

Abstract
Synucleinopathies are clinically and pathologically heterogeneous disorders characterized by pathologic aggregates of α-synuclein in neurons and glia, in the form of Lewy bodies, Lewy neurites, neuronal cytoplasmic inclusions, and glial cytoplasmic inclusions. Synucleinopathies can be divided into two major disease entities: Lewy body disease and multiple system atrophy (MSA). Common clinical presentations of Lewy body disease are Parkinson’s disease (PD), PD with dementia, and dementia with Lewy bodies (DLB), while MSA has two major clinical subtypes, MSA with predominant cerebellar ataxia and MSA with predominant parkinsonism. There are currently no disease-modifying therapies for the synucleinopathies, but information obtained from molecular genetics and models that explore mechanisms of α-synuclein conversion to pathologic oligomers and insoluble fibrils offer hope for eventual therapies. It remains unclear how α-synuclein can be associated with distinct cellular pathologies (e.g., Lewy bodies and glial cytoplasmic inclusions) and what factors determine neuroanatomical and cell type vulnerability. Accumulating evidence from in vitro and in vivo experiments suggests that α-synuclein species derived from Lewy body disease and MSA are distinct “strains” having different seeding properties. Recent advancements in in vitro seeding assays, such as real-time quaking-induced conversion (RT-QuIC) and protein misfolding cyclic amplification (PMCA), not only demonstrate distinct seeding activity in the synucleinopathies, but also offer exciting opportunities for molecular diagnosis using readily accessible peripheral tissue samples. Cryogenic electron microscopy (cryo-EM) structural studies of α-synuclein derived from recombinant or brain-derived filaments provide new insight into mechanisms of seeding in synucleinopathies. In this review, we describe clinical, genetic and neuropathologic features of synucleinopathies, including a discussion of the evolution of classification and staging of Lewy body disease. We also provide a brief discussion on proposed mechanisms of Lewy body formation, as well as evidence supporting the existence of distinct α-synuclein strains in Lewy body disease and MSA.
Keywords: Lewy body disease, Parkinson’s disease, Dementia with Lewy bodies, Multiple system atrophy, cryo-EM structures, RT-QuIC, PMCA, Biomarkers, AlphaFold
1. Introduction
Synucleinopathies are a group of neurodegenerative disorders characterized by neuronal or glial inclusions, or both, composed of aggregated α-synuclein. Pathologically, synucleinopathies can be divided into two major disease groups: Lewy body disease and multiple system atrophy (MSA) [1–3]. Lewy body disease is a pathologic term for neurodegenerative disorders characterized by aggregated α-synuclein in perikarya and neurites of neurons (Lewy bodies and Lewy neurites) [4]. Lewy body pathology is not restricted to Parkinson’s disease (PD), PD dementia (PDD), and dementia with Lewy bodies (DLB), but is also observed in several neurodevelopmental and neurometabolic disorders, such as PLA2G6-associated neurodegeneration (i.e., infantile neuroaxonal dystrophy, atypical neuroaxonal dystrophy, adult-onset dystonia-parkinsonism, and autosomal recessive early-onset parkinsonism), POLG-associated neurodegeneration, Niemann-Pick type C1, and Krabbe disease [5]. Although these diseases have α-synuclein aggregates at a much younger age than incidental Lewy bodies observed in other neurodegenerative disorders or healthy elderly individuals, these disorders are not included in this review. We herein describe clinical, genetic, and neuropathologic features of synucleinopathies, including a historical evolution of the disease concept, classification, and staging of Lewy body disease. We also review evidence supporting the existence of distinct α-synuclein strains in Lewy body disease and MSA, with a specific focus on in vitro seeding assays and cryogenic electron microscopy (cryo-EM) structural studies of α-synuclein.
2. Background
2.1 History of synucleinopathies
James Parkinson published a short monograph, “An Essay on Shaking Palsy,“ in 1817 [6], in which he described six patients with tremors, slow movements, and falls. His work received little attention for decades until Jean-Martin Charcot introduced it in his lectures in the late 1800’s. Charcot added rigidity as a prominent symptom and pointed out that the disability is not caused by palsy; he proposed to call the disease PD. In 1912, Friedrich H. Lewy reported the presence of intracellular inclusion bodies in the dorsal nucleus of the vagus nerve, nucleus basalis of Meynert, and the thalamic paraventricular nucleus of the brains of PD patients. Gonzalo Rodriguez Lafora also examined one patient with PD and confirmed the existence of neuronal inclusions in 1913. Konstantin Tretiakoff reported neuronal loss and neuronal inclusions in the substantia nigra in 1919, and it was proposed that these inclusions be called Lewy bodies [7–9]. In 1961, Haruo Okazaki reported two patients with progressive dementia without parkinsonism who had abundant Lewy bodies in the neocortex [10]. Kenji Kosaka subsequently found similar inclusions in the neocortex of patients with cognitive impairment and extrapyramidal symptoms and proposed the term diffuse Lewy body disease for this disorder [11]. Subsequently, an international consortium led by Ian McKeith coined the term DLB to refer to this clinicopathologic disorder [12]. Lewy bodies are now considered a pathological hallmark of PD, DLB, and PDD.
MSA is a distinctive synucleinopathy that includes two major clinicopathologic subtypes previously thought to be separate disorders: olivopontocerebellar atrophy (OPCA) and striatonigral degeneration (SND). The term Shy-Drager syndrome was originally used in 1960 to describe a neurological disorder associated with orthostatic hypotension of unknown etiology, but the pathologic descriptions by Shy and Drager clearly indicated that it was a disorder affecting multiple systems beyond the autonomic nervous system [13]. Accumulating evidence indicates that the majority of patients with SND have some degree of OPCA and vice versa. Moreover, progressive autonomic failure corresponding to Shy-Drager syndrome is common in both SND and OPCA. Therefore, Graham and Oppenheimer considered these variants to be the same disorder and coined the term MSA in 1969 [14]. Although there had been some debate about whether MSA was a single disease, the discovery of argyrophilic glial cytoplasmic inclusions (GCI), also known as Papp-Lantos bodies [15, 16], in SND and OPCA provided strong evidence that they were part of a disease spectrum [16].
Seminal studies in the early 1990’s in the laboratory of Tsunao Saitoh discovered a non-amyloid component in senile plaques (NACP) [17] using biochemical methods, and later showed that NACP was a synaptic protein with genetic locus on chromosome 4q21.3-q22 identical to α-synuclein [18]. Previous studies had defined α-synuclein as one member of a family of presynaptic proteins (α-, β- and γ-synuclein) with a role in membrane-associated processes at the presynaptic terminal (reviewed in [19]). In 1997, Polymeropoulos discovered a mutation in the gene for α-synuclein (SNCA) in Southern Italian families of Greek heritage with PD [20]. Subsequently, Spillantini and colleagues reported that α-synuclein was the major component of Lewy bodies in sporadic PD and DLB patients [21]. These findings combined pathological and genetic insight and established the disease concept of “synucleinopathy.” Wakabayashi and colleagues reported that the major protein component in GCI was also α-synuclein, linking MSA and Lewy body disease as the two major clinicopathologic subtypes of synucleinopathy [22].
2.2 Clinical spectrum
Lewy body disease falls into three major clinicopathologic subtypes: PD, PDD, and DLB. PD is the most common neurodegenerative movement disorder characterized by bradykinesia, rigidity, rest tremor, and postural instability, as well as various non-motor symptoms, such as rapid eye movement sleep behavior disorder (RBD), hyposmia, depression, and autonomic failure [23]. Levodopa-responsiveness is a supporting feature of PD, and a feature that is useful in differentiating PD from atypical parkinsonian syndromes. Although there are differences between studies, up to 83% of patients with PD eventually develop dementia later in the disease course, which is termed as PDD [24–26]. DLB is the second most common cause of neurodegenerative dementia, after Alzheimer’s disease. The cardinal features of DLB include dementia, fluctuating cognition, visual hallucinations, RBD, and parkinsonism [27]. Motor symptoms may be absent in up to 25% of autopsy-confirmed DLB patients [28]. A clinical diagnosis of PDD or DLB requires information about not only the nature of clinical findings, but also the timing of dementia relative to motor signs and symptoms. Patients with PD who develop dementia later in the disease course (“more than one-year” after onset of parkinsonism) are considered to have PDD. On the other hand, patients who develop dementia, with (or without) parkinsonism after one year of cognitive or psychiatric symptoms are diagnosed with DLB [27]. In rare cases, patients with Lewy body disease present with focal cortical syndromes [29], such as corticobasal syndrome [30], progressive aphasia [31–33], or Capgras syndrome [34]. Although dysphagia is a common clinical feature of PD and atypical parkinsonian disorders [35–37], there are only a few reports of isolated dysphagia without extrapyramidal syndrome in patients with Lewy body disease, which is referred to as Lewy body dysphagia [38, 39].
MSA is a movement disorder characterized by a variable combination of autonomic failure, levodopa-unresponsive parkinsonism, cerebellar ataxia, pyramidal signs, and non-motor symptoms [37]. MSA is subclassified into two clinical subtypes depending upon the predominant motor symptom: MSA with predominant cerebellar ataxia (MSA-C) and MSA with predominant parkinsonism (MSA-P) [37]. Autonomic failure, such as urinary incontinence and orthostatic hypotension, is required for the clinical diagnosis of MSA. Besides autonomic failure, non-motor symptoms, such as RBD, sleep-disordered breathing, dysphagia, severe dysphonia, and dysarthria, are also common in MSA [37, 40, 41]. Unlike PDD and DLB, dementia is not a typical feature of MSA [37]; however, a subset of MSA patients develops cognitive deficits, particularly executive dysfunction [42–44].
RBD is a parasomnia characterized by dream-enacting behaviors due to lack of muscle atonia during REM sleep (“dream sleep”) [45]. This results in the common description of patients “acting out their dreams.” RBD is one of the most frequent nonmotor symptoms in both Lewy body disease and MSA. The absence of RBD after a 5-year disease duration is considered a “red flag” for the diagnosis of PD [23]. In the diagnostic criteria for DLB, RBD is one of the four core clinical features [27]. Although RBD is not included in current clinical criteria for MSA [37], several studies have supported its usefulness for the diagnosis of MSA [46–48]. In synucleinopathies, RBD often precedes motor or other nonmotor symptoms. Several longitudinal studies have demonstrated that up to 80% of patients with isolated RBD eventually develop clinical features of synucleinopathies, including PD, DLB, or MSA [49–51]. Therefore, isolated RBD has been considered a prodromal phase of synucleinopathies.
2.3 Pathogenesis of Lewy body formation
α-Synuclein is a 140 amino acid protein that is encoded by the SNCA gene on chromosome 4q21 [52]. α-Synuclein consists of 3 modular domains (Fig. 1). The N-terminal amphipathic region (1-60 residues) of α-synuclein has lipid-binding properties [53]. The central region of α-synuclein contains a highly amyloidogenic domain, known as the non-amyloid-β component (NAC) domain (61-95 residues), which is critical for α-synuclein aggregation [17, 54]. The C-terminal acidic domain (96-140 residues) contributes to its natively unfolded structure. Truncation of this region results in increased potential for aggregation [55].
Fig. 1.

Structure of α-synuclein. Missense mutation sites linked to familial Parkinson’s disease and major post-translational modification sites are shown
α-Synuclein is a neuronal protein, predominantly located in the presynaptic terminal [17]. Although the function of α-synuclein has not been fully elucidated, it is thought to play a role in regulating neurotransmitter release, synaptic function, and synaptic plasticity [56]. Under physiological conditions, α-synuclein in the cytoplasm of the neurons exists in a dynamic equilibrium between soluble monomers and a variety of oligomers, including helically folded tetramers (Fig. 2) [57, 58]. Although the process of Lewy body formation is not fully understood, binding of α-synuclein to lipid membranes is thought to be a key mediator of oligomerization and aggregation of α-synuclein [59–62]. Lipid membranes can promote α-synuclein aggregation, and α-synuclein oligomer species result in membrane permeabilization, which can disrupt membrane integrity [60]. Destabilization of stable α-synuclein tetramers is also considered an early step in aggregation of α-synuclein [63]. Indeed, mutations in SNCA associated with familial PD or exposure to unsaturated fatty acids, such as oleic acid, decrease the tetramer-monomer ratio and induce cytoplasmic inclusions in human cellular models [64, 65]. Loss of nuclear membrane integrity may also trigger α-synuclein aggregation through interaction between α-synuclein and nuclear proaggregant factors, such as histone [66, 67]. A recent study using stimulated emission depletion (STED)-based super-resolution microscopy revealed that Lewy bodies contain not only aggregates of α-synuclein, but also crowded organelles and lipid membranes, including damaged lysosomes, mitochondria, and various vesicles (Fig. 2) [68].
Fig. 2.

Hypothetical scheme of Lewy body formation in neurons. α-Synuclein exists in equilibrium between monomers and tetramers in the cytoplasm. Under pathologic conditions, the tetramer-monomer ratio decreases, and monomers bind to vesicle membranes. On the surface of membrane, α-synuclein tend to oligomerize, and toxic oligomers can disrupt lipid membranes. Monomers and oligomers form insoluble fibrils in the cytoplasm. Disrupted membranes, organelles, α-synuclein oligomers and fibrils are involved in the Lewy body formation
α-Synuclein undergoes various post-translational modifications, including phosphorylation, ubiquitination, acetylation, nitration, O-GlcNAcylation and SUMOylation (Fig. 1) [69, 70]. Post-translational modifications of α-synuclein may influence its toxicity and propensity to aggregate. Phosphorylation of α-synuclein is the most investigated post-translational modification of α-synuclein, and it is considered a marker of pathologic α-synuclein [71]. Up to 90% of α-synuclein is phosphorylated at serine-129 in Lewy bodies, while only 4% of soluble α-synuclein is phosphorylated at this residue [71]. Multicolor STED microscopy revealed the subcellular arrangement of α-synuclein in Lewy body [72], with serine-129-phosphorylated α-synuclein at the periphery of Lewy body with cytoskeletal proteins, while α-synuclein truncated at aspartate-119 or asparagine-122 was condensed in the Lewy body core [72].
3. Genetics
3.1 Genetics of Lewy body disease
A direct causal relationship between α-synuclein and PD was first established in 1997 with the discovery of a single point mutation in the SNCA gene resulting in a non-synonymous amino acid substitution, p.A53T, causing familial early-onset PD [20]. Subsequently, several other substitutions in the α-synuclein protein (e.g., p.A18T, p.A29S, p.A30P, p.E46K, p.G46K, p.H50Q, p.G51D, and p.A53E) have been linked to autosomal dominant familial PD [73–78]. Furthermore, genomic multiplications (i.e., duplication and triplication) of SNCA cause familial PD with extramotor features, including dementia [79, 80]. These genetic studies suggested that overexpression of wild-type α-synuclein could be pathogenic and that the severity of the phenotype may be dose-dependent. For example, the clinical phenotypes tended to be more severe in families with SNCA triplication than SNCA duplication (e.g., non-motor features were more frequent in triplication families). Patients with SNCA triplication had greater severity of oligodendroglial and extra-perikaryal α-synuclein pathologies than in sporadic PD [81]. This finding suggests that high α-synuclein expression is associated with pathologic α-synuclein deposition in both neurons and oligodendrocytes. Importantly, this observation also nominated reduction of α-synuclein levels as a possible therapeutic strategy for synucleinopathies.
In addition to SNCA, pathogenic mutations in other genes have been associated with familial PD, including LRRK2, VPS35, PRKN, PINK1, DJ-1, and VPS13C [82, 83]. As DNA sequencing technologies have revolutionized population genetics over the last decade, an increasing number of other genes have been nominated for PD (e.g., CHCHD2, LRP10, TMEM230, DNAJC13), but these genetic loci lack definitive confirmation [83]. Even though the discovery of familial PD-related genes has contributed to dissecting the etiology of the disease (e.g., dysfunction of vesicle trafficking, lysosomal, and mitochondrial pathways) [84], they still only account for a relatively small proportion of the genetic risk in PD. A genetic hypothesis for PD is that the disease develops through a complex interplay of low penetrant genetic risk variants and unknown environmental determinants. Notably, only SNCA and a subset of LRRK2 mutations are indisputably associated with Lewy body pathology. Based on data from the Mayo Clinic brain bank, 59% (30/51) of cases with LRRK2 mutations had Lewy body disease, while the remaining LRRK2 cases had tau or TDP-43 pathology or nonspecific substantia nigra degeneration (unpublished data). The majority of the genes nominated for familial PD (see above) do not have α-synuclein pathology, or there are inconsistent reports of Lewy body pathology in the small number of patients who have come to autopsy [83]. This fact indicates that “familial PD” in the context of most genetic studies may include a variety of disorders, including some that are not associated with Lewy body disease.
Population-based approaches to resolve the genetic architecture of PD have focused on common variant associations at candidate genes, including SNCA and MAPT loci. Large unbiased studies characterizing patterns of linkage disequilibrium (e.g., the HapMap project) across the genome and high throughput genome-wide genotyping platforms, which simultaneously genotype hundreds of thousands of common single nucleotide variants have been performed. Early genome-wide association studies (GWAS) confirmed associations at the SNCA and MAPT loci, and highlighted novel associations at LRRK2 and PARK16 loci as risk factors for PD in both European and Japanese clinical cohorts [85, 86]. The only GWAS in autopsy-confirmed Lewy body-associated PD nominated a locus in PARK11 [87], a locus on chromosome 2q associated with familial PD [88].
Of note, the MAPT locus was identified as a risk locus for PD only in European cohorts, while the BST1 locus was identified as a risk locus only in Japanese cohorts [85, 86]. Additional loci have been nominated in Eastern Asian populations with GWAS methods [89]. The latest PD GWAS in Caucasian populations nominated 90 independent loci covering 78 genomic regions [90], with the SNCA locus showing the strongest signal. Although less well-characterized or studied, recent GWAS and whole-genome sequencing efforts in Lewy body dementia (i.e., PDD and DLB) in Caucasian populations have nominated at least five loci, including three that are also risk loci for PD (SNCA, GBA, and TMEM175) and two other (APOE and BIN1) known risk loci for Alzheimer’s disease [91, 92]. These findings indicate that Lewy body dementia shares genetic risk factors of both Alzheimer’s disease and PD, as may be expected from its mixed amyloid and α-synuclein pathology.
Another important genetic determinant of susceptibility to both PD and Lewy body dementia is variation in the GBA gene [93, 94]. Recessive mutations in GBA result in a lysosomal storage disorder known as Gaucher’s disease; however, astute clinical observations by Sidransky and co-workers noted that heterozygous carriers were at increased risk of PD and Lewy body dementia [95]. GBA is a GWAS locus for both disorders, but also is an example of a gene that harbors variants of differing penetrance, with rare variants also driving disease risk. GBA is the only significant finding in a gene burden analysis in a whole-genome sequence study of Lewy body dementia [39]. The role that GBA susceptibility variants may play in MSA remains controversial [96, 97].
