

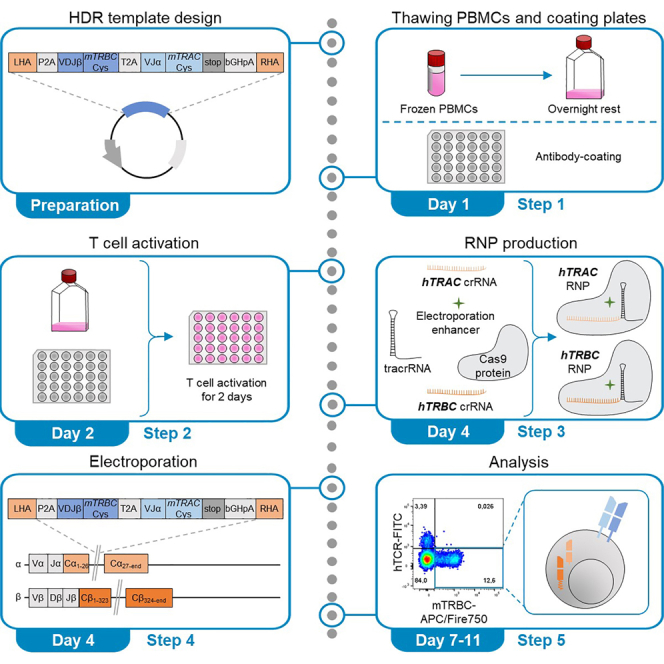
Summary
Adoptive T cell therapy using T-cell receptor (TCR)-engineered T cells allows to redirect T cell specificity and to target any antigen of interest. Here, we apply advanced genetic engineering using clustered regularly interspaced short palindromic repeats (CRISPR)-associated protein 9 (Cas9) for simultaneous editing of TCR α- and β-chains in primary human T cells. Together with non-virally delivered template DNA, this CRISPR-Cas9-system allows for elimination of the endogenous TCR and orthotopic placement of TCR α- and β-chains.
For complete details on the use and execution of this protocol, please refer to Schober et al. (2019) and Müller et al. (2021).
Subject areas: Health Sciences, Immunology, CRISPR, Biotechnology and bioengineering
Graphical Abstract
Highlights
-
•
OTR protocol generates advanced TCR-transgenic T cells within a week
-
•
Targeted CRISPR-Cas9-mediated large gene knock-in is completely non-viral
-
•
OTR T cells are closer to physiological T cells than conventionally engineered T cells
-
•
Protocol can also be adapted for other applications, e.g., gene tagging
Adoptive T cell therapy using T-cell receptor (TCR)-engineered T cells allows to redirect T cell specificity and to target any antigen of interest. Here, we apply advanced genetic engineering using clustered regularly interspaced short palindromic repeats (CRISPR)-associated protein 9 (Cas9) for simultaneous editing of TCR α- and β-chains in primary human T cells. Together with non-virally delivered template DNA, this CRISPR-Cas9-system allows for elimination of the endogenous TCR and orthotopic placement of TCR α- and β-chains.
Before you begin
This protocol describes the application of the CRISPR-Cas9-system for orthotopic TCR replacement (OTR) in primary human T cells using ribonucleoproteins (RNPs) to mediate homology-directed repair (HDR). Linearized double-stranded DNA (dsDNA) serves as HDR template for the knock-in (KI) and is designed to encode the full αβ TCR. T cells are activated by surface-bound anti-CD3 and anti-CD28 antibodies for two days. RNPs create double-strand breaks in the endogenous human TCR α-chain constant (hTRAC) and human TCR β-chain constant (hTRBC) loci. Both RNPs comprise the respective guide RNA (gRNA) and the Cas9 protein. To produce these hTRAC and hTRBC RNPs, trans-activating crRNA (tracrRNA) is incubated with hTRAC or hTRBC CRISPR RNA (crRNA), respectively, to form gRNAs before electroporation enhancer and Cas9 protein are added. Along with the DNA template, both RNPs are electroporated into the activated T cells. The resulting OTR leads to changes at the TCR DNA, RNA and protein level (Figure 1).
Figure 1.

Schematic of orthotopic TCR replacement through CRISPR-Cas9-mediated gene editing
On the TCR gene level, hTRAC and hTRBC RNPs create double-strand breaks (illustrated by gray double line) leading to homology-directed repair (HDR)-mediated integration of the DNA template into to the TRAC exon and non-homologous end-joining (NHEJ) in the TRBC locus. On the transcript level, the knock-in (KI) of the HDR template results in the knock-out (KO) of endogenous TRAC transcription. For TRBC, NHEJ leads to a frameshift which introduces a stop codon and results in the KO of endogenous TCR β-chain. On the protein level, only truncated endogenous but full transgenic TCR α-chain and β-chains are expressed. On the T cell surface, only the transgenic αβ TCR is expressed.
For this ‘single KI’ strategy, the HDR template is inserted into the endogenous TRAC locus, with concomitant knock-out (KO) of endogenous TRBC. A ‘dual KI’ strategy with individual insertion of transgenic α-chain into endogenous TRAC and transgenic β-chain into endogenous TRBC is also possible, albeit with lower overall αβ TCR re-expression rate. In both cases, OTR generates engineered T cells expressing only the transgenic TCR on their surface.
DNA template design
Timing: ∼1 week
Directed by a specific hTRAC gRNA, Cas9 nuclease creates a double-strand break in the first TRAC exon. For recombinant Streptococcus pyogenes Cas9 nuclease (used in this protocol) the necessary protospacer adjacent motif (PAM) sequence is 5′-NGG-3′ (here the PAM sequence is 3′-GGC-5′ on the antisense strand, and the hTRAC gRNA is complementary to the sense strand). The nuclease cuts 3 bp downstream of the PAM sequence. After the double-strand break, HDR enables insertion of the linearized dsDNA template into the genomic DNA.
