

Abstract
The role of colony stimulating factor-1 receptor (CSF-1R) in macrophage and organismal development has been extensively studied in mouse. Within the last decade mutations in the CSF1R have been shown to cause rare diseases of both pediatric (Brain Abnormalities, Neurodegeneration, and Dysosteosclerosis (BANDDOS), OMIM #618476) and adult [CSF1R-related leukoencephalopathy (CRL), OMIM #221820] onset. Here we review the genetics, penetrance and histopathological features of these diseases and discuss to what extent the animal models of Csf1r deficiency currently available provide systems in which to study the underlying mechanisms involved.
Keywords: CSF-1R, ALSP, CRL, HDLS, POLD, BANDDOS, leukodystrophy, microglia, neurodegeneration, dysosteosclerosis
Graphical Abstract

Colony-stimulating factor-1 receptor (CSF-1R) is a receptor tyrosine kinase that controls the development of most tissue macrophages and of bone-resorbing osteoclasts. Mutations in the CSF1R cause rare diseases of both pediatric and adult-onset that share a strong neurological component and, depending on the gene dosage, can also involve osteosclerosis. The defining features of these diseases, their reproducibility in animal models of Csf1r deficiency and insights prompting emerging therapies are discussed.
1. Introduction
Colony-stimulating factor-1 receptor (CSF-1R) is a receptor tyrosine kinase that controls the development of most tissue macrophages and, in combination with receptor activator of nuclear factor κB, of bone-resorbing osteoclasts. The CSF-1R is activated by two homodimeric glycoprotein ligands, CSF-1 and interleukin-34 (IL-34), which exhibit complementary spatio-temporal patterns of expression in vivo and thereby regulate different populations of tissue macrophages (reviewed in [1, 2]). Studies in homozygous Csf1r- and Csf1- null mice have shown that the CSF-1/CSF-1R pathway regulates the differentiation and maintenance of most tissue macrophages and osteoclasts, contributing to the development and remodeling of bones and soft tissues [1, 3–5]. Indeed, on the same outbred background, the phenotypic differences between Csf1-null and Csf1r-null mice were barely perceptible and only fully revealed when the two mutants were compared on single strain backgrounds [1, 4, 5]. In contrast, Il34-null mice have no gross phenotype, except for decreased numbers of Langerhans cells and microglia [6, 7]. In humans, bi-allelic mutations of the CSF1R are variably associated with osteopetrosis, yet consistently associated with epilepsy and structural brain anomalies, including ventriculomegaly and cerebellar atrophy, as well as leukodystrophy [8–12]. Furthermore, as detailed below, heterozygous CSF1R kinase-inactivating mutations and haploinsufficiency lead to a dominantly inherited adult-onset progressive dementia, currently designated CSF-1R-related leukoencephalopathy (CRL) [13, 14]. These data highlight the importance of CSF-1R signaling for brain development and homeostasis.
In the nervous system, CSF-1R is necessary for the development, survival and homeostatic function of microglia and regulates the differentiation of neural progenitor cells (reviewed in [15]). The functional complementarity of its ligands is also evident in the nervous system, where CSF-1 and IL-34 are expressed in largely non-overlapping areas of the brain and regulate the development and maintenance of different populations of microglia [6, 7, 16–19]. CSF-1 predominantly contributes to microglial development in the corpus callosum, pons and cerebellum [16–18], while IL-34 contributes to the maintenance of microglia in forebrain structures, but not in the cerebellum, or brainstem [6, 7]. Antibody-mediated depletion experiments in adult mice showed that CSF-1 is necessary for the maintenance of white matter microglia, while IL-34 predominantly controls grey matter microglia [20]. In addition to microglia, in the brain, CSF-1R is also expressed by neural progenitor cells, and subpopulations of neuronal cells [21–24]. While the functional significance of CSF-1R expression in neurons at steady state remains unclear, studies in vitro suggest that CSF-1R signaling in neural progenitor cells suppresses proliferation, enhances their neuronal differentiation and promotes the survival of their early progeny [22].
2. Clinical manifestations of bi-allelic CSF1R mutations in humans
Brain Abnormalities, Neurodegeneration, and Dysosteosclerosis (BANDDOS, OMIM #618476) is an autosomal recessive disorder caused by bi-allelic mutations in the CSF1R gene. To date, 12 cases have been reported harboring homozygous or compound heterozygous combinations of 9 different CSF1R mutations (Fig. 1A) [8–12]. Seven of the 9 mutations have been characterized as amorphic or hypomorphic (Table 1). The disorder is characterized by structural brain abnormalities, progressive neurologic deterioration and sclerotic bone dysplasia. Early onset patients can present with congenital hydrocephalus, hypotonia, seizures and developmental delay, while the delayed onset cases show normal development followed by progressive loss of motor and language skills, cognitive impairment, spasticity, seizures and psychiatric symptoms. Brain imaging shows periventricular white matter abnormalities and calcifications [8–11]. Calcifications can also be found in the cerebellar white matter [11]. Brain structural abnormalities vary, with ventriculomegaly reported in nine out of ten cases, agenesis/hypogenesis of the corpus callosum in five out of ten, and cerebellar atrophy in seven out of eight patients examined [8–11]. Skeletal anomalies include osteosclerosis of the craniofacial bones and skull, optic canal narrowing, kyphosis, platyspondyly and abnormalities of the long bones, e.g. undermodeling, widened metaphyses, and constricted and sclerotic diaphysis [8–11]. The extent of skeletal involvement may depend on CSF1R gene dosage with marked osteosclerosis reported in patients carrying two amorphic [8] or a combination of hypomorphic and amorphic alleles [10], while either no skeletal changes [9, 11], or mild osteosclerosis [10], were reported in homozygous carriers of hypomorphic mutations. Brain developmental deficits, e.g. callosal agenesis/hypogenesis, seem to follow a similar pattern, being present in all individuals carrying amorphic mutations and absent in those carrying bi-allelic hypomorphic mutations. In contrast, other features such as cerebellar atrophy and ventriculomegaly are present in most BANDDOS cases.
Fig. 1.

CSF1R mutations identified in CRL and BANDDOS. (A) Position of BANDDOS mutations in relation to the functional domains of CSF-1R. SP, signal peptide; Ig, immunoglobulin-like domain; TM, transmembrane domain; JMD, juxtamembrane domain; KID, kinase insert domain; C, catalytic loop; AL, activation loop. The tyrosine kinase domain is shown in dark blue. The numbers at the top indicate the amino acid position where each domain ends. Color coding (orange, blue) indicates the combinations of compound heterozygous mutations in two different BANDDOS cases. Bold characters indicate the BANDDOS cases carrying bi-allelic mutations. a Different mutations producing the same change in amino acid sequence were identified in CRL [13, 59] and BANDDOS [58] patients. bThe mutation decreases, but does not eliminate, normal splicing. The resulting alternatively spliced RNA undergoes nonsense-mediated decay (NMD). c Note that mutations leading to substitutions of the same threonine residue with either methionine or lysine have been reported as recessive in BANDDOS and dominantly inherited in CRL. (B) Position of CRL mutations. Light blue, variants for which asymptomatic carriers older than 60 years of age have been reported. Asymptomatic carriers of an additional mutation, c.1858+1G>T, that causes exon 13 skipping and the production of an uncharacterized protein variant have also been reported [126] (not shown). Boxes, protein product not translated, mRNA undergoes NMD. An additional mutation, that causes NMD, c2319+1C>A [50], is not shown. Green, possibly non-pathogenic mutations: p.E573K and p.G747R, hypomorphic and silent mutations identified in stroke patients that did not fulfill the CRL diagnostic criteria [27]. The c.1085G>A biallelic mutation encoding p.H362R was identified in a patient with probable CRL. The high frequency of this allele in the Eastern Asian population [54] suggests that at least in the heterozygous state, it may be non-pathogenic. List of mutations and references are provided in Table S1. (C) Type of mutation and the age of onset in patients with CRL. Left panel, truncating mutations and mutations affecting RNA stability. The substitution p.G17C in the signal peptide might affect CSF-1R translation, post-translational modification and/or its transport to the cell surface. Right panel, single amino acid substitutions. Each bar represents one patient. (D) Statistical comparison of the data in (C) showing earlier onset in patients with truncating mutations or mutations causing NMD. Each dot represents one patient.
Table 1.
Functional characterization of CSF1R mutations in BANDDOS
| gDNA mutation | Protein mutation | Protein location | Effect on signalling | Type of mutation | Reference |
|---|---|---|---|---|---|
| c.395C>T | p.P132L | Ig 2 | 50% reduction in JNK phosphorylation | Hypomorph | [10] |
| c.1441C>T | p.Q481* | Ig 5 | none expected | Amorph (NMD) | [10] |
| c.1620T>A | p.Y540* | interdomain | none expected (TKD absent) | Amorph | [8] |
| c.1754–1G>C | p.G585_K619delinsA | TKD | n/a | n/a | [58] |
| c.1859–119G>A | p.S620delins40 | TKD | 90% reduction in JNK phosphorylation | Amorph | [10] |
| c.1879_1881del | p.K627del | TKD | 50% reduction in JNK phosphorylation | Hypomorph | [10] |
| c.1929C>A | p.H643Q | TKD | n/a | Possibly hypomorph | [58] |
| c.1969+115_1969+116del | p.P658Sfs*24 | TKD | n/a | Hypomorpha | [10] |
| c.2498C>T | p.T833M | TKD | 20% phosphorylation retained at Y723 | Hypomorph | [11] |
The alternatively spliced transcript undergoes nonsense-mediated RNA decay (NMD), however ~50% normal splicing is retained
n/a, data not available
The age of onset of neurological symptoms is variable, ranging from infancy [8–12] up to the third decade of life [10] and does not seem to depend on CSF1R gene dosage. Carriers of two different combinations of hypomorphic and amorphic alleles (c.395C>T; c.1441C>T and c.1859–119G>A; c.1879_1881del) showed signs of neurological dysfunction perinatally and at 31 years of age, respectively [10] and among carriers of bi-allelic hypomorphic mutations the time of onset of neurological disease also varied between 2–23 years [9–11]. The data suggests that there is a spectrum of phenotypic expression of bi-allelic CSF1R mutations, ranging from null mutations leading to congenital malformations and early lethality, to hypomorphic mutations whose phenotypic expression could be compensated or exacerbated by other genetic components.
3. Clinical manifestations of mono-allelic CSF1R mutations in humans
3.1. HDLS, POLD, ALSP, ALAS, CRL - What’s in a name?
The involvement of dominantly-inherited, mono-allelic, CSF1R mutations in an adult-onset hereditary leukoencephalopathy was first recognized in 2012 [13]. This disease has been termed Hereditary Diffuse Leukoencephalopathy with Spheroids (HDLS), or adult-onset leukoencephalopathy/leukodystrophy with axonal spheroids (ALAS), to reflect its main histopathological features; or adult-onset leukodystrophy with axonal spheroids and pigmented glia (ALSP), to reflect that it encompasses both HDLS and Pigmented Orthochromatic Leukodystrophy (POLD) and more recently, CSF1R-related leukoencephalopathy (CRL), to distinguish it from another, similar leukoencephalopathy caused by recessive mutations in the gene encoding mitochondrial alanyl-transfer RNA synthetase 2 (AARS2) [14, 25].
3.2. Genetics and penetrance of CRL
CRL (OMIM #221820) is an autosomal dominant disease associated with cognitive impairment, psychiatric disorders, motor dysfunction and seizures, with typical onset between 10–60 years of age [26]. As of May 2021, 106 different CSF1R mutations were reported in CRL patients, most of which reside in the kinase domain (Fig 1B, Table S1). Aside from the earlier onset of disease in patients carrying truncating mutations or mutations that trigger nonsense-mediated mRNA decay (NMD) (Fig. 1C, D), no clear genotype-phenotype correlation is apparent. The incidental identification of mutations causing single amino acid substitutions in the juxtamembrane (p.E573K) and kinase insert (p.G747R) domains in stroke patients who did not fulfill CRL diagnostic criteria, suggested that unless they are truncating, mutations occurring outside the kinase domain are non-pathogenic [27]. However, the recent identification of an early onset case carrying a non-truncating mutation in the juxtamembrane domain (p.T576M) [28] contradicts this interpretation.
In a retrospective study of 122 ALSP patients, Konno et al [14] have shown that the penetrance of ALSP is age-dependent, increasing from 10% at age 27 years to 50%, at age 43 years and 95%, at age 60 years. However, familial case reports that identify healthy carriers of ten different CSF1R mutations that are pathogenic in their progeny, siblings or other relatives (Fig. 1B; Table 2) raise the possibility of incomplete penetrance even in aged individuals. The majority of neurologically healthy carriers were either 60 years of age and older [29–34], or expected to be well over the average age of onset for CRL (43 years) [14] as parents of 24–42-year-old probands [35–37]. This marked intra-familial variability of the phenotypic expression of CSF1R mutations raises the possibility that, similar to BANDDOS, the development and severity of clinical CRL might be influenced by other genetic, or environmental factors. Indeed, a protective role of pre-symptomatic immunosuppression in CRL has recently been suggested [32]. In contrast, other reports indicate that surgical tissue damage might precipitate the onset of cognitive decline [38, 39]. Furthermore, an unusual initial presentation of a CRL patient with bilateral lower extremity paresthesias and extensive spinal cord abnormalities, occurring after a bout of mononucleosis, has also been reported [40]. These findings are consistent with recent reports from studies in human postmortem brain tissue [41] and in the Csf1r+/− mouse model [42, 43], both of which indicate that CRL pathology is mediated by dyshomeostatic microglial responses.
Table 2.
Intra-familial variability of the phenotypic expression of CSF1R mutations
| Family | Case/ relation to proband | Sex | Age of onset | Age of healthy carrier | gDNA mutation | Protein Mutation | Location | Cell surface expression | Signaling | Type of mutation | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Proband II.3 | M | 37 | c.1858+1G>T | Unc | TKD | n/a | n/a | n/a | [126] | |
| 1 | II.1 (brother) | F | 45 | c.1858+1G>T | Unc | TKD | n/a | n/a | n/a | [126] | |
| 1 | II.2 (sister) | M | 43 | c.1858+1G>T | Unc | TKD | n/a | n/a | n/a | [126] | |
| 1 | I.2 (mother) | F | 68 | c.1858+1G>T | Unc | TKD | n/a | n/a | n/a | [126] | |
| 2 | Proband II.1 | F | 35 | c.1990G>A | p.E664K | TKD, AB | n/a | n/a | n/a | [29] | |
| 2 | II.2 (brother) | M | 56 | c.1990G>A | p.E664K | TKD, AB | n/a | n/a | n/a | [29] | |
| 2 | II.3 (sister) | F | 54 | c.1990G>A | p.E664K | TKD, AB | n/a | n/a | n/a | [29] | |
| 2 | II.3 (sister) | F | 55 | c.1990G>A | p.E664K | TKD, AB | n/a | n/a | n/a | [29] | |
| 2 | I.2 (mother)a | F | 83 | c.1990G>A | p.E664K | TKD, AB | n/a | n/a | n/a | [29] | |
| 3 | Proband III.1 | F | 33 | c.2344C>G | p.R782G | TKD, CL | n/a | n.e. | Amorph | [34] | |
| 3 | II.1 (mother) | F | 60 | c.2344C>G | p.R782G | TKD, CL | n/a | n.e. | Amorph | [34] | |
| 4 | Proband III.1 | F | 43 | c.2344C>G | p.R782G | TKD, CL | n/a | n.e. | Amorph | [34] | |
| 4 | II.4 (mother) | F | 67 | c.2344C>G | p.R782G | TKD, CL | n/a | n.e. | Amorph | [34] | |
| 5 | Proband II.2 | F | 39 | c.2350G>A | p.V784M | TKD | n/a | n/a | n/a | [30] | |
| 5 | I.1 (mother) | F | 79 | c.2350G>A | p.V784M | TKD | n/a | n/a | n/a | [30] | |
| 6 | Proband II.1 | F | 42 | c.2381T>C | p.I794T | TKD | normal | absent | Amorph | [36] | |
| 6 | I.2 (mother) | F | n/a | c.2381T>C | p.I794T | TKD | normal | absent | Amorph | [36] | |
| 7 | Proband III.1 | F | 35 | c.2381T>C | p.I794T | TKD | normal | absent | Amorph | [36] | |
| 7 | II.1 (father) | M | n/a | c.2381T>C | p.I794T | TKD | normal | absent | Amorph | [36] | |
| 8 | Proband III.1 | F | 28 | c.2552T>C | p.L851P | TKD | normal | absent | Amorph | [31, 36] | |
| 8 | III.3 (sister) | F | 25 | c.2552T>C | p.L851P | TKD | normal | absent | Amorph | [31, 36] | |
| 8 | II.2 (mother) | F | 64 | c.2552T>C | p.L851P | TKD | normal | absent | Amorph | [31, 36] | |
| 8 | II.3 (uncle) | M | 58 | c.2552T>C | p.L851P | TKD | normal | absent | Amorph | [31, 36] | |
| 9 | Proband II.1 | F | 40 | c.2625G>A | p.M875I | TKD | n/a | n/a | n/a | [32] | |
| 9 | I.1 (mother)b | F | 71 | c.2625G>A | p.M875I | TKD | n/a | n/a | n/a | [32] | |
| 10 | Proband II.1 | M | 28 | c.2629C>T | p.Q877* | TKD | n/a | n/a | n/a | [33] | |
| 10 | I.1 (father) | M | 69 | c.2629C>T | p.Q877* | TKD | n/a | n/a | n/a | [33] | |
| 11 | Proband II.1 | M | 24 | c.2645delC | p.P882Pfs*70 | TKD | n/a | n/a | n/a | [37] | |
| 11 | I.1 (mother) | F | n/a | c.2645delC | p.P882Pfs*70 | TKD | n/a | n/a | n/a | [37] | |
| 12 | Proband II.1 | M | 36 | c.2717T>C | p.I906T | TKD | n/a | n/a | n/a | [35] | |
| 12 | I.1 (mother) | F | n/a | c.2717T>C | p.I906T | TKD | n/a | n/a | n/a | [35] |
CSF1R mosaicism in saliva and blood;
Long-term immunosuppression;
n/a, data not available; Unc, uncharacterized; AB, ATP binding site; CL, catalytic loop; n.e., none expected;
