

Abstract
Background
In population genomics, polymorphisms that are highly differentiated between geographically separated populations are often suggestive of Darwinian positive selection. Genomic scans have highlighted several such regions in African and non-African populations, but only a handful of these have functional data that clearly associates candidate variations driving the selection process. Fine-Mapping of Adaptive Variation (FineMAV) was developed to address this in a high-throughput manner using population based whole-genome sequences generated by the 1000 Genomes Project. It pinpoints positively selected genetic variants in sequencing data by prioritizing high frequency, population-specific and functional derived alleles.
Results
We developed a stand-alone software that implements the FineMAV statistic. To graphically visualise the FineMAV scores, it outputs the statistics as bigWig files, which is a common file format supported by many genome browsers. It is available as a command-line and graphical user interface. The software was tested by replicating the FineMAV scores obtained using 1000 Genomes Project African, European, East and South Asian populations and subsequently applied to whole-genome sequencing datasets from Singapore and China to highlight population specific variants that can be subsequently modelled. The software tool is publicly available at https://github.com/fadilla-wahyudi/finemav.
Conclusions
The software tool described here determines genome-wide FineMAV scores, using low or high-coverage whole-genome sequencing datasets, that can be used to prioritize a list of population specific, highly differentiated candidate variants for in vitro or in vivo functional screens. The tool displays these scores on the human genome browsers for easy visualisation, annotation and comparison between different genomic regions in worldwide human populations.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12859-021-04506-9.
Keywords: Adaption, Evolutionary genomics, Human evolution, Population differentiation, Selective sweep
Background
Human whole-genome sequencing projects have contributed to the advancement of population genomics, specifically the unbiased detection of positive selection in human populations. In comparison to genotyping, sequencing mitigates ascertainment bias and captures greater genomic variation [1] making it suitable for selection scans. The rise in population-based sequencing initiatives has garnered an interest in the study of positive selection, because identifying genetic variants that are positively selected can provide insight into new molecular functions that come with adaptation. These selective scans have provided vast lists of genes and variants and except for a few classical examples [2], it has been difficult to identify potentially functional variants that should be followed up in vitro or in vivo models.
Fine-Mapping of Adaptation Variation (FineMAV) is a statistical method that prioritizes functional SNP candidates under selection and depends upon population differentiation [3]. It pinpoints candidate positively selected variants at putative loci in a high-throughput manner, thus, enabling the modelling of such variants in vitro or in vivo [3]. FineMAV was developed to overcome a challenge that existing positive selection statistical methods face in that they are unable to distinguish between neutral, hitchhiked variants and true positively selected variants [3]. FineMAV does this by incorporating methods that detect regions showing signatures of positive selection (population differentiation and high frequency of derived alleles) and subject these regions to functional annotation under the assumption that it is unlikely for a deleterious or functional variant to reach high frequency in a given randomly mating population unless it confers some sort of functional advantage [3].
To measure population differentiation, FineMAV employs a derived allele purity (DAP) equation to describe the disparate spread of derived alleles across populations [3]. The derived allele frequency (DAF) equation is used to determine sites with high frequency of derived alleles [3]. To annotate functionality, FineMAV uses the Combined Annotation-Dependent Depletion (CADD) method which takes into account multiple variant annotations and condenses it into a single score called the C score [4]. The C scores predict whether a SNP or indel in the human genome is functional, deleterious or pathogenic [4]. The phred-scaled C scores (CADD_PHRED) are expressed as rankings relative to all possible substitutions of the human genome and range from 1 to 99 [4]. For example, a variant that scores more than 10 would be within the top 10% of potentially deleterious substitutions. A score of 20 would indicate the top 1% and 30 would be 0.1% and so on [4]. Incorporating CADD scores can, therefore, differentiate between neutral alleles, which are predicted as non-deleterious, and true positively selected alleles, which are predicted as effectively functional or deleterious [3]. In this article, we introduce a stand-alone application that can perform FineMAV calculations on whole-genome sequencing data and can output bigWig files which can be used to graphically visualise the scores on genome browsers. We test the software using the 1000 Genomes Project phase 3 dataset [5] and whole-genome sequencing datasets from Singapore and China [6–8].
Implementation
Pipeline
The Python-based FineMAV software works with high-throughput, massively parallel, sequencing data and relies on the information that can be extracted from variant call format (VCF) files (version 4.2 and above) (Table 1). The Python script can be found in Additional file 1. The pipeline for the software is illustrated in Fig. 1. We recommend users to use jointly-called, multi-sample genomic VCF (gVCF) as it reports every site in the genome regardless of whether they carry variation or not. This is preferable for FineMAV analysis as it can distinguish between sites that are homozygous for the reference allele and those with missing data.
Table 1.
Information needed for the tab-delimited input files
| Information needed from the VCF file | Description | Mandatory VCF column |
|---|---|---|
| CHROM:POS | Chromosome number:position | Yes |
| ID | Identifier | Yes |
| REF | Reference base | Yes |
| ALT | Alternative base | Yes |
| AA | Ancestral allele | No |
| CADD_PHRED | Phred-scaled Combined Annotation Dependent Depletion (CADD) score | No |
| AF | Allele frequency (AF) for the alternative base. The AF should be reported for each population | No |
This information can be extracted from the VCF file and provided in a tab-delimited format for the software to calculate the FineMAV scores
Fig. 1.

