

Abstract
All life forms require nicotinamide adenine dinucleotide, NAD+, and its reduced form NADH. They are redox partners in hundreds of cellular enzymatic reactions. Changes in the intracellular levels of total NAD (NAD+ + NADH) and the (NAD+/NADH) ratio can cause cellular dysfunction. When not present in protein complexes, NADH and its phosphorylated form NADPH degrade through intricate mechanisms. Replenishment of a declining total NAD pool can be achieved with biosynthetic precursors that include one of the reduced forms of nicotinamide riboside (NR+), NRH. NRH, like NADH and NADPH, is prone to degradation via oxidation, hydration, and isomerization and, as such, is an excellent model compound to rationalize the nonenzymatic metabolism of NAD(P)H in a biological context. Here, we report on the stability of NRH and its propensity to isomerize and irreversibly degrade. We also report the preparation of two of its naturally occurring isomers, their chemical stability, their reactivity toward NRH-processing enzymes, and their cell-specific cytotoxicity. Furthermore, we identify a mechanism by which NRH degradation causes covalent peptide modifications, a process that could expose a novel type of NADH-protein modifications and correlate NADH accumulation with “protein aging.” This work highlights the current limitations in detecting NADH’s endogenous catabolites and in establishing the capacity for inducing cellular dysfunction.
Graphical Abstract
INTRODUCTION
Nicotinamide adenine dinucleotide (NAD+), along with its phosphorylated and reduced forms (NAD(P)(H)), is a vitamin B3-containing dinucleotide (Figure 1). Together, the NAD(P)(H) cofactors catalyze most known enzymatic hydride transfers.1–5 Unlike NAD+, NADH (Figure 1) is not a substrate for nonredox NAD+ consuming enzymes, such as sirtuins, adenosine diphosphoribosyl transferases (ARTs), and glycohydrolases.6–9 Critically, it has been observed that in the process of aging and several metabolic disease models (e.g., type II diabetes, cardiovascular diseases, cardiomyopathy, etc.), the NADH to NAD+ ratio increases, with, on occasion, an overall decrease of the (NADH + NAD+) pool.8,10–14
Figure 1.

Ribosylated precursors to NAD(P)(H), the associated metabolic and NAD+ consuming enzymes, and redox equilibrium. AK, adenosine kinase; NRK, nicotinamide riboside kinase; NMNAT, nicotinamide mononucleotide adenylyltransferase; NADK, NAD kinase; ARTs, adenosine diphosphoribosyl transferases.
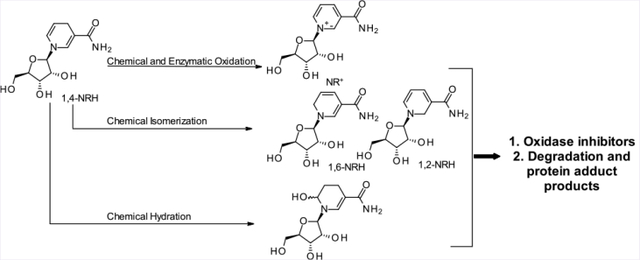
1,4-NAD(P)H, more commonly known as NAD(P)H, is most conspicuous for its redox capacity in enzyme-catalyzed reactions. The 1,4-nomenclature in 1,4-NAD(P)H indicates the position of the double bonds in the dihydronicotinamide moiety. While it is common knowledge that 1,4-NAD(P)H gets readily oxidized when exposed to air in the presence of moisture,15–17 the fact that NAD(P)H is also prone to isomerization and hydroxylation (Figure 2) under these conditions is less known. The products generated by this class of chemistry are the 1,2- and 1,6-isomers of NAD(P)H18,19 and the 6-OH-NAD(P)H,20–22 a compound known in the literature as NAD(P)HX (e.g., Figure 2; NADH(OH)). In a humid environment, 1,4-NADH powder isomerizes to the 1,2- and 1,6-NADH isomers.23,24 On average, a commercial source of NADH will contain up to 4% of these isomers if stored under nonanhydrous conditions for extended periods of time. The identity of these isomers was confirmed when 1,2- and 1,6-NAD(P)H were generated by reducing NAD(P)+ using NaBH4 in aqueous media.25 In solid-form storage, the 1,2- and 1,6-isomers of NAD(P)H have been proposed to form through a bimolecular reaction in which an apparent double bond rearrangement that lacks chemo-selectivity occurs via a hydride transfer from NAD(P)H to NAD(P)+.23 A particularly well-suited biochemical environment for this mismatched hydride transfer and endogenous production of isomers is the mitochondrial NAD(P)+ transhydrogenase (nicotinamide nucleotide transhydrogenase; NNT), which is responsible for the generation of NADPH and NAD+ from NADP+ and NADH in the mitochondrion.26 However, this suggestion is speculative as the mechanism of NNT remains under intense scrutiny. Finally, direct hydroxylation at the 6-position of the dihydropyridinyl ring of NAD(P)H generates NAD(P)H(OH) and occurs preferentially under mildly acidic phosphate buffer (pH 6.8) conditions.22,20
Figure 2.

Nonenzymatic routes to the derivatives of 1,4-NRH and NADH.
Together, these isomers and adducts are potent inhibitors of NAD(P)(H) dependent redox enzymes with Ki ranging from micromolar to low nanomolar values,24,27,28 and they contribute to the degradation processes of NAD(P)H.29 Still, the actual intracellular abundance of 1,2- and 1,6-isomers of NAD(P)H remains unknown, as is that of NAD(P)H(OH). One could assume that given the relatively low abundance of the 1,2- and 1,6-NAD(P)H isomers compared to the 1,4-parents, their physiological significance should be dismissed. Yet, these isomers are so detrimental to cellular homeostasis that their removal evokes specific conserved repair pathways.18–20,30–32 Two different classes of enzymes using either FAD (renalase) or FMN (pyridoxamine-phosphate oxidase and pyridoxamine-phosphate oxidase-related proteins) as a cofactor remove damaged forms of NAD(P)H as they oxidize 1,2- and 1,6-NAD(P)H to NAD(P)+.30,33 Similarly, the epimerase/dehydratase repair pathways convert the hydroxylated species back to NAD(P)+. Both enzymes are critical to cellular homeostasis and lethal if absent.32,34 This speaks to the physiological relevance of these NAD(P)H isomers.
Another critical point is that since these derivatives are chemically formed and degraded within the cells’ compartments where they are generated, the equilibrium between these derivatives and 1,4-NAD(P)H is likely to be permanently shifted away from the functional 1,4-NAD(P)H forms. As a result and over time, the damage these derivatives endogenously create could be much more severe than a simple chemical ratio might indicate.35 Therefore, the complexity of the solution chemistry of the reduced forms of NAD(P)+ and derivatives is likely to explain why these derivatives have not received their due attention in cell/tissues/organism-based assays, even though physiologically critical repair pathways are in place to prevent their accumulation.
Our inability to detect the presence of toxic NAD(P)H derivatives and quantify their relative abundance limits our efforts in ascertaining their physiological relevance and their role in cellular homeostasis and disease. Moreover, characterizing the correlation between abundance and function requires the challenging task of manipulating their intracellular levels in a physiologically meaningful manner. Being phosphorylated species, isomers and hydroxylated derivatives of NAD(P)H cannot be supplemented to cells directly with the expectation that they will be intracellularized intact.
