

Abstract
Background
Exome sequencing (ES) has become the most powerful and cost‐effective molecular tool for deciphering rare diseases with a diagnostic yield approaching 30%–40% in solo‐ES and 50% in trio‐ES. We applied an innovative parental DNA pooling method to reduce the parental sequencing cost while maintaining the diagnostic yield of trio‐ES.
Methods
We pooled six (Agilent‐CRE‐v2–100X) or five parental DNA (TWIST‐HCE–70X) aiming to detect allelic balance around 8–10% for heterozygous status. The strategies were applied as second‐tier (74 individuals after negative solo‐ES) and first‐tier approaches (324 individuals without previous ES).
Results
The allelic balance of parental‐pool variants was around 8.97%. Sanger sequencing uncovered false positives in 1.5% of sporadic variants. In the second‐tier approach, we evaluated than two thirds of the Sanger validations performed after solo‐ES (41/59–69%) would have been saved if the parental‐pool segregations had been available from the start. The parental‐pool strategy identified a causative diagnosis in 18/74 individuals (24%) in the second‐tier and in 116/324 individuals (36%) in the first‐tier approaches, including 19 genes newly associated with human disorders.
Conclusions
Parental‐pooling is an efficient alternative to trio‐ES. It provides rapid segregation and extension to translational research while reducing the cost of parental and Sanger sequencing.
Keywords: cost effectiveness, exome sequencing, rare diseases, trio‐like strategy; parental‐pool strategy
We applied an innovative parental DNA pooling method to reduce the parental sequencing cost while maintaining the diagnostic yield of trio‐ES. Parental‐pooling is an efficient alternative to trio‐ES. It provides rapid segregation and extension to translational research while reducing the cost of parental and Sanger sequencing.

1. BACKGROUND
Rare diseases represent clinically and genetically heterogeneous conditions. Approximately 8,000 rare diseases have been described, resulting in a heterogeneous group of disorders with or without clinical overlap (Dawkins et al., 2018). Next‐generation sequencing––especially exome sequencing (ES)––has become the first‐tier strategy to identify the molecular etiologies of these disorders (Stark et al., 2016; Tan et al., 2017), especially many ultra‐rare disorders (Ng et al., 2009, 2010), and is facilitated by international collaboration (Boycott et al., 2017). ES was therefore rapidly transferred in routine diagnosis. In developmental disorders and/or intellectual disability (DD/ID), the ES strategy has shifted toward trio rather than solo approaches, mostly because of the high rate of de novo variants (Hamdan et al., 2017; Vissers et al., 2010). Indeed, a trio strategy facilitates interpretation thanks to the information regarding familial segregation (Hartley et al., 2020). In addition, solo‐ES provides a mean diagnostic yield of approximately 25% to 58% (Clark et al., 2018; Snoeijnen‐Schouwenaars et al., 2019; Stark et al., 2016) while trio‐ES diagnostic yield ranges from 24% to 68% (Clark et al., 2018; Tarailo‐Graovac et al., 2016; Zhu et al., 2015). Though these values may seem similar, the chance of positive results is doubled with a trio strategy (95% CI 1.62–2.56; p < 0.0001; Clark et al., 2018). Indeed, one study reports a 36.5% diagnostic rate with trio‐ES applied in individuals with previous negative solo‐ES (Eldomery et al., 2017). Despite a lower diagnostic yield of the solo‐ES strategy, it is cheaper than trio‐ES, meaning that it can be offered to a larger number of individuals.
Various strategies can be used to optimize a solo‐ES analysis. The prospective reanalysis of solo‐ES data after a defined period of time using updated pipelines and databases increases the number of genes involved in human disorder available for interpretation. Our genomic laboratory has applied this strategy in routine diagnosis, resulting in the identification of a molecular cause in 24/156 individuals (15.4%) who were negative after first‐tier solo‐ES (Nambot et al., 2018). We also obtained a diagnosis in 48/313 individuals (15%) using a re‐analysis strategy in a research setting for 313 individuals who were negative after solo‐ES (Bruel et al., 2019). Several publications have highlighted the interest of ES re‐analysis, reporting an additional diagnostic yield ranging from 10.5% to 32% (Baker et al., 2019; Ewans et al., 2018; Li et al., 2019; Schmitz‐Abe et al., 2019). The implementation of second‐tier trio‐ES after negative solo‐ES also appears to be an efficient strategy to increase diagnostic yield and decipher molecular bases in developmental disorders (Tran Mau‐Them et al., 2020). In 18/70 individuals (25.8%), we identified a positive molecular result with nine variants in genes already implicated in human disorders and nine others in genes newly implicated in human disorders. Another publication also reports the interest of this strategy, with a diagnostic yield of 36.5% when known or novel genes involved in human diseases are considered (Eldomery et al., 2017). Interestingly, these two studies report variants (especially missense variants) in genes already involved in human disorders that were not considered pathogenic in the first‐tier solo analysis, highlighting the difficulty obtaining accurate interpretation of solo‐ES data. However, it also emphasizes the interest of this strategy in translational research, leading to an accelerated discovery of genes involved in novel human disorders.
Very few pooled DNA strategies have been published. They focused on different human disorders such as inflammatory bowel disorder, depression, maturity‐onset diabetes of the young, mitochondrial complex I deficiency, and DD/ID (Bansal et al., 2017; Calvo et al., 2010; Popp et al., 2017; Zhu et al., 2020). The number of individuals per pool ranged from 12 to 35 and the mean depth was above 100X (324X‐491X for ES and 3360X for targeted sequencing). The diagnostic rate ranged from 22% to 28% (Table 1).
TABLE 1.
Next generation sequencing and pooling methods in the literature
| Disorder | Individuals | NGS method | Pooling methods | Number of individual per pool | Mean depth | Diagnostic rate (number) | |
|---|---|---|---|---|---|---|---|
| Zhu et al | IBD, depression | 70 | ES | index case | 35 | NR | NA |
| Bansal et al | Diabetes | 6058 | Targeted sequencing | index case | 20–32 | NR | 0.6% (40) |
| Calvo et al | Mitochondrial | 60 | Targeted sequencing | index case | 20–21 | 3360X | 22% (13) |
| Popp et al | DD/ID | 96 | ES | index case | 12 | 324 to 491X | 28% (27) |
| Ryu et al | NA | 1125 | Targeted sequencing | index case | 25 | 1068X | NA |
Abbreviations: DA, developmental anomaly; IBD, inflammatory bowel disease; ID, intellectual disability, NA, not applicable; NGS, next generation sequencing.
Because the pooling included the index cases, Sanger sequencing was systematically required to identify the individual carrying the candidate variant before family segregation. In addition, the laboratory guidance regarding individual pooling in clinical use, concerns index cases (not parents), and disease‐targeted gene panels (not ES; Rehm et al., 2013).
In order to preserve the advantage of trio‐ES strategy for rapid parental segregation and decrease the costs of parental sequencing, we chose to apply a pooling strategy to parental rather than patient DNA samples. We present this parental‐pooled trio‐like ES strategy as a second‐tier approach (after negative solo‐ES) and as a first‐tier approach in individuals with rare diseases.
2. METHODS
2.1. Affected individuals
Three hundred and ninety‐eight individuals with various rare diseases and their parents were referred to our clinical genetics center (Figure 1a).
FIGURE 1.
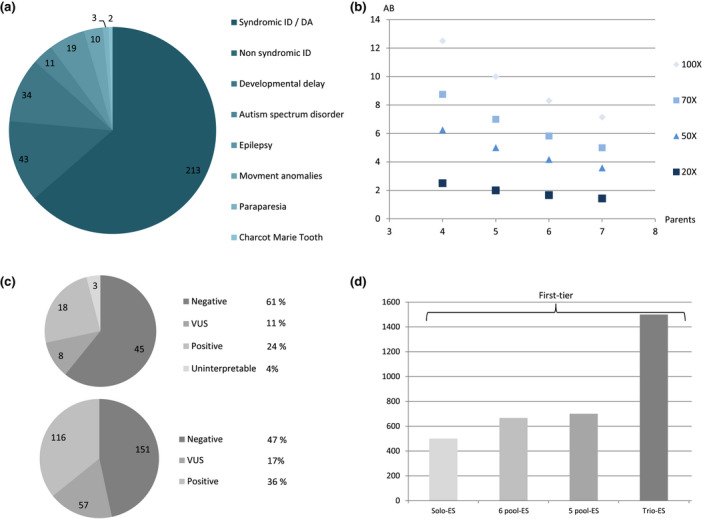
(a) Number of individuals per rare disease (neurologic and developmental anomaly focus). DA: developmental anomaly; ID: intellectual disability. (b) Expected allelic balance (AB) in %, depending on the sequencing depth (from 20X to 100X) and number of parents per pool (from 4 to 7 parents). (c) Molecular results in the second‐tier (top) and first‐tier cohorts (bottom) VUS: variants of unknown significance. (d) Sequencing cost in $ depending on the solo, trio, or first‐tier parental‐pool strategies (5 or 6 parents) and based on a sequencing cost of solo‐ES at 500$. In gray shades: prices of first‐tier strategies
As a proof‐of‐concept, we first selected six already solved individuals (positive controls) with different known variants and mode of inheritance (Table 1, Figure 2). Parental segregation of these variants had previously been confirmed by Sanger sequencing.
