

Abstract
Synthetic indole cannabinoids characterized by a 2′,2′-dimethylindan-5′-oyl group at the indole C3 position constitute a new class of ligands possessing high affinity for human CB2 receptors at a nanomolar concentration and a good selectivity index. Starting from the neutral antagonist 4, the effects of indole core modification on the pharmacodynamic profile of the ligands were investigated. Several N1 side chains afforded potent and CB2-selective neutral antagonists, notably derivatives 26 (R1 = n-propyl, R2 = H) and 35 (R1 = 4-pentynyl, R2 = H). Addition of a methyl group at C2 improved the selectivity for the CB2 receptor. Moreover, C2 indole substitution may control the CB2 activity as shown by the functionality switch in 35 (antagonist) and 49 (R1 = 4-pentynyl, R2 = CH3, partial agonist).
Graphical Abstract
INTRODUCTION
The endocannabinoid system (ECS) is implicated in a large spectrum of physiological and pathological processes, especially ones related to the central and peripheral nervous systems.1-10 Part of the complex intercellular communicating system are two types of heptahelical G-protein coupled receptors identified as cannabinoid receptor type 1 (CB1) and cannabinoid receptor type 2 (CB2).11-14 CB1 is expressed primarily in the central nervous system14,15 but can also be found in locations outside the brain.16-19 CB2, which is sometimes called the peripheral cannabinoid receptor, is expressed mainly in the cells associated with the immune system.11,14 CB2 receptors are also found in low concentrations in activated microglia and infiltrated macrophages in the brain,20,21 gastrointestinal tract tissues and other cells such as vascular endothelial cells, cardiomyocytes, and bone cells.22
Small-molecule modulators of CB2 receptor activity are of interest for therapeutic indications including neurological disorders, pain, and inflammation. CB2 agonists have potential utility for treating neurological disorders such as reperfusion brain injury;33 amyotrophic lateral34 and multiple sclerosis;35,36 and Alzheimer’s,37,38 Parkinson’s,37,38 and Huntington’s diseases.37-39 CB2 agonists also exhibit anti-inflammatory effects and antinociceptive effects in acute pain, persistent inflammatory pain, postoperative pain, cancer pain, and neuropathic pain,6,40-42 and they may aid in cardioprotection and treatment of gliomas.20,43-45 Interestingly, CB2 inverse agonists are also of interest for modulating inflammatory nociception.46,47 This seemingly contradictory potential for agonists and inverse agonists in similar indications stems from the ability of the CB2 receptors to mediate different processes depending on the conditions: recruitment of immune cells or interference with the action of other chemoattractants.46 Neutral antagonists would similarly be of great interest in this regard. CB2 neutral antagonists are relatively rare48 and as such have received less attention. However, there is some evidence that CB2 neutral antagonists may be promising in the treatment of obesity-associated inflammation, insulin resistance, and nonalcoholic fatty liver disease.49 Selective CB2 receptor modulators are needed across the full spectrum of functional activities for therapeutic targeting of the ECS.
Medicinal chemistry efforts targeting the ECS have been complicated by psychoactivity of CB1-receptor agonists. These side effects are associated with drugs of abuse. Many CB1 agonists developed in academic and pharmaceutical research labs have been hijacked for abuse in the recreational drug market.23-26 The illicit demand for cannabinoid agonists has been met with regulatory controls on their supply, which incidentally also restricts legitimate pharmaceutical research.27 CB2-selective ligands are attractive for chemotherapeutic development due in part to the absence of known psychoactive side effects,31,32 but many compounds designed to target CB2 receptors50 have nonetheless ended up as Schedule I controlled substances,51 presumably due to (off-target) CB1 interactions.
While marijuana-like psychoactivity is a primary factor behind the potential for abuse of many ECS modulators, ease of synthesis is an auxiliary supply-side factor behind the illicit production and distribution. For example, the simple two-step synthesis of JWH-018 (Scheme 1)28 increases its potential for abuse.29,52 Many 1,3-disubstituted indoles like JWH-018 can be made from indole and two simple electrophiles (e.g., acyl and alkyl halides), and many similarly have emerged as drugs of abuse.53 Conversely, less synthetically accessible cannabinoid ligands are less subject to abuse. Such compounds enable one to explore the chemotherapeutic potential of ECS modulators without the same risk of fueling the recreational drug market.
Scheme 1. Synthesis of JWH-018a.
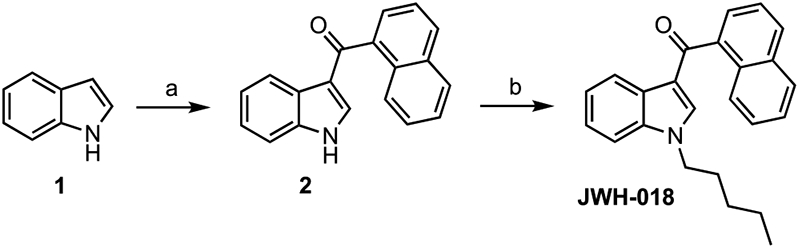
aReagents and conditions: (a) MeMgBr, Et2O/THF, 0 °C, then 1-naphthoyl Cl, reflux. (b) n-Pentyl Cl, KOH, DMSO, 80 °C.
There is a need for novel ECS modulators, particularly ones having low potential for abuse. To this end, we designed ligands that incorporate 2,2-dimethylindane into the known indole cannabinoid framework. 2,2-Dimethylindanes blend the benzenoid ring system of the traditional Huffman-(JWH)-type compounds, with the bulky cycloalkane motifs of influential54 CB2-selective indole cannabinoids from Abbott.30 2,2-Dimethylindane carboxylates can be crafted using recent synthetic methodologies,55,56 but they are less accessible than the commercially available building blocks needed to prepare many indole cannabinoid drugs of abuse. We expect our designs to support development of novel ECS modulators specifically for legitimate academic and pharmaceutical research endeavors and without risk of “significant diversion”52 to illicit markets.
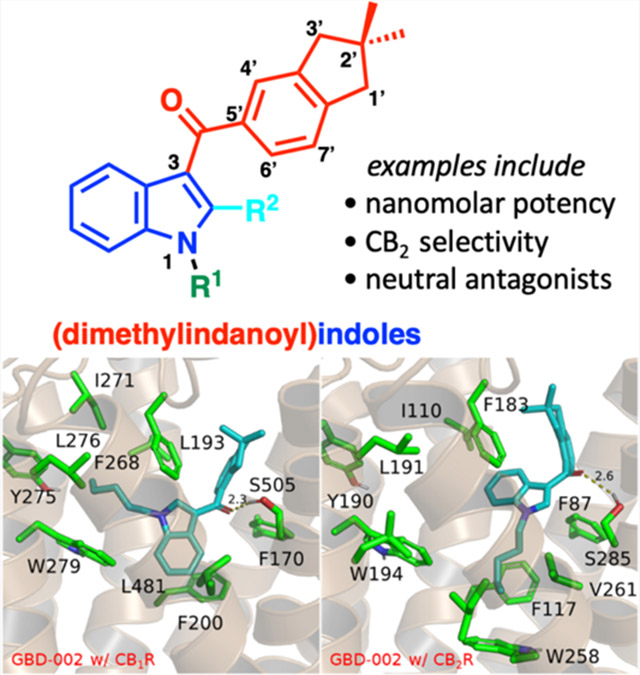
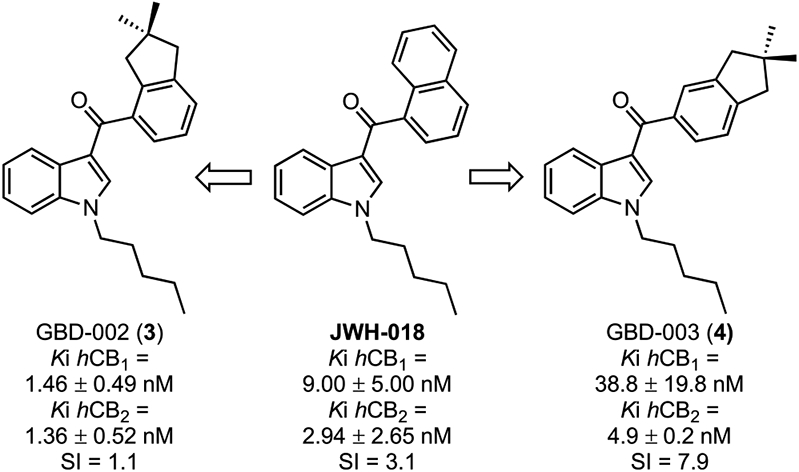
Indanoylindoles have not been examined previously as cannabinoid ligands. The first two indanoylindoles we considered—GBD-002 and GBD-003, Figure 1—indeed exhibit high affinities for both CB receptors, with GBD-003 showing selectivity for CB2 (Figure 1). Functional assays showed that GBD-002, like JWH-018, is a full agonist, whereas positional isomer GBD-003 is a neutral antagonist (Table 2, vide infra). Considering that CB2 antagonists are promising but rare, we focused on preparing a series of indanoylindoles based on GBD-003 for preliminary molecular pharmacology and structure–activity relationship (SAR) studies. Several CB2-selective neutral antagonists are described herein.
Figure 1.
Rational design of a novel class of potent cannabinoids.
Table 2.
Calculated Binding Free Energies (kcal/mol) Compared to Experimental Binding Free Energy (kcal/mol)
| rCB1 |
rCB2 |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ligand | Ki (nM) | ΔGPBa | ΔGexpb | ΔGcorrc | Ki (nM) | ΔGPBa | ΔGexpb | ΔGcorrc | ΔΔGexpe | ΔΔGcorra,d |
| GBD-002 | 1.46 | −41.92 | −12.13 | −12.65 | 1.36 | −40.79 | −12.17 | −12.28 | 0.04 | 0.37 |
| GBD-003 | 38.80 | −34.00 | −10.17 | −10.07 | 4.90 | −35.81 | −11.41 | −10.66 | 1.23 | 0.59 |
| GBD-005 | 2559.00 | −27.79 | −7.68 | −8.04 | 130.00 | −31.10 | −9.45 | −9.12 | 1.78 | 1.08 |
| GBD-013 | 152.00 | −32.25 | −9.36 | −9.50 | 15.60 | −35.26 | −10.72 | −10.48 | 1.36 | 0.98 |
| GBD-014 | 325.00 | −31.22 | −8.91 | −9.16 | 24.00 | −35.93 | −10.46 | −10.70 | 1.55 | 1.54 |
| GBD-029 | 1904.00 | −27.89 | −7.85 | −8.07 | 62.50 | −34.65 | −9.89 | −10.28 | 2.04 | 2.21 |
| rmsdf | 0.23 | rmsdf | 0.43 | |||||||
Calculated binding free energies obtained from the MM-PBSA calculations without any empirical correction.
The experimental binding free energy was converted from the experimental Ki using the thermodynamic equation ΔGexp = −RT ln (Ki).
Calculated binding free energies after empirical correction using eq 1.
ΔΔGcorr is the corrected binding free-energy difference [ΔG(CB1R) − ΔG(CB2R)].
ΔΔGexp = −RT ln (Ki (CB1R)/Ki(CB2R)) = ΔGexp(CB1R) − ΔGexp(CB2R).
The root-mean-square deviation (RMSD) of the corrected ΔG binding energy values versus the experimental ΔG binding energy values.
RESULTS AND DISCUSSION
Chemistry.
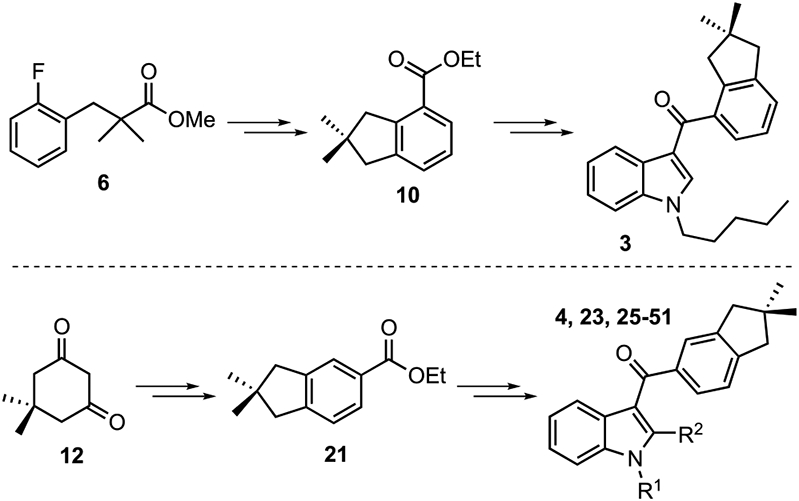
We prepared indanoylindoles from carboxylates 10 and 21 (Scheme 2). The synthesis of GBD-002 is outlined in Scheme 3. Starting from methyl isobutyrate (5), α-substitution with 2-fluorobenzyl bromide generates ester 6 (81%), which on reduction with DIBAL-H is converted to alcohol 7 in 88% yield. The alkyl iodide precursor (9) of indane 10 is obtained through a modified Finkelstein reaction in 81% overall yield. The two-step sequence involves mesylation of the 1° alcohol (7), followed by nucleophilic displacement with NaI-saturated N-methylpyrrolidone (NMP) to provide iodide 9.
Scheme 2. Overview of the Synthetic Approach.
Scheme 3. Synthesis of Benzoate 10, the Precursor to 3-(Indan-4-oyl)indole GBD-002a.

aReagents and conditions: (a) 2-fluorobenzyl bromide, LDA, THF, −78 °C to rt, 5 h, 81%. (b) DIBAL-H, THF, −78 °C to rt, 1 h, 88%. (c) MsCl, Et3N, CH2Cl2, 0 °C, 1 h, 91%. (d) NaI, NMP, 140 °C, 8 h, 89%. (e) (i) t-BuLi, n-pentane-Et2O (4:1 by vol), −78 °C, 15 min; (ii) THF, −78 °C; (iii) −78 °C to rt to −78 °C; (iv) ClCO2Et, −78 °C (15 min) to rt (15 min).
Indane-4-carboxylate 10 is prepared by treating 9 with tert-butyllithium (t-BuLi), followed by ethyl chloroformate under carefully controlled conditions.57 This transformation is envisioned to proceed by a five-step, one-pot process, starting with lithium–iodine exchange and lithiation ortho to the fluoro substituent to produce a dilithio intermediate. Release of lithium fluoride generates a benzyne-tethered alkyllithium that undergoes 5-exo-dig cyclization to the 4-lithiated indane, which is then trapped with ethyl chloroformate to produce 10.
Meanwhile, our lab was developing a synthesis of diyne monoester 16 (Scheme 4).55 Dimedone (12) is condensed with triflic anhydride and reacted with the lithium enolate of ethyl acetate for a Claisen-type tandem addition/fragmentation to generate β-keto ester 14, which is then dehydrated to give 16. Rhodium-catalyzed cyclotrimerization of 16 with TMS-acetylene provides a mixture of regioisomers (17 and 18), which can be hydrolyzed to 10 and then to 11. Acid 11 is converted to the acid chloride and then coupled with N-pentylindole to complete the synthesis of GBD-002 (3).
Scheme 4. Synthesis of the 3-(Indan-4-oyl)indole GBD-002a.
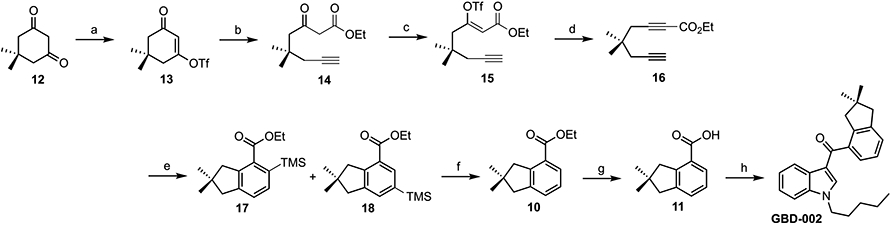
aReagents and conditions: (a) Tf2O, pyridine, CH2Cl2, −78 °C to rt, 20 min, 97%. (b) (i) lithio ethyl acetate, THF, −78 °C, 20 min; (ii) LHMDS, 0 °C (10 min) to rt (1 h), 63%. (c) Tf2O, aq. LiOH, hexanes, 0 °C to rt, 30 min, 88%. (d) LHMDS, THF, 0 °C to rt, 1 h, 63%. (e) (i) [Rh(cod)2]BF4, (±)-BINAP, H2, CH2Cl2, rt, 1 h; (ii) TMS-acetylene, CH2Cl2, rt, 1 h, 90%. (f) KI, TMSCl, H2O, CH3CN, 60 °C, 12 h, 68%. (g) KOH, H2O, EtOH, rt, 8 h, 95%. (h) (i) SOCl2, DMF, CH2Cl2, rt, 8 h; (ii) N-pentylindole, HFIP, rt, 24 h, 68%.
The synthesis of 3-(indan-5-oyl)indoles also begins with dimedone (12, Scheme 5) and builds on prior synthetic methodologies from the Dudley lab.56,58 Vinylogous acyl triflate 13 is reduced with DIBAL-H to produce hydroxycycloalkenyl triflate 19, which undergoes tandem fragmentation/olefination to furnish dienyne 20. Rhodium-catalyzed cycloisomerization of 20, followed by oxidation with DDQ, provides 21 in a one-pot process. Saponification of 21 gives acid 22, which is converted to the acid chloride for Friedel–Crafts acylation of indole and 2-methylindole (Scheme 4, R2 = H, Me). The resulting 3-acylindoles (23 and 24) were then N-alkylated to produce a small collection of indanoylindoles, designated as GBD-003 thru GBD-031.