3.2 Genetics of MSA
MSA is considered a sporadic disease; only a few familial MSA cases have been reported [98]. Several small multiplex families from Japan nominated mutations of the COQ2 gene for MSA [99]. In addition, a common substitution in COQ2, p.V393A, and several rare variants were associated with sporadic MSA [99]. The COQ2 gene encodes an enzyme involved in synthesis of coenzyme Q10, and the p.V393A variant results in lower production of coenzyme Q10. Several studies from non-Asian countries, however, failed to replicate the findings [100, 101]. A meta-analysis of Eastern Asian populations confirmed an association of COQ2 p.V393A variant with MSA (odds ratio [OR] 2.05; 95% CI 1.29–3.25, p = 0.002) [102]. Interestingly, a subgroup analysis revealed that the association was significant for MSA-C (OR 2.75, 95% CI 1.98–3.84, p <0.001), but not for MSA-P (OR 1.25, 95% CI 0.64–2.46, p = 0.51). This finding may partially explain population variance since the predominant subtypes of MSA are different in Asian and Western countries. MSA-C is the predominant phenotype in Asia, while MSA-P is the predominant subtype in Western countries [103, 104]. Some studies reported decreased levels of coenzyme Q10 in the cerebellum of MSA, suggesting a potential association between alterations of coenzyme Q10 activity and cerebellar pathology [105, 106].
The largest GWAS, which enrolled 918 MSA patients of European ancestry and 3,864 controls, did not find any common genetic association [107]. This GWAS identified four potential risk loci, including single nucleotide polymorphisms in the genes FBXO47, ELOVL7, EDN1, and MAPT; however, these findings could not be replicated in a GWAS that included 906 MSA patients and 941 unrelated healthy controls of the Han Chinese population [108]. These discrepancies could be partially explained by ethnic differences, as GWAS demonstrated some differences in the genetic contribution to PD between the European and Asian populations [89]. Large-scale whole-genome sequencing efforts are underway in MSA, PD and Lewy body dementia, and it is likely that efforts of global consortia will be needed to fully resolve the role of genetics in synucleinopathies.
4. Pathology of synucleinopathies
4.1 Neuropathologic features of Lewy body disease
In most cases of Lewy body disease without cognitive deficits, the macroscopic findings are comparable to age- and sex-matched controls, except for loss of neuromelanin pigment in the substantia nigra and locus coeruleus (Fig. 3). Dopaminergic neuronal loss in the substantia nigra, particularly in the ventrolateral part, is a pathologic hallmark of PD [109, 110]. The severity of neurodegeneration of the substantia nigra correlates with severity of extrapyramidal motor symptoms and the degree of striatal dopaminergic deficiency [111, 112]. Neuronal loss is moderate to marked in PD and PDD, but more variable in DLB. In fact, a subset of DLB patients lack parkinsonism and have preserved neuronal population in the substantia nigra. The locus coeruleus is a major noradrenergic nucleus, and neuronal loss leads to deficiency of noradrenaline, which may contribute to various symptoms, including cognitive impairment, affective symptoms, RBD, and gait difficulties [113].
Fig. 3.
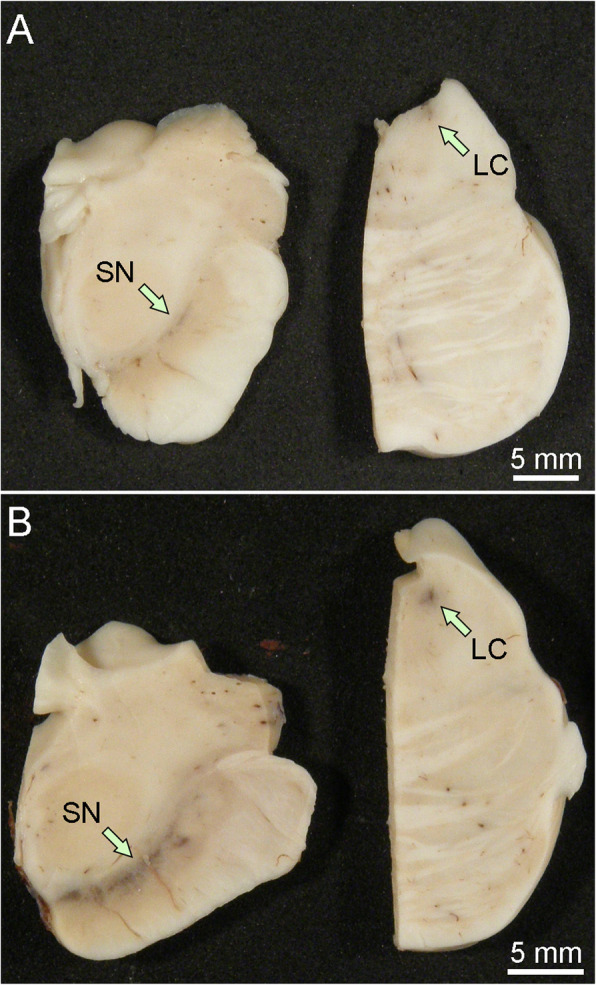
Macroscopic findings of Lewy body disease brains. The midbrain and pons from Lewy body disease (A) and control (B) brains. Loss of neuromelanin pigment in the substantia nigra and locus coeruleus is observed in Lewy body disease. Abbreviations: LC, locus coeruleus; SN, substantia nigra
Lewy bodies are round, eosinophilic inclusions in neuronal perikarya. There are two types of Lewy bodies: classical (or brainstem) type and cortical type. Classical-type Lewy bodies have a dense hyaline appearance with a peripheral clear halo and are easily visible on hematoxylin and eosin stained sections (Fig. 4 A). Cortical type Lewy bodies have a less compact appearance and are more difficult to detect with histologic methods (Fig. 4B) [10]. Both types of Lewy bodies are strongly immunoreactive with antibodies to α-synuclein (Fig. 4 C, D) [71]. In addition to α-synuclein, more than 90 components of Lewy bodies have been reported, based mostly upon immunohistochemical colocalization with brainstem type Lewy bodies, including sequestration of neurotransmitter enzymes of cholinergic and dopaminergic neurons [114–116]. Accumulation of phosphorylated α-synuclein also occurs within cell processes (mostly axonal), so-called Lewy neurites (Fig. 4E). Lewy neurites in the CA2/3 sectors of the hippocampus are a characteristic histopathologic finding in many cases of PD and most cases of PDD and DLB [117]. Deposits of phosphorylated α-synuclein are observed less frequently in oligodendroglia, and rarely in astrocytes in the midbrain and basal ganglia [118].
Fig. 4.

Representative images of histopathology of Lewy body disease. A, B, F-H hematoxylin and eosin staining, C-E immunohistochemistry for α-synuclein (NACP antibody). A, C Brainstem type Lewy body in the substantia nigra. B, D cortical type Lewy body (arrow) in the superior temporal cortex. E Lewy neurites in the CA2 sector of the hippocampus. F-G Lewy body disease shows neuronal loss with extracellular neuromelanin pigment in the substantia nigra (F), while it is minimum in a control case (G). H Spongiform change in the entorhinal cortex. Scale 100 μm in (A, B, and H); 50 μm in (C); 20 μm in (D-G, and I)
In addition to Lewy bodies and neuronal loss in the substantia nigra (Fig. 4 F, G), a subset of cases have spongiform change or neuropil microvacuolation that is often most severe in the amygdala, but also seen in limbic and superior temporal cortices (Fig. 4 H) [119–121]. The most important co-pathology in Lewy body disease is Alzheimer-type pathology. In initial reports of diffuse Lewy body disease, the main neuropathologic features included not only numerous cortical Lewy bodies, but also numerous senile plaques and neurofibrillary tangles in the cerebral cortex [11]. The majority of Lewy body disease cases, not exclusively diffuse Lewy body disease, have some degree of Alzheimer-type pathology (i.e., neocortical senile plaques and neurofibrillary tangles) [122–125]. Senile plaques in the cerebral cortex are common, and they often are characterized by non-neuritic diffuse amyloid deposits [126]. Up to 50% of PDD cases and 28-89% of DLB cases have sufficient pathology for a secondary neuropathologic diagnosis of Alzheimer’s disease [127–129].
Lewy bodies are also found frequently in cases with advanced Alzheimer’s disease, particularly in the amygdala [130–134]. Hamilton screened α-synuclein pathology in 145 cases of Alzheimer’s disease and found that 88 cases (61%) had Lewy bodies [131]. Of regions screened, the amygdala was the most frequently affected region. Interestingly, some of the cases had numerous Lewy bodies in the amygdala, but minimal or no brainstem Lewy bodies (amygdala-predominant Lewy bodies). Uchikado and colleagues also screened Lewy-related pathology in 347 cases of Alzheimer’s disease and found that 62 cases (18%) were consistent with amygdala-predominant Lewy bodies [133]. Of those, Lewy bodies were only found in the amygdala (“amygdala-only” Lewy body disease) in 32 cases (9%). The clinical significance of amygdala-predominant Lewy bodies in Alzheimer’s disease remains uncertain [135], but evidence suggests that they may be associated with increased frequency of visual hallucinations compared to Alzheimer’s disease without amygdala Lewy bodies [136].
Unlike Lewy-related pathology, α-synuclein oligomers have not been possible to confidently detected with routine histologic methods. Roberts and colleagues applied proximity ligation assay to detect α-synuclein oligomers in histologic sections and showed α-synuclein oligomers in the cingulate cortex and reticular formation of the medulla in PD [137]. They also detected α-synuclein oligomers in morphologically intact neurons and in the periphery of Lewy bodies.
4.2 Classification and staging of Lewy body disease
The term “Lewy body disease” was coined by Kosaka and colleagues to refer to neurodegenerative diseases with numerous Lewy bodies in the central nervous system [4]. He classified Lewy body disease into three groups: diffuse, transitional and brainstem-predominant types [138]. Brainstem-predominant Lewy body disease corresponded to PD in their scheme, and diffuse Lewy body disease was thought to be an extension of PD pathology into the limbic lobe and the neocortex. Diffuse Lewy body disease was separated into two forms: a common form and pure form. The common form had not only numerous Lewy bodies, including neocortical Lewy bodies, but also many senile plaques and variable neurofibrillary tangles. In contrast, the pure form had few or no Alzheimer-type changes [122]. Kosaka later added a “cerebral type” of Lewy body disease, which had numerous Lewy bodies in the cerebral cortex and amygdala, but minimal or no Lewy bodies in the brainstem and diencephalon [139].
The First International Consortium for Lewy Body Dementia (ICDLB) proposed criteria for clinical and pathologic diagnosis of DLB [12]. Subtypes of Lewy-related pathology were categorized based upon the severity and topographical distribution of Lewy bodies [140]: diffuse neocortical, limbic (transitional), and brainstem-predominant, which corresponded roughly to Kosaka’s classification of diffuse, transitional and brainstem-predominant Lewy body disease. The Third ICDLB report developed a diagnostic scheme that was devised to predict the likelihood that the pathology would be associated with DLB. The criteria took into account both the extent of Lewy-related pathology and Alzheimer’s-type pathology to assign a probability that the pathology would be associated with the clinical presentation (Table 1). The severity of Lewy-related pathology was semi-quantitatively assessed on a five-point scale: 0 = none, 1 = mild, 2 = moderate, 3 = severe, and 4 = very severe. Recommended brain regions for assessment included the dorsal motor nucleus of vagus, locus coeruleus and substantia nigra in the brainstem, as well as nucleus basalis of Meynert, amygdala, transentorhinal cortex, cingulate cortex, temporal, frontal and parietal cortices. The likelihood of DLB clinical syndrome was directly related to severity of Lewy body pathology and indirectly related to severity of Alzheimer pathology. It was recognized from studies of prospective cohorts that when Alzheimer pathology was severe, most patients had Alzheimer type dementia, rather than DLB [141]. A recent study validates this approach in that the diagnostic sensitivity for probable DLB was significantly higher in limbic Lewy body disease and diffuse neocortical Lewy body disease without neocortical tangles than in those with neocortical tangles [142]. These findings indicate that the phenotypic expression of DLB is associated directly related to the extent of Lewy bodies and inversely related to the extent of neurofibrillary tangles [142].
Table 1.
Likelihood of a typical clinical presentation of dementia with Lewy bodies
| Lewy-related pathology | ADNC based on NIA-AA | ||
|---|---|---|---|
| None/low | Intermediate | High | |
| Diffuse neocortical | High | High | Intermediate |
| Limbic (transitional) | High | Intermediate | Low |
| Brainstem-predominant | Low | Low | Low |
| Amygdala-predominant | Low | Low | Low |
| Olfactory bulb only | Low | Low | Low |
Abbreviations: ADNC Alzheimer’s disease neuropathological change, NIA-AA National Institute on Aging–Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer disease
Braak and colleagues proposed a staging scheme for Lewy-related pathology in PD [143] that was anatomically more detailed and specified than Kosaka’s classification of Lewy body disease. Lewy-related pathology initially occurred in the medulla oblongata (dorsal motor nucleus of vagus and the glossopharyngeal nucleus), and in the anterior olfactory nucleus of stage 1. In stage 2, α-synuclein pathology ascends to the pontine tegmentum, while stage 3 is associated with involvement of midbrain, stage 4 with limbic regions, and stages 5 and 6 with neocortical involvement. This staging scheme has largely been confirmed in several studies, but some exceptions have also been pointed out, such as cases with Lewy bodies restricted to the olfactory bulb or to the amygdala, particularly in cases with advanced Alzheimer’s disease (i.e., amygdala-predominant Lewy bodies). [133, 144, 145]. Following these studies, the ICDLB criteria were revised in 2017 to include amygdala-predominant and olfactory bulb types [27].
emi-quantitative evaluation has been used for diagnosing and classifying neurodegenerative disorders, but there are inherent weaknesses in semi-quantitative measures when it comes to inter-rater reliability [146]. Recently, Attems and colleagues proposed neuropathological consensus criteria for Lewy pathology, which they named “Lewy pathology consensus criteria” [147]. This new system is based on the ICDLB criteria modified to use a dichotomous approach for assessing Lewy-related pathology (i.e., present vs. absent), rather than semi-quantitative scores. In this scheme, a case that had very sparse Lewy bodies or Lewy neurites (i.e., only one Lewy neurite in a neocortical section) without Lewy-related pathology in other brain regions, such as medial temporal lobe, would be still assigned neocortical subtype. Nevertheless, in the select and relatively small study cohort, all cases with neocortical Lewy pathology had dementia. They also demonstrated that all cases were successfully classified, and that there was high inter-rater reliability. In parallel, they assessed other criteria on the same sections, and up to 30% of cases were unclassifiable with Braak staging or ICDLB criteria. Although this simple approach for Lewy pathology classification might be useful in routine diagnostic practice, clinicopathologic correlations of this classification, particularly neocortical type, need to be validated. They need to be compared critically to ICDLB neuropathologic criteria, which have been shown to “predict” the DLB clinical syndrome [148, 149].
4.3 Underlying pathology of PD, PDD, and DLB
PD, PDD, and DLB are considered distinct clinical entities that have varying degrees and distributions of Lewy body pathology. PD and PDD are disorders associated with extrapyramidal clinical syndrome of parkinsonism, which correlate less well with distribution of Lewy bodies, and better with nigrostriatal dopaminergic deficiency. Given this fact, ICDLB criteria were revised to capture this important clinical correlate, namely, moderate-to-severe neuronal loss in the ventrolateral substantia nigra [109]. The specification of the ventrolateral region was driven by the fact that ventrolateral dopaminergic neurons are the origin of the nigrostriatal dopaminergic pathway (projecting to the putamen and caudate nucleus), while ventromedial substantia nigra dopaminergic neurons are the origin of the mesolimbic pathway (projecting to the ventral striatum and nucleus accumbens).
While most patients with PD have brainstem-predominant Lewy body disease (or limbic Lewy body disease) at autopsy, a “pure form” of diffuse Lewy body disease can also be found in a few patients with PD [122, 150]. In contrast, most patients with PDD and DLB have diffuse Lewy body disease. It has proven challenging to differentiate DLB from PDD based on the neuropathologic findings alone [128]. Although DLB patients tend to have more severe Alzheimer-type pathology, and PDD patients tend to have a more severe neuronal loss in the substantia nigra [151], there are no clear neuropathologic distinctions between DLB and PDD, especially for patients with dementia and significant extrapyramidal signs [152].
4.4 Neuropathologic features and diagnosis of MSA
The current diagnostic criteria for MSA define three levels of certainty: possible, probable, and definite. The two former diagnoses are based on clinical features, while a definite diagnosis is based on neuropathologic evaluation [37]. A definite neuropathological diagnosis of MSA is established when there is evidence of widespread and abundant α-synuclein-positive GCI in association with neurodegenerative changes in striatonigral or olivopontocerebellar structures [153]. GCI are argyrophilic inclusions (positive on Gallyas silver stain) in the cytoplasm of oligodendrocytes [16]. They are visible on routine hematoxylin and eosin stains, but immunohistochemistry for phosphorylated α-synuclein is far more sensitive for visualization of GCI [22]. The pathogenesis of GCI is unknown, particularly the cellular origin of α-synuclein. Although oligodendrocytes express α-synuclein mRNA less than neurons [154, 155], mounting evidence from in vitro studies suggests that oligodendrocytes can take up α-synuclein monomers, oligomers, and fibrils, which are putatively involved in GCI formation [156–158].
Macroscopic findings in MSA vary with the subtype of MSA. In MSA-P, there is loss of neuromelanin pigment in the ventrolateral substantia, as well as atrophy and discoloration of the posterolateral putamen. In contrast, MSA-C has atrophy of the pontine base and the middle cerebellar peduncle with attenuation and discoloration of the cerebellar white matter (Fig. 5). The neocortex and limbic structures are usually macroscopically unremarkable.
Fig. 5.