The structure of the HDR template is illustrated in Figure 2. The construct is flanked by left and right homology arms (LHA and RHA) required for HDR and comprises the full α- and β-chains of the TCR to be introduced, self-cleaving peptides (P2A and T2A) to ensure separation and polycistronic expression of both TCR-chains, and a poly-A tail (bGHpA). The left homology arm (LHA) consists of 396 bp including a 5′-region with a primer site and is located upstream of the hTRAC gRNA cut site. The LHA ends 2 bp upstream of the hTRAC gRNA cut site (at base 26 of TRAC) to be in-frame with the TCR J-segment after splicing. The LHA is followed by the self-cleaving peptide P2A which allows the separation of the following β-chain segments from the LHA. The β-chain consists of the human variable parts and the murine constant region. The variable part comprises the VDJ-region starting from the respective leader sequence of the V-gene until the end of the respective J-gene of the chosen TCR β-chain. Transgenic VDJ β-transcription is therefore initiated with endogenous VJ-transcription without the use of extrinsic promoters. Here, the hTRBC is exchanged for its murine counterpart which includes an additional cysteine bridge (mTRBC-Cys) (Cohen et al., 2007). This modification allows for improved surface expression of the TCR and facilitates identification of re-expressed transgenic TCR and clear distinction from endogenous human TCR (Cohen et al., 2007). In short-term in vitro assays, transgenic TCRs with a fully human TCR constant region did not show any functional differences to TCRs with a murine constant region after OTR (Schober et al., 2019). The β-chain is followed by another self-cleaving peptide (T2A) to ensure polycistronic expression of β- and α-chain. The subsequent α-chain is designed according to the same principles as the β-chain: The variable part (VJα) is followed by the murine TRAC (mTRAC-Cys). The stop codon (TGA) concludes the transgenic TCR (α-chain) sequence. The following bovine growth hormone polyA signal (bGHpA) is not strictly necessary but included in this construct to stabilize the mRNA transcript. When human TCR constant regions are included in the construct design, the transgenic hTRAC region only encompasses the part that lies upstream of the editing site (in the first TRAC exon) and then seamlessly continues into the RHA. This approach additionally requires the introduction of a silent mutation in the PAM sequence to avoid editing once the transgenic TCR is integrated into the endogenous TRAC locus. The RHA concludes the HDR template and consists of 330 bp including a 3′ region with a primer site and is located downstream of the cut site.
-
1.
The HDR template is assembled in silico by combining the segments as described above. All sequences of these building blocks (updated from Schober et al., 2019) can be found in the supplemental information (Table S1).
-
2.
The DNA construct can then be ordered as a sequence-verified plasmid gene via commercial providers.
-
3.
After delivery, lyophilized plasmids are reconstituted to 60 ng/μL for HDR template amplification by PCR (and potentially vector amplification by Midiprep to generate back-up stocks).
Alternatives: The addition of Cas9 Target Sequences (CTS) – 20 bp gRNA recognition sequences – at the 5′-end of the DNA template has been described to improve TCR re-expression rates as these facilitate the delivery of the HDR template to the correct position in the genome (Nguyen et al., 2020; Shy et al., 2021). A single CTS at the 5′-end of the DNA template was shown to be sufficient for this effect. Constructs with 4–8 mismatched base pairs at the 5′-end of this CTS (allowing the Cas9 nuclease to bind, but not to cut) showed the greatest improvement in re-expression rates. Additionally, increased re-expression rates and cell viability have been demonstrated to result from the use of hybrid DNA templates. These hybrid DNA templates comprise single-stranded DNA (ssDNA) template including a CTS at the 5′-end with an annealing oligonucleotide covering the gRNA recognition site, the PAM sequence, and approximately 20 bp of the downstream homology arm of the HDR template) (Shy et al., 2021).
Figure 2.

DNA template design for TCR α- and β-chain integration into human TRAC locus via homology-directed repair
LHA, left homology arm; RHA, right homology arm; VDJβ, variable parts of β-chain; mTRBC-Cys, murine TCRβ constant region with additional cysteine bridge; VJα, variable parts of α-chain; mTRAC-Cys, murine TCRα constant region with additional cysteine bridge; bGHpA, bovine growth hormone poly-A tail; T2A and P2A, self-cleaving peptide inserts, TRAC exon 1 is illustrated in orange and includes the PAM sequence (yellow). For sequences of these segments see Table S1.
Reconstitute reagents
In preparation for the experiment, some reagents require reconstitution at their respective concentrations. Prior to electroporation, tracrRNA, crRNAs and electroporation enhancer are reconstituted. Additionally, interleukins (IL-2, IL-7 and IL-15) used for T-cell activation are also reconstituted and aliquoted.
-
4.
Reconstitute tracrRNA, hTRAC crRNA and hTRBC crRNA to 80 μM in Nuclease-Free Duplex Buffer (IDT). Aliquot and store at −20°C.
-
5.
Reconstitute electroporation enhancer to 400 μM in Nuclease-Free Duplex Buffer (IDT). Aliquot and store at −20°C.
-
6.
Reconstitute IL-2 to 10 μg/mL (100,000 U/mL) and IL-7 and IL-15 to 25 μg/mL by following manufacturer's instructions. Aliquot and store at −20°C.
Note: For the IL-2 used here, the International Unit value was 2.18 IU/ng. 10 μg/μL is therefore equivalent to 21,800 IU/mL.
Double-stranded DNA production
-
7.Perform PCR for amplification of linearized double-stranded HDR template.
-
a.Prepare 8 × 100 μL PCR master mix (see materials and equipment) for each HDR construct (without plasmid DNA).
-
b.Distribute 99 μL master mix into PCR-tubes.
-
c.Add 1 μL of plasmid DNA (pre-diluted to 60 ng/μL) per tube.Optional: Include negative control by adding nuclease-free PCR grade water instead of template DNA to the sample.
-
d.Mix by vortexing.
-
e.Spin down tubes to ensure the entire reaction mixture is at the bottom of the tube.
-
f.Ensure all the lids are properly sealed and place PCR tubes into thermocycler.
-
g.Run PCR with the following conditions:
PCR cycling conditions
Steps Temperature Time Cycles Initial Denaturation 95 °C 3 min 1 Denaturation 95°C 30 s 34 cycles Annealing 62 °C 30 s Extension 72 °C 3 min Final extension 72 °C 3 min 1 Hold 4 °C Unlimited Optional: Depending on the thermocycler, PCR cycling conditions (e.g., temperatures) may have to be adjusted. -
h.Keep samples at 4°C until further processing (running agarose gel or purification).
-
a.
-
8.Run agarose gel to ensure successful amplification of the HDR template (see problem 1).
-
a.Prepare 1% agarose gel including peqGREEN (1:20,000) as DNA/RNA dye.Optional: Other DNA/RNA dyes can be also used.
-
b.Prepare marker and samples by mixing 6 × TriTrack DNA loading Dye with GeneRuler 1 kb DNA Ladder or PCR product, respectively.
-
c.Mix samples by pipetting and load them onto the agarose gel.
-
d.Run gel at 120 V, 400 mA, 100 W for 20–45 min.
 Pause point: If the gel cannot be imaged immediately after electrophoresis is completed, reduce voltage (to 10 V) to avoid diffusion of the bands.
Pause point: If the gel cannot be imaged immediately after electrophoresis is completed, reduce voltage (to 10 V) to avoid diffusion of the bands.
-
a.
-
9.
Image gel under UV light to check if the PCR was successful (Figure 3).
-
10.
Replicates showing only one clear band of the expected size can be pooled for further processing (purification).
-
11.
After amplification, the pooled remaining double-stranded HDR template of the PCR reaction is isolated using the MinElute PCR Purification Kit.
-
12.
Follow manufacturer's protocol: https://www.qiagen.com/us/products/discovery-and-translational-research/dna-rna-purification/dna-purification/dna-clean-up/minelute-pcr-purification-kit/ (as of Sep. 22, 2021).
-
13.
Elute in 10 μL per column.
