

Abstract
Cancer cells migrating in confined microenvironments exhibit plasticity of migration modes. Confinement of contractile cells in a nonadhesive environment drives “leader bleb–based migration” (LBBM), morphologically characterized by a long bleb that points in the direction of movement separated from a cell body by a contractile neck. Although cells undergoing LBBM have been visualized within tumors, the organization of organelles and actin regulatory proteins mediating LBBM is unknown. We analyzed the localization of fluorescent organelle-specific markers and actin-associated proteins in human melanoma and osteosarcoma cells undergoing LBBM. We found that organelles from the endolysosomal, secretory, and metabolic systems as well as the vimentin and microtubule cytoskeletons localized primarily in the cell body, with some endoplasmic reticulum, microtubules, and mitochondria extending into the leader bleb. Overexpression of fluorescently tagged actin regulatory proteins showed that actin assembly factors localized toward the leader bleb tip, contractility regulators and cross-linkers in the cell body cortex and neck, and cross-linkers additionally throughout the leader bleb. Quantitative analysis showed that excess filamin-A and fascin-1 increased migration speed and persistence, while their depletion by small interfering RNA indicates a requirement in promoting cortical tension and pressure to drive LBBM. This indicates a critical role of specific actin crosslinkers in LBBM.
INTRODUCTION
Cancer metastasis is mediated by cell migration, wherein cells move out of the primary tumor and invade surrounding tissue, enter blood or lymphatic vessels, traverse the circulation, and leave the vasculature at a distant site where they colonize and form additional tumors (Martin et al., 2013). It has long been known that the acquisition of migratory function by tumor cells is mediated by epithelial-to-mesenchymal transition (EMT), whereby cells reduce intercellular attachment, increase extracellular matrix (ECM) attachment, and move via integrin-ECM–mediated migration (Hamidi and Ivaska, 2018). Mesenchymal migration is a well-characterized process in which cells polarize in the direction of movement, generate an actin polymerization–driven protrusion, adhere the leading edge to the ECM through integrins, and contract the cell rear to advance (Parsons et al., 2010). However, recently it has become clear that cancer cell migration is a much more plastic process. In addition to EMT, cancer cells can migrate in groups via collective migration (Friedl and Gilmour, 2009; Trepat and Fredberg, 2011; Yang et al., 2019) or individually by adhesion-independent ameboid migration (Sabeh et al., 2009). It is thought that the ability of cancer cells to switch between different modes of motility is an important factor in metastasis, as disseminating tumor cells must navigate a range of tissue microenvironments to escape points of origin and spread to distant sites (Friedl and Wolf, 2010).
The conversion between mesenchymal and ameboid motility modes, known as mesenchymal-to-ameboid transition (MAT), is thought to occur when highly contractile cells migrate in tight confinement and is mediated by loss of adhesion, which terminates the signals that drive actin polymerization–dependent protrusion (Friedl and Wolf, 2010; Huang et al., 2013; Tozluogˇlu et al., 2013; Bergert et al., 2015; Liu et al., 2015; Ruprecht et al., 2015). Instead of the lamellipodial or filopodial protrusions and integrin-based adhesions with the ECM typical of mesenchymal migration, ameboid cell migration is mediated by blebbing protrusions and nonspecific friction with the microenvironment (Lämmermann et al., 2008; Renkawitz et al., 2009; Ridley, 2011; Bergert et al., 2012; Paluch and Raz, 2013). Bleb formation is driven by cytosolic pressure overcoming the strength of the linkage between the plasma membrane and the cortical cytoskeleton, which results in a hydrostatically driven bubble of plasma membrane protruding from the cortex (Charras et al., 2005; Charras and Paluch, 2008). Blebbing is driven by increase in the intracellular hydrostatic pressure that can be induced by high contractility via nucleus-mediated signaling pathways or externally by confinement (Sahai and Marshall, 2003; Paluch et al., 2006; Gadea et al., 2007; Charras and Paluch, 2008; Maugis et al., 2010; Logue et al., 2015; Lomakin et al., 2020; Venturini et al., 2020). Following bleb formation, ameboid cell advance is mediated by a contractile cell rear that drives either retrograde flow of blebs entangled in the microenvironment by an interdigitation and swimming-based mechanism (Paluch and Raz, 2013) or of transmembrane proteins that interact nonspecifically with the confining microenvironment (Heuzé et al., 2013; Paluch and Raz, 2013). Thus, although mesenchymal and ameboid migration modes differ in their fundamental mechanisms of protrusion and adhesion, they share the common feature of being driven by retrograde cortical flow produced by a cellular asymmetry in actomyosin contractility.
Recently, a novel form of bleb-based motility was found in cells with high intrinsic contractility in a tightly confined and nonadhesive environment (Bergert et al., 2015; Liu et al., 2015; Logue et al., 2015; Ruprecht et al., 2015). Cells migrating under such conditions adopt an extremely polarized morphology, characterized by a single large stable bleb that leads the direction of migration in what has been termed “leader bleb–based migration” (LBBM) (Logue et al., 2015, 2018). The leader bleb is sausage-shaped and separated from the trailing cell body by a contractile neck (Bergert et al., 2015; Liu et al., 2015; Logue et al., 2015; Ruprecht et al., 2015). Leader blebs spontaneously generate from an actively blebbing cell when a single, very large bleb becomes stabilized by a still undefined mechanism. Once formed, the cell body and leader bleb behave as nearly distinct compartments, with extremely limited diffusion of cytosolic components between them (Logue et al., 2015), likely due to steric hindrance at the narrow neck. This compartmentalization leads to highly polarized signaling that dictates distinct cytoskeletal organization in the cell body and leader bleb (Logue et al., 2015). The cell body is surrounded by a contractile actomyosin cortex and often exhibits continuous small blebbing (Bergert et al., 2015; Liu et al., 2015; Logue et al., 2015; Ruprecht et al., 2015). Within the leader bleb, actin assembles into bundles near the bleb tip and forms a dense meshwork that undergoes rapid retrograde flow and disassembles near the bleb neck, where myosin II is concentrated (Bergert et al., 2015; Logue et al., 2015). The fast cortical flow drives rapid motility of the bleb, which pulls along the attached cell body. Although a similar migration morphology has been observed in vivo for melanoma cells in tumors (Tozluogˇlu et al., 2013) and primordial germ cells in zebrafish embryos (Goudarzi et al., 2012; Ruprecht et al., 2015), little is known about how cells achieve and maintain such extreme cellular compartmentalization and polarization of the cytoskeleton and whether this morphology affects basic cellular organization and function.
Here we approached the question of how the unusual morphology adopted by cells undergoing LBBM in nonadhesive confinement affects subcellular organization of organelles and cytoskeletal systems and the role of actin regulatory proteins in mediating LBBM. We performed a localization screen of organelle-specific fluorescent fusion proteins to survey the subcellular “anatomy” of A375M melanoma and U2OS osteosarcoma cells during LBBM. This revealed extreme compartmentalization between the cell body and leader bleb, with most membranous organelles and the bulk of the vimentin and microtubule (MT) cytoskeletons in the cell body, but with some MTs, mitochondria, and large endoplasmic reticulum (ER) cisternae additionally extending into the leader bleb. A localization and overexpression screen of fluorescently tagged actin regulatory proteins showed that excess filament cross-linking promotes large leader blebs and rapid LBBM. The roles of the bundler fascin-1 and cross-linker filamin-A were examined by small interfering RNA (siRNA) knockdown, indicating a requirement for these proteins in promoting cortical tension and LBBM. Together, these results reveal the striking polarization of organelles in cancer cells undergoing LBBM in nonadhesive confinement and support the role of actin bundling by fascin-1 and cross-linking by filamin-A in the cortex in this process.
RESULTS
We sought to characterize the distribution of organelle systems and the role of actin regulatory proteins in cancer cells undergoing LBBM in nonadhesive confinement. We first validated methods for investigating the effects of adhesion and confinement on the migration and distribution of the actomyosin and focal adhesion (FA) systems within A375M melanoma cells. Cells were plated on a coverslip coated with integrin ligand (10 μg/ml fibronectin [FN]) or nonadhesive bovine serum albumin (BSA; 1 µg/ml) or were placed in confinement between a polydimethylsiloxane (PDMS) pad and a similarly treated coverslip held apart by 3 µm beads as spacers (Figure 1, A–D) (Logue et al., 2018). The difference in cell adhesion between these conditions was verified by immunostaining for paxillin or expression of mEmerald-paxillin and confocal or total internal reflection fluoresecnce (TIRF) imaging, which showed FA staining at the ventral surface of cells plated on FN and diffuse paxillin in the cytoplasm with no dense plaques visible at the ventral surface of cells under nonadhesive confinement (Figure 1, L and M; Supplemental Figure 1, A and B). Time-lapse phase-contrast microscopy showed that on BSA-coated coverslips, cells remained rounded, exhibited small blebs, and failed to migrate (0.36 ± 0.03 µm/min) (Figure 1, A, A′, F, and G; Supplemental Movie S1). Cells adhered to FN in either nonconfined or confined conditions spread on the substrate and exhibited a spindle-shaped, mesenchymal-like morphology (Figure 1, B and B′–C) and underwent nondirectional migration (as indicated by a mean squared displacement (MSD) over time with a low slope, with confined adherent cells migrating significantly faster than nonconfined adherent cells (Figure 1, F and G; 1.13 ± 0.09 and 0.81 ± 0.71 µm/min, respectively). This suggests that confinement may enhance adhesion-mediated A375M cell migration. In contrast, cells under nonadhesive confinement between PDMS and BSA-coated coverslips often exhibited a “leader bleb” morphology, characterized by a sausage-shaped bleb separated from a rounded cell body by a thin neck (Figure 1, D, D′, E, H, and I; Supplemental Movie S1). Cells with this morphology underwent migration with the bleb leading the cell body at speeds similar to those of cells under adhesive confinement, but with more directional persistence compared with migration in the other three conditions, in agreement with previous studies (Figure 1, F and G) (Blaser et al., 2006; Tozluogˇlu et al., 2013; Liu et al., 2015; Logue et al., 2015, 2018; Ruprecht et al., 2015).