To address these limitations, we sought to understand better the properties of 1,4-dihydronicotinamide riboside (Figure 1; 1,4-NRH, 1) as a model compound for NAD(P)H and explore its chemical and biochemical space and that of its less common metabolites, the 1,2-NRH (2) and 1,6-NRH (3) isomers and the hydroxylated parents 4 (Figure 2). NRH 1 (Figure 1) has been recently characterized as an effective precursor of NAD+.36–38 While the conversion of exogenous NRH to NMNH by adenosine kinase rather than its oxidation to nicotinamide riboside by NRH:quinone oxidoreductase 2 (NQO2) is thought to be the first step toward its conversion to NAD,36,39 the biosynthetic mechanisms explaining this process remain under scrutiny.40–42 Critically, the physiological prevalence of intracellular NRH on the total NAD pool maintenance remains to be explored.
Here, we report on the stability of NRH and its propensity to isomerize and irreversibly degrade. We also report the preparation of two of its naturally occurring isomers, their chemical stability, their reactivity toward NRH-processing enzymes, and their cytotoxicity when administered exogenously. Finally, we identify a mechanism by which NRH degradation causes peptidic conjugation via the aglycone unit of NRH, a mechanism that differs from the well-studied ribose-derived glycation processes.
RESULTS
Production and Handling of 1,4-NRH.
As previously described, 1,4-NRH is readily generated from nicotinamide and tetraacetate riboside.43 We applied mechanochemical conditions to the Vorbrüggen glycosylation of nicotinamide with the tetraacetate riboside to generate nicotinamide riboside triacetate (Scheme 1, 6) as the synthetic intermediate to the NRH series. 1,4-NRH 1 is generated by reducing intermediate 6 using sodium dithionite under aqueous conditions in a biphasic system to allow the facile extraction of the triacetylated intermediate 7. This step is followed by the removal of the acetate moieties under methanolic potassium carbonate conditions using the molar equivalency of methanol under milling conditions.44 When in nondeoxygenated aqueous solutions, 1,4-NRH (1) decomposes over time. Therefore, NRH requires careful handling when in solution. Importantly, 1,4-NRH oxidizes upon storage, even at −80 °C, when stored in aqueous solutions. Freeze–thaws are particularly discouraged. As per NADH, UV spectroscopy measurements at 340 nm of 1,4-NRH solutions are particularly well-suited to monitor whether the 1,4-NRH integrity is maintained.29 However, the loss of 1,4-NRH is not due to simple oxidation of NRH to its oxidized form, nicotinamide riboside, NR+ (Figure 2, 5), and the formation of NR+’s hydrolytic products, nicotinamide (Nam) and ribose. Rather, 1,4-NRH decomposes as a complex mixture of materials, which varies upon the conditions under which this decomposition takes place (Figure 2). Consequently, when used for biological work, 1,4-NRH must be handled with care; otherwise, its use brings discrepancies to the studies’ outcomes. Therefore, we sought to better characterize the conditions under which NRH decomposes and its decomposition products.
Scheme 1. Synthetic Sequence to 1,4-NRH and Its 1,2- and 1,6-Isomers.

Production and Handling of 1,2- and 1,6-NRH.
With the view of better characterizing the 1,2- and the 1,6-isomers of NRH, we then endeavored to synthesize these species from the nicotinamide riboside triacetate (6). Unlike the dithionitecatalyzed reduction of NR+, reduction of NR+ salts with NaBH445 yields a mixture of 1,2; 1,4; and 1,6-NRH (Scheme 1; 2, 1, and 3) that is difficult to separate. The proportion of isomers thus generated varies greatly as a function of solvents, reagent equivalency, and temperature. Therefore, conditions that offered the highest levels of 1,2- and 1,6-isomers were sought. In the absence of a catalyst, the generation of 1,2-, 1,4-, and 1,6-dihydropyridines from N-alkylated nicotinamide salts is best achieved by using anhydrous DMF at 0 °C (Scheme 1).46 We applied this approach to the triacetate-protected NR-OTf (6) to enable the purification of the reduction products by silica or alumina chromatography. Following the NaBH4 reduction in DMF and an extensive extraction sequence, a solid foam containing a mixture of the triacetylated form of 1,2-NRH (8, 1,2-NRH TA), 1,4-NRH (7, 1,4-NRH TA), and 1,6-NRH (9, 1,6-NRH TA) in a respective ratio of 1.00:0.44:1.00 (67% yield) was obtained. To isolate the 1,2- and 1,6-isomers of NRH TA, the mixture was subjected to silica gel column chromatography using EtOH/EtOAc as the eluent system. While 1,2-triacetate NRH (8) eluted first, the 1,6-NRH triacetate (9) was extremely difficult to separate from the 1,4-NRH triacetate (7). Following multiple purification iterations, each isomer was isolated independently. However, the purification process consistently led to a loss of material, with the best recovery achieved in 35% total yield. Attempts to conduct this purification on alumina failed.
In summary, 1,2-NRH and 1,6-NRH triacetate are extremely labile entities that readily react with either ethanol or water or with the solid support itself throughout the purification process. While the trans-esterification process could be viewed as a reasonable mechanism leading to the loss of materials, 1,4-NRH triacetate 7 generated from the dithionite-method is readily purified using this chromatography method, and the recovery yields are good to excellent. The reactivity of 1,2-NRH and 1,6-NRH triacetate (8 and 9) toward mild reagents such as those employed in the purification process is indicative of the difficulties that one encounters in seeking to isolate, purify, or even detect derivatives of such nucleosides in cell extracts. Yet, having generated and purified the triacetylated form of 1,2-NRH and 1,6-NRH, careful deprotection was then undertaken using NaOMe in methanol at low temperatures (Scheme 1). Each fully deprotected isomer was then isolated, but no further purification could be performed without product decomposition. Characterization of each isomer was conducted using COSY, HMQC, and MS and HRMS data, while the LC-MS2 fragmentation pattern for each one of these isomers was characterized.
Stability of 1,4-NRH in Aqueous Media.
The broader stability of 1,4-NRH as a function of pH and temperature was systematically investigated using 1H NMR spectroscopy (Figures 3 and 4). Because NRH is used under conditions where the temperature is maintained at 37 °C, either in cell-based assays or animal studies, we discuss here the results observed at 37 °C. First, the mild conditions developed by Margolis to generate the hydroxylated forms of NAD(P)H were adapted and applied to 1,4-NRH (50 mM KH2PO4, pH 6.8, 37 °C) and progression monitored by 1H NMR at 1, 3, 12, and 24 h. The 1H NMR spectra (Figure 3) provided evidence that 1,4-NRH oxidizes to NR+ in addition to showing the formation of new species including one that matched the hydrated form of NRH NRH(OH) (Figure 2, 4, and Figure 4) with characteristic peaks at 7.35 and 5.64 ppm. The 1,6-NRH isomer could also be detected with characteristic peaks observed at 5.03, 6.10, and 7.24 ppm after just 3 h, consistent with what is known for NADH multicomponent compositions.23,24,44 Crucially, the carbohydrate and aliphatic regions became a lot more complex. The aliphatic region (1–2.5 ppm) reveals a –(CH2–CH2–CH)– sequence that can be attributed to hydrated catabolites described in Figure 4. It must be noted that only the 6-hydroxylated byproduct is shown in Figure 4, even though the 2-hydroxylated byproducts can also be envisaged as contributing to the mixture. Crucially, this chemistry is not observed to occur that rapidly in water, as when NRH was kept in water for 24 h at 37 °C, mainly NR+ formation was observed (NRH/NR+ = 454:1; Figure 4, panel A). At lower pH, pH 6.0, and in 500 mM PBS buffer, the decomposition of 1,4-NRH is even more rapid as evidenced by the 1H NMR after just 30 min of incubation (Figure 4, panel B).
