo, A. S. , Lanaro, C. , Breyer, C. A. , Ferreira, R. A. , Franco‐Penteado, C. , Costa, F. F. , Malavazi, I. , Netto, L. E. S. , de Oliveira, M. A. , & Cunha, A. F. (2018). Global analysis of erythroid cells redox status reveals the involvement of Prdx1 and Prdx2 in the severity of beta thalassemia. PLoS One, 13(12), 208–316. 10.1371/journal.pone.0208316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabloff, M. , Chandy, M. , Wang, Z. , Logan, B. R. , Ghavamzadeh, A. , Li, C. K. , & Hale, G. A. (2011). HLA‐matched sibling bone marrow transplantation for β‐thalassemia major. Blood, 117(5), 1745–1750. 10.1182/blood-2010-09-306829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagar, C. S. , Kumar, R. , Sharma, D. C. , & Kishor, P. (2015). Alpha hemoglobin stabilizing protein: Its causal relationship with the severity of beta thalassemia. Blood Cells, Molecules, and Diseases, 55(2), 104–107. 10.1016/j.bcmd.2015.05.005 [DOI] [PubMed] [Google Scholar]
- Sangkhae, V. , & Nemeth, E. (2017). Regulation of the iron homeostatic hormone hepcidin. Advance Nutrition, 8(1), 126–136. 10.3945/an.116.013961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarker, N. R. , Ghosh, A. K. , Saha, S. K. , & Shahriar, A. (2014). Recent advances in the management of Thalassaemia: A Review Update. Journal of ShaheedSuhrawardy Medical College, 6(1), 31–37. 10.3329/jssmc.v6i1.31490 [DOI] [Google Scholar]
- Shariati, L. , Modaress, M. , Khanahmad, H. , Hejazi, Z. , Tabatabaiefar, M. A. , Salehi, M. , & Modarressi, M. H. (2016). Comparison of different methods for erythroid differentiation in the K562 cell line. Biotechnology Letters, 38(8), 1243–1250. 10.1007/s10529-016-2101-8 [DOI] [PubMed] [Google Scholar]
- Shawkat, A. J. , Jwaid, A. H. , & Awad, G. M. (2019). Evaluating health‐related quality of life (HRQoL) in Iraqi adult and pediatric patients with beta‐thalassemia major using two different iron chelation therapies. Iraqi Journal of Pharmaceutical Sciences, 28(1), 44–52. 10.31351/vol28iss1pp44-52 [DOI] [Google Scholar]
- Sirachainan, N. , Chuansumrit, A. , Kadegasem, P. , Sasanakul, W. , Wongwerawattanakoon, P. , & Mahaklan, L. (2016). Normal hemostatic parameters in children and young adults with α‐thalassemia diseases. Thrombosis Research, 146, 35–42. 10.1016/j.thromres.2016.08.024 [DOI] [PubMed] [Google Scholar]
- Soliman, A. , Yassin, M. , Al Yafei, F. , Al‐Naimi, L. , Almarri, N. , Sabt, A. , & De Sanctis, V. (2014). Longitudinal study on liver functions in patients with thalassemia major before and after deferasirox (DFX) therapy. Mediterranean Journal of Hematology and Infectious Diseases, 6(1), 322–330. 10.4084/mjhid.2014.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stauder, R. , Valent, P. , & Theurl, I. (2018). Anemia at older age: Etiologies, clinical implications, and management. Blood Journal of American Society and Hematology, 131(5), 505–514. 10.1182/blood-2017-07-746446 [DOI] [PubMed] [Google Scholar]
- Taher, A. T. , & Cappellini, M. D. (2018). How I manage medical complications of β‐thalassemia in adults. Journal of American Society and Hematology, 32(17), 1781–1791. 10.1182/blood-2018-06-818187 [DOI] [PubMed] [Google Scholar]
- Taher, A. T. , Musallam, K. M. , Cappellini, M. D. , & Weatherall, D. J. (2011). Optimal management of β thalassaemiaintermedia. British Journal of Haematology, 152(5), 512–523. 10.1111/j.1365-2141.2010.08486.x [DOI] [PubMed] [Google Scholar]
- Tari, K. , ValizadehArdalan, P. , Abbaszadehdibavar, M. , Atashi, A. , Jalili, A. , & Gheidishahran, M. (2018). Thalassemia an update: Molecular basis, clinical features and treatment. International Journal of Biomedicine and Public Health, 1(1), 48–58. 10.22631/ijbmph.2018.56102 [DOI] [Google Scholar]
- Terpos, E. , & Voskaridou, E. (2010). Treatment options for thalassemia patients with osteoporosis. Annals of the New York Academy of Sciences, 1202(1), 237–243. 10.1111/j.1749-6632.2010.05542.x [DOI] [PubMed] [Google Scholar]
- Thein, S. L. (2018). Molecular basis of β thalassemia and potential therapeutic targets. Blood Cells Molecular Diseases, 70, 54–65. 10.1016/j.bcmd.2017.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vichinsky, E. P. (2005). Changing patterns of thalassemia worldwide. Annals of the New York Academy of Sciences, 1054(1), 18–24. [DOI] [PubMed] [Google Scholar]
- Wang, C. Y. , Jenkitkasemwong, S. , Duarte, S. , Sparkman, B. K. , Shawki, A. , Mackenzie, B. , & Knutson, M. D. (2012). ZIP8 is an iron and zinc transporter whose cell‐surface expression is up‐regulated by cellular iron loading. Journal of Biological Chemistry, 287(41), 34032–34043. 10.1074/jbc.M112.367284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weatherall, D. J. (2010). The inherited diseases of hemoglobin are an emerging global health burden. Blood, 115(22), 4331–4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilber, A. , Hargrove, P. W. , Kim, Y.‐S. , Riberdy, J. M. , Sankaran, V. G. , Papanikolaou, E. , Georgomanoli, M. , Anagnou, N. P. , Orkin, S. H. , Nienhuis, A. W. , & Persons, D. A. (2011). Therapeutic levels of fetal hemoglobin in erythroid progeny of β‐thalassemic CD34+ cells after lentiviral vector‐mediated gene transfer. Blood, 117(10), 2817–2826. 10.1182/blood-2010-08-300723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, J. , Li, S. , He, X. B. , Cheng, C. , & Le, W. (2016). Induced pluripotent stem cells in Alzheimer’s disease: Applications for disease modeling and cell‐replacement therapy. Molecular Neurodegeneration, 11(1), 39. 10.1186/s13024-016-0106-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatim, N. , Rahim, M. , Menon, K. , Al‐Hassan, F. , Ahmad, R. , Manocha, A. , Saleem, M. , & Yahaya, B. (2014). Molecular characterization of α‐and β‐thalassaemia among Malay patients. International Journal of Molecular Sciences, 15(5), 8835–8845. 10.3390/ijms15058835 [DOI] [PMC free article] [PubMed] [Google Scholar]