FIGURE 1:

Validation of the effects of low adhesion and confinement on the morphology and migration of A375M melanoma cells. A375M melanoma cells cultured on BSA (1 µg/ml; Low Adhesion [LA] [A, A′, D, D′–V] or fibronectin [10 µg/ml]; Adhesion [A] [B, B′, C, C′, E–G, J, L, N, Q])-treated coverslips with (C, C′, D, D′, E–V; Adhesion + Confinement, A + C,or Adh. + Conf.; and Low Adhesion + Confinement, LA + C, or L. Adh. + Conf.) or without (A, A′, B, B′, E–G, J, L, N, Q; Low Adhesion, LA, or Adhesion, A) confinement by a PDMS pad resting on 3 µm beads to define the confinement height (schematic representations shown in A–D, first column). (A–D) Right: Phase-contrast images from time-lapse movies at 0, 2, and 4 h. Scale bar = 100 µm. (A′–D′) Rose plots of representative cell migration tracks; cells were tracked for 8 h at 10 min intervals; the number of cells tracked (n) is shown on each plot. (E) Percentage of cells exhibiting leader bleb morphology (n = minimum of 50 cells per condition from N = 3 experiments). (F, G) Average cell migration speed (F) and mean squared displacement (MSD) vs. time (G). n (cells) = 50, 52, 55, 58 cells, respectively, N = 3 experiments. Statistical significance was determined by one-way ANOVA. **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001, NS not significant. (H) Phase image depicting example of measurements of length (yellow) and width (blue) of cell body and bleb in a cell undergoing LBBM. Scale bar = 10 µm. (I) Average length, width, and area of cell body and leader bleb; n (cells) shown below categories. (J, L, M, N, Q, R) Spinning-disk confocal (J, L, N, Q, R) or TIRFM (M) images of expressed fluorescent proteins: mEmerald (J, top row); FusionRed-F-Tractin (J, bottom row; L, M, N, middle column; N, Q, R,middle column); mEmerald-paxillin (L, M, first column); mEmerald-myosin IIA (N, first column); or mEmerald-histone H2B. Black dashed lines in schematic representations in L, M, and N depict Z-depth of the confocal image plane at the cell center or ventral surface. Scale bars = 10 µm. (K, O, P) Top: Schematic representation of midline (K, O) or peripheral line scans used for measuring fluorescence intensity distributions. Bottom: Average normalized (to maximum intensity [y-axis] and maximum length from the position of the neck [x-axis]) line scan analysis of the fluorescence intensity along the long axis of cells undergoing leader bleb–based motility and expressing mEmerald (K, green), FusionRed-F-Tractin (K, O, P) or mEmerald-myosin IIA (O, P, green); number of cells analyzed (n) shown on each plot. (S–V) Analysis of nuclear position and migration in cells undergoing LBBM from time-lapse spinning-disk confocal movies of cells coexpressing FusionRed-F-Tractin and mEmerald-H2B. (S) Nuclear position, n = 146 cells. (T) Rose plots of representative cell migration tracks when the nucleus is in the cell body (top) vs. when the nucleus is in the bleb (bottom); cells were tracked for 8 h at 10 min intervals; the number of cells tracked (n) is shown on each plot. (U, V) Average migration speed (U) and mean squared displacement (MSD) vs. time (V) when nucleus is in the cell body (n = 20) and when nucleus is in the bleb (n = 16). All data are representative of at least three independent experiments. Error is SEM. Statistical significance was determined by two-tailed Student’s t tests. **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001, NS not significant.
Movie S1.
Migration dynamics of A375M2 cells. Cells were plated on a coverslip coated with integrin ligand (10 μg/ml Fibronectin (FN), “Adhesion,” upper right) or non-adhesive bovine serum albumin (BSA, 1 μg/ml, “Low Adhesion,” upper left), or were placed in confinement between a PDMS pad and a FN-coated (“Adhesion + Confinement,” lower right), or BSA-coated (“Low Adhesion + Confinement,” lower left) coverslip held apart by 3 μm beads as spacers Beads can be seen as refractile points visible in lower panels. Colored lines highlight the position of the cell over time. Phase contrast time-lapse image sequences taken every 10 s (elapsed time shown in min:sec) on an inverted microscope system (Nikon Ti) using a 20× (0.75 NA, Plan Apo PH) objective lens and 0.9 NA condenser. Scale bar= 100 μm.
To confirm the previously characterized organization of the actomyosin cytoskeleton in cells undergoing LBBM (Liu et al., 2015; Logue et al., 2015, 2018; Ruprecht et al., 2015), we expressed FusionRed-F-Tractin (as a marker of actin filaments; Figure 1, L, J, N, Q, and R) or mEmerald-myosin IIA (Figure 1N). This showed that in adherent cells, actin and myosin IIA localized to peripheral and ventral contractile bundles. In contrast, in cells migrating under nonadhesive confinement, actin was concentrated in the cortex of the cell body and the bleb neck and formed a meshwork of thin bundles throughout the leader bleb, often with a prominent bundle a few microns behind and parallel to the leader bleb tip (Figure 1, J, L, N, and R) (Logue et al., 2015). Myosin IIA was present in the cell body cortex, absent in the central and distal portion of the leader bleb, and exhibited a sharp gradient to its highest concentration in the neck (Figure 1N). These observations were confirmed by averaging line scans taken along the midline or perimeter of the cells’ long axis, followed by length and intensity normalization (Figure 1, K, O, and P). This showed that actin and myosin IIA distributions were distinct from that of soluble mEmerald (Figure 1, J and K), which was reduced in the neck, and somewhat concentrated in the leader bleb compared with the cell body, possibly due to limited diffusion across the contractile neck (Logue et al., 2015).
It was previously observed in LBBM that the nucleus can localize in either the cell body or leader bleb (Liu et al., 2015; Logue et al., 2015). As the largest and stiffest organelle in the cell, it is possible that nuclear position could affect the cortical dynamics or friction driving LBBM. To test this hypothesis, we transiently transfected mEmerald-H2B as a chromatin marker to visualize nuclear position in A375M cells (Figure 1, Q and R). Analysis of cells exhibiting leader bleb morphology showed that the nucleus was present in the cell body or leader bleb or spanned the two in 55, 15, and 30% of all cells imaged, respectively (Figure 1S). Analysis of time-lapse movies with frames sorted according to nuclear position showed that when the nucleus was in the leader bleb, cells migrated more than three times faster and with much more directional persistence compared with when the nucleus was in the cell body (Figure 1, T–V; Supplemental Movie S2). Together, these data corroborate previous studies of the effects of nonadhesive confinement on melanoma cells and their actomyosin cytoskeleton morphology (Logue et al., 2015) and show that nuclear position in the leader bleb promotes rapid LBBM.
Movie S2.
Dynamics of nuclear position during LBBM. Spinning disk confocal time-lapse image sequences of A375M cells expressing mEmerald Histone H2B (green) and FusionRed F-Tractin (red) while undergoing LBBM in non-adhesive confinement. Images were taken every 30sec-1min (elapsed time shown in min:sec) on an inverted microscope system (Nikon Ti) using a 60× (1.4 NA, Plan Apo PH) oil immersion objective lens. Scale bar= 10μm.
Membranous organelles are predominantly localized in the cell body, with ER cisternae extending into the leader bleb
We next sought to map the distribution and organization of organelle systems in cells undergoing LBBM in nonadhesive confinement. We transiently expressed mEmerald-tagged markers of various organelles (Figure 2) together with FusionRed-F-Tractin and determined their distribution in adherent and nonadherent, confined A375M melanoma cells by spinning-disk confocal microscopy. For cells undergoing LBBM, we performed average normalized line scan analysis along both the midline and cortical margin of the cell’s long axis and also determined the fraction of total organelle marker fluorescence in the leader bleb versus the cell body.