Figure 3.

Decomposition of 1,4-NRH in aqueous solution over time. 1H NMR of a 1,4-NRH solution in phosphate buffer, 50 mM, pH 6.8 after 1, 3, 12, and 24 h at 37 °C.
Figure 4.

Stability of 1,4-NRH in aqueous solutions. Panel A: 1H NMR of a 1,4-NRH solution in D2O after 24 h at 37 °C. Panel B: 1H NMR of a 1,4-NRH solution in phosphate buffer, 500 mM, pH 6.0 after 30 min at 37 °C.
Reactivity of 1,4-NRH with NR+ in Aqueous Media.
We then investigated whether 1,4-NRH when in the presence of the oxidized form NR+ could isomerize to 1,2-NRH (Figure 2; 2) and 1,6-NRH (Figure 2; 3). 1,4-NRH was coincubated with NR+ in D2O for 100 h at room temperature. Under these conditions, the 1,6-NRH isomer accounted for 1 in 20 mol of (NRH+NR+), but the 1,2-NRH was not detectable (experimental materials, ESI-NMR1). Of note, the hydrated species were not observed; in particular, the aliphatic region remained free of signals even after 100 h of incubation in water. The salt composition and pH of the aqueous solution appear to be the limiting factors leading to the hydroxylation of 1,4-NRH.
Stability of 1,4-NRH in Culture Media.
Having each isomer at hand, we then examined the chemical stability of 1,4-NRH when in solution in cell growth media containing a high purity 10% FBS and established how resilient the reduced nucleoside was to oxidation, isomerization, and hydrolysis under cell-based assay conditions. As anticipated, 1,4-NRH slowly converted to NR+ and Nam over a 48 h period when incubated in standard nonheat-inactivated FBS-containing cell-growth media at 37 °C and 5% CO2, standard cell-based assay conditions. Importantly, in the absence of cells, 1,4-NRH was removed from solution through protein interactions (no longer detectable by 1H NMR), with only 80% of the initial 1,4-NRH remaining in solution after 6 h. Longer incubation periods resulted in the production of Nam through NR+ hydrolysis and further reduction of the total NRH pool. This observation indicates that when in the presence of cells, prolonged supplementation with 1,4-NRH must be monitored to assign physiological outcomes to the interaction of NRH with the cells, rather than with the growth media and NRH byproducts. Critically, when FBS is supplemented with 10 mM 1,4-NRH, the 1H NMR spectrum indicates the formation of the 1,6-NRH isomer, in proportions similar to those as observed in PBS. The stability of 1,2-NRH and 1,6-NRH in the same growth media was thus investigated. While 1,6-NRH proved relatively stable in the growth media for up to 6 h, the 1,2-NRH isomer decomposed in a matter of hours.
Cytotoxicity of 1,2-, 1,4-, and 1,6-NRH.
As 1,4-NRH is a precursor to 1,4-NAD(P)(H),38 1,2- and 1,6-NRH are putative precursors of 1,2- and 1,6-NAD(P)H, respectively. If this is the case, the formation of such isomers of NAD(P)H would be detrimental to cells.30 Therefore, to evaluate whether 1,2-NRH, 1,4-NRH, and 1,6-NRH could modulate cellular function, we evaluated their effect as cytotoxic agents using the Human Embryonic Kidney (HEK293T) and Hepatoma (HepG3) immortalized cell lines as cell-based models. The cytotoxicity of 100 μM 1,2-NRH, 1,4-NRH, and 1,6-NRH was determined by CellTiter-Fluor (Promega), which unlike the standard and widely used MTT assay47 measures cell viability independent of metabolic markers related to NAD(P)(H). The cells were exposed to the NRH isomers for 6 h, consistent with our stability studies. The 6 h media did not contain residual NRH isomers (confirmed by 1H NMR), indicating that each isomer was either transported into the growing cells or was bound to media proteins or degraded and thus no longer detectable by 1H NMR. The cell survival after 6 h exposure to NRH isomers is shown in Figure 5. At this concentration and over this 6 h incubation period, the 1,4- and the 1,6-NRH isomers reduced cell survival by at least 10%, and this change was statistically significant. 1,6-NRH was found to be the most cytotoxic isomer (Figure 5), and HEK293T cells are more sensitive to this isomer than HepG3 cells. 1,2-NRH was found to be less cytotoxic to these cells. This difference could be attributed to the reduced stability of 1,2-NRH in the media.
Figure 5.

Cytotoxicity of NRH derivatives (100 μM) on HEK293T and HepG3 cell lines. Values are reported as the mean percentage survival ± standard error of the mean (SEM) of three biological replicates (values in the experimental section).
Metabolism of 1,2-, 1,4-, and 1,6-NRH to NR+.
Renalase and pyridoxamine-phosphate oxidase are riboflavin-dependent oxidases, using FAD and FMN, respectively. NAD(P)H quinone dehydrogenase (NQO1) is one of the most ubiquitous riboflavin-dependent oxidases and one of the two major quinone reductases in mammalian systems. Its homologue NQO2, known as ribosyl dihydronicotinamide dehydrogenase, quinone, is the only enzyme known to oxidize 1,4-NRH to NR+ in the presence of a co-oxidant like O2 or menadione (Figure 1). NQO2 is expressed in both HEK293T and HepG3 cells, and its levels remain constant following the 6 h exposure to 100 μM 1,4-NRH. Considering the role of NQO2 as a detoxifying enzyme, we sought to establish whether the 1,2- and 1,6-NRH isomers affected the NQO2 function, as a possible route to cell toxicity. Under 1H NMR-enabled monitoring conditions, we examined whether NQO2 could oxidize all three nucleosides. First, we sought to establish whether these isomers were stable to the enzymatic reaction conditions. We rapidly realized that both BSA and FAD significantly modified the relative amount to NRH derivatives in solution over time (Figure S2), including 1,4-NRH (Figure 6). Under these conditions, we observed more than 20% material loss over 24 h in the 1,6-NRH and 1,4-NRH containing reaction solutions and up to 65% material loss over that same period for the 1,2-NRH containing solution (Figure S2). In the case of 1,2-NRH, the addition of the 1,2-NRH to the buffer containing BSA and FAD promoted an almost instantaneous change in the 1H NMR spectrum of 1,2-NRH, which over time lost signal-to-noise resolution, a clear indication that 1,2-NRH decomposed materials were taken out of the soluble phase. Such materials are likely to be protein adducts.
Figure 6.

Panel A: % of loss over time. Panel B: Relative products’ distribution over time.
Critically, FAD alone was able to facilitate the oxidation of 1,4-NRH (Figure 6) and 1,6-NRH to NR+ (Figure S3). However, in HEPES buffer containing BSA and FAD at pH 7.2 in the presence of the cosubstrate menadione, the rate of oxidation of 1,4-NRH to NR+ was greatly increased by NQO2 (Sigma-Aldrich; Figure 6). Under the selected conditions for measuring NQO2 activity, menadione was the rate-limiting substrate, being present at 1/2 the concentration of the NRHs. The residual 1,4-NRH present in 1,2-NRH and 1,4-NRH samples was readily oxidized to NR+ when NQO2 was present (Figure S4). Compared to 1,4-NRH, the oxidation of 1,6-NRH by NQO2 was significantly slower (Figure 7), and this oxidation could be accounted for by the presence of free FAD. Crucially, 1,6-NRH appeared to be a relatively stable isomer of 1,4-NRH under the reaction conditions.