Scheme 5. Synthesis of 3-(Indan-5-oyl)indolesa.
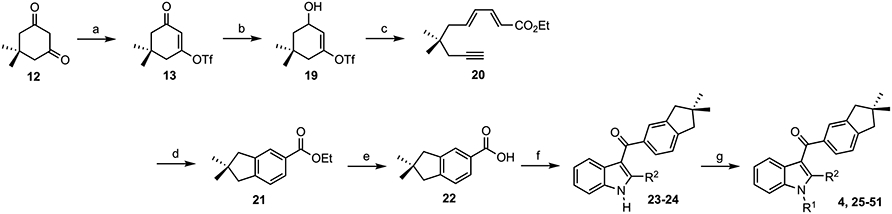
aReagents and conditions: (a) Tf2O, pyridine, CH2Cl2, −78 °C to rt, 20 min, 97%. (b) DIBAL-H, THF, −78 °C, 1–2 h, 90%. (c) Ethyl-E-4-(diethoxyphosphoryl)but-2-enoate, LHMDS, THF, −78 °C (15 min) to rt to 60 °C (1 h), 82%. (d) (i) [Rh(nbd)Cl]2, AgSbF6, CH2Cl2, rt, 1 h; (ii) DDQ, rt, 2 h, 85%. (e) KOH, H2O, EtOH, rt, 8 h, 96%. (f) (i) SOCl2, DMF, CH2Cl2, rt, 8 h; (ii) indole or 2-methylindole, HFIP, rt, 24 h, 53–58%. (g) Alkyl halide, NaH, DMSO, rt, 8 h.
We routinely produce gram quantities of indane-5-carboxylate 21 as a synthetic building block for other projects in the lab, and thus, it serves for us as a readily available and convenient starting point for the synthesis of various indanoylindoles. However, the four-step synthesis effectively ensures that indole cannabinoids derived from 21 will not be cost-competitive with ones that have been co-opted by the recreational drug market (e.g., JWH-018) and thus are unlikely to be subject to widespread abuse as recreational drugs.
Receptor Binding Affinity and SAR.
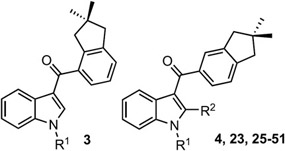
The indanoylindoles thus prepared are high-affinity cannabinoid ligands. Initially, a screen to detect compounds with affinity for human CB1 (hCB1 and human CB2 (hCB2) receptors was conducted by determining the ability of a relatively high concentration (1 μM) of each compound to displace a 0.2 nM concentration of the well-characterized nonselective CB1/CB2 cannabinoid radioligand [3H]CP-55,940 from its binding site (Table 1). Under these conditions, the concentration of a compound producing 50% displacement would approximate the affinity (e.g., Ki) for the receptor being examined, with higher displacement associated with greater affinity for the CB receptors. Compounds producing less than 50% displacement are predicted to exhibit low affinity (less than 1 μM). Selected compounds showing promising affinity and/or selectivity were selected for determination of Ki and selectivity index (SI) (Figure 2) and/or were screened for functional activity (vide infra; Table 3).
Table 1.
Screen of (Dimethylindanoyl)indole Derivatives for Binding to hCB1 and/or hCB2 Receptors
|
||||
|---|---|---|---|---|
| % displacement of [3H]CP-55,940 (0.2 nM) (1 μM of each compound) |
||||
| compound | R1 | R2 | hCB1 | hCB2 |
| GBD-002 (3)a,b | Pentyl | H | 85.7 ± 5.2 | 94.1 ± 2.3 |
| GBD-003 (4)a,b | pentyl | H | 93.4 ± 5.0 | 99.8 ± 0.9 |
| GBD-001 (23) | H | H | 9.4 ± 3.1 | 22.2 ± 6.0 |
| GBD-004 (25) | methyl | H | 10.8 ± 1.4 | 23.9 ± 13.0 |
| GBD-005 (26)a,b | propyl | H | 47.4 ± 0.3 | 90.5 ± 2.5 |
| GBD-006 (27)b | butyl | H | 82.7 ± 0.6 | 92.9 ± 1.3 |
| GBD-007 (28)b | hexyl | H | 79.1 ± 3.0 | 83.6 ± 4.9 |
| GBD-008 (29) | isobutyl | H | 42.2 ± 4.9 | 83.1 ± 2.4 |
| GBD-009 (30)b | benzyl | H | 69.2 ± 2.3 | 84.6 ± 3.3 |
| GBD-010 (31)b | 4-fluorobenzyl | H | 88.0 ± 1.4 | 92.3 ± 2.5 |
| GBD-011 (32)b | E-2-pentenyl | H | 84.2 ± 1.2 | 86.9 ± 3.9 |
| GBD-012 (33)b | Z-2-pentenyl | H | 60.7 ± 4.8 | 86.4 ± 5.0 |
| GBD-013 (34)a,b | 4-pentenyl | H | 80.0 ± 3.8 | 95.2 ± 2.9 |
| GBD-014 (35)a,b | 4-pentynyl | H | 71.0 ± 6.4 | 92.8 ± 1.6 |
| GBD-015 (36) | 2-pentynyl | H | 55.6 ± 5.7 | 65.9 ± 2.8 |
| GBD-016 (37) | allyl | H | 16.0 ± 5.3 | 60.6 ± 5.4 |
| GBD-017 (38) | propargyl | H | 33.4 ± 3.3 | 56.0 ± 3.6 |
| GBD-018 (39) | CH2CO2CH2CH3 | H | 19.4 ± 7.7 | 18.0 ± 10.9 |
| GBD-020 (40) | propyl | methyl | 44.4 ± 1.9 | 73.9 ± 3.4 |
| GBD-021 (41) | butyl | methyl | 43.5 ± 0.6 | 85.4 ± 3.9 |
| GBD-022 (42) | pentyl | methyl | 53.8 ± 0.8 | 83.6 ± 3.1 |
| GBD-023 (43) | hexyl | methyl | 26.2 ± 1.6 | 55.0 ± 5.9 |
| GBD-024 (44) | benzyl | methyl | 18.1 ± 13.3 | 51.2 ± 11.1 |
| GBD-025 (45) | 4-fluorobenzyl | methyl | 38.4 ± 5.0 | 48.8 ± 0.0 |
| GBD-026 (46) | E-2-pentenyl | methyl | 35.1 ± 3.3 | 73.7 ± 3.8 |
| GBD-027 (47) | Z-2-pentenyl | methyl | 43.0 ± 2.0 | 82.9 ± 3.4 |
| GBD-028 (48) | 4-pentenyl | methyl | 38.6 ± 1.8 | 86.4 ± 3.3 |
| GBD-029 (49)a,b | 4-pentynyl | methyl | 41.9 ± 2.2 | 93.1 ± 3.6 |
| GBD-030 (50) | 2-pentynyl | methyl | 16.4 ± 3.1 | 30.2 ± 2.7 |
| GBD-031 (51) | propargyl | methyl | 51.9 ± 0.8 | 63.0 ± 6.8 |
This compound was selected for determination of Ki.
This compound was selected for screening of functional activity.
Figure 2.
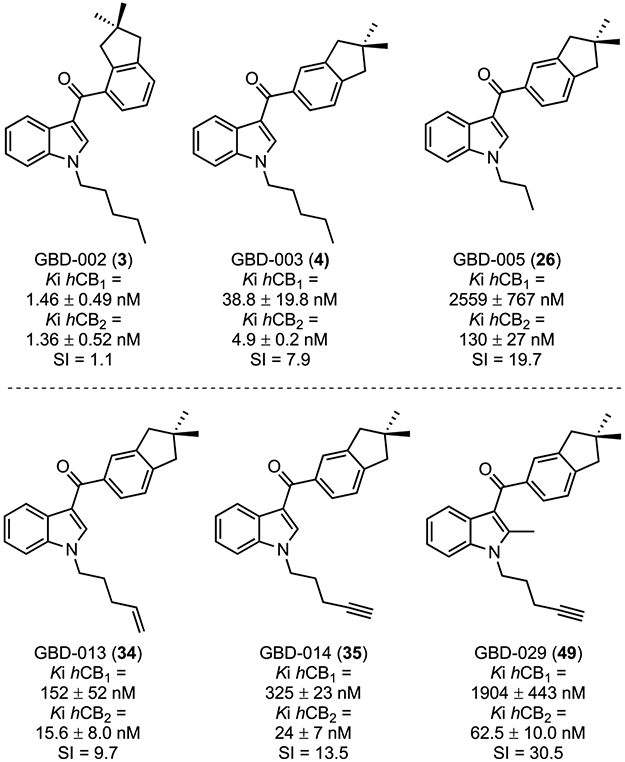
Affinities (Ki) of selected derivatives for hCB1 and hCB2 receptors.
Table 3.
[35S]-GTPγS Binding Assays of Selected and Reference Compounds for Efficacy at the hCB2 Cannabinoid Receptors
| compound | [35S]-GTPγS specific binding (control = 100%)a |
|---|---|
| CP-55,950 | 131 ± 6** |
| AM-630 | 67 ± 4** |
| GBD-002 (3) | 134 ± 5* |
| GBD-003 (4) | 108 ± 9 |
| GBD-005 (26) | 99 ± 3 |
| GBD-006 (27) | 102 ± 4 |
| GBD-007 (28) | 102 ± 9 |
| GBD-009 (30) | 87 ± 8 |
| GBD-010 (31) | 90 ± 3 |
| GBD-011 (32) | 81 ± 2** |
| GBD-012 (33) | 106 ± 7 |
| GBD-013 (34) | 92 ± 2* |
| GBD-014 (35) | 96 ± 5 |
| GBD-029 (49) | 121 ± 8 |
The results are expressed as the percentages of stimulation of [35S]-GTPγS binding (the basal value set at 100%) obtained for a concentration of ligands of 10 μM. Data are the mean ± standard error of the mean of three experiments. Statistical significance from 100% (basal) as assessed by one-sample t-test
P < 0.05
P < 0.01.
GBD-002 and GBD-003 produced almost 100% displacement of the radioligand from both hCB1 and 1CB2 receptors in initial binding screens (Table 1) and (as predicted from these initial screens) were subsequently determined to exhibit high affinity for cannabinoid receptors (CBRs) (Figure 2). Specifically, GBD-002 exhibited a Ki (hCB1) of 1.46 ± 0.49 nM and a Ki (hCB2) of 1.36 ± 0.52 nM; however, with an SI of only 1.1 (nonselective). The positional isomer GBD-003 is also a high-affinity ligand for both receptors (hCB1 Ki = 38.8 ± 19.8 nM and hCB2 Ki = 4.9 ± 0.2 nM), with improved selectivity for CB2 (SI = 7.9). Having determined that GBD-003 binds to CB2 receptors in a relatively selective manner, we investigated the broader series of indanoylindoles for binding to CB1 and CB2 receptors (Table 1). The N1-pentyl chain was first replaced with different lipophilic alkyl moieties including chains based on other JWH cannabinoids28,30,50,53,59 and novel N-alkyl groups among reported indole cannabinoids such as Z-2-pentenyl, 2-pentynyl, 4-pentynyl, and propargyl. The analogue lacking the N-substituent (23) showed little affinity for either receptor. A similar activity profile was found when the H was substituted with a methyl group (25). Replacement with longer straight alkyl chains afforded compounds with good to excellent CBR affinities, consistent with previously reported links between chain length and receptor binding in other synthetic indole cannabinoids.59 Compounds 26 (propyl), 27 (butyl), and 4 (pentyl) exhibited increasing CBR affinity with increasing chain length until lower affinity was observed for the 6-carbon (28, hexyl) variant.
Introduction of an isobutyl substituent (29) led to poor affinity for CB1 (42% displacement) and good affinity and selectivity for CB2 (83% displacement). Comparison of isobutyl 29 with its unbranched isomer (27, n-butyl) and desmethyl analogue (26, n-propyl) suggests that this branching disfavors CB1 receptor binding and enhances CB2 selectivity.
Compounds 30 (benzyl) and 31 (4-fluorobenzyl) bearing aromatic rings in the N-alkyl substituents displayed CBR affinities comparable to those of the aliphatic counterparts 27 (butyl) and 28 (hexyl) but less than that of 4 (pentyl), suggesting some steric tolerance of the receptors. Selectivity for binding to either CB1 or CB2 receptors was modest at best in these benzyl variants.
Replacement of the alkyl chain with a more hydrophilic group has a profound effect on the pharmacology of the ligands. Compound 39, punctuated with an ester group of a similar length [−CH2C(O)OCH2CH3] as compared with n-pentyl, showed a complete loss of affinity and selectivity (only 19 and 18% displacement at CB1 and CB2, respectively). This observation is consistent with the results reported by Hynes et al.,60 Frost et al.,30 and Longworth et al.61 for fenchyl amide indoles, cyclopropanecarbonyl indoles, and adamantyl amide indoles, respectively, in which polar N1 side chains generally cause a decrease in CB2 receptor binding relative to the n-pentyl analogue.
Another set of compounds was synthesized to evaluate the effect of unsaturation in the alkyl chain, starting with unsaturated versions of the three- and five-carbon straight alkyl chains that appear to provide good CB2 affinity and selectivity. Addition of double bonds to the pentyl chain yielded compounds 32–34, bearing the E-2-pentenyl, Z-2-pentenyl, and 4-pentenyl moieties, respectively. Although less potent than the n-pentyl analogue 4, the pentenyl compounds still retained good receptor affinities, particularly at CB2 (Table 1). The Z-isomer (33) was more selective than the E-isomer (32), and terminal alkene isomer 34 was more potent and CB2-selective. The alkynyl analogues 35 (4-pentynyl) and 36 (2-pentynyl) showed slightly lower affinities than the alkenyl analogues, although terminal alkyne 35 showed promising selectivity for CB2. In the three-carbon unsaturated series, the allyl (37) and propargyl (38) analogues exhibited modest CB2 affinities displacing only 61 and 56% of [3H]-CP-55,940, respectively, compared to the corresponding saturated counterpart 26 (propyl, 90% displacement).
We next examined N-substitution in the 2-methylindole series, inspired by the potent CBR ligand WIN-55,212-2. The 2-methylindoles generally showed lower affinity for both receptors but better CB2 selectivity compared to the analogues devoid of the additional substituent. Like the trend in the indole series, CB2 binding increased with the length of the N1-alkyl chain up to 5 carbons (40–43), whereas CB1 affinity was largely unaffected by these modifications in chain length. Aung et al.59 reported that the presence of a methyl group at C2 of the N1-propyl analogue enhanced CB2 selectivity (JWH-072, SI = 6 vs JWH-015, SI = 24). However, in this indanoylindole scaffold, 2-methylation has a negative impact (26 vs 40, Table 1). Neither of the N-arylmethyl variants (44 and 45) offered any apparent advantages in these initial assays, nor did the 2-pentynyl (50) or propargyl (51) variants. On the other hand, the pentenyl derivatives (46–48) showed an increase in CB2 affinity without a substantive effect on CB1 affinity, leading to enhanced selectivity, and the 4-pentynyl side chain of 49 showed good affinity and selectivity for CB2.
Guided by preliminary screening data from Table 1, we chose a small set of ligands and determined their binding affinities (Ki) for both CB receptors to determine an SI, as outlined in Figure 2. Data for JWH-018 were reported previously59 and are provided for reference in Figure 1. The N-pentyl indanoylindoles (3 and 4) were the most potent, and N-(4-pentynyl)-2-methylindole 49 was the most CB2-selective, having an hCB2 Ki of 62.5 ± 10.0 nM and SI = 30.5.
Molecular Modeling.
The six compounds highlighted in Figure 2 were also examined computationally in combined molecular docking/molecular dynamics (MD) simulations (Figure 3), which correlated with experimentally measured binding affinities (Ki, cf. Table 2 and Figure S1). The good correlation between experimental and computational data suggests that this computational protocol provides the opportunity to test future designs in silico.
Figure 3.

Binding modes of selected derivatives in CB1 (A1–F1) and CB2 (A2–F2) receptors. (A1) GBD-002 (3), CB1; (A2) GBD-002 (3), CB2; (B1) GBD-003 (4), CB1; (B2) GBD-003 (4), CB2; (C1) GBD-005 (26), CB1; (C2) GBD-005 (26), CB2; (D1) GBD-013 (34), CB1; (D2) GBD-013 (34), CB2; (E1) GBD-014 (35), CB1; (E2) GBD-014 (35), CB2; (F1) GBD-029 (49), CB1; (F2) GBD-029 (49), CB2. Original crystal structures of CB1R (5XR8) and CB2R (5ZTY) were obtained from the Protein Data Bank for modeling. Pivotal to the difference in binding modes of these compounds between CB1R and CB2R are the residue changes L193/I110 and L481/V261 (in CB1R and CB2R, respectively). These substitutions allow for the central indole scaffold of the GBD compounds to rotate, allowing for the dimethylindane moiety and its connecting ketone to engage in hydrogen bonding with S285. Modifications to the aliphatic chain such as truncation (C1-2) and dehydrogenation (D1-2, E1-2) reduce hydrophobic interactions with the nearby hydrophobic pockets in CB1R and CB2R. The resulting indole moiety rotation due to the residue changes between CB1R and CB2R also allows for substitutions (F1–F2) on the indole moiety with CB2R that are otherwise unavailable in CB1R.