Pipeline for calculating the genome-wide FineMAV scores. The boxed region highlighted in grey are the parts of the workflow that are automated by the software. The intermediate output files are deleted when the pipeline is complete. AA: ancestral allele, CADD_PHRED: phred-scaled Combined Annotation Dependent Depletion scores, VEP: Variant Effect Predictor, AF: allele frequency of the alternative allele
Input files and dataset
The FineMAV software requires the user to provide the input data from the VCF file in a tab-delimited file format (Table 1). We recommend extracting the information utilising the BCFtools [9] query command. Some of the information mentioned in Table 1 can be found in the INFO column and are not mandatory in VCF files.
In instances where the allele frequency (AF) for each population is not annotated in the INFO column, we suggest using the BCFtools fill-tags plugin to determine the AF first, and then piping it to the BCFtools query command for extraction. If the ancestral allele and/or the CADD_PHRED are not available in the VCF file, the software allows the user to supplement this information using the Ensembl Variant Effect Predictor (VEP) [10]. The output file must be tab-delimited and the first column must be the “Location”, which indicates the position of the variant using the standard coordinate format (i.e. chromosome:start).
We tested our implementation code by generating genome-wide FineMAV scores using the 1000 Genomes Project low coverage whole-genome sequences from African, European, East and South Asian continental populations to replicate the published results [3]. Subsequently, we used the pipeline to calculate FineMAV scores for high-coverage whole-genome sequencing datasets generated by the Singapore Sequencing Indian Project (SSIP) [6], the Singapore Sequencing Malay Project (SSMP) [7] and a dataset of 90 Han Chinese individuals (90HC), that included 83 samples from the 1000 Genomes Project [8].
Calculating the genome-wide FineMAV score
The FineMAV score was estimated for genome-wide single nucleotide polymorphisms (SNPs) as only the ancestral allele could be unambiguously determined only for this class of variations. The derived allele for each SNP was calculated by multiplying three metrics: DAP, DAF and the CADD_PHRED score [3]. DAP was calculated for each variant using the following equations: , and respectively, where is the number of populations, is the derived allele count in one population where and is the penalty parameter used to penalize allele sharing between the populations [3]. The aforementioned DAP equation is used when the population sizes are equal. In this software, the DAP is calculated using the derived allele frequency instead of counts in order to take into account population sizes that might be different.
The penalty parameter was determined empirically by Szpak et al. [3] for varying number of populations, ranging from 2 to 7 (Table 2). However, should the user intend to analyse more than 7 populations or decide on another value for , they are able to change it.
Table 2.
Recommended minimal value of the penalty parameter (), rounded off to two decimal places, for a given as determined by Szpak et al. [3]
| Number of populations () | Penalty parameter () |
|---|---|
| 2 | 4.96 |
| 3 | 3.50 |
| 4 | 2.98 |
| 5 | 2.71 |
| 6 | 2.53 |
| 7 | 2.41 |
FineMAV calculations are done by splitting the file(s) into smaller chunks to optimise the random access memory (RAM) usage (Fig. 2A). The default size of the chunk is 200,000 lines. However, the user can specify the chunk size they require. We also tested the performance of the chunk size option using 66,236,516 biallelic SNPs across four population groups. Computational experiments were run on Ubuntu 16.04 LTS with a 3.60 GHz 8-core Intel Core i7-4790 processor with 31.3 GB RAM and 950.6 GB of hard disk memory. The size of the input file, which contains the data extracted from the VCF file, and the VEP-generated file was 2.0 GB and 2.1 GB respectively. Figure 2B illustrates the maximum RAM usage and the time taken when different chunk sizes are utilised. As expected, the larger the chunk size, the faster the run time, up to a certain point. The optimal chunk size would vary depending on the size of the input files and the computing power.
Fig. 2.