FIGURE 2:

Distribution of organelle systems in A375M melanoma cells undergoing LBBM under nonadhesive confinement. A375M melanoma cells cultured on fibronectin (10 µg/ml)-coated coverslips (Adhesion, left three columns in A–E) or BSA (1 µg/ml)-coated coverslips with confinement by a PDMS pad resting on 3 µm beads to define the confinement height (L Adhesion + Confinement, right three columns in A–E; data analysis in A′–E′′ and F–H). (A–E) Spinning-disk confocal images of transiently coexpressed mEmerald (mEM)-tagged fusion protein markers of various organelles (green) together with FusionRed-F-Tractin (red). Images and analysis of low adhesion and confinement are of cells in which the nucleus was localized in the bleb; see Supplemental Figure 2 for images of cells with the nucleus in the cell body. (A–E) mEmerald tagged to (A) the prenylation signal sequence of Ras (CAAX), (B) clathrin heavy chain and caveolin, (C) Rab5 and LAMP1, (D) calreticulin and SIT, or (E) PDHA or PXMP2. (A′–E′) Average normalized (to maximum intensity [y-axis] and maximum length from the position of the neck [x-axis]) line scan analysis of the fluorescence intensity along the long axis midline of cells undergoing leader bleb–based motility and expressing the corresponding mEmerald-tagged fusion proteins noted in A–E; the number of cells analyzed (n) is shown on each plot. (F–H). Fraction of total cellular fluorescence intensity localized in cell body (red) vs. the leader bleb (green) for cells in which the nucleus was located in the cell body (G), or leader bleb (H), or agnostic to nuclear localization (F); the number of cells analyzed (n) is shown above each category. (I) Schematic representation of organelle distribution in a cell undergoing LBBM in nonadhesive confinement. Scale bars (A–E) = 10 µm.
We first examined the localization of membranous components of the cell (Supplemental Movie S3). We expressed A375M mEmerald tagged with the prenylation signal sequence of Ras (CAAX) as a marker of the plasma membrane in A375M cells. This showed an even distribution along the perimeter of both adherent and nonadherent confined cells exhibiting leader bleb morphology, as expected (Figure 2, A and A′). To investigate the organization of the endolysosomal membrane trafficking system, we expressed mEmerald-tagged fusions of either clathrin heavy chain, caveolin, Rab5, or LAMP1 as markers of clathrin-dependent endocytosis, clathrin-independent endocytosis, the endocytic recycling compartment, or lysosomes, respectively (Figure 2, B, B′, B′′, C, C′, and C′′). In adherent cells, these markers labeled many punctate vesicles throughout the cell as well as a cluster of vesicles toward the cell center, as expected. In cells exhibiting LBBM in nonadhesive confinement, vesicles labeled by all of these markers were primarily concentrated in the cell body, with a small fraction in the proximal portion of the leader bleb near the neck. Quantitative analysis largely confirmed the concentration of endolysosomal organelles in the cell body relative to the leader bleb, although a relatively high, even (nonpunctate) distribution of clathrin heavy chain, likely representing soluble protein, was also present throughout the leader bleb.
Movie S3.
Dynamics of MTOC position relative to the nucleus during LBBM. Spinning disk confocal time-lapse image sequences of A375M2 cells in non-adhesive confinement expressing mEmerald gtubulin (green) and FusionRed F-Tractin (red) while undergoing LBBM in non-adhesive confinement. The position of the nucleus could be seen from its exclusion of FusionRed F-Tractin. Left: nucleus in the cell body. Right: nucleus in the leader bleb. Images were taken every 30 sec-1min (elapsed time shown in min:sec) on an inverted microscope system (Nikon Ti) using a 60× (1.4 NA, Plan Apo PH) oil immersion objective lens. Scale bar= 10μm.
We next examined the organization of organelles making up the secretory system by expressing mEmerald-tagged calreticulin or sialyltransferase-1 (SIT) to label the ER and Golgi apparatus, respectively (Figure 2, D, D′. and D′′). In adherent cells, calreticulin localized to the nuclear envelope and a dense network of tubules and small cisternae that extended throughout the cell, while SIT labeled a cluster of vesicles adjacent to the nucleus near the cell center, as expected. In leader bleb cells in nonadhesive confinement, although the Golgi exhibited the expected cluster of vesicles, most of which resided in the cell body, the ER had some very unusual morphological features. In the cell body the ER marker labeled the nuclear envelope and a dense tubulo-cisternal network; however, the leader bleb lacked ER tubules and instead contained only large cisternae that protruded more than halfway into its interior.
To determine the organization of metabolic organelles, we expressed mEmerald-tagged pyruvate dehydrogenase A (PDHA) or peroxisomal membrane protein 2 (PXMP2) as markers of mitochondria and peroxisomes, respectively (Figure 2E). In adherent cells, PDHA labeled a network of worm-like mitochondria throughout the cell, while PXMP2 labeled a cluster of vesicles near the cell center. In leader bleb cells in nonadhesive confinement, both mitochondria and peroxisomes were localized in the cell body and at the base of the leader bleb, with about half the mitochondrial marker in each compartment and a greater fraction of peroxisome marker in the cell body than in the leader bleb (Figure 2, E′ and E′′).
Because we found that the position of the nucleus had an impact on LBBM speed, we also determined the effect of nuclear position on the distribution of organelles in these cells. We sorted the images based on nuclear position (visible due to its exclusion of fluorescent F-tractin; Supplemental Figure 2, A–F) and determined the fraction of fluorescent organelle marker in the cell body versus the bleb. This showed that nuclear position in leader bleb cells had little effect on organelle marker distribution (Figure 2, F–H).
To determine whether the distribution of organelles observed in A375M cells was common in other cancer cells undergoing LBBM, we examined the localization of several organelle markers in U2OS human osteosarcoma cells in nonadhesive confinement. This showed that similar fractions of U2OS and A375M cells exhibited leader bleb morphology when confined to 3 µm under PDMS. In addition, markers of actin filaments (FusionRed-F-Tractin), lysosomes (mEmerald-LAMP1), the nucleus (mEmerald-H2B), and ER (mEmerald-calnexin) all exhibited similar distributions between the cell body and leader bleb as that seen in A375M cells; however, mitochondria (mEmerald-PDHA) in U2OS cells were almost exclusively concentrated in the cell body and mostly absent from the leader bleb (Supplemental Figure 3F). Together, these results show that in cancer cells undergoing LBBM in nonadhesive confinement, membranous organelles are predominantly concentrated in the cell body with little extending into the leader bleb, and ER morphology is drastically altered and polarized with a tubular reticulum in the cell body and large cisternae extending into the bleb (Figure 2I).
MT and vimentin intermediate filament cytoskeletons are highly polarized during LBBM
We next sought to characterize the MT and vimentin intermediate filament (VIF) cytoskeletal systems in cells undergoing LBBM (Supplemental Movies S4 and S5). To elucidate the organization of the MT cytoskeleton, we expressed either mEmerald-gamma-tubulin (mEmerald-gT), three tandem green fluorescent proteins (GFPs) fused to the MT-binding domain of ensconsin (GFP-3X-EMTB) (Salbreux et al., 2012), or mEmerald-CLIP-170 as markers of the MT-organizing center (MTOC), MTs, and growing MT plus ends, respectively (Figure 3, A–C). In adherent cells, these markers localized to a perinuclear spot, a dense network, or small comet-shaped streaks throughout the cell with a concentration in cell protrusions, respectively, as expected. Examination of mEmerald-gT in cells undergoing LBBM showed a similar perinuclear spot localization (Supplemental Movie S4). Because polarization of the MTOC between the nucleus and leading edge is a hallmark of migrating mesenchymal cells, we quantified MTOC position relative to nuclear position in leader bleb cells under nonadhesive confinement. This showed that the MTOC generally localized between the nucleus and leader bleb tip, independent of whether the nucleus was in the cell body or leader bleb (Figure 3A′ Supplemental Movie S3). Examination of GFP-3X-EMTB as well as immunolocalization of endogenous tubulin (Supplemental Figure 3C) showed a dense network of cortical MT bundles surrounding the cell body. MTs also penetrated the contractile neck into the leader bleb, with bundles near the bleb base and single MTs extending nearer to the bleb tip where they often bent and looped back toward the bleb center (Figure 3B; Supplemental Figure 3, B and B′; Supplemental Movie S5). Similarly, CLIP-170 comets were present in the peripheral cell body and concentrated near the bleb base. Quantitative analysis showed that the bulk of MTs resided in the cell body, while growing MT plus ends formed a gradient within the leader bleb with a high concentration near the neck and fewer in the distal bleb region (Figure 3, C and C′).
