

Abstract
Aims
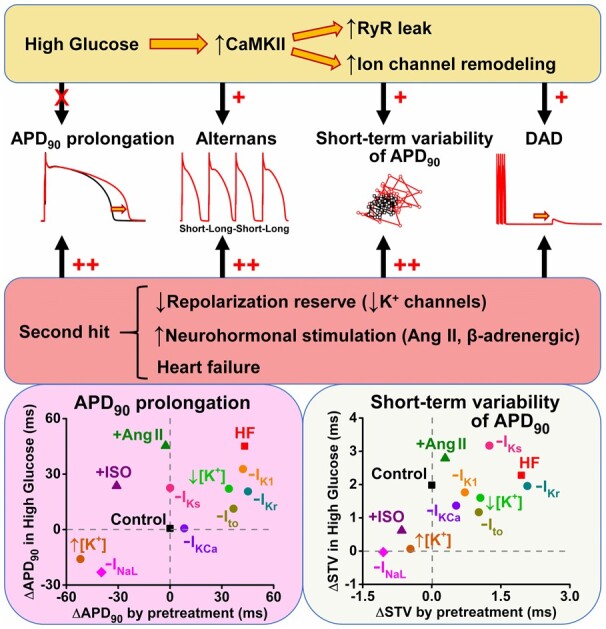
Diabetic hyperglycaemia is associated with increased arrhythmia risk. We aimed to investigate whether hyperglycaemia alone can be accountable for arrhythmias or whether it requires the presence of additional pathological factors.
Methods and results
Action potentials (APs) and arrhythmogenic spontaneous diastolic activities were measured in isolated murine ventricular, rabbit atrial, and ventricular myocytes acutely exposed to high glucose. Acute hyperglycaemia increased the short-term variability (STV) of action potential duration (APD), enhanced delayed afterdepolarizations, and the inducibility of APD alternans during tachypacing in both murine and rabbit atrial and ventricular myocytes. Hyperglycaemia also prolonged APD in mice and rabbit atrial cells but not in rabbit ventricular myocytes. However, rabbit ventricular APD was more strongly depressed by block of late Na+ current (INaL) during hyperglycaemia, consistent with elevated INaL in hyperglycaemia. All the above proarrhythmic glucose effects were Ca2+-dependent and abolished by CaMKII inhibition. Importantly, when the repolarization reserve was reduced by pharmacological inhibition of K+ channels (either Ito, IKr, IKs, or IK1) or hypokalaemia, acute hyperglycaemia further prolonged APD and further increased STV and alternans in rabbit ventricular myocytes. Likewise, when rabbit ventricular myocytes were pretreated with isoproterenol or angiotensin II, hyperglycaemia significantly prolonged APD, increased STV and promoted alternans. Moreover, acute hyperglycaemia markedly prolonged APD and further enhanced STV in failing rabbit ventricular myocytes.
Conclusion
We conclude that even though hyperglycaemia alone can enhance cellular proarrhythmic mechanisms, a second hit which reduces the repolarization reserve or stimulates G protein-coupled receptor signalling greatly exacerbates cardiac arrhythmogenesis in diabetic hyperglycaemia.
Keywords: Diabetic hyperglycaemia, Cardiac electrophysiology, Cardiac action potential, Delayed afterdepolarizations, Alternans
Graphical Abstract
1. Introduction
A two-hit mechanism for disease development was first proposed by Alfred G. Knudson in cancer biology.1 Later, this concept has been extended to other research fields, including cardiac arrhythmia genetics.2,3 It is well-established that the risk of arrhythmias in those carrying a genetic/acquired predisposition [e.g. ion channel mutation in long QT syndrome or remodelling in heart failure (HF)] is significantly increased when an additional stress occurs (e.g. sympathetic, ionic or metabolic imbalance, or certain drug treatments). Arrhythmia mechanisms usually require a trigger (e.g. unstable Ca2+ cycling) and a vulnerable substrate (e.g. repolarization abnormality, fibrosis, ischaemic/necrotic islands) which again imply that two (or more) factors may enhance arrhythmogenic propensity.2
Diabetes mellitus (DM) is associated with increased risk of cardiac arrhythmias and sudden cardiac death.4 Diabetic hyperglycaemia and glucose-variability correlate with arrhythmia risk.5 Moreover, diabetes at least doubles the risk of HF which further enhances arrhythmia susceptibility.6 Furthermore, hyperglycaemic-clamp to a blood glucose level of 15 mM for 2 h slightly prolonged QTc interval (by 31 ms) and markedly increased QTc dispersion (by 70%) even in healthy individuals.7 Hyperglycaemia is strongly associated with increased risk of early-onset ventricular tachycardia in non-diabetic patients following myocardial infarction.8
At the level of molecular signalling, Ca2+/calmodulin-dependent kinase II (CaMKII) has been implicated in cardiac arrhythmogenesis in DM, and up-regulation of CaMKII impairs ion channel function and Ca2+ regulation.9 Hyperglycaemia has been shown to induce post-translational modification of CaMKII by O-linked β-N-acetylglucosamine (O-GlcNAc) at the serine 280 site which results in autonomous activation of the kinase.10 In line with this, inhibition of CaMKII prevented arrhythmias in various diabetic animal models.9 However, it is still unclear whether hyperglycaemia alone can be accountable for arrhythmias or whether it requires the presence of an additional pathological factor (i.e. a second hit). Here, we tested the effect of acute hyperglycaemia on action potential (AP) stability in isolated atrial and ventricular myocytes from murine and rabbit hearts in healthy control and conditions with reduced repolarization reserve and neurohormonal stimulation.
2. Methods
All animal handling and laboratory procedures were in accordance with the approved protocols of the Institutional Animal Care and Use Committee at University of California, Davis (#19721 and #21064) conforming to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (8th edition, 2011).
2.1 Animal model and cell isolation
Atrial and ventricular cardiomyocytes were isolated from 27 C57BL/6J mice (male, 10- to 12-week-old, Jackson Laboratory, Stock No. 000664) and 60 New Zealand White rabbits (male, 3- to 4-month-old, Charles River Laboratories) using a standard enzymatic technique as previously described.11 Briefly, animals were injected with heparin (5000 U/kg body weight) and were subjected to general anaesthesia [induction with one time intravenous injection of 10 mg/kg body weight propofol (Rapanofal®, Ivaoes Animal Health, Miami, FL, USA) followed by 2–5% isoflurane inhalation in 100% oxygen throughout the procedure]. Deep surgical anaesthesia was confirmed by abolished pain reflexes. All animals were euthanized by surgical excision of the heart while in deep anaesthesia. Immediately after excision, the heart was rinsed in cold nominally Ca2+-free Minimal Essential Medium. The aorta was cannulated and retrograde perfused on constant flow Langendorff apparatus at 37°C with Ca2+-free normal Tyrode’s solution, gassed with 100% O2. Then, ventricular myocytes were digested using collagenase type II (Worthington Biochemical Co., Lakewood, NJ, USA) and protease type XIV (Sigma-Aldrich). Ventricular myocytes were dispersed mechanically and filtered through a nylon mesh and allowed to sediment for 10 min. The sedimentation was repeated three times using increasing [Ca2+] from 0.125 to 0.25 then 0.5 mmol/L. Finally, ventricular myocytes were kept in Tyrode’s solution (0.5 mmol/L [Ca2+]) at room temperature until use.