Macroscopic findings of multiple system atrophy (MSA). The brains from MSA (A, C, E, and G) and control (B, D, F, and H) cases. The pons and cerebellar white matter are atrophic in MSA (A). Loss of neuromelanin pigment in the locus coeruleus (C) and substantia nigra (E) is observed in MSA. The discoloration and atrophy of the lateral putamen in MSA (G). Abbreviations: CWM, cerebellar white matter; LC, locus coeruleus; SN, substantia nigra
System-specific neuronal loss and gliosis are observed in both striatonigral and olivopontocerebellar systems (Fig. 6 A-D). In addition to these macroscopically affected brain regions, there are more widespread α-synuclein pathology, characterized by GCI (Fig. 6E, F) and variable neuronal cytoplasmic inclusions. The distribution of neuronal cytoplasmic inclusions is distinct from that of Lewy bodies, and they are most often observed in the putamen, pontine nuclei (Fig. 6G) and inferior olivary nuclei (Fig. 6 H), which are not susceptible to Lewy bodies. Neuronal cytoplasmic inclusions can also be observed in the substantia nigra, cingulate cortex, amygdala, hippocampus, entorhinal cortex, hypothalamus and neocortex [159, 160]. Purkinje cells in the cerebellum do not have neuronal cytoplasmic inclusions, but abundant α-synuclein oligomers can be detected by proximity ligation assay [161]. Some MSA cases may have concurrent Lewy body disease [148]. In such cases, Gallyas silver staining is useful in distinguishing neuronal cytoplasmic inclusions from Lewy bodies; the former is positive, but the latter is not [162]. The clinicopathologic significance of neuronal cytoplasmic inclusions has been reviewed by Cykowski and colleagues in a large cohort of MSA cases. They found that neuronal cytoplasmic inclusions in neocortex were associated with cognitive impairment [160]. Other studies have reported that the burden of neuronal cytoplasmic inclusions is associated with cognitive impairment or memory loss in MSA [43, 44, 163].
Fig. 6.

Representative images of histopathology of MSA. A-D Hematoxylin and eosin staining. A Severe neuronal loss with gliosis in the lateral putamen. B Neurodegeneration is minimal in the same region in a case of minimal change MSA. C A typical MSA shows neuronal loss with extracellular neuromelanin pigment (arrows) in the substantia nigra, while it is well preserved in minimal change MSA (D). E-J Immunohistochemistry for α-synuclein (NACP antibody). Numerous GCIs in the putamen (E) and substantia nigra (F). Arrows indicate extracellular neuromelanin pigment (F). Neuronal cytoplasmic inclusions in the pontine nucleus (G, arrows) and inferior olivary nucleus (H, arrow). Abundant neuronal cytoplasmic inclusions in the dentate fascia in FTLD-synuclein (I) and hippocampal MSA (J). Scale bars: 50 μm in all images
4.5 Atypical subtypes of MSA
Although the diagnostic criteria for MSA require neurodegeneration in striatonigral or olivopontocerebellar systems, or both, some MSA cases do not have significant neurodegeneration even though they have widespread GCI. This rare subtype is referred to as “minimal change” MSA [164–168]. Cases of minimal change MSA suggest that GCI formation precedes neuronal loss. Clinical presentations of minimal change MSA vary. Some patients are asymptomatic (“preclinical MSA”) [165, 168], but in a case series from the UK, all minimal change MSA patients had respiratory dysfunction and early orthostatic hypotension [166]. Rarely, minimal change MSA has been reported with a limbic-predominant distribution of α-synuclein pathology [167].
MSA was previously considered three distinct disorders. Clinical presentations of each disorder have been considered typical features of MSA (i.e., autonomic dysfunction [Shy-Drager syndrome], parkinsonism [MSA-P], and cerebellar ataxia [MSA-C]). Several case series, however, have reported atypical clinical presentations of MSA [169–172]. Aoki and colleagues coined the term “frontotemporal lobar degeneration (FTLD)-synuclein” for a rare subtype of MSA, based on a series of four patients with atypical MSA [173]. These patients had clinical features consistent with frontotemporal dementia, including corticobasal syndrome, progressive nonfluent aphasia, or behavioral variant frontotemporal dementia. FTLD-synuclein cases had widespread GCI as well as striatonigral degeneration of MSA. Furthermore, abundant neuronal cytoplasmic inclusions, including Pick body-like inclusions, and severe neuronal loss were observed in limbic structures and the neocortex (Fig. 6I) [173].
More recently, Ando and colleagues reported that 12 of 146 (8%) of MSA had abundant neuronal cytoplasmic inclusions in the hippocampus (Fig. 6 J) and parahippocampal gyrus (“hippocampal MSA”) [174]. Severe neuronal loss with gliosis in the hippocampus and the medial temporal atrophy were also observed. Patients with hippocampal MSA had a longer disease duration and higher prevalence of cognitive impairment compared to typical MSA, but they lacked clinical features of FTLD-synuclein [174]. Despite differences in clinical presentations, hippocampal MSA and FTLD-synuclein share pathologic features; therefore, a subset of MSA may have a vulnerability to limbic structures, and these subtypes can be considered to belong to the same subtype of MSA.
4.6 α-Synuclein pathology in the peripheral nervous system
Lewy bodies are widely distributed not only in the central nervous system, but also in the peripheral autonomic nervous system, including peripheral nerves and neurons in the autonomic ganglia of the heart, submandibular glands, enteric nervous system, adrenal glands, skin and skin adnexa [175–183]. Therefore, detection of Lewy bodies in the peripheral tissues has been investigated as a promising diagnostic tool for Lewy body disease. Immunohistochemistry for phosphorylated-α-synuclein has detected Lewy bodies in biopsy samples of skin, enteric nervous system and other organs, although the sensitivity and specificity vary among studies probably due to different protocols (e.g., biopsy sites, antibodies, thickness of the sections, etc.) [184, 185]. One of the earliest studies by Ikemura and colleagues performed immunohistochemistry using skin samples from the arm and abdomen, showing 70% sensitivity in PD and PDD, and 40% in DLB with 100% specificity [177]. In contrast, Beach and colleagues did not detect phosphorylated-α-synuclein histopathology of the abdominal skin and scalp in patients with PD, DLB, incidental Lewy body disease, or Alzheimer’s disease with Lewy bodies, as well as healthy individuals (0% sensitivity and 100% specificity) [180]. Doppler and colleagues reported phosphorylated-α-synuclein-positive fibers of the subepidermal plexus and dermal nerve bundles not only in PD (73%), but also in MSA (75%) with 100% specificity [186]. Notably, the distribution of α-synuclein deposits was different between PD and MSA [186–188]; phosphorylated-α-synuclein was detected in autonomic fibers in PD; whereas, it was mainly detected in unmyelinated somatosensory fibers in MSA. Therefore, skin biopsy with phosphorylated-α-synuclein immunohistochemistry can be used for distinguishing two diseases. Interestingly, phosphorylated-α-synuclein deposits in cutaneous autonomic nerves can be detected by immunohistochemistry in 56–82% of patients with isolated RBD, a prodromal phase of synucleinopathies [189–193]. The presence of α-synuclein was associated with greater autonomic dysfunction in isolated RBD [192]. The results suggest that evaluation of peripheral nervous system involvement might be a feasible biomarker even in the prodromal phase of synucleinopathies. Increasingly sensitive and specific methods to detect abnormal α-synuclein in peripheral tissues (e.g., proximity ligation assay, real-time quaking-induced conversion [RT-QuIC], or protein misfolding cyclic amplification [PMCA]; see Sect. 5.4) offer hope for peripheral biomarkers for the disease [194–196].
Lewy-related pathology in the peripheral nervous system is not only useful as a promising diagnostic marker, but also important in considering the pathogenesis of Lewy body disease. Braak and colleagues hypothesized that Lewy-related pathology begins in the enteric nervous system and retrogradely propagates to the dorsal motor nucleus of the vagus through the vagal nerve [176]. Several studies using rodent models support this hypothesis [197–199]. Uemura and colleagues inoculated α-synuclein preformed fibrils into the stomach wall of wild-type mice and were able to detect Lewy body-like α-synuclein-positive aggregates in the dorsal motor nucleus of the vagus after variable time intervals [199]. This vagal nucleus α-synuclein pathology was abolished by vagotomy performed before inoculation of α-synuclein fibrils. This provides strong support for retrograde transport of inoculated α-synuclein via the vagus nerve. On the other hand, a study of 466 whole-body autopsies did not find any cases with Lewy-related pathology in the peripheral nervous system without concomitant involvement of the central nervous system, arguing against Braak’s hypothesis [200]. The relation of Lewy-related pathology in the peripheral and central nervous systems remains elucidated [28].
5. Distinct α-synuclein strains in Lewy body disease and MSA
5.1 Distinct seeding activity
As noted above, synucleinopathies are clinically and pathologically heterogeneous, but it remains unknown how the same protein can be associated with distinct pathologies (e.g., Lewy bodies and GCI) and what factors determine neuroanatomical and cell type vulnerability. Accumulating evidence suggests that distinct α-synuclein strains may be associated with different disorders. Several studies using α-synuclein extracted from MSA and Lewy body disease showed distinct seeding activities in vitro and in vivo [201–204]. Prusiner and colleagues reported that extracts from MSA brains, but not from PD brains, induced aggregation of α-synuclein in cultured cells expressing YFP-tagged A53T-mutated human α-synuclein [201]. Similarly, Woerman and colleagues demonstrated that α-synuclein from MSA postmortem brains induced protein aggregations in cultured cells, whereas α-synuclein isolated from Lewy body disease had no effect [202]. Yamasaki and colleagues also demonstrated distinct biochemical properties of α-synuclein from MSA and PD using a FRET biosensor assay based on expression of A53T α-synuclein-CFP/YFP [203]. Both soluble and insoluble fractions of MSA had robust seeding activity, while only insoluble fractions of PD had seeding activity. Moreover, the morphology of induced cellular inclusions was different in MSA and PD. Peng and colleagues demonstrated distinct seeding activity of brain-derived α-synuclein fibrils from GCI and Lewy bodies [204]. They treated primary oligodendrocytes that expressed α-synuclein with GCI-derived α-synuclein and Lewy body-derived α-synuclein and found that GCI-derived α-synuclein was 1,000 times more potent at seeding compared with Lewy body-derived α-synuclein. Injection of GCI-derived α-synuclein induced abundant neuronal inclusions in wild-type mice, but Lewy body-derived α-synuclein did not induce neuronal inclusions at 3 months after the injection. These experiments indicated that α-synuclein derived from GCI had more potent seeding activity in vitro and in vivo than that derived from Lewy bodies, which may correlate with a more aggressive disease course in MSA compared to Lewy body disease [204]. Disease-specific seeding activity may reflect conformational differences of α-synuclein fibrils [204].
5.2 Distinct conformation of α-synuclein with cryo-EM
Cryo-EM has emerged in the last decade as an effective tool for structure determination [205]. The advancement of transmission electron microscope optics and software for data analysis enables three-dimensional reconstruction of macromolecular assemblies at near-atomic resolution. Several studies using cryo-EM have revealed multiple polymorphs from recombinant α-synuclein [206–210]. Li and colleagues investigated full-length recombinant human α-synuclein and determined two predominant polymorphs, which they termed “rod” and “twister.” Both polymorphs were composed of two protofilaments with highly conserved kernel structures, but they differed in inter-protofilament interfaces [207]. The interface between the two protofilaments in the rod polymorph contained residues H50-E57 from the preNAC region (Fig. 7 A), while the interface in the twister polymorph contained residues V66-A78 from the NACore. Notably, six missense mutations (p.E46K, p.H50Q, p.G51D, p.A53E, p.A53T and p.A53V) that cause familial PD are located in the preNAC steric zipper (i.e., residues 46-56) in the rod polymorph. This suggests that PD-associated mutations may disrupt the preNAC zipper of fibril cores in the rod polymorph, while having little impact on the stability of the twister polymorph.
Fig. 7.

The cryo-EM structures of recombinant α-synuclein fibrils. A Two polymorphs of filaments from wild type fibrils: rod (Protein Data Bank [PDB] ID: 6CU7) and twister (PDB ID: 6CU8). B E46K mutant fibrils (PDB ID: 6L43). C H50Q mutant fibrils (PDB ID: 6PES). D A53T mutant fibrils (PDB ID: 6LRQ). Dotted boxes indicate the inter-protofilament interfaces of each α-synuclein fibril
Recent cryo-EM studies using recombinant full-length α-synuclein fibrils with missense mutations have provided more direct evidence on how these mutations affect the conformation of α-synuclein fibrils. N-terminally acetylated p.E46K mutant α-synuclein fibrils had conformational changes in the N-terminal region of the fibril core [211]. The protofilament interface of p.E46K mutant fibril covered V74-Q79, which differed from the interface covering H50-E57 in type 1 polymorph (Fig. 7B). The p.H50Q mutation was associated with two new polymorphs: narrow fibrils and wide fibrils [209]. The narrow fibrils were formed from a single protofilament (protofilament A), whereas the wide fibrils were composed of two protofilaments (protofilaments A and B). The inter-protofilament interface of the wide fibrils had only T59 and K60 (Fig. 7 C). N-terminally acetylated p.A53T mutant α-synuclein fibrils also had a small protofilament interface, consisting of T59 and K60 with no obvious interaction (Fig. 7D) [212]. The protofilament interface of the mutant fibril was less stable than the wild type. These results indicate that missense mutations associated with familial PD can alter the conformation of protofilaments and inter-protofilament interfaces, resulting in variable α-synuclein fibrils with distinct aggregation kinetics, seeding activity, and cytotoxicity.
Although many studies have used recombinant α-synuclein fibrils, evidence suggests that structures of recombinant filaments assembled in vitro may be different from those derived from human brains, as has been observed with tau protein [213–215]. Only a few studies have been reported on α-synuclein derived from human brains. One such study determined the atomic structures of α-synuclein fibrils isolated from MSA brains [216], in which two different types of filaments (type I and type II filaments) were observed (Fig. 8). The ratio of type I to type II differed among MSA patients. Both filaments had two different protofilaments, which consisted of an extended N-terminal arm and a compact C-terminal body. Unlike recombinant α-synuclein fibrils, both filaments were asymmetric. The inter-protofilament interface of type I fibrils contained V37-A53, while that of type II contained V40-A53. Mutations in p.G51D and p.A53E, which cause atypical synucleinopathies with features of PD and MSA [77, 78, 217], are located in this interface. These mutations increase the negative charge around the central cavity, which may lead to their different molecular composition [216]. The interface between the two different protofilaments forms a large cavity surrounded by side chains of K43, K45 and H50 from each protofilament (Fig. 8). This cavity encloses non-proteinaceous molecules. Studies by Puentes and colleagues using computational chemistry hypothesized that non-proteinaceous density in α-synuclein fibrils may be poly(ADP-ribose) (PAR), a negatively charged polymer generated by PAR polymerase-1 [218]. Previous studies have shown that PAR binds to α-synuclein and accelerates α-synuclein fibrillization, which results in cell death via parthanatos [219]. Using proximity ligation assay, they demonstrated PAR-α-synuclein interactions in postmortem brain tissue from PD, PDD, and MSA [218]. Furthermore, they confirmed that PAR and α-synuclein interact via electrostatic forces involving positively charged lysine residues in α-synuclein [218].
Fig. 8.

The cryo-EM structures of α-synuclein fibrils from MSA brains. Two distinct filaments: type I (PDB ID: 6XYO) and type II (PDB ID: 6XYP). Dotted boxes indicate the inter-protofilament interfaces. 43 K, 45 K, and 50 H are shown in red
Interestingly, the same group who reported the cryo-EM structures of α-synuclein filaments from MSA brains investigated whether seeded assemblies of α-synuclein had the same structures as brain-derived seeds [220]. They seeded the in vitro assembly of recombinant wild-type human α-synuclein with α-synuclein derived from MSA brains using PMCA. The resultant filaments showed distinct conformations from the original MSA brain-derived seed, indicating that the products from in vitro seeding do not necessarily replicate the atomic structure of the seed.
5.3 Machine learning-based protein structure prediction
Although advances in cryo-EM enable structural determination at near-atomic resolution, this technique is labor-intensive and not high throughput. As a potential alternative approach, machine learning algorithms can contribute to protein structure predictions [221, 222]. One such algorithm, AlphaFold2, has been used to accurately predict the structures of proteins. These state-of-the-art technologies hold great promise to provide predictions of the atomic-level structure of proteins; however, limitations must be addressed in predicting the structures of amyloid proteins, such as α-synuclein. As noted above, α-synuclein has multiple polymorphs and various intermediates and aggregates; therefore, the same amino acid sequence can produce different structures, which hinder in silico sequence-based predictions [223]. Nevertheless, further understanding of the atomic structure of α-synuclein may elucidate mechanisms of seeding activity and contribute to designing drugs targeting specific structural features of α-synuclein.
5.4 α-Synuclein seeding assays as potential biomarkers
A number of studies have sought fluid biomarkers for synucleinopathies [224–230], but reliable biomarkers are not yet widely available. Recently, biomarkers directly assessing α-synuclein have been developed using RT-QuIC or PMCA. RT-QuIC is an ultrasensitive biochemical assay used to detect self-templating amyloidogenic proteins in brain tissue and cerebrospinal fluid (CSF). It has also been explored to various peripheral tissue types. It was originally developed to detect pathogenic seeding of prions [231, 232], and is currently used in diagnosis of Creutzfeldt-Jakob disease [233]. PMCA is a technology that was originally established to detect misfolded prion aggregates through sonication-based amplification of misfolding and aggregation [234, 235]. In contrast to RT-QuIC, PMCA relies on long-duration shaking instead of more rapid sonication. Both techniques have been successful as biomarkers for aggregation-prone proteins, including α-synuclein [236].
Studies using RT-QuIC or PMCA to diagnose synucleinopathies with CSF samples are summarized in Table 2 [237–249]. Early studies used brain and CSF to develop and validate the RT-QuIC assays. Fairfoul and colleagues demonstrated α-synuclein seeding activity using a small number of CSF samples, and positive reactions were observed in 11/12 autopsy-confirmed cases of pure DLB (92% sensitivity), 11/17 cases of DLB with Alzheimer’s disease (65% sensitivity), 2/13 cases of Alzheimer’s disease with incidental Lewy bodies, and 2/2 cases of PD (100% sensitivity) [237]. None of the healthy controls, 5 cases of tauopathies, and 30 cases of pure Alzheimer’s disease showed positive RT-QuIC. Thus, the overall specificity was 100%. They also investigated CSF from 20 patients with clinically diagnosed PD (95% sensitivity) and 3 patients with isolated RBD (100% sensitivity). Interestingly, α-synuclein positivity with seeding assays was detected in individuals at risk of subsequent diagnosis of PD or DLB, indicating that seeding assays may be useful in detecting prodromal α-synucleinopathies [245].
Table 2.