Optional: It is possible to load two PCR samples (of the same construct, approx. 180 μL) onto the same column: Mix buffer PB and PCR reaction mix according to manufacturer's instruction, load mixture onto column and spin down. After discarding flow-through, load the remaining mixture before continuing with the next step of the protocol. Since still only 10 μL are used for elution, this increases the concentration of purified DNA per eluate.
-
14.
Measure concentration of purified PCR product using a NanoDrop 1000 spectrophotometer blanked with elution buffer.
Optional: Other machines for determining nucleic acid concentration can also be used.
Note: Concentration should range from 0.5–1.0 μg/μL. Lower concentrations will dilute the DNA/RNP-mix as higher volumes of DNA template are required per electroporation. This might reduce re-expression rates. If possible, it is recommended to adjust DNA concentration to 1.0 μg/μL.
-
15.
Store purified HDR template DNA at −20°C.
Figure 3.

Exemplary 1% agarose gel showing successful amplification of HDR template DNA
M: GeneRuler 1 kb DNA Ladder; Full αβ TCR construct (2.8 kb); TCR α-chain (1.8 kb); TCR β-chain (1.9 kb) and W: water control (negative control).
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Armenian hamster anti-mouse TCR-β chain, APC/Fire750-conjugated | BioLegend | H57-597; Cat#109246; RRID: AB_2629697 |
| Mouse anti-human CD45, ECD-conjugated | Beckman Coulter | J.33; Cat#A07784; |
| Mouse monoclonal anti-human CD8a, APC-eFluor® 780-conjugated | Thermo Fisher Scientific | OKT8; Cat#47-0086-42; RRID: AB_2573945 |
| Mouse monoclonal anti-human CD8a, PE-conjugated | Thermo Fisher Scientific | OKT8; Cat#12-0086-42; RRID: AB_10732344 |
| Mouse monoclonal anti-human TCR α/β, FITC-conjugated | BioLegend | IP26; Cat#306706; RRID: AB_314644 |
| Purified monoclonal mouse anti-human CD28 | BioLegend | CD28.2; Cat#302902; RRID: AB_314304 |
| Purified monoclonal mouse anti-human CD3 | BioLegend | OKT3; Cat#317302; RRID: AB_571927 |
| Biological samples | ||
| Human blood/PMBCs | Healthy volunteers | Transfusionsmedizin, UK Erlangen |
| Chemicals, peptides, and recombinant proteins | ||
| Albumin from bovine serum (BSA) | VWR Life Science | Cat#A3294 CAS: 9048-46-8 |
| Alt-R® Cas9 Electroporation Enhancer | Integrated DNA Technologies | Cat#1075916 |
| Alt-R® S.p. Cas9 Nuclease V3 | Integrated DNA Technologies | Cat#1081059 |
| ß-Mercaptoethanol (50 mM) | Thermo Fisher Scientific | Cat#31350010 |
| DNA/RNA dye, peqGREEN | VWR Peqlab | Cat#732-3196 |
| dNTP Mix (10 mM each) | Thermo Fisher Scientific | Cat#R01922 |
| Dulbecco’s Phosphate Buffered Saline (PBS) | Sigma-Aldrich | Cat#D8537 |
| GeneRuler 1 kb DNA Ladder | Thermo Fisher Scientific | Cat#SM0311 |
| Gentamicin (50 mg/mL) | Thermo Fisher Scientific | Cat#15750060 |
| Heat-Inactivated Fetal Bovine Serum (South America) | Anprotec | Cat#AC-SM-0027 |
| HEPES | Carl Roth | Cat#HN77.3; CAS: 7365-45-9 |
| Herculase II Fusion DNA Polymer | Agilent Technologies | Cat#600677 |
| L-Glutamine | Sigma-Aldrich | Cat#G8540; CAS: 56-85-9 |
| Penicillin-Streptomycin (10,000 U/mL) | Thermo Fisher Scientific | Cat#15140122; |
| Poly-L-glutamic acid sodium salt, mol wt 15,000–50,000 (PGA) | Sigma-Aldrich | Cat#P4761-100mg; CAS: 26247-79-0 |
| Propidium Iodide | Thermo Fisher Scientific | Cat#P1304MP; CAS: 25535-16-4 |
| Recombinant Human IL-15 | PeproTech | Cat#200-15; Accession Number: P40933 |
| Recombinant Human IL-2 | PeproTech | Cat#200-02; Accession Number: P60568 |
| Recombinant Human IL-7 | PeproTech | Cat#200-07; Accession Number: P13232 |
| RPMI 1640 Medium | Thermo Fisher Scientific | Cat#21875034 |
| RT-PCR Grade Water | Thermo Fisher Scientific | Cat#AM9935 |
| Critical commercial assays | ||
| MinElute PCR Purification Kit | Qiagen | Cat#28004 |
| P3 Primary Cell 4D-NucleofectorTM X Kit S | Lonza | Cat#V4XP-3032 |
| P3 Primary Cell 96-well NucleofectorTM Kit (960 RCT) | Lonza | Cat#V4SP-3960 |
| Oligonucleotides | ||
| Alt-R® CRISPR-Cas9 tracrRNA | Integrated DNA Technologies | Cat#1072534 |
|
hTRAC crRNA sequence (on antisense strand): 5′-AGAGTCT CTCAGCTGGTACA-3′ |
Ren et al. (2017) | n/a |
|
hTRAC HDR genomic forward primer: 5′-CTGCCTTTACTCTGCCAGAG-3′ |
Müller et al. (2021) | n/a |
|
hTRAC HDR genomic reverse primer: 5′- CATCATTGACCAGAGCTCTG-3′ |
Müller et al. (2021) | n/a |
|
hTRBC crRNA sequence: 5′-GGAGAATGACGAGTGGACCC-3′ |
Schober et al. (2019) | n/a |
| Recombinant DNA | ||
| HDR DNA template sequence | Schober et al. (2019) | n/a |
| Software and algorithms | ||
| FlowJo, Version 10.7.2 | Becton Dickinson & Company (BD) |
https://www.flowjo.com/solutions/flowjo; RRID: SCR_008520 |
| GraphPad Prism 8.3.0 (538) | GraphPad Software | https://www.graphpad.com/; RRID: SCR_002798 |
| Other | ||
| 4D-NucleofectorTM Core Unit | Lonza | Cat#AAF-1002B |
| 4D-NucleofectorTM X Unit | Lonza | Cat#AAF-1002X |
| BD LSRFortessa™ Cell Analyzer | BD Biosciences | n/a |
Materials and equipment
PCR master mix
| Reagent | Final concentration | Amount (for 100 μL reaction) |
|---|---|---|
| PCR grade water | n/a | 65.0 μL |
| 5× Herculase II Reaction Buffer | 1× | 20 μL |
| hTRAC HDR genomic forward primer (10 mM) | 0.4 mM | 4.0 μL |
| hTRAC HDR genomic reverse primer (10 mM) | 0.4 mM | 4.0 μL |
| dNTPs (10 mM) | 0.5 mM | 5.0 μL |
| Herculase II Fusion DNA Polymerase | 1.0% | 1.0 μL |
| DNA (prediluted to 60 ng/μL) | 600 ng/mL | 1.0 μL |
| Total | n/a | 100 μL |
PCR master mix is freshly prepared on ice for direct use.