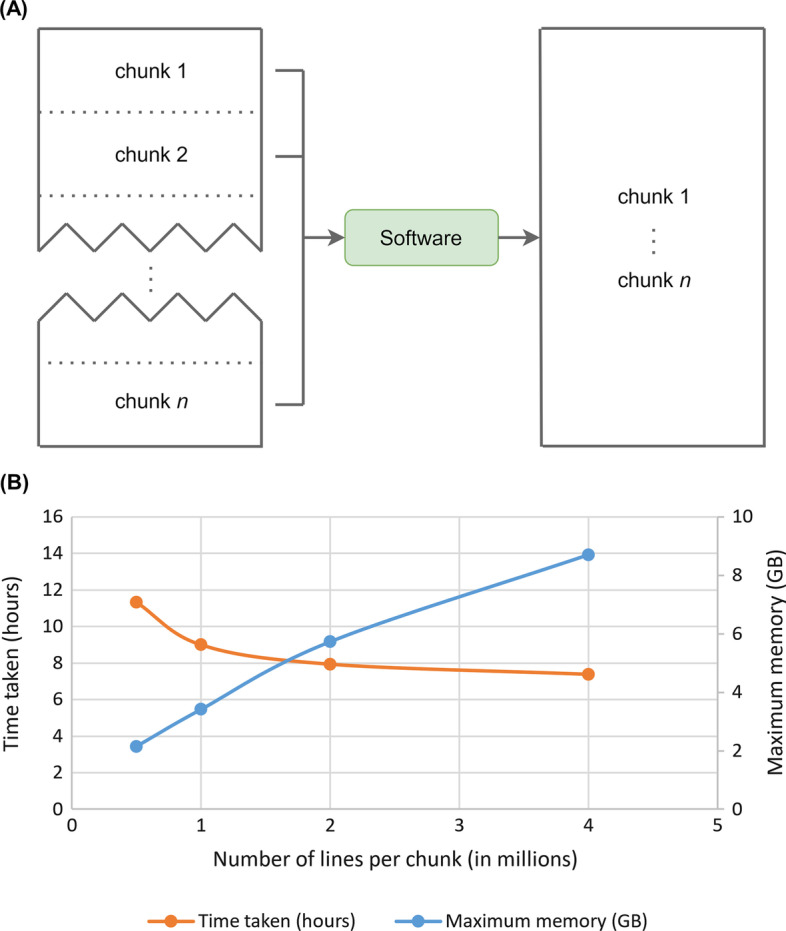
Utilising the chunk size option. A Diagram illustrating how the software separates the input files into chunks and iterates through them when performing the FineMAV calculations. It proceeds to merge them into one output file. B Utilising the chunk size option. A graph that compares the time taken and the maximum random access memory (RAM) when different chunk sizes for a dataset of 66,236,516 biallelic SNPs is used
Generating the output files
The software produces three kinds of output files: a log file, a tab-delimited file containing the genome-wide FineMAV scores along with the intermediate calculations and a bigWig file. The bigWig [11] is a common file format used for graphical visualisation on downloadable and online genome browsers such as Ensembl or the UCSC genome browsers [12, 13].
Results and discussion
We initially tested our implementation code by generating genome-wide FineMAV scores using the 1000 Genomes Project African, European, East and South Asian continental populations [5] and obtained FineMAV scores that were significantly correlated with the published data (Spearman’s correlations ≥ 0.9999 and the p-values < 0.00001) for all four continental populations [3]. When comparing the top 100 FineMAV outliers across all four continental populations with the published data, only 5/300 variants did not overlap with the published results and all five of these variants were missing, because they did not pass our data filtering criteria.
Next, we used the pipeline to calculate FineMAV scores for three high-coverage whole-genome sequencing datasets: the SSIP [6], SSMP [7] and 90HC [8]. These datasets were mapped to the GRCh37/hg19 reference genome. The VCF files for the autosomal and the X chromosome were merged and filtered to select high-quality biallelic sites that were variable in all three populations. This resulted in a final VCF file containing 5,748,704 SNPs in which the CHROM:POS, ID, REF, ALT and AF for each population were retrieved and stored in a tab-delimited file. As the VCF files did not contain the CADD_PHRED nor the ancestral alleles, a separate tab-delimited file containing this information for these SNPs was generated by leveraging the Ensembl VEP’s plugins. For CADD_PHRED annotation, we used the CADD version (v1.4) for the reference genome GRCh37/hg19 (https://krishna.gs.washington.edu/download/CADD/v1.4/GRCh37/whole_genome_SNVs.tsv.gz) [4, 14] The FASTA files containing the ancestral sequences were downloaded from the Ensembl webpage using the following URL: ftp://ftp.ensembl.org/pub/release75/fasta/ancestral_alleles/homo_sapiens_ancestor_GRCh37_e71.tar.bz2 [15, 16]. These two files were then fed to the software to produce bigWig files which can be visualised in genome browsers as presented in Fig. 3.
Fig. 3.

Annotated screenshot of the bigWig files of the genome-wide FineMAV scores. A FineMAV scores for Han Chinese (90HC, orange), Singaporean Indian (SSIP, blue) and Singaporean Malay (SSMP, grey) populations displayed on the Integrative Genomics Viewer (IGV). The genomic regions on display are the autosomal and the X chromosomes and the horizontal line depicts the 99th percentile. B A multi-locus view of two regions where the left panel displays a locus with a well-known positively selected missense variant in EDAR (rs3827760) in East Asians that also stands out in the SSMP population. The right panel displays a novel locus with two high scoring variants in SSIP: rs151233, a synonymous variant in APOBR and rs151234, an intronic variant in CLN3 that stand out in the SSIP
The FineMAV statistic was able to replicate known positively selected variants as well as pinpoint novel ones from the three populations (Table 3, Additional file 2). Examples of known variants included the derived alleles for rs3827760 (NC_000002.12:g.108897145A > G, ENSP00000258443.2:p.Val370Gly) in ectodysplasin A receptor (EDAR) and rs2293766 (NC_000007.13:g.100371358G > A, ENSP00000423579.1:p.Trp1883Ter) in zonadhesin (ZAN). These variants have also been highlighted previously in several genomic scans for selection in East Asian populations [3, 17, 18] and in this study, they were high scoring in the 90HC and SSMP (Table 3). Several studies that have looked at the missense variant rs3827760 in EDAR have confirmed its pleiotropic effects. The non-synonymous mutation has been found to be associated with hair thickness [19–21], shovel-shaped incisors [22–24], ear morphology [25, 26], increased density of eccrine sweat glands, reduced mammary fat pad and increased mammary ductal gland branching [21] and despite extensive research, it still remains unclear as to why this allele is positively selected in the region. Some have theorised that increased sweat gland density resulted in better thermoregulation during warmer climates or that perhaps male sexual preference may have played a role in its selection [21]. Others hypothesise that selection for increased mammary gland branching would lead to better mother-to-child nutrient transfer, especially for vitamin D, to prevent vitamin D deficiency in regions with lower ultraviolet (UV) levels [27]. ZAN encodes an acrosomal protein in the sperm called zonadhesin. A study employing Zan knockout mice found that their sperms remained fertile and had increased adhesion to the jelly-like coating of the egg (zona pellucida) of other species like pig, cow and rabbit [28]. As ZAN is responsible for species-specific binding, it can be speculated that a truncation, as a result of the nonsense mutation in rs2293766, could have mediated interbreeding between archaic humans and modern humans in Asia [29].