HF was induced in New Zealand White rabbits (male, 3- to 4-month-old) by aortic insufficiency and 4 weeks later by aortic constriction.12 Data here were obtained from three HF and three age-matched control rabbits. HF progression was monitored periodically by echocardiography. Cardiomyocytes were isolated from rabbits at 2–2.5 years of age when left ventricular end-systolic dimension exceeded 1.55 cm as previously described.12 HF animals exhibited significant cardiac hypertrophy, ventricular dilation, pulmonary congestion, and abdominal ascites fluid accumulation, all similar to our prior studies on this rabbit model.12,13 Cardiomyocytes isolated from healthy age-matched rabbits were used in control experiments.
2.2 Electrophysiology
Isolated cardiomyocytes were transferred to a temperature-controlled chamber (Warner Instruments, Hamden, CT, USA) mounted on a Leica DMI3000 B inverted microscope (Leica Microsystems, Buffalo Grove, IL, USA) and continuously perfused (2 mL/min) with Tyrode solution containing (in mmol/L): NaCl 140, KCl 4, CaCl2 1.8, MgCl2 1, HEPES 5, Na-HEPES 5, glucose 5.5, and mannitol 24.5; pH = 7.40 and osmolality = 320 ± 2 mOsm/L. High glucose effects were assessed by switching the bathing medium to a Tyrode solution containing 30 mmol/L glucose and 0 mannitol (osmolality and pH matched). Electrodes were fabricated from borosilicate glass (World Precision Instruments, Sarasota, FL, USA) having tip resistances of 2–2.5 MΩ when filled with internal solution containing (in mmol/L): K-aspartate 100, KCl 30, NaCl 8, Mg-ATP 5, phosphocreatine-K2 10, HEPES 10, EGTA 0.01, cAMP 0.002, and calmodulin 0.0001; pH = 7.20 (with KOH). Using this internal solution, the intracellular Ca2+ transient and contraction of the cardiomyocyte are preserved.14 Axopatch 200B amplifier (Axon Instruments Inc., Union City, CA, USA) was used for recordings, and the signals were digitized at 50 kHz by a Digidata 1322 A A/D converter (Axon Instruments) under software control (pClamp10.4). The series resistance was typically 3–5 MΩ, and it was compensated by 90%. Experiments were discarded when the series resistance was high or increased by >10%. All experiments were conducted at 37 ± 0.1°C.
APs were evoked by 2-ms-long supra-threshold depolarizing pulses delivered via the patch pipette. Fifty consecutive APs were recorded to examine the average behaviour at each pacing frequency. AP duration (APD) at 90% repolarization (APD90) was determined. Series of 50 consecutive APs were analysed to estimate short-term variability of APD90 (STV) according to the following formula: STV = Σ(|APDn+1 − APDn|)/[(nbeats − 1) × √2], where APDn and APDn+1 indicate the durations of the nth and (n + 1)th APs, and nbeats denotes the total number of consecutive beats analysed. Changes in STV are presented as Poincaré plots of 50 consecutive APD90. APD alternans magnitude was calculated as the difference between the average APD90 of odd and even numbered beats during 50 consecutive APs recorded. Diastolic arrhythmogenic activities were elicited by cessation of 1-min tachypacing, and membrane potential was recorded for additional 3 min. Delayed afterdepolarizations (DADs) were defined as >1 mV depolarization within 0.5 s. Spontaneous APs (sAPs) were defined as depolarizations showing overshoot with a fast upstroke phase.
Chemicals and reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA), if not specified otherwise. E-4031 and HMR-1556 were from Tocris (Bristol, UK).
2.3 Statistical analysis
Data are presented as mean ± SEM. The number of cells/animals in each experimental group is reported in the figure legends. Because multiple cells may come from one animal, we performed hierarchical statistical analysis in line with the previously outlined principles taking into account inter-subject variability.15,16 Normality of the data and the equality of group variance were assessed by D'Agostino–Pearson and Brown–Forsythe tests, respectively. Statistical significance of differences for continuous variables was tested using nested, two-tailed t-test to assess the effect of hyperglycaemia in paired experiments, and the effects of pretreatments with ion channel inhibitors or GPCR agonists were assessed using nested one-way analysis of variance followed by post hoc Dunnett multiple comparison test. Categorical outcomes were evaluated using Fisher’s exact test. GraphPad Prism 8.0 was used for data analysis. Differences were considered statistically significant if P < 0.05.
3. Results
3.1 Acute hyperglycaemia induces action potential changes that depend on CaMKII
AP responses to acute high glucose treatment (30 mM, 6 min) are shown in Figure 1. In murine ventricular myocytes, high glucose significantly prolonged APD at 1 Hz steady-state pacing, and significant APD alternans was induced by tachypacing at 10 Hz (Figure 1A). In rabbit atrial myocytes, high glucose prolonged APD but without affecting APD alternans magnitude (Figure1A, C). In contrast, high glucose in rabbit ventricular myocytes did not change APD, but increased APD alternans magnitude (Figure 1C) and also lowered the threshold for alternans appearance (from 5 to 4 Hz; Supplementary material online, Figure SIA). The AP peak and maximal rate of rise (dV/dtmax) were both reduced by high glucose in all cell types tested (Supplementary material online, Figure SIB). Importantly, CaMKII inhibition by AIP (1 μM) prevented all of these arrhythmogenic AP changes induced by high glucose (Figure1B, C) in all three cell types. Moreover, high glucose enhanced the STV, an effect that was also abolished by CaMKII inhibition (Figure 1D). Thus, changes in APD, APD alternans, STV, peak AP Vm and dv/dtmax induced acutely by high glucose all required CaMKII activity. However, under these baseline conditions in healthy myocytes, some of these perturbations were modest, or undetectable (e.g. APD in rabbit ventricular myocytes).
Figure 1.

Acute hyperglycaemia induces action potential changes dependent on CaMKII. (A) High glucose (30 mM, 6 min) induced action potential (AP) changes in murine ventricular and rabbit atrial and ventricular cardiomyocytes. Representative traces are shown at 1 Hz steady-state pacing. Paired data on AP duration at 90% repolarization (APD90) are shown in insets. Tachypacing-induced APD90 alternans is shown below (S and L refer to short and long APD90, respectively). (B) CaMKII inhibition using the selective inhibitor AIP (1 μM) attenuated high glucose effects. (C) APD90 at 1 Hz steady-state pacing and the magnitude of the tachypacing-induced APD90 alternans (at 10 Hz in mouse and at 5 Hz in rabbit). (D) Representative Poincaré plots of 50 consecutive APD values. Insets show short-term variability (STV) of APD90 at 1 Hz pacing. (Murine ventricular cells, control: n = 16 cells from nine animals at 1 Hz and n = 15 cells from eight animals at 10 Hz; AIP: n = 13 cells from six animals at both 1 Hz and 10 Hz pacing. Rabbit atrial cells, control: n = 12 cells from six animals at 1 Hz and n = 7 cells from four animals at 5 Hz; AIP: n = 8 cells from three animals at 1 Hz and n = 6 cells from three animals at 5 Hz pacing. Rabbit ventricular cells, control: n = 15 cells from seven animals at both 1 Hz and 5 Hz pacing; AIP: n = 12 cells from five animals at both 1 Hz and 5 Hz pacing.) Nested t-test; NS, non-significant; *P < 0.05, **P < 0.01, ***P < 0.001.