3.3. Is CRL the consequence of gain-of-function, loss-of-function, or haploinsufficiency?
Out of the 106 different CSF1R pathogenic mutations that were reported in 275 cases of CRL, 34 have been characterized in terms of expression and signaling [36, 44–49] (Table 3). Three mutations cause NMD and are therefore expected to produce haploinsufficiency [46, 47, 50]. Functional characterization of the most frequently reported CRL variant, p.I794T, in 293T cells, showed loss of kinase activity [44, 47, 51] and a drastic reduction of the proteolytic processing of the CSF-1R by ADAM and γ-secretase compared to wild type [52]. Further investigation is required to determine the generality of the latter finding for other CRL-associated missense mutations and its diagnostic significance. Aside from one notable exception, all of the remaining mutations are loss-of-function. The exception is the c.2527_2530delinsGGCA mutation (p.I843_L844delinsGI), reported to cause a slight increase in CSF-1R expression and signaling in monocytes [48]. However, this interpretation should be taken with caution, since it is based exclusively on ex vivo flow cytometric analysis of CSF-1R phosphorylation at tyrosine residue 723 in total CD14+ monocytes obtained from one heterozygous CRL patient. The levels of CSF-1R surface expression and phosphorylation in vivo depend on ligand availability, which was not explored. Further functional characterization of the mutant protein under controlled conditions in vitro is necessary to establish whether this mutation indeed causes a gain of function.
Table 3.
Functional characterization of CSF1R mutations in CRL
| gDNA mutation | gDNA Location | Predicted protein structural change | Location | Type of mutation | Experimental evidence | Reference | |
|---|---|---|---|---|---|---|---|
| Cell surface expression | Mutant chain signaling | ||||||
| c.310delC | Exon 4 | p.P104fs*8 | Ig 1 | Amorph | n/a (NMD) | n/a | [47] |
| c.2060_2061insT | Exon 15 | p.S688Efs*13 | KID | Amorph | n/a (NMD) | n/a | [51] |
| c.2319+1C>A | IVS17 | ASV | TKD | Amorph | n/a (NMD) | n/a | [50] |
| c.1699delA | Exon 12 | p.T567fs*44 | JMD | Amorph | unknown | n.e | [127] |
| c.1985T>C | Exon 15 | p.I662T | TKD | Amorph | unknown | absent | [47] |
| c.2294G>A | Exon17 | p.G765D | TKD | Amorph | unknown | absent | [47, 51] |
| c.2345G>A | Exon 18 | p.R782H | TKD, CL | Amorph | unknown | absent | [45] |
| c.2380A>T | Exon 18 | p.I794F | TKD | Amorph | unknown | absent | [47] |
| c.2442+1G>T | IVS18 | ASV1 | TKD | Amorph | unknown | absenta | [51] |
| c.2442+1G>T | IVS18 | ASV2 | TKD | Amorph | unknown | absent | [51] |
| c.2442+1G>T | IVS18 | ASV3 | TKD | Amorph | unknown | absent | [51] |
| c.2632C>G | Exon 20 | p.P878A | TKD | Amorph | unknown | absent | [47] |
| c.2632C>T | Exon 20 | p.P878S | TKD | Amorph | unknown | absent | [47] |
| c.2655_2656delAT | Exon 21 | p.Q886fs*55 | TKD | Amorph | unknown | absent | [47] |
| c. 1897G>A | Exon 14 | p.E633K | TKD | Amorph | decreased | absent | [44] |
| c.1766G>A | Exon 13 | p.G589E | TKD, AB | Amorph | normal | absent | [44] |
| c.1907T>A | Exon 14 | p.I636N | TKD | Amorph | normal | absent | [36] |
| c.2297T>C | Exon17 | p.M766T | TKD | Amorph | decreased | absent | [44, 51] |
| c.2308G>C | Exon17 | p.A770P | TKD | Amorph | decreased | absent | [44] |
| c.2324T>A | Exon 18 | p.I775N | TKD, CL | Amorph | decreased | absent | [44] |
| c.2334C>A | Exon 18 | p.D778E | TKD, CL | Amorph | unknown | absent | [47] |
| c.2342C>A | Exon 18 | p.A781E | TKD, CL | Amorph | normal | absent | [51] |
| c.2368C>A | Exon 19 | p.A823D | TKD | Amorph | decreased | absent | [36] |
| c.2381T>C | Exon 18 | p.I794T | TKD | Amorph | decreased | absenta | [44, 47, 51] |
| c.2470C>T | Exon 19 | p.P824S | TKD | Amorph | unknown | absent | [51] |
| c.2509G>T | Exon 19 | p.D837Y | TKD | Amorph | decreased | absent | [44] |
| c.2546T>C | Exon 19 | p.F849S | TKD | Amorph | decreased | absent | [44] |
| c.2552T>C | Exon 19 | p.L851P* | TKD | Amorph | normal | absent | [36] |
| c.2603T>C | Exon 20 | p.L868P | TKD | Amorph | decreased | absent | [44] |
| c.2624T>C | Exon 20 | p.M875T | TKD | Amorph | decreased | absent | [44, 45, 51] |
| c.2632C>A | Exon 20 | p.P878T | TKD | Amorph | decreased | absent | [44] |
| c2026C>T | Exon 15 | p.R676* | TKD | Amorph | normal | absent | [36] |
| c.2527_2530delinsGGCA | Exon 19 | p.I843_L844delinsGI | TKD | Hypermorph | Slightly increased | Increasedb | [48] |
| c.2342_2350del | Exon 18 | p.A781_N783del | TKD, CL | Hypomorph | decreased | decreasedc | [49] |
| c.2471C>G | Exon 19 | p.P824R | TKD | Hypomorph | decreased | decreasedc | [49] |
| c.2342C>T | Exon 18 | p.A781V | TKD, CL | Hypomorph | normal | decreased | [36] |
NMD, nonsense-mediated mRNA decay
ASV, alternatively spliced variant
Lack of dominant negative effect on CSF-1-induced phosphorylation of the wt chain
Based on ex vivo immunodetection of CSF-1R tyrosine phosphorylation a residue 723
Lack of dominant negative effect on the CSF-1-induced phosphorylation of Erk1/2; slight impairment of the response to IL-34.
IVS, intervening sequence; AVS, alternatively spliced variant; TKD, tyrosine kinase domain; AB, ATP binding site; CL catalytic loop
Importantly, functional characterization of CSF-1R mutations can produce conflicting results depending on the experimental system used. The most common approach of transfection of plasmids encoding the mutant chains into HEK293T cells and stimulation of the transfected cells with CSF-1, provides information concerning the kinase activity of the mutant chain. However, to understand how mutations impact CSF-1R function in CRL, both wild-type and mutant chains should be co-expressed. Thus far, only two studies [49, 51] report the results of co-expression experiments. Furthermore, the cellular context is also important. For example, characterization of the p.H362R CSF-1R mutation, located in Ig4 dimerization domain [53], has produced conflicting results, showing no impact on kinase activity when the mutant alone was expressed in HEK293T cells [47] and reductions in kinase activity, receptor internalization and CSF-1-induced M2 polarization in monocyte-derived macrophages of heterozygous carriers [54]. Thus, exploring the functional impact of CSF1R mutations in the correct allelic and cellular context is important. The development and characterization of iPSC-derived microglia should address this problem and contribute to a better understanding of the mechanisms contributing to disease development.
In a recent review article [55], Hume et al. claim that CRL is due to a “gain-of-function in CSF1R”, actually referring to a dominant-negative, inhibitory function. This assumption is based on the functional analysis of 4 mouse Csf1r mutants (E631K, M764T, I792T and M873T) equivalent to those found in CRL (E633K, M766T, I794T and M875T) by transfection into the murine IL-3-dependent pro-B cell line, Ba/F3 [56]. Based on the finding that in the presence of CSF-1, the mutant receptors can bind CSF-1, but fail to support proliferation, the authors conclude that in heterozygous individuals, wild type/mutant CSF-1R heterodimers must be inactive. In doing so, they ignore the evidence provided by co-transfection experiments of wild type and mutant CSF1R constructs, in which the mutant chain did not significantly inhibit the phosphorylation of wild type CSF-1R in response to CSF-1 binding [49, 51], indicating that a dominant-negative mechanism is unlikely. Indeed, unless overexpressed, the mutant receptor chains may not significantly impair signaling. Furthermore, to reinforce their “gain-of-function” hypothesis, Hume at al., [55, 57] also claim that haploinsufficiency per se is not likely to cause CRL. However, as shown above, three independent studies of human disease identify mutations in the CSF1R that lead to NMD and lack of a translated protein product, supporting haploinsufficiency as a contributing mechanism [46, 47, 50]. The lack of evidence of CRL in BANDDOS family members with heterozygous complete loss of function alleles (i.e. c.1441C>T and c.1620T>A encoding p.Q481* and p.Y540*, respectively) is cited by Hume as another compelling argument against haploinsufficiency as an underlying cause of CRL. However, it should be noted that this statement refers to carriers who were either still too young to exclude presentation of CRL later in life (parents of infants must still be of reproductive age, i.e <45 years old) or, if older, exhibited early signs of disease such as memory loss and brain calcification (70-year-old proband grandfather carrying p.Q481*). Furthermore, reports of two different mutations that encode the same protein product, p.G585_K619delinsA, one (c.1754–1G>C) characterized as recessive in BANDDOS [58], and the other (c.1754–2A>G) as dominant in CRL [13, 59], emphasize the dangers of postulating mechanisms based on a limited sample size. Thus, while we cannot a priori exclude that some of the uncharacterized CRL mutations exhibit dominant-negative functions, haploinsufficiency cannot be discounted as an underlying mechanism.
3.4. Radiologic features of CRL
The hallmark features of CRL are bilateral cerebral white matter abnormalities, enlargement of lateral ventricles and thinning of the corpus callosum (reviewed in [14]). White matter changes can occur pre-symptomatically and longitudinal studies show that they become more severe as the disease progresses (reviewed in [14]). Earlier studies suggested that neuropsychiatric symptoms precede the appearance of motor dysfunction. The spectrum of neuropsychiatric symptoms, including personality changes, executive dysfunction, social disinhibition and depression observed in patients, that could be mistaken for frontotemporal dementias, was explained by the involvement of the prefrontal deep white matter [60, 61]. Freeman et al, [60] correlated the selective disruption of corpus callosum sectors with the clinical presentations of patients. They proposed that slower degeneration of sensorimotor association fibers in sector 3 of the corpus callosum was responsible for the relative delay in the presentation of motor symptoms. Although the study of Freeman et al. lacks genetic confirmation of CRL, similar findings were subsequently reported in other studies of CRL patients [28, 61–63]. These studies point to the preferential vulnerabilities of subsets of association and projection fibers in CRL-associated neurodegeneration, although they no longer support the order in which the clinical symptomatology manifests. A summary of the reported initial symptoms and structural changes is presented in Figure 2.
Fig. 2.
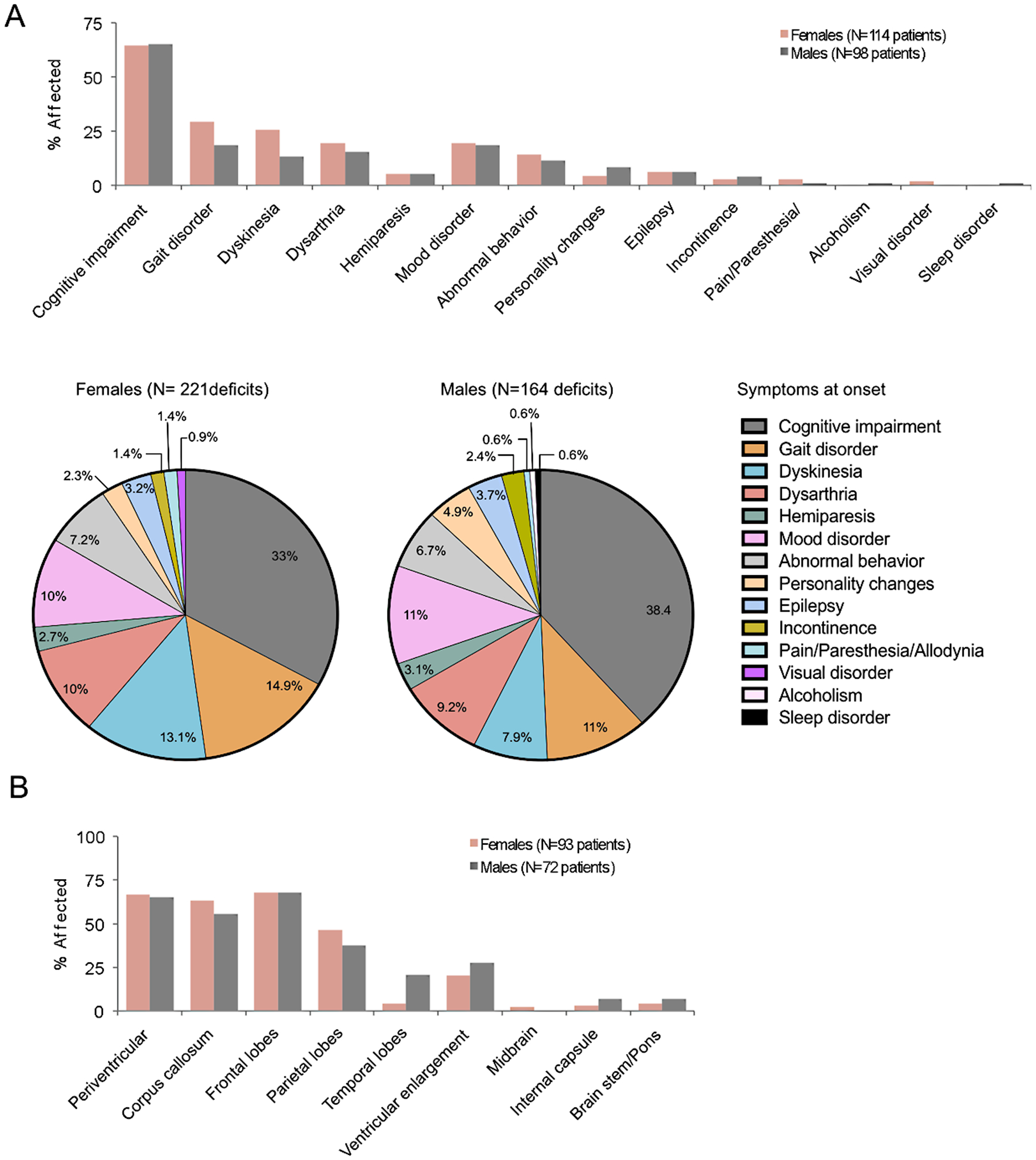
Typical clinical features of CRL. (A) Initial symptoms of CRL. Gait disorders and dyskinesia are more common in females than in males. (B) Similar distribution of MRI-detected brain lesions in male and female patients.