Studies on α-synuclein RT-QuIC using CSF
| Author | Autopsy | Disease | N | N Control | Sensitivity | Specificity |
|---|---|---|---|---|---|---|
| Fairfoul [237] | Yes | Pure DLB | 12 | 20 | 92% | 100% |
| DLB with AD | 17 | 20 | 65% | 100% | ||
| AD with ILBD | 13 | 20 | 15% | 100% | ||
| PD | 2 | 20 | 100% | 100% | ||
| No | PD | 20 | 15 | 95% | 100% | |
| IRBD | 3 | 15 | 100% | 100% | ||
| Manne [248] | No | PD | 15 | 11 | 94% | 100% |
| Shahnawaz [238] | No | PD | 76 | 65 | 88% | 94% |
| DLB | 10 | 65 | 100% | 94% | ||
| MSA | 10 | 65 | 80% | 94% | ||
| Groveman [239] | Yes | PD | 12 | 31 | 92% | 100% |
| DLB | 17 | 32 | 94% | 100% | ||
| Bongianni [240] | Yes | DLB | 7 | 49 | 100% | 96% |
| MSA | 1 | 50 | 100% | 96% | ||
| Mixed LBD *2 | 20 | 51 | 90% | 96% | ||
| No | DLB | 26 | 10 | 65% | 100% | |
| Kang [241] | No | PD | 105 | 79 | 96% | 82% |
| Van Rumund [242] | Yes *1 | PD | 53 | 52 | 84% | 98% |
| MSA | 17 | 52 | 35% | 98% | ||
| DLB | 1 | 52 | 100% | 98% | ||
| Garrido [243] | No | PD | 10 | 10 | 90% | 80% |
| LRRK2-PD | 15 | 10 | 40% | 80% | ||
| LRRK2-NMC | 16 | 10 | 19% | 80% | ||
| Rossi [244] | Yes | DLB | 14 | 101 | 100% | 98% |
| Mixed LBD *3 | 7 | 102 | 86% | 98% | ||
| No | DLB | 34 | 166 | 97% | 94% | |
| PD | 71 | 167 | 94% | 94% | ||
| IRBD | 18 | 168 | 100% | 94% | ||
| PAF | 28 | 169 | 93% | 94% | ||
| Iranzo [245] | No | IRBD | 52 | 40 | 90% | 90% |
| Shahnawaz [246] | No | PD | 94 | 56 | 94% | 100% |
| MSA | 75 | 56 | 85% | 100% | ||
| PD vs. MSA | 88 | 65 *4 | 97% | 94% | ||
| Rossi [257] | No | MCI-LB | 81 | 58 | 95% | 97% |
| Perra [249] | No | DLB | 16 | 32 | 100% | 90% |
*1: 98% of cases are not autopsy-confirmed. *2 This includes LBD with AD (n = 15), LBD with PART (n = 2), and CJD with LBD (n = 3). *3 This includes CJD with DLB (n = 2), CJD with brainstem LBD (n = 3), and other primary diagnoses with limbic LBD (n = 1) or brainstem LBD (n = 1). *4 This number indicates MSA patients. The sensitivity and specificity are for PD against MSA. Abbreviations: AD Alzheimer’s disease, CJD Creutzfeldt-Jakob disease, DLB Dementia with Lewy bodies, ILBD Incidental Lewy body disease, IRBD Isolated rapid eye movement sleep behavior disorder, LBD Lewy body disease, MCI Mild cognitive impairment, MSA Multiple system atrophy, PD Parkinson’s disease
In addition to studies comparing synucleinopathies to non-synucleinopathies [238], Shahnawaz and colleagues used PMCA to compare CSF from patients with PD and MSA [246]. They found distinct aggregation profiles in PD and MSA. Based on different kinetics of aggregation, they correctly identified PD in 85 of 88 samples (97% sensitivity) and 61 of 65 samples from MSA patients (94% sensitivity). Combining all samples, they correctly distinguished PD from MSA in 146 of the 153 samples for an overall accuracy of 95%.
Recent studies have applied RT-QuIC to biopsies of skin, submandibular gland, and olfactory mucosa (Table 3) [241, 247–252]. Abdominal skin from autopsy-confirmed patients with PD, MSA, Lewy body dementia, and non-synucleinopathies showed positive α-synuclein seeding activity in 44 of 47 PD (94% sensitivity), 2 of 3 MSA (67% sensitivity), all 7 Lewy body dementia (100% sensitivity), and 6 of 73 control group (92% specificity) [252]. This study also evaluated skin biopsy samples from living patients with PD and non-PD subjects, which showed 95% sensitivity and 100% specificity. Manne and colleagues sampled skin tissues of the scalp from patients with PD and control individuals and showed 96% sensitivity and 96% specificity [253]. More recently, a study compared the diagnostic accuracy based on RT-QuIC with α-synuclein immunofluorescence on skin biopsies from C7 paravertebral region and thigh, demonstrating that both RT-QuIC and immunofluorescence showed high diagnostic accuracy in differentiating synucleinopathies from non-synucleinopathies [256]. The submandibular gland was also used for RT-QuIC [251]. Positive seeding activity was present in all 13 cases of PD and 3 cases of incidental Lewy body disease (100% sensitivity), but only 1/16 control cases (94% specificity). Despite the high sensitivity and specificity, the invasiveness of submandibular gland biopsy procedure may limit its clinical application.
Table 3.
Studies on α-synuclein RT-QuIC using peripheral tissue samples
| Author | Tissue | Autopsy | Disease | N | N Control | Sensitivity | Specificity |
|---|---|---|---|---|---|---|---|
| De Luca [250] | OM | No | PD | 18 | 18 | 56% | 83% |
| MSA | 11 | 18 | 82% | 83% | |||
| Manne [251] | SMG | Yes | PD | 13 | 16 | 100% | 94% |
| ILBD | 3 | 16 | 100% | 94% | |||
| Wang [252] | Abdominal skin | Yes | PD | 47 | 73 | 94% | 93% |
| Lewy body dementia | 7 | 73 | 100% | 93% | |||
| MSA | 3 | 73 | 67% | 93% | |||
| Biopsy skin *1 | No | PD | 20 | 21 | 95% | 100% | |
| Manne [253] | Frozen skin *2 | Yes | PD | 25 | 25 | 96% | 96% |
| FFPE skin *2 | PD | 12 | 12 | 75% | 83% | ||
| Mammana [254] | Skin *3 | Yes | PD/DLB | 2 | 40 | 100% | 98% |
| ILBD | 7 | 40 | 86% | 98% | |||
| No | DLB | 15 | 41 | 100% | 95% | ||
| PD | 13 | 41 | 77% | 95% | |||
| Stefani [255] | OM | No | IRBD | 63 | 59 | 44% | 90% |
| PD | 41 | 59 | 46% | 90% | |||
| Perra [249] | OM | No | DLB | 43 | 38 | 81% | 92% |
*1 Biopsy skin samples are obtained from the leg or posterior cervical region. *2 The scalp skin is collected from the posterior lower occipital region along the midline. *3 Skin punches are collected from the cervical region and/or thigh. Abbreviations: FFPE Formalin-fixed paraffin-embedded, ILBD Incidental Lewy body disease, IRBD Isolated rapid eye movement sleep behavior disorder, MSA Multiple system atrophy, PD Parkinson’s disease, OM Olfactory mucosa, SMG Submandibular glands
Olfactory mucosa has also been investigated as a potential sampling site for RT-QuIC [249, 250, 254]. De Luca and colleagues reported that 10/18 cases of PD (56% sensitivity) and 9/11 cases of MSA (82% sensitivity) showed RT-QuIC seeding activity, while 16% of primary tauopathies showed positive (84% specificity) [250]. Perra and colleagues explored the diagnostic accuracy of α-synuclein RT-QuIC using the olfactory mucosa of patients with DLB, which showed 81% sensitivity and 92% specificity [249]. CSF was also analyzed in a subset of patients, resulting in 100% sensitivity and 90% specificity. Stefani and colleagues demonstrated positive α-synuclein RT-QuIC seeding activity in the olfactory mucosa in 44% of isolated RBD patients and 46% in PD patients with an overall specificity of 90% [254]. Interestingly, isolated RBD patients with positive α-synuclein seeding activity had olfactory dysfunction more frequently than those without seeding activity (79% vs. 23%). Although the sensitivity for detecting α-synuclein seeding activity was moderate in these studies [249, 250, 254], olfactory mucosa sampling by nasal swabbing is less invasive than a lumbar puncture, skin biopsy, or submandibular gland biopsy.
Although current diagnostic criteria do not include in vitro seeding assays as supportive biomarkers, these new methods may be involved in future criteria [23, 27, 37, 48, 257]. These assays will not only assist detection of synucleinopathies at an early stage, which is often a clinical challenge [258, 259], but also help recruit patients for future clinical trials of disease-modifying therapies targeting α-synuclein aggregation and propagation. Nevertheless, there are still some obstacles that need to be overcome in order for RT-QuIC to be a routine screening technique. First, the methods of these seeding assays, including a defined purification protocol for the substrate and sampling sites of peripheral tissues, must be standardized [260, 261]. Second, the majority of studies lack autopsy-confirmed diagnosis. Lewy body pathology can coexist with non-synucleinopathies, particularly Alzheimer’s disease; therefore, a positive result in non-synucleinopathies might not be a false-positive, but rather detected copathology. In addition, RT-QuIC and PMCA are more expensive assays compared to phosphorylated-α-synuclein immunofluorescence, which is also a promising approach to diagnose synucleinopathies as discussed in Sect. 4.6. Further studies are needed to confirm the usefulness of seeding assays and immunohistochemistry using the peripheral tissue samples for the diagnosis of synucleinopathies.
6. Conclusions
Synucleinopathies are clinically and pathologically heterogeneous neurodegenerative disorders for which there is currently no cure. To develop disease-modifying therapies for synucleinopathies, it is essential to elucidate how α-synuclein converts to pathologic oligomers and fibrils, which form Lewy bodies in Lewy body disease and GCI in MSA. Interaction between α-synuclein and lipid membranes (e.g., mitochondria, lysosome, synaptic vesicles, etc.) has gained traction as a part of the pathomechanism of Lewy body formation, whereas that of GCI formation remains to be elucidated. Classification and staging schemes are important to understand the clinicopathological heterogeneity of the disease. The current staging schemes of Lewy body disease are based upon evaluation only of the central nervous system. Given that Lewy body pathology is present in the peripheral nervous system, even in prodromal stages of the disease, a more comprehensive staging approach that samples peripheral tissues may provide more comprehensive staging.
The hypothesis that clinicopathologic heterogeneity of synucleinopathies is linked to different strains of α-synuclein is supported by mounting experimental evidence from seeding assays and advanced structural biology. As distinct strains of α-synuclein are increasingly used as biomarkers, diagnostic accuracy will likely improve. Although seeding assays using peripheral tissue samples hold promise, the methodologies need to be standardized for future clinical use. Accurate and early clinical diagnosis will be increasingly crucial for intervention early in the disease for future disease-modifying clinical trials.
Acknowledgements
The authors would like to thank the patients and their families who donated brains to Mayo Clinic brain bank. Figures were created with BioRender.com. Molecular graphics were performed with UCSF Chimera, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco [262].
Abbreviations
- cryo-EM
Cryogenic electron microscopy
- CSF
Cerebrospinal fluid
- DLB
Dementia with Lewy bodies
- FTLD
Frontotemporal lobar degeneration
- GCI
Glial cytoplasmic inclusion
- GWAS
Genome-wide association study
- ICDLB
International Consortium for Lewy Body Dementia
- MSA
Multiple system atrophy
- MSA-C
MSA with predominant cerebellar ataxia
- MSA-P
MSA with predominant parkinsonism
- NAC
Non-amyloid-β component
- NACP
Non-amyloid component in senile plaques
- OPCA
Olivopontocerebellar atrophy
- OR
Odds ratio
- PAR
Poly (ADP-ribose)
- PD
Parkinson’s disease
- PDD
Parkinson’s disease with dementia
- PMCA
Protein misfolding cyclic amplification
- RBD
Rapid eye movement sleep behavior disorder
- RT-QuIC
Real-time quaking-induced conversion
- SND
Striatonigral degeneration
- STED
Stimulated emission depletion
Authors’ contributions
SK, HS, and DWD conceived the manuscript. SK and HS wrote and NK, OAR, and DWD edited the manuscript. SK and HS prepared tables and figures. All authors read and approved the final manuscript.
Funding
This study was supported, in part, by National Institutes of Health grants (P50 NS072187 and U54-NS110435), a Jaye F. and Betty F. Dyer Foundation Fellowship in progressive supranuclear palsy research, CurePSP, and the Center for Individualized Medicine Seed Grant, Mayo Clinic Florida. The Mayo Clinic brain bank is supported by Rainwater Charitable Foundation. Lewy body disease research at the Mayo Clinic is supported by The Little Family Foundation, the Functional Genomics of Lewy body disease Program supported by Ted Turner and family, and the Mangurian Foundation Lewy Body Dementia Program. Mayo Clinic is an American Parkinson Disease Association (APDA) Center for Advanced Research and a Lewy Body Dementia Association (LBDA) Research Center of Excellence.
Availability of data and materials
Not applicable.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Shunsuke Koga, Email: koga.shunsuke@mayo.edu.
Hiroaki Sekiya, Email: sekiya.hiroaki@mayo.edu.
Naveen Kondru, Email: kondru.naveenchandra@mayo.edu.
Owen A. Ross, Email: ross.owen@mayo.edu
Dennis W. Dickson, Email: dickson.dennis@mayo.edu
References
- 1.Hardy J, Gwinn-Hardy K. Genetic classification of primary neurodegenerative disease. Science. 1998;282:1075–1079. doi: 10.1126/science.282.5391.1075. [DOI] [PubMed] [Google Scholar]
- 2.Dickson DW, Lin W, Liu WK, Yen SH. Multiple system atrophy: a sporadic synucleinopathy. Brain Pathol. 1999;9:721–732. doi: 10.1111/j.1750-3639.1999.tb00553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goedert M, Spillantini MG. Lewy body diseases and multiple system atrophy as alpha-synucleinopathies. Mol Psychiatry. 1998;3:462–465. doi: 10.1038/sj.mp.4000458. [DOI] [PubMed] [Google Scholar]
- 4.Kosaka K, Matsushita M, Oyanagi S, Mehraein P. A clinico-neuropathological study of “Lewy body disease.” Psy-chiatry Neurol Jpn. 1980;82:292–311. [PubMed]
- 5.Erskine D, Attems J. Insights into Lewy body disease from rare neurometabolic disorders. J Neural Transm (Vienna) 2021;128(10):1567–1575. doi: 10.1007/s00702-021-02355-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Parkinson J. An essay on the shaking palsy. 1817. J Neuropsychiatry Clin Neurosci. 2002;14:223–236. doi: 10.1176/jnp.14.2.223. [DOI] [PubMed] [Google Scholar]
- 7.Obeso JA, Stamelou M, Goetz CG, Poewe W, Lang AE, Weintraub D, Burn D, Halliday GM, Bezard E, Przedborski S, et al. Past, present, and future of Parkinson’s disease: A special essay on the 200th Anniversary of the Shaking Palsy. Mov Disord. 2017;32:1264–1310. doi: 10.1002/mds.27115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Engelhardt E. Lafora and Tretiakoff: the naming of the inclusion bodies discovered by Lewy. Arq Neuropsiquiatr. 2017;75:751–753. doi: 10.1590/0004-282X20170116. [DOI] [PubMed] [Google Scholar]
- 9.Rodrigues e Silva AM, Geldsetzer F, Holdorff B, Kielhorn FW, Balzer-Geldsetzer M, Oertel WH, Hurtig H, Dodel R. Who was the man who discovered the “Lewy bodies". Mov Disord. 2010;25:1765–1773. doi: 10.1002/mds.22956. [DOI] [PubMed] [Google Scholar]
- 10.Okazaki H, Lipkin LE, Aronson SM. Diffuse intracytoplasmic ganglionic inclusions (Lewy type) associated with progressive dementia and quadriparesis in flexion. J Neuropathol Exp Neurol. 1961;20:237–244. doi: 10.1097/00005072-196104000-00007. [DOI] [PubMed] [Google Scholar]
- 11.Kosaka K, Oyanagi S, Matsushita M, Hori A. Presenile dementia with Alzheimer-, Pick- and Lewy-body changes. Acta Neuropathol. 1976;36:221–233. doi: 10.1007/BF00685366. [DOI] [PubMed] [Google Scholar]
- 12.McKeith IG, Galasko D, Kosaka K, Perry EK, Dickson DW, Hansen LA, Salmon DP, Lowe J, Mirra SS, Byrne EJ, et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology. 1996;47:1113–1124. doi: 10.1212/wnl.47.5.1113. [DOI] [PubMed] [Google Scholar]
- 13.Shy GM, Drager GA. A neurological syndrome associated with orthostatic hypotension: a clinical-pathologic study. Arch Neurol. 1960;2:511–527. doi: 10.1001/archneur.1960.03840110025004. [DOI] [PubMed] [Google Scholar]
- 14.Graham JG, Oppenheimer DR. Orthostatic hypotension and nicotine sensitivity in a case of multiple system atrophy. J Neurol Neurosurg Psychiatry. 1969;32:28–34. doi: 10.1136/jnnp.32.1.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Papp MI, Lantos PL. The distribution of oligodendroglial inclusions in multiple system atrophy and its relevance to clinical symptomatology. Brain. 1994;117(Pt 2):235–243. doi: 10.1093/brain/117.2.235. [DOI] [PubMed] [Google Scholar]
- 16.Papp MI, Kahn JE, Lantos PL. Glial cytoplasmic inclusions in the CNS of patients with multiple system atrophy (striatonigral degeneration, olivopontocerebellar atrophy and Shy-Drager syndrome) J Neurol Sci. 1989;94:79–100. doi: 10.1016/0022-510x(89)90219-0. [DOI] [PubMed] [Google Scholar]
- 17.Iwai A, Masliah E, Yoshimoto M, Ge N, Flanagan L, de Silva HA, Kittel A, Saitoh T. The precursor protein of non-A beta component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron. 1995;14:467–475. doi: 10.1016/0896-6273(95)90302-x. [DOI] [PubMed] [Google Scholar]