3.2 Acute hyperglycaemia induces spontaneous diastolic activities that depend on CaMKII
Arrhythmogenic diastolic activities including DADs and spontaneous action potentials (sAPs) were measured following 1 min of tachypacing in normal and high glucose conditions (Figure 2A). High glucose increased the number of ventricular cells exhibiting DADs or sAPs in both mouse and rabbit (Supplementary material online, Figure SIIA). These glucose effects were prevented by the CaMKII inhibitor AIP (Figure 2B), and all DADs and sAPs were abolished by intracellular Ca2+ buffering using EGTA (Supplementary material online, Figure SIII). The frequency of diastolic events was also increased by high glucose in both cell types, and again was dependent on CaMKII signalling (Figure 2B). However, the average magnitude of DADs was unaltered with a tendency for shorter latency period before the first event occurring following cessation of tachypacing, which was prominent in mouse ventricular myocytes (Supplementary material online, Figure SIIB). We infer that CaMKII is also essential in mediating these Ca2+-dependent arrhythmogenic activities.
Figure 2.

Acute hyperglycaemia induces delayed afterdepolarizations dependent on CaMKII. (A) High glucose (30 mM, 6 min) enhanced spontaneous diastolic activities (delayed afterdepolarization, DAD; spontaneous action potential, sAP) in murine ventricular and rabbit atrial and ventricular cardiomyocytes following a tachypacing protocol shown above. (B) DAD and sAP frequencies were increased by high glucose, which were prevented using the CaMKII inhibitor AIP (1 μM). (Murine ventricular cells, control: n = 21 cells from nine animals; AIP: n = 18 cells from six animals. Rabbit atrial cells, control: n = 9 cells from five animals; AIP: n = 6 cells from three animals. Rabbit ventricular cells, control: n = 18 cells from eight animals; AIP: n = 12 cells from five animals.) Nested t-test; NS, non-significant; *P < 0.05, **P < 0.01, ***P < 0.001.
3.3 Reduced repolarization reserve enhances arrhythmogenic action potentials in hyperglycaemia
Next, we systematically tested how reduced repolarization reserve may potentiate high glucose effects in rabbit ventricular myocytes (our focus from here on) because of their higher relevance to human ventricular myocytes.17 The pre-drug APD and STV values were well-matched to control in each treatment group (Supplementary material online, Figure SIV).
First, we tested the impact of each K+ current on baseline APD in normal glucose, then we tested the effect of elevated glucose (Figure 3). Inhibiting transient outward K+ current (Ito) using 5 mM 4-aminopyridine (4-AP) affected mainly phase 1 AP repolarization, slightly prolonged APD and increased STV in normal glucose. Unlike the high glucose rabbit ventricular APD results in Figure 1, after Ito inhibition, high glucose tended to further prolong APD, significantly increased STV but had similar effects on APD alternans (Figure 3).
Figure 3.

Hyperglycaemia-induced arrhythmogenic action potentials are exacerbated by reduced repolarization reserve. (A) High glucose (30 mM, 6 min) induced action potential (AP) changes are enhanced in rabbit ventricular cardiomyocytes pretreated with either 4-aminopyridine (4-AP, 5 mM) to inhibit the transient outward K+ current (Ito), pentamidine-analogue 6 (PA-6, 200 nM) to inhibit the inward rectifier K+ current (IK1), E-4031 (1 μM) to inhibit rapid delayed rectifier K+ current (IKr), HMR-1556 (1 μM) to inhibit slow delayed rectifier K+ current (IKs), or apamin (100 nM) to inhibit the small-conductance Ca2+-activated K+ current (IKCa). Representative traces are shown at 1 Hz steady-state, and insets show tachypacing-induced alternans (S and L refer to short and long AP durations, respectively). (B) AP duration at 90% repolarization (APD90) at 1 Hz steady-state pacing and the magnitude of the tachypacing-induced APD90 alternans at 5 Hz pacing. (C) Representative Poincaré plots of 50 consecutive APD90 values. Statistics on short-term variability (STV) of APD90 at 1 Hz pacing. (At 1 Hz pacing, control: n = 15 cells from seven animals; 4-AP: n = 13 cells from six animals; PA-6: n = 11 cells from five animals; E-4031: n = 10 cells from five animals; HMR-1556: n = 10 cells from five animals; apamin: n = 8 cells from four animals. At 5 Hz pacing, control: n = 15 cells from seven animals; 4-AP: n = 7 cells from four animals; PA-6: n = 11 cells from five animals; E-4031: n = 10 cells from five animals; HMR-1556: n = 9 cells from five animals; apamin: n = 7 cells from four animals.) Nested t-test; NS, non-significant; *P < 0.05, **P < 0.01 vs. 5.5 mM glucose. Nested ANOVA, #P < 0.05, ##P < 0.01, ###P < 0.001 vs. control.
Inhibition of the inward rectifier K+ current (IK1) using pentamidine-analogue 6 (PA-6, 200 nM) slowed late repolarization especially, induced AP triangulation and prolonged APD. High glucose significantly exacerbated the increased APD and led to a pronounced APD alternans following IK1 inhibition, and a significant further increase in APD-STV (Figure 3A–C).
Inhibition of the rapid delayed rectifier K+ current (IKr) using E-4031 (1 μM) induced pronounced APD prolongation, increased STV and large APD alternans in normal glucose (Figure 3). High glucose further increased APD, STV and alternans following IKr inhibition. Inhibition of the slow delayed rectifier K+ current (IKs) using HMR-1556 (1 μM) did not change baseline APD in normal glucose but significantly increased APD alternans and STV (Figure3B, C). However, high glucose significantly prolonged APD and markedly increased STV and APD alternans following IKs inhibition (Figure 3C).
Inhibition of the small-conductance (SK) Ca2+-activated K+ current (IKCa) using apamin (100 nM) did not change baseline APD and STV but did promote APD alternans (Figure 3). High glucose still did not change APD following IKCa inhibition but did increase STV and tended to further enhance APD alternans. The effects of Ito, IK1, and IKr inhibitions on APD were more pronounced at slow pacing rates in normal glucose, and similar reverse-rate dependent APD prolongation by high glucose was found following the inhibition of these K+ channels (Supplementary material online, Figure SV). On the contrary, high glucose effect on APD following IKs and IKCa inhibitions became significant only at fast pacing rates (Supplementary material online, Figure SV), which may reflect the Ca2+-dependent up-regulation of these currents at higher Ca2+ levels.