Utilizing advanced MRI and functional MRI techniques, Zhan et. al. recently reassessed differential vulnerabilities of various brain structures to CRL-mediated neurodegeneration [64]. They showed that cognitive changes observed in CRL patients cannot be exclusively explained by the damage in the frontal and parietal subcortical white matter. They detailed the preferential damage to the association fibers between caudate nuclei and hippocampi, as well as between frontotemporal, hippocampal and thalamic connections, accounting for differential impairment of attention, learning and memory functions and emotional disturbances. This study also highlights an association between symptoms, including apraxia, agnosia, anomia and mild alexia and the disconnection of the two cerebral hemispheres, due to the preferential loss of connection fibers in the posterior area of the corpus callosum. Using diffusion kurtosis imaging, they showed selective damage of subcortical U-fibers that are critical for maintenance of consciousness, attention, cognitive processing, emotional stability and possibly in the development of personality traits. Interestingly, they also report enhanced functional connectivity, mainly between the precuneus, paracentral lobules and inferior frontal gyrus suggesting that ongoing regeneration and synaptic plasticity compensate for neuronal loss and degeneration in other areas of the brain.
Small spotty calcifications that are preferentially distributed in the frontal white matter adjacent to the horns of the lateral ventricles have been frequently reported in CT scans of CRL patients (35 out of 57 investigated cases, Table 4). The calcifications have been detected in presymptomatic CRL-associated mutation carriers as early as perinatally and either remain stable or decrease in size over time, suggesting that they may not be related to clinical symptoms or to the extent of white matter damage (reviewed in [14]). Of interest, Konno et al noted that the pericallosal distribution of calcifications is very similar to the distribution of mononuclear phagocytes in the human fetal brain, and suggested a causal relation between microglial dysfunction and the formation of calcifications [14].
Table 4.
Calcifications in CRL patients
| gDNA mutation | Sex | gDNA Location | Protein Mutation | Location | Type of mutation | Brain calcifications | Reference |
|---|---|---|---|---|---|---|---|
| c.1766G>A | M | Exon 13 | p.G589E | TKD, AB | Amorph | Fr | [13, 44, 128, 129] |
| c.2294G>A | F | Exon17 | p.G765D | TKD | Amorph | Fr | [51] |
| c.2297T>C | F | Exon17 | p.M766T | TKD | Amorph | Fr | [13, 44, 129] |
| c.2381T>C | F | Exon 18 | p.I794T | TKD | Amorph | Fr | [130] |
| c.2381T>C | F | Exon 18 | p.I794T | TKD | Amorph | Fr | [14, 131] |
| c.2442+1G>A | F | IVS18 | n/a | TKD | n/a | Fr | [132, 133] |
| c.2442+1G>T | M | IVS18 | 3 splice variants | TKD | Amorph | Fr | [51] |
| c.2442+5G>A | F | IVS18 | n/a | TKD | n/a | Fr | [129] |
| c.2470C>T | F | Exon 19 | p.P824S | TKD | Amorph | Fr | [51] |
| c.1765G>A | F | Exon 13 | p.G589R | TKD, AB | n/a | Fr, Pa | [129] |
| c.1765G>A | F | Exon 13 | p.G589R | TKD, AB | n/a | Fr, Pa | [134] |
| c.1766G>Aa | F | Exon 13 | p.G589E | TKD, AB | Amorph | Fr, Pa | [26] |
| c.1954G>C | F | Exon 14 | p.A652P | TKD | n/a | Fr, Pa | [129] |
| c.2342C>A | F | Exon 18 | p.A781E | TKD, AB | Amorph | Fr, Pa | [51] |
| c.2345G>T | F | Exon 18 | p.R782L | TKD, AB | Amorph | Fr, Pa | [135] |
| c.2381T>C | M | Exon 18 | p.I794T | TKD | Amorph | Fr, Pa | [51] |
| c.2442+5G>A | M | IVS18 | n/a | TKD | n/a | Fr, Pa | [129] |
| c.2442+5G>C | F | IVS18 | p.C774_N814delins SQGLQSHVCPSLPSSSPQAQ | TKD, CL | n/a | Fr, Pa | [13, 129, 136] |
| n/a | F | Exon17 | p.F758S | KID | n/a | Fr, Pa | [14] |
| c.310delC | F | Exon 4 | p.P104fs*8 | Ig 1 | Amorph (NMD) | Fr, Pa | [47] |
| c.1766G>A | F | Exon 13 | p.G589E | TKD, AB | Amorph | Fr, SC | [13, 44, 128, 129] |
| c.1700C>T | F | Exon 12 | p.T567M | JMD | PV | [28] | |
| c.1958G>A | F | Exon 14 | p.C653Y | TKD | PV | [137] | |
| c.2375C>A | M | Exon 18 | p.A792D | TKD | n/a | PV | [78] |
| c.2466G>A | F | Exon 19 | p.M822I | TKD | n/a | PV | [61] |
| c.2468C>T | F | Exon 19 | p.A823V | TKD | n/a | PV | [138] |
| c.2540A>T | F | Exon 19 | p.E847V | TKD | n/a | PV | [139] |
| c.2541G>C | F | Exon 19 | p.E847D | TKD | n/a | PV | [140] |
| c.2552T>C | F | Exon 19 | p.L851P* | TKD | PV | [36] | |
| c.2552T>C | F | Exon 19 | p.L851P* | TKD | Amorph | PV | [31, 36] |
| c.2552T>C | F | Exon 19 | p.L851P* | TKD | Amorph | PV | [31, 36] |
| c.2375C>A | M | Exon 18 | p.A792D | TKD | n/a | PV, BG | [141] |
| c.2909_2910insATCA | M | Exon 22 | p.F971Sfs*7 | C-terminus | n/a | Fr, PV | [14, 142] |
| c.1958G>A, inferred | F | Exon 14 | p.C653Y | TKD | SC | [143] | |
| c.2330G>A | F | Exon 18 | p.R777Q | TKD, CL | SC | [144] | |
| c.1907T>A | M | Exon 14 | p.I636N | TKD | Amorph | none | [36] |
| c.2026C>T | F | Exon 15 | p.R676* | TKD | Amorph | none | [36] |
| c.2026C>T | F | Exon 15 | p.R676* | TKD | Amorph | none | [36, 64] |
| c.2026C>T | F | Exon 15 | p.R676* | TKD | Amorph | none | [36, 64] |
| c.2294G>A | M | Exon17 | p.G765D | TKD | Amorph | none | [47, 51, 145] |
| c.2319+1C>A | M | IVS17 | n/a | TKD | Amorph (NMD) | none | [50] |
| c.2319+1C>A | M | IVS17 | n/a | TKD | Amorph (NMD) | none | [50] |
| c.2319+1C>A | M | IVS17 | n/a | TKD | Amorph (NMD) | none | [50] |
| c.2381T>C | F | Exon 18 | p.I794T | TKD | Amorph | none | [36] |
| c.2381T>C | F | Exon 18 | p.I794T | TKD | Amorph | none | [36] |
| c.2381T>C | F | Exon 18 | p.I794T | TKD | Amorph | none | [146] |
| c.2342C>T | F | Exon 18 | p.A781V | TKD, CL | Hypomorph | none | [36, 147] |
| c.2342C>T | F | Exon 18 | p.A781V | TKD, CL | Hypomorph | none | [36] |
| c.2368C>A | M | Exon 19 | p.A823D | TKD | Amorph | none | [36] |
| c.2442+1G>A | F | IVS18 | n/a | TKD | n/a | none | [146] |
| c.2442+1G>A | F | IVS18 | n/a | TKD | n/a | none | [146] |
| c.2446_2548delTCT | F | Exon 19 | p.F849del | TKD | n/a | none | [148] |
| c.2539G>A | F | Exon 19 | p.E847K | TKD | n/a | none | [110] |
| c.2539G>A | F | Exon 19 | p.E847K | TKD | n/a | none | [110] |
| c.2552T>C | F | Exon 19 | p.L851P* | TKD | Amorph | none | [36] |
| c.2563C>A | M | Exon 20 | p.P855T | TKD | n/a | none | [148] |
| c.2625G>A | F | Exon 20 | p.M875I | TKD | n/a | none | [146] |
Heathy carrier, 23 years old
Fr, frontal; Pa, parietal; PV, periventricular; BG, basal ganglia; SC, subcortical
IVS, intervening sequence; AVS, alternatively spliced variant; TKD, tyrosine kinase domain; AB, ATP binding site; CL catalytic loop
Decreased cortical glucose metabolism, increased levels of choline indicative of demyelination and decreased N-acetylaspartate levels, indicative of axonal damage, can also be detected even prior to the development of symptoms (reviewed in [14]).
3.5. CRL histopathology
Defining histological features include white matter degeneration, the presence of axonal spheroids and of lipid-laden and pigmented phagocytes in the affected white matter (reviewed in [14]). In contrast to primary oligodendrogliopathies, the U fibers are spared until later in the disease [14]. A study of autopsied brain tissue from 10 patients with CRL including two carriers of the p.C653Y mutation, one of which was asymptomatic and the other with advanced disease, permitted a chronological modeling of disease progression [65]. Based on the severity of axonal loss, Oyanagi et al. identified four pathological stages [65]. Stage I was characterized by small patchy loss of myelinated fibers in the centrum semiovale and frontal cerebral white matter, without atrophy. The areas of myelinated fiber loss increased in Stage II cerebral white matter, when slight atrophy of the white matter and dilation of the lateral ventricles became evident. In Stage III, the degeneration of frontal white matter was extensive and accompanied by atrophy of the thalamus and moderate dilation of the lateral and third ventricles. Finally, Stage IV was characterized by devastation of the cerebral white matter, degeneration of the pontine base and cerebellar white matter, severe dilation of the lateral ventricle and cortical thinning. Importantly, microglial densities in the affected white matter vary in a stage-dependent fashion, being maximal in stage II and becoming sparse in advanced stages of degeneration. Areas with different stages of white matter pathology can be observed simultaneously in the same patient [25]. Consistent with this, other studies showed a patchy distribution of Iba1 and P2ry12 positive homeostatic microglia, which were more abundant in relatively preserved areas, but sparse in devastated areas [66, 67]. In contrast, cells immunopositive for Iba1 and/or CD68 but negative for P2ry12, possibly infiltrated macrophages or dyshomeostatic microglia, tended to accumulate in the damaged white matter [66, 67].
A comparison of the morphology of microglia between the corpus callosum and the centrum semiovale [68] showed that at Stage I, while microglia in both areas had increased size, those in the corpus callosum were larger. The frequency of large amoeboid CD163+ microglia was maximal at Stage II, corresponding to the severe degeneration of axons. Also, at Stage II, large, rounded microglia possessing short, thick processes were evident at the site of a focal lesion in the splenium of the corpus callosum connecting with centrum semiovale lesions. The increased microglial activation in the corpus callosum was interpreted to contribute to the earlier and greater atrophy there, compared to the centrum semiovale. Overall, these observations support the concept that CSF1R mutations impair the homeostatic functions of microglia and suggest a selective vulnerability of microglia in the corpus callosum.
3.5.1. Dyshomeostatic microglia as mediators of neuropathology
Immunohistochemistry revealed that the gradual anatomic changes in CRL were accompanied by sequential changes in axonal and glial structure. One hallmark feature of the disease is the presence of axonal dilations (i.e. spheroids), an early and transient structural change occurring in transected or degenerating CNS axons [69, 70]. Intriguingly, phenotyping of these spheroid-containing axons suggests that axons that arise from parvalbumin and choline acetyl transferase expressing interneurons may be more vulnerable to injury [71]. The presence of spheroids was detected in the white matter, prior to changes in myelination, suggesting that axonal damage precedes the loss of myelin [25, 65]. The density of spheroids in the white matter was maximal in stage II and declined with subsequent progression of the disease. In the cortex, spheroids could also be detected in early disease in the deep layers [25] and their abundance increased in Stage IV, concomitant with the loss of neurons in the frontal cortex [65].
The mechanisms that give rise to disease-associated axonal spheroids are unclear. Interestingly, the dynamic changes in abundance of spheroids were paralleled, or even preceded by, changes in microglial numbers, morphology and expression of activation markers, e.g. CD68 and CD163 [65, 68]. As there is growing indication that CRL is a primary microgliopathy [14, 43], it remains to be determined how altered microglial activation triggers axonal dysfunction and degeneration. An emerging mechanism involves oxidative stress, a common denominator in a number of neurological diseases in which the presence of axonal spheroids was documented [25, 65, 72]. Studies in adult mouse primary neurons have shown that reactive oxygen species (ROS) promote actin aggregation, focal accumulations of the reverse Na+/Ca2+ exchanger 1 and voltage-gated N-type Ca2+ channel, resulting in axoplasmic Ca2+ increase and spheroid formation [73]. Oxidative stress may play an important role in CRL pathology as evidenced by the identification of an end product of oxidative damage, ceroid, as the source of pigmentation in CRL microglia, as well as of other markers of oxidative damage, such as 4-hydroxynonenal, malondialdehyde and nitrotyrosine [71]. In addition to being an indicator of neurotoxic activation [74], the characteristic accumulation of iron by microglia in CRL [71] may contribute to oxidative damage by enhancing the formation of reactive oxygen species via the Fenton reaction. Increased uptake and storage of iron by microglia would also diminish iron availability to oligodendrocytes, which in turn, is essential for (re)myelination [75, 76]. The possibility that microglial-generated oxidative stress has a role in CRL is further supported by studies in the Csf1r+/− mouse model, which identify gene expression changes in microglia that could promote a pro-oxidant phenotype [42].
3.5.2. Recurrent hypoxic-ischemic injuries as potential triggers of microglial activation
As components of the innate immune system, microglia become activated and proliferate in response to various forms of tissue injury, and it is possible that CSF1R mutations could modify their responses from protective to neurotoxic. However, aside from sporadic reports of surgical [38, 39], or viral [40] tissue injury occurring just prior to the onset of CRL, there is little evidence of an external precipitating factor. Extensive sampling from macroscopically normal white matter revealed a multifocal distribution of early stage lesions containing axonal spheroids that is consistent with recurring tissue injury [25]. Several studies [25, 49, 71] suggest that recurring hypoxic-ischemic injuries might play a role. This hypothesis is supported by observations that myelin loss sometimes follows a perivascular rather than a diffuse, pattern [71], as well as reports of vascular changes, including small vessel adventitial fibrosis [25, 71] and severe arteriosclerosis [49], in the subcortical white matter. In addition, Delaney et al. [49] report widespread cerebral amyloid angiopathy and perivascular accumulation of phosphorylated tau in the meninges and parenchyma of three post-mortem CRL cases and suggest that disruption of phagocyte-endothelial cell cross talk might result in altered blood brain barrier (BBB) function. It is not clear which phagocytic component, i.e. activated resident microglia, perivascular macrophages or circulating monocytes, might be involved in the disruption of the BBB. Other studies [48, 77, 78] have shown that the subset of nonclassical monocytes is decreased in CRL patients. However, one of the nine patients tested, who had an early onset and was in the most advanced disease state at the time of testing, showed the polar opposite distribution of monocyte subsets [78], raising the possibility that the changes in monocyte subsets may depend on the stage of disease. In mouse, the equivalent monocyte subset develops in a CSF-1R-dependent manner [79], act as housekeepers of vasculature and elicit tolerogenic responses [80]. These monocytes can also infiltrate the brain and differentiate into perivascular macrophages, which maintain the blood–brain barrier [81] and have neuroprotective functions following injury [82]. Further exploration of the relationship between the loss of nonclassical monocytes and vascular and BBB fitness in CRL is needed.
3.5.3. Dynamic changes in astrocytes and oligodendrocytes in CRL
Astrocytosis is a frequent finding in advanced white matter lesions of CRL [65] and sometimes evident in normal-appearing or mildly-affected areas [71]. Similar to microglia, astrocytes contain ceroid and markers of oxidative stress and damage [71].
A decrease in the density of oligodendrocytes is apparent in the frontal white matter at stage III and becomes marked at Stage IV [65].
Since neither astrocytes nor oligodendrocytes express the CSF-1R, the changes in their density and activation states are likely to reflect their responses to ongoing pathology.
4. Modeling CSF1R deficiency in the mouse
4.1. The Csf1r-null mutation as a model of BANDDOS:
The phenotypes of Csf1r−/− mice on the FVB/NJ background strikingly resemble the early onset, more severe, forms of BANDDOS, including perinatal lethality, osteopetrosis, cerebellar atrophy and ventriculomegaly [5, 22]. In addition, the failure of midline crossing of the corpus callosum is apparent in 80% of Csf1r-null mice. Furthermore, the severe depletion of microglia documented in a BANDDOS patient [58] is mimicked in Csf1r−/− mice [83, 84]. Similar to BANDDOS, there is a spectrum of phenotypic expression in Csf1r-null mutant mice across different genetic backgrounds. Csf1r−/− mice were originally characterized on the mixed 129SvJ-C57BL/6J-C3Heb/FeJ-a/a background on which ~70% survived beyond 3 weeks of age [4]. In contrast, on the FVB/NJ background they did not survive beyond 1 month of age [5] and on the C57BL6/J and C3H/Hej backgrounds, they exhibited embryonic/newborn lethality (unpublished observations). Although no radiologic data assessing the structure of white matter and presence of calcifications is available, the strong anatomic similarities and the perinatal lethality render the Csf1r-null FVB/NJ mouse a good model of severe BANDDOS.