Binding of GBD-002 with CB1R showed a strong hydrogen bond with the nearby S505 via the carbonyl oxygen and π−π stacking interactions between the indole ring with F200 and the indane ring with F268 (Figure 3A1). In the CB2R model, the indane ring interacted similar to the CB1 receptor in the lipophilic pocket formed by F183, S285, and F87 resulting in the retention of hydrogen bond (Figure 3A2). Meanwhile, the nonconserved residues V261 and I110 (substituted with L482 and L193 in the CB1 receptor, respectively) allowed more room for the indole core to relax and rotate, changing the binding angle in the central transmembrane binding site. The indane ring of the positional isomer GBD-003 induced a steric strain in CB1R, shifting the moiety closer to the ECL2 domain (Figure 3B1). The shift rotated the carbonyl group away from S505 which makes hydrogen bonding unfavorable. In CB2R however, the loss in affinity was not observed as I110 and V261 still allowed strong π−π stacking interactions with F183 and hydrogen bonds with S285 (Figure 3B2).
A >60-fold reduction in CB1R affinity was observed in GBD-005, with the propyl tail being too short to occupy the binding pocket delimited by I271, L276, and Y275 and create effective van der Waals interactions (Figure 3C1). The truncation of the lipophilic tail is not as deleterious within CB2R as the scaffold is still able to retain the hydrogen bond (Figure 3C2). The greater loss of CB1 binding affinity translates into greater CB2 selectivity. GBD-013 and GBD-014 have similar binding modes to GBD-003. Although the three compounds have N-substituents with similar lengths, the double bond in GBD-013 weakens the hydrophobic interactions in CB2R (Figure 3D2) and more so in CB1R (Figure 3D1), resulting in a 10-fold selectivity. Hydrophobic interactions of the 5 C chain with the hydrophobic channel in CB1R further reduced with the substitution of a triple bond (GBD-014, Figure 3E1,E2).
2-Methylation of the indole core (GBD-029) positioned the ligand such that the additional methyl group clashes with L193 in CB1R (Figure 3F1). This crowding disrupts the preferred binding mode and substantially weakens the binding affinity. In CB2R (Figure 3F2), on the other hand, the steric constraint is less prominent as the rotation of the indole ring places the methyl group in an open space between transmembrane helices 5 and 6, thereby limiting the steric strain between the ligand and protein, affording a higher potency and selectivity against CB2R.
The experimental and computational binding data are compiled in Table 2 and depicted graphically in Figure S1. Experimental binding affinities (Ki) were converted into free energies of binding (ΔGexp), and absolute magnitudes of raw calculated protein binding energies (ΔGPB) were adjusted using a standard correction (ΔGcorr) to align with the experimental data using eq 1
| (1) |
The difference between the CB1R and CB2R binding energies defines selectivity as measured experimentally (ΔΔGexp) and estimated computationally (ΔΔGcorr). We see good correlation (R2 = 0.94) between the calculated (ΔGPB) and measured (ΔGexp) binding energies (Figure S1, top), suggesting that SARs can be used to estimate binding affinities (and selectivities, Figure S1, bottom) with a reasonable degree of confidence.
CB2R Functional Activity.
Several compounds in the indole series and 49, the most selective in the 2-methylindole series, were subjected to in vitro pharmacological evaluation in a [35S]GTPγS binding assay to evaluate the intrinsic functional activity. The CBR-mediated G-protein activities of the selected compounds at CB2R as well as reference ligands CP-55,940 (CB2 full agonist) and AM-620 (CB2 inverse agonist) were measured by examining the ability of a single, high (near receptor-saturating) 10 μM concentration to modulate [35S]GTPγS binding in hCB2-transfected CHO-cells as previously described.62
The molecular geometry of the indanoyl moiety has a striking effect on hCB2R-mediated G-protein activation (Table 3). Consistent with other indole cannabinoids, GBD-002 increased [35S]GTPγS binding, indicative of the full agonist property (134 ± 5% basal). When the indane point of attachment was moved from C4 (GBD-002) to C5 (GBD-003), the indanoylindane evoked little if any agonist response (108 ± 9% basal) compared to the reference full agonist CP-55,940 (131 ± 6% basal). This lack of significant agonist activity suggests that GBD-003 may act as a neutral hCB2R antagonist. Co-incubation of GBD-003 in CHO-hCB2 membranes produced a concentration-dependent reduction in [35S]GTPγS binding elicited by CP-55,940 (Figure 4). The ability to reverse the response of a full agonist (CP-55,940, IC50 = 1.8 μM, Kb = 9.7 nM) indicates that GBD-003 is best characterized as a CB2R antagonist.
Figure 4.
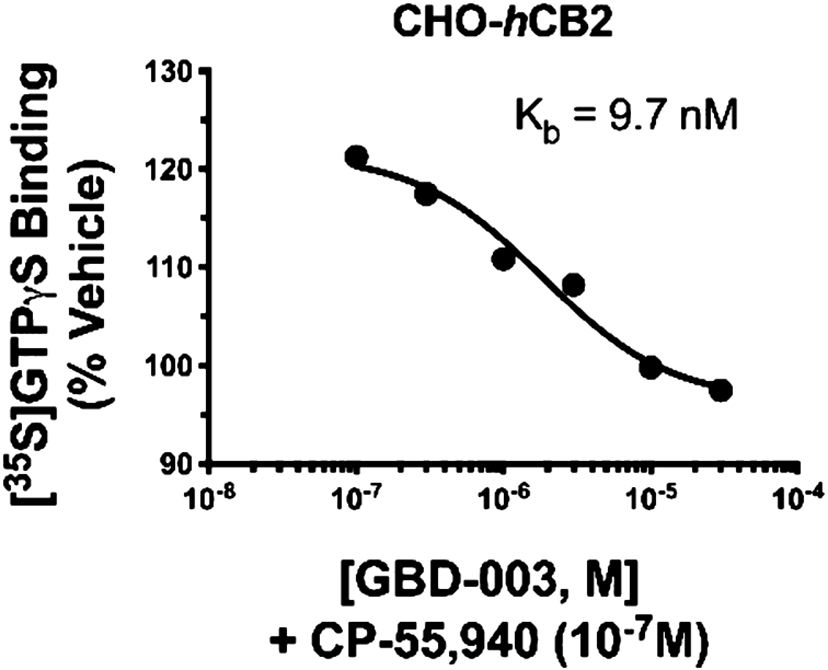
GBD-003 acts as an antagonist at CHO-hCB2 receptors, producing concentration-dependent reduction of G-protein activation produced by the hCB2 receptor agonist CP-55,940.
Other indanoylindoles examined are best broadly characterized functionally as neutral antagonists or perhaps weak inverse agonists. The N-alkyl derivatives (26–28), similar to the results from receptor binding studies, suggest that the length of the alkyl chain does not influence functional activity. Arylmethyl derivatives (30, benzyl and 31, p-fluorobenzyl) reduced [35S]GTPγS binding to 87 ± 8% and 90 ± 3% of basal, respectively, consistent with weak inverse agonism. Two of the N-pentenyl isomers (32 and 34) behaved as weak inverse agonists (81 ± 2% basal and 92 ± 2% basal, respectively), whereas Z-isomer 33 registered as a weak agonist/antagonist (106 ± 7% basal) in our screening, and N-pentynyl 35 insignificantly altered the basal binding (96 ± 5% basal), consistent with an antagonist profile. Interestingly, the addition of a methyl group to C2 of 35 to produce compound 49 shifted the CB2R activity from the antagonist to the agonist (121 ± 8% basal).
CONCLUSIONS
We identify (2,2-dimethylindanoyl)indoles as a promising new class of indole cannabinoids comprising potent and CB2-selective ligands and presumably having lower potential for abuse as recreational drugs compared to other indole cannabinoids. Similar to the reference compound JWH-018, GBD-002 (3) and GBD-003 (4) displayed excellent affinities for both CB receptors. The latter (GBD-003) is moderately selective for CB2 and, in contrast to most indole cannabinoid ligands, can be characterized as a neutral antagonist. We developed a preliminary SAR profile for these new (2,2-dimethylindanoyl)indoles. The observed patterns on affinities and selectivity in both the indole and methylindole series are generally in accordance with a number of previously reported SAR investigations of indole-derived cannabinoids, with the major exception of their unusual antagonist functional activity. The N-alkyl substituent provides opportunities to alter affinity and selectivity while broadly retaining antagonist activity. Other changes to the indanoylindole core structure had larger effects on functional activity, with GBD-029 (2-methylindole) and GBD-002 (indanoyl positional isomer) exhibiting agonist activity. The indanoylindole derivatives reported here provide a new template for future development of CB2-selective neutral antagonists.
EXPERIMENTAL SECTION
Chemistry.
All the chemicals were used as received unless otherwise stated. Diethyl ether (Et2O), tetrahydrofuran (THF), methylene chloride (DCM), and toluene were dried over a column of molecular sieves under argon. All reactions were carried out under an inert nitrogen atmosphere unless otherwise stated. Crude products were purified in a Biotage Isolera One Flash Purification System using Biotage prepacked cartridges (50 μm irregular silica). Yields refer to isolated material following silica gel chromatography. The purity of the compounds tested was determined by quantitative 1H NMR using the absolute internal calibrant method (see the Supporting Information). 1H NMR and 13C NMR spectra were recorded on a JEOL 400 MHz spectrometer using CDCl3 and dimethyl sulfoxide-d6 (DMSO-d6) as the deuterated solvent [≥99.8 atom % D, contains 0.03% (v/v) TMS]. The chemical shifts (δ) were reported in parts per million (ppm) relative to the internal standard TMS. High-resolution mass spectroscopy (HRMS) data were obtained on a UHR-TOF maXis 4G instrument (Bruker Daltonics, Bremen, Germany) using electrospray ionization (ESI). Melting points (mp) were taken in open capillaries on a Stuart melting point apparatus SMP11 and are uncorrected.
Methyl 3-(2-Fluorophenyl)-2,2-dimethylpropanoate (6).63
A mixture of diisopropylamine (3.0 mL, 21.5 mmol) and THF (98 mL) was first cooled and stirred at −78 °C for 15 min. Next, n-BuLi (1.6 M in hexanes, 13.5 mL, 21.5 mmol) was added dropwise via a syringe. The mixture was stirred for another 15 min at −78 °C, 25 min at 0 °C, and 15 min at −78 °C. Methyl isobutyrate (2.2 mL, 19.6 mmol) was then added at the same temperature. After 30 min, 2-fluorobenzyl bromide (2.7 mL, 21.5 mmol) was finally added. The mixture was stirred at −78 °C for 1 h and then at ambient temperature for 5 h. After the reaction, the yellow solution was extracted with ether (3 × 50 mL). The organic layer was washed with brine, dried over Na2SO4, and concentrated under reduced pressure. Purification by automatic flash column chromatography on silica gel (gradient elution from 2 to 20% EtOAc-hexanes) afforded a colorless, viscous liquid (84% yield). 1H NMR (400 MHz, CDCl3): δ 1.19 (s, 6H), 2.92 (s, 2H), 3.67 (s, 3H), 6.98–7.06 (m, 2H), 7.07–7.13 (m, 1H), 7.16–7.23 (m, 1H). 19FNMR (376 MHz, CDCl3): δ −115.90. 13C{1H}NMR (100 MHz, CDCl3): δ 24.71, 38.64, 38.65, 43.64, 51.80, 115.21 (d, JC–F = 20 Hz), 123.64 (d, JC–F = 4 Hz), 124.88 (d, JC–F = 16 Hz), 128.30 (d, JC–F = 8 Hz), 132.21 (d, JC–F = 5 Hz), 161.46 (d, JC–F = 244 Hz), 177.79. HRMS (ESI, m/z): for C12H16FO2 [M + H]+ calcd, 211.1129; found, 211.1124.
3-(2-Fluorophenyl)-2,2-dimethylpropan-1-ol (7).63
A mixture of 6 (2.0 g, 9.5 mmol) and THF (43 mL) was first cooled and stirred at −78 °C for 15 min. At the same temperature, DIBAL-H (1.0 M in toluene, 20.9 mL, 20.9 mmol) was then added dropwise. After stirring for 1 h, the mixture was warmed to ambient temperature and ether (21 mL) was added. The solution was quenched in an ice bath with water (0.8 mL), then 15% NaOH (0.8 mL), and finally water (2.1 mL). The resulting heterogeneous mixture was removed from the bath, dried over MgSO4, filtered, and concentrated under reduced pressure. Purification by automatic flash column chromatography on silica gel (gradient elution from 5 to 40% EtOAc-hexanes) afforded a white, waxy solid (71% yield). 1H NMR (400 MHz, CDCl3): δ 0.90 (s, 6H), 1.69 (br s, 1H), 2.63 (s, 2H), 3.31 (d, J = 8 Hz, 2H), 6.99–7.08 (m, 2H), 7.15–7.22 (m, 2H). 19FNMR (376 MHz, CDCl3): δ −116.17. 13C{1H}NMR (100 MHz, CDCl3): δ 24.03, 36.92, 36.93, 37.03, 71.02, 115.25 (d, JC-F = 23 Hz), 123.67 (d, JC–F = 3 Hz), 125.77 (d, JC–F = 16 Hz), 127.96 (d, JC–F = 8 Hz), 133.13 (d, JC–F = 5 Hz), 161.68 (d, JC–F = 242 Hz). HRMS (ESI, m/z): for C11H14FO [M − H]− calcd, 181.1034; found, 181.1030.
3-(2-Fluorophenyl)-2,2-dimethylpropyl Methanesulfonate (8).63
Triethylamine (4.6 mL, 32.9 mmol) was added to a solution of 7 (4.0 g, 22.0 mmol) and dichloromethane (73 mL). The mixture was cooled to 0 °C, and then, methanesulfonyl chloride (2.1 mL, 26.3 mmol) was added. After stirring for 1 h, the mixture was warmed to ambient temperature and diluted with dichloromethane. The mixture was successively washed with cold water, chilled 10% HCl, saturated NaHCO3, and brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure, producing a colorless, viscous liquid (91% yield). The product was used immediately in the next step without purification. 1H NMR (400 MHz, CDCl3): δ 1.00 (s, 6H), 2.68 (s, 2H), 3.03 (s, 3H), 3.92 (s, 2H), 7.01–7.11 (m, 2H), 7.13–7.25 (m, 2H). 19FNMR (376 MHz, CDCl3): δ −115.22. 13C{1H}NMR (100 MHz, CDCl3): δ 23.84, 35.82, 37.00, 37.11, 76.86, 115.39 (d, JC–F = 23 Hz), 123.75 (d, JC–F = 4 Hz), 124.39 (d, JC–F = 16 Hz), 128.38 (d, JC–F = 8 Hz), 132.80 (d, JC–F = 4 Hz), 161.43 (d, JC–F = 244 Hz).
1-Fluoro-2-(3-iodo-2,2-dimethylpropyl)benzene (9).63
NaI (16.9 g, 113.0 mmol) was added to a solution of 8 (5.9 g, 22.6 mmol) and NMP (45 mL). The mixture was heated to 140 °C overnight. After cooling to ambient temperature, the orange mixture was diluted with water, followed by extraction with pentane. The organic layer was then successively washed with saturated NaS2O3, saturated CuSO4, water, and brine. Purification of the crude product by automatic flash column chromatography on silica gel (gradient elution from 2 to 20% EtOAc-hexanes) afforded a colorless, viscous liquid (89% yield). 1H NMR (400 MHz, CDCl3): δ 1.05 (s, 6H), 2.72 (s, 2H), 3.20 (s, 2H), 6.99–7.10 (m, 2H), 7.19–7.28 (m, 2H). 19FNMR (376 MHz, CDCl3): δ −114.86. 13C{1H}NMR (100 MHz, CDCl3): δ 24.14, 26.79, 26.80, 35.28, 38.66, 115.33 (d, JC–F = 23 Hz), 123.64 (d, JC–F = 4 Hz), 125.25 (d, JC–F = 15 Hz), 128.22 (d, JC–F = 8 Hz), 132.44 (d, JC–F = 5 Hz), 161.43 (d, JC–F = 244 Hz).
Ethyl 2,2-Dimethyl-2,3-dihydro-1H-indene-4-carboxylate (10).57
Method 1:
Compound 9 (4.5 g, 15.5 mmol) was first dissolved in 155 mL of anhydrous n-pentane-diethyl ether solution (4:1 by volume). The solution was cooled to −78 °C, after which t-BuLi (1.9 M solution in pentane, 26.1 mL, 49.5 mmol) was added dropwise. The solution was stirred at the same temperature for 15 min, and then, dry THF (7.8 mL) was added, turning the blue solution to a yellow, heterogeneous mixture. Next, the reaction mixture was warmed to room temperature (rt), stirred for 30 min, and cooled back to −78 °C. Ethyl chloroformate (15.3 mL, 154.7 mmol) was added dropwise, and the mixture was stirred for 15 min before warming up to rt and stirring for another 15 min. The mixture was quenched with water and extracted with diethyl ether (3 × 100 mL). The organic layer pool was dried over Na2SO4 and concentrated under reduced pressure, producing a brown, viscous liquid which was used in the next step without purification and characterization.