In summary, Ito, IK1, and IKr inhibitions each prolonged APD, STV, and except for Ito promoted alternans. In addition, high glucose exacerbated these APD, STV and alternans effects. This suggests that the high glucose-induced reduction in repolarization reserve, APD, STV and alternans is not mediated by only one of these three currents; otherwise the high glucose effect would have been lost after that current’s inhibition. IKs inhibition alone did not increase APD but did raise APD alternans and STV, and again high glucose exacerbated all three APD measures even after IKs block. IKCa block was most like IKs in these regards, but effects were smaller and closer to those without K+ channel inhibition (Figure 1).
3.4 Changes in serum K+ level alter action potential response to hyperglycaemia
Alterations in serum K+ levels, both hypo- and hyperkalaemia, frequently occur in diabetes and are implicated in arrhythmogenesis. Hypokalaemia is known to reduce repolarization reserve by decreasing IK1 and IKr conductances.18 In cardiac myocytes, hypokalaemia (2 mM [K+]o) induced ∼-20 mV shift in resting membrane potential (vs. control 4 mM [K+]o in line with the change in K+ equilibrium potential) and significantly prolonged APD (Figure 4). High glucose further prolonged APD and increased alternans and STV in hypokalaemia (Figure 4). The high glucose-induced APD prolongation in hypokalaemia was more pronounced at fast pacing rates (Supplementary material online, Figure SVI).
Figure 4.

Hyperglycaemia-induced arrhythmogenic action potentials are dependent on serum K+ levels. (A) High glucose (30 mM, 6 min) induced action potential (AP) changes in hypokalaemia and hyperkalaemia (2 and 8 mM extracellular K+ concentrations, respectively). Representative traces are shown at 1 Hz steady-state, and insets show tachypacing-induced alternans (S and L refer to short and long AP durations, respectively). (B) AP duration at 90% repolarization (APD90) at 1 Hz pacing and the magnitude of the tachypacing-induced APD90 alternans at 5 Hz pacing. (C) Representative Poincaré plots of 50 consecutive APD90 values. Statistics on short-term variability (STV) of APD90 at 1 Hz pacing. (At 1 Hz pacing, normokalaemia: n = 15 cells from seven animals; hypokalaemia: n = 12 cells from five animals; hyperkalaemia: n = 13 cells from four animals. At 5 Hz pacing, normokalaemia: n = 15 cells from seven animals; hypokalaemia: n = 8 cells from five animals; hyperkalaemia: n = 12 cells from four animals.) Nested t-test; NS, non-significant; *P < 0.05, **P < 0.01 vs. 5.5 mM glucose. Nested ANOVA; #P < 0.05, ##P < 0.01, ###P < 0.001 vs. control.
Hyperkalaemia (8 mM [K+]o) induced ∼+18 mV shift in resting membrane potential (vs. control 4 mM [K+]o) and significantly shortened APD. High glucose slightly shortened APD and reduced alternans in hyperkalaemia (Figure 4). The high glucose-induced APD shortening in hyperkalaemia showed reverse-rate dependence (Supplementary material online, Figure SVI). We conclude that high glucose exacerbated the directional effects on APD of both hypo- and hyperkalaemia, which could aggravate the arrhythmogenic consequences of either change in [K+].
3.5 Acute hyperglycaemia enhances action potential changes by increasing late Na+ current
Late Na+ current (INaL) is critically regulated by CaMKII and contributes to APD prolongation and arrhythmias in various heart diseases.9,19 Therefore, we tested the contribution of INaL to the proarrhythmic effects of hyperglycaemia using the selective INaL inhibitor GS-967 (1 μM). GS-967 tended to slightly shorten APD and decrease STV in normal glucose (Figure 5). However, high glucose in the presence of GS-967 significantly shortened APD in a reverse-rate dependent manner (Supplementary material online, Figure SVII) and failed to increase APD alternans and STV (Figure 5). These data demonstrate the important role of increased INaL in the arrhythmogenic effects of hyperglycaemia.
Figure 5.

Hyperglycaemia-induced action potential changes following inhibition of late Na+ current. (A) High glucose (30 mM, 6 min) induced action potential (AP) shortening in rabbit ventricular cardiomyocytes following late Na+ current (INaL) inhibitor GS-967 (1 μM) treatment. Representative traces are shown at 1 Hz steady-state, and insets show tachypacing-induced alternans (S and L refer to short and long AP durations, respectively). (B) Representative Poincaré plots of 50 consecutive APD90 values. (C) AP duration at 90% repolarization (APD90), the magnitude of the tachypacing-induced APD90 alternans and the short-term variability (STV) of APD90. (Control: n = 15 cells from seven animals; GS-967: n = 10 cells from five animals.) Nested t-test; NS, non-significant; *P < 0.05, **P < 0.01 vs. 5.5 mM glucose.
3.6 Acute hyperglycaemia enhances arrhythmogenic action potential changes following neurohumoral stimulation
Increased neurohumoral tone is frequently reported in diabetes. Therefore, we tested the effects of two key neurohormonal mediators stimulating G-protein coupled receptor (GPCR) signalling and Ca2+ cycling in myocytes. Angiotensin II (Ang-II, 100 nM) did not alter baseline APD but increased the magnitude of tachypacing-induced alternans (Figure6A, B). However, hyperglycaemia markedly prolonged APD and further increased alternans and STV following Ang-II (Figure6B, C). These high glucose effects in Ang-II were larger at rapid pacing rates (Supplementary material online, Figure SVIII). Thus, Ang-II as a second hit appears to worsen the high glucose effects on APD, alternans and STV.
Figure 6.

Hyperglycaemia-induced arrhythmogenic action potentials are exacerbated by neurohormonal stimulation. (A) High glucose (30 mM, 6 min) induced action potential (AP) changes are enhanced in rabbit ventricular cardiomyocytes following angiotensin II (Ang II, 100 nM) or isoproterenol (ISO, 30 nM) treatments. Representative traces are shown at 1 Hz steady-state, and insets show tachypacing-induced alternans (S and L refer to short and long AP durations, respectively). (B) AP duration at 90% repolarization (APD90) at 1 Hz pacing, the magnitude of the tachypacing-induced APD90 alternans at 5 Hz pacing. (C) Representative Poincaré plots of 50 consecutive APD90 values. Statistics on short-term variability (STV) of APD90 at 1 Hz pacing. (At 1 Hz pacing, control: n = 15 cells from seven animals; Ang II: n = 10 cells from six animals; ISO: n = 10 cells from five animals. At 5 Hz pacing, control: n = 15 cells from seven animals; Ang II: n = 9 cells from five animals; ISO: n = 6 cells from four animals.) Nested t-test; NS, non-significant; *P < 0.05, **P < 0.01, ***P < 0.001 vs. 5.5 mM glucose. Nested ANOVA; #P < 0.05, ##P < 0.01 vs. control.
The β-adrenergic receptor agonist isoproterenol (ISO, 30 nM) significantly shortened APD and reduced both STV and APD alternans (Figure 6A–C). However, these effects of ISO were abolished in hyperglycaemia, which may negatively affect the repolarization stability induced by sympathetic stimulation.