4.2. The Csf1r heterozygous mouse model of CRL:
4.2.1. Rationale for the Csf1r heterozygous mouse model:
Three mutations causing CSF1R haploinsufficiency have been described in CRL patients [47, 50, 51], the description of the first of these prompting an investigation of whether Csf1r+/− mice could model CRL. The C57BL6/J background was chosen because it permitted the expression of a more severe Csf1r deficiency phenotype and because of the availability of mutant and transgenic strains on this background that could aid in the investigation of the mechanisms underlying CRL development.
4.2.2. Penetrance and progression of behavioral deficits in Csf1r+/− mice:
Behavioral studies showed that Csf1r+/− mice developed cognitive deficits, motor coordination deficits (more marked in females), depression- and anxiety- like behavior [42, 43, 85], all characteristic of CRL [14](Figs. 2 & 3A). A small percentage of mice exhibit seizure-like activity (unpublished observations). The penetrance of disease (2 or more deficits) in 15-month-old mice was 58% (Fig. 3B), corresponding to a similar degree of penetrance in humans at the equivalent age (43 years) [26, 86]. Interestingly, the course of disease progression in mice is accelerated by a higher fat-containing diet (Fig. 3B).
Fig. 3.
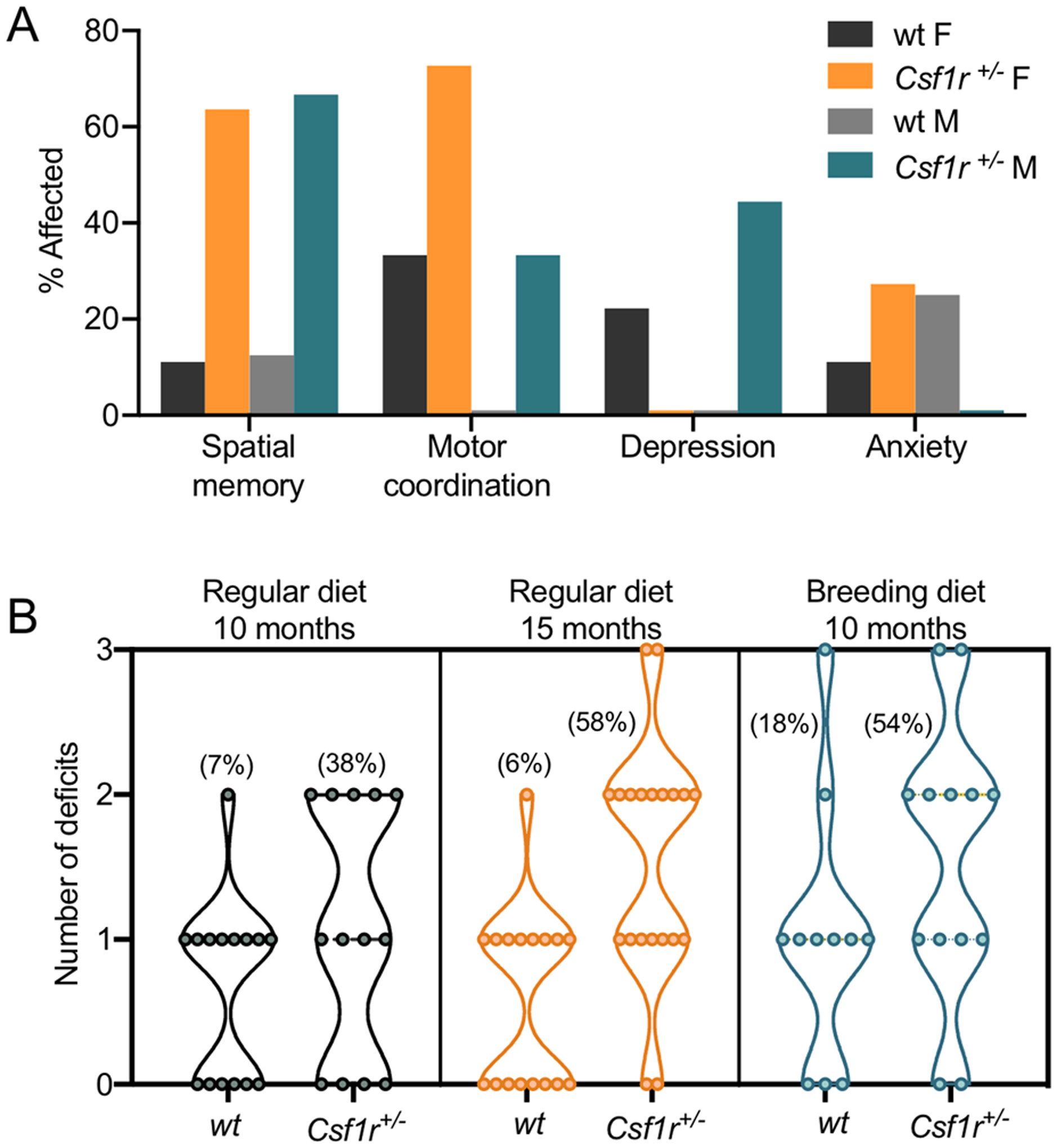
Features of CRL-like disease in mice. (A) Prevalence of behavioral deficits at 15–18 months of age in female (F) and male (M) mice maintained on a regular diet. Their behavior was assessed at the indicated age using a battery of tests that evaluate spatial memory (object placement), motor coordination (balance beam), depression- (forced swim) and anxiety- (elevated zero or plus maze) like behaviors [42]. Motor deficit and depression-like behavior were defined as any score falling above the average scores of wild type (wt) mice plus one standard deviation. Anxiety-like behavior was defined as any score falling below the average % time spent in the open zone of the maze by wt mice minus one standard deviation. The analysis includes only the mice that performed in all four tests. (B) Penetrance of CRL-like disease in Csf1r+/− mice at different ages and effect of dietary fat content. Mice were maintained on either a regular diet (PicoLab Rodent diet 5053 containing 6.3%fat) [42] or a breeding diet (PicoLab Rodent diet 5058 containing 10.2% fat) [85]. Only mice exhibiting two or more behavioral deficits were considered to be affected. Note that 50% penetrance is reached 5 months earlier in the group maintained on the higher fat-containing diet. Percentage affected in each category is shown in brackets.
4.2.3. Gross pathology:
Radiological investigations consistently show callosal atrophy. However, neither white matter hyperintensities nor calcifications were consistently found. Nevertheless, increases in the callosal G ratio and Cystatin 7 expression, indicative of active demyelination coupled with remyelination, accompanied by decreased myelin basic protein staining, axonal degeneration and the presence of axonal spheroids, were reproduced in the mouse model [42, 43].
4.2.4. Changes in glial cell populations of Csf1r+/− mice:
Microglial densities are slightly increased throughout the brain, even during the asymptomatic stages [85]. These increases are secondary to elevation of cerebral Csf2 expression, which is also elevated in CRL [42] (section 4.2.5). It is not clear whether this early pathology is evident in CRL, as most analyses are post-mortem. However, compared with mouse, human mononuclear phagocytes have significantly less in vitro and in vivo proliferative responses to CSF-1 and CSF-2 [87–89](Stanley ER, personal observations). Thus, while the activating effect of CSF-2 on macrophages is preserved across species [90], a proliferative response might not occur in human microglia. In symptomatic 18-month-old Csf1r+/− mice, white matter microglia are unevenly distributed with frequent accumulations found in the periventricular area of the corpus callosum and occasionally in the cerebellar white matter (Fig. 4A, B) [42]. Within the corpus callosum, but not in the cortex, the microglia exhibit decreased ramification, consistent with an activated phenotype (Fig. 4C). These features are similar to those reported in early (Stage II) CRL (Fig. 4D) [65]. Furthermore, similar to CRL [65, 71, 91], these microglial patches contain increased GFAP staining, indicative of astrocytosis, while astrocyte density in the unaffected white matter areas and the cortex is unchanged (Fig. 4B) [43, 85]. In CRL, oligodendrocytes do not substantially decrease until advanced (Stage IV) disease. However, in the mouse, CC1+ oligodendrocytes are slightly increased in the callosal white matter [42]. The elevation in astrocytes and oligodendrocytes in Csf1r+/− mice may be driven by the expansion of microglia [22, 92, 93]. Overall, the available data indicate that symptomatic 18-month-old mice exhibit histological features similar to those of Stage II CRL.
Fig. 4.

Changes in the distribution and morphology of microglia in mouse and human CRL. (A) Periventricular accumulation of microglia in the corpus callosum of 18-month-old Csf1r+/− mice (arrow). Lower panels, small clusters of microglia are occasionally found in the cerebellar white matter (arrow), but not in the fimbria. (B) Astrocytosis in areas of callosal microglial accumulation. (C) Morphological changes (e.g. shortening of the processes) selectively occur in the white matter. (D) Changes in microglia density and expression of CD163 in the frontal cortex (Cx) and white matter (WM) at various stages of human CRL (reprinted with permission from [65]). CC, corpus callosum; Cb, cerebellum; Fb, fimbria; Cx, cortex; WM, white matter.
4.2.5. Mechanistic insights from the mouse - possible contributions of CSF-2 and CSF-3 to pathology:
Studies in the mouse have shown that Csf1r monoallelic deletion in microglia but not in the neural lineage is sufficient to reproduce all aspects of Csf1r+/− disease, including the loss in layer V neurons, supporting the notion that CRL is a primary microgliopathy [42, 43]. Asymptomatic 11-week-old Csf1r+/− mice possessed slightly elevated microglial densities in multiple brain regions that remain elevated up to 18-months of age [42]. The early accumulation of microglia was counter-intuitive, given the reduced expression of the CSF-1R in Csf1r+/− mice [85]. Furthermore, there was no compensatory increase in mRNA or protein for either of the CSF-1R ligands, CSF-1 or IL-34, in the same brain regions. An unbiased mRNA screen for inflammatory cytokines, chemokines and receptors in young Csf1r+/− mice identified increased expression of two myeloid growth factors, Csf2, encoding GM-CSF and Csf3, encoding G-CSF [85]. CSF-2 is a strong microglial mitogen [94, 95], is proinflammatory and promotes a demyelinating phenotype in microglia [96], while CSF-3 induces a pro-oxidant phenotype in microglia [97]. Interestingly, both Csf2 [42] and Csf3 (Biundo, F., Chitu, V. and Stanley, E.R., unpublished) expression are elevated in CRL patient brains, in which alterations in CSF-3 signaling have also been reported [67].
Studies in the mouse model suggest that CSF-2 plays an important role in CRL development. Normalization of cerebral Csf2 expression by monoallelic deletion prevented the forebrain microgliosis and attenuated most behavioral deficits and histopathological changes [42]. Furthermore, microglial monoallelic deletion of Csf2rb, encoding the signaling subunit of the CSF-2 receptor, also prevented the increase in forebrain microglial densities, indicating that the CSF-2-induced microgliosis was due to direct signaling in microglia. RNAseq of Csf1r+/− forebrain microglia revealed a dyshomeostatic state, indicating maladaptive functions and activation of pathways resulting in oxidative stress, oligodendrocyte death and demyelination. These predictions were validated by immunohistochemistry. Csf2 heterozygosity attenuated the dyshomeostasis and the related histopathological changes [42]. Biundo et al have established that the cells exhibiting increased Csf2 expression in the corpus callosum were astrocytes, oligodendrocytes and to a lesser extent, microglia, while oligodendrocytes were the major contributors in the cortex [43]. Given that Csf1r heterozygosity in microglia is sufficient for the increased expression of Csf2, it is of interest to establish how the dysregulated microglia instruct neuroglial Csf2 expression.
Preliminary studies indicate that the increased cerebral expression of Csf3 contributes in a non-overlapping manner with Csf2 to the behavioral deficits and microglial activation in Csf1r+/− mice (Biundo, F., Chitu, V. and Stanley, E.R., unpublished).
4.2.6. Additional mouse models:
Two additional models of Csf1r deficiency have recently been developed that are potentially relevant for investigation of the pathological mechanisms of BANDDOS and CRL. A knock-in mouse harboring the E631K mutation, corresponding to the CRL mutation E633K, has been developed. Homozygous Csf1rE631K/E631K mice resemble Csf1r−/−mice, with significant embryonic macrophage loss, severe postnatal growth retardation and pre-weaning mortality [98]. Future detailed characterization of Csf1rE631K/+ mice is necessary to determine their suitability as a model of CRL. In addition, mice bearing a homozygous deletion of a Csf1r super-enhancer, the fms-intronic regulatory element (FIRE), have been developed on the C57BL/6J/CBA background. Csf1rΔFIRE/ΔFIRE mice exhibit a selective loss of several populations of tissue resident macrophages (skin, heart, kidney and peritoneum), while other populations of macrophages (liver, spleen and lung), monocytes and osteoclasts are unaffected. Within the brain, there is loss of microglia and ventricular macrophages and a decrease in stromal choroid plexus macrophages, while perivascular and brain-associated macrophages are produced normally [98, 99]. In contrast to Csf1r−/− mice, Csf1rΔFIRE/ΔFIRE mice are healthy, fertile, with normal teeth and bones and without anatomical abnormalities in the brain. However, when backcrossed onto the C57BL/6 background there is significantly increased perinatal mortality due to hydrocephalus. Although the original report found no detectable change in microglia in Csf1rΔFIRE/+ mice on a mixed genetic background [99], there is some evidence that on the C57BL/6 background Csf1rΔFIRE/+ mice reproduce the low-grade microgliosis observed in Csf1r+/− mice (David A. Hume, personal communication). While the FIRE enhancer is conserved in humans [99], to date, no mutations have been reported in patients with BANDDOS or CRL. Nevertheless, the Csf1rΔFIRE/+ mice could be useful to explore the long-term consequences of Csf1r insufficiency in specific macrophage populations, including microglia, in different experimental settings.
5. Other models of Csf1r deficiency:
Models of Csf1r deficiency have also been developed in zebrafish and rats.
5.1. Zebrafish:
Due to gene duplication, zebrafish express two homologs of the human CSF1R: csf1ra and csf1rb. Together, both homologs regulate microglial density in the adult zebrafish brain - csf1ra is required for the establishment of primitive macrophage-derived microglia during development, whereas csf1rb has a selective role in controlling the ontogeny of definitive macrophages, including adult microglia [100, 101]. Csf1r double mutant (csf1ra−/−; csf1rb−/− or csf1rDM) fish exhibit the microglial deficiency and osteopetrosis seen in severe BANDDOS [9, 58]. However, studies of callosal development or degeneration cannot be performed in zebrafish because they lack structural homologs of the corpus callosum [102]. Nevertheless, proteomic analysis of csf1rDM brains revealed downregulation of Cux1a [58], a transcription factor required for the formation of layer II/III callosal projections in mice [103]. This finding might be relevant to the mechanisms contributing to the agenesis of the corpus callosum seen in BANDDOS, because downregulation of Cux1 was also documented in the cerebral cortex of a BANDDOS patient [58], as well as in Csf1r-null mice [22]. Thus, csf1rDM zebrafish are a useful model for some investigations of BANDOOS. However, studies of aspects of commissural development and degeneration require mammalian models.
Based on microglial density measurements and the importance of csf1rb for adult microglia, Oosterhof et al. reasoned that csf1ra−/−;b+/− mutant zebrafish would mimic the CSF1R haploinsufficiency in CRL patient microglia [9]. However, partial or even complete csf1r deficiency did not result in obvious myelin-related pathology or degeneration at 8 months of age, possibly because the fish analyzed were too young to develop CRL-like pathology (the lifespan of captive zebrafish can extend to 5.5 years). Furthermore, studies of CRL in zebrafish are not only complicated by their lack of the corpus callosum, but also by the lack of the gene homologue of CSF-2 [104], that contributes to disease development in the Csf1r+/− mouse [42]. Thus, at present, it is unlikely that the csf1ra−/−;b+/− mutants will prove to be a useful model of human CRL.
5.2. Rat:
As a model of human disease, the rat offers several advantages over the mouse, including the ability to learn a wider variety of tasks and the larger size of the brain, which facilitates experimental manipulations [105]. The disadvantages of the rat in aging studies are that the lifespan of a laboratory rat is longer than that of the mouse [106, 107] and that the development of technologies to manipulate genes in rats has been slower, although this is changing [108].