Figure 7.

1,2-NRH, 1,4-NRH, and 1,6-NRH are oxidized to NR by NQO2 in the presence of FAD and menadione. 1,2-NRH and 1,6-NRH are oxidized to NR+ more slowly than 1,4-NRH. Slow oxidation of all three isomers is observed in the presence of FAD only, with 1,4-NRH being the most reactive toward FAD.
Inhibition of NQO2 by 1,2- and 1,6-NRH.
With this perspective that the rate of oxidation of 1,2- and 1,6-NRH to NR+ was slower than that of 1,4-NRH to NR+, we decided to investigate whether 1,2-NRH and 1,6-NRH could be inhibitors of NQO2. Co-incubation of 1,4-NRH and NQO2 in the presence of molar equivalents of 1,6-NRH led to a decrease in the rate of production of NR+ (Figure 8). The loss of NRH-derived species was also less when 1,6-NRH was used (Figure 8, panel B). This observation is consistent with 1,6-NRH behaving as a stable isomer of 1,4-NRH in solution.
Figure 8.

Inhibition of NQO2 catalyzed oxidation of 1,4-NRH to NR+ by 1,2-NRH and 1,6-NRH: Panel A: 1,6-NRH reduces the rate of 1,4-NRH oxidation by NQO2 more effectively than 1,2-NRH. Panel B: The loss of NRH isomers present in solution occurs over time under NQO2 incubation conditions in the presence of 1,6-NRH.
Modeling of 1,2-NRH and 1,6-NRH in the Binding Site of NQO2.
We sought a rationale for the more pronounced inhibitory effect of 1,6-NRH than of 1,2-NRH on NQO2. Using the computational model of FAD-bound NQO2 based upon PDB entry 2QX8,48 we have found dock poses for the two isomers at the hydride-exchange site of FAD-bound NQO2. Our quantum-based computational modeling indicates that the ring system of 1,6-NRH stacks favorably with the central ring of the flavin moiety and overlaps the expected binding mode of 1,4-NRH (Figure S5). This inhibitory binding position of 1,6-NRH is itself unlikely to lead to hydride transfer with r(H6–NFAD) = 3.2 Å versus r(H4–NFAD) = 2.7 Å for 1,4-NRH. For the 1,2-NRH isomer (not shown), we find a dock pose similar to that of 1,6-NRH; r(H2–NFAD) is likewise too far to engender hydride transfer, and weaker inhibitory activity is expected because 1,2-NRH’s amide abuts Trp195 of NQO2.
NRH and Its Degradation Products React with Peptidic Lysines and Modify Proteins.
Protein modifications occur widely in cells and in vivo. For instance, protein glycation by reductive sugars like glucose is common and occurs via a reaction between the aldehydic form of glucose and a free amine residue, either an N-terminus or a lysine side chain, leading to a Schiff-base rearrangement. These modifications are not reversible. This type of modification occurs with other types of glycating species such as glyoxal etc. The chemical commonality in these types of modifications is the reactive aldehyde. NAD is also known to modify proteins via parylation and modification of the peptidic sequence by the introduction of the ribosyl moiety. Unlike glycation, this process is often reversible and does not include the formation of a Schiff base. Having observed a sustained loss (irreversible) of the three NRH isomers in the presence of the buffer protein BSA over time, we decided to explore whether conjugation to proteins could be detected upon incubation of BSA with either of these NRH isomers. Considering that 1,4-NRH is the most abundant form of NRH and a representative of functional 1,4-NADH, we selected it for this investigation. Following a 24 h incubation process in the same buffer as that used for NQO2 reactions, but in the absence of NQO2, the protein content of this reaction mixture was analyzed following simple tryptic digest followed by LC-MS2 analyses to determine whether glycation other than the one anticipated from a residual riboside conjugation could be detected. The conjugation of a ribosylated aldehydic intermediate such as the aldehyde described in Figure 4 with a lysine residue was thought to cause a C11H18N2O5 addition. We observed this increase in the occurrence of adducts in several peptidic sequences, all rich in lysine residues.
Furthermore, upon the loss of the ribose, the anticipated mass shift stemming from a monoconjugation of a dialdehyde (Figure 4) with a lysine residue of BSA would be (125 = −2 + 127)(−2H + C6H9NO2) with one positive charge loss. Many peptidic sequences with this type of modification were also detected. The remaining aldehyde can be readily oxidized to the carboxylic acid upon sample handling, and a mass shift of 141 (−2H + C6H9NO3) can be rationalized. Critically, some of these modifications are already present on the BSA before its exposure to 1,4-NRH, indicating that BSA modifications by NRH-like entities occur endogenously.
DISCUSSION
The universally agreed correlation between the maintenance of NAD levels and (NAD+/NADH) ratio, and cellular homeostasis, has led to a wide range of nutritional approaches seeking to increase NAD bioavailability in humans to improve health, ward off aging, and mitigate diseases. A range of products to increase NAD bioavailability are now available (e.g., ref 49). While changes in circulating NAD levels can be monitored, the effects of a given supplementation on the NADH levels and thus the NAD+/NADH ratio are often overlooked. A sustained or even transient unregulated increase of NADH abundance associates with reductive stress that leads to reactive oxygen species generation and oxidative stress outcomes.38 The possibility of increasing the levels of NADH as total NAD levels rise should be of concern. Pyridoxamine-phosphate oxidase and renalase are two mammalian enzymes that are known to oxidize endogenous 1,2- and 1,6-NAD(P)H to NAD(P)+.30,33 Crucially, renalase is known to oxidize 1,2- and 1,6-NAD(P)H preferentially over 1,2- and 1,6-NRH.19,50 It has low expression levels in HEK293T but increases upon oxidative stress in hepatocytes.51,52 Similarly, pyridoxamine-phosphate oxidase is primarily expressed in the liver. The toxicity observed for the two isomers of 1,4-NRH in HEK293T cells could stem from their conversion to 1,2- and 1,6-NADH, respectively, going unchallenged by low renalase and pyridoxamine-phosphate oxidase expression, and the subsequent inhibition of redox enzymes. Alternatively, these nucleosides could compromise cellular function through mechanisms not requiring conversion to dinucleotides, but rather are associated with their ability to decompose to aldehydes and glycating derivatives (Figure 4) affecting HEK293T more than HepG3 cells.
With the present work, we explored the chemical stability of the dihydropyridine moiety using 1H NMR spectroscopy to evaluate the chemical changes that 1,4-NRH undergoes while in solution. Other analytical methods, more prevalent in cell biology and molecular biology, which include mass spectrometry coupled to liquid chromatography, UV spectroscopy, as well as fluorescence, do not allow the characterization of the changes as they happen; 1H NMR spectroscopy provides this flexibility. Having explored the stability of 1,4-NRH and found that it correlates quite closely to that of NAD(P)H with regard to its reactivity to oxygen and ability to isomerize to 1,6-NRH, we chemically synthesized and isolated the pure form of the other isomers of 1,4-NRH and explored the integrity of the heterocycle. We demonstrated here that 1,4-NRH’s isomers inhibited NQO2 and that 1,4-NRH can also generate glycating entities. We observed that the addition of water on 1,4-NRH was rapid in phosphate-containing buffers, and that decomposition led to intermediates which readily react forming Shiff bases, those of solvent-exposed lysine residues of proteins of the like of BSA. While 1,4-NRH might not be overly abundant, its chemical reactivity toward protein residues could provide some evidence that intracellular NADH might promote protein aging, especially in organelles where it is the most abundant, the mitochondrion. Such conjugation is particularly relevant to cellular regulation for which nonreversible protein modifications could lead to protein loss of function and over time to the aging of the proteome. Therefore, NQO2’s role in the maintenance of cellular homeostasis might be less about the detoxification of xenobiotics and more about the removal of endogenous excess 1,4-NRH, generated from excess NADH.