3.7 Acute hyperglycaemia enhances arrhythmogenic action potentials in heart failure
HF is characterized by electrophysiological remodelling with reduced repolarization reserve and increased neurohormonal tone. APD was slightly prolonged, and STV was increased in HF already at normal glucose (Figure 7). Importantly, high glucose further increased both AP parameters in HF leading to severely impaired AP repolarization (Figure 7). These data indicate the markedly increased proarrhythmic potency of hyperglycaemia in the failing heart.
Figure 7.

Hyperglycaemia-induced arrhythmogenic action potential changes are exacerbated in heart failure. (A) High glucose (30 mM, 6 min) further prolonged action potential duration (APD) in heart failure (HF) vs. age-matched (AM) control. (B) AP duration at 90% repolarization (APD90) and short-term variability (STV) of APD90. (AM control: n = 8 cells from three animals; HF: n = 8 cells from three animals.) Nested t-test; NS, non-significant; *P < 0.05 vs. 5.5 mM glucose; #P < 0.05 vs. AM control.
4. Discussion
Diabetes and metabolic syndrome are associated with increased arrhythmia risk, but effective therapies are still lacking.4 Given that and their steadily increasing prevalence, the need for better understanding of cellular and molecular arrhythmia mechanisms in diabetes is warranted. Moving towards this goal, our knowledge on signalling networks and molecular mechanisms in diabetic cardiomyopathy has significantly improved in recent years.20 One of these key signalling pathways that are up-regulated in DM is CaMKII, which regulates several ion channels and Ca2+ handling proteins to increase arrhythmia susceptibility.9,10 Moreover, the CaMKII-induced electrophysiological changes19,21 closely resemble the remodelling that occurs in DM22–24; and importantly, CaMKII inhibition was found to be protective against arrhythmias in various diabetic animal models.9,10,25–27 CaMKII is also subject to post-translational modifications, including O-GlcNAcylation of a key CaMKIIδ site (Ser280) that promotes chronic activation of the kinase10 and subsequent phosphorylation of its targets that include numerous ion channels.9
Na+ channel regulation by CaMKII was shown to reduce the fast transient Na+ current (INaT) by a negative shift in steady-state inactivation, which decreases the fraction of available Na+ channels at a given membrane potential, but CaMKII also markedly increases INaL.19 Consistent with these CaMKII effects, reduced INaT23 and increased INaL24 in diabetic mice contributed to impaired impulse propagation and APD prolongation. These reports are in line with our data showing that acute hyperglycaemia induced CaMKII-dependent reduction of dV/dt (a surrogate of INaT function) and APD prolongation in mouse ventricular and rabbit atrial myocytes (Figure 1). Moreover, acute hyperglycaemia shortened APD following INaL inhibition in rabbit ventricular myocytes (Figure 5). This suggests that the CaMKII-dependent increase in INaL in both HF and diabetes contributes to the additional stress on repolarization reserve observed in high glucose here. The hyperglycaemic-induced increase in INaL can shift the balance between repolarizing and depolarizing currents under the AP plateau phase,28 which may also explain why inhibition of any K+ channel could not suppress the APD effects of high glucose (Figure 3). Importantly, the INaL inhibitor ranolazine provided benefits to diabetic patients enrolled in the Combination Assessment of Ranolazine In Stable Angina (CARISA) trial.29
Several K+ channels are regulated by CaMKII with differential temporal dynamics. Acutely, CaMKII was shown to reduce Ito inactivation, enhance Ito recovery, and increase IK1 and IKs.12,21,30 Thus, acute CaMKII activation increases repolarization capacity, which may counterbalance the increased depolarizing fluxes through INaL, L-type Ca2+ current and Na+/Ca2+ exchanger. On the contrary, chronic CaMKII activation down-regulates the functional expression of K+ channels demonstrated in genetic CaMKII overexpression21 and in HF.12,30 A similar mechanism may also occur in chronic diabetes, and down-regulation of various K+ channels have been reported in type 1 diabetic mice,30 rats,31,32 rabbits,33,34 and dogs.22 Thus, the chronic and acute effects of hyperglycaemia may be additive. Moreover, insulin substitution in type 1 DM prevented the reduction in IKr33 and IKs,22 and partially restored Ito.22 In line with this, the fast component of Ito mediated by KV4.2 and KChIP2 proteins were significantly reduced in cardiac-specific insulin receptor knockout (CIRKO) mice.35 However, insulin treatment did not affect the reduction in KV1.5 channels in atrial myocytes from diabetic Akita mice, but increased INaT.36 High glucose may also regulate the activity of ATP-sensitive K+ channels37 and play a role in ischaemia-related arrhythmias,8 which require further investigation.
Intracellular Ca2+ cycling is critically regulated by CaMKII via phosphorylation of the ryanodine receptor (RyR), phospholamban and L-type Ca2+ channel. RyRs can get phosphorylated at serine 2814 by CaMKII which leads to increased spontaneous Ca2+ leak from the sarcoplasmic reticulum (SR)38 and increased DADs, both of which are characteristic of HF39 and diabetic hyperglycaemia.10,27 In line with this, acute hyperglycaemia was shown to increase Ca spark and wave frequencies through enhanced CaMKII activity.10 In the present study, we found that CaMKII inhibition prevented this form of arrhythmogenic spontaneous DADs and sAPs in hyperglycaemia (Figure 2). It was previously shown that acute hyperglycaemia did not affect Ca2+ transient amplitude but led to diastolic [Ca2+] elevation and enhanced premature ventricular complexes in Langendorff-perfused rat hearts.10 On the contrary, intracellular Ca2+ cycling is significantly impaired in chronic diabetes, characterized by reductions in Ca2+ transient amplitude and rates of rise and decay, diminished SR Ca2+ load, and further increased SR Ca2+ leak.40 Moreover, the SR Ca2+ threshold at which depolarizing inward currents are generated is also significantly reduced (below the actual SR Ca2+ load) in diabetes.27 These changes in Ca2+ cycling can further promote afterdepolarizations in diabetic hyperglycaemia.
These molecular ionic channel (sarcolemmal and SR) mechanisms may account for the atrial and ventricular AP changes that leads to impaired repolarization and temporal heterogeneity in the diabetic heart. However, the electrophysiological effects of acute hyperglycaemia were only modest in healthy myocytes (Figure 1). In agreement with our rabbit data, acute hyperglycaemia induced modest changes in QTc but markedly increased QTc dispersion in healthy human volunteers, independent of insulin action.7 Similarly, STV but not QTc was increased in patients with impaired glucose tolerance.41 But importantly, we found here that AP repolarization became severely impaired when hyperglycaemia was applied to a cell in the presence of an additional pathological factor (second hit) including reduced repolarization reserve (specific K+ channel inhibitors in Figure 3 and hypokalaemia in Figure 4), GPCR stimulation (Figure 6) and HF (Figure 7).