Method 2:
A mixture of TMS-esters 17 and 18 (20.3 mg, 0.07 mmol), KI (58.0 mg, 0.35 mmol), H2O (6 μL, 0.35 mmol), and acetonitrile (0.7 mL) was mixed in a vial. Trimethylsilyl chloride (44 μL, 0.35 mmol) was then added dropwise, and the mixture was stirred at 60 °C. After 12 h, the mixture was quenched with saturated NaHCO3 and extracted with ethyl acetate. The organic layer pool was washed brine, dried over Na2SO4, and concentrated under reduced pressure. The crude product was purified by automatic flash column chromatography on silica gel (isocratic elution: 5% EtOAc-hexanes) to afford 10 as a colorless, viscous liquid (10.4 mg, 68% yield). 1H NMR (400 MHz, CDCl3): δ 1.15 (s, 6H), 1.39 (t, 3H), 2.74 (s, 2H), 3.10 (s, 2H), 4.35 (q, 2H), 7.19 (t, 1H), 7.33 (d, 1H), 7.81 (d, 1H). 13C{1H}NMR (100 MHz, CDCl3): δ 14.38, 28.85, 39.52, 47.27, 48.79, 60.56, 126.08, 127.04, 127.86, 128.86, 145.10, 145.95, 167.25. HRMS (ESI, m/z): for C14H9O2 [M + H]+ calcd, 219.1375; found, 219.1380.
2,2-Dimethyl-2,3-dihydro-1H-indene-4-carboxylic acid (11).
Ester 10 (crude from method 1 or purified from method 2) was first dissolved in ethanol (0.2 M). KOH (2.0 M solution in H2O, 3.0 eq) was then added, and the mixture was stirred overnight at ambient temperature. The mixture was then cooled in an ice bath and acidified with concentrated HCl until pH 1. The solids were filtered and washed with cold water. The resulting white powder (Method 1: 17% over two steps, Method 2: 95%) was used in the next step without further purification. 1H NMR (400 MHz, CDCl3): δ 1.16 (s, 6H), 2.76 (s, 2H), 3.16 (s, 2H), 7.23 (t, 1H), 7.39 (d, 1H), 7.90 (d, 1H). 13C{1H}NMR (100 MHz, CDCl3): δ 28.95, 39.69, 47.34, 49.04, 125.90, 126.36, 128.81, 129.99, 145.42, 147.17, 172.84. HRMS (ESI, m/z): for C12H13O2 [M − H]− calcd, 189.0921; found, 189.0914. mp decomposes at 277 °C.
(2,2-dimethyl-2,3-dihydro-1H-inden-4-yl) (1-pentyl-1H-indol-3-yl)methanone (GBD-002, 3).64
A mixture containing 11 (164.8 mg, 0.87 mmol) and dichloromethane (5.1 mL) was cooled to 0 °C. Thionyl chloride (0.16 mL, 2.2 mmol) was added dropwise, followed by a catalytic amount of DMF (7 μL, 0.09 mmol). After stirring at ambient temperature for 8 h, the mixture was concentrated under reduced pressure and redissolved in 0.6 mL of 1,1,1,3,3,3-hexafluoroisopropanol (HFIP). The solution was added dropwise to a mixture of N-pentylindole (486.6 mg, 2.6 mmol) and 0.6 mL of HFIP. The mixture was stirred at ambient temperature for 24 h and concentrated under reduced pressure. Purification by automatic flash column chromatography on silica gel (gradient elution from 5 to 40% EtOAc-hexanes) afforded a colorless oil (68% yield). 1H NMR (400 MHz, CDCl3): δ 0.88 (t, 3H), 1.13 (s, 6H), 1.26–1.40 (m, 4H), 1.86 (quint, 2H), 2.78 (s, 2H), 2.91 (s, 2H), 4.12 (t, 2H), 7.19–7.24 (m, 1H), 7.29–7.35 (m, 3H), 7.36–7.42 (m, 2H), 7.46 (s, 1H), 8.39–8.44 ppm (m, 1H). 13C{1H}NMR (100 MHz, CDCl3): δ 13.91, 22.21, 28.69, 28.97, 29.56, 40.03, 47.05, 47.14, 47.53, 109.83, 116.34, 122.60, 122.79, 123.41, 125.68, 126.09, 126.56, 127.05, 136.86, 137.32, 137.61, 142.78, 144.92, 191.88. HRMS (ESI, m/z): for C25H30NO [M + H]+ calcd, 360.2322; found, 360.2317. Purity (qNMR), 100.4%.
5,5-Dimethyl-3-oxocyclohex-1-en-1-yl trifluoromethanesulfonate, VAT (13).56
Pyridine (11.9 mL, 146.8 mmol) was added to a suspension of dimedone (10.3 g, 73.4 mmol) in dry dichloromethane (367 mL) at ambient temperature. The mixture was cooled to −78 °C and stirred for 10 min. Triflic anhydride (13.6 mL, 80.7 mmol) was then added slowly. After 20 min, the mixture was removed from the bath and warmed to ambient temperature. The mixture was quenched with 1 M HCl after complete consumption of the starting material and extracted with diethyl ether (3 × 50 mL). The organic layer pool was washed with saturated Na2CO3 and brine, dried over Na2SO4, and concentrated under reduced pressure. The crude product was purified by automatic flash column chromatography on silica gel (gradient elution from 2% to 20% EtOAc-hexanes) to afford VAT as a colorless, viscous liquid (19.3 g, 97% yield). 1H NMR (400 MHz, CDCl3): δ 1.14 (s, 6H), 2.32 (s, 2H), 2.55 (s, 2H), 6.08 (s, 1H). 13C{1H}NMR (100 MHz, CDCl3): δ 28.03, 33.42, 42.37, 50.58, 116.84, 118.38, 166.09, 197.55.
Ethyl 5,5-Dimethyl-3-oxooct-7-ynoate (14).
LHMDS (1.0 M solution in THF, 9.1 mL, 9.1 mmol) was first cooled to −78 °C for 10 min. Ethyl acetate (0.9 mL, 9.1 mmol) was then added, and the mixture was stirred for 15 min. In a separate flask, a mixture of 13 (2.1 g, 7.6 mmol) in THF (30 mL) was also cooled to −78 °C. To this mixture, the ethyl lithioacetate solution was added slowly. After 20 min, the mixture was warmed to 0 °C, and then, LHMDS (1.0 M solution in THF, 22.7 mL, 22.7 mmol) was added dropwise over a period of 20 min. The resulting red solution was warmed to ambient temperature and stirred for 1 h. Saturated NH4Cl was added, and the mixture was extracted with ether (3 × 20 mL). The organic layer was washed with brine, dried over Na2SO4, and concentrated under reduced pressure. The crude product was purified by automatic flash column chromatography on silica gel (isocratic elution: 5% EtOAc-hexanes) to afford the β-ketoester as a yellow, viscous liquid (1.0 g, 63% yield). 1H NMR (400 MHz, CDCl3): δ 1.08 (s, 6H), 1.26 (t, 3H), 1.99 (t, 1H), 2.26 (d, 2H), 2.57 (s, 2H), 3.41 (s, 2H), 4.17 (q, 2H). 13C{1H}NMR (100 MHz, CDCl3): δ 14.09, 26.93, 31.14, 33.42, 50.95, 51.55, 61.32, 70.54, 81.90, 167.12, 201.92.
Ethyl (Z)-5,5-Dimethyl-3-(((trifluoromethyl)sulfonyl)oxy)oct-2-en-7-ynoate (15).
A mixture of 14 (0.97 g, 4.6 mmol) and hexanes (23 mL) was cooled in an ice bath. Saturated aqueous LiOH (1.5 mL/mmol β-ketoester) was added, and the resulting suspension was stirred for 5 min. Triflic anhydride (1.9 mL, 11.5 mmol) was then added slowly. The mixture was removed from the bath and stirred for 30 min. After the reaction, the mixture was diluted with water and extracted with ether (3 × 10 mL). The organic layer was washed with brine, dried over Na2SO4, and concentrated under reduced pressure. The crude product was purified by automatic flash column chromatography on silica gel (isocratic elution: 5% EtOAc-hexanes) to afford the triflate as a light-yellow, viscous liquid (1.39 g, 88% yield). 1H NMR (400 MHz, CDCl3): δ 1.09 (s, 6H), 1.32 (t, 3H), 2.09 (t, 1H), 2.16 (d, 2H), 2.40 (s, 2H), 4.26 (q, 2H), 5.84 (s, 1H). 13C{1H}NMR (100 MHz, CDCl3): δ 14.00, 26.72, 31.61, 34.82, 45.00, 61.33, 71.51, 80.85, 115.01, 116.70, 119.88, 156.43, 162.40.
Ethyl 5,5-Dimethylocta-2,7-diynoate (16).
To a mixture of 15 (1.2 g, 3.5 mmol) in THF (17 mL) cooled at 0 °C, LHMDS (1.0 M solution in THF, 3.8 mL, 3.8 mmol) was added. The solution was warmed to ambient temperature and stirred for 1 h. After the reaction, saturated NH4Cl was added, and the mixture was extracted with ether (3 × 10 mL). The organic layer was washed with brine, dried over Na2SO4, and concentrated under reduced pressure. The crude product was purified by automatic flash column chromatography on silica gel (gradient elution from 2 to 20% EtOAc-hexanes) to afford the diyne as a light-yellow oil (0.42 g, 63% yield). 1H NMR (400 MHz, CDCl3): δ 1.11 (s, 6H), 1.31 (t, 3H), 2.03 (t, 1H), 2.22 (s, 2H), 2.37 (s, 2H), 4.22 (q, 2H). 13C{1H}NMR (100 MHz, CDCl3): δ 14.04, 26.39, 30.87, 31.13, 34.09, 61.83, 70.76, 75.14, 81.29, 86.70, 153.72.
Ethyl 2,2-Dimethyl-5-(trimethylsilyl)-2,3-dihydro-1H-indene-4-carboxylate (17/18).
In a vial, [Rh(COD)2]BF4 (16.2 mg, 0.04 mmol), (±)-BINAP (24.6 mg, 0.04 mmol), and CH2Cl2 (7.9 mL) were stirred for 1 h at ambient temperature under a H2 atmosphere. The mixture was concentrated and redissolved in CH2Cl2 (2.0 mL). TMS-acetylene (0.25 mL, 1.7 mmol) was added, followed by a solution of diyne 16 (152.0 mg, 0.79 mmol) in CH2Cl2 (7.9 mL). After stirring for 1 h at ambient temperature, the mixture was concentrated and purified by automatic flash column chromatography on silica gel (isocratic elution: 2% EtOAc-hexanes) to afford a mixture of TMS-esters as a colorless, viscous liquid (206 mg, 90% yield). 1H NMR (400 MHz, CD3CO): ortho isomer: δ 0.29 (s, 9H), 1.14 (s, 6H), 1.40 (t, 3H), 2.73 (s, 2H), 2.91 (s, 2H), 4.36 (q, 2H), 7.25 (d, 1H), 7.42 (d, 1H). Meta isomer: δ 0.28 (s, 9H), 1.15 (s, 6H), 1.40 (t, 3H), 2.74 (s, 2H), 3.09 (s, 2H), 4.36 (q, 2H), 7.47 (d, 1H), 7.95 (d, 1H). 13C{1H}NMR (100 MHz, CDCl3): δ 1.38, 15.37, 15.45, 29.90, 29.96, 40.42, 40.43, 48.28, 48.49, 49.30, 49.99, 61.58, 61.90, 127.60, 127.67, 133.91, 134.32, 134.65, 135.22, 138.57, 139.19, 144.47, 145.51, 146.68, 147.62, 168.62, 170.96. HRMS (ESI, m/z): for C17H27O2Si [M + H]+ calcd, 291.1768; found, 291.1775.
3-Hydroxy-5,5-dimethyl-cyclohex-1-en-1-yl Trifluoromethane Sulfonate, VHAT (19).58
A mixture of 13 (19.3 g, 70.9 mmol) and dry THF (322 mL) was cooled to −78 °C. DIBAL-H (1.0 M solution in toluene, 92.2 mL, 92.2 mmol) was added slowly, and the resulting mixture was stirred for 10 min. The mixture was warmed to ambient temperature until complete consumption of the starting material and then diluted with ether (~160 mL) before cooling in an ice bath. Water (3.7 mL) was added slowly, followed by 15% aqueous NaOH (3.7 mL) and water (9.2 mL). The mixture was removed from the bath and dried over MgSO4. After 15 min of stirring, the mixture was filtered. The filtrate was concentrated under reduced pressure and finally purified by automatic flash column chromatography on silica gel (gradient elution from 5% to 40% EtOAc-hexanes) to afford VHAT as a colorless, viscous liquid (14.5 g, 75% yield). 1H NMR (400 MHz, CDCl3): δ 0.99 (s, 3H), 1.09 (s, 3H), 1.33 (dd, 1H), 1.83 (dd, 1H), 1.96 (broad, s, 1H), 2.01 (d, 1H), 2.27 (d, 1H), 4.43–4.46 (m, 1H), 5.80 (s, 1H). 13C{1H}NMR (100 MHz, CDCl3): δ 25.95, 30.55, 32.30, 41.08, 44.17, 65.57, 116.91, 120.35, 149.82.
Ethyl-E-4-(diethoxyphosphoryl)but-2-enoate.65
Triethyl phosphite (10.0 g, 38.8 mmol) and ethyl 4-bromocrotonate (7.9 g, 46.6 mmol) were combined in a pressure flask and heated to 140 °C overnight. The mixture was cooled to ambient temperature, transferred to a flask, and distilled until complete removal of ethyl bromide. The crude product was a brown, viscous liquid and used in the next step without purification. The 1H NMR is consistent with the literature report.
Ethyl (2E, 4E)-7,7-Dimethyldeca-2,4-dien-9-ynoate (20).56
A mixture of 19 (5.3 g, 19.5 mmol), ethyl-E-4-(diethoxyphosphoryl)-but-2-enoate (7.3 g, 29.2 mmol), and dry THF (97 mL) was cooled to −78 °C. LHMDS (1.0 M solution in THF, 42.8 mL, 42.8 mmol) was added slowly, and the resulting mixture was stirred for 15 min. The mixture was warmed to ambient temperature and then at 60 °C for 1 h or until complete consumption of the starting material. After cooling to ambient temperature, 1 M HCl (~97 mL) was added, and the mixture was extracted with ethyl acetate (3 × 25 mL). The organic layer was washed with water and brine, dried over Na2SO4, and concentrated under reduced pressure. The crude product was purified by automatic flash column chromatography on silica gel (gradient elution from 2 to 20% EtOAc-hexanes) to afford dienyne as a light-yellow, viscous liquid (3.2 g, 75% yield). 1H NMR (400 MHz, CDCl3): δ 0.10 (s, 6H), 1.30 (t, 3H), 2.03 (t, 1H), 2.08 (d, 2H), 2.19 (d, 2H), 4.21 (q, 2H), 5.82 (d, 1H) 6.10–6.16 (m, 1H), 6.22 (dd, 1H), 7.28 (dd, 1H). 13C{1H}NMR (100 MHz, CDCl3): δ 14.30, 26.66, 31.51, 34.31, 44.43, 60.19, 70.32, 81.99, 119.83, 131.13, 140.44, 144.57, 167.19.
Ethyl 2,2-Dimethyl-2,3-dihydro-1H-indene-5-carboxylate (21).56
To a mixture of 20 (3.2 g, 14.6 mmol) in dry dichloromethane (291 mL), di-μ-chlorobis(norbornadiene)dirhodium [Rh(nbd)Cl]2 (0.14 g, 0.29 mmol) was added, followed by silver hexafluoroantimonate (AgSbF6, 0.43 g, 1.24 mmol). The resulting mixture was stirred at ambient temperature for 1 h. DDQ (4.1 g, 17.5 mmol) was added, and stirring was continued until complete oxidation (1–2 h). After the reaction, the brown mixture was filtered in basic alumina. The filtrate was concentrated under reduced pressure and purified by automatic flash column chromatography on silica gel (gradient elution from 1 to 10% EtOAc-hexanes) to afford the ester as a colorless, viscous liquid (2.7 g, 85% yield). 1H NMR (400 MHz, CDCl3): δ 1.15 (s, 6H), 1.38 (t, 3H), 2.75 (s, 4H), 4.35 (q, 2H), 7.21 (d, 1H), 7.81–7.86 (m, 2H). 13C{1H}NMR (100 MHz, CDCl3): δ 14.38, 28.60, 40.51, 47.31, 47.74, 60.70, 124.53, 125.79, 127.88, 128.52, 143.84, 149.27, 167.09.
2,2-Dimethyl-2,3-dihydro-1H-indene-5-carboxylic acid (22).
Compound 21 (3.7 g, 17.1 mmol) was first dissolved in ethanol (82 mL). Aqueous KOH (9.1 mL) was then added, and the mixture was stirred overnight at ambient temperature. After the reaction, the mixture was neutralized with 3 M HCl. The solids were filtered and washed with cold water. The crude product was white, fluffy crystals (2.8 g, 85%) and used in the next step without further purification. 1H NMR (400 MHz, CDCl3): δ 1.16 (s, 6H), 2.77 (s, 4H), 7.25 (d, 1H), 7.89–7.93 (m, 2H). 13C{1H}NMR (100 MHz, CDCl3): δ 28.69, 40.65, 47.36, 47.93, 124.80, 126.56, 127.40, 128.73, 144.16, 150.62, 172.73 HRMS (ESI, m/z): for C12H13O2 [M − H]− calcd, 189.0921; found, 189.0915. mp 158–160 °C.