FIGURE 2.
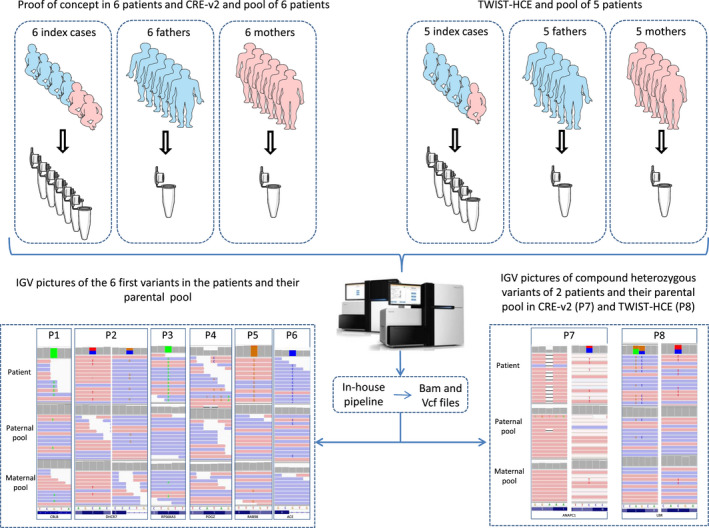
Workflow of the parental‐pool ES strategy with IGV pictures of the variants. On the left side are positive controls with six parental pools and CRE‐v2 kit. On the right side are compound heterozygous examples of five parental pools with TWIST‐HCE kit
From August 2018 to December 2019, first‐tier parental‐pool ES was proposed to all individuals with a rare disease and available parental samples, and a second‐tier parental‐pool ES was systematically proposed when the physician requested solo‐ES re‐analysis if parental samples were available.
2.2. Pooling design
Because one variant with heterozygous status in a parent would be seen in a pool at a percentage of N = 1/(n parents × 2 alleles), we first calculated that a variant would be seen at 1/(6 × 2) = 8.3% (i.e., 8–9 reads with the variant) with a sequencing depth of 100X and a pooling of six parental DNA samples (Figure 1b). This threshold of expected allelic balance (AB) at 8.3% was chosen a priori because ES was currently produced with a mean depth of 100X. This hypothesis was initially tested with the Agilent‐CRE (Clinical Research Exome) v2 enrichment kit. One year later, we switched to an enriched version of the TWIST‐HCE (Human Core Exome) enrichment kit with an expected mean depth of 70X. Because the novel expected AB with six parents would be only 5.83% (considered insufficient), we decreased the number of parents pooled from 6 to 5 to obtain an expected AB of 7% (Figure 1b). We did not investigate if the AB of 5.83% (six parents pooled together and a mean sequencing depth of 70X) was adequate to correctly call the presence/absence of variant in the parental‐pool.
DNA samples were extracted from peripheral whole‐blood samples with QIAcube DNA Blood kit (QIAGEN) according to the supplier's protocol. Each parental DNA sample was quantified by fluorimetry with a Qubit (Thermofisher) according to the supplier protocol diluted and pooled at equimolar concentration, according to the initial DNA concentration. We created two independent parental pools by mixing 6 or 5 maternal DNA samples and 6 or 5 paternal DNA samples (Figure 2), depending on the enrichment kit utilized (Table 3). We did not barcode each individual parent in the pools because of inherent additional costs and also because segregation of identified variants have been performed in all cases.
TABLE 3.
List of variants used as positive controls with their AB in parental pools (1 to 6) and two examples (7 and 8) of compound heterozygous variants with the 5 and 6 parents parental‐pool strategies
| Individual | Gene | MIM number | Genotype | Genomic position | c.DNA | Protein | Paternal‐pool allelic balance | Maternal‐pool allelic balance | Sanger segregation |
|---|---|---|---|---|---|---|---|---|---|
| 1 | CBLB | 604491 | hm | chr3:g.105572421G>A | NM_170662.3:c.256C>T | p.(Arg86*) | 0.09 | 0.08 | Maternally and paternally inherited |
| 2 | DHCR7 | 602858 | htc | chr11:g.71146710C>T | NM_001360.2:c.1139G>A | p.(Cys380Tyr) | 0 | 0.09 | Maternally inherited |
| chr11:g.71146886C>G | NM_001360.2:c.964‐1G>C | p.? | 0.08 | 0 | Paternally inherited | ||||
| 3 | RPS6KA3 | 300075 | hmi | chrX:g.20206617G>A | NM_004586.2:c.629C>T | p.(Thr210Ile) | 0 | 0.11 | Maternally inherited |
| 4 | POGZ | 614787 | ht | chr1:g.151378304_151378305del | NM_015100.3:c.3206_3207del | p.(Tyr1069*) | 0 | 0 | De novo |
| 5 | PLP1 | 300401 | hmi | chrX:g.103043401T>G | NM_001128834.1:c.658T>G | p.(Cys220Gly) | 0 | 0.7 | Maternally inherited |
| 6 | ACE | 106180 | hm | chr17:g.61573878G>C | NM_000789.2:c.3503+1G>C | p.? | 0.10 | 0 | Paternally inherited |
| 7 | ANAPC1 | 608473 | htc | chr2:g.112541514C>T | NM_022662.3:c.5023G>A | p.(Glu1675Lys) | 0 | 0.09 | Maternally inherited |
| chr2:g.112621271delA | NM_022662.3:c.1033delT | p.(Ser345Profs*6) | 0.07 | 0 | Paternally inherited | ||||
| 8 | LBR | 600024 | htc | chr1:g.225591096C>T | NM_002296.2:c.1757G>A | p.(Arg586His) | 0 | 0.07 | Maternally inherited |
| chr1:g.225594482_225594483delinsGC | NM_002296.3:c.1366_1367delinsGC | p.(Leu456Ala) | 0.08 | 0 | Paternally inherited |
Abbreviations: Hm, homozygous; hmi, hemizygous; ht, heterozygous; htc, compound heterozygous.
2.3. Exome sequencing
Exome enrichment and sequencing were performed on a HiSeq4000 or a NovaSeq6000 according to supplier protocol, reads alignment, and bioinformatics analyses were carried out as previously described (Methods S1; Nambot et al., 2018). Variants and CNV calling were only performed in the index case. Our pipeline then extracted the depth, AB, and genotype, at all SNV detected positions, in both paternal and maternal pools. Variant calling was performed with GATK HaplotypeCaller v3.8. CNV detection in the index case was performed using eXome Mendelian Markov Model (XHMM) based on exome read depth normalization (Fromer et al., 2012; Fromer & Purcell, 2014).
2.4. Variant interpretation
For all individuals, rare variant's interpretation was focused first, on de novo variants, homozygous, or compound heterozygous variants within OMIM‐morbid genes (genes described in the OMIM database and involved in human disorders), and second, extended to all other genes (non‐OMIM morbid genes described in the OMIM database but not linked to a human disorder––and non‐OMIM––genes not described in the OMIM database). Based on our laboratory experience, variants with AB >0.01 in a parental‐pool were considered inherited. Variants with AB = 0.01 or allelic depth for the alternative allele = 1 were manually checked on the Integrative Genomics Viewer (IGV) to discriminate sequencing errors from possible presence of the variant in the parental‐pool.
Candidate variants were confirmed with a second independent method. Candidate variants in genes not involved in human disorders were also shared through international collaborative system (MatchMaker Exchange; Philippakis et al., 2015) to gather additional individuals and to strengthen genotype–phenotype correlations.
3. RESULTS
3.1. Sequencing results
In the 74 individuals who underwent second‐tier parental‐pool ES, five different enrichment kits were used. Only 18/74 individuals (24%) benefited from the same enrichment kits used for the parental‐pool (Table 2).