A Csf1r-null mutation has also been described in the rat [109]. On outbred backgrounds, the gross phenotypes of the Csf1r−/− rat and Csf1r−/− mouse closely resemble one another [4, 109]. Like Csf1r-null mice, Csf1r-null rat models reproduce features of BAANDOS, including the osteopetrosis and ventriculomegaly, but fail to reproduce the callosal agenesis/dysgenesis, and the cerebellar atrophy. Thus, the Csf1r-null DA rat does not appear to offer an advantage over the Csf1r-null FVB/NJ mouse in modeling BANDDOS.
While on the outbred background a significant proportion of both Csf1r−/− rats and mice survive post-weaning [4, 109], most rats obtained by backcrossing the original mutation for 7 generations to the dark agouti (DA) inbred background [57] either die or must be euthanized before 10–12 weeks, due to breathing difficulties. Given the longer lifespan of the rat, the survival of these rats is comparable to the survival of Csf1r−/− mice on the FVB/NJ background (3–4 weeks of age). Thus, on single strain backgrounds, neither model offers an advantage over the other for studying the role of CSF-1R in neurodegenerative disease or in aging.
Phenotypic characterization of Csf1r heterozygous DA rats has revealed significant differences from the Csf1r heterozygous C57BL6/J mouse [57]. In contrast to Csf1r +/− mice [42, 85], in Csf1r +/− rats Patkar et al. did not observe elevated microglial densities in any brain area and did not detect ventricular enlargement. However, it should be noted that ventricular enlargement is only apparent in advanced disease. In Csf1r +/− mice ventricular enlargement was only observed in mice with severe sensorimotor deficits [85] and longitudinal MRI studies in humans show that ventricular enlargement occurs concomitantly with white matter loss and cortical atrophy [51, 65, 110]. Using unspecified tests, the authors report the lack of a motor deficit in 18-month-old Csf1r +/− rats. However, in view of the 50% penetrance of disease at equivalent ages observed in humans and mice and of the small sample size (5 male rats/genotype), this conclusion is premature. Furthermore, testing of motor function in males is not likely to be useful as motor coordination deficits occur preferentially in females [42](Fig. 3A). Other behavioral tests were not reported. Thus, given the limited behavioral testing and small animal numbers used, it is premature to conclude that the Csf1r+/− rat does not model CRL.
6.0. Emerging therapies for CRL:
Observations in human and mouse indicate that CRL is a primary microgliopathy and suggest that CRL patients would benefit from microglial replacement therapy. However, given the yolk sac origin of microglia [83] and its importance for microglial identity in the transplantation setting [111], this approach is challenging. Nevertheless, the study of a family in which 5 members possessed a CRL mutant allele indicated that expression of wild type CSF-1R in some cells, via mosaicism or chimerism, was beneficial, suggesting hematopoietic stem cell transplantation as a therapeutic approach [112]. More recently, allogeneic stem cell transplantation of ALSP patients has been reported. The outcomes were variable, ranging from no benefit [31], to short-term cognitive stabilization [113, 114], to long-term stabilization [112]. Results of long-term follow-up in these cases are needed to validate allogeneic bone marrow transplantation as a therapeutic approach. An alternative approach, of transplantation of genetically-engineered autologous hematopoietic stem cells, has proven to be beneficial in X-linked adrenoleukodystrophy [115, 116]. Recently strategies for efficient microglial replacement therapy have been developed in mouse [117–119]. Transplantation experiments in the CRL mouse model are warranted to determine the effectiveness of therapy with both hematopoietic stem cells and induced pluripotent stem cell-derived microglial progenitors.
An alternative to microglial replacement is to reestablish microglial homeostasis. As indicated earlier, CRL microglia accumulate ceroid, a lipopigment that might impair mitochondrial and lysosomal functions [120, 121] and iron, an indicator of neurotoxic activation that can contribute to the generation of ROS. Short-term administration of a CSF-1R inhibitor that efficiently crosses the blood brain barrier, such as PLX5622 [122] resulting in the removal of these toxic microglia and their replacement with newly generated, more homeostatic versions might be, at least temporarily, beneficial in CRL. Indeed, transient depletion of microglia using CSF-1R inhibitor treatment has been shown to be beneficial in mouse models of neurodegenerative disease. In addition, experiments with various microglial depletion models suggest that while microglia are essential for normal brain development, in normal healthy adult mice not subject to imposed conditions they can be depleted for short periods without inducing behavioral or cognitive deficits or specific pathologies (reviewed in [122]). Another approach to reestablishing microglial homeostasis is to target CSF-2 signaling. Studies in the mouse model [42] indicate that the elevation of CSF-2 plays a causal role in CRL development and suggest that targeting of CSF-2 signaling in early disease would be beneficial.
7.0. Conclusions:
Depending on the gene dosage, Csf1r deficiency in mice recapitulates most features of BANDDOS and CRL and, as discussed, the Csf1r-null DA rat and zebrafish models do not appear to offer an advantage. Studies of the Csf1r+/− mouse have led to a better understanding of the underlying mechanisms involved in CRL development. They provided support to the notion that CRL is a primary microgliopathy [43], identified molecular pathways that could contribute to pathology [42] and offer a platform for further investigations. Along these lines, it is remarkable that both clinical and animal model studies indicate that females tend to be more severely affected by CRL. It is now recognized that microglia are sexually differentiated during development and maintain sexually-dimorphic features independently of their environment [123]. Furthermore, with aging, females experience earlier and/or more pronounced changes in brain metabolism and microglia activation than males [124, 125]. These naturally-occurring changes might interact with Csf1r insufficiency to precipitate the onset and accelerate the progression of CRL in females. In addition, the higher prevalence of gait dysfunction and dyskinesia in CRL female patients raises the possibility that estrogens and/or androgens might specifically regulate subpopulations of microglia such as the cerebellar microglia or those interacting with motor neurons. The mouse model reproduces the sexually dimorphic features of CRL and provides a system for genetic and hormonal interventions aimed at elucidating the underlying mechanisms. Most importantly, this model permits the identification of causal relationships in disease development through detailed investigation of the activities of dyshomeostatic microglia and their interaction with neural lineage cells during the presymptomatic stage.
Supplementary Material
Acknowledgments:
We thank Dr. Fabrizio Biundo for reviewing the manuscript. We also thank David and Ruth Levine for their support. This work was supported by grants from the National Institutes of Health: Grant R01NS091519 (to ERS), U54 HD090260 (support for the Rose F. Kennedy IDDRC) and the P30CA013330 NCI Cancer Center Grant. The funding sources had no involvement in study design; in the collection, analysis and interpretation of data; in the writing of the report and in the decision to submit the article for publication.
Abbreviations:
- BANDDOS
Brain Abnormalities, Neurodegeneration, and Dysosteosclerosis
- CRL
CSF1R-related leukoencephalopathy
- CSF-1
Colony stimulating factor-1
- CSF-1R
Colony stimulating factor-1 receptor
- IL-34
Interleukin-34
- NMD
nonsense-mediated mRNA decay
- ROS
reactive oxygen species
Footnotes
Conflicts of Interest:
The authors declare that they have no conflict of interest.
Supporting Information:
Table S1. CSF1R mutations in CRL.
References:
- 1.Chitu V & Stanley ER (2017) Regulation of Embryonic and Postnatal Development by the CSF-1 Receptor, Curr Top Dev Biol. 123, 229–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stanley ER & Chitu V (2014) CSF-1 receptor signaling in myeloid cells, Cold Spring Harb Perspect Biol. 6, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cecchini MG, Dominguez MG, Mocci S, Wetterwald A, Felix R, Fleisch H, Chisholm O, Hofstetter W, Pollard JW & Stanley ER (1994) Role of colony stimulating factor-1 in the establishment and regulation of tissue macrophages during postnatal development of the mouse, Development. 120, 1357–72. [DOI] [PubMed] [Google Scholar]
- 4.Dai XM, Ryan GR, Hapel AJ, Dominguez MG, Russell RG, Kapp S, Sylvestre V & Stanley ER (2002) Targeted disruption of the mouse colony-stimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies, and reproductive defects, Blood. 99, 111–20. [DOI] [PubMed] [Google Scholar]
- 5.Dai XM, Zong XH, Akhter MP & Stanley ER (2004) Osteoclast deficiency results in disorganized matrix, reduced mineralization, and abnormal osteoblast behavior in developing bone, J Bone Miner Res. 19, 1441–51. [DOI] [PubMed] [Google Scholar]
- 6.Wang Y, Szretter KJ, Vermi W, Gilfillan S, Rossini C, Cella M, Barrow AD, Diamond MS & Colonna M (2012) IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia, Nat Immunol. 13, 753–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Greter M, Lelios I, Pelczar P, Hoeffel G, Price J, Leboeuf M, Kundig TM, Frei K, Ginhoux F, Merad M & Becher B (2012) Stroma-derived interleukin-34 controls the development and maintenance of langerhans cells and the maintenance of microglia, Immunity. 37, 1050–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Monies D, Maddirevula S, Kurdi W, Alanazy MH, Alkhalidi H, Al-Owain M, Sulaiman RA, Faqeih E, Goljan E, Ibrahim N, Abdulwahab F, Hashem M, Abouelhoda M, Shaheen R, Arold ST & Alkuraya FS (2017) Autozygosity reveals recessive mutations and novel mechanisms in dominant genes: implications in variant interpretation, Genet Med. [DOI] [PubMed] [Google Scholar]
- 9.Oosterhof N, Kuil LE, van der Linde HC, Burm SM, Berdowski W, van Ijcken WFJ, van Swieten JC, Hol EM, Verheijen MHG & van Ham TJ (2018) Colony-Stimulating Factor 1 Receptor (CSF1R) Regulates Microglia Density and Distribution, but Not Microglia Differentiation In Vivo, Cell Rep. 24, 1203–1217 e6. [DOI] [PubMed] [Google Scholar]
- 10.Guo L, Bertola DR, Takanohashi A, Saito A, Segawa Y, Yokota T, Ishibashi S, Nishida Y, Yamamoto GL, Franco J, Honjo RS, Kim CA, Musso CM, Timmons M, Pizzino A, Taft RJ, Lajoie B, Knight MA, Fischbeck KH, Singleton AB, Ferreira CR, Wang Z, Yan L, Garbern JY, Simsek-Kiper PO, Ohashi H, Robey PG, Boyde A, Matsumoto N, Miyake N, Spranger J, Schiffmann R, Vanderver A, Nishimura G, Passos-Bueno M, Simons C, Ishikawa K & Ikegawa S (2019) Bi-allelic CSF1R Mutations Cause Skeletal Dysplasia of Dysosteosclerosis-Pyle Disease Spectrum and Degenerative Encephalopathy with Brain Malformation, Am J Hum Genet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tamhankar PM, Zhu B, Tamhankar VP, Mithbawkar S, Seabra L, Livingston JH, Ikeuchi T & Crow YJ (2020) A Novel Hypomorphic CSF1R Gene Mutation in the Biallelic State Leading to Fatal Childhood Neurodegeneration, Neuropediatrics. 51, 302–306. [DOI] [PubMed] [Google Scholar]
- 12.Helman G, Lajoie BR, Crawford J, Takanohashi A, Walkiewicz M, Dolzhenko E, Gross AM, Gainullin VG, Bent SJ, Jenkinson EM, Ferdinandusse S, Waterham HR, Dorboz I, Bertini E, Miyake N, Wolf NI, Abbink TEM, Kirwin SM, Tan CM, Hobson GM, Guo L, Ikegawa S, Pizzino A, Schmidt JL, Bernard G, Schiffmann R, van der Knaap MS, Simons C, Taft RJ & Vanderver A (2020) Genome sequencing in persistently unsolved white matter disorders, Ann Clin Transl Neurol. 7, 144–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rademakers R, Baker M, Nicholson AM, Rutherford NJ, Finch N, Soto-Ortolaza A, Lash J, Wider C, Wojtas A, DeJesus-Hernandez M, Adamson J, Kouri N, Sundal C, Shuster EA, Aasly J, MacKenzie J, Roeber S, Kretzschmar HA, Boeve BF, Knopman DS, Petersen RC, Cairns NJ, Ghetti B, Spina S, Garbern J, Tselis AC, Uitti R, Das P, Van Gerpen JA, Meschia JF, Levy S, Broderick DF, Graff-Radford N, Ross OA, Miller BB, Swerdlow RH, Dickson DW & Wszolek ZK (2011) Mutations in the colony stimulating factor 1 receptor (CSF1R) gene cause hereditary diffuse leukoencephalopathy with spheroids, Nat Genet. 44, 200–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Konno T, Kasanuki K, Ikeuchi T, Dickson DW & Wszolek ZK (2018) CSF1R-related leukoencephalopathy: A major player in primary microgliopathies, Neurology. 91, 1092–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chitu V, Gokhan S, Nandi S, Mehler MF & Stanley ER (2016) Emerging Roles for CSF-1 Receptor and its Ligands in the Nervous System, Trends Neurosci. 39, 378–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kondo Y & Duncan ID (2009) Selective reduction in microglia density and function in the white matter of colony-stimulating factor-1-deficient mice, J Neurosci Res. 87, 2686–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sasaki A, Yokoo H, Naito M, Kaizu C, Shultz LD & Nakazato Y (2000) Effects of macrophage-colony-stimulating factor deficiency on the maturation of microglia and brain macrophages and on their expression of scavenger receptor, Neuropathology. 20, 134–142. [DOI] [PubMed] [Google Scholar]
- 18.Kana V, Desland FA, Casanova-Acebes M, Ayata P, Badimon A, Nabel E, Yamamuro K, Sneeboer M, Tan IL, Flanigan ME, Rose SA, Chang C, Leader A, Le Bourhis H, Sweet ES, Tung N, Wroblewska A, Lavin Y, See P, Baccarini A, Ginhoux F, Chitu V, Stanley ER, Russo SJ, Yue Z, Brown BD, Joyner AL, De Witte LD, Morishita H, Schaefer A & Merad M (2019) CSF-1 controls cerebellar microglia and is required for motor function and social interaction, J Exp Med. 216, 2265–2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Badimon A, Strasburger HJ, Ayata P, Chen X, Nair A, Ikegami A, Hwang P, Chan AT, Graves SM, Uweru JO, Ledderose C, Kutlu MG, Wheeler MA, Kahan A, Ishikawa M, Wang YC, Loh YE, Jiang JX, Surmeier DJ, Robson SC, Junger WG, Sebra R, Calipari ES, Kenny PJ, Eyo UB, Colonna M, Quintana FJ, Wake H, Gradinaru V & Schaefer A (2020) Negative feedback control of neuronal activity by microglia, Nature. 586, 417–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Easley-Neal C, Foreman O, Sharma N, Zarrin AA & Weimer RM (2019) CSF1R Ligands IL-34 and CSF1 Are Differentially Required for Microglia Development and Maintenance in White and Gray Matter Brain Regions, Front Immunol. 10, 2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Y, Berezovska O & Fedoroff S (1999) Expression of colony stimulating factor-1 receptor (CSF-1R) by CNS neurons in mice, Journal of neuroscience research. 57, 616–32. [PubMed] [Google Scholar]