- 18.Chen X, de Silva HA, Pettenati MJ, Rao PN, St George-Hyslop P, Roses AD, Xia Y, Horsburgh K, Ueda K, Saitoh T. The human NACP/alpha-synuclein gene: chromosome assignment to 4q21.3-q22 and TaqI RFLP analysis. Genomics. 1995;26:425–427. doi: 10.1016/0888-7543(95)80237-g. [DOI] [PubMed] [Google Scholar]
- 19.George JM. The synucleins. Genome Biol. 2002;3:REVIEWS3002. doi: 10.1186/gb-2001-3-1-reviews3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 21.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 22.Wakabayashi K, Yoshimoto M, Tsuji S, Takahashi H. Alpha-synuclein immunoreactivity in glial cytoplasmic inclusions in multiple system atrophy. Neurosci Lett. 1998;249:180–182. doi: 10.1016/s0304-3940(98)00407-8. [DOI] [PubMed] [Google Scholar]
- 23.Postuma RB, Berg D, Stern M, Poewe W, Olanow CW, Oertel W, Obeso J, Marek K, Litvan I, Lang AE, et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov Disord. 2015;30:1591–1601. doi: 10.1002/mds.26424. [DOI] [PubMed] [Google Scholar]
- 24.Emre M, Aarsland D, Brown R, Burn DJ, Duyckaerts C, Mizuno Y, Broe GA, Cummings J, Dickson DW, Gauthier S, et al. Clinical diagnostic criteria for dementia associated with Parkinson’s disease. Mov Disord. 2007;22:1689–1707. doi: 10.1002/mds.21507. [DOI] [PubMed] [Google Scholar]
- 25.Szeto JYY, Walton CC, Rizos A, Martinez-Martin P, Halliday GM, Naismith SL, Chaudhuri KR, Lewis SJG. Dementia in long-term Parkinson’s disease patients: a multicentre retrospective study. NPJ Parkinsons Dis. 2020;6:2. doi: 10.1038/s41531-019-0106-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hely MA, Reid WG, Adena MA, Halliday GM, Morris JG. The Sydney multicenter study of Parkinson’s disease: the inevitability of dementia at 20 years. Mov Disord. 2008;23:837–844. doi: 10.1002/mds.21956. [DOI] [PubMed] [Google Scholar]
- 27.McKeith IG, Boeve BF, Dickson DW, Halliday G, Taylor JP, Weintraub D, Aarsland D, Galvin J, Attems J, Ballard CG, et al. Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology. 2017;89:88–100. doi: 10.1212/WNL.0000000000004058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Outeiro TF, Koss DJ, Erskine D, Walker L, Kurzawa-Akanbi M, Burn D, Donaghy P, Morris C, Taylor JP, Thomas A, et al. Dementia with Lewy bodies: an update and outlook. Mol Neurodegener. 2019;14:5. doi: 10.1186/s13024-019-0306-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kon T, Tomiyama M, Wakabayashi K. Neuropathology of Lewy body disease: Clinicopathological crosstalk between typical and atypical cases. Neuropathology. 2020;40:30–39. doi: 10.1111/neup.12597. [DOI] [PubMed] [Google Scholar]
- 30.Kasanuki K, Josephs KA, Ferman TJ, Murray ME, Koga S, Konno T, Sakae N, Parks A, Uitti RJ, Van Gerpen JA, et al. Diffuse Lewy body disease manifesting as corticobasal syndrome: A rare form of Lewy body disease. Neurology. 2018;91:e268–e279. doi: 10.1212/WNL.0000000000005828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Buciuc M, Whitwell JL, Kasanuki K, Graff-Radford J, Machulda MM, Duffy JR, Strand EA, Lowe VJ, Graff-Radford NR, Rush BK, et al. Lewy Body Disease is a Contributor to Logopenic Progressive Aphasia Phenotype. Ann Neurol. 2021;89:520–533. doi: 10.1002/ana.25979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koga S, Murakami A, Josephs KA, Dickson DW: Diffuse Lewy body disease presenting as Parkinson disease with progressive aphasia. Neuropathology 2021. In Press [DOI] [PubMed]
- 33.Kapasi A, Brosch JR, Nudelman KN, Agrawal S, Foroud TM, Schneider JA. A novel SNCA E83Q mutation in a case of dementia with Lewy bodies and atypical frontotemporal lobar degeneration. Neuropathology. 2020;40:620–626. doi: 10.1111/neup.12687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Josephs KA. Capgras syndrome and its relationship to neurodegenerative disease. Arch Neurol. 2007;64:1762–1766. doi: 10.1001/archneur.64.12.1762. [DOI] [PubMed] [Google Scholar]
- 35.Kalf JG, de Swart BJ, Bloem BR, Munneke M. Prevalence of oropharyngeal dysphagia in Parkinson’s disease: a meta-analysis. Parkinsonism Relat Disord. 2012;18:311–315. doi: 10.1016/j.parkreldis.2011.11.006. [DOI] [PubMed] [Google Scholar]
- 36.Londos E, Hanxsson O, Alm Hirsch I, Janneskog A, Bulow M, Palmqvist S. Dysphagia in Lewy body dementia - a clinical observational study of swallowing function by videofluoroscopic examination. BMC Neurol. 2013;13:140. doi: 10.1186/1471-2377-13-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gilman S, Wenning GK, Low PA, Brooks DJ, Mathias CJ, Trojanowski JQ, Wood NW, Colosimo C, Durr A, Fowler CJ, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology. 2008;71:670–676. doi: 10.1212/01.wnl.0000324625.00404.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jackson M, Lennox G, Balsitis M, Lowe J. Lewy body dysphagia. J Neurol Neurosurg Psychiatry. 1995;58:756–758. doi: 10.1136/jnnp.58.6.756-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kovari E, Burkhardt K, Lobrinus JA, Bouras C. Lewy body dysphagia. Acta Neuropathol. 2007;114:295–298. doi: 10.1007/s00401-007-0233-6. [DOI] [PubMed] [Google Scholar]
- 40.Shimohata T, Shinoda H, Nakayama H, Ozawa T, Terajima K, Yoshizawa H, Matsuzawa Y, Onodera O, Naruse S, Tanaka K, et al. Daytime hypoxemia, sleep-disordered breathing, and laryngopharyngeal findings in multiple system atrophy. Arch Neurol. 2007;64:856–861. doi: 10.1001/archneur.64.6.856. [DOI] [PubMed] [Google Scholar]
- 41.Calandra-Buonaura G, Alfonsi E, Vignatelli L, Benarroch EE, Giannini G, Iranzo A, Low PA, Martinelli P, Provini F, Quinn N, et al. Dysphagia in multiple system atrophy consensus statement on diagnosis, prognosis and treatment. Parkinsonism Relat Disord. 2021;86:124–132. doi: 10.1016/j.parkreldis.2021.03.027. [DOI] [PubMed] [Google Scholar]
- 42.Stankovic I, Krismer F, Jesic A, Antonini A, Benke T, Brown RG, Burn DJ, Holton JL, Kaufmann H, Kostic VS, et al. Cognitive impairment in multiple system atrophy: A position statement by the neuropsychology task force of the MDS multiple system atrophy (MODIMSA) study group. Mov Disord. 2014;29:857–867. doi: 10.1002/mds.25880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koga S, Parks A, Uitti RJ, van Gerpen JA, Cheshire WP, Wszolek ZK, Dickson DW. Profile of cognitive impairment and underlying pathology in multiple system atrophy. Mov Disord. 2017;32:405–413. doi: 10.1002/mds.26874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miki Y, Foti SC, Hansen D, Strand KM, Asi YT, Tsushima E, Jaunmuktane Z, Lees AJ, Warner TT, Quinn N, et al. Hippocampal alpha-synuclein pathology correlates with memory impairment in multiple system atrophy. Brain. 2020;143:1798–1810. doi: 10.1093/brain/awaa126. [DOI] [PubMed] [Google Scholar]
- 45.Schenck CH, Bundlie SR, Patterson AL, Mahowald MW. Rapid eye movement sleep behavior disorder. A treatable parasomnia affecting older adults. JAMA. 1987;257:1786–1789. [PubMed] [Google Scholar]
- 46.Kaufmann H, Norcliffe-Kaufmann L, Palma JA, Biaggioni I, Low PA, Singer W, Goldstein DS, Peltier AC, Shibao CA, Gibbons CH, et al. Natural history of pure autonomic failure: A United States prospective cohort. Ann Neurol. 2017;81:287–297. doi: 10.1002/ana.24877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koga S, Cheshire WP, Tipton PW, Driver-Dunckley ED, Wszolek ZK, Uitti RJ, Graff-Radford NR, van Gerpen JA, Dickson DW. Clinical features of autopsy-confirmed multiple system atrophy in the Mayo Clinic Florida brain bank. Parkinsonism Relat Disord. 2021;89:155–161. doi: 10.1016/j.parkreldis.2021.07.007. [DOI] [PubMed] [Google Scholar]
- 48.Stankovic I, Quinn N, Vignatelli L, Antonini A, Berg D, Coon E, Cortelli P, Fanciulli A, Ferreira JJ, Freeman R, et al. A critique of the second consensus criteria for multiple system atrophy. Mov Disord. 2019;34:975–984. doi: 10.1002/mds.27701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schenck CH, Boeve BF, Mahowald MW. Delayed emergence of a parkinsonian disorder or dementia in 81% of older men initially diagnosed with idiopathic rapid eye movement sleep behavior disorder: a 16-year update on a previously reported series. Sleep Med. 2013;14:744–748. doi: 10.1016/j.sleep.2012.10.009. [DOI] [PubMed] [Google Scholar]
- 50.Iranzo A, Tolosa E, Gelpi E, Molinuevo JL, Valldeoriola F, Serradell M, Sanchez-Valle R, Vilaseca I, Lomena F, Vilas D, et al. Neurodegenerative disease status and post-mortem pathology in idiopathic rapid-eye-movement sleep behaviour disorder: an observational cohort study. Lancet Neurol. 2013;12:443–453. doi: 10.1016/S1474-4422(13)70056-5. [DOI] [PubMed] [Google Scholar]
- 51.Postuma RB, Iranzo A, Hu M, Hogl B, Boeve BF, Manni R, Oertel WH, Arnulf I, Ferini-Strambi L, Puligheddu M, et al. Risk and predictors of dementia and parkinsonism in idiopathic REM sleep behaviour disorder: a multicentre study. Brain. 2019;142:744–759. doi: 10.1093/brain/awz030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jakes R, Spillantini MG, Goedert M. Identification of two distinct synucleins from human brain. FEBS Lett. 1994;345:27–32. doi: 10.1016/0014-5793(94)00395-5. [DOI] [PubMed] [Google Scholar]
- 53.Davidson WS, Jonas A, Clayton DF, George JM. Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem. 1998;273:9443–9449. doi: 10.1074/jbc.273.16.9443. [DOI] [PubMed] [Google Scholar]
- 54.Li HT, Du HN, Tang L, Hu J, Hu HY. Structural transformation and aggregation of human alpha-synuclein in trifluoroethanol: non-amyloid component sequence is essential and beta-sheet formation is prerequisite to aggregation. Biopolymers. 2002;64:221–226. doi: 10.1002/bip.10179. [DOI] [PubMed] [Google Scholar]
- 55.Crowther RA, Jakes R, Spillantini MG, Goedert M. Synthetic filaments assembled from C-terminally truncated alpha-synuclein. FEBS Lett. 1998;436:309–312. doi: 10.1016/s0014-5793(98)01146-6. [DOI] [PubMed] [Google Scholar]
- 56.Lashuel HA, Overk CR, Oueslati A, Masliah E. The many faces of alpha-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci. 2013;14:38–48. doi: 10.1038/nrn3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bartels T, Choi JG, Selkoe DJ. alpha-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature. 2011;477:107–110. doi: 10.1038/nature10324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang W, Perovic I, Chittuluru J, Kaganovich A, Nguyen LT, Liao J, Auclair JR, Johnson D, Landeru A, Simorellis AK, et al. A soluble alpha-synuclein construct forms a dynamic tetramer. Proc Natl Acad Sci U S A. 2011;108:17797–17802. doi: 10.1073/pnas.1113260108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Killinger BA, Melki R, Brundin P, Kordower JH. Endogenous alpha-synuclein monomers, oligomers and resulting pathology: let’s talk about the lipids in the room. NPJ Parkinsons Dis. 2019;5:23. doi: 10.1038/s41531-019-0095-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Suzuki M, Sango K, Wada K, Nagai Y. Pathological role of lipid interaction with alpha-synuclein in Parkinson’s disease. Neurochem Int. 2018;119:97–106. doi: 10.1016/j.neuint.2017.12.014. [DOI] [PubMed] [Google Scholar]
- 61.Musteikyte G, Jayaram AK, Xu CK, Vendruscolo M, Krainer G, Knowles TPJ. Interactions of alpha-synuclein oligomers with lipid membranes. Biochim Biophys Acta Biomembr. 2021;1863:183536. doi: 10.1016/j.bbamem.2020.183536. [DOI] [PubMed] [Google Scholar]
- 62.Burre J, Sharma M, Sudhof TC. Cell Biology and Pathophysiology of alpha-Synuclein. Csh Perspect Med. 2018;8(3):a024091. doi: 10.1101/cshperspect.a024091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nuber S, Rajsombath M, Minakaki G, Winkler J, Muller CP, Ericsson M, Caldarone B, Dettmer U, Selkoe DJ. Abrogating Native alpha-Synuclein Tetramers in Mice Causes a L-DOPA-Responsive Motor Syndrome Closely Resembling Parkinson’s Disease. Neuron. 2018;100:75-90 e75. doi: 10.1016/j.neuron.2018.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dettmer U, Newman AJ, Soldner F, Luth ES, Kim NC, von Saucken VE, Sanderson JB, Jaenisch R, Bartels T, Selkoe D. Parkinson-causing alpha-synuclein missense mutations shift native tetramers to monomers as a mechanism for disease initiation. Nat Commun. 2015;6:7314. doi: 10.1038/ncomms8314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fanning S, Haque A, Imberdis T, Baru V, Barrasa MI, Nuber S, Termine D, Ramalingam N, Ho GPH, Noble T, et al. Lipidomic Analysis of alpha-Synuclein Neurotoxicity Identifies Stearoyl CoA Desaturase as a Target for Parkinson Treatment. Mol Cell. 2019;73:1001-1014 e1008. doi: 10.1016/j.molcel.2018.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jiang P, Gan M, Yen SH, Moussaud S, McLean PJ, Dickson DW. Proaggregant nuclear factor(s) trigger rapid formation of alpha-synuclein aggregates in apoptotic neurons. Acta Neuropathol. 2016;132:77–91. doi: 10.1007/s00401-016-1542-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jiang P, Gan M, Yen SH, McLean PJ, Dickson DW. Histones facilitate alpha-synuclein aggregation during neuronal apoptosis. Acta Neuropathol. 2017;133:547–558. doi: 10.1007/s00401-016-1660-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shahmoradian SH, Lewis AJ, Genoud C, Hench J, Moors TE, Navarro PP, Castano-Diez D, Schweighauser G, Graff-Meyer A, Goldie KN, et al. Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat Neurosci. 2019;22:1099–1109. doi: 10.1038/s41593-019-0423-2. [DOI] [PubMed] [Google Scholar]
- 69.Zhang J, Li X, Li JD. The Roles of Post-translational Modifications on alpha-Synuclein in the Pathogenesis of Parkinson’s Diseases. Front Neurosci. 2019;13:381. doi: 10.3389/fnins.2019.00381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Savyon M, Engelender S. SUMOylation in alpha-Synuclein Homeostasis and Pathology. Front Aging Neurosci. 2020;12:167. doi: 10.3389/fnagi.2020.00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, Iwatsubo T. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol. 2002;4:160–164. doi: 10.1038/ncb748. [DOI] [PubMed] [Google Scholar]
- 72.Moors TE, Maat CA, Niedieker D, Mona D, Petersen D, Timmermans-Huisman E, Kole J, El-Mashtoly SF, Spycher L, Zago W, et al. The subcellular arrangement of alpha-synuclein proteoforms in the Parkinson’s disease brain as revealed by multicolor STED microscopy. Acta Neuropathol. 2021;142:423–448. doi: 10.1007/s00401-021-02329-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- 74.Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atares B, et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55:164–173. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- 75.Appel-Cresswell S, Vilarino-Guell C, Encarnacion M, Sherman H, Yu I, Shah B, Weir D, Thompson C, Szu-Tu C, Trinh J, et al. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov Disord. 2013;28:811–813. doi: 10.1002/mds.25421. [DOI] [PubMed] [Google Scholar]
- 76.Hoffman-Zacharska D, Koziorowski D, Ross OA, Milewski M, Poznanski JA, Jurek M, Wszolek ZK, Soto-Ortolaza A, Awek JAS, Janik P, et al. Novel A18T and pA29S substitutions in alpha-synuclein may be associated with sporadic Parkinson’s disease. Parkinsonism Relat Disord. 2013;19:1057–1060. doi: 10.1016/j.parkreldis.2013.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kiely AP, Asi YT, Kara E, Limousin P, Ling H, Lewis P, Proukakis C, Quinn N, Lees AJ, Hardy J, et al. alpha-Synucleinopathy associated with G51D SNCA mutation: a link between Parkinson’s disease and multiple system atrophy? Acta Neuropathol. 2013;125:753–769. doi: 10.1007/s00401-013-1096-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pasanen P, Myllykangas L, Siitonen M, Raunio A, Kaakkola S, Lyytinen J, Tienari PJ, Poyhonen M, Paetau A. Novel alpha-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol Aging. 2014;35:2180 e2181-2185. doi: 10.1016/j.neurobiolaging.2014.03.024. [DOI] [PubMed] [Google Scholar]
- 79.Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 80.Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, Levecque C, Larvor L, Andrieux J, Hulihan M, et al. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet. 2004;364:1167–1169. doi: 10.1016/S0140-6736(04)17103-1. [DOI] [PubMed] [Google Scholar]
- 81.Fujishiro H, Imamura AY, Lin WL, Uchikado H, Mark MH, Golbe LI, Markopoulou K, Wszolek ZK, Dickson DW. Diversity of pathological features other than Lewy bodies in familial Parkinson’s disease due to SNCA mutations. Am J Neurodegener Dis. 2013;2:266–275. [PMC free article] [PubMed] [Google Scholar]