TABLE 2.
Version of the exome enrichment kits used in individuals and parental pools in first and second‐tier strategies
| Capture kit version | Second‐tier parental pool | First‐tier parental pool | |||
|---|---|---|---|---|---|
| Agilent‐CRE‐v2 | TWIST‐HCE | Agilent‐CRE‐v2 | TWIST_HCE | ||
| Index case | Agilent‐v5‐51Mb | 3 | 8 | ||
| Agilent‐v6 | |||||
| Agilent‐CRE | 13 | ||||
| Agilent‐CRE‐v2 | 6 | 26 | |||
| TWIST‐HCE | 12 | 317 | |||
Abbreviations: CRE, Clinical Research Exome; HCE, Human Core Exome.
In three individuals, the results were not interpretable because too many de novo variants (mean of 255 [80–354]) were detected. In 2/3 individuals, ES was performed with an Agilent‐v5‐51Mb enrichment kit, whereas the parental‐pool was performed with Agilent‐CRE‐v2 or an enriched version of the TWIST‐HCE enrichment kit. In the third individual, the proband and parental‐pool enrichment kit were similar (Agilent‐CRE‐v2), but the de novo variants consisted of retrogene‐like pictures on IGV (increased depth on exons and misalignment at the intron–exon junction possibly due to a cDNA/RNA contamination). In order to investigate this anomaly, genome sequencing (GS) was performed on another sample and did not confirm the ES results (Figure 3e).
FIGURE 3.

IGV examples of false positive and negative variants. (a) IGV pictures of the BRD7 (left) and GPDP4 (right) strand bias. (b) IGV pictures of the OTOA poor quality mapping. (c and d) IGV pictures of the TADAD2B and ALG8 variants. Note the absence of strand bias. (e) IGV picture of the possible cDNA contamination with misaligned read at the exon‐intron junction in the individual solo‐ES (top) but not in solo‐GS (bottom; two distinct samples from the same individual). (f) IGV picture of the HNRNPAB variant. Note the absence of variant at this position. (g) IGV picture of the SHANK3 variant in the individual (top) and the parental pools (middle and bottom). Note the presence of the variant in both parental pools
In the 324 individuals with first‐tier parental‐pool ES, two enrichment kits were used: seven individuals benefited from the Agilent‐CRE‐v2 kit and 317 from the enriched TWIST‐HCE kit (Table 2).
3.2. Allelic balance, sensitivity, and specificity
In the proof‐of‐concept cohort, the mean depth was 106X (81–122), and it was 104X and 101X in the paternal and maternal pool, respectively. The parental‐pool ES strategy identified the correct segregation of variants in all 6 controls. De novo variants were absent in both parental pools, while inherited ones (X‐linked or autosomal recessive) were present with an AB ranging from 7% to 11% (expected value 8.3%; Table 3; Figure 2).
The distribution of AB of rare variants in all the parental pools was 8.97%, which is concordant with the excepted value of 8.3% and 10% in the 6 and 5 parental pools, respectively (Figure 4a). But when considering the same enrichment kits in the index cases and their parental pools (Agilent‐CRE‐v2 and enriched TWIST‐HCE kits), the number of de novo variants in parental pools was higher than in the individuals with trio‐ES strategy for both kits (Figure 4b). Interestingly, there was no significant difference in rare de novo variants between 295 pools and 281 trios (Figure 4c). Lastly, there was no significant difference in the number of de novo variants between 5 and 6‐sample parental pools (Figure 4d).
FIGURE 4.
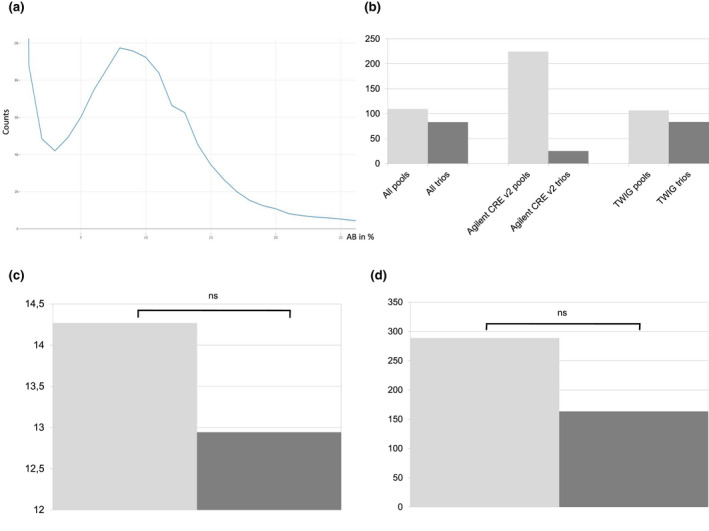
(a) Distribution of the AB of rare variants in the entire parental pools. Note the Gaussian curve with a maximum around 9%. (b) Comparison of the mean number of rare de novo variants in 295 pools (light gray; 288 TWIST‐HCE and 7 CRE‐v2) and 281 trios (dark gray; 280 TWIST‐HCE; and 1 CRE‐v2) with two different enrichment kits (multiallelic, SnpCluster and outliers >500 de novo excluded; individual depth ≥10). (c) Comparison of the mean number of rare de novo variants 281 in pools (light gray) and 291 trios (dark gray; multiallelic, SnpCluster and outliers >50 de novo excluded; individual depth ≥10; rare <1%; NSSI only). NS, not significant. (d) Comparison of the mean number of de novo variants in six (265; light gray) and five (50; dark gray) parental pools. NS: not significant
In the second‐tier parental‐pool cohort, the sensitivity (Se) and specificity (Sp) were 100% and 93%, respectively. The positive predictive value (PPV) and negative predictive value (NPV) were 89% and 100%, respectively. In the first‐tier parental‐pool cohort the Se and Sp were 99% and 98%, and the PPV and NPV were 98% and 99%, respectively. Overall, the Se and Sp were 99% and 96%, and the PPV and NPV were 94% and 99%, respectively.
3.3. Molecular results
In the second‐tier parental‐pool strategy, after excluding 3/74 individuals (4%) with non‐interpretable data, we identified 22 de novo, 2 homozygous, 1 biallelic, 1 hemizygous, 2 inherited rare variants, and 1 CNV of clinical interest in 29/71 individuals (41%). In the 22 individuals with de novo variants identified with the parental‐pool strategy, Sanger sequencing found three variants inherited from an unaffected parent (BRD7, GDPD4, and TADA2B ‐ 13%). The three individuals were reclassified as negative.
Overall, a causative diagnosis was identified in 18/71 individuals (25%; Table S1, Figure 1c), and nonconclusive results were obtained in 8/71 individuals (11%; Table S2). In 13/18 individuals with causative diagnosis (72%), the strategy identified genes newly associated with human disorders. In the 5/18 other individuals (28%), causative variants (CRAT, PHIP, ASCL1, and TRAPPC11) or CNVs ((10:27702089_29599990)x1 including WAC) were identified in known OMIM‐morbid genes (Table S1). These variants were not considered causative in the initial solo‐ES strategy.
We retrospectively looked at Sanger validations performed for candidate variants after solo‐ES and before implementing the parental‐pool strategy. Among the 71 individuals with interpretable parental‐pool data, 59 Sanger validations were performed in 36 individuals, with a mean of 1.5 variations per individual (1–6). Two thirds of the Sanger validations (41/59– 69%) would have been saved if the parental‐pool segregations had been available. In 37/41 (90%) Sanger validations, a heterozygous variant was inherited from an asymptomatic parent and in the other 4/41 (10%), two variants were not compound heterozygous but inherited from the same parent.
In the first‐tier parental‐pool strategy, all the data were interpretable. We identified variants of clinical interest in 176/324 individuals (54%), namely: 105 heterozygous (80 de novo), 22 biallelic, 12 homozygous, 3 hemizygous rare variants, and 22 CNVs in 143/324 individuals (44%). Eight individuals also presented with possible double diagnostic: 6 with SNV and 2 with CNVs and heterozygous SNV (Table 4). Two individuals presented a double hit in GPR98 and STRC (SNVs + CNVs; Table S1; Table 4).
TABLE 4.