FIGURE 3:

Distribution of MT and vimentin cytoskeletons in A375M melanoma cells undergoing LBBM under nonadhesive confinement. A375M melanoma cells cultured on fibronectin (10 µg/ml)-coated coverslips (Adhesion, top rows in A–D) or BSA (1 µg/ml)-coated coverslips with confinement by a PDMS pad resting on 3 µm beads to define the confinement height (Low Adhesion + Confinement, bottom two rows in A–D; data analysis in A′–D′ and E–G). (A–D) Spinning-disk confocal images of transiently coexpressed mEmerald (mEm)-tagged fusion protein markers of cytoskeletal structures (green; see also Table 1) together with FusionRed-F-Tractin (red). Images and analysis of low adhesion and confinement are of cells in which the nucleus was localized in the bleb; see Supplemental Figure 3 for images of cells with the nucleus in the cell body. Bottom rows show high-contrast zooms (scale bars = 5 µm) of the regions in the yellow boxes in the second rows (scale bars = 10 µm). (A) mEmerald-gamma-tubulin (gtubulin). (B) Three tandem GFPs fused to the MT-binding domain of ensconsin (GFP-3X-EMTB). (C) mEmerald-CLIP-170. (D) mEmerald-vimentin. (A′) Analysis of the position of the centrosome relative to the nucleus (toward the cell body rear [red] or toward the bleb tip [green]) from images of cells coexpressing mEmerald-gamma-tubulin and FusionRed-F-Tractin for cells in which the nucleus was located in the cell body (cell body), leader bleb (cell bleb), or agnostic to nuclear localization (total); the number of cells analyzed (n) is shown above each category. Schematics of categorization scheme shown in right column. (B′–D′) Average normalized (to maximum intensity [y-axis] and maximum length from the position of the neck [x-axis]) line scan analysis of the fluorescence intensity along the long axis midline of cells undergoing leader bleb–based motility and expressing the corresponding mEmerald-tagged fusion proteins noted in B–D; the number of cells analyzed (n) is shown on each plot. (F–H). Fraction of total cellular fluorescence intensity localized in cell body (red) vs. the leader bleb (green) for cells in which the nucleus was located in the cell body (G), or leader bleb (H), or agnostic to nuclear localization (F); the number of cells analyzed (n) is shown above each category. (H) Schematic representation of organelle distribution in a cell undergoing LBBM in nonadhesive confinement.
Movie S4.
Dynamics of the microtubule and vimentin cytoskeletons during LBBM. Spinning disk confocal time-lapse image sequences of A375M2 cells in non-adhesive confinement expressing either mEmerald fused of three tandem GFPs fused to the microtubule binding domain of ensconsin (GFP-3X-EMTB, left, green), mEmerald-CLIP-170 (center, green) or mEmerald-vimentin (right, green), together with FusionRed F-Tractin (red) while undergoing LBBM in non-adhesive confinement. In all three examples the nucleus is positioned in the leader bleb (seen by exclusion of Fusion Red-F-Tractin). Images were taken every 30s-1min (elapsed time shown in min:sec) on an inverted microscope system (Nikon Ti) using a 60× (1.4 NA, Plan Apo PH) oil immersion objective lens. Scale bar= 10μm.
Movie S5.
Dynamics of membranous organelles during LBBM. Spinning disk confocal time-lapse image sequences of A375M2 cells in non-adhesive confinement expressing expressed mEmerald-tagged fusions of either caveolin (green, marking clathrin-independent endocytosis, upper far left), clathrin heavy chain (green, marking clathrin-dependent endocytosis, upper middle left), Rab5 (green, marking the endocytic recycling compartment, upper middle right), LAMP1 (green, marking lysosomes, upper far right), calreticulin (green, marking the endoplasmic reticulum, lower far left), sialyltransferase-1 (SiT, green, marking the Golgi apparatus, lower middle left), pyruvate dehydrogenase A (PDHA, green, marking mitochondria, lower middle right) or peroxisomal membrane protein 2 (PXMP2, green, marking peroxisomes, lower far right) together with FusionRed F-Tractin (red) while undergoing LBBM in non-adhesive confinement. In all six examples the nucleus is positioned in the leader bleb (seen by exclusion of Fusion Red-F-Tractin). Images were taken every 30sec-1 min (elapsed time shown in min:sec) on an inverted microscope system (Nikon Ti) using a 60× (1.4 NA, Plan Apo PH) oil immersion objective lens. Scale bar= 10μm.
We next examined VIF organization by expression of mEmerald-vimentin (Figure 3, D and D′, Supplemental Movie S5). In adherent cells, VIF formed a network throughout the cell with a concentration in cell protrusions. In contrast, in cells undergoing LBBM, VIF were extremely polarized. Here, they formed a dense network in the cell body and were most often completely absent from the leader bleb, except when the nucleus with a few small associated VIF fragments was in the bleb (Figure 3D and Supplemental Figure 3E), consistent with previous observations (Lavenus et al., 2020). Analysis of the effect of nucleus location in the cell body or leader bleb on the distribution of MT and VIF cytoskeletal markers showed that nuclear position had little effect on overall cytoskeletal localization (Figure 3, E–G; Supplemental Figure 3, A–E). In addition, examination of the organization of MTs (GFP-3X-EMTB) and VIFs (mEmerald-vimentin) in U2OS cells undergoing LBBM showed cells organizations similar to that seen in A375M cells (Supplemental Figure 3F). Together these results show that the MT and VIF cytoskeletons are highly polarized during LBBM, localized primarily in the cell body with the centrosome oriented in the direction of migration (Figure 3H).
Actin regulatory proteins display distinct polarized distributions between the cell body and neck and within the leader bleb
We next sought to probe the role of actin-associated regulatory proteins in LBBM. We overexpressed a selection of fluorescent protein–tagged actin regulatory proteins whose expression in A375M melanoma cells was confirmed from published RNAseq data (Kapushesky et al., 2010) and analyzed their localizations and the effects of their overexpression on cell morphology and migration in nonadhesive confinement (Figure 4).
FIGURE 4:

Distribution of actin-associated regulatory proteins in A375M melanoma cells undergoing LBBM under nonadhesive confinement. A375M melanoma cells cultured on fibronectin (10 µg/ml)-coated coverslips (Adhesion, right three columns in A–C) or BSA (1 µg/ml)-coated coverslips with confinement by a PDMS pad resting on 3 µm beads to define the confinement height (Low Adhesion + Confinement, left three columns in A–C; data analysis in A′–C′′′′). (A–C) Spinning-disk confocal images of transiently coexpressed mEmerald (mEm)-tagged actin regulatory proteins (green; see also Table 1) together with FusionRed-F-Tractin (red). Images and analysis of low adhesion and confinement are of cells in which the nucleus was localized in the bleb; see Supplemental Figure 4 for images of cells with the nucleus in the cell body. Scale bars = 10 µm. (A) Actin assembly-regulating proteins. Top row: mEmerald-ARP3; second row: mEmerald-cortactin; third row: mEmerald-mDia1; fourth row: mEmerald-VASP. (B) Actomyosin contraction-regulating proteins. Top row: mEmerald-non-muscle myosin IIA; second row: mEmerald-non-muscle-myosin IIB; third row: mEmerald-tropomyosin 2; fourth row: mEmerald-tropomyosin 4. (C) Actin cross-linking proteins. Top row: mEmerald-α2-spectrin: second row: mEmerald-α-actinin-1; third row: mEmerald-fascin; fourth row: mEmerald-filamin-A. Scale bars = 10 µm. (A′–C′′′′) Average normalized (to maximum intensity [y-axis] and maximum length from the position of the neck [x-axis]) line scan analysis of the fluorescence intensity along the long axis midline (left column) or periphery (right column) of cells undergoing leader bleb–based motility and expressing the corresponding mEmerald-tagged fusion proteins noted in A–C; the number of cells analyzed (n) is shown on each plot.
We examined the localization of proteins representing different classes of actin regulators, including those involved in filament nucleation and elongation, contractility, and crosslinking (Supplemental movie S6). To characterize the organization of actin filament nucleation and elongation factors, we expressed mEmerald-tagged fusions of Arp3 as a marker of the Arp2/3 complex, the Arp2/3 recruiter cortactin, the barbed-end elongation factor VASP, and the formin family nucleation and elongation factor mDia1 (Figure 4A). In adherent cells, Arp3, cortactin, and mDia1 were soluble with some concentration in protrusions, while VASP was localized to protrusions and focal adhesions. In leader bleb cells in nonadhesive confinement, all four proteins showed strong soluble pools with some enrichment toward the bleb tip (Figure 4, A′–A′′′′; Supplemental Figure 4). This result indicates that actin assembly-promoting factors may be polarized to the bleb tip, where they could contribute to actin assembly to power retrograde flow in the leader bleb.
Movie S6.
Dynamics of actin regulatory proteins during LBBM. Spinning disk confocal time-lapse image sequences of A375M2 cells in non-adhesive confinement expressing expressed mEmerald-tagged fusions of either Arp3 (green, marking the Arp2/3 complex, top row far left), cortactin (green, top row middle left), mDia1 (green, top row middle right), VASP (green, top row far right), nonmuscle myosins (NM) IIA (green, middle row far left) and IIB (green, middle row, middle left), tropomyosin 2 (TM2, green, middle row middle right), tropomyosin 4 (TM4, green, middle row far right), aII (A2) spectrin (green, bottom row far left) ?-actinin1 (A-Actinin-1, green, bottom rown middle left), fascin1 (green, bottom row middle right) or filamin A (green, bottom row far right) together with FusionRed F-Tractin (red) while undergoing LBBM in non-adhesive confinement. In all twelve examples the nucleus is positioned in the leader bleb (seen by exclusion of Fusion Red-F-Tractin). Images were taken every 30sec-1min (elapsed time shown in min:sec) on an inverted microscope system (Nikon Ti) using a 60× (1.4 NA, Plan Apo PH) oil immersion objective lens. Scale bar= 10μm.