- 82.Konno T, Ross OA, Puschmann A, Dickson DW, Wszolek ZK. Autosomal dominant Parkinson’s disease caused by SNCA duplications. Parkinsonism Relat Disord. 2016;22(Suppl 1):S1-6. doi: 10.1016/j.parkreldis.2015.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Blauwendraat C, Nalls MA, Singleton AB. The genetic architecture of Parkinson’s disease. Lancet Neurol. 2020;19:170–178. doi: 10.1016/S1474-4422(19)30287-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Vazquez-Velez GE, Zoghbi HY. Parkinson’s Disease Genetics and Pathophysiology. Annu Rev Neurosci. 2021;44:87–108. doi: 10.1146/annurev-neuro-100720-034518. [DOI] [PubMed] [Google Scholar]
- 85.Satake W, Nakabayashi Y, Mizuta I, Hirota Y, Ito C, Kubo M, Kawaguchi T, Tsunoda T, Watanabe M, Takeda A, et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet. 2009;41:1303–1307. doi: 10.1038/ng.485. [DOI] [PubMed] [Google Scholar]
- 86.Simon-Sanchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, Paisan-Ruiz C, Lichtner P, Scholz SW, Hernandez DG, et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet. 2009;41:1308–1312. doi: 10.1038/ng.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Beecham GW, Dickson DW, Scott WK, Martin ER, Schellenberg G, Nuytemans K, Larson EB, Buxbaum JD, Trojanowski JQ, Van Deerlin VM, et al. PARK10 is a major locus for sporadic neuropathologically confirmed Parkinson disease. Neurology. 2015;84:972–980. doi: 10.1212/WNL.0000000000001332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lautier C, Goldwurm S, Durr A, Giovannone B, Tsiaras WG, Pezzoli G, Brice A, Smith RJ. Mutations in the GIGYF2 (TNRC15) gene at the PARK11 locus in familial Parkinson disease. Am J Hum Genet. 2008;82:822–833. doi: 10.1016/j.ajhg.2008.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Foo JN, Chew EGY, Chung SJ, Peng R, Blauwendraat C, Nalls MA, Mok KY, Satake W, Toda T, Chao Y, et al. Identification of Risk Loci for Parkinson Disease in Asians and Comparison of Risk Between Asians and Europeans: A Genome-Wide Association Study. JAMA Neurol. 2020;77:746–754. doi: 10.1001/jamaneurol.2020.0428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chang D, Nalls MA, Hallgrimsdottir IB, Hunkapiller J, van der Brug M, Cai F, International Parkinson’s Disease Genomics C, and Me Research T. Kerchner GA, Ayalon G, et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat Genet. 2017;49:1511–1516. doi: 10.1038/ng.3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chia R, Sabir MS, Bandres-Ciga S, Saez-Atienzar S, Reynolds RH, Gustavsson E, Walton RL, Ahmed S, Viollet C, Ding J, et al. Genome sequencing analysis identifies new loci associated with Lewy body dementia and provides insights into its genetic architecture. Nat Genet. 2021;53:294–303. doi: 10.1038/s41588-021-00785-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Guerreiro R, Ross OA, Kun-Rodrigues C, Hernandez DG, Orme T, Eicher JD, Shepherd CE, Parkkinen L, Darwent L, Heckman MG, et al. Investigating the genetic architecture of dementia with Lewy bodies: a two-stage genome-wide association study. Lancet Neurol. 2018;17:64–74. doi: 10.1016/S1474-4422(17)30400-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, Bar-Shira A, Berg D, Bras J, Brice A, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med. 2009;361:1651–1661. doi: 10.1056/NEJMoa0901281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Do J, McKinney C, Sharma P, Sidransky E. Glucocerebrosidase and its relevance to Parkinson disease. Mol Neurodegener. 2019;14:36. doi: 10.1186/s13024-019-0336-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lopez G, Kim J, Wiggs E, Cintron D, Groden C, Tayebi N, Mistry PK, Pastores GM, Zimran A, Goker-Alpan O, Sidransky E. Clinical course and prognosis in patients with Gaucher disease and parkinsonism. Neurol Genet. 2016;2:e57. doi: 10.1212/NXG.0000000000000057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wernick AI, Walton RL, Koga S, Soto-Beasley AI, Heckman MG, Gan-Or Z, Ren Y, Rademakers R, Uitti RJ, Wszolek ZK, et al. GBA variation and susceptibility to multiple system atrophy. Parkinsonism Relat Disord. 2020;77:64–69. doi: 10.1016/j.parkreldis.2020.06.007. [DOI] [PubMed] [Google Scholar]
- 97.Mitsui J, Matsukawa T, Sasaki H, Yabe I, Matsushima M, Durr A, Brice A, Takashima H, Kikuchi A, Aoki M, et al. Variants associated with Gaucher disease in multiple system atrophy. Ann Clin Transl Neurol. 2015;2:417–426. doi: 10.1002/acn3.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hara K, Momose Y, Tokiguchi S, Shimohata M, Terajima K, Onodera O, Kakita A, Yamada M, Takahashi H, Hirasawa M, et al. Multiplex families with multiple system atrophy. Arch Neurol. 2007;64:545–551. doi: 10.1001/archneur.64.4.545. [DOI] [PubMed] [Google Scholar]
- 99.Multiple-System Atrophy Research C. Mutations in COQ2 in familial and sporadic multiple-system atrophy. N Engl J Med. 2013;369:233–244. doi: 10.1056/NEJMoa1212115. [DOI] [PubMed] [Google Scholar]
- 100.Sharma M, Wenning G, Kruger R. European Multiple-System Atrophy Study G: Mutant COQ2 in multiple-system atrophy. N Engl J Med. 2014;371:80–81. doi: 10.1056/NEJMc1311763. [DOI] [PubMed] [Google Scholar]
- 101.Schottlaender LV, Houlden H, Multiple-System Atrophy Brain Bank C. Mutant COQ2 in multiple-system atrophy. N Engl J Med. 2014;371:81. doi: 10.1056/NEJMc1311763. [DOI] [PubMed] [Google Scholar]
- 102.Zhao Q, Yang X, Tian S, An R, Zheng J, Xu Y. Association of the COQ2 V393A variant with risk of multiple system atrophy in East Asians: a case-control study and meta-analysis of the literature. Neurol Sci. 2016;37:423–430. doi: 10.1007/s10072-015-2414-8. [DOI] [PubMed] [Google Scholar]
- 103.Ozawa T, Paviour D, Quinn NP, Josephs KA, Sangha H, Kilford L, Healy DG, Wood NW, Lees AJ, Holton JL, Revesz T. The spectrum of pathological involvement of the striatonigral and olivopontocerebellar systems in multiple system atrophy: clinicopathological correlations. Brain. 2004;127:2657–2671. doi: 10.1093/brain/awh303. [DOI] [PubMed] [Google Scholar]
- 104.Ozawa T, Tada M, Kakita A, Onodera O, Ishihara T, Morita T, Shimohata T, Wakabayashi K, Takahashi H, Nishizawa M. The phenotype spectrum of Japanese multiple system atrophy. Journal of neurology, neurosurgery, and psychiatry. 2010;81:1253–1255. doi: 10.1136/jnnp.2009.182576. [DOI] [PubMed] [Google Scholar]
- 105.Schottlaender LV, Bettencourt C, Kiely AP, Chalasani A, Neergheen V, Holton JL, Hargreaves I, Houlden H. Coenzyme Q10 Levels Are Decreased in the Cerebellum of Multiple-System Atrophy Patients. PLoS One. 2016;11:e0149557. doi: 10.1371/journal.pone.0149557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Barca E, Kleiner G, Tang G, Ziosi M, Tadesse S, Masliah E, Louis ED, Faust P, Kang UJ, Torres J, et al. Decreased Coenzyme Q10 Levels in Multiple System Atrophy Cerebellum. J Neuropathol Exp Neurol. 2016;75:663–672. doi: 10.1093/jnen/nlw037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sailer A, Scholz SW, Nalls MA, Schulte C, Federoff M, Price TR, Lees A, Ross OA, Dickson DW, Mok K, et al. A genome-wide association study in multiple system atrophy. Neurology. 2016;87:1591–1598. doi: 10.1212/WNL.0000000000003221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gu X, Chen Y, Zhou Q, Lu YC, Cao B, Zhang L, Kuo MC, Wu YR, Wu RM, Tan EK, et al. Analysis of GWAS-linked variants in multiple system atrophy. Neurobiol Aging. 2018;67:201 e201–201 e204. doi: 10.1016/j.neurobiolaging.2018.03.018. [DOI] [PubMed] [Google Scholar]
- 109.Dickson DW, Braak H, Duda JE, Duyckaerts C, Gasser T, Halliday GM, Hardy J, Leverenz JB, Del Tredici K, Wszolek ZK, Litvan I. Neuropathological assessment of Parkinson’s disease: refining the diagnostic criteria. Lancet Neurol. 2009;8:1150–1157. doi: 10.1016/S1474-4422(09)70238-8. [DOI] [PubMed] [Google Scholar]
- 110.Fearnley JM, Lees AJ. Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain. 1991;114(Pt 5):2283–2301. doi: 10.1093/brain/114.5.2283. [DOI] [PubMed] [Google Scholar]
- 111.Greffard S, Verny M, Bonnet AM, Beinis JY, Gallinari C, Meaume S, Piette F, Hauw JJ, Duyckaerts C. Motor score of the Unified Parkinson Disease Rating Scale as a good predictor of Lewy body-associated neuronal loss in the substantia nigra. Arch Neurol. 2006;63:584–588. doi: 10.1001/archneur.63.4.584. [DOI] [PubMed] [Google Scholar]
- 112.Kasanuki K, Heckman MG, Diehl NN, Murray ME, Koga S, Soto A, Ross OA, Dickson DW. Regional analysis and genetic association of nigrostriatal degeneration in Lewy body disease. Mov Disord. 2017;32:1584–1593. doi: 10.1002/mds.27184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Oertel WH, Henrich MT, Janzen A, Geibl FF. The locus coeruleus: Another vulnerability target in Parkinson’s disease. Mov Disord. 2019;34:1423–1429. doi: 10.1002/mds.27785. [DOI] [PubMed] [Google Scholar]
- 114.Wakabayashi K, Tanji K, Odagiri S, Miki Y, Mori F, Takahashi H. The Lewy body in Parkinson’s disease and related neurodegenerative disorders. Mol Neurobiol. 2013;47:495–508. doi: 10.1007/s12035-012-8280-y. [DOI] [PubMed] [Google Scholar]
- 115.Dugger BN, Dickson DW. Cell type specific sequestration of choline acetyltransferase and tyrosine hydroxylase within Lewy bodies. Acta Neuropathol. 2010;120:633–639. doi: 10.1007/s00401-010-0739-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Pollanen MS, Dickson DW, Bergeron C. Pathology and biology of the Lewy body. J Neuropathol Exp Neurol. 1993;52:183–191. doi: 10.1097/00005072-199305000-00001. [DOI] [PubMed] [Google Scholar]
- 117.Dickson DW, Ruan D, Crystal H, Mark MH, Davies P, Kress Y, Yen SH. Hippocampal degeneration differentiates diffuse Lewy body disease (DLBD) from Alzheimer’s disease: light and electron microscopic immunocytochemistry of CA2-3 neurites specific to DLBD. Neurology. 1991;41:1402–1409. doi: 10.1212/wnl.41.9.1402. [DOI] [PubMed] [Google Scholar]
- 118.Wakabayashi K, Hayashi S, Yoshimoto M, Kudo H, Takahashi H. NACP/alpha-synuclein-positive filamentous inclusions in astrocytes and oligodendrocytes of Parkinson’s disease brains. Acta Neuropathol. 2000;99:14–20. doi: 10.1007/pl00007400. [DOI] [PubMed] [Google Scholar]
- 119.Sherzai A, Edland SD, Masliah E, Hansen L, Pizzo DP, Sherzai A, Corey-Bloom J. Spongiform change in dementia with Lewy bodies and Alzheimer disease. Alzheimer Dis Assoc Disord. 2013;27:157–161. doi: 10.1097/WAD.0b013e318256d507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Iseki E, Li F, Kosaka K. Close relationship between spongiform change and ubiquitin-positive granular structures in diffuse Lewy body disease. J Neurol Sci. 1997;146:53–57. doi: 10.1016/s0022-510x(96)00282-1. [DOI] [PubMed] [Google Scholar]
- 121.Hansen LA, Masliah E, Terry RD, Mirra SS. A neuropathological subset of Alzheimer’s disease with concomitant Lewy body disease and spongiform change. Acta Neuropathol. 1989;78:194–201. doi: 10.1007/BF00688209. [DOI] [PubMed] [Google Scholar]
- 122.Kosaka K. Diffuse Lewy body disease in Japan. J Neurol. 1990;237:197–204. doi: 10.1007/BF00314594. [DOI] [PubMed] [Google Scholar]
- 123.Jellinger KA, Seppi K, Wenning GK, Poewe W. Impact of coexistent Alzheimer pathology on the natural history of Parkinson’s disease. J Neural Transm (Vienna) 2002;109:329–339. doi: 10.1007/s007020200027. [DOI] [PubMed] [Google Scholar]
- 124.Marui W, Iseki E, Kato M, Akatsu H, Kosaka K. Pathological entity of dementia with Lewy bodies and its differentiation from Alzheimer’s disease. Acta Neuropathol. 2004;108:121–128. doi: 10.1007/s00401-004-0869-4. [DOI] [PubMed] [Google Scholar]
- 125.Dickson DW, Heckman MG, Murray ME, Soto AI, Walton RL, Diehl NN, van Gerpen JA, Uitti RJ, Wszolek ZK, Ertekin-Taner N, et al. APOE epsilon4 is associated with severity of Lewy body pathology independent of Alzheimer pathology. Neurology. 2018;91:e1182–e1195. doi: 10.1212/WNL.0000000000006212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Dickson DW, Crystal H, Mattiace LA, Kress Y, Schwagerl A, Ksiezak-Reding H, Davies P, Yen SH. Diffuse Lewy body disease: light and electron microscopic immunocytochemistry of senile plaques. Acta Neuropathol. 1989;78:572–584. doi: 10.1007/BF00691284. [DOI] [PubMed] [Google Scholar]
- 127.Irwin DJ, Lee VM, Trojanowski JQ. Parkinson’s disease dementia: convergence of alpha-synuclein, tau and amyloid-beta pathologies. Nat Rev Neurosci. 2013;14:626–636. doi: 10.1038/nrn3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Walker L, Stefanis L, Attems J. Clinical and neuropathological differences between Parkinson’s disease, Parkinson’s disease dementia and dementia with Lewy bodies - current issues and future directions. J Neurochem. 2019;150:467–474. doi: 10.1111/jnc.14698. [DOI] [PubMed] [Google Scholar]
- 129.Dugger BN, Adler CH, Shill HA, Caviness J, Jacobson S, Driver-Dunckley E, Beach TG. Arizona Parkinson’s Disease C: Concomitant pathologies among a spectrum of parkinsonian disorders. Parkinsonism Relat Disord. 2014;20:525–529. doi: 10.1016/j.parkreldis.2014.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lippa CF, Fujiwara H, Mann DM, Giasson B, Baba M, Schmidt ML, Nee LE, O’Connell B, Pollen DA, St George-Hyslop P, et al. Lewy bodies contain altered alpha-synuclein in brains of many familial Alzheimer’s disease patients with mutations in presenilin and amyloid precursor protein genes. Am J Pathol. 1998;153:1365–1370. doi: 10.1016/s0002-9440(10)65722-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hamilton RL. Lewy bodies in Alzheimer’s disease: a neuropathological review of 145 cases using alpha-synuclein immunohistochemistry. Brain Pathol. 2000;10:378–384. doi: 10.1111/j.1750-3639.2000.tb00269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Popescu A, Lippa CF, Lee VM, Trojanowski JQ. Lewy bodies in the amygdala: increase of alpha-synuclein aggregates in neurodegenerative diseases with tau-based inclusions. Arch Neurol. 2004;61:1915–1919. doi: 10.1001/archneur.61.12.1915. [DOI] [PubMed] [Google Scholar]
- 133.Uchikado H, Lin WL, DeLucia MW, Dickson DW. Alzheimer disease with amygdala Lewy bodies: a distinct form of alpha-synucleinopathy. J Neuropathol Exp Neurol. 2006;65:685–697. doi: 10.1097/01.jnen.0000225908.90052.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Twohig D, Nielsen HM. alpha-synuclein in the pathophysiology of Alzheimer’s disease. Mol Neurodegener. 2019;14:23. doi: 10.1186/s13024-019-0320-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Roudil J, Deramecourt V, Dufournet B, Dubois B, Ceccaldi M, Duyckaerts C, Pasquier F, Lebouvier T, Brainbank Neuro CEBNN. Influence of Lewy Pathology on Alzheimer’s Disease Phenotype: A Retrospective Clinico-Pathological Study. J Alzheimers Dis. 2018;63:1317–1323. doi: 10.3233/JAD-170914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ferman TJ, Arvanitakis Z, Fujishiro H, Duara R, Parfitt F, Purdy M, Waters C, Barker W, Graff-Radford NR, Dickson DW. Pathology and temporal onset of visual hallucinations, misperceptions and family misidentification distinguishes dementia with Lewy bodies from Alzheimer’s disease. Parkinsonism Relat Disord. 2013;19:227–231. doi: 10.1016/j.parkreldis.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Roberts RF, Wade-Martins R, Alegre-Abarrategui J. Direct visualization of alpha-synuclein oligomers reveals previously undetected pathology in Parkinson’s disease brain. Brain. 2015;138:1642–1657. doi: 10.1093/brain/awv040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kosaka K, Yoshimura M, Ikeda K, Budka H. Diffuse type of Lewy body disease: progressive dementia with abundant cortical Lewy bodies and senile changes of varying degree--a new disease? Clin Neuropathol. 1984;3:185–192. [PubMed] [Google Scholar]
- 139.Kosaka K, Iseki E, Odawara T, Yamamoto T. Cerebral type of Lewy body disease. Neuropathology. 1996;16:32–35. [Google Scholar]
- 140.McKeith IG, Dickson DW, Lowe J, Emre M, O’Brien JT, Feldman H, Cummings J, Duda JE, Lippa C, Perry EK, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology. 2005;65:1863–1872. doi: 10.1212/01.wnl.0000187889.17253.b1. [DOI] [PubMed] [Google Scholar]
- 141.Lopez OL, Hamilton RL, Becker JT, Wisniewski S, Kaufer DI, DeKosky ST. Severity of cognitive impairment and the clinical diagnosis of AD with Lewy bodies. Neurology. 2000;54:1780–1787. doi: 10.1212/wnl.54.9.1780. [DOI] [PubMed] [Google Scholar]
- 142.Ferman TJ, Aoki N, Boeve BF, Aakre JA, Kantarci K, Graff-Radford J, Parisi JE, Van Gerpen JA, Graff-Radford NR, Uitti RJ, et al. Subtypes of dementia with Lewy bodies are associated with alpha-synuclein and tau distribution. Neurology. 2020;95:e155–e165. doi: 10.1212/WNL.0000000000009763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 144.Dickson DW, Uchikado H, Fujishiro H, Tsuboi Y. Evidence in favor of Braak staging of Parkinson’s disease. Mov Disord. 2010;25(Suppl 1):S78-82. doi: 10.1002/mds.22637. [DOI] [PubMed] [Google Scholar]