Individuals with double hits and/or double diagnosis
| Individual | OMIM‐morbid | Gene | Genotype | Segregation | Genomic position | c.DNA | protein | CADD | polyphen | GERP | misZ | pLI |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | + | ADNP | ht | De novo | chr20:g.49508824_49508827del | NM_015339.2:c.2424_2427del | p.(Lys809Serfs*19) | 2.22 | 1 | |||
| + | USP27X | hmi | Maternally inherited | chrX:g.49645630C>A | NM_001145073.1:c.720C>A | p.(Phe240Leu) | 3.28 | 0.93 | ||||
| 2 | + | ASL | hm | Paternally and maternally inherited | chr7:g.65548091C>T | NM_000048.3:c.376C>T | p.(Arg126Trp) | 14.6 | 1 | 3.98 | 0.95 | 0 |
| ‐ | KANSL3 | hm | Paternally and maternally inherited | chr2:g.97278215C>T | NM_001115016.2:c.998G>A | p.(Arg333Lys) | 23.9 | 0.77 | 5.29 | 2.58 | 1 | |
| 3 | + | CBL | ht | De novo | chr11:g.119148891T>C | LRG_608t1:c.1111T>C | p.(Tyr371His) | 25.3 | 1 | 5.65 | 1.16 | 0 |
| + | PTPN11 | ht | Maternally inherited | chr12:g.112926909A>G | LRG_614t1:c.1529A>G | p.(Gln510Arg) | 23.9 | 1 | 5.13 | |||
| 4 | + | BRCA1 | ht | NR | chr17:g.41245479_41245482del | LRG_292t1:c.2066_2069del | p.(Ser689Lysfs*11) | 0.85 | 0 | |||
| Deletion 16q24.3 | ht | Maternally inherited | chr16:g.89590412–89599046 | |||||||||
| 5 | + | BRCA2 | ht | NR | chr13:g.32971125_32971126del | LRG_293t1:c.9592_9593 | p.(Cys3198Tyrfs*23) | |||||
| Duplication 16p11.2 | Maternally inherited | chr16:g.8836685–29001335 | ||||||||||
| 6 | + | BRCA2 | ht | NR | chr13:g.32914699_32914702del | LRG_293t1:c.6207_6210del | p.(Glu2070Valfs*10) | |||||
| ‐ | CYFIP1 | ht | De novo | chr15:g.22962436C>G | NM_014608.2:c.2160‐4C>G | p.? | 2.73 | 0.97 | ||||
| 7 | + | A2ML1 | ht | Maternally inherited | chr12:g.9000236G>T | NM_144670.4:c.1775G>T | p.(Arg592Leu) | 28.1 | 0.97 | 2.74 | 0.46 | 0 |
| + | TRIO | ht | Maternally inherited | chr5:g.14482785G>A | NM_007118.2:c.6560G>A | p.(Arg2187His) | 24 | 0.94 | 5 | 5.6 | 1 | |
| 8 | + | PGM1 | htc | Paternally inherited | chr1:g.64100539T>G | NM_002633.2:c.722T>G | p.(Leu241Arg) | 18.9 | 1 | 5.13 | −0.06 | 0 |
| Maternally inherited | chr1:g.64114218T>G | NM_002633.2:c.1175T>G | p.(Leu392Arg) | 32 | 1 | 6.06 | ||||||
| ‐ | KIAA0368 | ht | De novo | chr9:g.114184413C>T | NM_001080398.1:c.1866+1G>A | p.? | 25.4 | 4.22 | 1.9 | 1 | ||
| 9 | + | GPR98** | ht | Paternally inherited | chr5:g.89949539A>G | NM_032119.3:c.4148A>G | p.(Tyr1383Cys) | 26.4 | 1 | 5.48 | 0.07 | 0 |
| Duplication 5q14.3 | Maternally inherited | chr5:89969869–89986860 | ||||||||||
| 10 | + | STRC | hm | Maternally inherited | chr15:g.43897539G>A | NM_153700.2:c.3853C>T | p.(Gln1285*) | 21 | 5.46 | 1.41 | 0 | |
| Deletion 15q15.3 | ht | Paternally inherited | chr5:g.43888604–43940261 |
Abbreviations, Hm, homozygous, hmi: hemizygous; ht, heterozygous; htc, compound heterozygous; NR, not reported.
In 3/324 (0.9%) individuals, parental‐pool ES strategy indicated a de novo variant that was found inherited from an unaffected parent after Sanger sequencing (ALG8, HNRNPAB, and OTOA ‐ Table 5). In 1/324 individuals, parental‐pool ES revealed an inherited SHANK3 variant that was found to be de novo after Sanger sequencing. The variant was a G duplication, present two times in each parental‐pool but located before an eight polyG track that can cause misalignment (Table 5).
TABLE 5.
False positive and false negative variants identified in the cohort
| Strategy | Gene | Genotype | genomic position | Index case allelic depth | Index case allelic balance | Paternal pool depth | Paternal pool allelic balance | Maternal pool depth | Maternal pool allelic balance | Sanger segregation |
|---|---|---|---|---|---|---|---|---|---|---|
| Second‐tier | TADA2B | ht | chr4:g.7056620C>T | 125 | 0.51 | 87 | 0 | 99 | 0 | Paternally inherited |
| BRD7 | ht | chr16:g.50354575T>C | 16 | 0.67 | 46 | 0 | 50 | 0 | Maternally inherited | |
| GDPD4 | ht | chr11:g.76979979C>A | 24 | 0.50 | 52 | 0 | 48 | 0 | Maternally inherited | |
| First‐tier | ALG8 | ht | chr11:g.77825024G>A | 45 | 0.33 | 54 | 0 | 37 | 0 | Maternally inherited |
| HNRNPAB | ht | chr5:g.177637237_177637274del | 107 | 0.40 | 132 | 0 | 131 | 0 | Maternally inherited | |
| OTOA | hm | chr16:g.21763731_21763732del | 6 | 1 | 2 | 0 | 11 | 0 | Maternally inherited | |
| SHANK3 | ht | chr22:g.51159940dup | 72 | 0.5 | 113 | 0.02 | 119 | 0.02 | De novo | |
| TANGO6 | ht | chr16:g.69056767A>C | 91 | 0.28 | 99 | 0 | 103 | 0 | Absent |
Abbreviations: Hm, homozygous, hmi, hemizygous; ht, heterozygous; htc, compound heterozygous.
A causative diagnosis was identified in 116/324 individuals (36%; Table S1, Figure 1c) and nonconclusive results in 57/324 individuals (17%). Among the 116 individuals with a causative diagnosis, variants, or CNVs were identified in known OMIM‐morbid genes in 106/116 individuals (91%) and also in nine genes newly involved in human disorders in 10/116 individuals (9%; Table S1).
In three cases of the first‐tier strategy, a parental germinal mosaïcism was highly suspected since siblings were affected and the causative variant was absent in parental pools and also not found by Sanger sequencing in healthy parental DNA blood samples (SCN1A, YWHAE, and ZMIZ1).
4. DISCUSSION
This novel study presents the feasibility and interest of a parental‐pool ES strategy for the diagnosis and molecular analysis of individuals with rare diseases.
Regarding the metrics for validity and accuracy, the overall Se and Sp for a variant to be de novo in the parental‐pool were 99% and 97%, respectively, and the PPV and NPV were of 97% and 99%, respectively, close to what is published from trio‐ES (Kong et al., 2018). These values indicate that a parental‐pool strategy is an effective and reliable strategy for the diagnosis of rare diseases, both after negative solo‐ES (yield 24%) and as a first‐tier approach (yield 37%). This strategy is easy to implement both for the laboratory technicians and for the bioinformatics pipelines. More specifically, the creation of a parental‐pool only requires dosage and precise dilution with the use of standard laboratory equipment. In addition, variant detection in the bioinformatics pipelines is barely modified since it still relies on individual rather than parental‐pool metrics. Only if a variant is detected in the individual will the pipeline report the depth and allelic balance from the parental‐pool. However, only the most recent version of GATK with HaplotypeCaller v4 can handle non‐diploid organisms and pooled data.
Nevertheless, pooling DNA complexify data analysis since the AB of one variant will be divided by two times the number of parents in the pool. Therefore, a heterozygous variant will have an expected AB of 1/12 (8.3%) in pools of 6 and 1/10 (10%) in pools of 5. These AB of less than 20% are challenging to pinpoint in pipelines devoted to constitutive variants with estimated AB of about 50% (20–70; Anand et al., 2016). One study reported a pooling strategy on 25 index cases but with a use of target enrichment sequencing and a mean depth of 1.068X (Ryu et al., 2018). The expected AB of a heterozygous variant in a pool with this depth is 0.2136% (21 reads with the variant), which was enough for the purpose of the study.