The role of each K+ channel in AP repolarization has already been established, and the major contributions of Ito, IKr, and IK1 to physiological repolarization are well agreed upon.42 Not surprisingly, their inhibitions caused significant APD prolongation and enhanced hyperglycaemia effects (Figure 3). More interestingly, even inhibition of IKs, which did not alter baseline APD, significantly promoted the proarrhythmic effects of high glucose (Figure 3). This is in agreement with the demonstrated role of IKs as a critical repolarization reserve current in human and large animals despite minimal impact of IKs block on APD in the absence of β-adrenergic agonists.43 Moreover, CaMKII acutely up-regulates IKs, which may counterbalance the depolarizing effects of hyperglycaemia, and such a role for calcium-dependent IKs up-regulation to limit APD prolongation was previously shown in HF.12 The contribution of IKCa to APD alternans but not baseline APD has only been studied under normoglycaemic conditions44; however, its contribution was further enhanced in the AP adaptation to hyperglycaemia (Figure 3). Hypokalaemia is known to reduce the repolarization reserve by decreasing IK1 and IKr,18 whereas the hyperpolarizing shift in the resting membrane potential increases Na+ channel availability and INaL. In line with that, hypokalaemia markedly enhanced the arrhythmogenic potential of hyperglycaemia (Figure 4).
Increased GPCR stimulation is a critical regulator of heart function, and increased activity of the renin-angiotensin-aldosterone system is commonly reported in diabetes as well as in HF.45 Ang-II markedly increased the arrhythmogenic AP changes in hyperglycaemia (Figure 6). Ang-II is known to increase the production of reactive oxygen species (ROS), which by itself can promote RyR activity46 and that of other channels, but an important additional effect of ROS is the promotion of autonomous CaMKII activation47 which further modulates several of these same targets.9 In diabetic rats, down-regulation of cardiac K+ channels was also shown to be dependent on Ang-II mediated ROS production.48 Moreover, hyperglycaemia prevented physiological APD shortening during β-adrenergic stimulation (Figure 6), which is a critical adaptation response to maintain diastolic filling and coronary perfusion at rapid heart rates.
HF is characterized by both reduced repolarization reserve, chronically increased GPCR stimulation and CaMKII activation.12,39 Hyperglycaemia significantly prolonged APD and impaired repolarization stability in HF (Figure 7). In line with this, HF patients with diabetes have worse cardiovascular outcomes and prognosis than those without diabetes, and the two diseases share a complex interrelated pathophysiology.49
Diabetes is also associated with increased risk of atrial fibrillation (AF).4,6 Moreover, a positive linear association was found between glycated haemoglobin (HbA1c) levels and the risk of AF in patients with and without DM.50 The molecular pathophysiology of AF is complex, but altered Na+ and K+ channel function, impaired intracellular Ca2+ handling, and enhanced CaMKII activity are among the principle mechanisms of AF.51 Acute hyperglycaemia significantly increased APD and STV (Figure 1), reduced dV/dtmax (Supplementary material online, Figure SI), and slightly enhanced DADs (Figure 2) in rabbit atrial myocytes, all of which were CaMKII-dependent. These proarrhythmic alterations can contribute to AF initiation. Smaller IK1 and IKs in atria vs. ventricle52 might explain the increased susceptibility of atrial cells to acute hyperglycaemia, and we speculate that atrial arrhythmogenesis can be further enhanced by a second hit that enhances SR Ca2+ leak, which requires further investigation.
The two-hit arrhythmia mechanism described here has important clinical implications for risk assessment and therapeutic considerations in diabetes. First, it highlights the increased arrhythmic risk of hyperglycaemia in patients with reduced repolarization reserve caused by congenital and acquired long QT syndromes, cardiac co-morbidities, and ionic and sympathetic imbalances. Several drugs used clinically have been shown to reduce repolarization reserve (e.g. off-target hERG inhibition) but do not affect baseline phenotype (referred to hidden cardiotoxicity53); however, they may contribute to disease under stress conditions, including hyperglycaemia. It also emphasizes the importance of tight control of blood glucose and K+ levels. Second, it suggests that the cure for two (or multiple) hits may require a combination therapy including glucose-lowering medications, angiotensin receptor blockers, and maybe in the future a targeted CaMKII inhibitor which may reverse arrhythmogenic AP remodelling.
Limitations
Here, we studied only the acute effects of hyperglycaemia in healthy and failing cardiomyocytes; however, chronic effects of diabetes also include remodelling, fibrosis, mitochondrial dysfunction, and other diabetic complications affecting cardiac arrhythmogenesis. Alterations in insulin signalling can also contribute to proarrhythmia and K+ channel remodelling. Further studies are required to elucidate the changes in intracellular Ca2+ cycling and the exact arrhythmia mechanisms in diabetic hyperglycaemia. Here, we used pharmacological agents to manipulate ion channel function, and these small-molecule inhibitors may have some off-target effects which have to be considered when interpreting the data.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Supplementary Material
Acknowledgements
We thank Benjamin W. Van, Erin Y. Shen, Sonya Baidar, and Maura Ferrero for their help in animal care, cell isolation, and laboratory tasks. We also thank William T. Ferrier, Linda Talken, Lynette M. Mendoza, and Kenneth S. Ginsburg for their help in surgical procedures and echocardiographic follow-up of heart failure rabbits.
Conflict of interest: none declared.
Funding
This work was supported by grants from the National Institutes of Health (USA) R01-HL030077 (to D.M.B.), P01-HL141084 (to D.M.B.), R01-HL142282 (to D.M.B. and J.B.), and F32-HL144017 (to C.Y.K.).
Data availability
The data underlying this article are available in the article and in its online supplementary material.
Translational perspective
Cardiac arrhythmia mechanisms in diabetes are incompletely understood. Here we show that in healthy cardiomyocytes, hyperglycaemia alone only moderately increased cellular proarrhythmia via CaMKII activation. However, a second hit which impairs K+ channel function or increases neurohormonal tone markedly enhanced arrhythmogenic action potentials (increased action potential prolongation, alternans, short-term variability, and afterdepolarizations). Our results may (i) help to identify patients at risk, (ii) suggest tight control of blood glucose and K+ levels in patients with long QT and heart failure, and (iii) propose a combination therapy including glucose-lowering medications, angiotensin receptor blockers, and potentially CaMKII inhibitors to prevent arrhythmia.