Alternatives: In addition to Herculase II Fusion DNA Polymerase, other polymerases are commercially available (e.g. Ampli Taq 360). Adjustments to the PCR conditions might be necessary.
SC+ supplement
| Reagent | Final concentration | Amount |
|---|---|---|
| ß-Mercaptoethanol (50 mM) | 1 mM | 10 mL |
| Gentamicin (50 mg/mL) | 1 mg/mL | 10 mL |
| HEPES | 23.83 g/L | 11.9 g |
| L-Glutamine | 4.0 g/L | 2.0 g |
| Penicillin-Streptomycin (10,000 U/mL) | 2,000 U/mL | 100 mL |
| RPMI 1640 Medium | n/a | 380 mL |
| Total | n/a | 500 mL |
Store at −20 °C for several months.
SC- supplement
| Reagent | Final concentration | Amount |
|---|---|---|
| ß-Mercaptoethanol (50 mM) | 1 mM | 2.4 mL |
| HEPES | 23.83 g/L | 2.86 mg |
| L-Glutamine | 4.0 g/L | 0.48 g |
| RPMI 1640 Medium | n/a | 116.8 mL |
| Total | n/a | 120 mL |
Store at −20 °C for several months.
Antibiotic mix (as supplement for SC- medium)
| Reagent | Final concentration | Amount |
|---|---|---|
| Gentamicin (50 mg/mL) | 1 mg/mL | 0.4 mL |
| Penicillin-Streptomycin (10,000 U/mL) | 2,000 U/mL | 4.0 mL |
| RPMI 1640 Medium | n/a | 15.6 mL |
| Total | n/a | 20 mL |
Store at −20 °C for several months.
Complete RPMI medium
| Reagent | Final concentration | Amount |
|---|---|---|
| RPMI 1640 Medium | n/a | 500 mL |
| Heat-Inactivated Fetal Bovine Serum (FBS) | 10% | 50 mL |
| SC+ supplement | 5% | 25 mL |
| Total | n/a | 575 mL |
Store at 4 °C for several weeks.
FACS buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| PBS buffer, pH 7.4 | n/a | 500 mL |
| BSA | 0.5% | 2.5 g |
| Total | n/a | 500 mL |
Store at 4 °C for 2–4 weeks.
Step-by-step method details
Thawing and resting PBMCs overnight
When working with frozen PBMCs, these steps prepare the cells for subsequent stimulation. The overnight rest is not necessary when working with freshly isolated PBMCs.
-
1.
Warm complete RPMI medium in water bath set at 37°C.
-
2.Thaw PBMCs as quickly as possible to reduce the time that the cells spend in freezing medium (containing DMSO).
-
a.Hold cryovials in water bath until cells start thawing. Then pipette thawing cell suspension up and down to speed up the thawing process.
-
b.Transfer thawed cells into 50 mL tube with pre-warmed complete RPMI medium.
-
a.
-
3.
Wash cells twice by centrifuging (480 × g, 7 min, 20°C–22°C), discarding supernatant and resuspending in pre-warmed complete RPMI medium.
-
4.
Count cells and seed at 2 × 106 cells/mL in complete RPMI medium supplemented with 50 U/mL IL-2.
-
5.
Incubate cells 18–24 h at 37°C, 5% CO2.
Optional: Instead of working with frozen PBMCs, PBMCs also can be freshly isolated (by density gradient centrifugation) from a healthy donor and directly activated (without overnight rest).
Coating of plates with anti-CD3/CD28 antibodies
This step prepares the tissue-culture plates and/or flasks for activation of PBMCs by coating them with anti-CD3 and anti-CD28 antibodies. The required volume of coating solution depends on the cell numbers to be activated and therefore on the number of wells (more precisely, the area) to be coated. Here, we describe the process for the 24-well plate format. To adapt activating antibody-coating and T cell activation to different formats (e.g., 48-well plates of T25-flasks), calculate the area in relation to the surface of a 24-well plate well and adjust required volume of coating solution and cell suspension accordingly.
Alternatives: Dynabeads® Human T-Activator CD3/CD28 which are covalently coupled to anti-CD3 and anti-CD28 antibodies can be used for polyclonal T cell activation (Roth et al., 2018).
-
6.
Dilute anti-CD3 and anti-CD28 antibodies to a final concentration of 1 μg/mL in sterile PBS buffer.
-
7.
Coat 24-well plate with 500 μL/well.
-
8.
Seal plates with Parafilm® to avoid evaporation and ensure sterile conditions.
-
9.
Keep plates at 4°C 18–24 h.
Optional: Antibody coating can also be performed for 2 h at 37°C.
T cell activation
For activation of T cells, PBMCs are incubated on tissue-culture plates with surface-bound anti-CD3 and anti-CD28-antibodies. Interleukins (IL-2, IL-7 and IL-15) are added to the culture medium.
Per CRISPR sample, 1 × 106 cells are electroporated. In addition to the OTR samples, control samples (TCR KO and mock) are included in the experiment. TCR KO (KO of endogenous α-chain and β-chain) samples are required to evaluate the efficiency endogenous TCR KO and while mock samples (unedited cells) serve as negative control.
-
10.
Count PBMCs.
-
11.
Resuspend PBMCs at 1 × 106 cells/mL in complete RPMI medium supplemented with 300 U/mL IL-2, 5 ng/mL IL-7 and 5 ng/mL IL-15.
CRITICAL: During the activation, usually up to half of the PBMCs do not survive and it is therefore important to account for this cell death at this activation step. When working with frozen PBMCs, activate at least 2.5 × 106 cells per CRISPR sample (2.5 × the amount of cells required for electroporation).
-
12.
Wash 24-well plates twice with PBS without letting the wells dry out.
-
13.
Add 1.5 mL of the PBMC suspension from step 11 per well to the coated 24-well plates.
-
14.
Incubate at 37°C, 5% CO2 for two days (48 h).
Optional: To improve T cell viability after electroporation and TCR re-expression rates, the addition of DNA-sensing inhibitors has been reported to be beneficial (Kath et al., 2021). DNA-sensing inhibitors like RU.521 (small-molecule inhibitor of cyclic GMP-AMP synthase (cGAS)) and the synthetic oligonucleotide ODN A151 (TLR9 antagonist and inhibitor of other cytosolic DNA-sensors) can be added 6 h before the end of incubation to increase survival and TCR re-expression rates (Kath et al., 2021).
Preparation of electroporation
T cell activation is stopped and cells are prepared for electroporation by transferring them into a 96-well plate (‘cells plate’).
Additionally, antibiotic-free SC- medium is prepared. The SC- medium comprises the same ingredients and supplements as complete RPMI medium except for antibiotics (penicillin-streptomycin, gentamicin). As each CRISPR sample is seeded at 1 × 106 cells/mL after electroporation, 24-well plates with 1 mL SC- medium per well are prepared and pre-warmed in the incubator.