There are additional potential difficulties in the parental‐pool strategy. In ES, exon enrichment is currently incomplete, and the sequencing depth is not homogeneous. Some exonic regions can have a depth lower than 20X, leading to a maximal AB of heterozygous variants of 1.6% and 2% in a pool of 6 and 5 parental DNA samples, respectively (1 or 2 reads supporting the variants). This can be confounded with sequencing errors or reads misalignment (Bansal et al., 2010). Moreover, even if the sequencing depth could be increased to improve the precision of the parental‐pool, it is not certain that the region poorly captured at 20X will be better captured with higher depth and it will increase the sequencing cost.
Moreover, the calculation of AB was based on the assumption that there would be an equal representation of each parental allele in the pool. This hypothesis requires an exact DNA assay with effective equimolar concentrations of pool DNA samples. Over or under representation of parental DNA in the pool can lead to biased AB, and to false positive or negative results, with suspected de novo variant in the individual. Indeed, Sanger sequencing validation identified inherited variants in 3/22 of the presumed de novo variants from the second‐tier parental‐pool cohort and in 3/80 from the first‐tier parental‐pool cohort. The six heterozygous variants were considered de novo since no read with these variants were detected in both parental pools despite appropriate depths (>45) in 9/12 pools (Table 5). With these depths, the minimal expected AB was 4%, which would have been enough to detect the variants. In two cases (BRD7 and GDPD4 variants), the IGV pictures were evocative of a strand bias (Figure 3a). The OTOA variant had both parental‐pool depths below 15X (expected AB <1.5%) and fell into a low mappability region (Figure 3b). The TADAD2B variant had appropriate depth in the individual and parental‐pool, with no strand bias or misalignment (Figure 3c). The ALG8 variant was inherited from the maternal pool where the depth was lower, with an expected AB of the variant at 37X of 3% (Figure 3d). The HNRNPAB frameshift variant was a deletion of 38 nucleotides, which was not present on the parental IGV (Figure 3f).
Our overall false positive rate (FPR; 1.5%, 6/395 individuals) was lower than what has been estimated elsewhere. Indeed, the FPR in pooled DNA is thought to be 3–6.3% (Bansal et al., 2010; Ryu et al., 2018), which was estimated by comparing randomly selected variants identified in pooled individuals and genotyping results [29]. However, with three variants (2 strand bias and 1 low mappability) that could not have been verified, the overall FPR rate would have been less than 1% (3/395 individuals), or 1.4% (1/71) and 0.6% (2/324) in the second‐tier and first‐tier parental‐pool cohorts, respectively. The use of distinct enrichment kits for individuals and parental pools may also have led to more errors, since only 24% of individuals were enriched with the same kit as the pools in the second‐tier parental‐pool cohort. The FPR may also be related to the increased number of parents in the pools, resulting in lower reliability for parental AB. We also observed a false negative result due to a misalignment in SHANK3 (Figure 3g, Table 5). Therefore, unlike classical trio‐ES, we advocate that Sanger sequencing remains mandatory to confirm the segregation of the candidate variants identified with our parental‐pool strategy. With additional costs (consumable, demultiplexing), barcoding each parent before pooling could help determining segregation and improve accuracy.
Moreover, in 3/74 (4%) individuals from the second‐tier parental‐pool cohort, the ES data could not be interpreted because of an excess of de novo variants. In two cases, this difference could be explained by a discrepancy between enrichment kits used for ES in the index case and the parental‐pool, resulting in too many de novo variant. We therefore encourage laboratories that might opt for a parental‐pool strategy to ensure that individuals and parents are sequenced with the same ES enrichment kits.
The parental‐pool strategy presents additional limits. ES detected candidate CNVs in 27/395 (6.8%) affected individuals (including double hit and double diagnosis), but contrary to current trio‐ES strategy, parental‐pool ES cannot be used to establish parental segregation. Our CNV detection strategy is based on XHMM, which relies on read depth difference between samples (Fromer et al., 2012; Fromer & Purcell, 2014). Because potential CNV depth is smoothed by the other parents’ reads prior to XHMM launch, the read depth difference between samples is not sufficient for the software to detect CNV in the parental pools. Therefore, compared to a solo strategy, parental‐pooling strategy does not appear helpful for CNV interpretation. Additional methods such as qPCR or FISH are still required to establish CNV segregation in the parental samples. In addition, a parental mosaïcism was highly suspected in three cases since the variant, which was absent in parental‐pool, was also found in an affected sibling. Parental mosaïcism, which is a challenge even in classical ES with average depth of 70X and often requires high depth sequencing, cannot be detected by parental‐pool sequencing.
The contribution of ES is now undisputed in the identification of causative variants in Mendelian disorders. Nevertheless, the best approach in terms of diagnostic yield and cost‐effectiveness is not clear‐cut for laboratories choosing to currently perform ES. Extending solo‐ES analysis to non‐OMIM morbid or non‐OMIM genes can be time consuming without information about inheritance. One could focus on truncating variant in genes with pLI >0.9 and/or o/e <0.3, but interpretation would be more difficult for missense variants. One of the major advantages of the trio‐ES strategy is that, thanks to availability of parental segregation, it speeds up the selection of candidate variants such as compound heterozygous or de novo variants [Deciphering Developmental Disorders Study, 2017; Rauch et al., 2012]. Therefore, non‐OMIM (morbid) genes can be analyzed more quickly with trio‐ES, accelerating translational research. The diagnostic rate is also slightly higher, but the likelihood of identifying a known or novel molecular cause either is doubled10. In the first‐tier parental‐pool cohort, the diagnostic yield (36%) is similar to the results obtained with current trio‐ES strategy. The major interest of the parental‐pool strategy after negative solo‐ES was therefore for translational research, considering the identification of a number of highly candidate genes susceptible to be involved in novel human disorders (13/18 individuals ‐ 72%; Table S1). These genes, mostly sporadic de novo missense variants (12/13 ‐ 61.5%), were suspected because of compatible familial segregation. Their implication was strengthened by the identification of identical types of variants in individuals with overlapping phenotypes (Salpietro et al., 2019; Snijders Blok et al., 2019). This approach also has an interest for routine diagnostics since four variants in well‐known OMIM‐morbid genes initially not considered as pathogenic in solo‐ES strategy were retained as causative (Table S1): (i) two genes (CRAT, PHIP) recently implicated in rare diseases were not related to OMIM‐disorders at the time of solo‐ES analysis [Drecourt et al., 2018; Jansen et al., 2018]; (ii) the phenotypic spectrum made variants difficult to interpret in the solo strategy, especially for missense variants. For example, a sporadic ASCL1 variant (p. Glu127Lys) was identified in an individual with apnea, bradycardia, vagal hypertonia, language delay, and Hirschprung disease when ASCL1 variants have mostly been associated with congenital hypoventilation or Haddad syndrome (OMIM: 209880; de Pontual et al., 2003).
The major obstacle for laboratories performing diagnostic trio‐ES is the additional cost of parental sequencing, which, despite falling costs, remains expensive. The parental‐pool ES strategy is thus a promising compromise considering the diagnostic yield and costs. The per‐individual sequencing costs for the 5 and 6‐sample parental‐pool ES first‐tier strategy (666 and 700 dollars, respectively) is drastically lower than a first‐tier trio‐ES approach (1,500 dollars; Figure 1d). In addition, parental‐pooling reduces the number and costs of Sanger sequencing performed for parental segregation to help variant interpretation, especially for missense variants. In the second‐tier parental‐pool cohort, two thirds of the Sanger validations performed for solo‐ES interpretation would not have been needed if ES were available for the parents. Even if parental‐pool ES strategy would be more cost‐efficient than trio strategy, the calculation of the cost‐saving cannot to be limited only to sequencing and Sanger costs reduction. It depends on multiple factors (biological time for interpretation and multidisciplinary meetings, technical time and consumables for primer design, and PCR analyses). Additional in‐depth medico‐economic studies would be useful to precisely evaluate the cost‐efficiency of both strategies.
In the coming years, ES might be replaced by GS, which detects exonic regions, intronic, and structural variants more efficiently [Belkadi et al., 2015; Redin et al., 2017]. Interestingly, this strategy could be transposed to GS, but with fewer parents per pool or increased sequencing depth. Since the mean depth of GS currently used in constitutional analysis is around 30X, the expected AB at this depth would be 7.5X by pooling two parents. However, GS does not require enrichment and its sequencing depth is more uniform than ES. Therefore, additional studies would be necessary to determine the AB threshold and the number of parents that could be pooled. Similar to ES, a parental‐pool GS strategy would decrease sequencing costs and facilitate the interpretation of variants since parental segregation would be immediately available. These advantages appear essential for the analysis of the vast amount of data produced by GS.