Table 3.
Top 10 FineMAV candidates from the Han Chinese (90HC), Singaporean Indian (SSIP) and Singaporean Malay (SSMP) populations
| Chr | Position a | SNP ID | Gene | Consequence b | DAF 90HC | DAF SSIP | DAF SSMP | FineMAV | Known or novel |
|---|---|---|---|---|---|---|---|---|---|
| 90HC | |||||||||
| 2 | 109513601 | rs3827760:A > G | EDAR | Missense (p.Val370Ala) | 0.922 | 0.029 | 0.490 | 4.661 | Known [3, 17, 18] |
| 5 | 176099727 | rs13186794:A > G | – | Intergenic | 0.494 | 0.057 | 0.047 | 4.114 | Novel |
| 5 | 176099728 | rs13186795:A > G | – | Intergenic | 0.494 | 0.057 | 0.057 | 4.096 | Novel |
| 4 | 31442427 | rs56345433:G > A | – | Intergenic | 0.528 | 0.086 | 0.021 | 3.211 | Novel |
| 3 | 98031307 | rs2316271:T > A | OR5H8 | Stop gained (p.Leu184Ter) | 0.767 | 0.314 | 0.599 | 3.102 | Novel |
| 16 | 31088347 | rs749671:G > A | ZNF646 | Synonymous (p.Glu234 =) | 0.906 | 0.043 | 0.776 | 3.053 | Known [18] |
| 5 | 76129053 | rs631465:T > C | F2RL1 | Synonymous (p.Ile207 =) | 0.522 | 0.014 | 0.208 | 3.008 | Novel |
| 2 | 109451118 | rs72627476:A > G | CCDC138 | Intronic | 0.917 | 0.029 | 0.484 | 2.961 | Known [3] |
| 12 | 132106717 | rs10794470:T > C | AC117500.3 | Intronic | 0.272 | 0.000 | 0.005 | 2.940 | Novel |
| 7 | 14587199 | rs10236893:G > A | DGKB | Intronic | 0.417 | 0.029 | 0.120 | 2.895 | Novel |
| SSIP | |||||||||
| 16 | 28506428 | rs151233:C > T | APOBR | Synonymous (p.Leu22 =) | 0.006 | 0.571 | 0.026 | 7.677 | Novel |
| 16 | 30936081 | rs35675346:G > A | FBXL19 | Missense (p.Glu10Lys) | 0.061 | 0.800 | 0.188 | 7.213 | Known [18, 43] |
| 16 | 28505660 | rs151234:G > C | CLN3 | Intronic | 0.006 | 0.571 | 0.031 | 6.839 | Novel |
| 16 | 31044683 | rs58726213:A > G | STX4 | Upstream gene | 0.089 | 0.871 | 0.214 | 6.686 | Known [18, 43] |
| 15 | 64592833 | rs114713921:T > C | CSNK1G1 | 5 prime UTR | 0.006 | 0.486 | 0.036 | 6.341 | Novel |
| 16 | 30666367 | rs3747481:C > T | PRR14 | Missense (p.Pro359Leu) | 0.100 | 0.857 | 0.245 | 6.090 | Known [18] |
| 19 | 49206674 | rs601338:G > A | FUT2 | Stop gained (p.Trp154Ter) | 0.011 | 0.186 | 0.016 | 6.033 | Known [44] |
| 15 | 91452595 | rs2106673:A > G | MAN2A2 | Missense (p.Gln412Arg) | 0.017 | 0.514 | 0.063 | 5.746 | Novel |
| 10 | 17407147 | rs729170:G > T | ST8SIA6 | Intronic | 0.006 | 0.343 | 0.005 | 5.736 | Novel |
| 15 | 64653984 | rs8026043:G > T | PCLAF | Downstream gene | 0.006 | 0.486 | 0.036 | 5.726 | Novel |
| SSMP | |||||||||
| 2 | 98272491 | rs2290123:A > G | ACTR1B | 3 prime UTR | 0.033 | 0.029 | 0.380 | 3.378 | Known [18] |
| 2 | 97613974 | rs114979404:C > G | FAM178B | Intronic | 0.022 | 0.029 | 0.375 | 2.806 | Known [18] |
| 17 | 2238152 | rs79597880:T > C | TSR1 | Missense (p.Lys199Glu) | 0.089 | 0.014 | 0.297 | 2.747 | Novel |
| 16 | 31088347 | rs749671:G > A | ZNF646 | Synonymous (p.Glu234 =) | 0.906 | 0.043 | 0.776 | 2.616 | Known [18] |
| 7 | 100371358 | rs2293766:G > A | ZAN | Stop gained (p.Trp1883Ter) | 0.528 | 0.257 | 0.557 | 2.531 | Known [3, 45] |
| 2 | 109513601 | rs3827760:A > G | EDAR | Missense (p.Val370Ala) | 0.922 | 0.029 | 0.490 | 2.474 | Known [3, 17, 18] |
| 3 | 98031307 | rs2316271:T > A | OR5H8 | Stop gained (p.Leu184Ter) | 0.767 | 0.314 | 0.599 | 2.424 | Novel |
| 11 | 62848487 | rs11231341:A > C | SLC22A24 | Stop gained (p.Tyr501Ter) | 0.867 | 0.757 | 0.792 | 2.421 | Novel |
| 12 | 57865558 | rs2229300:G > T | GLI1 | Missense (p.Gly1012Val) | 0.050 | 0.014 | 0.224 | 2.402 | Novel |
| 16 | 31075175 | rs2303223:G > A | ZNF668 | Synonymous (p.Gly225 =) | 0.911 | 0.043 | 0.781 | 2.290 | Known [3] |
aThe genomic position according to the GRCh37/hg19 reference genome
bThe most severe variant consequence according to Ensembl
Chr: chromosome, DAF: derived allele frequency, UTR: untranslated region