Such a paradox highlights a new role for NQO2 activity, as its enzymatic activity has dual functions: reduction of quinones in a detoxification process and oxidation of NRH to NR+ to maintain low NRH intracellular levels. This work provides some insights as to how NAD(P)H:quinone oxidoreductase 1 (NQO1), the homologue of NQO2 which uses NADH and NADPH as substrates, might be interacting with the respective isomers of its dinucleotide substrates. Enzymes that reduce the levels of unbound NADH, like NQO1, would prevent the occurrence of these derivatives. While the cellular mechanisms underpinning the observed outcomes remain to be investigated, the role of excess NADH and its ability to chemically isomerize and generate endogenous 1,2- and 1–6-NADH isomers as well as Schiff-base adducts could be a link between metabolic dysfunction and intracellular NADH overabundance.
METHODS
Full details of the synthetic and analytical experiments reported in this manuscript can be found in the Supporting Information. The full characterization spectra of the reported molecules can be also found in the Supporting Information.
Stability of NRH in Water and in Phosphate Buffer.
Figure 3: 50 mg of 1,4-NRH was dissolved in 2.5 mL of sodium phosphate buffer (50 mM, pH = 6.8). A total of 450 μL of the 1,4-NRH + buffer solution was transferred to an NMR tube. A total of 50 μL of D2O was added to the NMR tube to yield a total volume of 500 μL. The NMR tube was vortexed to achieve a homogeneous solution and then placed in a 37 °C hot water bath for 1, 3, 12, and 24 h, at which time a 1H NMR spectrum was taken.
Figure 4, Panel A: 30 mg of 1,4-NRH was dissolved in 1.5 mL of deionized degassed water (pH= 7.3). A total of 450 μL of the 1,4-NRH + H2O solution was transferred to an NMR tube. A total of 50 μL of D2O was added to the NMR tube to yield a total volume of 500 μL. The NMR tube was vortexed to achieve a homogeneous solution and then placed in a 37 °C hot water bath for 24 h, after which time a 1H NMR spectrum was taken.
Figure 4, Panel B: 50 mg of 1,4-NRH was dissolved in 2.5 mL of sodium phosphate buffer (500 mM, pH = 6.0). A total of 450 μL of the 1,4-NRH + buffer solution was transferred to an NMR tube. A total of 50 μL of D2O was added to the NMR tube to yield a total volume of 500 μL. The NMR tube was vortexed to achieve a homogeneous solution and then placed in a 37 °C hot water bath for 30 min, after which time a 1H NMR spectrum was taken.
Isomerism of NRH and NR+.
1,4-NRH was coincubated with NR+ in D2O (final concentration 10 mM of each component). Under these conditions, the 1,6-NRH isomer formed and could be detected after 100 h. Products that can readily be identified include alpha and beta-ribose, nicotinamide, NR+, and 1,6-NRH. The appearance of products with aliphatic hydrogens consistent with structures proposed by Margoli via the intermediacy of NR(OH)H, which can also generate decomposition products, remain to be characterized.
Cell Culture.
Human Embryonic Kidney (HEK293T) and Hepatoma (HepG3) cells were purchased from American Type Culture Collection (ATCC). Cells were grown at 37 °C in a 5% CO2 incubator in Dulbecco’s modified Eagle’s medium (DMEM; Hyclone) supplemented with glutamine (Gibco), 10% fetal bovine serum (FBS; Atlanta Biologicals Premium Select), and 1% sodium pyruvate (Gibco). Cells were routinely tested and found to be free of mycoplasma contamination using Lonza MycoAlert Assay.
Cytotoxicity Studies.
The cytotoxicity of NRH and its derivatives was determined by CellTiter-Fluor (Promega), which measures cell viability independent of metabolic markers related to NAD(P)(H). HEK293T cells were seeded at a density of 5000 cells/well in a 96-well clear bottomed black plate coated with poly-L-lysine (Corning). HepG3 cell lines were seeded at a density of 15 000 cells/well in an uncoated 96-well clear bottomed black plate (Greiner Bio). Cell plating density was optimized to ensure ~75% confluency at the end of the assay. The following day, 100 mM stock solutions of 1,4-NRH, 1,2-NRH, and 1,6-NRH were prepared in a phosphate buffered saline solution without calcium or magnesium (PBS, VWR). Cells were exposed to 100 μM of each compound in the medium for 6 h. After 6 h, supplements were washed once in PBS, and fresh medium was replaced. The cells were placed back at 37 °C in a 5% CO2 incubator and allowed to grow for another 66 h (total 72 h). At the end of 72 h, 100 μL of CellTiter-Fluor reagent was added to all wells and incubated for at least 30 min at 37 °C. Fluorescence intensity was measured using a Tecan Infinite M1000 Pro plate reader (380 nm Ex/505 nm Em). The fluorescent signal is then expressed as a percentage relative to the control to reflect cell survival. Values are reported as the mean percentage survival ± standard error of the mean (SEM) of three biological replicates.
Recombinant Human NQO2 Enzyme Activity.
Recombinant human NQO2 was obtained from Sigma and diluted in 50 mM phosphate buffer. All one-dimensional proton NMR spectra were obtained at 300 K on a Bruker Ascend 400 MHz ultrashielded spectrometer (Bruker Biospin) operating at 400.13 MHz for protons and used to determine enzyme activity. TopSpin 3.2 (Bruker BioSpin) was used for all NMR spectral acquisition (ns = 128) and preprocessing, and the automation of sample submission was performed using ICON-NMR (Bruker BioSpin). All samples were automatically shimmed, and their acquisition time was 10 min and 8 s. All experiments were performed at 25 °C. The 1,2-NRH, 1,4-NRH, and 1,6-NRH used were synthesized according to the procedure described in the experimental supplementary file. The rates of the NQO2 enzyme were determined by NMR spectroscopy by recording the rate of oxidation of 1,2-NRH, 1,4-NRH, and 1,6-NRH to NR at t = 0, 6, and 24 h (ns = 128) at 25 °C in the NMR tube, made up of borosilicate glass (7 in. × 5 mm) containing a final volume of 5.10 mL: 450 μL of phosphate buffer (50.0 mM, pH 7.1), 10 μL of recombinant NQO2 (1 mg dissolved in 500 μL HEPES buffer), 50.0 μL of NRH (10.0 mM in D2O). All the experiments were performed in a single day. For each independent experiment, freshly prepared solutions of NRHs in D2O and 450 μL of phosphate buffer, containing 125 mM NaCl, 5 μM FAD, BSA (1 mg Ml−1), and 10 μL/mL menadione (93 mM solution in 1 mL ethanol) were used.
Identification of Modified Lysine Residues in BSA.