5. CONCLUSIONS
Parental‐pool ES provides a promising alternative to trio‐ES by combining decreased parental sequencing costs, rapid parental segregation, and straightforward application to non‐OMIM (morbid) genes analysis. It is an efficient strategy for increasing diagnostic yield and accelerating translational research. This strategy could be applicable in GS, but additional feasibility and validation studies are required.
DISCLOSURES
On behalf of all authors, the corresponding author states that there is no conflict of interest.
ETHICS STATEMENT
Written consent was obtained (and archived) from all participants, to perform ES in clinical settings and to use data for research purposes. I attest that the Institutional Review Board Comité de Protection des Personnes (number GAD: DC2011‐1332) approved this study.
CONSENT FOR PUBLICATION
Informed consent was obtained from all individual participants (or from the parents) included in the study.
Supporting information
Method S1
Table S1
Table S2
ACKNOWLEDGMENTS
The authors thank the patients and their families for their participation. The authors thank the Integragen society (Evry) and Centre National de Recherche en Génomique Humaine (Atomic Energy Commission ‐ Evry) for exome sequencing. The authors also thank the University of Burgundy Centre de Calcul (CCuB) for technical support and management of the informatics platform, and the Genematcher plateform for datasharing.
Tran Mau‐Them, F. , Duffourd, Y. , Vitobello, A. , Bruel, A.‐L. , Denommé‐Pichon, A.‐S. , Nambot, S. , Delanne, J. , Moutton, S. , Sorlin, A. , Couturier, V. , Bourgeois, V. , Chevarin, M. , Poe, C. , Mosca‐Boidron, A.‐L. , Callier, P. , Safraou, H. , Faivre, L. , Philippe, C. , & Thauvin‐Robinet, C. ; Orphanomix Physician’s Group (2021). Interest of exome sequencing trio‐like strategy based on pooled parental DNA for diagnosis and translational research in rare diseases. Molecular Genetics & Genomic Medicine, 9, e1836. 10.1002/mgg3.1836
Funding information
This work was supported by grants from: the Regional Council of Burgundy (Plan d’Actions Régional pour l’Innovation ‐ PARI), the Dijon University Hospital, the ISITE‐BFC (PIA ANR), and the “Fonds Européen de DEveloppement Regional” (FEDER). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. One of the authors of this publication is a member of the European Reference Network for Developmental Anomalies and Intellectual Disability (ERN‐ITHACA)”.
DATA AVAILABILITY STATEMENT
The datasets generated during and/or analyzed during the current study are not publicly available due to size of the data and individual privacy but are available from the corresponding author upon reasonable request.
REFERENCES
- Anand, S. , Mangano, E. , Barizzone, N. , Bordoni, R. , Sorosina, M. , Clarelli, F. , Corrado, L. , Martinelli Boneschi, F. , D’Alfonso, S. , & De Bellis, G. (2016). Next Generation sequencing of pooled samples: Guideline for variants’ filtering. Scientific Reports, 6, 33735. 10.1038/srep33735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker, S. W. , Murrell, J. R. , Nesbitt, A. I. , Pechter, K. B. , Balciuniene, J. , Zhao, X. , Yu, Z. , Denenberg, E. H. , DeChene, E. T. , Wilkens, A. B. , Bhoj, E. J. , Guan, Q. , Dulik, M. C. , Conlin, L. K. , Abou Tayoun, A. N. , Luo, M. , Wu, C. , Cao, K. , Sarmady, M. , … Santani, A. B. (2019). Automated clinical exome reanalysis reveals novel diagnoses. The Journal of Molecular Diagnostics, 1, 38–48. 10.1016/j.jmoldx.2018.07.008 [DOI] [PubMed] [Google Scholar]
- Bansal, V. , Bansal, V. , & Libiger, O. (2010). A statistical method for the detection of variants from next‐generation resequencing of DNA pools. Bioinformatics, 26(12), i318–i324. 10.1093/bioinformatics/btq214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal, V. , Gassenhuber, J. , Phillips, T. , Oliveira, G. , Harbaugh, R. , Villarasa, N. , Topol, E. J. , Seufferlein, T. , & Boehm, B. O. (2017). Spectrum of mutations in monogenic diabetes genes identified from high‐throughput DNA sequencing of 6888 individuals. BMC Medicine, 15(1), 213. 10.1186/s12916-017-0977-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkadi, A. , Bolze, A. , Itan, Y. , Cobat, A. , Vincent, Q. B. , Antipenko, A. , Shang, L. , Boisson, B. , Casanova, J.‐L. , & Abel, L. (2015). Whole‐genome sequencing is more powerful than whole‐exome sequencing for detecting exome variants. Proceedings of the National Academy of Sciences of the United States of America, 112(17), 5473–5478. 10.1073/pnas.1418631112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boycott, K. M. , Rath, A. , Chong, J. X. , Hartley, T. , Alkuraya, F. S. , Baynam, G. , Brookes, A. J. , Brudno, M. , Carracedo, A. , den Dunnen, J. T. , Dyke, S. O. M. , Estivill, X. , Goldblatt, J. , Gonthier, C. , Groft, S. C. , Gut, I. , Hamosh, A. , Hieter, P. , Höhn, S. , … Lochmüller, H. (2017). International cooperation to enable the diagnosis of all rare genetic diseases. American Journal of Human Genetics, 100(5), 695–705. 10.1016/j.ajhg.2017.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruel, A.‐L. , Nambot, S. , Quéré, V. , Vitobello, A. , Thevenon, J. , Assoum, M. , Moutton, S. , Houcinat, N. , Lehalle, D. , Jean‐Marçais, N. , Chevarin, M. , Jouan, T. , Poë, C. , Callier, P. , Tisserand, E. , Philippe, C. , Them, F. T. M. , Duffourd, Y. , Faivre, L. , & Thauvin‐Robinet, C. (2019). Increased diagnostic and new genes identification outcome using research reanalysis of singleton exome sequencing. European Journal of Human Genetics, 27(10), 1519–1531. 10.1038/s41431-019-0442-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo, S. E. , Tucker, E. J. , Compton, A. G. , Kirby, D. M. , Crawford, G. , Burtt, N. P. , Rivas, M. , Guiducci, C. , Bruno, D. L. , Goldberger, O. A. , Redman, M. C. , Wiltshire, E. , Wilson, C. J. , Altshuler, D. , Gabriel, S. B. , Daly, M. J. , Thorburn, D. R. , & Mootha, V. K. (2010). High‐throughput, pooled sequencing identifies mutations in NUBPL and FOXRED1 in human complex I deficiency. Nature Genetics, 42(10), 851–858. 10.1038/ng.659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark, M. M. , Stark, Z. , Farnaes, L. , Tan, T. Y. , White, S. M. , Dimmock, D. , & Kingsmore, S. F. (2018). Meta‐analysis of the diagnostic and clinical utility of genome and exome sequencing and chromosomal microarray in children with suspected genetic diseases. NPJ Genomic Medicine, 3, 16. 10.1038/s41525-018-0053-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawkins, H. J. S. , Draghia‐Akli, R. , Lasko, P. , Lau, L. P. L. , Jonker, A. H. , Cutillo, C. M. , Rath, A. , Boycott, K. M. , Baynam, G. , Lochmüller, H. , Kaufmann, P. , Le Cam, Y. , Hivert, V. , & Austin, C. P. (2018). Progress in rare diseases research 2010–2016: An IRDiRC perspective. Clinical and Translational Science, 11(1), 11–20. 10.1111/cts.12501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Pontual, L. , Népote, V. , Attié‐Bitach, T. , Al Halabiah, H. , Trang, H. , Elghouzzi, V. , Levacher, B. , Benihoud, K. , Auge, J. , Faure, C. , & Laudier, B. (2003). Noradrenergic neuronal development is impaired by mutation of the proneural HASH‐1 gene in congenital central hypoventilation syndrome (Ondine's curse). Human Molecular Genetics, 12(23), 3173–3180. 10.1093/hmg/ddg339 [DOI] [PubMed] [Google Scholar]