As seen in Fig. 3, the Singaporean Indian population have more population-specific signals than the Han Chinese and Singaporean Malay populations. This is because the Han Chinese and Singaporean Malays are genetically more closely related to each other than the Singaporean Indian population [18], and FineMAV penalizes allele sharing between populations and highlights high frequency population-specific mutations. Some of the highest-scoring SNPs observed in SSIP are located in chromosome 16 (Fig. 3, Table 3). We suspected that this could be due to the effect of genetic hitchhiking, driven by the selection of rs201075024 (NC_000016.9:g.31099000C > T, ENSP00000280606.6:p.Gly34Ser) (PRSS53), a SNP that has been reported to be positively selected in South Asian populations and was missing in our dataset [3]. PRSS53 encodes a serine protease and is expressed in hair follicles [30] and rs201075024 lies 10 base pairs away from rs11150606 (NC_000016.9:g.31099011 T > C, ENSP00000280606.6:p.Gln30Arg), another SNP in the same gene that is positively selected in East Asians [3, 30] and has been associated with hair shape in Latin Americans [30]. The functional effects of the rs201075024 missense mutation on the serine protease is still unknown. However, based on previous publications, it can be hypothesised that this variant also influences hair shape in South Asians. rs201075024 (PRSS53) was excluded from this selection scan because it was not polymorphic in all three datasets. Within the top 10 FineMAV candidates in SSIP, three SNPs are in linkage disequilibrium with rs201075024 (PRSS53): rs35675346 (NC_000016.9:g.30936081G > A, ENSP00000369666.2:p.Glu10Lys) (FBXL19), rs58726213 (NC_000016.9:g.31044683A > G) (STX4) and rs3747481 (NC_000016.9:g.30666367C > T, ENSP00000300835.4:p.Pro359Leu) (PRR14), with r2 values of 0.45, 0.26 and 0.16, respectively. This suggests that the three SNPs may be neutral and tagging the PRSS53 rs201075024 variant. Interestingly, according to the Genotype-Tissue Expression (GTEx) database (V8 release), these SNPs were reported as expression and splicing quantitative trait loci (eQTL/sQTL) for PRSS53 in various tissues [31].
Examples of novel SNPs that were identified in this study included two missense mutations: rs79597880 (NC_000017.10:g.2238152 T > C, ENSP00000301364.4:p.Lys199Glu) in the pre-rRNA-processing protein TSR1 homolog (TSR1) and rs2229300 (NC_000012.11:g.57865558G > T, ENSP00000228682.2:p.Gly1012Val) in glioma-associated oncogene family zinc finger 1 (GLI1) in the Singaporean Malay population (Table 3). So far, the effects of these SNPs on their respective proteins are unknown. The exact function of TSR1 is yet to be elucidated, but it plays a role in ribosome maturation [32]. It was reported that several rare (minor allele frequency < 1%) mutations of this gene, including missense mutations, may be associated with spontaneous coronary artery dissection (SCAD), a condition where the coronary artery tears resulting in two lumens: the true lumen and a false one [33]. However, there are no functional studies to confirm this association. GLI1, on the other hand, is a well-established oncogene and its protein is a drug target for several anti-cancer medications [34]. According to the Catalogue of Somatic Mutations in Cancer (COSMIC), 65.55% of mutations that are observed in GLI1 are missense substitutions [35]. The missense mutation rs2229300 is listed as an entry in COSMIC (COSV57366104) and was found in 14 tissue samples: 12 in the prostate [36], one in the large intestine and one in the lung [35].
It should be noted that admixture, or shared ancestry, can result in less population-specific signals [3]. This could mean that non-population-specific variants can be ranked high if the variants are highly deleterious and, therefore, have a high CADD_PHRED score. This is true for the derived allele in rs11231341 (NC_000011.9:g.62848487A > C, ENSP00000396586.1:p.Tyr501Ter) (SLC22A24) in which the global allele frequency is 0.75 [5] but was the 8th highest scoring allele in Singaporean Malays (Table 3), a highly admixed population.