We next examined the localization of proteins involved in generating and regulating actomyosin contractility, including myosins IIA and IIB, the high-molecular-weight tropomyosin 2 (also called tropomyosin 1.6), and the low-molecular-weight tropomyosin 4 (also called tropomyosin 4.2) (Figure 4B). mEmerald fusions expressed in adherent cells showed that these proteins all localized to stress fibers, as expected. In leader bleb cells under nonadhesive confinement, myosins IIA and IIB showed strong enrichment in the cell body cortex and very low levels in the leader bleb, with IIA more concentrated in the neck than IIB (Figure 4, B′ and B′′). Both tropomyosins also concentrated in the cell body cortex, with tropomyosin 4 additionally concentrating in the bleb neck and base and on actin bundles near the bleb tip (Figure 4, B′′′ and B′′′′). Thus, proteins involved in contractility all concentrate in the cell body cortex, suggesting an important role here, possibly in generating cortical tension to maintain pressure into the leader bleb. In addition, these proteins show distinct subcellular distributions, suggesting specific roles for myosin IIA in maintaining the contractile neck and tropomyosin 4 in regulating actin filaments near the bleb tip.
To determine the distribution of actin filament cross-linking, we expressed mEmerald-tagged fusions of the large flexible cortical cross-linkers αII spectrin and filamin-A, the midlength cross-linker α-actinin-1, and the short filopodial cross-linker fascin-1 (Figure 4C). In adherent cells, all four cross-linkers were distributed throughout the cortex, with α-actinin-1 and filamin-A additionally concentrated on stress fibers and fascin-1 additionally concentrated on filopodia. In leader bleb cells under nonadhesive confinement, while all four cross-linking proteins localized to the cell body cortex, they additionally exhibited quite distinct localizations in the leader bleb. The flexible cross-linker αII spectrin was additionally concentrated in the neck and largely absent from the leader bleb (Figure 4C′), while filamin-A and α-actinin-1 localized to actin in the leader bleb, forming strong gradients with the highest concentration toward the neck (Figure 4, C′′′ and C′′′′). In contrast, fascin-1 tended to concentrate on actin bundles near the leader bleb tip, similar to the localization of tropomyosin 4. Expression of mEmerald fusions of filamin-A or fascin-1 in U2OS cells undergoing LBBM showed localizations similar to those seen in A375M cells (Supplemental Figure 3). These results suggest that actin filament cross-linking may play a role in maintaining the cell body and suggest specific roles for different types of cross-linkers in mediating the polarized filament organization and dynamics in the neck and leader bleb.
Overexpression screen indicates a critical role of actin cross-linking proteins in LBBM
To determine the role of actin regulatory proteins in mediating leader bleb formation and morphology, we analyzed the effects of overexpressing these proteins (Supplemental Figure 5) on morphology parameters in A375M cells in nonadhesive confinement (Figure 5, A and B). Analysis of the length and width of the cell body and leader bleb showed that overexpressing mEmerald-tagged actin nucleation/elongation regulators did not affect cell body morphology compared with mEmerald-control, although overexpression of mEmerald-mDia1 significantly reduced leader bleb length (Figure 5A). In addition, overexpression of contractile regulators had no significant effect on morphology metrics, except tropomyosin 4, which also reduced leader bleb length (Figure 5, A and B). In contrast, overexpression of α-actinin-1 and filamin-A both caused striking increases in leader bleb length and width (Figure 5, A and B), while overexpression of αII spectrin and fascin-1 had no effect on cell morphometrics. These results confirm the critical role of α-actinin-1–mediated filament cross-linking in promoting leader bleb formation (Logue et al., 2015) and additionally implicate filamin-A, another filament cross-linker, in this process. They further suggest that mDia1 and tropomyosin 4 could negatively regulate leader bleb elongation.
FIGURE 5:

Analysis of the effects of the overexpression of actin regulatory proteins on leader bleb formation and cell migration. A375M melanoma cells cultured on BSA (1 µg/ml)-treated coverslips with confinement by a PDMS pad resting on 3 µm beads to define the confinement height and induce leader bleb–based motility. Cells were transiently coexpressing various mEmerald (mEm)-tagged actin regulatory proteins or GFP control together with FusionRed-F-Tractin. Analysis (A–I) presented for cells in which the nucleus was localized in the bleb; see Supplemental Figure 5 for analysis of cells with the nucleus in the cell body. (A–I) Fusion proteins expressed are color coded: GFP control (white); actin assembly-regulating proteins (red, mEmerald-ARP3, mEmerald-cortactin, mEmerald-mDia1, mEmerald-VASP); actomyosin contraction-regulating proteins (green, mEmerald-non-muscle myosin IIA, mEmerald non-muscle-myosin IIB, mEmerald-tropomyosin 2, mEmerald-tropomyosin 4); actin cross-linking proteins (blue, mEmerald-α2-spectrin, mEmerald-α-actinin-1, mEmerald-fascin-1, mEmerald-filamin-A. Average length (A) and width (B) of leader bleb and cell motility speed (E); n (cells) is shown below categories. Error is SEM. Statistical significance was determined by one-way ANOVA. **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001, NS not significant. (C) Rose plots of representative migration tracks (8 h, 10 min intervals) of cells overexpressing the noted fusion protein and undergoing LBBM; the number of cells is noted on each plot. (D) Fraction of cells undergoing leader bleb–based motility with the nucleus localized in the cell body vs. the bleb. The number of cells analyzed (n) is shown below each category. Average mean squared displacement over time for cells undergoing LBBM and overexpressing (F) aII spectrin (G) a-actinin (H) fascin-1 or (I) filamin-A; n (cells) = 29 (a-2-spectrin), 42 (α-actinin-1, 34 (fascin-1), and 37 (filamin-A) cells.
We next determined how actin regulatory proteins contribute to parameters of LBBM in nonadhesive confinement. We analyzed the speed and MSD over time from time-lapse movies of cells overexpressing fluorescently labeled actin regulatory proteins (Figure 5C; Supplemental Figure 5, A–C). This showed that overexpression of neither actin nucleation/elongation factors nor contractility regulators had any significant effect on LBBM. In contrast, overexpression of either fascin-1 or filamin-A caused significantly faster, more directionally persistent migration, α-actinin-1 overexpression increased speed but had little effect on persistence, while excess αII spectrin strongly reduced directional persistence with no effect on speed (Figure 5, G–I). These effects were independent of nuclear position (Figure 5D; Supplemental Figure 5D). Collectively, these results show that high levels of specific actin cross-linking proteins induce larger leader blebs and/or promote rapid, directional LBBM, suggesting that α-actinin-1, αII spectrin, fascin-1, and filamin-A could be critical to leader bleb formation and migration in nonadhesive confinement.
Actin cross-linking proteins promote LBBM
We next sought to determine the requirement of the actin cross-linking proteins filamin-A and fascin-1 in leader bleb formation and LBBM (Figure 6A ; Supplemental Figure 6, A and B). Superresolution imaging of immunolocalized endogenous filamin-A and fascin-1 showed localizations similar to those of expressed fluorescently labeled fusion proteins in adherent spread cells and nonadherent confined leader bleb cells, with both proteins concentrated in the cell body cortex and, additionally, filamin-A in the leader bleb base and fascin-1 toward the leader bleb tip (Figure 6C). To test the role of filamin-A and fascin-1 in LBBM, we suppressed their expression in A375M cells by siRNA and assayed their morphology and motility behavior under nonadhesive confinement. Western blot analysis indicated suppression of protein levels, which could be partially restored by re-expression of fluorescently tagged fusion proteins (Figure 6B). Analysis of cells in nonadhesive confinement showed that suppression of filamin-A strongly reduced the percentage of cells exhibiting leader bleb morphology and strongly reduced the size of the largest cell bleb (Figure 6, D–F). In contrast, suppression of fascin-1 had no effect on the percentage of cells forming leader blebs and actually increased the size of the largest cell bleb (Figure 6, D–F; Supplemental Figure 6, C and D). Importantly, re-expression of mEmerald-tagged fusion proteins rescued the effects of fascin-1 depletion, while filamin-A was partially rescued with mEmerald-tagged protein. Our data suggest opposing roles of the actin cross-linking proteins, with filamin-A promoting leader bleb formation and size and fascin-1 limiting leader bleb size.