Note: The viability of KI cells is higher when the cells are cultivated in antibiotic-free SC- medium for 12–16 h after electroporation. After 12–16 h, cell culture is continued in complete (antibiotics-containing) RPMI medium (SC+).
-
15.
Check for cell cluster formation under an inverse microscope.
-
16.
Recover the activated cells from the wells and pool them into a 50 mL tube.
-
17.
Wash cells by centrifuging (480 × g, 7 min, 20°C–22°C), discarding the supernatant and resuspending in complete RPMI medium.
-
18.
Count PBMCs.
Note: Cell numbers are usually lower than before activation as only T cells among PBMCs are being activated and some T cells may also undergo activation-induced cell death. Apoptotic bodies were removed during the washing steps and cell number calculations for the subsequent steps were based on the cell count of live cells. No further steps (e.g. FACS sorting) were performed to separate dead from live cells.
-
19.
Centrifuge (480 × g, 7 min, 20°C–22°C), discard supernatant and resuspend at a density of 5 × 106 cells/mL in complete RPMI medium.
-
20.
Seed 1 × 106 cells (200 μL of cell suspension) per CRISPR sample into a 96-well V-bottom ‘cells plate' well.
Note: At this step, the plate layout is important. Make sure to choose a plate layout that is appropriate for the use of a multi-channel pipette, which allows a fast transfer to NucleocuvettesTM (e.g., when working with 16-well NucleocuvetteTM Strips, prepare the cells in rows of 8).
-
21.
Keep the 96-well plate at 37°C, 5% CO2 until further use.
-
22.
Cells of the mock samples can be resuspended to a density of 1 × 106 cells/mL in complete RMPI medium supplemented with 180 U/mL IL-2 and directly transferred into the 24-well plate.
Note: Mock samples are not being electroporated in this protocol as previous data have shown that electroporation itself (without the addition of DNA or RNPs) has no considerable effect on the endogenous TCR expression.
-
23.
Per CRISPR sample, prepare 1 mL of SC- medium for cell cultivation after electroporation (at 1 × 106 cells/mL) by supplementing RPMI 1640 medium with 10% heat-inactivated FBS, 5% SC- (see materials and equipment) and 180 U/mL IL-2.
-
24.
Distribute 1 mL SC- medium per well into 24-well plates and keep the plate at 37°C, 5% CO2 until further use to pre-warm the medium.
RNP production
CRISPR-Cas9-mediated OTR requires hTRAC and hTRBC RNPs for simultaneous editing of TCR α- and β-chains. Both RNPs are complexes of Cas9 protein and gRNA. Respective gRNAs consist of target-specific hTRAC or hTRBC crRNA sequences, directing Cas9 nuclease to the respective genetic loci, as well as of tracrRNA, serving as scaffold for the Cas9 protein.
For final electroporation, 3 μL of each RNP (final concentration 20 μM) are needed per sample. Here, we first produce 40 μM gRNAs by mixing equimolar amounts of tracrRNA with hTRAC or hTRBC crRNA. After the addition of electroporation enhancer (to enhance editing efficiency), the RNP production is concluded by mixing and then incubating equal volumes of gRNA and Cas9 protein (see Figure 4).
Optional: The addition of poly-L-glutamic acid (PGA) to RNPs has been described to improve editing efficiency and cell survival rates (Kath et al., 2021; Nguyen et al., 2020).
Note: Due to working with small volumes, pipetting errors and inaccuracies can have a considerable effect. We therefore advise to prepare additional 10% of RNPs to account for pipetting errors.
-
25.
Mix equal volumes of tracrRNA (80 μM stock) with hTRAC or hTRBC crRNA (80 μM stock), respectively, to prepare 40 μM hTRAC and 40 μM hTRBC gRNA. Mix well by pipetting.
-
26.
If necessary, shortly spin down to ensure the complete reaction mixture is at the bottom of the tube.
-
27.
Heat at 95°C for 5 min in PCR cycler and then allow RNPs to cool to 20°C–22°C on bench top.
Alternatives: Run gRNA production protocol by 95°C for 5 min, followed by hold at 21°C. Using a 1.3 °C/s ramp (maximal ramp for this specific PCR cycler) we were able to produce gRNA for the RNPs which yielded very high TCR KO efficiencies.
-
28.
Store gRNA on ice if you do not continue directly.
-
29.Dilute Cas9 nuclease to 6 μM with commercial PBS buffer.CRITICAL: Minimize the time that Cas9 nuclease is at room temperature (20°C–22°C) and return to -20°C as quickly as possible. Take from stock only what you need for the experiment. Do not dilute the stock.
-
a.Calculate required volumes of Cas9 protein (62 μM stock) and commercial PBS buffer.
-
b.Prepare tube with required volume of PBS buffer.
-
c.Set up pipette for the required volume of Cas9 nuclease.
-
d.Quickly take Cas9 stock from −20°C. To avoid foaming, do not mix the Cas9 stock before taking out the required volume to prepare the dilution.
-
e.Slowly add Cas9 with prepared pipette and swirl slowly while pipetting.Note: When adding the nuclease, you will see streaks of the Cas9 stock in the PBS buffer. After having added the required volume of Cas9, make sure to thoroughly mix by pipetting until all streaks have disappeared.
-
a.
-
30.
Keep diluted Cas9 on ice if you do not continue directly, otherwise bring it to 20°C–22°C for RNP assembly.
-
31.
Add electroporation enhancer (400 μM stock) to gRNAs for a final concentration of 20 μM.
-
32.
To mix equal volumes of diluted Cas9 nuclease (6 μM) and gRNA (40 μM), very slowly add Cas9 to the gRNA while moving pipette tip in cycles.
-
33.
Mix by pipetting.
-
34.
Check if RNP solution is clear and does not contain any precipitate.
-
35.
Incubate RNPs for 15 min at 20°C–22°C.
-
36.
Keep RNPs on ice until further use if they are processed the same day.
Optional: Store RNPs at −80°C if they are not processed the same day.
Figure 4.

Workflow of hTRAC and hTRBC ribonucleoproteins (RNPs) production
tracrRNA (black) is mixed with respective crRNA (hTRAC crRNA, light orange or hTRBC crRNA, dark orange) to produce gRNA. Electroporation enhancer (green asterisk) and then Cas9 protein (grey) are added to conclude RNP production.
Prepare electroporation buffer
Prepare 20 μL P3 electroporation buffer (Lonza) per condition. As this buffer is toxic for cells, the time cells spend in this buffer should be kept to a minimum.
-
37.
Per sample, 20 μL P3 buffer are prepared by mixing 16.4 μL P3 with 3.6 μL supplement.
-
38.
Keep at 20°C–22°C or on ice, if needed later.
Optional: To reduce costs, Lonza NucleocuvettesTM can be washed with ethanol and reused. When working with reused NucleocuvettesTM, the self-made buffers such as ‘1M’ (Chicaybam et al., 2013) can be used instead of commercial P3 electroporation buffer. However, reuse of NucleocuvettesTM increases the risk of error codes during electroporation.