- 22.Nandi S, Gokhan S, Dai XM, Wei S, Enikolopov G, Lin H, Mehler MF & Stanley ER (2012) The CSF-1 receptor ligands IL-34 and CSF-1 exhibit distinct developmental brain expression patterns and regulate neural progenitor cell maintenance and maturation, Dev Biol. 367, 100–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luo J, Elwood F, Britschgi M, Villeda S, Zhang H, Ding Z, Zhu L, Alabsi H, Getachew R, Narasimhan R, Wabl R, Fainberg N, James ML, Wong G, Relton J, Gambhir SS, Pollard JW & Wyss-Coray T (2013) Colony-stimulating factor 1 receptor (CSF1R) signaling in injured neurons facilitates protection and survival, J Exp Med. 210, 157–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clare AJ, Day RC, Empson RM & Hughes SM (2018) Transcriptome Profiling of Layer 5 Intratelencephalic Projection Neurons From the Mature Mouse Motor Cortex, Front Mol Neurosci. 11, 410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alturkustani M, Keith J, Hazrati LN, Rademakers R & Ang LC (2015) Pathologic staging of white matter lesions in adult-onset leukoencephalopathy/leukodystrophy with axonal spheroids, J Neuropathol Exp Neurol. 74, 233–40. [DOI] [PubMed] [Google Scholar]
- 26.Konno T, Yoshida K, Mizuno T, Kawarai T, Tada M, Nozaki H, Ikeda SI, Nishizawa M, Onodera O, Wszolek ZK & Ikeuchi T (2017) Clinical and genetic characterization of adult-onset leukoencephalopathy with axonal spheroids and pigmented glia associated with CSF1R mutation, Eur J Neurol. 24, 37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Konno T, Miura T, Harriott AM, Mezaki N, Edwards ES, Rademakers R, Ross OA, Meschia JF, Ikeuchi T & Wszolek ZK (2018) Partial loss of function of colony-stimulating factor 1 receptor in a patient with white matter abnormalities, Eur J Neurol. 25, 875–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen J, Luo S, Li N, Li H, Han J & Ling L (2020) A Novel Missense Mutation of the CSF1R Gene Causes Incurable CSF1R-Related Leukoencephalopathy: Case Report and Review of Literature, Int J Gen Med. 13, 1613–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eichler FS, Li J, Guo Y, Caruso PA, Bjonnes AC, Pan J, Booker JK, Lane JM, Tare A, Vlasac I, Hakonarson H, Gusella JF, Zhang J, Keating BJ & Saxena R (2016) CSF1R mosaicism in a family with hereditary diffuse leukoencephalopathy with spheroids, Brain. 139, 1666–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.La Piana R, Webber A, Guiot MC, Del Pilar Cortes M & Brais B (2014) A novel mutation in the CSF1R gene causes a variable leukoencephalopathy with spheroids, Neurogenetics. 15, 289–94. [DOI] [PubMed] [Google Scholar]
- 31.Wang M & Zhang X (2019) A novel CSF-1R mutation in a family with hereditary diffuse leukoencephalopathy with axonal spheroids misdiagnosed as hydrocephalus, Neurogenetics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tipton PW, Stanley ER, Chitu V & Wszolek ZK (2021) Is Pre-Symptomatic Immunosuppression Protective in CSF1R-Related Leukoencephalopathy?, Mov Disord. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karle KN, Biskup S, Schule R, Schweitzer KJ, Kruger R, Bauer P, Bender B, Nagele T & Schols L (2013) De novo mutations in hereditary diffuse leukoencephalopathy with axonal spheroids (HDLS), Neurology. 81, 2039–44. [DOI] [PubMed] [Google Scholar]
- 34.Abe T, Kawarai T, Fujita K, Sako W, Terasawa Y, Matsuda T, Sakai W, Tsukamoto-Miyashiro A, Matsui N, Izumi Y, Kaji R & Harada M (2017) MR Spectroscopy in Patients with Hereditary Diffuse Leukoencephalopathy with Spheroids and Asymptomatic Carriers of Colony-stimulating Factor 1 Receptor Mutation, Magnetic resonance in medical sciences : MRMS : an official journal of Japan Society of Magnetic Resonance in Medicine. 16, 297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Battisti C, Di Donato I, Bianchi S, Monti L, Formichi P, Rufa A, Taglia I, Cerase A, Dotti MT & Federico A (2014) Hereditary diffuse leukoencephalopathy with axonal spheroids: three patients with stroke-like presentation carrying new mutations in the CSF1R gene, J Neurol. 261, 768–72. [DOI] [PubMed] [Google Scholar]
- 36.Tian WT, Zhan FX, Liu Q, Luan XH, Zhang C, Shang L, Zhang BY, Pan SJ, Miao F, Hu J, Zhong P, Liu SH, Zhu ZY, Zhou HY, Sun S, Liu XL, Huang XJ, Jiang JW, Ma JF, Wang Y, Chen SF, Tang HD, Chen SD & Cao L (2019) Clinicopathologic characterization and abnormal autophagy of CSF1R-related leukoencephalopathy, Transl Neurodegener. 8, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Du Q, Chen H, Shi Z, Zhang Y, Wang J & Zhou H (2019) A novel mutation in the CSF1R gene causes hereditary diffuse leukoencephalopathy with axonal spheroids, Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical Neurophysiology. 40, 1287–1290. [DOI] [PubMed] [Google Scholar]
- 38.Kortvelyessy P, Krageloh-Mann I, Mawrin C, Heinze HJ, Bittner D, Wieland I, Zenker M, Nestor P, Lim JP, Gosavi P, Mintern JD, Ross EM & Gleeson PA (2015) Hereditary diffuse leukoencephalopathy with spheroids (HDLS) with a novel CSF1R mutation and spinal cord involvement, J Neurol Sci. 358, 515–7. [DOI] [PubMed] [Google Scholar]
- 39.Kim EJ, Shin JH, Lee JH, Kim JH, Na DL, Suh YL, Hwang SJ, Lee JH, Lee YM, Shin MJ, Lee MJ, Kim SJ, Yoon U, Park do Y, Jung DS, Ahn JW, Sung S & Huh GY (2015) Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia linked CSF1R mutation: Report of four Korean cases, J Neurol Sci. 349, 232–8. [DOI] [PubMed] [Google Scholar]
- 40.Ho VM, Hovsepian DA & Shieh PB (2019) Myelopathy in a patient with leukodystrophy due to CSF1R mutation, Neurol Genet. 5, e376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kempthorne L, Yoon H, Madore C, Smith S, Wszolek ZK, Rademakers R, Kim J, Butovsky O & Dickson DW (2020) Correction to: Loss of homeostatic microglial phenotype in CSF1R-related Leukoencephalopathy, Acta Neuropathol Commun. 8, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chitu V, Biundo F, Shlager GGL, Park ES, Wang P, Gulinello ME, Gokhan S, Ketchum HC, Saha K, DeTure MA, Dickson DW, Wszolek ZK, Zheng D, Croxford AL, Becher B, Sun D, Mehler MF & Stanley ER (2020) Microglial Homeostasis Requires Balanced CSF-1/CSF-2 Receptor Signaling, Cell Rep. 30, 3004–3019 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Biundo F, Chitu V, Shlager GGL, Park ES, Gulinello ME, Saha K, Ketchum HC, Fernandes C, Gokhan S, Mehler MF & Stanley ER (2021) Microglial reduction of colony stimulating factor-1 receptor expression is sufficient to confer adult onset leukodystrophy, Glia. 69, 779–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hiyoshi M, Hashimoto M, Yukihara M, Bhuyan F & Suzu S (2013) M-CSF receptor mutations in hereditary diffuse leukoencephalopathy with spheroids impair not only kinase activity but also surface expression, Biochem Biophys Res Commun. 440, 589–93. [DOI] [PubMed] [Google Scholar]
- 45.Nicholson AM, Baker MC, Finch NA, Rutherford NJ, Wider C, Graff-Radford NR, Nelson PT, Clark HB, Wszolek ZK, Dickson DW, Knopman DS & Rademakers R (2013) CSF1R mutations link POLD and HDLS as a single disease entity, Neurology. 80, 1033–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Konno T, Tada M, Tada M, Nishizawa M & Ikeuchi T (2014) [Hereditary Diffuse Leukoencephalopathy with Spheroids (HDLS): A Review of the Literature on its Clinical Characteristics and Mutations in the Colony-Stimulating Factor-1 Receptor Gene], Brain Nerve. 66, 581–90. [PubMed] [Google Scholar]
- 47.Miura T, Mezaki N, Konno T, Iwasaki A, Hara N, Miura M, Funayama M, Unai Y, Tashiro Y, Okita K, Kihara T, Ito N, Kanatsuka Y, Jones DT, Hara N, Ishiguro T, Tokutake T, Kasuga K, Nozaki H, Dickson DW, Onodera O, Wszolek ZK & Ikeuchi T (2018) Identification and functional characterization of novel mutations including frameshift mutation in exon 4 of CSF1R in patients with adult-onset leukoencephalopathy with axonal spheroids and pigmented glia, J Neurol. 265, 2415–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kraya T, Quandt D, Pfirrmann T, Kindermann A, Lampe L, Schroeter ML, Kohlhase J, Stoevesandt D, Hoffmann K & Villavicencio-Lorini P (2019) Functional characterization of a novel CSF1R mutation causing hereditary diffuse leukoencephalopathy with spheroids, Mol Genet Genomic Med. 7, e00595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Delaney C, Farrell M, Doherty CP, Brennan K, O’Keeffe E, Greene C, Byrne K, Kelly E, Birmingham N, Hickey P, Cronin S, Savvides SN, Doyle SL & Campbell M (2020) Attenuated CSF-1R signalling drives cerebrovascular pathology, EMBO Mol Med, e12889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leng C, Lu L, Wang G, Zhang Y, Xu Y, Lin X, Shen N, Xu X, Qun S, Sun M & Ge W (2019) A novel dominant-negative mutation of the CSF1R gene causes adult-onset leukoencephalopathy with axonal spheroids and pigmented glia, Am J Transl Res. 11, 6093–6101. [PMC free article] [PubMed] [Google Scholar]
- 51.Konno T, Tada M, Tada M, Koyama A, Nozaki H, Harigaya Y, Nishimiya J, Matsunaga A, Yoshikura N, Ishihara K, Arakawa M, Isami A, Okazaki K, Yokoo H, Itoh K, Yoneda M, Kawamura M, Inuzuka T, Takahashi H, Nishizawa M, Onodera O, Kakita A & Ikeuchi T (2014) Haploinsufficiency of CSF-1R and clinicopathologic characterization in patients with HDLS, Neurology. 82, 139–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wei Y, Ma M, Lin S, Li X, Shu Y, Wang Z, Zhou Y, Hu B, Cheng B, Duan S, Huang X, Xu H, Zhang YW & Zheng H (2021) Proteolytic Shedding of Human Colony-Stimulating Factor 1 Receptor and its implication, J Cell Mol Med. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Elegheert J, Desfosses A, Shkumatov AV, Wu X, Bracke N, Verstraete K, Van Craenenbroeck K, Brooks BR, Svergun DI, Vergauwen B, Gutsche I & Savvides SN (2011) Extracellular complexes of the hematopoietic human and mouse CSF-1 receptor are driven by common assembly principles, Structure. 19, 1762–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yeh YM, Hsu SJ, Lin PC, Hsu KF, Wu PY, Su WC, Chang JY & Shen MR (2017) The c.1085A>G Genetic Variant of CSF1R Gene Regulates Tumor Immunity by Altering the Proliferation, Polarization, and Function of Macrophages, Clin Cancer Res. 23, 6021–6030. [DOI] [PubMed] [Google Scholar]
- 55.Hume DA, Caruso M, Ferrari-Cestari M, Summers KM, Pridans C & Irvine KM (2020) Phenotypic impacts of CSF1R deficiencies in humans and model organisms, J Leukoc Biol. 107, 205–219. [DOI] [PubMed] [Google Scholar]
- 56.Pridans C, Sauter KA, Baer K, Kissel H & Hume DA (2013) CSF1R mutations in hereditary diffuse leukoencephalopathy with spheroids are loss of function, Sci Rep. 3, 3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Patkar OL, Caruso M, Teakle N, Keshvari S, Bush SJ, Pridans C, Belmer A, Summers KM, Irvine KM & Hume DA (2021) Analysis of homozygous and heterozygous Csf1r knockout in the rat as a model for understanding microglial function in brain development and the impacts of human CSF1R mutations, Neurobiol Dis. 151, 105268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Oosterhof N, Chang IJ, Karimiani EG, Kuil LE, Jensen DM, Daza R, Young E, Astle L, van der Linde HC, Shivaram GM, Demmers J, Latimer CS, Keene CD, Loter E, Maroofian R, van Ham TJ, Hevner RF & Bennett JT (2019) Homozygous Mutations in CSF1R Cause a Pediatric-Onset Leukoencephalopathy and Can Result in Congenital Absence of Microglia, Am J Hum Genet. 104, 936–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sundal C, Fujioka S, Van Gerpen JA, Wider C, Nicholson AM, Baker M, Shuster EA, Aasly J, Spina S, Ghetti B, Roeber S, Garbern J, Tselis A, Swerdlow RH, Miller BB, Borjesson-Hanson A, Uitti RJ, Ross OA, Stoessl AJ, Rademakers R, Josephs KA, Dickson DW, Broderick D & Wszolek ZK (2013) Parkinsonian features in hereditary diffuse leukoencephalopathy with spheroids (HDLS) and CSF1R mutations, Parkinsonism Relat Disord. 19, 869–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Freeman SH, Hyman BT, Sims KB, Hedley-Whyte ET, Vossough A, Frosch MP & Schmahmann JD (2009) Adult onset leukodystrophy with neuroaxonal spheroids: clinical, neuroimaging and neuropathologic observations, Brain Pathol. 19, 39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Coomans C, Sieben A, Lammens M, Ceuterick-de Groote C, Vandenbroecke C, Goethals I, Van Melkebeke D & Hemelsoet D (2018) Early-onset dementia, leukoencephalopathy and brain calcifications: a clinical, imaging and pathological comparison of ALSP and PLOSL/Nasu Hakola disease, Acta Neurol Belg. 118, 607–615. [DOI] [PubMed] [Google Scholar]
- 62.Kim M, Lee H, Cho HJ, Young Chun S, Shin JH, Kim EJ, Woo Ahn J, Huh GY, Baek SY & Lee JH (2017) Pathologic Correlation of Paramagnetic White Matter Lesions in Adult-Onset Leukoencephalopathy With Axonal Spheroids and Pigmented Glia, J Neuropathol Exp Neurol. 76, 924–928. [DOI] [PubMed] [Google Scholar]
- 63.Adams SJ, Kirk A & Auer RN (2018) Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP): Integrating the literature on hereditary diffuse leukoencephalopathy with spheroids (HDLS) and pigmentary orthochromatic leukodystrophy (POLD), J Clin Neurosci. 48, 42–49. [DOI] [PubMed] [Google Scholar]
- 64.Zhan FX, Zhu ZY, Liu Q, Zhou HY, Luan XH, Huang XJ, Liu XL, Tian WT, Wang SG, Song XX, Chen G, Zhao ML, Wang Y, Tang HD, Hu J, Chen SD, Li BY & Cao L (2020) Altered structural and functional connectivity in CSF1R-related leukoencephalopathy, Brain Imaging Behav. [DOI] [PubMed] [Google Scholar]
- 65.Oyanagi K, Kinoshita M, Suzuki-Kouyama E, Inoue T, Nakahara A, Tokiwai M, Arai N, Satoh JI, Aoki N, Jinnai K, Yazawa I, Arai K, Ishihara K, Kawamura M, Ishizawa K, Hasegawa K, Yagisita S, Amano N, Yoshida K, Terada S, Yoshida M, Akiyama H, Mitsuyama Y & Ikeda SI (2017) Adult onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP) and Nasu-Hakola disease: lesion staging and dynamic changes of axons and microglial subsets, Brain Pathol. 27, 748–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tada M, Konno T, Tada M, Tezuka T, Miura T, Mezaki N, Okazaki KI, Arakawa M, Itoh K, Yamamoto T, Yokoo H, Yoshikura N, Ishihara K, Horie M, Takebayashi H, Toyoshima Y, Naito M, Onodera O, Nishizawa M, Takahashi H, Ikeuchi T & Kakita A (2016) Characteristic microglial features in patients with hereditary diffuse leukoencephalopathy with spheroids, Ann Neurol. [DOI] [PubMed] [Google Scholar]
- 67.Kempthorne L, Yoon H, Madore C, Smith S, Wszolek ZK, Rademakers R, Kim J, Butovsky O & Dickson DW (2020) Loss of homeostatic microglial phenotype in CSF1R-related Leukoencephalopathy, Acta Neuropathol Commun. 8, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kinoshita M, Oyanagi K, Kondo Y, Ishizawa K, Ishihara K, Yoshida M, Inoue T, Mitsuyama Y, Yoshida K, Yamada M, Sekijima Y & Ikeda SI (2021) Pathologic basis of the preferential thinning of thecorpus callosum in adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP), eNeurologicalSci. 22, 100310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Coleman M (2005) Axon degeneration mechanisms: commonality amid diversity, Nat Rev Neurosci. 6, 889–98. [DOI] [PubMed] [Google Scholar]