References
- 1. Knudson AG Jr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA 1971;68:820–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Keating MT, Sanguinetti MC.. Molecular and cellular mechanisms of cardiac arrhythmias. Cell 2001;104:569–580. [DOI] [PubMed] [Google Scholar]
- 3. Bezzina CR, Lahrouchi N, Priori SG.. Genetics of sudden cardiac death. Circ Res 2015;116:1919–1936. [DOI] [PubMed] [Google Scholar]
- 4. Cosentino F, Grant PJ, Aboyans V, Bailey CJ, Ceriello A, Delgado V, Federici M, Filippatos G, Grobbee DE, Hansen TB, Huikuri HV, Johansson I, Jüni P, Lettino M, Marx N, Mellbin LG, Östgren CJ, Rocca B, Roffi M, Sattar N, Seferović PM, Sousa-Uva M, Valensi P, Wheeler DC; ESC Scientific Document Group. 2019 ESC Guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD. Eur Heart J 2020;41:255–323. [DOI] [PubMed] [Google Scholar]
- 5. Jouven X, Lemaitre RN, Rea TD, Sotoodehnia N, Empana JP, Siscovick DS.. Diabetes, glucose level, and risk of sudden cardiac death. Eur Heart J 2005;26:2142–2147. [DOI] [PubMed] [Google Scholar]
- 6. Kannel WB, McGee DL.. Diabetes and cardiovascular disease. The Framingham study. JAMA 1979;241:2035–2038. [DOI] [PubMed] [Google Scholar]
- 7. Marfella R, Nappo F, De Angelis L, Siniscalchi M, Rossi F, Giugliano D.. The effect of acute hyperglycaemia on QTc duration in healthy man. Diabetologia 2000;43:571–575. [DOI] [PubMed] [Google Scholar]
- 8. Tran HV, Gore JM, Darling CE, Ash AS, Kiefe CI, Goldberg RJ.. Hyperglycemia and risk of ventricular tachycardia among patients hospitalized with acute myocardial infarction. Cardiovasc Diabetol 2018;17:136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hegyi B, Bers DM, Bossuyt J.. CaMKII signaling in heart diseases: emerging role in diabetic cardiomyopathy. J Mol Cell Cardiol 2019;127:246–259. [DOI] [PubMed] [Google Scholar]
- 10. Erickson JR, Pereira L, Wang L, Han G, Ferguson A, Dao K, Copeland RJ, Despa F, Hart GW, Ripplinger CM, Bers DM.. Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature 2013;502:372–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hegyi B, Bányász T, Izu LT, Belardinelli L, Bers DM, Chen-Izu Y.. β-adrenergic regulation of late Na+ current during cardiac action potential is mediated by both PKA and CaMKII. J Mol Cell Cardiol 2018;123:168–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hegyi B, Bossuyt J, Ginsburg KS, Mendoza LM, Talken L, Ferrier WT, Pogwizd SM, Izu LT, Chen-Izu Y, Bers DM.. Altered repolarization reserve in failing rabbit ventricular myocytes: calcium and beta-adrenergic effects on delayed- and inward-rectifier potassium currents. Circ Arrhythm Electrophysiol 2018;11:e005852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hegyi B, Morotti S, Liu C, Ginsburg KS, Bossuyt J, Belardinelli L, Izu LT, Chen-Izu Y, Banyasz T, Grandi E, Bers DM.. Enhanced depolarization drive in failing rabbit ventricular myocytes: calcium-dependent and beta-adrenergic effects on late sodium, L-type calcium, and sodium-calcium exchange currents. Circ Arrhythm Electrophysiol 2019;12:e007061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hegyi B, Bossuyt J, Griffiths LG, Shimkunas R, Coulibaly Z, Jian Z, Grimsrud KN, Sondergaard CS, Ginsburg KS, Chiamvimonvat N, Belardinelli L, Varro A, Papp JG, Pollesello P, Levijoki J, Izu LT, Boyd WD, Banyasz T, Bers DM, Chen-Izu Y.. Complex electrophysiological remodeling in postinfarction ischemic heart failure. Proc Natl Acad Sci USA 2018;115:E3036–E3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Coppini R, Ferrantini C, Yao L, Fan P, Del Lungo M, Stillitano F, Sartiani L, Tosi B, Suffredini S, Tesi C, Yacoub M, Olivotto I, Belardinelli L, Poggesi C, Cerbai E, Mugelli A.. Late sodium current inhibition reverses electromechanical dysfunction in human hypertrophic cardiomyopathy. Circulation 2013;127:575–584. [DOI] [PubMed] [Google Scholar]
- 16. Sikkel MB, Francis DP, Howard J, Gordon F, Rowlands C, Peters NS, Lyon AR, Harding SE, MacLeod KT.. Hierarchical statistical techniques are necessary to draw reliable conclusions from analysis of isolated cardiomyocyte studies. Cardiovasc Res 2017;113:1743–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pogwizd SM, Bers DM.. Rabbit models of heart disease. Drug Discov Today Dis Models 2008;5:185–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Weiss JN, Qu Z, Shivkumar K.. Electrophysiology of hypokalemia and hyperkalemia. Circ Arrhythm Electrophysiol 2017;10:e004667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wagner S, Dybkova N, Rasenack EC, Jacobshagen C, Fabritz L, Kirchhof P, Maier SK, Zhang T, Hasenfuss G, Brown JH, Bers DM, Maier LS.. Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J Clin Invest 2006;116:3127–3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jia G, Hill MA, Sowers JR.. Diabetic cardiomyopathy: an update of mechanisms contributing to this clinical entity. Circ Res 2018;122:624–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wagner S, Hacker E, Grandi E, Weber SL, Dybkova N, Sossalla S, Sowa T, Fabritz L, Kirchhof P, Bers DM, Maier LS.. Ca/calmodulin kinase II differentially modulates potassium currents. Circ Arrhythm Electrophysiol 2009;2:285–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lengyel C, Virag L, Biro T, Jost N, Magyar J, Biliczki P, Kocsis E, Skoumal R, Nanasi PP, Toth M, Kecskemeti V, Papp JG, Varro A.. Diabetes mellitus attenuates the repolarization reserve in mammalian heart. Cardiovasc Res 2007;73:512–520. [DOI] [PubMed] [Google Scholar]
- 23. Stables CL, Musa H, Mitra A, Bhushal S, Deo M, Guerrero-Serna G, Mironov S, Zarzoso M, Vikstrom KL, Cawthorn W, Pandit SV.. Reduced Na+ current density underlies impaired propagation in the diabetic rabbit ventricle. J Mol Cell Cardiol 2014;69:24–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lu Z, Jiang YP, Wu CY, Ballou LM, Liu S, Carpenter ES, Rosen MR, Cohen IS, Lin RZ.. Increased persistent sodium current due to decreased PI3K signaling contributes to QT prolongation in the diabetic heart. Diabetes 2013;62:4257–4265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sommese L, Valverde CA, Blanco P, Castro MC, Rueda OV, Kaetzel M, Dedman J, Anderson ME, Mattiazzi A, Palomeque J.. Ryanodine receptor phosphorylation by CaMKII promotes spontaneous Ca(2+) release events in a rodent model of early stage diabetes: the arrhythmogenic substrate. Int J Cardiol 2016;202:394–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Federico M, Portiansky EL, Sommese L, Alvarado FJ, Blanco PG, Zanuzzi CN, Dedman J, Kaetzel M, Wehrens XHT, Mattiazzi A, Palomeque J.. Calcium-calmodulin-dependent protein kinase mediates the intracellular signalling pathways of cardiac apoptosis in mice with impaired glucose tolerance. J Physiol 2017;595:4089–4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Popescu I, Yin G, Velmurugan S, Erickson JR, Despa F, Despa S.. Lower sarcoplasmic reticulum Ca2+ threshold for triggering afterdepolarizations in diabetic rat hearts. Heart Rhythm 2019;16:765–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hegyi B, Chen-Izu Y, Izu LT, Rajamani S, Belardinelli L, Bers DM, Bányász T.. Balance between rapid delayed rectifier K+ current and late Na+ current on ventricular repolarization: an effective antiarrhythmic target? Circ Arrhythm Electrophysiol 2020;13:e008130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Timmis AD, Chaitman BR, Crager M.. Effects of ranolazine on exercise tolerance and HbA1c in patients with chronic angina and diabetes. Eur Heart J 2006;27:42–48. [DOI] [PubMed] [Google Scholar]