Electroporation
For electroporation, activated T cells are resuspended in electroporation buffer and then mixed with HDR template and RNPs (Figure 5). After transfer into the 16-well NucleocuvetteTM Strip, cells are electroporated in the Lonza 4D-NucleofectorTM. By electroporation, RNPs and template DNA are delivered into the nucleus of T cells where genetic editing occurs.
Figure 5.

Genetic engineering of T cells via CRISPR-Cas9-mediated TCR KI
Activated T cells with endogenous TCRs (orange) are required for successful editing. In preparation for electroporation, the linearized double-stranded DNA template is mixed with hTRAC and hTRBC RNPs. T cells resuspended in electroporation buffer are mixed with the DNA-RNPs mixture and electroporated. Respective crRNAs direct the Cas9 nuclease either to the TRAC or TRBC locus and allow for CRISPR-Cas9-mediated DNA double-strand breaks (right). At the TRAC locus, homology-directed repair (HDR) results in the targeted KI of the HDR template DNA encoding the full transgenic αβ TCR which leads to a simultaneous KO of the endogenous TRAC (black triangle). At the TRBC locus, the double-strand break is resolved by non-homologous end-joining leading to the KO of the endogenous TRBC (black cross). On the surface of edited T cells only the transgenic TCR (blue) is expressed.
When working with 16-well NucleocuvetteTM Strips, cells are prepared in the ‘cells plate’ (Figure 6) in rows of 8 (see step 20). To minimize the time that cells spend in the electroporation buffer, only (up to) 8 samples are electroporated at a time. In preparation for electroporation, respective cells are transferred to the ‘centrifugation plate’ (Figure 6) and spun down. After centrifugation, cells are resuspended in P3 electroporation buffer, mixed with DNA/RNP-mix in the ‘pipetting plate’ (Figure 6) and transferred to NucleocuvetteTM Strip.
-
39.Switch on and set up NucleofectorTM.
-
a.Select wells to be electroporated.
-
b.Select electroporation program using the pulse sequence EH100.
-
a.
Alternatives: Different pulse sequences can be used (e.g., the use of EH115 has been successfully applied (Roth et al., 2018). After testing various pulse settings, we found that EH100 yields the best combination of editing efficiency and cell viability in our setting.
-
40.
Prepare electroporation buffer for samples of the first electroporation (up to 8 samples). Distribute 20 μL of P3 electroporation buffer per well into a V-bottom 96-well plate (‘pipetting plate’) following the layout of the ‘cells plate’ (Figure 6).
-
41.Prepare targeting construct mix for the first OTR samples in the ‘pipetting plate’:
-
a.First, add 0.5 μg template DNA to the wells of the ‘pipetting plate’. Volume of DNA depends on the concentration after purification. Values should range between 0.5 and 1.5 μL.
-
b.Add 3.0 μL hTRAC RNP + 3.0 μL hTRBC RNP to the template DNA (see problem 2).
-
a.
-
42.
For the TCR KO sample, mix only 3.0 μL hTRAC RNP + 3.0 μL hTRBC RNP in the ‘pipetting plate’ to prepare targeting construct mix. Afterwards, treat TCR KO sample exactly as the OTR samples.
Alternatives: When working with ssDNA templates, higher amounts of template can be used since ssDNA demonstrates less toxicity (Roth et al., 2018). However, the use of ssDNA templates increases the complexity of the protocol as templates need to be singularized.
-
43.
Incubate at 20°C–22°C for at least 30 s.
-
44.Transfer cells to be electroporated to the 96-well V-bottom ‘centrifugation plate’ and spin down.
-
a.Centrifuge for 3 min at 480 × g, 20°C–22°C.
-
b.Swiftly discard remaining supernatant.
-
a.
-
45.
Set multichannel to 20 μL and take up 20 μL P3 buffer ‘pipetting plate’ (see problem 3).
-
46.
Resuspend cell pellet in 20 μL P3 buffer. Avoid air bubbles and be cautious not to create any foam.
-
47.
Increase pipetting volume to 25 μL. This 5 μL-increase in volume accounts for the left-over supernatant in the well.
-
48.
Take 25 μL of resuspended cells of the ‘centrifugation plate’ and transfer to DNA/RNP-wells of the ‘pipetting plate’.
-
49.
Mix by carefully pipetting up and down. Avoid air bubbles or foam.
-
50.
Increase pipetting volume to 32 μL. This 7 μL-increase in volume accounts for the 2 × 3 μL of RNPs and the approx. 1 μL of DNA.
-
51.
Quickly transfer cells/DNA/RNP-mix to fresh 16-well NucleocuvetteTM Strip.
-
52.
Place lid on top and tap NucleocuvetteTM Strip on the bench to remove any air bubbles (see problem 3).
-
53.
Place NucleocuvetteTM Strip into NucleofectorTM.
-
54.
Start electroporation program: EH100 (see problem 4)
-
55.Transfer electroporated cells to SC- medium.
-
a.Add 80 μL of pre-warmed SC- medium (from the 24-well plate (step 24) to the NucleocuvetteTM well and mix (to dilute P3 buffer).
-
b.Transfer cells to 24-well plate with 1 mL of pre-warmed SC- medium per well to seed the cells at a density of 1 × 106 cells/mL.
-
c.Take some SC- medium from the same well to flush NucleocuvetteTM well to collect any left-over cells.
-
a.
-
56.
Incubate cells at 37°C, 5% CO2 for 12–16 h.
Figure 6.

Workflow of preparing samples for electroporation
Step 1: The targeting construct mix is prepared in the ‘pipetting plate’ (blue plate) by mixing DNA template and RNPs (light blue wells). Additionally, 20 μL P3 buffer is added to the plate (light green wells). Cells to be electroporated are transferred from the ‘cells plate’ (orange plate) to the ‘centrifugation plate’ (grey plate). Step 2: After centrifugation, the supernatant in the ‘centrifugation plate’ is discarded. Step 3: Cell pellets are resuspended in 20 μL of P3 electroporation buffer. Step 4: 25 μL of resuspended cells (dark green) are transferred to the DNA/RNP-mix wells in the ‘pipetting plate’ and mixed. Step 5: 32 μL of cells/DNA/RNPs-mix (dark blue) is transferred to the 16-well NucleocuvetteTM Strip. Step 6: NucleocuvetteTM Strip is placed in the NucleofectorTM and cells are electroporated. Afterwards, cells are transferred to the 24-well plate with pre-warmed SC- medium (not shown). Step 7: Process is repeated for remaining samples.
Addition of antibiotics
Viability of KI cells is higher when they are cultivated in antibiotic-free SC- medium for 12–16 h after electroporation. Afterwards, SC- medium can be replaced by complete RPMI medium (including antibiotics) or the SC- medium can simply be supplemented with an antibiotic mix to produce complete RPMI medium.
-
57.Here, the SC- medium is supplemented with 5% antibiotics mix (see materials and equipment) 12–16 h after electroporation.
-
a.Per well, add 50 μL of antibiotics mix to the 1 mL SC- medium.