Conclusions
We developed a user-friendly command line and graphical user interface (Fig. 4) platform to enable determination of genome-wide FineMAV scores using whole-genome sequencing datasets and to subsequently display these scores on genome browsers. This allows for easy, visual comparison between different genomic regions and human populations. It is designed to leverage on familiar bioinformatics tools and genome browsers, to be memory-efficient in anticipation of larger worldwide population sequencing datasets [37–42].
Fig. 4.
Screenshot of the FineMAV software as a graphical user interface
Availability and requirements
Project name: FineMAV.
Project home page: https://github.com/fadilla-wahyudi/finemav.
Operating systems: Linux.
Programming language: Python.
Other requirements: None.
License: MIT License.
Any restrictions to use by non-academics: None.
Supplementary Information
Additional file 1. Source code for the FineMAV program.
Additional file 2. Table containing the top 50 FineMAV candidates from the Han Chinese (90HC), Singaporean Indian (SSIP) and Singaporean Malay (SSMP) populations.
Acknowledgements
We would like to thank Chris Tyler-Smith and Yali Xue for their helpful comments. This research was supported by the School of Science, Monash University Malaysia and carried out as part of a M.Sc. project by FW.
Abbreviations
- 90HC
Whole-genome sequencing dataset of 90 Han Chinese individuals
- AF
Allele frequency
- CADD
Combined Annotation-Dependent Depletion
- CADD_PHRED
Phred-scaled C score
- DAF
Derived allele frequency
- DAP
Derived allele purity
- FineMAV
Fine-Mapping of Adaptive Variation
- gVCF
Genome variant call format
- RAM
Random access memory
- SNP
Single nucleotide polymorphism
- SSIP
Singapore Sequencing Indian Project
- SSMP
Singapore Sequencing Malay Project
- VCF
Variant call format
- VEP
Variant Effect Predictor
Authors' contributions
FW and JD designed the pipeline. MS conceived the statistical method. QA, JD and SR supervised this study. YT provided the Singapore datasets. FW implemented the pipeline, with assistance from FA, and wrote the draft. All authors read and approved the final manuscript.
Funding
FW was funded by a graduate research scholarship awarded by the School of Science, Monash University Malaysia.
Availability of data and materials
The software tool is publicly available on GitHub (https://github.com/fadilla-wahyudi/finemav). The datasets analysed during the current study are available from the following links: 1000 Genomes Project Phase 3 dataset (ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502/), Sequencing of 90 Han Chinese genomes (90HC) (https://www.ebi.ac.uk/ena/data/view/PRJEB20820), Singapore Sequencing Indian Project (SSIP) (https://blog.nus.edu.sg/sshsphphg/singapore-sequencing-indian/), and Singapore Sequencing Malay Project (SSMP) (https://blog.nus.edu.sg/sshsphphg/singapore-sequencing-malay/).
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Jasbir Dhaliwal, Email: jasbirkaur.dhaliwal@monash.edu.
Qasim Ayub, Email: qasim.ayub@monash.edu.
References
- 1.Lachance J, Tishkoff SA. SNP ascertainment bias in population genetic analyses: why it is important, and how to correct it. Ann Med Surg. 2013;35(9):780–786. doi: 10.1002/bies.201300014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Szpak M, Xue Y, Ayub Q, Tyler-Smith C. How well do we understand the basis of classic selective sweeps in humans? FEBS Lett. 2019;593(13):1431–1448. doi: 10.1002/1873-3468.13447. [DOI] [PubMed] [Google Scholar]
- 3.Szpak M, Mezzavilla M, Ayub Q, Chen Y, Xue Y, Tyler-Smith C. FineMAV: prioritizing candidate genetic variants driving local adaptations in human populations. Genome Biol. 2018;19(5):1–18. doi: 10.1186/s13059-017-1380-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.The 1000 Genomes Project Consortium A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wong LP, Lai JKH, Saw WY, Ong RTH, Cheng AY, Pillai NE, Liu X, Xu W, Chen P, Foo JN, et al. Insights into the genetic structure and diversity of 38 South Asian Indians from deep whole-genome sequencing. PLoS Genet. 2014;10(5):e1004377–e1004377. doi: 10.1371/journal.pgen.1004377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wong LP, Ong RTH, Poh WT, Liu X, Chen P, Li R, Lam KKY, Pillai NE, Sim KS, Xu H, et al. Deep whole-genome sequencing of 100 Southeast Asian Malays. Am J Hum Genet. 2013;92(1):52–66. doi: 10.1016/j.ajhg.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lan T, Lin H, Zhu W, Laurent TCAM, Yang M, Liu X, Wang J, Wang J, Yang H, Xu X, et al. Deep whole-genome sequencing of 90 Han Chinese genomes. GigaScience. 