- 70.Beirowski B, Nogradi A, Babetto E, Garcia-Alias G & Coleman MP (2010) Mechanisms of axonal spheroid formation in central nervous system Wallerian degeneration, J Neuropathol Exp Neurol. 69, 455–72. [DOI] [PubMed] [Google Scholar]
- 71.Ali ZS, Van Der Voorn JP & Powers JM (2007) A comparative morphologic analysis of adult onset leukodystrophy with neuroaxonal spheroids and pigmented glia--a role for oxidative damage, J Neuropathol Exp Neurol. 66, 660–72. [DOI] [PubMed] [Google Scholar]
- 72.Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, Raman R, Davies P, Masliah E, Williams DS & Goldstein LS (2005) Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease, Science. 307, 1282–8. [DOI] [PubMed] [Google Scholar]
- 73.Barsukova AG, Forte M & Bourdette D (2012) Focal increases of axoplasmic Ca2+, aggregation of sodium-calcium exchanger, N-type Ca2+ channel, and actin define the sites of spheroids in axons undergoing oxidative stress, J Neurosci. 32, 12028–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Thomsen MS, Andersen MV, Christoffersen PR, Jensen MD, Lichota J & Moos T (2015) Neurodegeneration with inflammation is accompanied by accumulation of iron and ferritin in microglia and neurons, Neurobiol Dis. 81, 108–18. [DOI] [PubMed] [Google Scholar]
- 75.Nnah IC & Wessling-Resnick M (2018) Brain Iron Homeostasis: A Focus on Microglial Iron, Pharmaceuticals (Basel). 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cheli VT, Correale J, Paez PM & Pasquini JM (2020) Iron Metabolism in Oligodendrocytes and Astrocytes, Implications for Myelination and Remyelination, ASN Neuro. 12, 1759091420962681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hofer TP, Zawada AM, Frankenberger M, Skokann K, Satzl AA, Gesierich W, Schuberth M, Levin J, Danek A, Rotter B, Heine GH & Ziegler-Heitbrock L (2015) Characterization of subsets of the CD16-positive monocytes: impact of granulomatous inflammation and M-CSF-receptor mutation, Blood. [DOI] [PubMed] [Google Scholar]
- 78.Hamatani M, Yamashita H, Ochi H, Ashida S, Hashi Y, Okada Y, Fujii C, Kawamura K, Kitazawa R, Nakagawa M, Mizuno T, Takahashi R & Kondo T (2020) Altered features of monocytes in adult onset leukoencephalopathy with axonal spheroids and pigmented glia: A clue to the pathomechanism of microglial dyshomeostasis, Neurobiol Dis, 104867. [DOI] [PubMed] [Google Scholar]
- 79.Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, Strauss-Ayali D, Viukov S, Guilliams M, Misharin A, Hume DA, Perlman H, Malissen B, Zelzer E & Jung S (2013) Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis, Immunity. 38, 79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Thomas G, Tacke R, Hedrick CC & Hanna RN (2015) Nonclassical patrolling monocyte function in the vasculature, Arterioscler Thromb Vasc Biol. 35, 1306–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Audoy-Remus J, Richard JF, Soulet D, Zhou H, Kubes P & Vallieres L (2008) Rod-Shaped monocytes patrol the brain vasculature and give rise to perivascular macrophages under the influence of proinflammatory cytokines and angiopoietin-2, J Neurosci. 28, 10187–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bellavance MA, Gosselin D, Yong VW, Stys PK & Rivest S (2015) Patrolling monocytes play a critical role in CX3CR1-mediated neuroprotection during excitotoxicity, Brain Struct Funct. 220, 1759–76. [DOI] [PubMed] [Google Scholar]
- 83.Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER, Samokhvalov IM & Merad M (2010) Fate mapping analysis reveals that adult microglia derive from primitive macrophages, Science. 330, 841–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Erblich B, Zhu L, Etgen AM, Dobrenis K & Pollard JW (2011) Absence of colony stimulation factor-1 receptor results in loss of microglia, disrupted brain development and olfactory deficits, PLoS One. 6, e26317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chitu V, Gokhan S, Gulinello M, Branch CA, Patil M, Basu R, Stoddart C, Mehler MF & Stanley ER (2015) Phenotypic characterization of a Csf1r haploinsufficient mouse model of adult-onset leukodystrophy with axonal spheroids and pigmented glia (ALSP), Neurobiol Dis. 74, 219–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang S, Lai X, Deng Y & Song Y (2020) Correlation between mouse age and human age in anti-tumor research: Significance and method establishment, Life Sci. 242, 117242. [DOI] [PubMed] [Google Scholar]
- 87.Bennett S, Por SB, Cooley MA & Breit SN (1993) In vitro replication dynamics of human culture-derived macrophages in a long term serum-free system, JImmunol. 150, 2364–2371. [PubMed] [Google Scholar]
- 88.Cheung DL & Hamilton JA (1992) Regulation of human monocyte DNA synthesis by colony-stimulating factors, cytokines, and cyclic adenosine monophosphate, Blood. 79, 1972–1981. [PubMed] [Google Scholar]
- 89.Li Y, Chen Q, Zheng D, Yin L, Chionh YH, Wong LH, Tan SQ, Tan TC, Chan JK, Alonso S, Dedon PC, Lim B & Chen J (2013) Induction of functional human macrophages from bone marrow promonocytes by M-CSF in humanized mice, J Immunol. 191, 3192–9. [DOI] [PubMed] [Google Scholar]
- 90.Hamilton JA (2020) GM-CSF in inflammation, J Exp Med. 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kawakami I, Iseki E, Kasanuki K, Minegishi M, Sato K, Hino H, Shibuya K, Fujisawa K, Higashi S, Akiyama H, Furuta A, Takanashi M, Li Y, Hattori N, Mitsuyama Y & Arai H (2016) A family with hereditary diffuse leukoencephalopathy with spheroids caused by a novel c.2442+2T>C mutation in the CSF1R gene, J Neurol Sci. 367, 349–55. [DOI] [PubMed] [Google Scholar]
- 92.Hagemeyer N, Hanft KM, Akriditou MA, Unger N, Park ES, Stanley ER, Staszewski O, Dimou L & Prinz M (2017) Microglia contribute to normal myelinogenesis and to oligodendrocyte progenitor maintenance during adulthood, Acta Neuropathol. 134, 441–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mohri I, Taniike M, Taniguchi H, Kanekiyo T, Aritake K, Inui T, Fukumoto N, Eguchi N, Kushi A, Sasai H, Kanaoka Y, Ozono K, Narumiya S, Suzuki K & Urade Y (2006) Prostaglandin D2-mediated microglia/astrocyte interaction enhances astrogliosis and demyelination in twitcher, J Neurosci. 26, 4383–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Schermer C & Humpel C (2002) Granulocyte macrophage-colony stimulating factor activates microglia in rat cortex organotypic brain slices, Neurosci Lett. 328, 180–4. [DOI] [PubMed] [Google Scholar]
- 95.Xiao BG, Xu LY & Yang JS (2002) TGF-beta 1 synergizes with GM-CSF to promote the generation of glial cell-derived dendriform cells in vitro, Brain Behav Immun. 16, 685–97. [DOI] [PubMed] [Google Scholar]
- 96.Smith ME (1993) Phagocytosis of myelin by microglia in vitro, J Neurosci Res. 35, 480–7. [DOI] [PubMed] [Google Scholar]
- 97.Basso L, Lapointe TK, Iftinca M, Marsters C, Hollenberg MD, Kurrasch DM & Altier C (2017) Granulocyte-colony-stimulating factor (G-CSF) signaling in spinal microglia drives visceral sensitization following colitis, Proc Natl Acad Sci U S A. 114, 11235–11240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Munro DAD, Bradford BM, Mariani SA, Hampton DW, Vink CS, Chandran S, Hume DA, Pridans C & Priller J (2020) CNS macrophages differentially rely on an intronic Csf1r enhancer for their development, Development. 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rojo R, Raper A, Ozdemir DD, Lefevre L, Grabert K, Wollscheid-Lengeling E, Bradford B, Caruso M, Gazova I, Sanchez A, Lisowski ZM, Alves J, Molina-Gonzalez I, Davtyan H, Lodge RJ, Glover JD, Wallace R, Munro DAD, David E, Amit I, Miron VE, Priller J, Jenkins SJ, Hardingham GE, Blurton-Jones M, Mabbott NA, Summers KM, Hohenstein P, Hume DA & Pridans C (2019) Deletion of a Csf1r enhancer selectively impacts CSF1R expression and development of tissue macrophage populations, Nat Commun. 10, 3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Herbomel P, Thisse B & Thisse C (2001) Zebrafish early macrophages colonize cephalic mesenchyme and developing brain, retina, and epidermis through a M-CSF receptor-dependent invasive process, Developmental biology. 238, 274–88. [DOI] [PubMed] [Google Scholar]
- 101.Ferrero G, Miserocchi M, Di Ruggiero E & Wittamer V (2021) A csf1rb mutation uncouples two waves of microglia development in zebrafish, Development. 148. [DOI] [PubMed] [Google Scholar]
- 102.Chinn GA, Hirokawa KE, Chuang TM, Urbina C, Patel F, Fong J, Funatsu N & Monuki ES (2015) Agenesis of the Corpus Callosum Due to Defective Glial Wedge Formation in Lhx2 Mutant Mice, Cereb Cortex. 25, 2707–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rodriguez-Tornos FM, Briz CG, Weiss LA, Sebastian-Serrano A, Ares S, Navarrete M, Frangeul L, Galazo M, Jabaudon D, Esteban JA & Nieto M (2016) Cux1 Enables Interhemispheric Connections of Layer II/III Neurons by Regulating Kv1-Dependent Firing, Neuron. 89, 494–506. [DOI] [PubMed] [Google Scholar]
- 104.Pazhakh V & Lieschke GJ (2018) Hematopoietic growth factors: the scenario in zebrafish, Growth Factors. 36, 196–212. [DOI] [PubMed] [Google Scholar]
- 105.Iannaccone PM & Jacob HJ (2009) Rats!, Dis Model Mech. 2, 206–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sengupta P (2013) The Laboratory Rat: Relating Its Age With Human’s, Int J Prev Med. 4, 624–30. [PMC free article] [PubMed] [Google Scholar]
- 107.Dutta S & Sengupta P (2016) Men and mice: Relating their ages, Life Sci. 152, 244–8. [DOI] [PubMed] [Google Scholar]
- 108.Meek S, Mashimo T & Burdon T (2017) From engineering to editing the rat genome, Mamm Genome. 28, 302–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Pridans C, Raper A, Davis GM, Alves J, Sauter KA, Lefevre L, Regan T, Meek S, Sutherland L, Thomson AJ, Clohisey S, Bush SJ, Rojo R, Lisowski ZM, Wallace R, Grabert K, Upton KR, Tsai YT, Brown D, Smith LB, Summers KM, Mabbott NA, Piccardo P, Cheeseman MT, Burdon T & Hume DA (2018) Pleiotropic Impacts of Macrophage and Microglial Deficiency on Development in Rats with Targeted Mutation of the Csf1r Locus, J Immunol. 201, 2683–2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kim SI, Jeon B, Bae J, Won JK, Kim HJ, Yim J, Kim YJ & Park SH (2019) An Autopsy Proven Case of CSF1R-mutant Adult-onset Leukoencephalopathy with Axonal Spheroids and Pigmented Glia (ALSP) with Premature Ovarian Failure, Exp Neurobiol. 28, 119–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bennett FC, Bennett ML, Yaqoob F, Mulinyawe SB, Grant GA, Hayden Gephart M, Plowey ED & Barres BA (2018) A Combination of Ontogeny and CNS Environment Establishes Microglial Identity, Neuron. 98, 1170–1183 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Eichler F, Duncan C, Musolino PL, Orchard PJ, De Oliveira S, Thrasher AJ, Armant M, Dansereau C, Lund TC, Miller WP, Raymond GV, Sankar R, Shah AJ, Sevin C, Gaspar HB, Gissen P, Amartino H, Bratkovic D, Smith NJC, Paker AM, Shamir E, O’Meara T, Davidson D, Aubourg P & Williams DA (2017) Hematopoietic Stem-Cell Gene Therapy for Cerebral Adrenoleukodystrophy, N Engl J Med. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gelfand JM, Greenfield AL, Barkovich M, Mendelsohn BA, Van Haren K, Hess CP & Mannis GN (2020) Allogeneic HSCT for adult-onset leukoencephalopathy with spheroids and pigmented glia, Brain. 143, 503–511. [DOI] [PubMed] [Google Scholar]
- 114.Mochel F, Delorme C, Czernecki V, Froger J, Cormier F, Ellie E, Fegueux N, Lehericy S, Lumbroso S, Schiffmann R, Aubourg P, Roze E, Labauge P & Nguyen S (2019) Haematopoietic stem cell transplantation in CSF1R-related adult-onset leukoencephalopathy with axonal spheroids and pigmented glia, J Neurol Neurosurg Psychiatry. 90, 1375–1376. [DOI] [PubMed] [Google Scholar]
- 115.Eichler F, Duncan C, Musolino PL, Orchard PJ, De Oliveira S, Thrasher AJ, Armant M, Dansereau C, Lund TC, Miller WP, Raymond GV, Sankar R, Shah AJ, Sevin C, Gaspar HB, Gissen P, Amartino H, Bratkovic D, Smith NJC, Paker AM, Shamir E, O’Meara T, Davidson D, Aubourg P & Williams DA (2017) Hematopoietic Stem-Cell Gene Therapy for Cerebral Adrenoleukodystrophy, N Engl J Med. 377, 1630–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mallack EJ, Turk B, Yan H & Eichler FS (2019) The Landscape of Hematopoietic Stem Cell Transplant and Gene Therapy for X-Linked Adrenoleukodystrophy, Curr Treat Options Neurol. 21, 61. [DOI] [PubMed] [Google Scholar]
- 117.Han J, Sarlus H, Wszolek ZK, Karrenbauer VD & Harris RA (2020) Microglial replacement therapy: a potential therapeutic strategy for incurable CSF1R-related leukoencephalopathy, Acta Neuropathol Commun. 8, 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hohsfield LA, Najafi AR, Ghorbanian Y, Soni N, Hingco EE, Kim SJ, Jue AD, Swarup V, Inlay MA & Green KN (2020) Effects of long-term and brain-wide colonization of peripheral bone marrow-derived myeloid cells in the CNS, J Neuroinflammation. 17, 279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Xu Z, Rao Y, Huang Y, Zhou T, Feng R, Xiong S, Yuan TF, Qin S, Lu Y, Zhou X, Li X, Qin B, Mao Y & Peng B (2020) Efficient Strategies for Microglia Replacement in the Central Nervous System, Cell Rep. 33, 108443. [DOI] [PubMed] [Google Scholar]
- 120.Perrotta I (2018) Occurrence and characterization of lipofuscin and ceroid in human atherosclerotic plaque, Ultrastruct Pathol. 42, 477–488. [DOI] [PubMed] [Google Scholar]
- 121.Terman A (2001) Garbage catastrophe theory of aging: imperfect removal of oxidative damage?, Redox Rep. 6, 15–26. [DOI] [PubMed] [Google Scholar]
- 122.Green KN, Crapser JD & Hohsfield LA (2020) To Kill a Microglia: A Case for CSF1R Inhibitors, Trends Immunol. 41, 771–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Villa A, Della Torre S & Maggi A (2019) Sexual differentiation of microglia, Front Neuroendocrinol. 52, 156–164. [DOI] [PubMed] [Google Scholar]