-
b.As the mock sample was already cultured in complete RPMI medium, no antibiotics need to be added here.
-
a.
Note: Since the mock sample is not electroporated, mock cells tend to proliferate faster than those of electroporated samples. Therefore, it might be necessary to split mock sample cells (or at least add some fresh medium) if the medium starts to turn yellow.
-
58.
Incubate the cells at 37°C, 5% CO2 for another 3 days until flow cytometry analysis.
Validation of TCR KI by flow cytometry analysis
Since RNPs are stable for around 72 h, flow cytometry analysis can be performed from day 3 after electroporation onwards. Transgene expression levels can be slightly higher at later time points, but are not expected to change substantially. To evaluate the efficiency of transgenic TCR re-expression and endogenous TCR KO, cells are stained for transgenic and endogenous TCR and analyzed by flow cytometry. As the transgenic TCR is equipped with a murine TCR β constant region (mTRBC), staining of mTRBC allows identification of transgenic TCRs and differentiation from endogenous TCRs (hTCR). Additionally, CD8 and/or CD4 expression (here: CD8) is analyzed. Our target population is the mTRBC+hTCR- population of (in this case) CD8+ cells. This population is made up of cells expressing the transgenic TCR (mTRBC+) with simultaneous KO of the endogenous TCR (hTCR-).
Alternatives: When working with an HDR template containing fully human TCR sequences, transgenic TCRs can be identified and differentiated by antigen-specific peptide-MHC (pMHC)-multimer staining. Another approach, albeit less precise, would be antibody staining for the specific Vβ-chain of the transgenic TCR.
Note: Staining is performed on ice.
-
59.
Resuspend cells in the cell culture well before taking an aliquot for staining.
-
60.
Recover 25–50 μL of cells per sample (including TCR KO and mock samples) and transfer sample to a 96-well V-bottom plate.
-
61.
Return the plate with the remaining cells to the incubator (at 37°C, 5% CO2) until further use.
-
62.
Additionally, recover mock cells for single color (S.C.) stainings and add them to the plate.
Note: 100,000 cells per S.C. are sufficient to set up the instrument and create compensation controls.
-
63.Wash 2.5 × with FACS buffer:
-
a.Fill wells to 200 μL with cold FACS wash buffer, centrifuge (3 min, 480 × g, 4°C), discard supernatant (= 0.5 × wash step).
-
b.Resuspend pellet in 200 μL cold FACS buffer, centrifuge, discard supernatant (= 1 × wash step). Repeat this washing step once.
-
a.
-
64.
Prepare antibody master mix for 50 μL staining volume per sample by adding antibodies (anti-CD8-PE, anti-hTCR-FITC and anti-mTRBC-APC/Fire750) in their respective dilution to the FACS buffer.
Optional: CD3-staining can be included to facilitate the identification of cells expressing the transgenic TCR as these cells should be CD3+mTRBC+ double positive.
-
65.
Resuspend cell pellets of samples in 50 μL antibody master mix.
-
66.
Resuspend cell pellets of S.C. in 50 μL FACS buffer and add one of the S.C. antibodies (anti-CD8-PE, anti-hTCR-FITC, anti-CD45-ECD and anti-mTRBC-APC-eFluor780) per well in the respective dilution.
-
67.
Mix samples by pipetting and incubate 20 min on ice in the dark.
-
68.
Fill wells to 200 μL with FACS buffer, centrifuge (3 min, 480 × g, 4°C) and discard supernatant.
-
69.
Resuspend CRISPR-samples (excluding S.C.) in 50 μL of Propidium Iodide (PI) solution (2 μg/mL) and incubate for 3 min on ice in the dark.
Optional: PI can be added to the CRISPR-samples 3 min before the end of the incubation. After 3 min of incubation with PI, samples can be washed 2.5 ×.
-
70.
Wash 1.5 × with FACS buffer (see step 63).
-
71.
Resuspend the sample in 200 μL FACS buffer and filter through a nylon mesh (mesh width: 100 μm).
-
72.
Acquire samples at flow cytometer.
-
73.
Quantify percentages and counts of CD8+ mTRBC+hTCR- cells (see problems 5 and 6).
Expected outcomes
The described DNA template generation and purification is expected to yield 0.5–1.0 μg/μL. Exemplary flow cytometry results after OTR are shown in Figure 7. TCR KO efficiencies are expected to range from 95-99% if only the KO is performed and 90%–95% if TCR KO and KI are performed simultaneously. TCR re-expression after KI of the full transgenic TCR into TRAC is expected to range from 5-15%. This value becomes considerably lower (i.e., 1%–2%) for dual TCR α- and β-chain editing, when the α-chain is targeted to the TRAC and the β-chain is simultaneously targeted to the TRBC. For both engineering approaches, a distinct mTRBC+hTCR- population is expected. Additionally, the absence of a double positive mTRBC+hTCR+ population indicates that no mispairing between transgenic and endogenous TCR-chains occurs (problem 6).
Figure 7.

Exemplary flow cytometry plots after OTR
Mock sample (not electroporated), TCR KO (KO of endogenous α-chain and β-chain), Single KI (Full TCR targeted into TRAC with concomitant β-chain KO) and Dual KI (α-chain targeted into TRAC and the β-chain into TRBC). All samples are pre-gated on living, single CD8+ lymphocytes.
Limitations
This protocol was optimized for TCR replacement in primary human T cells. When working with other cell types and different receptors, experimental conditions and settings most likely need adaption. This protocol is limited to the electroporation of 1 × 106 cells per NucleocuvetteTM well. Upscaling of the electroporation process can be achieved by adapting this protocol to the use of 100 μL NucleocuvetteTM Vessels. This allows for the electroporation of up to 30 × 106 cells per cartridge.
TCR re-expression rates of 5–15% are expected. DNA-sensing inhibitors (see T cell activation), PGA (see RNP production), HDR-enhancers (Kath et al., 2021; Nguyen et al., 2020) have been described to enhance re-expression rates. The combination of DNA-dependent protein kinase (DNA-PK) inhibitor M3814 and HDAC class I/II Inhibitor Trichostatin A has been shown to increase re-expression rates especially in combination the hybrid DNA templates described above (see DNA template design) (Shy et al., 2021).
Typically, the TCR re-expression rates (that is the transgenic protein surface expression rates) are lower for TCRs than they are for chimeric antigen receptors (CARs). However, if there are still B cells present in the culture system (often in the first 1–2 weeks after electroporation), CAR-expression levels can be very low to non-existent as B cells induce downregulation of CARs on edited cells.
Troubleshooting
Problem 1
PCR was not successful as there is more or less than one band showing on agarose gel.
Potential solution
If no bands are visible, make sure the primer sequence is correct and DNA is added (in the correct concentration).
If more than one band appears one the agarose gel, the primers may have bound at more than the intended sites on the plasmid. To fine-tune the PCR, adjust the annealing temperature. Depending on the polymerase, extension time and number of cycles might also need adjustment.
Problem 2
Prepared RNPs are not sufficient to prepare all samples.