- 30. Hegyi B, Borst JM, Bailey LRJ, Shen EY, Lucena AJ, Navedo MF, Bossuyt J, Bers DM.. Hyperglycemia regulates cardiac K+ channels via O-GlcNAc-CaMKII and NOX2-ROS-PKC pathways. Basic Res Cardiol 2020;115:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Magyar J, Rusznak Z, Szentesi P, Szucs G, Kovacs L.. Action potentials and potassium currents in rat ventricular muscle during experimental diabetes. J Mol Cell Cardiol 1992;24:841–853. [DOI] [PubMed] [Google Scholar]
- 32. Shimoni Y, Firek L, Severson D, Giles W.. Short-term diabetes alters K+ currents in rat ventricular myocytes. Circ Res 1994;74:620–628. [DOI] [PubMed] [Google Scholar]
- 33. Zhang Y, Xiao J, Wang H, Luo X, Wang J, Villeneuve LR, Zhang H, Bai Y, Yang B, Wang Z.. Restoring depressed HERG K+ channel function as a mechanism for insulin treatment of abnormal QT prolongation and associated arrhythmias in diabetic rabbits. Am J Physiol Heart Circ Physiol 2006;291:H1446–H1455. [DOI] [PubMed] [Google Scholar]
- 34. Lengyel C, Virag L, Kovacs PP, Kristof A, Pacher P, Kocsis E, Koltay ZM, Nanasi PP, Toth M, Kecskemeti V, Papp JG, Varro A, Jost N.. Role of slow delayed rectifier K+-current in QT prolongation in the alloxan-induced diabetic rabbit heart. Acta Physiol (Oxf )2008;192:359–368. [DOI] [PubMed] [Google Scholar]
- 35. Lopez-Izquierdo A, Pereira RO, Wende AR, Punske BB, Abel ED, Tristani-Firouzi M.. The absence of insulin signaling in the heart induces changes in potassium channel expression and ventricular repolarization. Am J Physiol Heart Circ Physiol 2014;306:H747–H754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Polina I, Jansen HJ, Li T, Moghtadaei M, Bohne LJ, Liu Y, Krishnaswamy P, Egom EE, Belke DD, Rafferty SA, Ezeani M, Gillis AM, Rose RA.. Loss of insulin signaling may contribute to atrial fibrillation and atrial electrical remodeling in type 1 diabetes. Proc Natl Acad Sci USA 2020;117:7990–8000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jovanovic S, Jovanovic A.. High glucose regulates the activity of cardiac sarcolemmal ATP-sensitive K+ channels via 1,3-bisphosphoglycerate: a novel link between cardiac membrane excitability and glucose metabolism. Diabetes 2005;54:383–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wehrens XH, Lehnart SE, Reiken SR, Marks AR.. Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ Res 2004;94:e61–e70. [DOI] [PubMed] [Google Scholar]
- 39. Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM.. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res 2005;97:1314–1322. [DOI] [PubMed] [Google Scholar]
- 40. Pereira L, Ruiz-Hurtado G, Rueda A, Mercadier J-J, Benitah J-P, Gómez AM.. Calcium signaling in diabetic cardiomyocytes. Cell Calcium 2014;56:372–380. [DOI] [PubMed] [Google Scholar]
- 41. Orosz A, Baczko I, Nyiraty S, Korei AE, Putz Z, Takacs R, Nemes A, Varkonyi TT, Balogh L, Abraham G, Kempler P, Papp JG, Varro A, Lengyel C.. Increased short-term beat-to-beat QT interval variability in patients with impaired glucose tolerance. Front Endocrinol (Lausanne) 2017;8:129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chiamvimonvat N, Chen-Izu Y, Clancy CE, Deschenes I, Dobrev D, Heijman J, Izu L, Qu Z, Ripplinger CM, Vandenberg JI, Weiss JN, Koren G, Banyasz T, Grandi E, Sanguinetti MC, Bers DM, Nerbonne JM.. Potassium currents in the heart: functional roles in repolarization, arrhythmia and therapeutics. J Physiol 2017;595:2229–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jost N, Virág L, Bitay M, Takács J, Lengyel C, Biliczki P, Nagy Z, Bogáts G, Lathrop DA, Papp JG, Varró A, Restricting excessive cardiac action potential and QT prolongation: a vital role for IKs in human ventricular muscle. Circulation 2005;112:1392–1399. [DOI] [PubMed] [Google Scholar]
- 44. Lee YS, Chang PC, Hsueh CH, Maruyama M, Park HW, Rhee KS, Hsieh YC, Shen C, Weiss JN, Chen Z, Lin SF, Chen PS.. Apamin-sensitive calcium-activated potassium currents in rabbit ventricles with chronic myocardial infarction. J Cardiovasc Electrophysiol 2013;24:1144–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lim HS, MacFadyen RJ, Lip GY.. Diabetes mellitus, the renin-angiotensin-aldosterone system, and the heart. Arch Intern Med 2004;164:1737–1748. [DOI] [PubMed] [Google Scholar]
- 46. Oda T, Yang Y, Uchinoumi H, Thomas DD, Chen-Izu Y, Kato T, Yamamoto T, Yano M, Cornea RL, Bers DM.. Oxidation of ryanodine receptor (RyR) and calmodulin enhance Ca release and pathologically alter, RyR structure and calmodulin affinity. J Mol Cell Cardiol 2015;85:240–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Erickson JR, Joiner ML, Guan X, Kutschke W, Yang J, Oddis CV, Bartlett RK, Lowe JS, O'Donnell SE, Aykin-Burns N, Zimmerman MC, Zimmerman K, Ham AJ, Weiss RM, Spitz DR, Shea MA, Colbran RJ, Mohler PJ, Anderson ME.. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 2008;133:462–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Shimoni Y, Hunt D, Chuang M, Chen KY, Kargacin G, Severson DL.. Modulation of potassium currents by angiotensin and oxidative stress in cardiac cells from the diabetic rat. J Physiol 2005;567:177–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kenny HC, Abel ED.. Heart failure in type 2 diabetes mellitus. Circ Res 2019;124:121–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Huxley RR, Alonso A, Lopez FL, Filion KB, Agarwal SK, Loehr LR, Soliman EZ, Pankow JS, Selvin E.. Type 2 diabetes, glucose homeostasis and incident atrial fibrillation: the Atherosclerosis Risk in Communities study. Heart 2012;98:133–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nattel S, Heijman J, Zhou L, Dobrev D.. Molecular basis of atrial fibrillation pathophysiology and therapy: a translational perspective. Circ Res 2020;127:51–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ravens U, Cerbai E.. Role of potassium currents in cardiac arrhythmias. Europace 2008;10:1133–1137. [DOI] [PubMed] [Google Scholar]
- 53. Ferdinandy P, Baczko I, Bencsik P, Giricz Z, Gorbe A, Pacher P, Varga ZV, Varro A, Schulz R.. Definition of hidden drug cardiotoxicity: paradigm change in cardiac safety testing and its clinical implications. Eur Heart J 2019;40:1771–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article are available in the article and in its online supplementary material.