A total of 50 μL of 1 mg mL−1 bovine serum albumin (BSA) was incubated with 50 μL of 1,4-NRH and 400 μL of a phosphate buffered saline solution at RT for 8 days. BSA was also incubated in the absence of NRH for comparison at the same concentration. A total of 100 μL of each solution was further diluted with 900 μL of 0.1% trifluoroacetic acid (TFA) and loaded onto separate washed and equilibrated C2 columns (Waters Sep-Pak Vac 1 cm3 C2 cartridge, WAT052710). After the entire sample was loaded, the column was washed with 3 mL (3 column volumes) of 0.1% TFA, followed by an additional wash with 3 mL of 10% ACN in 0.1% TFA. The protein was eluted from the column with 2 mL of 60% ACN in 0.1% TFA collected into one fraction and brought to dryness in a speed-vac concentrator. The dried sample was resuspended into 50 μL of 50 mM ammonium bicarbonate/10 mM tris(2-carboxye thyl)phosphine hydrochloride and digested for 7 h with 1 μL sequencing grade trypsin at 37 °C. High resolution mass spectrometry was performed on the resulting peptide solution using a Q-Exactive Plus mass spectrometer (Thermo Fisher Scientific) coupled with a Thermo Easy-nLC 1000 liquid chromatograph. A 0.5 μL injection volume was used for each sample, loaded onto an ES800 C18 column equipped with a C18 guard column. Solvent A consisted of 3% acetonitrile (ACN) in H2O with 0.2% formic acid (FA), and solvent B consisted of 3% water in ACN with 0.2% FA. The flow rate was set to 300 nL per minute. To provide chromatographic separation, a linear solvent gradient was used from 2% to 30% B over a period of 36 min, as well as a linear gradient from 30% B to 90% B for an additional 15 min. A 5 min wash at 90% B was used to elute any remaining material from the column at the end of each run. Blanks were also run between samples to minimize carryover. The EASY-spray source was used, in positive ion mode, to introduce the sample into the MS using electrospray ionization. A spray voltage of 2500 and a capillary temperature of 250 °C was used. One full scan from 400 to 2000 m/z was performed at 70,000 resolution, followed by 6 dd-ms2 scans at 17 500 resolution of the top 6 features identified in the full scan. Features with a charge of no less than 2 and no more than 8 were selected for HCD fragmentation with a 2.0 m/z isolation window, using a normalized collision energy of 27. A dynamic exclusion of 15 s was utilized to maximize coverage. Total run time for each sample analyzed was 60 min. The resulting raw files were uploaded into Thermo Proteome Discoverer software v2.2 to provide peptide identification. The sequest processing mode was used to search a custom fasta file containing the sequence of bovine serum albumin, with a precursor mass tolerance of 5 ppm and a fragment mass tolerance of 0.05 Da. Semitryptic peptides were allowed with a maximum of two missed cleavage sites.
The following custom dynamic modifications specific to lysine residues were included in the search: (1) NRH Adduct C11H18N2O5/+258.122 Da (K); (2) NRH/+256.106 Da (K); (3) NRH Adduct C11H18N2O6/+274.117 Da (K); (4) NRH Adduct C6H9NO2/+127.063 Da (K); (5) NRH Adduct C6H9NO3/+143.058 Da (K).
A maximum of two equal modifications and four dynamic modifications were allowed per peptide. The spectrum confidence filter node was used to filter out any spectra that did not have high confidence. The percolator node was used with a target FDR (strict) set to 0.01 and 0.05 for relaxed, with validation based on q value. Precursor quantification was performed using both unique and razor peptides, with abundance based on area. No normalization or scaling was applied.
Quantum-Based Computational Modeling NQO2.
A truncated model of the NQO2(FAD) enzyme (17 amino acid residues) was built from PDB entry 2QX8 [48] (res 1.60 Å) using Gaussview 5.0 (Gaussian Inc., Wallingford, CT). The FAD molecule was truncated at the first phosphate to produce a model system with a net charge of −1. In the search for favorable docking poses of the three NRH isomers on the face of FAD, eight classes of poses of each isomer were considered through manual positioning: two positions for the amide moiety × two positions for the glycosidic bond × two positions for the ribose moiety (left versus right of FAD). Manual docking was followed by semiempirical PM7 energy optimizations in which only the NRH ligand was permitted to move. The resulting lowest energy structures were selected for follow-up ONIOM hybrid calculations (high-level:B3LYP[3,4]/6–31G-(d):low-level:PM7) in order to distinguish them by using an all-electron density functional model (B3LYP) for the high-level region. The ligand positions were reoptimized, ultimately using the ONIOM(wB97XD/6–31G(d):PM7) model in the presence of water using the default SCRF model. The wB97XD model is a density functional method that includes the effects of dispersion. Optimizations were deemed complete when the forces met the default convergence criteria. All calculations were carried out using Gaussian16.
CONCLUSION
Intracellular 1,4-NAD(P)H, just like 1,4-NRH in solution, not only oxidizes but also generates isomers and hydroxylated adducts. Accumulation of NAD(P)H in cells53,54 would lead to increased formation of these isomeric and hydrated species, and since these species possess irreversible chemical reactivities to protein conjugating agents, the equilibrium is constantly displaced toward their formation and thus could deplete the NAD(P)(H) pools. The work highlights some of the processes that such endogenously generated species are likely to disrupt. Unfortunately, quantification of these NAD(P)H catabolites from cell culture or tissues is not yet possible as they “disappear” from the quantifiable environment. Additionally, protein modifications by such species could be substantial and be mistaken for enzyme-catalyzed ADP ribosylation, in a way reminiscent of the chemically driven mitochondrial protein acetylation.55 It is this nonenzymatic intrinsic biochemistry that NRH helps expose. Critically, this work brings up possible unwanted effects of boosting not only NAD+ but also NAD(P) H in cells and in tissues, as these reduced forms could potentially be detrimental to cellular homeostasis and promote cellular dysfunction.
Supplementary Material
Table 1.
Coverage and Relative Abundance of Selected Lysine Modified BSA Peptides
| sequence | modifications | abundances in BSA | abundance in BSA treated with 1,4-NRH |
|---|---|---|---|
| LHEKTPVSEKVTK | NRH adduct C6H9NO2 [K4] | 2.61 × 1008 | 1.1 × 1009 |
| LHEKTPVSEK | NRH adduct C6H9NO2 [K4] | 32 803 514 | 88 550 358 |
| CTKPESERMPCTEDYLSLILNR | NRH adduct C6H9NO3 [K3] | BDL | 6.46 × 1008 |
| YQEAKDAFLGSFLYEYSR | NRH adduct C11H18N2O5 [K5] | 8 007 322 | 16 282 244 |
ACKNOWLEDGMENTS
We thank The University of South Alabama Mass Spectrometry Core Facility and the valuable technical contributions from L. Schambeau and S. Tadi. We thank the Alabama Supercomputer Authority for providing computational support and resources.
Funding
This work was supported by R21AT009908 from the National Institutes of Health, National Center for Complementary and Integrative Health. M.E.M. is partially supported by Elysium Health. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.0c00757.
Fully synthetic experimental details; full characterization spectra; 1H NMR, 13C NMR, 2D COSY, and HMQC, HRMS, and comparative fragmentation patterns of the 1,2-NRH, 1,6-NRH, and 1,4-NRH compounds reported in the Methods section (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acschembio.0c00757
Contributor Information
Mikhail V. Makarov, University of South Alabama, Department of Pharmacology, Mobile, Alabama 36688-0002, United States.
Faisal Hayat, University of South Alabama, Department of Pharmacology, Mobile, Alabama 36688-0002, United States.
Briley Graves, University of South Alabama, Department of Pharmacology, Mobile, Alabama 36688-0002, United States.
Manoj Sonavane, University of South Alabama, Department of Physiology and Cell Biology, Mobile, Alabama 36688-0002, United States.