2017;6(9):1–7. doi: 10.1093/gigascience/gix067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. Genome project data processing S: the sequence alignment/map (SAM) format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McLaren W, Gil L, Hunt S, Riat HS, Ritchie G, Thormann A, Flicek P, Cunningham F. The ensembl variant effect predictor. Genome Biol. 2016;17(1):122. doi: 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kent WJ, Zweig AS, Barber G, Hinrichs AS, Karolchik D. BigWig and BigBed: enabling browsing of large distributed datasets. Bioinformatics. 2010;26(17):2204–2207. doi: 10.1093/bioinformatics/btq351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002;12(6):996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stalker J, Gibbins B, Meidl P, Smith J, Spooner W, Hotz H-R, Cox AV. The Ensembl Web site: mechanics of a genome browser. Genome Res. 2004;14(5):951–955. doi: 10.1101/gr.1863004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2018;47(D1):D886–D894. doi: 10.1093/nar/gky1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paten B, Herrero J, Beal K, Fitzgerald S, Birney E. Enredo and pecan: genome-wide mammalian consistency-based multiple alignment with paralogs. Genome Res. 2008;18(11):1814–1828. doi: 10.1101/gr.076554.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Paten B, Herrero J, Fitzgerald S, Beal K, Flicek P, Holmes I, Birney E. Genome-wide nucleotide-level mammalian ancestor reconstruction. Genome Res. 2008;18(11):1829–1843. doi: 10.1101/gr.076521.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sabeti PC, Varilly P, Fry B, Lohmueller J, Hostetter E, Cotsapas C, Xie X, Byrne EH, McCarroll SA, Gaudet R, et al. Genome-wide detection and characterization of positive selection in human populations. Nature. 2007;449(7164):913–918. doi: 10.1038/nature06250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu D, Dou J, Chai X, Bellis C, Wilm A, Shih CC, Soon WWJ, Bertin N, Lin CB, Khor CC, et al. Large-scale whole-genome sequencing of three diverse asian populations in Singapore. Cell. 2019;179(3):736–749.e715. doi: 10.1016/j.cell.2019.09.019. [DOI] [PubMed] [Google Scholar]
- 19.Fujimoto A, Kimura R, Ohashi J, Omi K, Yuliwulandari R, Batubara L, Mustofa MS, Samakkarn U, Settheetham-ishida W, Ishida T, et al. A scan for genetic determinants of human hair morphology: EDAR is associated with Asian hair thickness. Hum Mol Genet. 2007;17(6):835–843. doi: 10.1093/hmg/ddm355. [DOI] [PubMed] [Google Scholar]
- 20.Fujimoto A, Ohashi J, Nishida N, Miyagawa T, Morishita Y, Tsunoda T, Kimura R, Tokunaga K. A replication study confirmed the EDAR gene to be a major contributor to population differentiation regarding head hair thickness in Asia. Hum Genet. 2008;124(4):179–185. doi: 10.1007/s00439-008-0537-1. [DOI] [PubMed] [Google Scholar]
- 21.Kamberov GK, Wang S, Tan J, Gerbault P, Wark A, Tan L, Yang Y, Li S, Tang K, Chen H, et al. Modeling recent human evolution in mice by expression of a selected EDAR variant. Cell. 2013;152(4):691–702. doi: 10.1016/j.cell.2013.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kimura R, Yamaguchi T, Takeda M, Kondo O, Toma T, Haneji K, Hanihara T, Matsukusa H, Kawamura S, Maki K, et al. A common variation in EDAR is a genetic determinant of shovel-shaped incisors. Am J Hum Genet. 2009;85(4):528. doi: 10.1016/j.ajhg.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Park J, Yamaguchi T, Watanabe C, Kawaguchi A, Haneji K, Takeda M, Kim Y, Tomoyasu Y, Watanabe M, Oota H, et al. Effects of an Asian-specific nonsynonymous EDAR variant on multiple dental traits. J Hum Genet. 2012;57(8):508–514. doi: 10.1038/jhg.2012.60. [DOI] [PubMed] [Google Scholar]
- 24.Tan J, Peng Q, Li J, Guan Y, Zhang L, Jiao Y, Yang Y, Wang S, Jin L. Characteristics of dental morphology in the Xinjiang Uyghurs and correlation with the EDARV370A variant. Sci China Life Sci. 2014;57(5):510–518. doi: 10.1007/s11427-014-4654-x. [DOI] [PubMed] [Google Scholar]
- 25.Adhikari K, Reales G, Smith AJ, Konka E, Palmen J, Quinto-Sanchez M, Acuña-Alonzo V, Jaramillo C, Arias W, Fuentes M, et al. A genome-wide association study identifies multiple loci for variation in human ear morphology. Nat Commun. 2015;6(1):1–10. doi: 10.1038/ncomms8500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shaffer JR, Li J, Lee MK, Roosenboom J, Orlova E, Adhikari K, Agee M, Alipanahi B, Auton A, Bell RK, et al. Multiethnic GWAS reveals polygenic architecture of earlobe attachment. Am J Hum Genet. 2017;101(6):913–924. doi: 10.1016/j.ajhg.2017.