FIGURE 6:

Effects of filamin-A and fascin-1 knockdown on cell morphometrics, mechanics, and migration in nonadhesive confinement. (A–L) A375M cells were treated with nontargeting siRNAs (siCtrl) or siRNA targeting human fascin-1 (si fascin-1) or filamin-A (si filamin-A) with or without the additional expression of mEmerald-fascin-1 (si fascin-1+rescue) or filamin-A (si fascin-1+rescue). (A) Western blot analysis of cell lysates. Blots were probed with antibodies specific to fascin-1, filamin-A, GFP, or GAPDH. (B) Quantitative analysis of relative protein level from Western blots normalized to GAPDH and compared with siControl, measured from three independent experiments. (C) Immunofluorescence of endogenous fascin-1 (top) or filamin-A (bottom) (green) together with phalloidin staining of F-actin (red) and DAPI staining of DNA (blue) in cells under nonadhesive confinement. Scale bars = 10 µm. (D) Percentage of cells exhibiting leader bleb morphology (n = minimum of 50 cells per condition from N = 3 experiments). (E, F, J–L) A375M melanoma cells cultured on BSA (1 µg/ml)-treated coverslips with confinement by a PDMS pad resting on 3 mm beads to define the confinement height and induce leader bleb–based motility. (E, F) Average length (E) and width (F) of leader bleb. (G–I). Schematic representation of the AFM-based assay used for determining cell cortex tension and intracellular pressure in A375M cells plated on nonadhesive or uncoated glass. kc, cantilever spring constant; d, cantilever deflection; Z, piezo Z displacement (G). Tukey box plots of cortex tension (H) and intracellular hydrostatic pressure (I), where the + and line denote the mean and median, respectively. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001, NS not significant p ≤ 0.05. (J). Rose plots of representative migration tracks (4 h, 10 min intervals) of cells undergoing LBBM; the number of cells is noted on each plot. (K) Average speed of cells undergoing LBBM. (L) Average mean squared displacement over time; n (cells) = 58 (siControl [nontargeting], 76 (sifascin-1), 38 (sifascin1+Rescue), and 51 (sifilamin-A) and 51 (sifilamin-A+Rescue). All data are representatives of at least three independent experiments. Error is SEM. The statistical significance was determined by two-tailed Student’s t tests, one-way ANOVA, and/or Mann–Whitney. **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001, NS not significant.
FIGURE 7:

Role of filamin-A and fascin-1 in leader bleb formation and migration in nonadhesive confinement. Model for actin filament cross-linking proteins; fascin-1 and filamin-A effects on leader bleb formation, mechanics and LBBM. Leader bleb cell showing reduction and rescue of fascin-1 (top arrows) and the effects on cell morphology, cortical tension, intracellular pressure, and cell migration speed and directionality in melanoma A375M cells. Excess fascin-1 shows no change in bleb size, while the reduction of fascin-1 results in an increase in leader bleb size and decreased motility. Leader bleb cell showing reduction and rescue of filamin-A (bottom arrows) and the effects on cell morphology, cortical tension, intracellular pressure, and cell migration speed and directionality in melanoma A375M2 cells. Excess filamin-A results in an increase in bleb size due to its linkage to cortex and contractility of actomyosin. Reduction of filamin-A shows an increase in overall blebs but no production of leader bleb and decrease in cell speed and directionality with partial rescue, due to lack of pressure production.
We next sought to determine whether filamin-A and fascin-1 contribute to leader bleb morphology through regulation of cell cortical mechanics. To test this, we used atomic force microscopy (AFM) coupled with confocal imaging on nonadherent A375M cells subjected to minimal deformation with a tipless cantilever (Logue et al., 2015, 2018) (Figure 6G). We extracted cortical tension and intracellular pressure from the resulting force-displacement curves and measurements of the cell radius (see Materials and Methods). Importantly, expression of neither GFP, FusionRed-F-Tractin, nor control siRNA caused any difference in cell radius, cortical tension, or intracellular pressure compared with untreated A375M cells (Supplemental Figure 6, E–G). While siRNA depletion of either filamin-A or fascin-1 did not induce changes in cell radius, it did cause significant decreases in both cortical tension and intracellular pressure (Figure 6, H and I; Supplemental Figure 6E). The effects of filamin-A or fascin-1 depletion on cell mechanical properties were rescued by reexpression of the respective mEmerald-fusion proteins (Figure 6, H and I; Supplemental Figure 6, E–G). Thus, filamin-A and fascin-1 promote cortical tension and intracellular pressure.
To determine the relative contributions of filamin-A or fascin-1 to LBBM, we examined the effect of their depletion on motility parameters from time-lapse movies of cells in nonadhesive confinement (Supplemental Movie S7). Speed and MSD over time analysis showed that depletion of filamin-A or fascin-1 resulted in significant decreases in cell speed and directionality (Figure 6, J–L). Expression of mEmerald-fascin-1 in fascin-1–depleted cells rescued the motility defects, while expression of filamin-A restored some motility but did not result in a complete rescue, likely due to the low level of fusion protein expression compared with the endogenous filamin-A level (Figure 6, J and K).
Movie S7.
Effects of filamin-A or Fascin1 depletion on migration dynamics of A375M2 in non-adhesive confinement. Cells were plated on a coverslip coated with integrin ligand (10 μg/ml Fibronectin (FN), “Adhesion,” upper right) or non-adhesive bovine serum albumin (BSA, 1 μg/ml, “Low Adhesion,” upper left), or were placed in confinement between a PDMS pad and a FN-coated (“Adhesion + Confinement,” lower right), or BSA-coated (“Low Adhesion + Confinement,” lower left) coverslip held apart by 3 μm beads as spacers Beads can be seen as refractile points visible in lower panels. Colored lines highlight the position of the cell over time. Phase contrast time-lapse image sequences taken every 10 s (elapsed time shown in min:sec) on an inverted microscope system (Nikon Ti) using a 20× (0.75 NA, Plan Apo PH) objective lens and 0.9 NA condenser. Scale bar= 100 μm.
Collectively, these results show that the cross-linking proteins filamin-A and fascin-1 both contribute to cortical mechanical properties and LBBM: Excess of these cross-linkers enhances LBBM and mechanics, while reduced levels inhibit these properties. Filamin-A, which localizes to the cell body cortex and base of the leader bleb, contributes to leader bleb formation, with overexpression increasing bleb size and diminished levels blocking bleb formation. In contrast, fascin-1, which concentrates in the cell body cortex and leader bleb tip, does not regulate bleb formation and may actually limit leader bleb size, yet is vital to LBBM. Thus, our results uncover critical roles for distinct actin cross-linkers in specific aspects of cell functions that contribute to LBBM in nonadhesive confinement.
DISCUSSION
We surveyed the localization and organization of organelles and actin regulatory proteins in human melanoma and osteosarcoma cells in nonadhesive confinement to provide a map of subcellular anatomy of cancer cells undergoing LBBM. We found that membranous organelles, including components of the endolysosomal, secretory, and metabolic pathways, exhibit a highly polarized distribution, predominantly concentrated in the cell body, with a minor fraction in the leader bleb. Similarly, the bulk of the vimentin (Lavenus et al., 2020) and MT cytoskeletons concentrate in the cell body. These results suggest that the cell body compartment maintains most of the general cellular functions of metabolism and protein processing and turnover, while the leader bleb compartment may be highly specialized exclusively for motility. As some mitochondria, cisternal ER, and growing MT plus ends also were found to extend into the leader bleb, this further implies that these organelles somehow support the motile function of this compartment. Mitochondria likely supply ATP for the massive actin treadmilling and myosin contractile activities, while the unusual large ER cisternae could be specialized for regulation of calcium within the leader bleb (Aoki et al., 2021). Growing MTs may help to maintain leader bleb polarity, as they do in adhesion-dependent, mesenchymal cell migration (Rodriguez et al., 2003). In support of the similar roles of the MT cytoskeleton in mesenchymal migration and LBBM, we also found that the MTOC positioned forward of the nucleus during LBBM as it does in adhesion-dependent migration (Lauffenburger and Horwitz, 1996; Nabi, 1999), suggesting a conserved role of the MTOC in maintaining polarity in different modes of migration. Additionally, our discovery that nuclear position in the leader bleb correlated with rapid directional migration highlights a critical role of the nucleus in LBBM, as has been shown for adhesion-dependent migration in three-dimensional microenvironments (Graham et al., 2018). We suggest that the large, incompressible nucleus could promote friction between cortically attached transmembrane proteins undergoing retrograde flow in the leader bleb and the tightly confined nonadhesive cellular environment (Liu et al., 2015) to facilitate LBBM.
Our survey of actin regulatory protein localization and the effects of their overexpression showed that filament nucleators and elongation factors had a high soluble fraction and appeared to concentrate toward the distal leader bleb tip where they likely contribute to actin assembly to power retrograde flow. The localization of myosin II isoforms and tropomyosins in the cell body cortex indicates an important role for contractility in this compartment, likely in maintaining cortical tension to generate intracellular pressure (Charras and Paluch, 2008; Paluch and Raz, 2013; Svitkina, 2018) to drive the formation and maintenance of the leader bleb. The concentration of these contractile components in the neck likely serves to focus their activity to narrow this region, similar to a cytokinetic furrow, and thereby preserve compartmentalization between the body and bleb (Logue et al., 2015). The distinct distribution of myosin II isoforms, with IIB exclusively in the body and neck and IIA additionally forming a gradient in the leader bleb, is reminiscent of the polarization of these isoforms in adhesion-dependent migration (Vicente-Manzanares et al., 2009; Beach et al., 2017), again highlighting conserved roles of proteins in different modes of migration.