Edward A. Salter, University of South Alabama, Department of Chemistry, Mobile, Alabama 36688-0002, United States
Andrzej Wierzbicki, University of South Alabama, Department of Chemistry, Mobile, Alabama 36688-0002, United States.
Natalie R. Gassman, University of South Alabama, Department of Physiology and Cell Biology, Mobile, Alabama 36688-0002, United States.
Marie E. Migaud, University of South Alabama, Department of Pharmacology, Mobile, Alabama 36688-0002, United States.
REFERENCES
- (1).Magni G, Amici A, Emanuelli M, Orsomando G, Raffaelli N, and Ruggieri S (2004) Enzymology of NAD+ homeostasis in man. Cell. Mol. Life Sci. 61 (1), 19–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Berger F, Ramirez-Hernandez MH, and Ziegler M (2004) The new life of a centenarian: signalling functions of NAD(P). Trends Biochem. Sci. 29 (3), 111–118. [DOI] [PubMed] [Google Scholar]
- (3).Chiarugi A, Dolle C, Felici R, and Ziegler M (2012) The NAD metabolome–a key determinant of cancer cell biology. Nat. Rev. Cancer 12 (11), 741–752. [DOI] [PubMed] [Google Scholar]
- (4).Magni G, Orsomando G, Raffelli N, and Ruggieri S (2008) Enzymology of mammalian NAD metabolism in health and disease. Front. Biosci., Landmark Ed. 13, 6135–6154. [DOI] [PubMed] [Google Scholar]
- (5).Stromland O, Niere M, Nikiforov AA, VanLinden MR, Heiland I, and Ziegler M (2019) Keeping the balance in NAD metabolism. Biochem. Soc. Trans. 47 (1), 119–130. [DOI] [PubMed] [Google Scholar]
- (6).Koch-Nolte F, Fischer S, Haag F, and Ziegler M (2011) Compartmentation of NAD+-dependent signalling. FEBS Lett. 585 (11), 1651–1656. [DOI] [PubMed] [Google Scholar]
- (7).Belenky P, Bogan KL, and Brenner C (2007) NAD+ metabolism in health and disease. Trends Biochem. Sci. 32 (1), 12–9. [DOI] [PubMed] [Google Scholar]
- (8).Chini CCS, Tarrago MG, and Chini EN (2017) NAD and the aging process: Role in life, death and everything in between. Mol. Cell. Endocrinol. 455, 62–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Croteau DL, Fang EF, Nilsen H, and Bohr VA (2017) NAD(+) in DNA repair and mitochondrial maintenance. Cell Cycle 16 (6), 491–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Chaturvedi P, and Tyagi SC (2018) NAD(+): A big player in cardiac and skeletal muscle remodeling and aging. J. Cell. Physiol. 233 (3), 1895–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Mendelsohn AR, and Larrick JW (2017) The NAD +/PARP1/SIRT1 Axis in Aging. Rejuvenation Res. 20 (3), 244–247. [DOI] [PubMed] [Google Scholar]
- (12).Lu M, Zhu XH, and Chen W (2016) In vivo (31) P MRS assessment of intracellular NAD metabolites and NAD(+) /NADH redox state in human brain at 4 T. NMR Biomed. 29 (7), 1010–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Katsyuba E, and Auwerx J (2017) Modulating NAD(+) metabolism, from bench to bedside. EMBO J. 36 (18), 2670–2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Cantó C, and Auwerx J (2011) NAD+ as a signaling molecule modulating metabolism. Cold Spring Harbor Symp. Quant. Biol. 76, 291–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).McComb RB, and Gay RJ (1968) A comparison of reduced-NAD preparations from four commercial sources. Clin. Chem. 14 (8), 754–763. [PubMed] [Google Scholar]
- (16).Wallace TC, and Coughlin RW (1978) Rate of catalytic oxidation and decomposition of NADH by graphite in aqueous solutions. Biotechnol. Bioeng. 20 (3), 403–420. [Google Scholar]
- (17).Morrison CS, Armiger WB, Dodds DR, Dordick JS, and Koffas MAG (2018) Improved strategies for electrochemical 1,4-NAD(P)H2 regeneration: A new era of bioreactors for industrial biocatalysis. Biotechnol. Adv. 36 (1), 120–131. [DOI] [PubMed] [Google Scholar]
- (18).Beaupre BA, Hoag MR, Roman J, Försterling FH, and Moran GR (2015) Metabolic function for human renalase: oxidation of isomeric forms of β-NAD(P)H that are inhibitory to primary metabolism. Biochemistry 54 (3), 795–806. [DOI] [PubMed] [Google Scholar]
- (19).Moran GR, and Hoag MR (2017) The enzyme: Renalase. Arch. Biochem. Biophys. 632, 66–76. [DOI] [PubMed] [Google Scholar]
- (20).Kremer LS, Danhauser K, Herebian D, Petkovic Ramadža D, Piekutowska-Abramczuk D, Seibt A, Müller-Felber W, Haack TB, Płoski R, Lohmeier K, Schneider D, Klee D, Rokicki D, Mayatepek E, Strom TM, Meitinger T, Klopstock T, Pronicka E, Mayr JA, Baric I, Distelmaier F, and Prokisch H (2016) NAXE Mutations Disrupt the Cellular NAD(P)HX Repair System and Cause a Lethal Neurometabolic Disorder of Early Childhood. Am. J. Hum. Genet. 99 (4), 894–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Zinner K, Faljoni A, and Cilento G (1973) On the structure of NADH-X. Biochem. Biophys. Res. Commun. 51 (1), 181–185. [DOI] [PubMed] [Google Scholar]
- (22).Regueiro Varela B, Amelunxen R, and Grisolia S (1970) Synthesis and degradation of monohydroxy-tetrahydronicotinamide adenine dinucleotide phosphate. Physiol. Chem. Phys. 2 (5), 445–454. [Google Scholar]
- (23).Godtfredsen SE, Ottesen M, and Andersen NR (1979) On the mode of formation of 1,6-dihydro-NAD in NADH preparations. Carlsberg Res. Commun. 44 (2), 65–75. [Google Scholar]
- (24).Yamauti J, Yoshimura S, Takagahara I, Fujii K, Tai A, Yamashita J, and Hprio T (1981) Isolation and characterization of two potent inhibitors of various NADH dehydrogenases formed during storage of NADH. J. Biochem. 90 (4), 941–955. [DOI] [PubMed] [Google Scholar]
- (25).Hoag MR, Roman J, Beaupre BA, Silvaggi NR, and Moran GR (2015) Bacterial Renalase: Structure and Kinetics of an Enzyme with 2- and 6-Dihydro-β-NAD(P) Oxidase Activity from Pseudomonas phaseolicola. Biochemistry 54 (24), 3791–3802. [DOI] [PubMed] [Google Scholar]
- (26).Metherell LA, Guerra-Assuncao JA, Sternberg MJ, and David A (2016) Three-Dimensional Model of Human Nicotinamide Nucleotide Transhydrogenase (NNT) and Sequence-Structure Analysis of its Disease-Causing Variations. Hum. Mutat. 37 (10), 1074–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Prabhakar P, Laboy JI, Wang J, Budker T, Din ZZ, Chobanian M, and Fahien LA (1998) Effect of NADH-X on cytosolic glycerol-3-phosphate dehydrogenase. Arch. Biochem. Biophys. 360 (2), 195–205. [DOI] [PubMed] [Google Scholar]
- (28).Margolis SA, Howell BF, and Schaffer R (1977) Lactate dehydrogenase inhibitors in NADH preparations. Clin. Chem. 23 (9), 1581–1584. [PubMed] [Google Scholar]