- 145.Beach TG, Adler CH, Lue L, Sue LI, Bachalakuri J, Henry-Watson J, Sasse J, Boyer S, Shirohi S, Brooks R, et al. Unified staging system for Lewy body disorders: correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol. 2009;117:613–634. doi: 10.1007/s00401-009-0538-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Attems J, Neltner JH, Nelson PT. Quantitative neuropathological assessment to investigate cerebral multi-morbidity. Alzheimers Res Ther. 2014;6:85. doi: 10.1186/s13195-014-0085-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Attems J, Toledo JB, Walker L, Gelpi E, Gentleman S, Halliday G, Hortobagyi T, Jellinger K, Kovacs GG, Lee EB, et al. Neuropathological consensus criteria for the evaluation of Lewy pathology in post-mortem brains: a multi-centre study. Acta Neuropathol. 2021;141:159–172. doi: 10.1007/s00401-020-02255-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Koga S, Li F, Zhao N, Roemer SF, Ferman TJ, Wernick AI, Walton RL, Faroqi AH, Graff-Radford NR, Cheshire WP, et al. Clinicopathologic and genetic features of multiple system atrophy with Lewy body disease. Brain Pathol. 2020;30:766–778. doi: 10.1111/bpa.12839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Fujishiro H, Ferman TJ, Boeve BF, Smith GE, Graff-Radford NR, Uitti RJ, Wszolek ZK, Knopman DS, Petersen RC, Parisi JE, Dickson DW. Validation of the neuropathologic criteria of the third consortium for dementia with Lewy bodies for prospectively diagnosed cases. J Neuropathol Exp Neurol. 2008;67:649–656. doi: 10.1097/NEN.0b013e31817d7a1d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Dickson DW. Neuropathology of Parkinson disease. Parkinsonism Relat Disord. 2018;46(Suppl 1):S30–S33. [DOI] [PMC free article] [PubMed]
- 151.Jellinger KA, Attems J. Does striatal pathology distinguish Parkinson disease with dementia and dementia with Lewy bodies? Acta Neuropathol. 2006;112:253–260. doi: 10.1007/s00401-006-0088-2. [DOI] [PubMed] [Google Scholar]
- 152.Tsuboi Y, Uchikado H, Dickson DW. Neuropathology of Parkinson’s disease dementia and dementia with Lewy bodies with reference to striatal pathology. Parkinsonism Relat Disord. 2007;13(Suppl 3):S221-224. doi: 10.1016/S1353-8020(08)70005-1. [DOI] [PubMed] [Google Scholar]
- 153.Trojanowski JQ, Revesz T. Proposed neuropathological criteria for the post mortem diagnosis of multiple system atrophy. Neuropathol Appl Neurobiol. 2007;33:615–620. doi: 10.1111/j.1365-2990.2007.00907.x. [DOI] [PubMed] [Google Scholar]
- 154.Miller DW, Johnson JM, Solano SM, Hollingsworth ZR, Standaert DG, Young AB. Absence of alpha-synuclein mRNA expression in normal and multiple system atrophy oligodendroglia. J Neural Transm (Vienna) 2005;112:1613–1624. doi: 10.1007/s00702-005-0378-1. [DOI] [PubMed] [Google Scholar]
- 155.Asi YT, Simpson JE, Heath PR, Wharton SB, Lees AJ, Revesz T, Houlden H, Holton JL. Alpha-synuclein mRNA expression in oligodendrocytes in MSA. Glia. 2014;62:964–970. doi: 10.1002/glia.22653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Konno M, Hasegawa T, Baba T, Miura E, Sugeno N, Kikuchi A, Fiesel FC, Sasaki T, Aoki M, Itoyama Y, Takeda A. Suppression of dynamin GTPase decreases alpha-synuclein uptake by neuronal and oligodendroglial cells: a potent therapeutic target for synucleinopathy. Mol Neurodegener. 2012;7:38. doi: 10.1186/1750-1326-7-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Reyes JF, Rey NL, Bousset L, Melki R, Brundin P, Angot E. Alpha-synuclein transfers from neurons to oligodendrocytes. Glia. 2014;62:387–398. doi: 10.1002/glia.22611. [DOI] [PubMed] [Google Scholar]
- 158.Kaji S, Maki T, Ishimoto T, Yamakado H, Takahashi R. Insights into the pathogenesis of multiple system atrophy: focus on glial cytoplasmic inclusions. Transl Neurodegener. 2020;9:7. doi: 10.1186/s40035-020-0185-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Nishie M, Mori F, Yoshimoto M, Takahashi H, Wakabayashi K. A quantitative investigation of neuronal cytoplasmic and intranuclear inclusions in the pontine and inferior olivary nuclei in multiple system atrophy. Neuropathol Appl Neurobiol. 2004;30:546–554. doi: 10.1111/j.1365-2990.2004.00564.x. [DOI] [PubMed] [Google Scholar]
- 160.Cykowski MD, Coon EA, Powell SZ, Jenkins SM, Benarroch EE, Low PA, Schmeichel AM, Parisi JE. Expanding the spectrum of neuronal pathology in multiple system atrophy. Brain. 2015;138:2293–2309. doi: 10.1093/brain/awv114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Sekiya H, Kowa H, Koga H, Takata M, Satake W, Futamura N, Funakawa I, Jinnai K, Takahashi M, Kondo T, et al. Wide distribution of alpha-synuclein oligomers in multiple system atrophy brain detected by proximity ligation. Acta Neuropathol. 2019;137:455–466. doi: 10.1007/s00401-019-01961-w. [DOI] [PubMed] [Google Scholar]
- 162.Uchihara T, Nakamura A, Mochizuki Y, Hayashi M, Orimo S, Isozaki E, Mizutani T. Silver stainings distinguish Lewy bodies and glial cytoplasmic inclusions: comparison between Gallyas-Braak and Campbell-Switzer methods. Acta Neuropathol. 2005;110:255–260. doi: 10.1007/s00401-005-1044-2. [DOI] [PubMed] [Google Scholar]
- 163.Homma T, Mochizuki Y, Komori T, Isozaki E. Frequent globular neuronal cytoplasmic inclusions in the medial temporal region as a possible characteristic feature in multiple system atrophy with dementia. Neuropathology. 2016;36:421–431. doi: 10.1111/neup.12289. [DOI] [PubMed] [Google Scholar]
- 164.Wenning GK, Quinn N, Magalhaes M, Mathias C, Daniel SE. "Minimal change” multiple system atrophy. Mov Disord. 1994;9:161–166. doi: 10.1002/mds.870090206. [DOI] [PubMed] [Google Scholar]
- 165.Kon T, Mori F, Tanji K, Miki Y, Wakabayashi K. An autopsy case of preclinical multiple system atrophy (MSA-C) Neuropathology. 2013;33:667–672. doi: 10.1111/neup.12037. [DOI] [PubMed] [Google Scholar]
- 166.Ling H, Asi YT, Petrovic IN, Ahmed Z, Prashanth LK, Hazrati LN, Nishizawa M, Ozawa T, Lang A, Lees AJ, et al. Minimal change multiple system atrophy: an aggressive variant? Mov Disord. 2015;30:960–967. doi: 10.1002/mds.26220. [DOI] [PubMed] [Google Scholar]
- 167.Koga S, Dickson DW. "Minimal change” multiple system atrophy with limbic-predominant alpha-synuclein pathology. Acta Neuropathol. 2019;137:167–169. doi: 10.1007/s00401-018-1901-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Fujishiro H, Ahn TB, Frigerio R, DelleDonne A, Josephs KA, Parisi JE, Eric Ahlskog J, Dickson DW. Glial cytoplasmic inclusions in neurologically normal elderly: prodromal multiple system atrophy? Acta Neuropathol. 2008;116:269–275. doi: 10.1007/s00401-008-0398-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Kato S, Nakamura H. Cytoplasmic argyrophilic inclusions in neurons of pontine nuclei in patients with olivopontocerebellar atrophy: immunohistochemical and ultrastructural studies. Acta Neuropathol. 1990;79:584–594. doi: 10.1007/BF00294235. [DOI] [PubMed] [Google Scholar]
- 170.Horoupian DS, Dickson DW. Striatonigral degeneration, olivopontocerebellar atrophy and “atypical” Pick disease. Acta Neuropathol. 1991;81:287–295. doi: 10.1007/BF00305870. [DOI] [PubMed] [Google Scholar]
- 171.Shibuya K, Nagatomo H, Iwabuchi K, Inoue M, Yagishita S, Itoh Y. Asymmetrical temporal lobe atrophy with massive neuronal inclusions in multiple system atrophy. J Neurol Sci. 2000;179:50–58. doi: 10.1016/s0022-510x(00)00364-6. [DOI] [PubMed] [Google Scholar]
- 172.Piao YS, Hayashi S, Hasegawa M, Wakabayashi K, Yamada M, Yoshimoto M, Ishikawa A, Iwatsubo T, Takahashi H. Co-localization of alpha-synuclein and phosphorylated tau in neuronal and glial cytoplasmic inclusions in a patient with multiple system atrophy of long duration. Acta Neuropathol. 2001;101:285–293. doi: 10.1007/s004010000292. [DOI] [PubMed] [Google Scholar]
- 173.Aoki N, Boyer PJ, Lund C, Lin WL, Koga S, Ross OA, Weiner M, Lipton A, Powers JM, White CL 3, Dickson DW. Atypical multiple system atrophy is a new subtype of frontotemporal lobar degeneration: frontotemporal lobar degeneration associated with alpha-synuclein. Acta Neuropathol. 2015;130:93–105. [DOI] [PMC free article] [PubMed]
- 174.Ando T, Riku Y, Akagi A, Miyahara H, Hirano M, Ikeda T, Yabata H, Koizumi R, Oba C, Morozumi S, et al: Multiple system atrophy variant with severe hippocampal pathology. Brain Pathol 2021:e13002. [DOI] [PMC free article] [PubMed]
- 175.Michell AW, Luheshi LM, Barker RA. Skin and platelet alpha-synuclein as peripheral biomarkers of Parkinson’s disease. Neurosci Lett. 2005;381:294–298. doi: 10.1016/j.neulet.2005.02.030. [DOI] [PubMed] [Google Scholar]
- 176.Braak H, de Vos RA, Bohl J, Del Tredici K. Gastric alpha-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci Lett. 2006;396:67–72. doi: 10.1016/j.neulet.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 177.Ikemura M, Saito Y, Sengoku R, Sakiyama Y, Hatsuta H, Kanemaru K, Sawabe M, Arai T, Ito G, Iwatsubo T, et al. Lewy body pathology involves cutaneous nerves. J Neuropathol Exp Neurol. 2008;67:945–953. doi: 10.1097/NEN.0b013e318186de48. [DOI] [PubMed] [Google Scholar]
- 178.Fujishiro H, Frigerio R, Burnett M, Klos KJ, Josephs KA, Delledonne A, Parisi JE, Ahlskog JE, Dickson DW. Cardiac sympathetic denervation correlates with clinical and pathologic stages of Parkinson’s disease. Mov Disord. 2008;23:1085–1092. doi: 10.1002/mds.21989. [DOI] [PubMed] [Google Scholar]
- 179.Miki Y, Tomiyama M, Ueno T, Haga R, Nishijima H, Suzuki C, Mori F, Kaimori M, Baba M, Wakabayashi K. Clinical availability of skin biopsy in the diagnosis of Parkinson’s disease. Neurosci Lett. 2010;469:357–359. doi: 10.1016/j.neulet.2009.12.027. [DOI] [PubMed] [Google Scholar]
- 180.Beach TG, Adler CH, Sue LI, Vedders L, Lue L, White Iii CL, Akiyama H, Caviness JN, Shill HA, Sabbagh MN, Walker DG. Multi-organ distribution of phosphorylated alpha-synuclein histopathology in subjects with Lewy body disorders. Acta neuropathologica. 2010;119:689–702. doi: 10.1007/s00401-010-0664-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Del Tredici K, Hawkes CH, Ghebremedhin E, Braak H. Lewy pathology in the submandibular gland of individuals with incidental Lewy body disease and sporadic Parkinson’s disease. Acta Neuropathol. 2010;119:703–713. doi: 10.1007/s00401-010-0665-2. [DOI] [PubMed] [Google Scholar]
- 182.Gelpi E, Navarro-Otano J, Tolosa E, Gaig C, Compta Y, Rey MJ, Marti MJ, Hernandez I, Valldeoriola F, Rene R, Ribalta T. Multiple organ involvement by alpha-synuclein pathology in Lewy body disorders. Mov Disord. 2014;29:1010–1018. doi: 10.1002/mds.25776. [DOI] [PubMed] [Google Scholar]
- 183.Tanei ZI, Saito Y, Ito S, Matsubara T, Motoda A, Yamazaki M, Sakashita Y, Kawakami I, Ikemura M, Tanaka S, et al. Lewy pathology of the esophagus correlates with the progression of Lewy body disease: a Japanese cohort study of autopsy cases. Acta Neuropathol. 2021;141:25–37. doi: 10.1007/s00401-020-02233-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Wakabayashi K. Where and how alpha-synuclein pathology spreads in Parkinson’s disease. Neuropathology. 2020;40:415–425. doi: 10.1111/neup.12691. [DOI] [PubMed] [Google Scholar]
- 185.Doppler K. Detection of Dermal Alpha-Synuclein Deposits as a Biomarker for Parkinson’s Disease. J Parkinsons Dis. 2021;11:937–947. doi: 10.3233/JPD-202489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Doppler K, Weis J, Karl K, Ebert S, Ebentheuer J, Trenkwalder C, Klebe S, Volkmann J, Sommer C. Distinctive distribution of phospho-alpha-synuclein in dermal nerves in multiple system atrophy. Mov Disord. 2015;30:1688–1692. doi: 10.1002/mds.26293. [DOI] [PubMed] [Google Scholar]
- 187.Donadio V, Incensi A, El-Agnaf O, Rizzo G, Vaikath N, Del Sorbo F, Scaglione C, Capellari S, Elia A, Stanzani Maserati M, et al. Skin alpha-synuclein deposits differ in clinical variants of synucleinopathy: an in vivo study. Sci Rep. 2018;8:14246. doi: 10.1038/s41598-018-32588-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Donadio V, Incensi A, Rizzo G, De Micco R, Tessitore A, Devigili G, Del Sorbo F, Bonvegna S, Infante R, Magnani M, et al. Skin Biopsy May Help to Distinguish Multiple System Atrophy-Parkinsonism from Parkinson’s Disease With Orthostatic Hypotension. Mov Disord. 2020;35:1649–1657. doi: 10.1002/mds.28126. [DOI] [PubMed] [Google Scholar]
- 189.Antelmi E, Donadio V, Incensi A, Plazzi G, Liguori R. Skin nerve phosphorylated alpha-synuclein deposits in idiopathic REM sleep behavior disorder. Neurology. 2017;88:2128–2131. doi: 10.1212/WNL.0000000000003989. [DOI] [PubMed] [Google Scholar]
- 190.Doppler K, Jentschke HM, Schulmeyer L, Vadasz D, Janzen A, Luster M, Hoffken H, Mayer G, Brumberg J, Booij J, et al. Dermal phospho-alpha-synuclein deposits confirm REM sleep behaviour disorder as prodromal Parkinson’s disease. Acta Neuropathol. 2017;133:535–545. doi: 10.1007/s00401-017-1684-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Antelmi E, Pizza F, Franceschini C, Ferri R, Plazzi G. REM sleep behavior disorder in narcolepsy: A secondary form or an intrinsic feature? Sleep Med Rev. 2020;50:101254. doi: 10.1016/j.smrv.2019.101254. [DOI] [PubMed] [Google Scholar]
- 192.Miglis MG, Zitser J, Schneider L, During E, Jaradeh S, Freeman R, Gibbons CH: Cutaneous Alpha-Synuclein is Correlated with Autonomic Impairment in Isolated REM Sleep Behavior Disorder. Sleep 2021. [DOI] [PMC free article] [PubMed]
- 193.Al-Qassabi A, Tsao TS, Racolta A, Kremer T, Canamero M, Belousov A, Santana MA, Beck RC, Zhang H, Meridew J, et al. Immunohistochemical Detection of Synuclein Pathology in Skin in Idiopathic Rapid Eye Movement Sleep Behavior Disorder and Parkinsonism. Mov Disord. 2021;36:895–904. doi: 10.1002/mds.28399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Donadio V, Incensi A, Rizzo G, Capellari S, Pantieri R, Stanzani Maserati M, Devigili G, Eleopra R, Defazio G, Montini F, et al. A new potential biomarker for dementia with Lewy bodies: Skin nerve alpha-synuclein deposits. Neurology. 2017;89:318–326. doi: 10.1212/WNL.0000000000004146. [DOI] [PubMed] [Google Scholar]
- 195.Mazzetti S, Basellini MJ, Ferri V, Cassani E, Cereda E, Paolini M, Calogero AM, Bolliri C, De Leonardis M, Sacilotto G, et al. alpha-Synuclein oligomers in skin biopsy of idiopathic and monozygotic twin patients with Parkinson’s disease. Brain. 2020;143:920–931. doi: 10.1093/brain/awaa008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Ruffmann C, Bengoa-Vergniory N, Poggiolini I, Ritchie D, Hu MT, Alegre-Abarrategui J, Parkkinen L. Detection of alpha-synuclein conformational variants from gastro-intestinal biopsy tissue as a potential biomarker for Parkinson’s disease. Neuropathol Appl Neurobiol. 2018;44:722–736. doi: 10.1111/nan.12486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Ulusoy A, Rusconi R, Perez-Revuelta BI, Musgrove RE, Helwig M, Winzen-Reichert B, Di Monte DA. Caudo-rostral brain spreading of alpha-synuclein through vagal connections. EMBO Mol Med. 2013;5:1119–1127. doi: 10.1002/emmm.201302475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Holmqvist S, Chutna O, Bousset L, Aldrin-Kirk P, Li W, Bjorklund T, Wang ZY, Roybon L, Melki R, Li JY. Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol. 2014;128:805–820. doi: 10.1007/s00401-014-1343-6. [DOI] [PubMed] [Google Scholar]
- 199.Uemura N, Yagi H, Uemura MT, Hatanaka Y, Yamakado H, Takahashi R. Inoculation of alpha-synuclein preformed fibrils into the mouse gastrointestinal tract induces Lewy body-like aggregates in the brainstem via the vagus nerve. Mol Neurodegener. 2018;13:21. doi: 10.1186/s13024-018-0257-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Adler CH, Beach TG. Neuropathological basis of nonmotor manifestations of Parkinson’s disease. Mov Disord. 2016;31:1114–1119. doi: 10.1002/mds.26605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Prusiner SB, Woerman AL, Mordes DA, Watts JC, Rampersaud R, Berry DB, Patel S, Oehler A, Lowe JK, Kravitz SN, et al. Evidence for alpha-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc Natl Acad Sci U S A. 2015;112:E5308-5317. doi: 10.1073/pnas.1514475112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Woerman AL, Stohr J, Aoyagi A, Rampersaud R, Krejciova Z, Watts JC, Ohyama T, Patel S, Widjaja K, Oehler A, et al. Propagation of prions causing synucleinopathies in cultured cells. Proc Natl Acad Sci U S A. 2015;112:E4949-4958. doi: 10.1073/pnas.1513426112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Yamasaki TR, Holmes BB, Furman JL, Dhavale DD, Su BW, Song ES, Cairns NJ, Kotzbauer PT, Diamond MI. Parkinson’s disease and multiple system atrophy have distinct alpha-synuclein seed characteristics. J Biol Chem. 2019;294:1045–1058. doi: 10.1074/jbc.RA118.004471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Peng C, Gathagan RJ, Covell DJ, Medellin C, Stieber A, Robinson JL, Zhang B, Pitkin RM, Olufemi MF, Luk KC, et al. Cellular milieu imparts distinct pathological alpha-synuclein strains in alpha-synucleinopathies. Nature. 2018;557:558–563. doi: 10.1038/s41586-018-0104-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Assaiya A, Burada AP, Dhingra S, Kumar J. An overview of the recent advances in cryo-electron microscopy for life sciences. Emerg Top Life Sci. 2021;5:151–168. doi: 10.1042/ETLS20200295. [DOI] [PubMed] [Google Scholar]