While previous work revealed a critical role for actin cross-linking by Eps8 and α-actinin in LBBM (Logue et al., 2015), our current study extends this and suggests that different filament cross-linkers with distinct properties and localizations may cooperate to mediate the specific cortical actin architecture required for LBBM. Our studies showed that Eps8 (Logue et al., 2015), the flexible cross-linkers filamin-A and αII spectrin, and the short bundlers α-actinin-1 and fascin-1 all localize to the cell body cortex, where they likely contribute to the actin meshwork integrity required for maintaining high cortical tension (Clark et al., 2013; Logue et al., 2015; Koenderink and Paluch, 2018). In addition to their common localization in the cell body, these cross-linkers exhibited distinct localizations in the leader bleb. The αII spectrin, filamin-A, and α-actinin-1 were all concentrated in the neck and at the base of the leader bleb, similar to myosin IIA, suggesting a role in supporting a contractile architecture of actin or assisting in segregating membranous organelles from the leader bleb (Ghisleni et al., 2020; Krueger et al., 2020). In contrast, the short cross-linker fascin-1 was co-localized with tropomyosin 4 on actin bundles in the leader bleb tip, suggesting a possible role in maintaining tight bundles to sterically inhibit Arp2/3-mediated branching (Bugyi et al., 2010; Hsiao et al., 2015; Brayford et al., 2016) and thereby preserve bundle architecture, perhaps to girdle the distal bleb and maintain the sausage-like shape. The distinct spatial distribution of actin cross-linkers could be regulated by localized signaling or by network architecture-mediated sorting, as has been observed in vitro (Winkelman et al., 2016).
The results of our protein overexpression and depletion experiments highlight the critical importance of distinct types of actin filament cross-linkers in mediation of LBBM (Figure 7). Our studies show that α-actinin-1, Eps8 (Logue et al., 2015), filamin-A, and fascin-1 (this study) all promote cortical tension and intracellular pressure, suggesting a general role for cross-linking in the mediation of these basic physical requirements for bleb formation. Despite their common roles in cell mechanics, our results suggest that these proteins contribute to distinct aspects of LBBM, which may be mediated by their distinct localizations. α-Actinin-1, Eps8 (Logue et al., 2015), and filamin-A all localize to the cell body cortex and the bleb base and neck, and the overexpression of either α-actinin-1 or filamin-A increases leader bleb size and promotes migration, while depletion of either filamin-A or Eps8 inhibits leader bleb formation. In contrast, fascin-1 localizes primarily toward the leader bleb tip, and its overexpression has no effect on leader bleb morphology but still enhances motility, while its depletion enhances bleb size yet inhibits motility. This highlights the critical role of cross-linking in the cell body, neck, and leader bleb base in promoting bleb formation and size and bundling at the bleb tip perhaps in regulating bleb length for optimal motility. Because LBBM speed is likely dependent on the rate of actin filament assembly at the bleb tip, retrograde flow, and disassembly at the bleb base, it is possible that cross-linkers in the contractile zone at the base may cooperate with cofilin-family filament severing proteins to enhance disassembly (Breitsprecher et al., 2011; Ullo and Logue, 2018; Wioland et al., 2019) and thus accelerate filament treadmilling to drive faster migration, or enhance retrograde flow by promoting actomyosin contractility (Kelley et al., 2020) at the bleb base. The increase in bleb size caused by fascin-1 depletion also suggests a possible role for the tight, parallel actin bundles near the bleb tip in limiting bleb size. These results together highlight the importance of specific spatial regulation of actin architecture, assembly, disassembly, and motion in the mediation of LBBM in nonadhesive confinement.
Melanoma and osteosarcoma spread from the skin and bones by entering the bloodstream intravenously or lymphatically, traversing and gaining access to diverse distant tissues (Wong and Hynes, 2006). This spread is mediated by cell migration through the dermis, or connective tissue, within the perivascular space and between smooth and/or skeletal muscle fibers, and through the endothelium into the circulation (Friedl and Wolf, 2010; Charras and Sahai, 2014). This diverse array of highly confining tissue microenvironments requires that cancer cells adapt to each by switching between migration modes to mediate the metastatic journey (Yamada and Sixt, 2019). Our experimental system of tight confinement of cancer cells between nonadhesive surfaces is designed to simulate one possible confining microenvironment that lacks ligands for cancer cell surface adhesion receptors. In such an environment in vitro, cultured and highly passaged cancer cells take on the highly polarized LBBM morphology that has also been observed for melanoma cells migrating in the dermis of living mice (Tozluogˇlu et al., 2013; Charras and Sahai, 2014). Our discovery here of the exceptional degree of organelle polarization during LBBM, which occurs in our in vitro system in the absence of gradients of extracellular cues, underscores the self-organization of this extreme cell asymmetry. Understanding the molecular mechanism of LBBM polarization will be an important focus in the future to allow development of anti-metastatic therapeutics.
MATERIALS AND METHODS
Cell culture and transfection
A375M and U2OS cells were obtained from the American Type Culture Collection (cat # CRL-1619; ATCC, Manassas, VA) and maintained for up to 15 passages in DMEM supplemented with 10% fetal bovine serum (FBS, cat # 508-540-7100 A3160401; Life Technologies, Carlsbad, CA), GlutaMAX (Life Technologies), antibiotic–antimycotic (Life Technologies), and 20 mM HEPES, pH 7.4. A Nucleofector 2b device (Kit V; Lonza, Basel, Switzerland) was used to transfect plasmids. Cells were plated on six-well glass-bottom plates (Cellvis, Mountain View, CA) either directly or after coating with 10 µg/ml human plasma fibronectin (cat # FC010; Millipore, Billerica, MA) or 10 µg/ml BSA (cat # A3059), as noted in each figure.
Nonadhesive confinement under PDMS
Cells were confined between a slab of PDMS and a #1.5 coverslip (Corning, Corning, NY), which were held a defined distance apart by polystyrene beads. PDMS (Dow Corning 184 SYLGARD) was purchased from Krayden (Westminster, CO). Two milliliters was cured overnight at 37°C in each well of a six- or 12-well glass-bottom plate (Cellvis). Using a hole punch (cat # 504535 World Precision Instruments, Sarasota, FL), an 8-mm hole was cut in the slab, and 3 ml of serum-free medium containing 1% BSA was added to each well and incubated overnight at 37°C. After aspirating away the serum-free media containing 1% BSA, 200 µl of complete media containing trypsinized cells (250,000 to 1 million) and 0.5 µl of beads (3.11 µm; Bangs Laboratories, Fishers, IN) was then pipetted into the hole in the PDMS. The vacuum created by briefly lifting one side of the opening with a 200 µl pipette tip was used to move cells and beads underneath the PDMS. Finally, 1–3 ml of complete media was added to each well and cells recovered in a tissue culture incubator for 30–60 min before imaging.
Plasmids
Most plasmids were a kind gift from Michael Davidson (Florida State University) and are available from Addgene, as noted.
FusionRed-F-Tractin was the gift of Michael Schell (Uniformed Services University of the Health Sciences) (Schell et al., 2001).
pEGFP-N1 (Addgene plasmid #13031; http://n2t.net/addgene:13031; RRID:Addgene 13031).
mEmerald-Calnexin-N-14 (Addgene plasmid #54021; https://n2t.net/addgene.54021/RRID:Addgene 54021).
pmEmerald-MyosinIIA-C-18 (Addgene plasmid #54190; http://n2t.net/addgene:54190; RRID:Addgene 54190).
pmEmerald-Paxillin-22 (Addgene plasmid #54219; http://n2t.net/addgene:54219; RRID:Addgene 54219).
pmEmerald-H2B-C-10 (Addgene plasmid #54112; http://n2t.net/addgene:54112; RRID:Addgene 54112).
EGFP-CAAX (Addgene plasmid #86056; http://n2t.net/addgene:86056; RRID:Addgene 86056).
pmEmerald-Clathrin-15 (Addgene plasmid #54040; http://n2t.net/addgene:54040; RRID:Addgene 54040).
pmEmerald-Caveolin-C-10 (Addgene plasmid #54025; http://n2t.net/addgene:54025; RRID:Addgene 54025).
pmEmerald-Rab5a-7 (Addgene plasmid #54243; http://n2t.net/addgene:54243; RRID:Addgene 54243).
pmEmerald-Lysosomes-20 (Addgene plasmid #54149; http://n2t.net/addgene:54149; RRID:Addgene 54149).
pmEmerald-Calreticulin-C-10 (Addgene plasmid #54022; http://n2t.net/addgene:54022; RRID:Addgene 54022).
pmEmerald-SiT (Addgene plasmid #54255; http://n2t.net/addgene:54255; RRID:Addgene 54255).
pmEmerald-PDHA (Addgene plasmid #54224; http://n2t.net/addgene:54224; RRID:Addgene 54224).
pmEmerald-PMP-C-10 (Addgene plasmid #54235; http://n2t.net/addgene:54235; RRID:Addgene 54235).
pmEmerald-Gamma-Tubulin (Addgene plasmid #54105; http://n2t.net/addgene:54105; RRID:Addgene 54105).