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hlusko L, Carlson J, Chaplin G, Elias S, Hoffecker J, Huffman M, Jablonski N, Monson T, O’rourke D, Pilloud M, et al. Environmental selection during the last ice age on the mother-to-infant transmission of vitamin D and fatty acids through breast milk. Proc Natl Acad Sci USA. 2018;115(19):E4426. doi: 10.1073/pnas.1711788115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tardif S, Wilson MD, Wagner R, Hunt P, Gertsenstein M, Nagy A, Lobe C, Koop BF, Hardy DM. Zonadhesin is essential for species specificity of sperm adhesion to the egg zona pellucida. J Biol Chem. 2010;285(32):24863–24870. doi: 10.1074/jbc.M110.123125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Skoglund P, Jakobsson M. Archaic human ancestry in East Asia. Proc Natl Acad Sci USA. 2011;108(45):18301–18306. doi: 10.1073/pnas.1108181108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adhikari K, Fontanil T, Cal S, Mendoza-Revilla J, Fuentes-Guajardo M, Chacón-Duque J-C, Al-Saadi F, Johansson J, Quinto-Sanchez M, Acuña-Alonzo V, et al. A genome-wide association scan in admixed Latin Americans identifies loci influencing facial and scalp hair features. Nat Commun. 2016;7:10815. doi: 10.1038/ncomms10815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.The GTEx Consortium Genetic effects on gene expression across human tissues. Nature. 2017;550(7675):204–213. doi: 10.1038/nature24277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Urszula MM, Uma J, Vadim S, Zhuo Angel C, Juri R, David T, Atlanta GC. Pre-40S ribosome biogenesis factor Tsr1 is an inactive structural mimic of translational GTPases. Nat Commun. 2016;7(1):11789. doi: 10.1038/ncomms11789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun Y, Chen Y, Li Y, Li Z, Li C, Yu T, Xiao L, Yu B, Zhao H, Tao M, et al. Association of TSR1 variants and spontaneous coronary artery dissection. J Am Coll Cardiol. 2019;74(2):167–176. doi: 10.1016/j.jacc.2019.04.062. [DOI] [PubMed] [Google Scholar]
- 34.Palle K, Mani C, Tripathi K, Athar M. Aberrant GLI1 activation in DNA damage response, carcinogenesis and chemoresistance. Cancers. 2015;7(4):2330–2351. doi: 10.3390/cancers7040894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, Boutselakis H, Cole CG, Creatore C, Dawson E, et al. COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019;47(D1):D941–D947. doi: 10.1093/nar/gky1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lv S-D, Wang H-Y, Yu X-P, Zhai Q-L, Wu Y-B, Wei Q, Huang W-H. Integrative molecular characterization of Chinese prostate cancer specimens. Asian J Androl. 2020;22(2):162–168. doi: 10.4103/aja.aja_36_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koch L. Exploring human genomic diversity with gnomAD. Nat Rev Genet. 2020;21(8):448. doi: 10.1038/s41576-020-0255-7. [DOI] [PubMed] [Google Scholar]
- 38.Rajagopal D. India to launch its 1st human genome cataloguing project. The Economic Times. 2019.
- 39.Cheung E. Tens of thousands in Hong Kong to have their DNA sequenced in government-backed genome project. South China Morning Post. 2019.
- 40.Lévy Y. Genomic medicine 2025: France in the race for precision medicine. The Lancet. 2016;388(10062):2872. doi: 10.1016/S0140-6736(16)32467-9. [DOI] [PubMed] [Google Scholar]
- 41.Department of Health and Social Care. Matt Hancock announces ambition to map 5 million genomes. 2018.
- 42.Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, Downey P, Elliott P, Green J, Landray M, et al. UK Biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 2015;12(3):e1001779. doi: 10.1371/journal.pmed.1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Teo YY, Sim X, Ong RTH, Tan AKS, Chen J, Tantoso E, Small KS, Ku CS, Lee EJD, Seielstad M, et al. Singapore genome variation project: a haplotype map of three South-East Asian populations. Genome Res. 2009;19(11):2154–2162. doi: 10.1101/gr.095000.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ferrer-Admetlla A, Sikora M, Laayouni H, Esteve A, Roubinet F, Blancher A, Calafell F, Bertranpetit J, Casals F. A natural history of FUT2 polymorphism in humans. Mol Biol Evol. 2009;26(9):1993–2003. doi: 10.1093/molbev/msp108. [DOI] [PubMed] [Google Scholar]
- 45.Yngvadottir B, Xue Y, Searle S, Hunt S, Delgado M, Morrison J, Whittaker P, Deloukas P, Tyler-Smith C. A genome-wide survey of the prevalence and evolutionary forces acting on human nonsense SNPs. Am J Hum Genet. 2009;84(2):224–234. doi: 10.1016/j.ajhg.2009.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. Source code for the FineMAV program.
Additional file 2. Table containing the top 50 FineMAV candidates from the Han Chinese (90HC), Singaporean Indian (SSIP) and Singaporean Malay (SSMP) populations.
Data Availability Statement
The software tool is publicly available on GitHub (https://github.com/fadilla-wahyudi/finemav). The datasets analysed during the current study are available from the following links: 1000 Genomes Project Phase 3 dataset (ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502/), Sequencing of 90 Han Chinese genomes (90HC) (https://www.ebi.ac.uk/ena/data/view/PRJEB20820), Singapore Sequencing Indian Project (SSIP) (https://blog.nus.edu.sg/sshsphphg/singapore-sequencing-indian/), and Singapore Sequencing Malay Project (SSMP) (https://blog.nus.edu.sg/sshsphphg/singapore-sequencing-malay/).