- (29).Margolis SA, Yap WT, Matthews B, and Schaffer R (1978) Reverse-phase liquid chromatographic analysis of the acid-catalyzed rearrangement of NADH, separation of new products and an analysis of the first two reactions of the rearrangement. J. Liq. Chromatogr. 1 (5), 669–691. [Google Scholar]
- (30).Marbaix AY, Chehade G, Noel G, Morsomme P, Vertommen D, Bommer GT, and Van Schaftingen E (2019) Pyridoxamine-phosphate oxidases and pyridoxamine-phosphate oxidase-related proteins catalyze the oxidation of 6-NAD(P)H to NAD(P)+. Biochem. J. 476 (20), 3033–3052. [DOI] [PubMed] [Google Scholar]
- (31).Marbaix AY, Tyteca D, Niehaus TD, Hanson AD, Linster CL, and Van Schaftingen E (2014) Occurrence and subcellular distribution of the NAD(P)HX repair system in mammals. Biochem. J. 460 (1), 49–58. [DOI] [PubMed] [Google Scholar]
- (32).Becker-Kettern J, Paczia N, Conrotte J-F, Zhu C, Fiehn O, Jung PP, Steinmetz LM, and Linster CL (2018) NAD(P)HX repair deficiency causes central metabolic perturbations in yeast and human cells. FEBS J. 285 (18), 3376–3401. [DOI] [PubMed] [Google Scholar]
- (33).Beaupre BA, Roman JV, Hoag MR, Meneely KM, Silvaggi NR, Lamb AL, and Moran GR (2016) Ligand binding phenomena that pertain to the metabolic function of renalase. Arch. Biochem. Biophys. 612, 46–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Marbaix AY, Tyteca D, Niehaus TD, Hanson AD, Linster CL, and Van Schaftingen E (2014) Occurrence and subcellular distribution of the NADPHX repair system in mammals. Biochem. J. 460 (1), 49–58. [DOI] [PubMed] [Google Scholar]
- (35).Margolis SA, Howell BF, and Schaffer R (1976) Purification and analysis of the purity of NADH. Clin. Chem. (Washington, DC, U. S.) 22 (8), 1322–1329. [PubMed] [Google Scholar]
- (36).Yang Y, Mohammed FS, Zhang N, and Sauve AA (2019) Dihydronicotinamide riboside is a potent NAD(+) concentration enhancer in vitro and in vivo. J. Biol. Chem. 294 (23), 9295–9307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Giroud-Gerbetant J, Joffraud M, Giner MP, Cercillieux A, Bartova S, Makarov MV, Zapata-Perez R, Sanchez-Garcia JL, Houtkooper RH, Migaud ME, Moco S, and Canto C (2019) A reduced form of nicotinamide riboside defines a new path for NAD(+) biosynthesis and acts as an orally bioavailable NAD(+) precursor. Mol. Metab. 30, 192–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Sonavane M, Hayat F, Makarov M, Migaud ME, and Gassman NR (2020) Dihydronicotinamide riboside promotes cell-specific cytotoxicity by tipping the balance between metabolic regulation and oxidative stress. PLoS One 15 (11), e0242174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Ziegler M, and Nikiforov AA (2020) NAD on the rise again. Nature Metabolism 2 (4), 291–292. [DOI] [PubMed] [Google Scholar]
- (40).Jamieson D, Wilson K, Pridgeon S, Margetts JP, Edmondson RJ, Leung HY, Knox R, and Boddy AV (2007) NAD(P)H:quinone oxidoreductase 1 and nrh:quinone oxidoreductase 2 activity and expression in bladder and ovarian cancer and lower NRH:quinone oxidoreductase 2 activity associated with an NQO2 exon 3 single-nucleotide polymorphism. Clin. Cancer Res. 13 (5), 1584–1590. [DOI] [PubMed] [Google Scholar]
- (41).Celli CM, Tran N, Knox R, and Jaiswal AK (2006) NRH:quinone oxidoreductase 2 (NQO2) catalyzes metabolic activation of quinones and anti-tumor drugs. Biochem. Pharmacol. 72 (3), 366–376. [DOI] [PubMed] [Google Scholar]
- (42).Nolan KA, Zhao H, Faulder PF, Frenkel AD, Timson DJ, Siegel D, Ross D, Burke TR, Stratford IJ, and Bryce RA (2007) Coumarin-based inhibitors of human NAD(P)H:quinone oxidoreductase-1. Identification, structure-activity, off-target effects and in vitro human pancreatic cancer toxicity. J. Med. Chem. 50 (25), 6316–6325. [DOI] [PubMed] [Google Scholar]
- (43).Makarov MV, Harris NW, Rodrigues M, and Migaud ME (2019) Scalable syntheses of traceable ribosylated NAD(+) precursors. Org. Biomol. Chem. 17 (38), 8716–8720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Makarov MV, and Migaud ME (2019) Syntheses and chemical properties of beta-nicotinamide riboside and its analogues and derivatives. Beilstein J. Org. Chem. 15, 401–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Lovesey AC (1969) Potential coenzyme inhibitors. 3. Some reactions of substituted nicotinamide and dihydronicotinamide derivatives. J. Med. Chem. 12 (6), 1018–1023. [DOI] [PubMed] [Google Scholar]
- (46).Saba T, Burnett JWH, Li J, Kechagiopoulos PN, and Wang X (2020) A facile analytical method for reliable selectivity examination in cofactor NADH regeneration. Chem. Commun. (Cambridge, U. K.) 56 (8), 1231–1234. [DOI] [PubMed] [Google Scholar]
- (47).Mosmann T (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 65 (1–2), 55–63. [DOI] [PubMed] [Google Scholar]
- (48).Calamini B, Santarsiero BD, Boutin JA, and Mesecar AD (2008) Kinetic, thermodynamic and X-ray structural insights into the interaction of melatonin and analogues with quinone reductase 2. Biochem. J. 413 (1), 81–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Braidy N, and Liu Y (2020) NAD+ therapy in age-related degenerative disorders: A benefit/risk analysis. Exp. Gerontol. 132, 110831. [DOI] [PubMed] [Google Scholar]
- (50).Beaupre BA, Roman JV, Hoag MR, Meneely KM, Silvaggi NR, Lamb AL, and Moran GR (2016) Ligand binding phenomena that pertain to the metabolic function of renalase. Arch. Biochem. Biophys. 612, 46–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Severina IS, Fedchenko VI, Veselovsky AV, and Medvedev AE (2015) The history of renalase from amine oxidase to a a-NAD(P)H-oxidase/anomerase. Biomed. Khim. 61 (6), 667–679. [DOI] [PubMed] [Google Scholar]
- (52).Zhang T, Gu J, Guo J, Chen K, Li H, and Wang J (2019) Renalase Attenuates Mouse Fatty Liver Ischemia/Reperfusion Injury through Mitigating Oxidative Stress and Mitochondrial Damage via Activating SIRT1. Oxid Med. Cell Longev 2019, 7534285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Xiao W, and Loscalzo J (2020) Metabolic Responses to Reductive Stress. Antioxid. Redox Signaling 32 (18), 1330–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Xiao W, Wang RS, Handy DE, and Loscalzo J (2018) NAD(H) and NADP(H) Redox Couples and Cellular Energy Metabolism. Antioxid. Redox Signaling 28 (3), 251–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Baeza J, Smallegan MJ, and Denu JM (2015) Site-specific reactivity of nonenzymatic lysine acetylation. ACS Chem. Biol. 10 (1), 122–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.