General Procedure for the Synthesis of Indoles 23 and 24.64
A mixture containing 22 (500.0 mg, 2.6 mmol) and dichloromethane (15.5 mL) was cooled to 0 °C. Thionyl chloride (0.4 mL, 5.3 mmol) was added dropwise, followed by a catalytic amount of DMF (0.02 mL, 0.3 mmol). After stirring at ambient temperature for 24 h, the mixture was concentrated under reduced pressure and redissolved in 1.5 mL of 1,1,1,3,3,3-HFIP. The acid chloride solution was added dropwise to a mixture of indole (2.8 mmol) and 2.0 mL of HFIP. The mixture was stirred at ambient temperature for 24 h, diluted with DCM, concentrated under reduced pressure, and purified by automatic flash column chromatography on silica gel (gradient elution from 1 to 10% MeOH─CH2Cl2).
(2,2-Dimethyl-2,3-dihydro-1H-inden-5-yl) (1H-indol-3-yl)-methanone (GBD-001, 23).
Obtained as white, powdery crystals (58% yield). 1H NMR (400 MHz, DMSO-d6): δ 1.15 (s, 6H), 2.77 (overlapping s, 4H), 7.19–7.27 (m, 2H), 7.31 (d, 1H), 7.50–7.53 (m, 1H), 7.55–7.58 (m, 1H), 7.59 (d, 1H), 7.93 (s, 1H), 8.22–8.25 (m, 1H), 12.00 ppm (s, 1H). 13C{1H}NMR (100 MHz, DMSO-d6): δ 28.45 40.12, 46.94, 47.10, 112.20, 115.13, 121.50, 121.75, 123.01, 124.40, 124.72, 126.36, 126.99, 135.40, 136.66, 138.84, 143.34, 146.69, 190.08. HRMS (ESI, m/z): for C20H18NO [M − H]− calcd, 288.1394; found, 288.1394. mp 224–225 °C. Purity (qNMR), 100.1%.
(2,2-Dimethyl-2,3-dihydro-1H-inden-5-yl) (2-methyl-1H-indol-3-yl)methanone (GBD-019, 24).
Obtained as white, powdery crystals (53% yield). 1H NMR (400 MHz, DMSO-d6): δ 1.13 (s, 6H), 2.72 (s, 2H), 2.76 (s, 2H), 7.00 (t, 1H), 7.10 (t, 1H), 7.27 (d, 1H), 7.24 (d, 1H), 7.36 (d, 1H), 7.38 (d, 1H), 7.43 (s, 1H), 11.87 (s, 1H). 13C{1H}NMR (100 MHz, DMSO-d6): δ 14.13, 28.30, 40.21, 46.83, 47.07, 111.18, 112.75, 120.01, 120.74, 121.67, 124.35, 124.51, 126.77, 127.35, 134.92, 139.72, 143.27, 143.80, 146.77, 191.84. HRMS (ESI, m/z): for C21H22NO [M + H]+ calcd, 304.1696; found, 304.1636. mp 239–241 °C.
General Procedure for the Synthesis of 4, 26–34, 36, 37, and 39.
NaH (27.6 mg, 0.69 mmol) and DMSO (1.0 mL) were mixed thoroughly in a round-bottom flask. In a separate pear-shaped flask, 23 (100 mg, 0.35 mmol) and DMSO (1.5 mL) were heated to ~40 °C to form a solution and then added slowly to the NaH suspension. After stirring at ambient temperature for 30 min, the alkyl halide (0.42 mmol) was added. The resulting mixture was stirred overnight at ambient temperature. The reaction mixture was quenched with water and extracted with ethyl acetate (2 × 10 mL). The organic layer was washed with brine, dried over Na2SO4, concentrated under reduced pressure, and purified by automatic flash column chromatography on silica gel (gradient elution from 5 to 40% EtOAc-hexanes).
(2,2-Dimethyl-2,3-dihydro-1H-inden-5-yl) (1-pentyl-1H-indol-3-yl)methanone (GBD-003, 4).
Obtained as white crystals (88% yield). 1H NMR (400 MHz, CDCl3) δ 0.89 (t, 3H), 1.19 (s, 6H), 1.26–140 (m, 4H), 1.87 (quintet, 2H), 2.79 (s, 4H), 4.15 (t, 2H), 7.22–7.27 (m, 1H), 7.28–7.35 (m, 2H), 7.36–7.41 (m, 1H), 7.57–7.62 (m, 2H), 7.64 (s, 1H), 8.39–8.45 (m, 1H). 13C{1H}NMR (100 MHz, CDCl3) δ 13.93, 22.22, 28.72, 28.98, 29.57, 40.48, 47.10, 47.52, 47.43, 109.77, 115.67, 122.40, 122.80, 123.31, 124.27, 125.15, 127.30, 127.45, 136.68, 136.75, 139.21, 143.88, 147.37, 191.20. HRMS (ESI, m/z) for C25H30NO [M + H]+ calcd, 360.2322, found 360.2318. mp 62–64 °C. Purity (qNMR), 99.9%.
(2,2-dimethyl-2,3-dihydro-1H-inden-5-yl) (1-propyl-1H-indol-3-yl)methanone (GBD-003, 26).
Obtained as white, powdery crystals (83% yield). 1H NMR (400 MHz, CDCl3): δ 0.95 (t, 3H), 1.19 (s, 6H), 1.91 (sextet, 2H), 2.79 (s, 4H), 4.13 (t, 2H), 7.23–7.27 (m, 1H), 7.29–7.35 (m, 2H), 7.36–7.42 (m, 1H), 7.58–7.62 (m, 2H), 7.64 (s, 1H), 8.39–8.45 (m, 1H). 13C{1H}NMR (100 MHz, CDCl3): δ 11.42, 23.21, 28.73, 40.48, 47.52, 47.73, 48.74, 109.79, 115.67, 122.42, 122.82, 123.32, 124.26, 125.13, 127.28, 127.46, 136.76, 136.78, 139.21, 143.90, 147.36, 191.22. HRMS (ESI, m/z): for C23H26NO [M + H]+ calcd, 332.2009; found, 332.2006. mp 91–93 °C. Purity (qNMR), 99.2%.
(2,2-Dimethyl-2,3-dihydro-1H-inden-5-yl) (1-butyl-1H-indol-3-yl)methanone (GBD-006, 27).
Obtained as white, powdery crystals (85% yield). 1H NMR (400 MHz, CDCl3): δ 0.93 (t, 3H), 1.19 (s, 6H), 1.36 (sextet, 2H), 1.86 (quintet, 2H), 2.80 (s, 4H), 4.17 (t, 2H), 7.23–7.27 (m, 1H), 7.29–7.35 (m, 2H), 7.36–7.41 (m, 1H), 7.58–7.62 (m, 2H), 7.64 (s, 1H), 8.39–8.44 ppm (m, 1H). 13C{1H}NMR (100 MHz, CDCl3) δ 13.63, 20.09, 28.73, 31.92, 40.48, 46.86, 47.52, 47.73, 109.77, 15.69, 122.41, 122.82, 123.31, 124.27, 125.14, 127.28, 127.45, 136.68, 136.76, 139.21, 143.90, 147.37, 191.20. HRMS (ESI, m/z): for C24H28NO [M + H]+ calcd, 346.2165; found, 346.2163. mp 70–72 °C. Purity (qNMR), 100.1%.
(2,2-Dimethyl-2,3-dihydro-1H-inden-5-yl) (1-hexyl-1H-indol-3-yl)methanone (GBD-007, 28).
Obtained as white, powdery crystals (82% yield). 1H NMR (400 MHz, CDCl3): δ 0.87 (t, 3H), 1.19 (s, 6H), 1.31 (m, 6H), 1.87 (quintet, 2H), 2.80 (s, 2H), 4.15 (t, 2H), 7.23–7.27 (m, 1H), 7.29–7.36 (m, 2H), 7.37–7.41 (m, 1H), 7.57–7.62 (m, 2H), 7.64 (s, 1H), 8.39–8.44 ppm (m, 1H). 13C{1H}NMR (100 MHz, CDCl3): δ 13.97, 22.50, 26.54, 28.73, 29.83, 31.28, 40.48, 47.12, 47.53, 47.74, 109.77, 115.68, 122.41, 122.82, 123.31, 124.27, 125.15, 127.30, 127.47, 136.67, 136.75, 139.23, 143.88, 147.36, 191.20. HRMS (ESI, m/z): for C26H32NO [M + H]+ calcd, 374.2478; found, 374.2477. mp 61–63 °C. Purity (qNMR), 99.7%.
(2,2-Dimethyl-2,3-dihydro-1H-inden-5-yl) (1-isobutyl-1H-indol-3-yl)methanone (GBD-008, 29).
Obtained as yellowish oil (53% yield). 1H NMR (400 MHz, CDCl3): δ 0.94 (d, 6H), 1.18 (s, 6H), 2.23 (m, 1H), 2.79 (s, 4H), 3.94 (d, 2H), 7.22–7.26 (m, 1H), 7.28–7.33 (m, 2H), 7.34–7.39 (m, 1H), 7.54–7.61 (m, 2H), 7.64 (s, 1H), 8.39–8.44 ppm (m, 1H). 13C{1H}NMR (100 MHz, CDCl3): δ 20.19, 28.72, 29.15, 40.46, 47.51, 47.71, 54.68, 109.97, 115.54, 122.37, 122.76, 123.29, 124.25, 125.16, 127.29, 127.40, 137.00, 137.16, 139.18, 143.89, 147.38, 191.23. HRMS (ESI, m/z): for C24H28NO [M + H]+ calcd, 346.2165; found, 346.2161. Purity (qNMR), 98.5%.
(1-Benzyl-1H-indol-3-yl) (2,2-dimethyl-2,3-dihydro-1H-inden-5-yl)methanone (GBD-009, 30).
Obtained as white, powdery crystals (86% yield). 1H NMR (400 MHz, CDCl3): δ 1.17 (s, 6H), 2.78 (s, 4H), 5.37 (s, 2H), 7.11–7.15 (m, 2H), 7.21–7.35 (m, 7H), 7.58–7.62 (app. d, 1H), 7.64 (s, 2H), 8.42–8.46 ppm (m, 1H). 13C{1H}NMR (100 MHz, CDCl3): δ 28.71, 40.47, 47.50, 47.71, 50.74, 110.14, 116.25, 122.64, 122.82, 123.63, 124.31, 125.16, 126.78, 127.31, 127.52, 128.10, 128.98, 135.92, 136.96, 137.02, 139.01, 143.91, 147.52, 191.24. HRMS (ESI, m/z): for C27H26NO [M + H]+ calcd, 380.2009; found, 380.2005. mp 111–114 °C. Purity (qNMR), 99.6%.
(2,2-Dimethyl-2,3-dihydro-1H-inden-5-yl) (1-(4-fluorobenzyl)-1H-indol-3-yl)methanone (GBD-010, 31).
Obtained as white, powdery crystals (90% yield). 1H NMR (400 MHz, CDCl3): δ 1.18 (s, 6H), 2.78 (s, 4H), 5.34 (s, 2H), 6.98–7.04 (m, 2H), 7.09–7.14 (m, 2H), 7.24 (m, 1H), 7.28–7.35 (m, 3H), 7.58–7.65 (m, 3H), 8.40–8.43 ppm (m, 1H). 13C{1H}NMR (100 MHz, CDCl3): δ 28.70, 40.49, 47.50, 47.71, 50.09, 110.02, 115.86, 116.06, 116.40, 122.72, 122.90, 123.72, 124.33, 125.15, 127.31, 127.55, 131.64, 128.50, 128.58, 136.68, 136.87, 138.95, 143.96, 147.62, 163.65, 191.20. 19FNMR (376 MHz, CDCl3): δ −113.65. HRMS (ESI, m/z): for C27H25FNO [M + H]+ calcd, 398.1915; found, 398.1911. mp 124–126 °C. Purity (qNMR), 100.5%.
(E)-(2,2-Dimethyl-2,3-dihydro-1H-inden-5-yl) (1-(pent-2-en-1-yl)-1H-indol-3-yl)methanone (GBD-011, 32).
Obtained as white, powdery crystals (88% yield). 1H NMR (400 MHz, CDCl3): δ 0.98 (t, 3H), 1.18 (s, 6H), 2.06 (quint, 2H), 2.79 (s, 4H), 4.69–4.74 (app. d, 2H), 5.56–5.65 (m, 1H, J = 15.3 Hz, 6.0 Hz, 1.4 Hz), 5.71–5.80 (m, 1H, J = 15.4 Hz, 6.2 Hz, 1.4 Hz), 7.22–7.27 (m, 1H), 7.29–7.35 (m, 2H), 7.36–7.42 (m, 1H), 7.58–7.62 (m, 2H), 7.65 (s, 1H), 8.39–8.44 (m, 1H). 13C{1H}NMR (100 MHz, CDCl3): δ 13.21, 25.18, 28.72, 40.48, 47.52, 47.72, 48.94, 110.05, 115.86, 122.46, 122.70, 122.77, 123.33, 124.28, 125.14, 127.30, 127.52, 136.47, 136.85, 137.27, 139.17, 143.87, 147.38, 191.20. HRMS (ESI, m/z): for C25H28NO [M + H]+ calcd, 358.2165; found, 358.2163. mp 80–82 °C. Purity (qNMR), 99.1%.
(Z)-(2,2-Dimethyl-2,3-dihydro-1H-inden-5-yl) (1-(pent-2-en-1-yl)-1H-indol-3-yl)methanone (GBD-012, 33).
Obtained as white, powdery crystals (68% yield). 1H NMR (400 MHz, CDCl3): δ 1.07 (t, 3H), 1.17 (s, 6H), 2.24 (quint, 2H), 2.78 (s, 4H), 4.78 (app. d, 2H), 5.51–5.59 (m, 1H, J = 10.8 Hz, 6.8 Hz, 0.8 Hz), 5.68–5.76 (m, 1H, J = 10.7 Hz, 7.4 Hz, 0.8 Hz), 7.22–7.26 (m, 1H), 7.29–7.34 (m, 2H), 7.35–7.40 (m, 1H), 7.56–7.62 (m, 2H), 7.63 (s, 1H), 8.37–8.43 (m, 1H). 13C{1H}NMR (100 MHz, CDCl3): δ 14.00, 20.96, 28.72, 40.48, 43.79, 47.52, 47.73, 109.83, 115.91, 122.54, 122.58, 122.82, 123.36, 124.28, 125.14, 127.29, 127.60, 136.18, 136.83, 136.85, 139.17, 143.87, 147.38, 191.18. HRMS (ESI, m/z): for C25H28NO [M + H]+ calcd, 358.2165; found, 358.2162. mp 70–71 °C. Purity (qNMR), 99.5%.
(2,2-Dimethyl-2,3-dihydro-1H-inden-5-yl) (1-(pent-4-en-1-yl)-1H-indol-3-yl)methanone (GBD-013, 34).
Obtained as light-yellow, powdery crystals (74% yield). 1H NMR (400 MHz, CDCl3): δ 1.18 (s, 6H), 1.97 (quint, 2H), 2.09 (q, 2H), 2.79 (s, 4H), 4.16 (t, 2H), 5.01–5.04 (m 1H), 5.05–5.09 (m, 1H), 5.72–5.84 (ddt, 1H), 7.22–7.26 (m, 1H), 7.29–7.34 (m, 2H), 7.35–7.40 (m, 1H), 7.56–7.61 (m, 2H), 7.63 (s, 1H), 8.39–8.44 ppm (m, 1H). 13C{1H}NMR (100 MHz, CDCl3): δ 28.72, 28.77, 30.67, 40.48, 46.24, 47.53, 47.73, 109.75, 115.80, 116.17, 122.46, 122.86, 123.37, 124.28, 125.12, 127.26, 127.48, 136.69, 136.71, 136.79, 139.18, 143.88, 147.39, 191.18. HRMS (ESI, m/z): for C25H28NO [M + H]+ calcd, 358.2165; found, 358.2162. mp 78–79 °C. Purity (qNMR), 99.3%.
(2,2-Dimethyl-2,3-dihydro-1H-inden-5-yl) (1-(pent-2-yn-1-yl)-1H-indol-3-yl)methanone (GBD-015, 36).
Obtained as light-yellow, powdery crystals (55% yield). 1H NMR (400 MHz, CDCl3): δ 1.13 (t, 3H), 1.19 (s, 6H), 2.18–2.25 (qt, 2H), 2.79 (s, 4H), 4.88 (t, 2H), 7.23–7.27 (m, 1H), 7.31–7.38 (m, 2H), 7.42–7.48 (m, 1H), 7.60–7.68 (m, 2H), 7.77 (s, 1H), 8.39–8.45 (m, 1H). 13C{1H}NMR (100 MHz, CDCl3): δ 12.37, 13.58, 28.71, 37.02, 40.49, 47.53, 47.73, 71.93, 88.93, 109.79, 116.00, 122.70, 122.88, 123.53, 124.31, 125.15, 127.33, 127.63, 136.07, 136.48, 139.05, 143.85, 147.47, 191.18. HRMS (ESI, m/z): for C25H26NO [M + H]+ calcd, 356.2009; found, 332.2007. mp 98–99 °C. Purity (qNMR), 96.8%.
(1-Allyl-1H-indol-3-yl) (2,2-dimethyl-2,3-dihydro-1H-inden-5-yl)-methanone (GBD-016, 37).