- 124.Mangold CA, Masser DR, Stanford DR, Bixler GV, Pisupati A, Giles CB, Wren JD, Ford MM, Sonntag WE & Freeman WM (2017) CNS-wide Sexually Dimorphic Induction of the Major Histocompatibility Complex 1 Pathway With Aging, J Gerontol A Biol Sci Med Sci. 72, 16–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zhao L, Mao Z, Woody SK & Brinton RD (2016) Sex differences in metabolic aging of the brain: insights into female susceptibility to Alzheimer’s disease, Neurobiol Aging. 42, 69–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Yang X, Huang P, Tan Y & Xiao Q (2019) A Novel Splicing Mutation in the CSF1R Gene in a Family With Hereditary Diffuse Leukoencephalopathy With Axonal Spheroids, Front Genet. 10, 491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Guerreiro R, Kara E, Le Ber I, Bras J, Rohrer JD, Taipa R, Lashley T, Dupuits C, Gurunlian N, Mochel F, Warren JD, Hannequin D, Sedel F, Depienne C, Camuzat A, Golfier V, Du Boisgueheneuc F, Schottlaender L, Fox NC, Beck J, Mead S, Rossor MN, Hardy J, Revesz T, Brice A & Houlden H (2013) Genetic analysis of inherited leukodystrophies: genotype-phenotype correlations in the CSF1R gene, JAMA Neurol. 70, 875–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Fujioka S, Broderick DF, Sundal C, Baker MC, Rademakers R & Wszolek ZK (2013) An adult-onset leukoencephalopathy with axonal spheroids and pigmented glia accompanied by brain calcifications: a case report and a literature review of brain calcifications disorders, J Neurol. 260, 2665–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Konno T, Broderick DF, Mezaki N, Isami A, Kaneda D, Tashiro Y, Tokutake T, Keegan BM, Woodruff BK, Miura T, Nozaki H, Nishizawa M, Onodera O, Wszolek ZK & Ikeuchi T (2017) Diagnostic Value of Brain Calcifications in Adult-Onset Leukoencephalopathy with Axonal Spheroids and Pigmented Glia, AJNR Am J Neuroradiol. 38, 77–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Meyer-Ohlendorf M, Braczynski A, Al-Qaisi O, Gessler F, Biskup S, Weise L, Steinbach JP, Wagner M, Mittelbronn M & Bahr O (2015) Comprehensive diagnostics in a case of hereditary diffuse leukodystrophy with spheroids, BMC neurology. 15, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lan MY, Liu JS, Chang CC, Chen YF, Su CS, Peng CH & Chang YY (2016) Clinicopathologic and Genetic Studies of 2 Patients With Hereditary Diffuse Leukoencephalopathy With Axonal Spheroids, Alzheimer Dis Assoc Disord. 30, 73–6. [DOI] [PubMed] [Google Scholar]
- 132.Lynch DS, Zhang WJ, Lakshmanan R, Kinsella JA, Uzun GA, Karbay M, Tufekcioglu Z, Hanagasi H, Burke G, Foulds N, Hammans SR, Bhattacharjee A, Wilson H, Adams M, Walker M, Nicoll JA, Chataway J, Fox N, Davagnanam I, Phadke R & Houlden H (2016) Analysis of Mutations in AARS2 in a Series of CSF1R-Negative Patients With Adult-Onset Leukoencephalopathy With Axonal Spheroids and Pigmented Glia, JAMA Neurol. 73, 1433–1439. [DOI] [PubMed] [Google Scholar]
- 133.Lakshmanan R, Adams ME, Lynch DS, Kinsella JA, Phadke R, Schott JM, Murphy E, Rohrer JD, Chataway J, Houlden H, Fox NC & Davagnanam I (2017) Redefining the phenotype of ALSP and AARS2 mutation-related leukodystrophy, Neurol Genet. 3, e135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Daida K, Nishioka K, Li Y, Nakajima S, Tanaka R & Hattori N (2017) CSF1R Mutation p.G589R and the Distribution Pattern of Brain Calcification, Intern Med. 56, 2507–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Bonvegna S, Straccia G, Golfre Andreasi N, Elia AE, Marucci G, Di Bella D, Cilia R & Eleopra R (2020) Parkinsonism and Nigrostriatal Damage Secondary to CSF1R-Related Primary Microgliopathy, Mov Disord. 35, 2360–2362. [DOI] [PubMed] [Google Scholar]
- 136.Mateen FJ, Keegan BM, Krecke K, Parisi JE, Trenerry MR & Pittock SJ (2010) Sporadic leucodystrophy with neuroaxonal spheroids: persistence of DWI changes and neurocognitive profiles: a case study, J Neurol Neurosurg Psychiatry. 81, 619–22. [DOI] [PubMed] [Google Scholar]
- 137.Mitsui J, Matsukawa T, Ishiura H, Higasa K, Yoshimura J, Saito TL, Ahsan B, Takahashi Y, Goto J, Iwata A, Niimi Y, Riku Y, Goto Y, Mano K, Yoshida M, Morishita S & Tsuji S (2012) CSF1R mutations identified in three families with autosomal dominantly inherited leukoencephalopathy, Am J Med Genet B Neuropsychiatr Genet. 159B, 951–7. [DOI] [PubMed] [Google Scholar]
- 138.Terasawa Y, Osaki Y, Kawarai T, Sugimoto T, Orlacchio A, Abe T, Izumi Y & Kaji R (2013) Increasing and persistent DWI changes in a patient with hereditary diffuse leukoencephalopathy with spheroids, J Neurol Sci. 335, 213–5. [DOI] [PubMed] [Google Scholar]
- 139.Gore E, Manley A, Dees D, Appleby BS & Lerner AJ (2016) A young-onset frontal dementia with dramatic calcifications due to a novel CSF1R mutation, Neurocase. 22, 257–62. [DOI] [PubMed] [Google Scholar]
- 140.Blume J & Weissert R (2017) Suspected Perinatal Depression Revealed to be Hereditary Diffuse Leukoencephalopathy with Spheroids, J Mov Disord. 10, 59–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ueda S, Yamashita H, Hikiami R, Sawamoto N, Yoshida K & Takahashi R (2015) A novel A792D mutation in the CSF1R gene causes hereditary diffuse leukoencephalopathy with axonal spheroids characterized by slow progression, eNeurologicalSci. 1, 7–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wu L, Liu J, Sha L, Wang X, Li J, Dong J & Jia J (2017) Sporadic Cases with Novel Mutations and Pedigree in Hereditary Leukoencephalopathy with Axonal Spheroids, J Alzheimers Dis. 56, 893–898. [DOI] [PubMed] [Google Scholar]
- 143.Riku Y, Ando T, Goto Y, Mano K, Iwasaki Y, Sobue G & Yoshida M (2014) Early pathologic changes in hereditary diffuse leukoencephalopathy with spheroids, J Neuropathol Exp Neurol. 73, 1183–90. [DOI] [PubMed] [Google Scholar]
- 144.Yokote A, Ouma S, Takahashi K, Hara F, Yoshida K & Tsuboi Y (2020) [A case of hereditary diffuse leukoencephalopathy with spheroids and pigmented glia presenting with long-term mild psychiatric symptoms], Rinsho Shinkeigaku. 60, 420–424. [DOI] [PubMed] [Google Scholar]
- 145.Funayama M, Sugihara M, Takata T, Mimura M & Ikeuchi T (2019) Remarkable behavioural signs and progressive non-fluent aphasia in a patient with adult-onset leucoencephalopathy with axonal spheroids and pigmented glia, Psychogeriatrics. 19, 282–285. [DOI] [PubMed] [Google Scholar]
- 146.Saitoh BY, Yamasaki R, Hayashi S, Yoshimura S, Tateishi T, Ohyagi Y, Murai H, Iwaki T, Yoshida K & Kira J (2013) A case of hereditary diffuse leukoencephalopathy with axonal spheroids caused by a de novo mutation in CSF1R masquerading as primary progressive multiple sclerosis, Mult Scler. 19, 1367–70. [DOI] [PubMed] [Google Scholar]
- 147.Prieto-Morin C, Ayrignac X, Ellie E, Tournier-Lasserve E & Labauge P (2016) CSF1R-related leukoencephalopathy mimicking primary progressive multiple sclerosis, J Neurol. 263, 1864–5. [DOI] [PubMed] [Google Scholar]
- 148.Mao C, Zhou L, Zhou L, Yang Y, Niu J, Li J, Huang X, Ren H, Zhao Y, Peng B & Gao J (2020) Biopsy histopathology in the diagnosis of adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP), Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical Neurophysiology. 41, 403–409. [DOI] [PubMed] [Google Scholar]
- 149.Chen Z, Tan YJ, Lian MM, Tandiono M, Foo JN, Lim WK, Kandiah N, Tan EK & Ng ASL (2021) High Diagnostic Utility Incorporating a Targeted Neurodegeneration Gene Panel With MRI Brain Diagnostic Algorithms in Patients With Young-Onset Cognitive Impairment With Leukodystrophy, Front Neurol. 12, 631407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Konno T, Broderick DF & Wszolek ZK (2017) Brain calcification in a CSF1R mutation carrier precedes white matter degeneration, Mov Disord. 32, 1493–1495. [DOI] [PubMed] [Google Scholar]
- 151.Lynch DS, Jaunmuktane Z, Sheerin UM, Phadke R, Brandner S, Milonas I, Dean A, Bajaj N, McNicholas N, Costello D, Cronin S, McGuigan C, Rossor M, Fox N, Murphy E, Chataway J & Houlden H (2016) Hereditary leukoencephalopathy with axonal spheroids: a spectrum of phenotypes from CNS vasculitis to parkinsonism in an adult onset leukodystrophy series, J Neurol Neurosurg Psychiatry. 87, 512–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Bayat M, Shekhrajka N & Bayat A (2019) Hereditary leukodystrophy with axonal spheroids (HDLS) presenting subacutely: a CNS-vasculitis mimic, Acta Neurol Belg. 119, 633–635. [DOI] [PubMed] [Google Scholar]
- 153.Lynch DS, Rodrigues Brandao de Paiva A, Zhang WJ, Bugiardini E, Freua F, Tavares Lucato L, Macedo-Souza LI, Lakshmanan R, Kinsella JA, Merwick A, Rossor AM, Bajaj N, Herron B, McMonagle P, Morrison PJ, Hughes D, Pittman A, Laura M, Reilly MM, Warren JD, Mummery CJ, Schott JM, Adams M, Fox NC, Murphy E, Davagnanam I, Kok F, Chataway J & Houlden H (2017) Clinical and genetic characterization of leukoencephalopathies in adults, Brain. 140, 1204–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Zhuang LP, Liu CY, Li YX, Huang HP & Zou ZY (2020) Clinical features and genetic characteristics of hereditary diffuse leukoencephalopathy with spheroids due to CSF1R mutation: a case report and literature review, Ann Transl Med. 8, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Du Q, Wang M & Zhou H (2021) A novel mutation in CSF1R associated with hereditary diffuse leukoencephalopathy with spheroids, Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical Neurophysiology. [DOI] [PubMed] [Google Scholar]
- 156.Jin C, Washimi Y, Yoshida K, Hashizume Y & Yazawa I (2015) Characterization of spheroids in hereditary diffuse leukoencephalopathy with axonal spheroids, J Neurol Sci. 352, 74–8. [DOI] [PubMed] [Google Scholar]
- 157.Codjia P, Ayrignac X, Mochel F, Mouzat K, Carra-Dalliere C, Castelnovo G, Ellie E, Etcharry-Bouyx F, Verny C, Belliard S, Hannequin D, Marelli C, Nadjar Y, Le Ber I, Dorboz I, Samaan S, Boespflug-Tanguy O, Lumbroso S & Labauge P (2018) Adult-Onset Leukoencephalopathy with Axonal Spheroids and Pigmented Glia: An MRI Study of 16 French Cases, AJNR Am J Neuroradiol. 39, 1657–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Inui T, Kawarai T, Fujita K, Kawamura K, Mitsui T, Orlacchio A, Kamada M, Abe T, Izumi Y & Kaji R (2013) A new CSF1R mutation presenting with an extensive white matter lesion mimicking primary progressive multiple sclerosis, J Neurol Sci. 334, 192–5. [DOI] [PubMed] [Google Scholar]
- 159.Makary MS, Awan U, Kisanuki YY & Slone HW (2019) Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia: Clinical and imaging characteristics, Neuroradiol J. 32, 139–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Lubomski M, Buckland ME, Sy J, Wei H, Tan IYL, Kane B & Spring PJ (2018) Adult-onset leukoencephalopathy with neuroaxonal spheroids and pigmented glia mimicking systemic lupus erythematosus cerebral vasculitis, J Neurol Sci. 395, 25–28. [DOI] [PubMed] [Google Scholar]
- 161.Ahmed R, Guerreiro R, Rohrer JD, Guven G, Rossor MN, Hardy J & Fox NC (2013) A novel A781V mutation in the CSF1R gene causes hereditary diffuse leucoencephalopathy with axonal spheroids, J Neurol Sci. 332, 141–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Foulds N, Pengelly RJ, Hammans SR, Nicoll JA, Ellison DW, Ditchfield A, Beck S & Ennis S (2015) Adult-Onset Leukoencephalopathy with Axonal Spheroids and Pigmented Glia Caused by a Novel R782G Mutation in CSF1R, Sci Rep. 5, 10042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Kinoshita M, Yoshida K, Oyanagi K, Hashimoto T & Ikeda S (2012) Hereditary diffuse leukoencephalopathy with axonal spheroids caused by R782H mutation in CSF1R: case report, J Neurol Sci. 318, 115–8. [DOI] [PubMed] [Google Scholar]
- 164.Shu Y, Long L, Liao S, Yang J, Li J, Qiu W, Yang Y, Bao J, Wu A, Hu X & Lu Z (2016) Involvement of the optic nerve in mutated CSF1R-induced hereditary diffuse leukoencephalopathy with axonal spheroids, BMC neurology. 16, 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Stoiloudis P, Parissis D, Smyrni N, Stardeli T, Afrantou T, Konstantinopoulou E, Grigoriadis N & Ioannidis P (2021) Hereditary diffuse leukoencephalopathy with spheroids mimicking primary progressive aphasia: report of a Greek case, Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical Neurophysiology. [DOI] [PubMed] [Google Scholar]
- 166.Zur-Wyrozumska K, Kaczmarska P & Mensah-Glanowska P (2021) Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia associated with an A792D mutation in the CSF1R gene in a Polish patient, Neurol Neurochir Pol. [DOI] [PubMed] [Google Scholar]
- 167.Kondo Y, Kinoshita M, Fukushima K, Yoshida K & Ikeda S (2013) Early involvement of the corpus callosum in a patient with hereditary diffuse leukoencephalopathy with spheroids carrying the de novo K793T mutation of CSF1R, Intern Med. 52, 503–6. [DOI] [PubMed] [Google Scholar]
- 168.Liu Q, Guo XN, Liu CY & Xu WH (2020) A proposed synergistic effect of CSF1R and NMUR2 variants contributes to binge eating in hereditary diffuse leukoencephalopathy with spheroids, Ann Transl Med. 8, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Sharma R, Graff-Radford J, Rademakers R, Boeve BF, Petersen RC & Jones DT (2019) CSF1R mutation presenting as dementia with Lewy bodies, Neurocase. 25, 17–20. [DOI] [PubMed] [Google Scholar]
- 170.Saitoh BY, Yamasaki R, Hiwatashi A, Matsushita T, Hayashi S, Mitsunaga Y, Maeda Y, Isobe N, Yoshida K, Ikeda SI & Kira JI (2019) Discriminative clinical and neuroimaging features of motor-predominant hereditary diffuse leukoencephalopathy with axonal spheroids and primary progressive multiple sclerosis: A preliminary cross-sectional study, Mult Scler Relat Disord. 31, 22–31. [DOI] [PubMed] [Google Scholar]
- 171.Saitoh B-Y, Yoshida K, Hayashi S, Yamasaki R, Sato S, Kamada T, Suzuki SO, Murai H, Iwaki T, Ikeda S-I & Kira J (2013) Sporadic hereditary diffuse leukoencephalopathy with axonal spheroids showing numerous lesions with restricted diffusivity caused by a novel splice site mutation in the CSF1R gene, Clinical and Experimental Neuroimmunology. 4, 76–81. [Google Scholar]
- 172.Kleinfeld K, Mobley B, Hedera P, Wegner A, Sriram S & Pawate S (2013) Adult-onset leukoencephalopathy with neuroaxonal spheroids and pigmented glia: report of five cases and a new mutation, J Neurol. 260, 558–71. [DOI] [PubMed] [Google Scholar]
- 173.Levin J, Tiedt S, Arzberger T, Biskup S, Schuberth M, Stenglein-Krapf G, Kreth FW, Hogen T, la Fougere C, Linn J, van der Knaap MS, Giese A, Kretzschmar HA & Danek A (2014) Diffuse leukoencephalopathy with spheroids: biopsy findings and a novel mutation, Clinical neurology and neurosurgery. 122, 113–5. [DOI] [PubMed] [Google Scholar]
- 174.Di Donato I, Stabile C, Bianchi S, Taglia I, Mignarri A, Salvatore S, Giorgio E, Brusco A, Simone I, Dotti MT & Federico A (2015) A Novel CSF1R Mutation in a Patient with Clinical and Neuroradiological Features of Hereditary Diffuse Leukoencephalopathy with Axonal Spheroids, J Alzheimers Dis. 47, 319–22. [DOI] [PubMed] [Google Scholar]
- 175.Sundal C, Baker M, Karrenbauer V, Gustavsen M, Bedri S, Glaser A, Myhr KM, Haugarvoll K, Zetterberg H, Harbo H, Kockum I, Hillert J, Wszolek Z, Rademakers R & Andersen O (2015) Hereditary diffuse leukoencephalopathy with spheroids with phenotype of primary progressive multiple sclerosis, Eur J Neurol. 22, 328–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Cheng X, Shen W, Zou H, Shen L, Gu X, Huang D, Sun Y, Wang B, Tian Q & Xu J (2015) [Analysis of CSF1R gene mutation in a Chinese family with hereditary diffuse leukoencephalopathy with neuroaxonal spheroids], Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 32, 208–12. [DOI] [PubMed] [Google Scholar]
- 177.Lapalme-Remis S, Warman Chardon J, Bourque PR & Oboudiyat C (2016) Diffuse leukoencephalopathy with spheroids presenting as primary progressive aphasia, Neurology. 86, 1464–1465. [DOI] [PubMed] [Google Scholar]
- 178.Shi T, Li J, Tan C & Chen J (2019) Diagnosis of hereditary diffuse leukoencephalopathy with neuroaxonal spheroids based on next-generation sequencing in a family: Case report and literature review, Medicine (Baltimore). 98, e15802. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.