- 206.Guerrero-Ferreira R, Taylor NM, Mona D, Ringler P, Lauer ME, Riek R, Britschgi M, Stahlberg H. Cryo-EM structure of alpha-synuclein fibrils. Elife. 2018;7:e36402. [DOI] [PMC free article] [PubMed]
- 207.Li B, Ge P, Murray KA, Sheth P, Zhang M, Nair G, Sawaya MR, Shin WS, Boyer DR, Ye S, et al. Cryo-EM of full-length alpha-synuclein reveals fibril polymorphs with a common structural kernel. Nat Commun. 2018;9:3609. doi: 10.1038/s41467-018-05971-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Li Y, Zhao C, Luo F, Liu Z, Gui X, Luo Z, Zhang X, Li D, Liu C, Li X. Amyloid fibril structure of alpha-synuclein determined by cryo-electron microscopy. Cell Res. 2018;28:897–903. doi: 10.1038/s41422-018-0075-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Boyer DR, Li B, Sun C, Fan W, Sawaya MR, Jiang L, Eisenberg DS. Structures of fibrils formed by alpha-synuclein hereditary disease mutant H50Q reveal new polymorphs. Nat Struct Mol Biol. 2019;26:1044–1052. doi: 10.1038/s41594-019-0322-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Guerrero-Ferreira R, Taylor NM, Arteni AA, Kumari P, Mona D, Ringler P, Britschgi M, Lauer ME, Makky A, Verasdonck J, et al. Two new polymorphic structures of human full-length alpha-synuclein fibrils solved by cryo-electron microscopy. Elife. 2019;8:e48907. [DOI] [PMC free article] [PubMed]
- 211.Zhao K, Li Y, Liu Z, Long H, Zhao C, Luo F, Sun Y, Tao Y, Su XD, Li D, et al. Parkinson’s disease associated mutation E46K of alpha-synuclein triggers the formation of a distinct fibril structure. Nat Commun. 2020;11:2643. doi: 10.1038/s41467-020-16386-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Sun Y, Hou S, Zhao K, Long H, Liu Z, Gao J, Zhang Y, Su XD, Li D, Liu C. Cryo-EM structure of full-length alpha-synuclein amyloid fibril with Parkinson’s disease familial A53T mutation. Cell Res. 2020;30:360–362. doi: 10.1038/s41422-020-0299-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Fitzpatrick AWP, Falcon B, He S, Murzin AG, Murshudov G, Garringer HJ, Crowther RA, Ghetti B, Goedert M, Scheres SHW. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature. 2017;547:185–190. doi: 10.1038/nature23002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Zhang w, Falcon B, Murzin AG, Fan J, Crowther RA, Goedert M, Scheres SH. Heparin-induced tau filaments are polymorphic and differ from those in Alzheimer’s and Pick’s diseases. Elife. 2019;8:e43584. [DOI] [PMC free article] [PubMed]
- 215.Scheres SH, Zhang W, Falcon B, Goedert M. Cryo-EM structures of tau filaments. Curr Opin Struct Biol. 2020;64:17–25. doi: 10.1016/j.sbi.2020.05.011. [DOI] [PubMed] [Google Scholar]
- 216.Schweighauser M, Shi Y, Tarutani A, Kametani F, Murzin AG, Ghetti B, Matsubara T, Tomita T, Ando T, Hasegawa K, et al. Structures of alpha-synuclein filaments from multiple system atrophy. Nature. 2020;585:464–469. doi: 10.1038/s41586-020-2317-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Kiely AP, Ling H, Asi YT, Kara E, Proukakis C, Schapira AH, Morris HR, Roberts HC, Lubbe S, Limousin P, et al. Distinct clinical and neuropathological features of G51D SNCA mutation cases compared with SNCA duplication and H50Q mutation. Mol Neurodegener. 2015;10:41. doi: 10.1186/s13024-015-0038-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Puentes LN, Lengyel-Zhand Z, Lee JY, Hsieh CJ, Schneider ME, Jr., Edwards KJ, Luk KC, Lee VM, Trojanowski JQ, Mach RH. Poly (ADP-ribose) Interacts With Phosphorylated alpha-Synuclein in Post Mortem PD Samples. Front Aging Neurosci. 2021;13:704041. doi: 10.3389/fnagi.2021.704041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Kam TI, Mao X, Park H, Chou SC, Karuppagounder SS, Umanah GE, Yun SP, Brahmachari S, Panicker N, Chen R, et al. Poly(ADP-ribose) drives pathologic alpha-synuclein neurodegeneration in Parkinson’s disease. Science. 2018;362:eaat8407. doi: 10.1126/science.aat8407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Lovestam S, Schweighauser M, Matsubara T, Murayama S, Tomita T, Ando T, Hasegawa K, Yoshida M, Tarutani A, Hasegawa M, et al. Seeded assembly in vitro does not replicate the structures of alpha-synuclein filaments from multiple system atrophy. FEBS Open Bio. 2021;11:999–1013. doi: 10.1002/2211-5463.13110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Baek M, DiMaio F, Anishchenko I, Dauparas J, Ovchinnikov S, Lee GR, Wang J, Cong Q, Kinch LN, Schaeffer RD, et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science. 2021;373:871–876. doi: 10.1126/science.abj8754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Zidek A, Potapenko A, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–589. doi: 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Pinheiro F, Santos J, Ventura S. AlphaFold and the amyloid landscape. J Mol Biol. 2021;433:167059. doi: 10.1016/j.jmb.2021.167059. [DOI] [PubMed] [Google Scholar]
- 224.Cong S, Xiang C, Wang H, Cong S. Diagnostic utility of fluid biomarkers in multiple system atrophy: a systematic review and meta-analysis. J Neurol. 2021;268:2703–2712. doi: 10.1007/s00415-020-09781-9. [DOI] [PubMed] [Google Scholar]
- 225.Laurens B, Constantinescu R, Freeman R, Gerhard A, Jellinger K, Jeromin A, Krismer F, Mollenhauer B, Schlossmacher MG, Shaw LM, et al. Fluid biomarkers in multiple system atrophy: A review of the MSA Biomarker Initiative. Neurobiol Dis. 2015;80:29–41. doi: 10.1016/j.nbd.2015.05.004. [DOI] [PubMed] [Google Scholar]
- 226.Parnetti L, Castrioto A, Chiasserini D, Persichetti E, Tambasco N, El-Agnaf O, Calabresi P. Cerebrospinal fluid biomarkers in Parkinson disease. Nat Rev Neurol. 2013;9:131–140. doi: 10.1038/nrneurol.2013.10. [DOI] [PubMed] [Google Scholar]
- 227.Lleo A, Cavedo E, Parnetti L, Vanderstichele H, Herukka SK, Andreasen N, Ghidoni R, Lewczuk P, Jeromin A, Winblad B, et al. Cerebrospinal fluid biomarkers in trials for Alzheimer and Parkinson diseases. Nat Rev Neurol. 2015;11:41–55. doi: 10.1038/nrneurol.2014.232. [DOI] [PubMed] [Google Scholar]
- 228.Emmanouilidou E, Papagiannakis N, Kouloulia S, Galaziou A, Antonellou R, Papadimitriou D, Athanasiadou A, Bozi M, Koros C, Maniati M, et al. Peripheral alpha-synuclein levels in patients with genetic and non-genetic forms of Parkinson’s disease. Parkinsonism Relat Disord. 2020;73:35–40. doi: 10.1016/j.parkreldis.2020.03.014. [DOI] [PubMed] [Google Scholar]
- 229.Folke J, Rydbirk R, Lokkegaard A, Hejl AM, Winge K, Starhof C, Salvesen L, Pedersen LO, Aznar S, Pakkenberg B, Brudek T. Cerebrospinal fluid and plasma distribution of anti-alpha-synuclein IgMs and IgGs in multiple system atrophy and Parkinson’s disease. Parkinsonism Relat Disord. 2021;87:98–104. doi: 10.1016/j.parkreldis.2021.05.001. [DOI] [PubMed] [Google Scholar]
- 230.Koga S, Dickson DW. Recent advances in neuropathology, biomarkers and therapeutic approach of multiple system atrophy. J Neurol Neurosurg Psychiatry. 2018;89:175–184. doi: 10.1136/jnnp-2017-315813. [DOI] [PubMed] [Google Scholar]
- 231.Atarashi R, Sano K, Satoh K, Nishida N. Real-time quaking-induced conversion: a highly sensitive assay for prion detection. Prion. 2011;5:150–153. doi: 10.4161/pri.5.3.16893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Atarashi R, Satoh K, Sano K, Fuse T, Yamaguchi N, Ishibashi D, Matsubara T, Nakagaki T, Yamanaka H, Shirabe S, et al. Ultrasensitive human prion detection in cerebrospinal fluid by real-time quaking-induced conversion. Nat Med. 2011;17:175–178. doi: 10.1038/nm.2294. [DOI] [PubMed] [Google Scholar]
- 233.Hermann P, Laux M, Glatzel M, Matschke J, Knipper T, Goebel S, Treig J, Schulz-Schaeffer W, Cramm M, Schmitz M, Zerr I. Validation and utilization of amended diagnostic criteria in Creutzfeldt-Jakob disease surveillance. Neurology. 2018;91:e331–8. [DOI] [PubMed]
- 234.Saborio GP, Permanne B, Soto C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature. 2001;411:810–813. doi: 10.1038/35081095. [DOI] [PubMed] [Google Scholar]
- 235.Soto C, Saborio GP, Anderes L. Cyclic amplification of protein misfolding: application to prion-related disorders and beyond. Trends Neurosci. 2002;25:390–394. doi: 10.1016/s0166-2236(02)02195-1. [DOI] [PubMed] [Google Scholar]
- 236.Caughey B, Kraus A. Transmissibility versus Pathogenicity of Self-Propagating Protein Aggregates. Viruses. 2019;11:1044. doi: 10.3390/v11111044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Fairfoul G, McGuire LI, Pal S, Ironside JW, Neumann J, Christie S, Joachim C, Esiri M, Evetts SG, Rolinski M, et al. Alpha-synuclein RT-QuIC in the CSF of patients with alpha-synucleinopathies. Ann Clin Transl Neurol. 2016;3:812–818. doi: 10.1002/acn3.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Shahnawaz M, Tokuda T, Waragai M, Mendez N, Ishii R, Trenkwalder C, Mollenhauer B, Soto C. Development of a Biochemical Diagnosis of Parkinson Disease by Detection of alpha-Synuclein Misfolded Aggregates in Cerebrospinal Fluid. JAMA Neurol. 2017;74:163–172. doi: 10.1001/jamaneurol.2016.4547. [DOI] [PubMed] [Google Scholar]
- 239.Groveman BR, Orrù CD, Hughson AG, Raymond LD, Zanusso G, Ghetti B, Campbell KJ, Safar J, Galasko D, Caughey B. Rapid and ultra-sensitive quantitation of disease-associated α-synuclein seeds in brain and cerebrospinal fluid by αSyn RT-QuIC. Acta Neuropathol Commun. 2018;6:7. doi: 10.1186/s40478-018-0508-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Bongianni M, Ladogana A, Capaldi S, Klotz S, Baiardi S, Cagnin A, Perra D, Fiorini M, Poleggi A, Legname G, et al. α-Synuclein RT-QuIC assay in cerebrospinal fluid of patients with dementia with Lewy bodies. Ann Clin Transl Neurol. 2019;6:2120–2126. doi: 10.1002/acn3.50897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Kang UJ, Boehme AK, Fairfoul G,Shahnawaz M, Ma TC, Hutten SJ, Green A, Soto C: Comparative study ofcerebrospinal fluid alpha-synuclein seeding aggregation assays fordiagnosis of Parkinson's disease. Mov Disord. 2019;34:536–544. [DOI] [PMC free article] [PubMed]
- 242.van Rumund A, Green AJE, Fairfoul G,Esselink RAJ, Bloem BR, Verbeek MM: α-Synuclein real-timequaking-induced conversion in the cerebrospinal fluid of uncertaincases of parkinsonism. Ann Neurol. 2019;85:777–81. [DOI] [PMC free article] [PubMed]
- 243.Garrido A, Fairfoul G, Tolosa ES, MartíMJ, Green A: α-synuclein RT-QuIC in cerebrospinal fluid ofLRRK2-linked Parkinson's disease. Ann Clin Transl Neurol. 2019;6:1024–32. [DOI] [PMC free article] [PubMed]
- 244.Rossi M, Candelise N, Baiardi S,Capellari S, Giannini G, Orru CD, Antelmi E, Mammana A, Hughson AG,Calandra-Buonaura G, et al: Ultrasensitive RT-QuIC assay with highsensitivity and specificity for Lewy body-associatedsynucleinopathies. Acta Neuropathol. 2020,;40:49–62. [DOI] [PMC free article] [PubMed]
- 245.Iranzo A, Fairfoul G, Ayudhaya ACN,Serradell M, Gelpi E, Vilaseca I, Sanchez-Valle R, Gaig C, SantamariaJ, Tolosa E, et al: Detection of α-synuclein in CSF by RT-QuIC inpatients with isolated rapid-eye-movement sleep behaviour disorder: alongitudinal observational study. Lancet Neurol. 2021;20:203–12. [DOI] [PubMed]
- 246.Shahnawaz M, Mukherjee A, Pritzkow S,Mendez N, Rabadia P, Liu X, Hu B, Schmeichel A, Singer W, Wu G, etal: Discriminating alpha-synuclein strains in Parkinson's disease andmultiple system atrophy. Nature 2020, 578:273–7. [DOI] [PMC free article] [PubMed]
- 247.Rossi M, Baiardi S, Teunissen CE,Quadalti C, van de Beek M, Mammana A, Maserati MS, Van der Flier WM,Sambati L, Zenesini C, et al: Diagnostic Value of the CSF α-SynucleinReal-Time Quaking-Induced Conversion Assay at the Prodromal MCI Stageof Dementia With Lewy Bodies. Neurology. 2021. [DOI] [PMC free article] [PubMed]
- 248.Manne S, Kondru N, Hepker M, Jin H,Anantharam V, Lewis M, Huang X, Kanthasamy A, Kanthasamy AG:Ultrasensitive Detection of Aggregated alpha-Synuclein in GlialCells, Human Cerebrospinal Fluid, and Brain Tissue Using the RT-QuICAssay: New High-Throughput Neuroimmune Biomarker Assay forParkinsonian Disorders. J Neuroimmune Pharmacol. 2019;14:423–35. [DOI] [PMC free article] [PubMed]
- 249.Perra D, Bongianni M, Novi G, Janes F,Bessi V, Capaldi S, Sacchetto L, Tagliapietra M, Schenone G, MorbelliS, et al: Alpha-synuclein seeds in olfactory mucosa and cerebrospinalfluid of patients with dementia with Lewy bodies. Brain Commun. 2021;3:fcab045. [DOI] [PMC free article] [PubMed]
- 250.De Luca CMG, Elia AE, Portaleone SM,Cazzaniga FA, Rossi M, Bistaffa E, De Cecco E, Narkiewicz J, SalzanoG, Carletta O, et al: Efficient RT-QuIC seeding activity forα-synuclein in olfactory mucosa samples of patients with Parkinson'sdisease and multiple system atrophy. Transl Neurodegener. 2019;8:24. [DOI] [PMC free article] [PubMed]
- 251.Manne S, Kondru N, Jin H, Anantharam V, Huang X, Kanthasamy A, Kanthasamy AG. α-Synuclein real-timequaking-induced conversion in the submandibular glands of Parkinson’sdisease patients. Mov Disord. 2020;35:268–78. [DOI] [PMC free article] [PubMed]
- 252.Wang Z, Becker K, Donadio V, Siedlak S,Yuan J, Rezaee M, Incensi A, Kuzkina A, Orrú CD, Tatsuoka C, et al:Skin α-Synuclein Aggregation Seeding Activity as a Novel Biomarkerfor Parkinson Disease. JAMA Neuro.l 2020;78:1–11. [DOI] [PMC free article] [PubMed]
- 253.Manne S, Kondru N, Jin H, Serrano GE,Anantharam V, Kanthasamy A, Adler CH, Beach TG, Kanthasamy AG:Blinded RT-QuIC Analysis of alpha-Synuclein Biomarker in Skin TissueFrom Parkinson's Disease Patients. Mov Disord. 2020;35:2230–9. [DOI] [PMC free article] [PubMed]
- 254.Mammana A, Baiardi S, Quadalti C, RossiM, Donadio V, Capellari S, Liguori R, Parchi P: RT-QuIC Detection ofPathological alpha-Synuclein in Skin Punches of Patients with LewyBody Disease. Mov Disord. 2021;36:2173–7. [DOI] [PMC free article] [PubMed]
- 255.Stefani A, Iranzo A, Holzknecht E,Perra D, Bongianni M, Gaig C, Heim B, Serradell M, Sacchetto L,Garrido A, et al: Alpha-synuclein seeds in olfactory mucosa ofpatients with isolated REM sleep behaviour disorder. Brain. 2021;144:1118–26. [DOI] [PubMed]
- 256.Donadio V, Wang Z, Incensi A, Rizzo G,Fileccia E, Vacchiano V, Capellari S, Magnani M, Scaglione C,Stanzani Maserati M, et al: In Vivo Diagnosis of Synucleinopathies: AComparative Study of Skin Biopsy and RT-QuIC. Neurology. 2021;96:e2513–24. [DOI] [PMC free article] [PubMed]
- 257.Berg D, Postuma RB, Adler CH, Bloem BR, Chan P, Dubois B, Gasser T, Goetz CG, Halliday G, Joseph L, et al. MDS research criteria for prodromal Parkinson’s disease. Mov Disord. 2015;30:1600–1611. doi: 10.1002/mds.26431. [DOI] [PubMed] [Google Scholar]
- 258.Koga S, Aoki N, Uitti RJ, van Gerpen JA, Cheshire WP, Josephs KA, Wszolek ZK, Langston JW, Dickson DW. When DLB, PD, and PSP masquerade as MSA: an autopsy study of 134 patients. Neurology. 2015;85:404–412. doi: 10.1212/WNL.0000000000001807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Miki Y, Foti SC, Asi YT, Tsushima E, Quinn N, Ling H, Holton JL. Improving diagnostic accuracy of multiple system atrophy: a clinicopathological study. Brain. 2019;142:2813–2827. doi: 10.1093/brain/awz189. [DOI] [PubMed] [Google Scholar]
- 260.Dong TT, Satoh K. The Latest Research on RT-QuIC Assays-A Literature Review. Pathogens. 2021;10:305. doi: 10.3390/pathogens10030305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Leclair-Visonneau L, Rouaud T, Derkinderen P. Skin biopSYN or how to predict Parkinson’s disease. Parkinsonism Relat Disord. 2021;86:105–107. doi: 10.1016/j.parkreldis.2021.04.030. [DOI] [PubMed] [Google Scholar]
- 262.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.