Obtained as yellowish oil (81% yield). 1H NMR (400 MHz, CDCl3): δ 1.18 (s, 6H), 2.79 (s, 4H), 4.76 (m, 2H), 5.10–5.17 (m, 1H), 5.23–5.28 (m, 1H), 5.94–6.04 (m, 1H), 7.22–7.26 (m, 1H), 7.29–7.38 (m, 3H), 7.57–7.61 (m, 2H), 7.64 (s, 1H), 8.41–8.45 ppm (m, 1H). 13C{1H}NMR (100 MHz, CDCl3): δ 28.67, 40.43, 47.46, 47.66, 49.33, 109.97, 116.01, 118.39, 122.50, 122.74, 123.42, 124.25, 125.04, 127.23, 127.39, 132.07, 136.60, 136.80, 139.04, 143.87, 147.40, 191.17. HRMS (ESI, m/z): for C23H24NO [M + H]+ calcd, 330.1852; found, 330.1848. Purity (qNMR), 98.3%.
Ethyl 2-(3-(2,2-Dimethyl-2,3-dihydro-1H-indene-5-carbonyl)-1H-indol-1-yl)acetate (GBD-018, 39).
Obtained as yellowish oil (28% yield). 1H NMR (400 MHz, CDCl3): δ 1.18 (s, 6H), 1.26 (t, 3H), 27.8, (s, 4H), 4.22 (q, 2H), 4.86 (s, 2H), 7.22–7.36 (m, 4H), 7.59–7.63 (m, 2H), 7.65 (s, 1H), 8.40–8.45 ppm (m, 1H). 13C{1H}NMR (100 MHz, CDCl3): δ 14.10, 28.69, 40.47, 47.48, 47.70, 48.15, 62.11, 109.20, 116.80, 122.76, 122.91, 123.85, 124.32, 125.11, 127.21, 127.34, 137.10, 137.32, 138.89, 143.90, 147.60, 167.49, 191.28. HRMS (ESI, m/z): for C24H26NO3 [M + H]+ calcd, 376.1907; found, 376.1904. Purity (qNMR), 96.5%.
(2,2-dimethyl-2,3-dihydro-1H-inden-5-yl) (1-methyl-1H-indol-3-yl)methanone (GBD-004, 25).
NaH (14.2 mg, 0.36 mmol) and DMSO (1.0 mL) were mixed thoroughly in a round-bottom flask. In a separate pear-shaped flask, 23 (51.4 mg, 0.18 mmol) and DMSO (1.5 mL) were heated to ~40 °C to form a solution and then added slowly to the NaH suspension. After stirring at ambient temperature for 30 min, (chloromethyl)trimethylsilane (0.03 mL, 0.21 mmol) was added. The resulting mixture was stirred overnight at ambient temperature. The reaction was quenched with water and extracted with ethyl acetate (2 × 5 mL). The organic layer was washed with brine, dried over Na2SO4, and concentrated under reduced pressure. Purification by automatic flash column chromatography on silica gel (gradient elution from 5 to 40% EtOAc-hexanes) afforded white, powdery crystals (64% yield). 1H NMR (400 MHz, CDCl3): δ 1.18 (s, 6H), 2.79, (s, 4H), 3.85 (s, 3H), 7.22–7.26 (m, 1H), 7.31–7.39 (m, 3H), 7.56 (s, 1H), 7.57–7.62 (app. d, 1H), 7.63 (s, 1H), 8.40–8.44 ppm (m, 1H). 13C{1H}NMR (100 MHz, CDCl3) δ 28.71, 33.53, 40.48, 47.51, 47.71, 109.53, 115.75, 122.51, 122.74, 123.47, 124.28, 125.06, 127.03, 127.28, 137.46, 137.61, 139.15, 143.86, 147.36, 191.18. HRMS (ESI, m/z): for C21H22NO [M + H]+ calcd, 304.1696; found, 304.1693. mp 92–95 °C. Purity (qNMR), 99.9%.
General Procedure for the Synthesis of 35 and 38.
To a mixture of 23 (100 mg, 0.35 mmol) and DMSO (2.0 mL) was added KOH (38.8 mg, 0.69 mmol). After stirring at ambient temperature for 1 h, the alkyl halide (0.52 mmol) was added. The resulting mixture was stirred for 2 h at ambient temperature. The reaction was quenched with water and extracted with ethyl acetate (2 × 10 mL). The organic layer was washed with brine, dried over Na2SO4, concentrated under reduced pressure, and purified by automatic flash column chromatography on silica gel (gradient elution from 5 to 40% EtOAc-hexanes).
(2,2-Dimethyl-2,3-dihydro-1H-inden-5-yl) (1-(pent-4-yn-1-yl)-1H-indol-3-yl)methanone (GBD-014, 35).
Obtained as light-yellow, powdery crystals (79% yield). 1H NMR (400 MHz, CDCl3): δ 1.18 (s, 6H), 2.03–2.12 (m, 3H), 2.17–2.22 (td, 2H, J = 6.4 Hz, 2.5 Hz), 2.79, (s, 4H), 4.34 (t, 2H), 7.22–7.28 (m, 1H), 7.29–7.36 (m, 2H), 7.39–7.45 (m, 1H), 7.58–7.63 (d, 1H), 7.65 (m, 2H), 8.39–8.46 ppm (m, 1H). 13C{1H}NMR (100 MHz, CDCl3): δ 15.64, 28.06, 28.71, 40.49, 45.29, 47.53, 47.73, 70.21, 82.30, 109.68, 115.84, 122.55, 122.91, 123.48, 124.32, 125.20, 127.33, 127.53, 136.59, 136.97, 139.06, 143.88, 147.47, 191.15. HRMS (ESI, m/z): for C25H26NO [M + H]+ calcd, 356.2009; found, 356.2006. mp 84–85 °C. Purity (qNMR), 100.4%.
(2,2-Dimethyl-2,3-dihydro-1H-inden-5-yl) (1-(prop-2-yn-1-yl)-1H-indol-3-yl)methanone (GBD-017, 38).
Obtained as light-yellow oil (66% yield). 1H NMR (400 MHz, CDCl3): δ 1.19 (s, 6H), 2.49 (t, 1H), 2.80 (s, 4H), 4.93 (d, 2H), 7.23–7.28 (m, 1H), 7.32–7.41 (m, 2H), 7.43–7.49 (m, 1H), 7.59–7.67 (m, 2H), 7.74 (s, 1H), 8.39–8.45 ppm (m, 1H). 13C{1H}NMR (100 MHz, CDCl3): δ 28.71, 36.53, 40.50, 47.50, 47.73, 75.02, 76.29, 109.64, 116.47, 122.87, 122.97, 123.76, 124.34, 125.14, 127.33, 127.56, 135.81, 136.41, 138.89, 143.96, 147.63, 191.21. HRMS (ESI, m/z): for C23H22NO [M + H]+ calcd, 328.1696; found, 328.1695. Purity (qNMR), 100.0%.
General Procedure for the Synthesis of 40–48 and 50.
NaH (26.4 mg, 0.66 mmol) and DMSO (1.0 mL) were mixed thoroughly in a round-bottom flask. In a separate pear-shaped flask, 24 (100 mg, 0.33 mmol) and DMSO (1.5 mL) were heated to ~40 °C to form a solution and then added slowly to the NaH suspension. After stirring at ambient temperature for 30 min, the alkyl halide (0.40 mmol) was added. The resulting mixture was stirred overnight at ambient temperature. The reaction mixture was quenched with water and extracted with ethyl acetate (2 × 10 mL). The organic layer was washed with brine, dried over Na2SO4, concentrated under reduced pressure, and purified by automatic flash column chromatography on silica gel (gradient elution from 5 to 40% EtOAc-hexanes).
(2,2-Dimethyl-2,3-dihydro-1H-inden-5-yl) (2-methyl-1-propyl-1H-indol-3-yl)methanone (GBD-020, 40).
Obtained as yellowish oil (72% yield). 1H NMR (400 MHz, CDCl3): δ 1.01 (t, 3H), 1.17 (s, 6H), 1.85 (sextet, 2H), 2.59 (s, 3H), 2.75 (s, 2H), 2.78 (s, 2H), 4.11 (t, 2H), 7.05 (app. t, 1H), 7.15–7.21 (m, 2H), 7.33 (app. t, 2H), 7.53 (app. d, 1H), 7.60 (s, 1H). 13C{1H}NMR (100 MHz, CDCl3): δ 11.48, 12.43, 22.99, 28.64, 40.50, 44.84, 47.40, 47.74, 109.36, 113.92, 121.02, 121.06, 121.70, 124.24, 125.41, 127.28, 127.86, 135.83, 139.56, 143.70, 143.79, 147.83, 193.28. HRMS (ESI, m/z): for C24H28NO [M + H]+ calcd, 346.2165; found, 346.2162. Purity (qNMR), 98.8%.
(2,2-Dimethyl-2,3-dihydro-1H-inden-5-yl) (1-butyl-2-methyl-1H-indol-3-yl)methanone (GBD-021, 41).
Obtained as yellowish oil (71% yield). 1H NMR (400 MHz, CDCl3): δ 0.98 (t, 3H), 1.16 (s, 6H), 1.43 (sextet, 2H), 1.78 (quintet, 2H), 2.59 (s, 3H), 2.74 (s, 2H), 2.78 (s, 2H), 4.13 (t, 2H), 7.04 (app. t, 1H), 7.15–7.21 (m, 2H), 7.32 (dd, 2H), 7.52 (app. d, 1H), 7.59 (s, 1H). 13C{1H}NMR (100 MHz, CDCl3): δ 12.42, 13.83, 20.31, 28.65, 31.81, 40.52, 43.15, 47.41, 47.76, 109.33, 113.94, 121.02, 121.09, 121.70, 124.25, 125.42, 127.31, 127.88, 135.79, 139.58, 143.72, 143.77, 147.84, 193.28. HRMS (ESI, m/z): for C25H30NO [M + H]+ calcd, 360.2322; found, 360.2318. Purity (qNMR), 97.3%.
(2,2-Dimethyl-2,3-dihydro-1H-inden-5-yl) (2-methyl-1-pentyl-1H-indol-3-yl)methanone (GBD-022, 42).
Obtained as yellowish oil (74% yield). 1H NMR (400 MHz, CDCl3): δ 0.92 (t, 3H), 1.17 (s, 6H), 1.34–1.43 (m, 4H), 1.80 (quintet, 2H), 2.59 (s, 3H), 2.75 (s, 2H), 2.78 (s, 2H), 4.12 (t, 2H), 7.05 (m, 1H), 7.15–7.21 (m, 2H), 7.33 (app. t, 2H), 7.53 (app. d, 1H), 7.61 (s, 1H). 13C{1H}NMR (100 MHz, CDCl3): δ 12.42, 13.95, 22.41, 28.65, 29.13, 29.43, 40.51, 43.34, 47.41, 47.75, 109.33, 113.92, 121.02, 121.08, 121.71, 124.24, 125.41, 127.31, 127.86, 135.78, 139.59, 143.70, 143.75, 147.82, 193.26. HRMS (ESI, m/z): for C26H32NO [M + H]+ calcd, 374.2478; found, 374.2477. Purity (qNMR), 96.2%.
(2,2-Dimethyl-2,3-dihydro-1H-inden-5-yl) (1-hexyl-2-methyl-1H-indol-3-yl)methanone (GBD-023, 43).
Obtained as yellowish oil (62% yield). 1H NMR (400 MHz, CDCl3): δ 0.89 (t, 3H), 1.17 (s, 6H), 1.28–1.44 (m, 6H), 1.79 (quintet, 2H), 2.59 (s, 3H), 2.74 (s, 2H), 2.78 (s, 2H), 4.12 (t, 2H), 7.04 (m, 1H), 7.15–7.21 (m, 2H), 7.32 (app. t, 2H), 7.52 (app. d, 1H), 7.59 (s, 1H). 13C{1H}NMR (100 MHz, CDCl3): δ 12.44, 14.00, 22.55, 26.72, 28.65, 29.70, 31.47, 40.52, 43.39, 47.42, 47.76, 109.32, 113.93, 121.02, 121.09, 121.71, 124.25, 125.43, 127.32, 127.87, 135.78, 139.60, 143.71, 143.75, 147.83, 193.28. HRMS (ESI, m/z): for C27H34NO [M + H]+ calcd, 388.2635; found, 388.2632. Purity (qNMR), 98.1%.
(2,2-Dimethyl-2,3-dihydro-1H-inden-5-yl) (1-benzyl-2-methyl-1H-indol-3-yl)methanone (GBD-024, 44).
Obtained as yellowish oil (56% yield). 1H NMR (400 MHz, CDCl3): δ 1.18 (s, 6H), 2.54 (s, 3H), 2.76 (s, 2H), 2.79 (s, 2H), 5.40 (s, 2H), 7.01–7.11 (m, 3H), 7.13–7.23 (m, 2H), 7.24–7.33 (m, 4H), 7.39–7.43 (app. d, 1H), 7.54–7.58 (app. d, 1H), 7.63 (br s, 1H). 13C{1H}NMR (100 MHz, CDCl3): δ 12.48, 28.65, 40.53, 46.62, 47.42, 47.77, 109.54, 114.45, 121.16, 121.36, 122.14, 124.30, 125.47, 125.99, 127.31, 127.69, 127.96, 128.97, 136.28, 136.31, 139.39, 143.79, 143.95, 148.06, 193.34. HRMS (ESI, m/z): for C28H28NO [M + H]+ calcd, 394.2165; found, 394.2162. Purity (qNMR), 97.4%.
(2,2-Dimethyl-2,3-dihydro-1H-inden-5-yl) (1-(4-fluorobenzyl)-2-methyl-1H-indol-3-yl)methanone (GBD-025, 45).
Obtained as yellowish oil (52% yield). 1H NMR (400 MHz, CDCl3): δ 1.18 (s, 6H), 2.53 (s, 3H), 2.76 (s, 2H), 2.79 (s, 2H), 5.36 (s, 2H), 6.98–7.02 (m, 4H), 7.06–7.11 (m, 1H), 7.14–7.27 (m, 3H), 7.39–7.43 (app. d, 1H), 7.54–7.57 (app. d, 1H), 7.63 (br s, 1H). 13C{1H}NMR (100 MHz, CDCl3): δ 12.44, 28.64, 40.54, 45.98, 47.41, 47.77, 109.40, 114.59, 115.83, 116.04, 121.23, 121.45, 122.24, 124.32, 125.47, 127.32, 127.96, 127.72, 127.64, 132.01, 136.14, 139.29, 143.86, 143.83, 148.16, 163.41, 193.29. 19FNMR (376 MHz, CDCl3): δ −114.40. HRMS (ESI, m/z): for C28H27FNO [M + H]+ calcd, 412.2071; found, 412.2068. Purity (qNMR), 96.8%.
(E)-(2,2-dimethyl-2,3-dihydro-1H-inden-5-yl) (2-methyl-1-(pent-2-en-1-yl)-1H-indol-3-yl)methanone (GBD-026, 46).
Obtained as yellowish oil (78% yield). 1H NMR (400 MHz, CDCl3): δ 0.94 (t, 3H), 1.17 (s, 6H), 2.02 (quint, 2H), 2.56 (s, 3H), 2.75 (s, 2H), 2.78 (s, 2H), 4.69–4.73 (m, 2H), 5.48–5.58 (m, 2H), 7.03–7.09 (m, 1H), 7.15–7.22 (m, 2H), 7.31 (dd, 1H), 7.37 (dd, 1H), 7.54 (d, 1H), 7.61 (s, 1H). 13C{1H}NMR (100 MHz, CDCl3): δ 12.32, 13.19, 25.07, 28.62, 40.49, 44.92, 47.38, 47.73, 109.41, 114.02, 121.02, 121.09, 121.79, 122.51, 124.23, 125.41, 127.24, 127.87, 135.38, 135.84, 139.51, 143.69, 143.88, 147.85, 193.27. HRMS (ESI, m/z): for C26H30NO [M + H]+ calcd, 372.2322; found, 372.2318. Purity (qNMR), 95.9%.
(Z)-(2,2-dimethyl-2,3-dihydro-1H-inden-5-yl) (2-methyl-1-(pent-2-en-1-yl)-1H-indol-3-yl)methanone (GBD-027, 47).
Obtained as white, powdery crystals (73% yield). 1H NMR (400 MHz, CDCl3): δ 1.11 (t, 3H), 1.17 (s, 6H), 2.29 (quint, 2H), 2.58 (s, 3H), 2.75 (s, 2H), 2.78 (s, 2H), 4.78–4.83 (m, 2H), 5.32–5.40 (m, 1H), 5.57–5.65 (m, 1H), 7.04–7.09 (m, 1H), 7.16–7.22 (m, 2H), 7.30 (dd, 1H), 7.36 (dd, 1H), 7.53 (d, 1H), 7.61 (s, 1H). 13C{1H}NMR (100 MHz, CDCl3): δ 12.39, 14.03, 21.07, 28.65, 40.52, 40.63, 47.41, 47.76, 109.24, 114.15, 121.12, 121.16, 121.81, 123.38, 124.26, 125.44, 127.37, 127.91, 134.90, 135.76, 139.50, 143.67, 143.73, 147.91, 193.26. HRMS (ESI, m/z) for C26H30NO [M + H]+: calcd, 372.2322; found, 372.2322. mp 65–66 °C. Purity (qNMR), 100.5%.
(2,2-Dimethyl-2,3-dihydro-1H-inden-5-yl) (2-methyl-1-(pent-4-en-1-yl)-1H-indol-3-yl)methanone (GBD-028, 48).