- Deciphering Developmental Disorders Study . (2017). Prevalence and architecture of de novo mutations in developmental disorders. Nature, 542(7642), 433–438. 10.1038/nature21062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drecourt, A. , Babdor, J. , Dussiot, M. , Petit, F. , Goudin, N. , Garfa‐Traoré, M. , Habarou, F. , Bole‐Feysot, C. , Nitschké, P. , Ottolenghi, C. , Metodiev, M. D. , Serre, V. , Desguerre, I. , Boddaert, N. , Hermine, O. , Munnich, A. , & Rötig, A. (2018). Impaired transferrin receptor palmitoylation and recycling in neurodegeneration with brain iron accumulation. American Journal of Human Genetics, 102(2), 266–277. 10.1016/j.ajhg.2018.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldomery, M. K. , Coban‐Akdemir, Z. , Harel, T. , Rosenfeld, J. A. , Gambin, T. , Stray‐Pedersen, A. , Küry, S. , Mercier, S. , Lessel, D. , Denecke, J. , Wiszniewski, W. , Penney, S. , Liu, P. , Bi, W. , Lalani, S. R. , Schaaf, C. P. , Wangler, M. F. , Bacino, C. A. , Lewis, R. A. , … Lupski, J. R. (2017). Lessons learned from additional research analyses of unsolved clinical exome cases. Genome Medicine, 9(1), 26. 10.1186/s13073-017-0412-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewans, L. J. , Schofield, D. , Shrestha, R. , Zhu, Y. , Gayevskiy, V. , Ying, K. , Walsh, C. , Lee, E. , Kirk, E. P. , Colley, A. , Ellaway, C. , Turner, A. , Mowat, D. , Worgan, L. , Freckmann, M.‐L. , Lipke, M. , Sachdev, R. , Miller, D. , Field, M. , … Roscioli, T. (2018). Whole‐exome sequencing reanalysis at 12 months boosts diagnosis and is cost‐effective when applied early in Mendelian disorders. Genetics in Medicine, 20(12), 1564–1574. 10.1038/gim.2018.39 [DOI] [PubMed] [Google Scholar]
- Fromer, M. , Moran, J. L. , Chambert, K. , Banks, E. , Bergen, S. E. , Ruderfer, D. M. , Handsaker, R. E. , McCarroll, S. A. , O’Donovan, M. C. , Owen, M. J. , Kirov, G. , Sullivan, P. F. , Hultman, C. M. , Sklar, P. , & Purcell, S. M. (2012). Discovery and statistical genotyping of copy‐number variation from whole‐exome sequencing depth. American Journal of Human Genetics, 91(4), 597–607. 10.1016/j.ajhg.2012.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromer, M. , & Purcell, S. M. (2014). Using XHMM software to detect copy number variation in whole‐exome sequencing data. Current Protocols in Human Genetics, 81(1), 1–21. 10.1002/0471142905.hg0723s81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdan, F. F. , Myers, C. T. , Cossette, P. , Lemay, P. , Spiegelman, D. , Laporte, A. D. , Nassif, C. , Diallo, O. , Monlong, J. , Cadieux‐Dion, M. , Dobrzeniecka, S. , Meloche, C. , Retterer, K. , Cho, M. T. , Rosenfeld, J. A. , Bi, W. , Massicotte, C. , Miguet, M. , Brunga, L. , … Michaud, J. L. (2017). High rate of recurrent de novo mutations in developmental and epileptic encephalopathies. American Journal of Human Genetics, 101(5), 664–685. 10.1016/j.ajhg.2017.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley, T. , Lemire, G. , Kernohan, K. D. , Howley, H. E. , Adams, D. R. , & Boycott, K. M. (2020). New diagnostic approaches for undiagnosed rare genetic diseases. Annual Review of Genomics and Human Genetics, 21, 351–372. 10.1146/annurev-genom-083118-015345 [DOI] [PubMed] [Google Scholar]
- Jansen, S. , Hoischen, A. , Coe, B. P. , Carvill, G. L. , Van Esch, H. , Bosch, D. G. M. , Andersen, U. A. , Baker, C. , Bauters, M. , Bernier, R. A. , van Bon, B. W. , Claahsen‐van der Grinten, H. L. , Gecz, J. , Gilissen, C. , Grillo, L. , Hackett, A. , Kleefstra, T. , Koolen, D. , Kvarnung, M. , … de Vries, B. B. A. (2018). A genotype‐first approach identifies an intellectual disability‐overweight syndrome caused by PHIP haploinsufficiency. European Journal of Human Genetics, 26, 54–63. 10.1038/s41431-017-0039-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong, S. W. , Lee, I. H. , Liu, X. , Hirschhorn, J. N. , & Mandl, K. D. (2018). Measuring coverage and accuracy of whole‐exome sequencing in clinical context. Genetics in Medicine, 20(12), 1617–1626. 10.1038/gim.2018.51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J. , Gao, K. , Yan, H. , Xiangwei, W. , Liu, N. , Wang, T. , Xu, H. , Lin, Z. , Xie, H. , Wang, J. , Wu, Y. E. , & Jiang, Y. (2019). Reanalysis of whole exome sequencing data in patients with epilepsy and intellectual disability/mental retardation. Gene, 700, 168–175. 10.1016/j.gene.2019.03.037 [DOI] [PubMed] [Google Scholar]
- Nambot, S. , Thevenon, J. , Kuentz, P. , Duffourd, Y. , Tisserant, E. , Bruel, A.‐L. , Mosca‐Boidron, A.‐L. , Masurel‐Paulet, A. , Lehalle, D. , Jean‐Marçais, N. , Lefebvre, M. , Vabres, P. , El Chehadeh‐Djebbar, S. , Philippe, C. , Tran Mau‐Them, F. , St‐Onge, J. , Jouan, T. , Chevarin, M. , Poé, C. , … Thauvin‐Robinet, C. (2018). Clinical whole‐exome sequencing for the diagnosis of rare disorders with congenital anomalies and/or intellectual disability: substantial interest of prospective annual reanalysis. Genetics in Medicine, 20(6), 645–654. 10.1038/gim.2017.162 [DOI] [PubMed] [Google Scholar]
- Ng, S. B. , Buckingham, K. J. , Lee, C. , Bigham, A. W. , Tabor, H. K. , Dent, K. M. , Huff, C. D. , Shannon, P. T. , Jabs, E. W. , Nickerson, D. A. , Shendure, J. , & Bamshad, M. J. (2010). Exome sequencing identifies the cause of a Mendelian disorder. Nature Genetics, 42(1), 30–35. 10.1038/ng.499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng, S. B. , Turner, E. H. , Robertson, P. D. , Flygare, S. D. , Bigham, A. W. , Lee, C. , Shaffer, T. , Wong, M. , Bhattacharjee, A. , Eichler, E. E. , Bamshad, M. , Nickerson, D. A. , & Shendure, J. (2009). Targeted capture and massively parallel sequencing of 12 human exomes. Nature, 461(7261), 272–276. 10.1038/nature08250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippakis, A. A. , Azzariti, D. R. , Beltran, S. , Brookes, A. J. , Brownstein, C. A. , Brudno, M. , Brunner, H. G. , Buske, O. J. , Carey, K. , Doll, C. , & Dumitriu, S. (2015). The Matchmaker Exchange: A platform for rare disease gene discovery. Human Mutation, 36(10), 915–921. 10.1002/humu.22858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp, B. , Ekici, A. B. , Thiel, C. T. , Hoyer, J. , Wiesener, A. , Kraus, C. , Reis, A. , & Zweier, C. (2017). Exome Pool‐Seq in neurodevelopmental disorders. European Journal of Human Genetics, 25(12), 1364–1376. 10.1038/s41431-017-0022-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch, A. , Wieczorek, D. , Graf, E. , Wieland, T. , Endele, S. , Schwarzmayr, T. , Albrecht, B. , Bartholdi, D. , Beygo, J. , Di Donato, N. , Dufke, A. , Cremer, K. , Hempel, M. , Horn, D. , Hoyer, J. , Joset, P. , Röpke, A. , Moog, U. , Riess, A. , … Strom, T. M. (2012). Range of genetic mutations associated with severe non‐syndromic sporadic intellectual disability: An exome sequencing study. Lancet, 380(9854), 1674–1682. 10.1016/S0140-6736(12)61480-9 [DOI] [PubMed] [Google Scholar]