Potential solution
Prepare new RNPs to continue the current experiment. To avoid this problem in future experiments, increase pipetting error margins considered in preparation of RNPs.
Problem 3
Air bubble is trapped in NucleocuvetteTM.
Potential solution
If there is a big air bubble underneath the liquid surface, firmly tab (avoid spilling) the NucleocuvetteTM on the bench. Tapping the cuvette angled to the side works best for this purpose. To avoid trapping air, carefully pipette the liquid to the bottom of the NucleocuvetteTM.
Smaller air bubbles on the surface can be burst (or pushed up until they burst) with a fresh pipette tip. To avoid small bubbles in the NucleocuvetteTM, it is crucial to prevent air bubbles or even foaming during the resuspension of the cells in P3 electroporation buffer and the subsequent mixing with RNPs.
Problem 4
NucleofectorTM displays warning (yellow) or error (red) code during electroporation.
Potential solution
Warning and error codes can be due to several reasons. By clicking on the respective well, the error code can be displayed. Explanation, possible causes and suggested procedures are listed in the manufacturer's manual (Chapter 4: Error Codes of the 4D-Nucleofector™ System, page 35–36) and on their website .
Generally, we would advise to continue with the experiment as TCR replacement can still be successful even after error codes, albeit often with lower TCR re-expression rates. If enough stimulated cells are left over, repeating sample preparation and electroporation for the respective well(s) in a fresh NucleocuvetteTM is the best solution.
Problem 5
Cell viability after electroporation is lower than expected.
Potential solution
Reduced cell viability might result from prolonged time which cells had to spend in P3 electroporation buffer. Although 16 samples can be electroporated at once, we would advise to only work with up to 8 samples at a time. This reduces the time before electroporated cells are transferred to the SC- medium. Additionally, introducing DNA-sensing inhibitors into the protocol has been described to increase cell viability (Kath et al., 2021).
Problem 6
Flow cytometry analysis does not show the expected results.
Potential solution
A larger, unedited (hTCR+mTRBC-) population indicates that too much DNA template was used. In contrast, using too little DNA template achieves high KO rates but correspondingly fewer KI cells.
Adjusting the amount of DNA template accordingly should solve these problems. TCR KO rates <90%–95% in the KO only control indicate problems with CRISPR-Cas9 editing (independent from HDR) and/or the protocol.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to the lead contact, Kilian Schober (kilian.schober@uk-erlangen.de).
Materials availability
This study did not generate new unique reagents.
Acknowledgments
The work of C.M. and K.S. was supported by the German Federal Ministry of Education and Research (BMBF; project 01KI2013). The work of T.R.M and D.H.B. was supported by German Center for Infection Research (DZIF) as well as by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) SFB 1321/TP17, SFB 1054/B09, and SFB 1371/TP04. We thank Ev-Marie Schuster for critical reading of the manuscript.
Author contributions
Conceptualization, K.S., T.R.M., and D.H.B.; Methodology, K.S., T.R.M., and D.H.B.; Validation, K.S., C.M., T.R.M., and D.H.B; Investigation, K.S., C.M., and T.R.M.; Writing – Original Draft, C.M. and K.S.; Writing – Review & Editing, K.S., C.M., T.R.M., and D.H.B.
Declaration of interests
D.H.B. is co-founder of STAGE Cell Therapeutics GmbH (now Juno Therapeutics, a Bristol-Myers Squibb Company) and T Cell Factory B.V. (now Kite, a Gilead Company). D.H.B. has a consulting contract with and receives sponsored research support from Juno Therapeutics. D.H.B. is member of the Scientific Advisory Board of Immatics. All other authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2021.101031.
Contributor Information
Carolin Moosmann, Email: carolin.moosmann@uk-erlangen.de.
Kilian Schober, Email: kilian.schober@uk-erlangen.de.
Supplemental information
Data and code availability
This study did not generate any new data sets or code. Data sets supporting the protocol and further examples of OTR are described by Schober et al. (2019) and Müller et al. (2021).
References
- Chicaybam L., Sodre A.L., Curzio B.A., Bonamino M.H. An efficient low cost method for gene transfer to T lymphocytes. PLoS One. 2013;8:e60298. doi: 10.1371/journal.pone.0060298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen C.J., Li Y.F., El-Gamil M., Robbins P.F., Rosenberg S.A., Morgan R.A. Enhanced antitumor activity of T cells engineered to express T-cell receptors with a second disulfide bond. Cancer Res. 2007;67:3898–3903. doi: 10.1158/0008-5472.CAN-06-3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kath J., Du W., Thommandru B., Turk R., Amini L., Stein M., Zittel T., Martini S., Ostendorf L., Wilhelm A., et al. Fast, efficient and virus-free generation of TRAC-replaced CAR T cells. BioRxiv. 2021 doi: 10.1101/2021.02.14.431017. [DOI] [Google Scholar]
- Müller T.R., Jarosch S., Hammel M., Leube J., Grassmann S., Bernard B., Effenberger M., Andrä I., Chaudhry M.Z., Käuferle T., et al. Targeted T cell receptor gene editing provides predictable T cell product function for immunotherapy. Cell Rep. Med. 2021;2:100374. doi: 10.1016/j.xcrm.2021.100374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen D.N., Roth T.L., Li P.J., Chen P.A., Apathy R., Mamedov M.R., Vo L.T., Tobin V.R., Goodman D., Shifrut E., et al. Polymer-stabilized Cas9 nanoparticles and modified repair templates increase genome editing efficiency. Nat. Biotechnol. 2020;38:44–49. doi: 10.1038/s41587-019-0325-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J., Liu X., Fang C., Jiang S., June C.H., Zhao Y. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin. Cancer Res. 2017;23:2255–2266. doi: 10.1158/1078-0432.CCR-16-1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth T.L., Puig-Saus C., Yu R., Shifrut E., Carnevale J., Li P.J., Hiatt J., Saco J., Krystofinski P., Li H., et al. Reprogramming human T cell function and specificity with non-viral genome targeting. Nature. 2018;559:405–409. doi: 10.1038/s41586-018-0326-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schober K., Müller T.R., Gökmen F., Grassmann S., Effenberger M., Poltorak M., Stemberger C., Schumann K., Roth T.L., Marson A., Busch D.H. Orthotopic replacement of T-cell receptor α- and β-chains with preservation of near-physiological T-cell function. Nat. Biomed. Eng. 2019;3:974–984. doi: 10.1038/s41551-019-0409-0. [DOI] [PubMed] [Google Scholar]
- Shy B.R., Vykunta V., Ha A., Roth T.L., Talbot A., Nguyen D.N., Chen Y.Y., Blaeschke F., Vedova S., Mamedov M.R., et al. Hybrid ssDNA repair templates enable high yield genome engineering in primary cells for disease modeling and cell therapy manufacturing. BioRxiv. 2021 doi: 10.1101/2021.09.02.458799. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any new data sets or code. Data sets supporting the protocol and further examples of OTR are described by Schober et al. (2019) and Müller et al. (2021).