Obtained as yellowish oil (48% yield). 1H NMR (400 MHz, CDCl3): δ 1.17 (s, 6H), 1.90 (quintet, 2H), 2.18 (q, 2H), 2.58 (s, 3H), 2.74 (s, 2H), 2.78 (s, 2H), 4.14 (t, 2H), 5.03–5.14 (m, 2H), 5.78–5.90 (ddt, 1H), 7.02–7.08 (m, 1H), 7.15–7.21 (m, 2H), 7.29–7.36 (app. t, 2H), 7.53 (app. d, 1H), 7.60 (s, 1H). 13C{1H}NMR (100 MHz, CDCl3): δ 12.41, 28.58, 28.64, 30.90, 40.50, 42.68, 47.41, 47.75, 109.27, 114.04, 115.95, 121.08, 121.11, 121.76, 124.25, 125.41, 127.31, 127.87, 135.75, 136.94, 139.52, 143.64, 143.72, 147.87, 193.25. HRMS (ESI, m/z): for C26H30NO [M + H]+ calcd, 372.2322; found, 372.2319. Purity (qNMR), 99.7%.
(2,2-Dimethyl-2,3-dihydro-1H-inden-5-yl) (2-methyl-1-(pent-2-yn-1-yl)-1H-indol-3-yl)methanone (GBD-030, 50).
Obtained as yellowish oil (22% yield). 1H NMR (400 MHz, CDCl3): δ 1.09 (t, 3H), 1.17 (s, 6H), 2.15 (qt, 2H), 2.62 (s, 3H), 2.75 (s, 2H), 2.78 (s, 2H), 4.84 (t, 2H), 7.05–7.11 (m, 1H), 7.17–7.25 (m, 2H), 7.36–7.42 (m, 2H), 7.54 (app. d, 1H), 7.62 (s, 1H). 13C{1H}NMR (100 MHz, CDCl3): δ 12.29, 12.33, 13.59, 28.64, 33.04, 40.51, 47.39, 47.75, 72.65, 86.73, 109.33, 114.42, 121.08, 121.31, 122.02, 124.27, 125.46, 127.26, 127.94, 135.57, 139.36, 143.38, 143.74, 148.01, 193.23. HRMS (ESI, m/z): for C26H28NO [M + H]+ calcd, 370.2165; found, 370.2163. Purity (qNMR), 99.4%.
General Procedure for the Synthesis of 49 and 51.
To a mixture of 24 (100 mg, 0.33 mmol) and DMSO (2.0 mL) was added KOH (37.0 mg, 0.66 mmol). After stirring at ambient temperature for 1 h, the alkyl halide (0.49 mmol) was added. The resulting mixture was stirred for 2 h at ambient temperature. The reaction mixture was quenched with water and extracted with ethyl acetate (2 × 10 mL). The organic layer was washed with brine, dried over Na2SO4, concentrated under reduced pressure, and purified by automatic flash column chromatography on silica gel (gradient elution from 5 to 40% EtOAc-hexanes).
(2,2-Dimethyl-2,3-dihydro-1H-inden-5-yl) (2-methyl-1-(pent-4-yn-1-yl)-1H-indol-3-yl)methanone (GBD-029, 49).
Obtained as yellowish oil (63% yield). 1H NMR (400 MHz, CDCl3): δ 1.17 (s, 6H), 2.02 (quintet, 2H), 2.10 (t, 1H), 2.26–2.32 (td, 2H), 2.61 (s, 3H), 2.75 (s, 2H), 2.78 (s, 2H), 4.29 (t, 2H), 7.03–7.09 (m, 1H), 7.16–7.22 (m, 2H), 7.33–7.40 (m, 2H), 7.53 (app. d, 1H), 7.60 (s, 1H). 13C{1H}NMR (100 MHz, CDCl3): δ 12.37, 15.95, 28.17, 28.62, 40.49, 41.83, 47.38, 47.73, 69.89, 82.75, 109.23, 114.16, 121.10, 121.17, 121.88, 124.25, 125.41, 127.29, 127.87, 135.79, 139.42, 143.65, 143.73, 147.94, 193.23. HRMS (ESI, m/z): for C26H28NO [M + H]+ calcd, 370.2165; found, 370.2163. Purity (qNMR), 100.5%.
(2,2-Dimethyl-2,3-dihydro-1H-inden-5-yl) (2-methyl-1-(prop-2-yn-1-yl)-1H-indol-3-yl)methanone (GBD-031, 51).
Obtained as colorless oil (32% yield). 1H NMR (400 MHz, CDCl3): δ 1.17 (s, 6H), 2.33 (t, 1H), 2.63 (s, 3H), 2.75 (s, 2H), 2.79 (s, 2H), 4.89 (d, 2H), 7.07–7.11 (m, 1H), 7.18–7.25 (m, 2H), 7.36–7.43 (m, 2H), 7.54 (app. d, 1H), 7.62 (s, 1H). 13C{1H}NMR (100 MHz, CDCl3): δ 12.25, 28.65, 32.61, 40.53, 47.40, 47.77, 73.17, 77.22, 109.12, 114.80, 121.23, 121.55, 122.27, 124.32, 125.50, 127.30, 127.99, 135.50, 139.17, 143.01, 143.81, 148.21, 193.22. HRMS (ESI, m/z): for C24H24NO [M+ H]+ calcd, 342.1852; found, 342.1849. Purity (qNMR), 97.2%.
Pharmacology.
CP-55,940, rimonabant, and AM-630 were obtained from Tocris Bioscience (Bristol, UK). All drugs were prepared as a stock solution in 100% DMSO at a concentration of 100 mM, divided into aliquots, and maintained at −4 °C until use. [3H]CP-55,950 (168 Ci/mmol) and [35S]GTPγS (1250 Ci/mmol) were purchased from Perkin Elmer (Boston, MA). All other reagents were obtained from Fisher Scientific Inc. (Pittsburgh, PA).
Competition Receptor Binding.
Increasing concentrations of the GBD compounds were incubated with 0.2 nM of the nonselective CB1/CB2 agonist [3H]CP-55,940 in a final volume of 1 mL of the binding buffer (50 mM Tris, 0.05% bovine serum albumin, 5 mM of MgCl2, pH 7.4) as described previously.66 Each binding assay contained 100 or 50 μg of membrane protein prepared from CHO-hCB1 or CHO-hCB2 cells, respectively. Reactions were incubated for 90 min at rt. Nonspecific binding was defined as binding observed in the presence of 10 μM of the nonselective CB1/CB2 ligand WIN-55,212-2. Reactions were terminated by rapid vacuum filtration through Whatman GF/B glass fiber filters, followed by three washes with the ice-cold binding buffer. Filters were then immediately placed into scintillation vials with 4 mL of the scintiverse™ BD cocktail scintillation fluid (Fisher Scientific, Fair Lawn, NJ). Samples were incubated overnight in the scintillation fluid, and vortexed and bound reactivity was determined by employing a liquid scintillation spectrophotometer (Tri Carb 2100 TR Liquid Scintillation Analyzer, Packard Instrument Company, Meriden, CT).
[35S]GTPγS Binding.
All [35S]GTPγS assays were conducted as described previously62 in a buffer containing 20 mM N-(2-hydroxyethyl)piperazine-N′-ethanesulfonic acid (HEPES), 100 mM NaCl, 10 mM MgCl2, 0.05% BSA, and 20 units/L of adenosine deaminase. Assays were performed in triplicate in a final volume of 1 mL with all reactions containing 0.1 nM [35S]GTPγS and an experimental drug or drug vehicle and 50 or 25 μg of membrane protein prepared from CHO-hCB2 cells, respectively. Nonspecific binding was defined by inclusion of 10 μM of nonradiolabeled GTPγS. Reactions were incubated at 30 °C for 30 min and terminated by rapid vacuum filtration through Whatman GF/B glass fiber filters and followed by four 1 mL washes with the ice-cold filtration buffer (20 mM HEPES, pH 7.4, 0.05% BSA). Filters were then immediately placed into scintillation vials with 4 mL of the scintiverse™ BD cocktail scintillation fluid (Fisher Scientific, Fair Lawn, NJ). Samples were incubated overnight in the scintillation fluid, and vortexed and bound reactivity was determined by employing a liquid scintillation spectrophotometer (Tri Carb 2100 TR Liquid Scintillation Analyzer, Packard Instrument Company, Meriden, CT).
Data Analysis.
Curve fitting was conducted utilizing GraphPad Prism v6.0b (GraphPad Software, Inc.; San Diego, CA). Nonlinear regression for one-site competition was used to determine the IC50 for competition receptor binding. The Cheng-Prusoff equation was employed to convert the experimental IC50 values obtained from competition receptor binding experiments to Ki values (a quantitative measure of receptor affinity). Statistical significance of values from 100% (basal) in the [35S]GTPγS assay was assessed by the one-sample t-test (*P < 0.05 and **P < 0.01).
Molecular Modeling.
To elucidate the binding modes of the GBD series of compounds, a hybrid docking/MD simulation approach was utilized. This methodology has been previously used accurately and precisely to estimate the binding affinity and selectivity of compounds toward the cannabinoid receptors.67 In summary, the selected ligands (Figure 2) GBD-002 and GBD-003 had their lowest energy conformation estimated using OpenEye OMEGA.68 These ligands were then prepared with Autodock Tools to the PDBQT format for use with Autodock Vina.69,70 CB1 and CB2 receptor structures (PDB ID: 5XR8 for CB1R and 5ZTY for CB2R) were used for the initial docking, and the grid center was placed upon the original crystal structure ligand.71,72 The resulting binding poses (Figure 3A1,A2,B1,B2) then had their atomic charges calculated using the AM1-BCC method in the Antechamber module of AMBER1673,74 and were subsequently energy-minimized using the SANDER module of AMBER16.73 The energy-minimized binding structures then had their binding free energy estimated using the MMPBSA.py75 module of AMBER16.73 The rest of the GBD series of compounds were then modeled using PyMol76 to revise the structure of GBD-003 within either CB1R or CB2R as the initial binding structure. The initial binding structures of these compounds were similarly energy-minimized and had their binding free energy estimated by using the same MMPBSA.py module.
Supplementary Material
ACKNOWLEDGMENTS
Prof Steven Kinsey (University of Connecticut) is gratefully acknowledged for helpful discussions and assistance with early development of this project.
Funding
This work was supported in part by the funding from the National Institutes of Health (R01 DA039143) for PLP, with additional support from West Virginia University and the Eberly Family Foundation. Molecular modeling studies were supported by the National Institutes of Health (P20 GM130456 and T32 DA016176).
ABBREVIATIONS
- ECS
endocannabinoid system
- HFIP
1,1,1,3,3,3-hexafluoroisopropanol
- BINAP
(2,2′-bis(diphenylphosphino)-1,1′-binaphthyl)
- SI
selectivity index
- nbd
norbornadiene
- n-BuLi
n-butyllithium
- t-BuLi
tert-butyllithium
- MsCl
mesyl chloride
- Tf2O
triflic anhydride
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.1c00442.
Molecular formula strings (CSV)
Graphs correlating experimental and calculated binding energies, copies of 1H and 13C NMR spectra and qNMR analysis, and PDB files for docking models (PDF)
The authors declare no competing financial interest.
Contributor Information
Harvey F. Fulo, C. Eugene Bennett Department of Chemistry, West Virginia University, Morgantown, West Virginia 26506, United States
Amal Shoeib, Department of Pharmacology and Toxicology, University of Arkansas for Medical Sciences, Little Rock, Arkansas 72205, United States.
Christian V. Cabanlong, Department of Pharmacology and Toxicology, University of Arkansas for Medical Sciences, Little Rock, Arkansas 72205, United States
Alexander H. Williams, Department of Pharmaceutical Sciences, University of Kentucky, Lexington, Kentucky 40536, United States
Chang-Guo Zhan, Department of Pharmaceutical Sciences, University of Kentucky, Lexington, Kentucky 40536, United States.
Paul L. Prather, Department of Pharmacology and Toxicology, University of Arkansas for Medical Sciences, Little Rock, Arkansas 72205, United States
Gregory B. Dudley, C. Eugene Bennett Department of Chemistry, West Virginia University, Morgantown, West Virginia 26506, United States.
REFERENCES
- (1).Di Marzo V Targeting the Endocannabinoid System: To Enhance or Reduce? Nat. Rev. Drug Discovery 2008, 7, 438–455. [DOI] [PubMed] [Google Scholar]
- (2).Finn DP Endocannabinoid-mediated Modulation of Stress Responses: Physiological and Pathophysiological Significance. Immunobiology 2010, 215, 629–646. [DOI] [PubMed] [Google Scholar]
- (3).Bab I; Zimmer A Cannabinoid Receptors and the Regulation of Bone Mass. Br. J. Pharmacol 2008, 153, 182–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Oesch S; Gertsch J Cannabinoid Receptor Ligands as Potential Anticancer Agents—High Hopes for New Therapies? J. Pharm. Pharmacol 2009, 61, 839–853. [DOI] [PubMed] [Google Scholar]
- (5).Karsak M; Gaffal E; Date R; Wang-Eckhardt L; Rehnelt J; Petrosino S; Starowicz K; Steuder R; Schlicker E; Cravatt B; Mechoulam R; Buettner R; Werner S; Di Marzo V; Tuting T; Zimmer A Attenuation of Allergic Contact Dermatitis Through the Endocannabinoid System. Science 2007, 316, 1494–1497. [DOI] [PubMed] [Google Scholar]
- (6).Guindon J; Hohmann AG Cannabinoid CB2 receptors: a therapeutic target for the treatment of inflammatory and neuropathic pain. Br. J. Pharmacol 2008, 153, 319–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Malfitano AM; Ciaglia E; Gangemi G; Gazzerro P; Laezza C; Bifulco M Update on the Endocannabinoid System as an Anticancer Target. Expert Opin. Ther. Targets 2011, 15, 297–308. [DOI] [PubMed] [Google Scholar]
- (8).Cianchi F; Papucci L; Schiavone N; Lulli M; Magnelli L; Vinci MC; Messerini L; Manera C; Ronconi E; Romagnani P; Donnini M; Perigli G; Trallori G; Tanganelli E; Capaccioli S; Masini E Cannabinoid Receptor Activation Induces Apoptosis Through Tumor Necrosis Factor Alpha-mediated Ceramide de novo Synthesis in Colon Cancer Cells. Clin. Cancer Res 2008, 14, 7691–7700. [DOI] [PubMed] [Google Scholar]
- (9).Velasco G; Sánchez C; Guzmán M Towards the Use of Cannabinoids as Antitumour Agents. Nat. Rev. Cancer 2012, 12, 436–444. [DOI] [PubMed] [Google Scholar]
- (10).Pertwee RG Targeting the Endocannabinoid System with Cannabinoid Receptor Agonists: Pharmacological Strategies and Therapeutic Possibilities. Philos. Trans. R. Soc. Lond. B Biol. Sci 2012, 367, 3353–3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Munro S; Thomas KL; Abu-Shaar M Molecular Characterization of a Peripheral Receptor for Cannabinoids. Nature 1993, 365, 61–65. [DOI] [PubMed] [Google Scholar]
- (12).Reggio P Endocannabinoid Binding to the Cannabinoid Receptors: What is Known and What Remains Unknown. Curr. Med. Chem 2010, 17, 1468–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Matsuda LA; Lolait SJ; Brownstein MJ; Young AC; Bonner TI Structure of a Cannabinoid Receptor and Functional Expression of the Cloned cDNA. Nature 1990, 346, 561–564. [DOI] [PubMed] [Google Scholar]
- (14).Svízenska I; Dubový P; Sulcova A Cannabinoid Receptors 1 and 2 (CB1 and CB2), Their Distribution, Ligands and Functional Involvement in Nervous System Structures — A Short Review. Pharmacol. Biochem. Behav 2008, 90, 501–511. [DOI] [PubMed] [Google Scholar]
- (15).Ameri A The Effects of Cannabinoids on the Brain. Prog. Neurobiol 1999, 58, 315–348. [DOI] [PubMed] [Google Scholar]
- (16).Das SK; Paria BC; Chakraborty I; Dey SK Cannabinoid Ligand-Receptor Signaling in the Mouse Uterus. Proc. Natl. Acad. Sci. U.S.A 1995, 92, 4332–4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Galiegue S; Mary S; Marchand J; Dussossoy D; Carriere D; Carayon P; Bouaboula M; Shire D; Fur G; Casellas P Expression of Central and Peripheral Cannabinoid Receptors in Human Immune Tissues and Leukocyte Subpopulations. Eur. J. Biochem 1995, 232, 54–61. [DOI] [PubMed] [Google Scholar]
- (18).Gerard CM; Mollereau C; Vassart G; Parmentier M Molecular Cloning of a Human Cannabinoid Receptor Which is Also Expressed in Testis. Biochem. J 1991, 279, 129–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Wenger T; Ledent C; Csernus V; Gerendai I The Central Cannabinoid Receptor Inactivation Suppresses Endocrine Reproductive Functions. Biochem. Biophys. Res. Commun 2001, 284, 363–368. [DOI] [PubMed] [Google Scholar]
- (20).Benito C; Tolón RM; Pazos MR; Núñez E; Castillo AI; Romero J Cannabinoid CB2 Receptors in Human Brain Inflammation. Br. J. Pharmacol 2008, 153, 277–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Onaivi ES; Ishiguro H; Gong J-P; Patel S; Perchuk A; Meozzi PA; Myers L; Mora Z; Tagliaferro P; Gardner E; Brusco A; Akinshola BE; Liu Q-R; Hope B; Iwasaki S; Arinami T; Teasenfitz L; Uhl GR Discovery of the Presence and Functional Expression of Cannabinoid CB2 Receptors in Brain. Ann. N.Y. Acad. Sci 2006, 1074, 514–536. [DOI] [PubMed] [Google Scholar]