- Redin, C. , Brand, H. , Collins, R. L. , Kammin, T. , Mitchell, E. , Hodge, J. C. , Hanscom, C. , Pillalamarri, V. , Seabra, C. M. , Abbott, M.‐A. , Abdul‐Rahman, O. A. , Aberg, E. , Adley, R. , Alcaraz‐Estrada, S. L. , Alkuraya, F. S. , An, Y. U. , Anderson, M.‐A. , Antolik, C. , Anyane‐Yeboa, K. , … Talkowski, M. E. (2017). The genomic landscape of balanced cytogenetic abnormalities associated with human congenital anomalies. Nature Genetics, 49(1), 36–45. 10.1038/ng.3720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehm, H. L. , Bale, S. J. , Bayrak‐Toydemir, P. , Berg, J. S. , Brown, K. K. , Deignan, J. L. , Friez, M. J. , Funke, B. H. , Hegde, M. R. , & Lyon, E. (2013). ACMG clinical laboratory standards for next‐generation sequencing. Genetics in Medicine, 15(9), 733–747. 10.1038/gim.2013.92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu, S. , Han, J. , Norden‐Krichmar, T. M. , Schork, N. J. , & Suh, Y. (2018). Effective discovery of rare variants by pooled target capture sequencing: A comparative analysis with individually indexed target capture sequencing. Mutation Research, 809, 24–31. 10.1016/j.mrfmmm.2018.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salpietro, V. , Malintan, N. T. , Llano‐Rivas, I. , Spaeth, C. G. , Efthymiou, S. , Striano, P. , Vandrovcova, J. , Cutrupi, M. C. , Chimenz, R. , David, E. , Di Rosa, G. , Marce‐Grau, A. , Raspall‐Chaure, M. , Martin‐Hernandez, E. , Zara, F. , Minetti, C. , Bello, O. D. , De Zorzi, R. , Fortuna, S. , … Houlden, H. (2019). Mutations in the neuronal vesicular SNARE VAMP2 affect synaptic membrane fusion and impair human neurodevelopment. American Journal of Human Genetics, 104(4), 721–730. 10.1016/j.ajhg.2019.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz‐Abe, K. , Li, Q. , Rosen, S. M. , Nori, N. , Madden, J. A. , Genetti, C. A. , Wojcik, M. H. , Ponnaluri, S. , Gubbels, C. S. , Picker, J. D. , O’Donnell‐Luria, A. H. , Yu, T. W. , Bodamer, O. , Brownstein, C. A. , Beggs, A. H. , & Agrawal, P. B. (2019). Unique bioinformatic approach and comprehensive reanalysis improve diagnostic yield of clinical exomes. European Journal of Human Genetics, 27(9), 1398–1405. 10.1038/s41431-019-0401-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijders Blok, L. , Kleefstra, T. , Venselaar, H. , Maas, S. , Kroes, H. Y. , Lachmeijer, A. M. A. , van Gassen, K. L. I. , Firth, H. V. , Tomkins, S. , Bodek, S. , Õunap, K. , Wojcik, M. H. , Cunniff, C. , Bergstrom, K. , Powis, Z. , Tang, S. , Shinde, D. N. , Au, C. , Iglesias, A. D. , … Fisher, S. E. (2019). De novo variants disturbing the transactivation capacity of POU3F3 cause a characteristic neurodevelopmental disorder. American Journal of Human Genetics, 105(2), 403–412. 10.1016/j.ajhg.2019.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snoeijen‐Schouwenaars, F. M. , van Ool, J. S. , Verhoeven, J. S. , van Mierlo, P. , Braakman, H. M. H. , Smeets, E. E. , Nicolai, J. , Schoots, J. , Teunissen, M. W. A. , Rouhl, R. P. W. , Tan, I. Y. , Yntema, H. G. , Brunner, H. G. , Pfundt, R. , Stegmann, A. P. , Kamsteeg, E.‐J. , Schelhaas, H. J. , & Willemsen, M. H. (2019). Diagnostic exome sequencing in 100 consecutive patients with both epilepsy and intellectual disability. Epilepsia, 60(1), 155–164. 10.1111/epi.14618 [DOI] [PubMed] [Google Scholar]
- Stark, Z. , Tan, T. Y. , Chong, B. , Brett, G. R. , Yap, P. , Walsh, M. , Yeung, A. , Peters, H. , Mordaunt, D. , Cowie, S. , Amor, D. J. , Savarirayan, R. , McGillivray, G. , Downie, L. , Ekert, P. G. , Theda, C. , James, P. A. , Yaplito‐Lee, J. , Ryan, M. M. , … White, S. M. (2016). A prospective evaluation of whole‐exome sequencing as a first‐tier molecular test in infants with suspected monogenic disorders. Genetics in Medicine, 18(11), 1090–1096. 10.1038/gim.2016.1 [DOI] [PubMed] [Google Scholar]
- Tan, T. Y. , Dillon, O. J. , Stark, Z. , Schofield, D. , Alam, K. , Shrestha, R. , Chong, B. , Phelan, D. , Brett, G. R. , Creed, E. , Jarmolowicz, A. , Yap, P. , Walsh, M. , Downie, L. , Amor, D. J. , Savarirayan, R. , McGillivray, G. , Yeung, A. , Peters, H. , … White, S. M. (2017). Diagnostic impact and cost‐effectiveness of whole‐exome sequencing for ambulant children with suspected monogenic conditions. JAMA Pediatrics, 171(9), 855–862. 10.1001/jamapediatrics.2017.1755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarailo‐Graovac, M. , Shyr, C. , Ross, C. J. , Horvath, G. A. , Salvarinova, R. , Ye, X. C. , Zhang, L.‐H. , Bhavsar, A. P. , Lee, J. J. Y. , Drögemöller, B. I. , Abdelsayed, M. , Alfadhel, M. , Armstrong, L. , Baumgartner, M. R. , Burda, P. , Connolly, M. B. , Cameron, J. , Demos, M. , Dewan, T. , … van Karnebeek, C. D. (2016). Exome sequencing and the management of neurometabolic disorders. New England Journal of Medicine, 374(23), 2246–2255. 10.1056/NEJMoa1515792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran Mau‐Them, F. , Moutton, S. , Racine, C. , Vitobello, A. , Bruel, A.‐L. , Nambot, S. , Kushner, S. A. , de Vrij, F. M. S. , Lehalle, D. , Jean‐Marçais, N. , Lecoquierre, F. , Delanne, J. , Thevenon, J. , Poe, C. , Jouan, T. , Chevarin, M. , Geneviève, D. , Willems, M. , Coubes, C. , … Thauvin‐Robinet, C. (2020). Second‐tier trio exome sequencing after negative solo clinical exome sequencing: An efficient strategy to increase diagnostic yield and decipher molecular bases in undiagnosed developmental disorders. Human Genetics, 139(11), 1381–1390. 10.1007/s00439-020-02178-8 [DOI] [PubMed] [Google Scholar]
- Vissers, L. E. L. M. , de Ligt, J. , Gilissen, C. , Janssen, I. , Steehouwer, M. , de Vries, P. , van Lier, B. , Arts, P. , Wieskamp, N. , del Rosario, M. , van Bon, B. W. M. , Hoischen, A. , de Vries, B. B. A. , Brunner, H. G. , & Veltman, J. A. (2010). A de novo paradigm for mental retardation. Nature Genetics, 42(12), 1109–1112. 10.1038/ng.712 [DOI] [PubMed] [Google Scholar]
- Zhu, S. , He, M. , Liu, Z. , Qin, Z. , Wang, Z. , & Duan, L. (2020). Shared genetic susceptibilities for irritable bowel syndrome and depressive disorder in Chinese patients uncovered by pooled whole‐exome sequencing. Journal of Advanced Research, 23, 113–121. 10.1016/j.jare.2020.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, X. , Petrovski, S. , Xie, P. , Ruzzo, E. K. , Lu, Y.‐F. , McSweeney, K. M. , Ben‐Zeev, B. , Nissenkorn, A. , Anikster, Y. , Oz‐Levi, D. , Dhindsa, R. S. , Hitomi, Y. , Schoch, K. , Spillmann, R. C. , Heimer, G. , Marek‐Yagel, D. , Tzadok, M. , Han, Y. , Worley, G. , … Goldstein, D. B. (2015). Whole‐exome sequencing in undiagnosed genetic diseases: Interpreting 119 trios. Genetics in Medicine, 17(10), 774–781. 10.1038/gim.2014.191 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Method S1
Table S1
Table S2
Data Availability Statement
The datasets generated during and/or analyzed during the current study are not publicly available due to size of the data and individual privacy but are available from the corresponding author upon reasonable request.