- (22).Maccarrone M; Bab I; Bíró T; Cabral GA; Dey SK; Di Marzo V; Konje JC; Kunos G; Mechoulam R; Pacher P; Sharkey KA; Zimmer A Endocannabinoid Signaling at the Periphery: 50 years After THC. Trends Pharmacol. Sci 2015, 36, 277–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Maxwell JC Psychoactive Substances — Some New, Some Old: A Scan of the Situation in the U.S. Drug Alcohol Depend. 2014, 134, 71–77. [DOI] [PubMed] [Google Scholar]
- (24).Ford BM; Tai S; Fantegrossi WE; Prather PL Synthetic Pot: Not Your Grandfather’s Marijuana. Trends Pharmacol. Sci 2017, 38, 257–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Banister SD; Wilkinson SM; Longworth M; Stuart J; Apetz N; English K; Brooker L; Goebel C; Hibbs DE; Glass M; Connor M; McGregor IS; Kassiou M The Synthesis and Pharmacological Evaluation of Adamantane-Derived Indoles: Cannabimimetic Drugs of Abuse. ACS Chem. Neurosci 2013, 4, 1081–1092 and the references cited therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Wiley JL; Marusich JA; Lefever TW; Grabenauer M; Moore KN; Thomas BF Cannabinoids in Disguise: Δ9-Tetrahydrocannabinol-like Effects of Tetramethylcyclopropyl Ketone Indoles. Neuropharmacology 2013, 75, 145–154 and the references cited therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Nutt DJ; King LA; Nichols DE Effects of Schedule I Drug Laws on Neuroscience Research and Treatment Innovation. Nat. Rev. Neurosci 2013, 14, 577–585. [DOI] [PubMed] [Google Scholar]
- (28).Huffman JW; Zengin G; Wu M-J; Lu J; Hynd G; Bushell K; Thompson ALS; Bushell S; Tartal C; Hurst DP; Reggio PH; Selley DE; Cassidy MP; Wiley JL; Martin BR Structure-activity relationships for 1-alkyl-3-(1-naphthoyl)indoles at the cannabinoid CB1 and CB2 receptors: steric and electronic effects of naphthoyl substituents. New highly selective CB2 receptor agonists. Bioorg. Med. Chem 2005, 13, 89–112. [DOI] [PubMed] [Google Scholar]
- (29).Drug Enforcement Administration. Schedules of Controlled Substances: Temporary Placement of Five Synthetic Cannabinoids into Schedule I. Fed. Regist 2011, 76, 11075–11078. [PubMed] [Google Scholar]
- (30).Frost JM; Dart MJ; Tietje KR; Garrison TR; Grayson GK; Daza AV; El-Kouhen OF; Yao BB; Hsieh GC; Pai M; Zhu CZ; Chandran P; Meyer MD Indol-3-ylcycloalkyl Ketones: Effects of N1 Substituted Indole Side Chain Variations on CB2Cannabinoid Receptor Activity. J. Med. Chem 2010, 53, 295–315. [DOI] [PubMed] [Google Scholar]
- (31).Malan TP; Ibrahim MM; Lai J; Vanderah TW; Makriyannis A; Porreca F CB2 cannabinoid receptor agonists: pain relief without psychoactive effects? Curr. Opin. Pharmacol 2003, 3, 62–67. [DOI] [PubMed] [Google Scholar]
- (32).Manera C; Tuccinardi T; Martinelli A Indoles and Related Compounds as Cannabinoid Ligands. Mini Rev. Med. Chem 2008, 8, 370–387. [DOI] [PubMed] [Google Scholar]
- (33).Zhang M; Adler MW; Abood ME; Ganea D; Jallo J; Tuma RF CB2 receptor activation attenuates microcirculatory dysfunction during cerebral ischemic/reperfusion injury. Microvasc. Res 2009, 78, 86–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Shoemaker JL; Seely KA; Reed RL; Crow JP; Prather PL The CB2 cannabinoid agonist AM-1241 prolongs survival in a transgenic mouse model of amyotrophic lateral sclerosis when initiated at symptom onset. J. Neurochem 2007, 101, 87–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Croxford JL; Miller SD Immunoregulation of a viral model of multiple sclerosis using the synthetic cannabinoid R(+)-WIN55,212. J. Clin. Invest 2003, 111, 1231–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Fernández-Ruiz J; Romero J; Velasco G; Tolón RM; Ramos JA; Guzmán M Cannabinoid CB2 receptor: a new target for controlling neural cell survival? Trends Pharmacol. Sci 2007, 28, 39–45. [DOI] [PubMed] [Google Scholar]
- (37).Benito C; Núñez E; Tolón RM; Carrier EJ; Rábano A; Hillard CJ; Romero J Cannabinoid CB2Receptors and Fatty Acid Amide Hydrolase Are Selectively Overexpressed in Neuritic Plaque-Associated Glia in Alzheimer’s Disease Brains. J. Neurosci 2003, 23, 11136–11141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Pacher P; Bátkai S; Kunos G The Endocannabinoid System as an Emerging Target of Pharmacotherapy. Pharmacol. Rev 2006, 58, 389–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Sagredo O; García-Arencibia M; de Lago E; Finetti S; Decio A; Fernández-Ruiz J Cannabinoids and Neuroprotection in Basal Ganglia Disorders. Mol. Neurobiol 2007, 36, 82–91. [DOI] [PubMed] [Google Scholar]
- (40).Whiteside G; Lee G; Valenzano K The Role of the Cannabinoid CB2 Receptor in Pain Transmission and Therapeutic Potential of Small Molecule CB2 Receptor Agonists. Curr. Med. Chem 2007, 14, 917–936. [DOI] [PubMed] [Google Scholar]
- (41).Cheng Y; Hitchcock SA Targeting Cannabinoid Agonists for Inflammatory and Neuropathic Pain. Expert Opin. Invest. Drugs 2007, 16, 951–965. [DOI] [PubMed] [Google Scholar]
- (42).Yao BB; Hsieh GC; Frost JM; Fan Y; Garrison TR; Daza AV; Grayson GK; Zhu CZ; Pai M; Chandran P; Salyers AK; Wensink EJ; Honore P; Sullivan JP; Dart MJ; Meyer MD In Vitro and In Vivo Characterization of A-796260: A Selective Cannabinoid CB2 Receptor Agonist Exhibiting Analgesic Activity in Rodent Pain Models. Br. J. Pharmacol 2008, 153, 390–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Ramirez BG; Blazquez C; Gomez del Pulgar T; Guzman M; de Ceballos ML Prevention of Alzheimer’s Disease Pathology by Cannabinoids: Neuroprotection Mediated by Blockade of Microglial Activation. J. Neurosci 2005, 25, 1904–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Body-Malapel M; El-Bakali J; Mérour E; Stern E; Millet R; Desreumaux P 741 4-Oxo-1,4-Dihydroquinoline-3-Carboxamides Derivatives As New Potent and Selective Cb2 Agonists with Anti-Inflammatory and Analgesic Properties in the Gut. Gastroenterology 2008, 134, A–107. [Google Scholar]
- (45).Benito C; Romero JP; Tolón RM; Clemente D; Docagne F; Hillard CJ; Guaza C; Romero J Cannabinoid CB1 and CB2 Receptors and Fatty Acid Amide Hydrolase are Specific Markers of Plaque Cell Subtypes in Human Multiple Sclerosis. J. Neurosci 2007, 27, 2396–2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Pasquini S; Ligresti A; Mugnaini C; Semeraro T; Cicione L; De Rosa M; Guida F; Luongo L; De Chiaro M; Cascio MG; Bolognini D; Marini P; Pertwee R; Maione S; Marzo VD; Corelli F Investigations on the 4-Quinolone-3-carboxylic acid Motif. 3. Synthesis, Structure-affinity Relationships, and Pharmacological Characterization of 6-Substituted 4-quinolone-3-carboxamides as Highly Selective Cannabinoid-2 Receptor Ligands. J. Med. Chem 2010, 53, 5915–5928 and the references cited therein. [DOI] [PubMed] [Google Scholar]
- (47).Lunn CA; Fine JS; Rojas-Triana A; Jackson JV; Fan X; Kung TT; Gonsiorek W; Schwarz MA; Lavey B; Kozlowski JA; Narula SK; Lundell DJ; Hipkin RW; Bober LA A Novel Cannabinoid Peripheral Cannabinoid Receptor-Selective Inverse Agonist Blocks Leukocyte Recruitment in Vivo. J. Pharmacol. Exp. Therapeut 2006, 316, 780–788. [DOI] [PubMed] [Google Scholar]
- (48).Savinainen JR; Kokkola T; Salo OMH; Poso A; Järvinen T; Laitinen JT Identification of WIN55212-3 as a competitive neutral antagonist of the human cannabinoid CB2 receptor. Br. J. Pharmacol 2005, 145, 636–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Deveaux V; Cadoudal T; Ichigotani Y; Teixeira-Clerc F; Louvet A; Manin S; Nhieu JT-V; Belot MP; Zimmer A; Even P; Cani PD; Knauf C; Burcelin R; Bertola A; Le Marchand-Brustel Y; Gual P; Mallat A; Lotersztajn S Cannabinoid CB2 Receptor Potentiates Obesity-Associated Inflammation, Insulin Resistance and Hepatic Steatosis. PloS One 2009, 4, No. e5844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Frost JM; Dart MJ; Tietje KR; Garrison TR; Grayson GK; Daza AV; El-Kouhen OF; Miller LN; Li L; Yao BB; Hsieh GC; Pai M; Zhu CZ; Chandran P; Meyer MD Indol-3-yl-tetramethylcyclopropyl Ketones: Effects of Indole Ring Substitution on CB2Cannabinoid Receptor Activity. J. Med. Chem 2008, 51, 1904–1912. [DOI] [PubMed] [Google Scholar]
- (51).Drug Enforcement Administration. Schedules of Controlled Substances: Placement of UR-144, XLR11, and AKB48 into Schedule I. Fed. Regist 2016, 81, 29142–29145. [PubMed] [Google Scholar]
- (52).Regulatory agencies including the US Drug Enforcement Agency consider “history and current pattern of abuse”. “evidence that individuals are taking the drug or other substance in amounts sufficient to create a hazard”; and “significant diversion of the drug or other substance from legitimate drug channels” as key “indicators that a drug or other substance has a potential for abuse”; see: Drugs of Abuse: A DEA Resource Guide, 2020 Edition, April 13, 2020. see https://www.dea.gov/documents/2020/04/13/drugs-abuse (accessed Apr 4 2021).
- (53).Wiley JL; Marusich JA; Huffman JW Moving Around the Molecule: Relationship Between Chemical Structure and in vivo Activity of Synthetic Cannabinoids. Life Sci 2014, 97, 55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Pasquini S; Mugnaini C; Ligresti A; Tafi A; Brogi S; Falciani C; Pedani V; Pesco N; Guida F; Luongo L; Varani K; Borea PA; Maione S; Di Marzo V; Corelli F Design, Synthesis, and Pharmacological Characterizationof Indol-3- ylacetamides, Indol-3-yloxoacetamides, and Indol-3-ylcarboxamides: Potent and Selective CB2 Cannabinoid Receptor Inverse Agonists. J. Med. Chem 2012, 55, 5391–5402. [DOI] [PubMed] [Google Scholar]
- (55).Tavakoli A; Dudley GB Synthesis of 4,4-Dimethyl-1,6-heptadiyne and Alcyopterosin O. Org. Lett 2020, 22, 8947–8951. [DOI] [PubMed] [Google Scholar]
- (56).Kramer NJ; Hoang TT; Dudley GB Reaction Discovery Using Neopentylene-Tethered Coupling Partners: Cycloisomerization/Oxidation of Electron-Deficient Dienynes. Org. Lett 2017, 19, 4636–4639. [DOI] [PubMed] [Google Scholar]
- (57).Bailey WF; Longstaff SC Generation and Cyclization of a Benzyne-Tethered Alkyllithium: Preparation of 4-Substituted Indans. J. Org. Chem 1998, 63, 432–433. [DOI] [PubMed] [Google Scholar]
- (58).Hoang TT; Dudley GB Synthesis of High-Value 1,6-Enynes by Tandem Fragmentation/Olefination. Org. Lett 2013, 15, 4026–4029. [DOI] [PubMed] [Google Scholar]
- (59).Aung MM; Griffin G; Huffman JW; Wu M-J; Keel C; Yang B; Showalter VM; Abood ME; Martin BR Influence of the N-1 Alkyl Chain Length of Cannabimimetic Indoles upon CB1 and CB2 Receptor Binding. Drug Alcohol Depend. 2000, 60, 133–140. [DOI] [PubMed] [Google Scholar]
- (60).Hynes J; Leftheris K; Wu H; Pandit C; Chen P; Norris DJ; Chen B-C; Zhao R; Kiener PA; Chen X; Turk LA; Patil-Koota V; Gillooly KM; Shuster DJ; McIntyre KW C-3 Amido-Indole Cannabinoid Receptor Modulators. Bioorg. Med. Chem. Lett 2002, 12, 2399–2402. [DOI] [PubMed] [Google Scholar]
- (61).Longworth M; Connor M; Banister SD; Kassiou M Synthesis and Pharmacological Profiling of the Metabolites of Synthetic Cannabinoid Drugs APICA, STS-135, ADB-PINACA, and 5FADB-PINACA. ACS Chem. Neurosci 2017, 8, 1673–1680. [DOI] [PubMed] [Google Scholar]
- (62).Brents LK; Gallus-Zawada A; Radominska-Pandya A; Vasiljevik T; Prisinzano TE; Fantegrossi WE; Moran JH; Prather PL Mono-hydroxylated Metabolites of the K2 Synthetic Cannabinoid JWH-073 Retain High Cannabinoid 1 Receptor (CB1R) Affinity and Exhibit Neutral Antagonist to Partial Agonist Activity. Biochem. Pharmacol 2012, 83, 952–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Klamo SB; Wendt OF; Henling LM; Day MW; Bercaw JE Dialkyl and Methyl-Alkyl Zirconocenes: Synthesis and Characterization of Zirconocene-Alkyls That Model the Polymeryl Chain in Alkene Polymerizations. Organometallics 2007, 26, 3018–3030. [Google Scholar]
- (64).Vekariya RH; Aubé J Hexafluoro-2-propanol-Promoted Intermolecular Friedel–Crafts Acylation Reaction. J. Org. Lett 2016, 18, 3534–3537. [DOI] [PubMed] [Google Scholar]
- (65).Hudlicky T; Frazier JO; Seoane G; Tiedje M; Seoane A; Kwart LD; Beal C Topological selectivity in the intramolecular [4 + 1] pyrroline annulation. Formal total stereospecific synthesis of (.+−.)-supinidine, (.+−.)-isoretronecanol, and (.+−.)-trachelanthamidine. J. Am. Chem. Soc 1986, 108, 3755–3762. [Google Scholar]
- (66).Brents LK; Reichard EE; Zimmerman SM; Moran JH; Fantegrossi WE; Prather PL Phase I Hydroxylated Metabolites of the K2 Synthetic Cannabinoid JWH-018 Retain in vitro and in vivo Cannabinoid 1 Receptor Affinity and Activity. PloS One 2011, 6, No. e21917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Yang J-F; Williams AH; Penthala NR; Prather PL; Crooks PA; Zhan C-G Binding Modes and Selectivity of Cannabinoid 1 (CB1) and Cannabinoid 2 (CB2) Receptor Ligands. ACS Chem. Neurosci 2020, 11, 3455–3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Hawkins PCD; Skillman AG; Warren GL; Ellingson BA; Stahl MT Conformer generation with OMEGA: algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model 2010, 50, 572–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Morris GM; Huey R; Lindstrom W; Sanner MF; Belew RK; Goodsell DS; Olson AJ AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem 2009, 30, 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Trott O; Olson AJ AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem 2010, 31, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Hua T; Vemuri K; Pu M; Qu L; Han GW; Wu Y; Zhao S; Shui W; Li S; Korde A; Laprairie RB; Stahl EL; Ho J-H; Zvonok N; Zhou H; Kufareva I; Wu B; Zhao Q; Hanson MA; Bohn LM; Makriyannis A; Stevens RC; Liu Z-J Crystal structure of the human cannabinoid receptor CB1. Cell 2016, 167, 750–762.e714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Li X; Hua T; Vemuri K; Ho J-H; Wu Y; Wu L; Popov P; Benchama O; Zvonok N; Locke K. a.; Qu L; Han GW; Iyer MR; Cinar R; Coffey NJ; Wang J; Wu M; Katritch V; Zhao S; Kunos G; Bohn LM; Makriyannis A; Stevens RC; Liu Z-J Crystal structure of the human cannabinoid receptor CB2. Cell 2019, 176, 459–467 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Wang J; Wang W; Kollman PA; Case DA Antechamber: an accessory software package for molecular mechanical calculations. J. Am. Chem. Soc 2001, 222, U403. [Google Scholar]
- (74).Pearlman DA; Case DA; Caldwell JW; Ross WS; Cheatham TE III; DeBolt S; Ferguson D; Seibel G; Kollman P AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput. Phys. Commun 1995, 91, 1–41. [Google Scholar]
- (75).Miller BR III; McGee TD Jr.; Swails JM; Homeyer N; Gohlke H; Roitberg AE MMPBSA py: an efficient program for end-state free energy calculations. J. Chem. Theory Comput 2012, 8, 3314–3321. [DOI] [PubMed] [Google Scholar]
- (76).DeLano WL Pymol: An open-source molecular graphics tool. CCP4 Newsl. protein crystallogr 2002, 40, 82–92. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.