3x-eGFP-Ensconsin (Addgene plasmid #26742; http://www.addgene.org/26742/; RRID:Addgene 26742).
pmEmerald-CLIP170-C-18 (Addgene plasmid #54043; http://n2t.net/addgene:54043; RRID:Addgene 54043).
pmEmerald-Vimentin-C-18 (Addgene plasmid #54051; http://n2t.net/addgene:54051; RRID:Addgene 54051).
pmEmerald-ARP3-N-12 (Addgene plasmid #53995; http://n2t.net/addgene:53995; RRID:Addgene 53995).
pmEmerald-Cortactin-C-12 (Addgene plasmid #54051; http://n2t.net/addgene:54051; RRID:Addgene 54051).
pmEmerald-mDia1-C-14 (Addgene plasmid #54156; http://n2t.net/addgene:54156; RRID:Addgene 54156).
pmEmerald-VASP-5 (Addgene plasmid #54296; http://n2t.net/addgene:54296; RRID:Addgene 54296).
pmEmerald-MyosinIIB-C-18 (Addgene plasmid #54192; http://n2t.net/addgene:54192; RRID:Addgene 54192).
pmEmerald-alpha-Actinin-C-14 (Addgene plasmid #53989; http://n2t.net/addgene:53989; RRID:Addgene 53989).
pmEmerald-Fascin-C-10 (Addgene plasmid #54094; http://n2t.net/addgene:54094; RRID:Addgene 54094).
pmEmerald-FilaminA-N-9 (Addgene plasmid #54098; http://n2t.net/addgene:54098; RRID:Addgene 54098).
Microscopy
Immunofluorescence or time-lapse live cell imaging was performed using a spinning-disk confocal digital microscope system as described previously in Shin et al. (2010) and Logue et al. (2015, 2018). Briefly, an automated Eclipse Ti microscope equipped with the Perfect Focus System (Nikon, Japan), a servomotor-driven X-Y stage with a piezo top plate (Applied Scientific Instruments, Eugene, OR), and a CSU-X1-A3 spinning-disk confocal scan head (Yokogawa, Japan) was used. Illumination was provided by a quartz-halogen lamp using a 546 nm bandpass filter for phase-contrast images. Images were collected with either a CoolSNAP HQ2 or MYO cooled CCD camera (Photometrics, Tucson, AZ) using a 20× (0.75 NA, Plan Apo PH) objective lens and 0.9 NA condensor. TIRF imaging illumination was provided by solid state lasers (100 mW, 488 nm for mEmerald; 500 mW, 561 nm for FusionRed) directed to the microscope by a custom-designed optical fiber-coupled laser combiner (Spectral Applied Research, Canada [ Shin et al., 2010]) with fiber-coupled output to either the confocal scanhead or an automated multiport epi-illuminator (Nikon). Images were also acquired using either a 40× or a 60× (1.4 NA, Plan Apo PH) oil immersion objective lens. Prior to TIRF imaging of mEmerald-paxillin and FusionRed-F-Tractin or immunostaining of paxillin, F-actin, and DNA with 4′,6-diamidino-2-phenylindole (DAPI), a confocal image through the bottom plane of the cell was first acquired using the 488 and 561 nm lasers in the red and green channels to image lack of stress fibers and focal adhesions in confined cells. Illumination, image acquisition, and microscope functions were controlled by Metamorph software (Molecular Devices, Sunnyvale, CA). For all experiments, a stage-top incubator (Tokai-Hit, Japan) was used to maintain samples at 37°C. Superresolution imaging was performed using a Zeiss 880 Airyscan confocal microscope system. Confocal Z-stacks at 0.21 mm step size were taken using the 488 and 561 nm lasers in the green (mEmerald-fascin-1 or filamin-A) and red (FusionRed-F-Tractin) channels to image structures in confined cells.
AFM
A375M melanoma cells were plated on a glass-bottom dish (Willco Wells) with culture media solution (Life Technologies). Cells were briefly incubated for ∼10 min to let them weakly adhere to the glass-bottom dish surface. AFM force spectroscopy experiments were performed using a Bruker BioScope Catalyst AFM system mounted on an inverted Axiovert 200M microscope (Zeiss) equipped with a confocal laser scanning microscope 510 Meta (Zeiss) and a 40× objective lens (0.95 NA, Plan-Apochromat; Zeiss). The combined microscope instrument was placed on an acoustic isolation table (Kinetic Systems). During AFM experiments, cells were maintained at the physiologically relevant temperature 37°C using a heated stage (Bruker). A soft silicon nitride tipless AFM probe (HQ:CSC38/tipless/Cr-Au; MikroMasch) was used for nonadherent A375M cell gentle compression. The AFM microcantilevers were precalibrated using the standard thermal noise fluctuations method, with estimated spring constants for microcantilevers used between 0.06 and 0.08 N/m. Immediately after calibration, the AFM tipless probe was moved on top of a rounded A375M cell. Five successive force curves were performed on each A375M cell. The deflection set point was set to 30 nm, yielding applied forces between 1.8 and 2.4 nN.
All AFM force-distance curve measurements were analyzed using a custom-written Matlab (The MathWorks) code to calculate the cellular actomyosin cortex tension and intracellular hydrostatic pressure. For curve fitting, indentation depths between 0 and 500 nm were relatively consistent in yielding good fits (R2 > 0.9). Curves with poor fits, R2 < 0.9, were discarded from the analysis. Additionally, we discarded noisy force curves and/or curves that presented jumps possibly due to cantilever and plasma membrane adhesion, slippage, or very weakly adherent moving cells.
The nonadherent A375M cell cellular actomyosin cortex tension (T; pN/µm) was calculated by fitting each recorded force-distance curve using the cortical tension formulas described in Cartagena-Rivera et al. (2016) that defines the force balance relating the applied cantilever force with the pressure excess inside the rounded cells and the corresponding actin cortex tension; 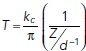 , where T is the cellular surface tension, kc is the AFM cantilever spring constant, Z is the Z-piezo extension, and d is the cantilever mean deflection. Additionally, the intracellular hydrostatic pressure (P; Pa) was calculated by using the Laplace’s law for spheres;
, where T is the cellular surface tension, kc is the AFM cantilever spring constant, Z is the Z-piezo extension, and d is the cantilever mean deflection. Additionally, the intracellular hydrostatic pressure (P; Pa) was calculated by using the Laplace’s law for spheres;  , where P is the intracellular hydrostatic pressure and R is the initial radius of the nonadherent cell.
, where P is the intracellular hydrostatic pressure and R is the initial radius of the nonadherent cell.
Immunofluorescence
Adherent cells or cells confined between a BSA-coated coverslip and an agarose pad (prepared similarly to that with PDMS) were fixed using 4% paraformaldehyde in tris-buffered saline (TBS, 20 mM Tris 150 mM NaCl pH 7.4) with 0.1% Tween 20 (TBS-T) for 20 min at 20 min at room temperature (RT) before blocking with TBS containing 1% BSA and 0.1% Triton X-100 for 1 h at RT. Antibodies for staining were used at 1:200 (paxillin, filamin, and fascin), respectively, in blocking buffer for 2 h at RT. After washing with TBS, Alexa Fluor 568–conjugated secondaries (Life Technologies) were used at 1:400 in blocking buffer for 2 h at RT. Alexa Fluor 647–conjugated phalloidin (Life Technologies) was used at 1:500 in blocking buffer for 2 h at RT.
Image analysis
Cell migration speed, MSD, and rose plot analysis.
Cell migration analysis was performed on time-lapse phase-contrast image series (10 h at 10 min intervals). Velocity, MSD over time. and rose plot analyses were performed using a Microsoft Excel plug-in, DiPer (Gorelik and Gautreau, 2014), and the Fiji plug-in, MTrackJ (Meijering et al., 2012). Images were checked to confirm that beads were not obstructing the path of a cell.
Percent of cells with leader bleb morphology.
The percentage of cell with leader bleb morphology was determined from time-lapse series (10 h at 10 min intervals) of either spinning-disk confocal images of FusionRed-F-Tractin or phase-contrast images. Cells were scored using the Fiji plug-in, Cell counter https://imagej.nih.gov/ij/plugins/cell-counter.html. From every frame, the total number of cells with leader bleb morphology, displaying a cell body, thin neck, and single large leader bleb simultaneously, and the total number of cells in the frame were recorded in Microsoft Excel (Redmond, WA). Cells were counted as having leader bleb morphology if they displayed it for more than three frames (>30 min).
Cell body and leader bleb morphometrics.
Cell body and leader bleb length and width were measured for cells under nonadhesive confinement from time-lapse series (10 min at 30 s intervals) of spinning-disk confocal images of either the expressed mEmerald fusion protein or FusionRed-F-Tractin. The multi-line tool from Metamorph software (Molecular Devices, Sunnyvale, CA) was used. For length, a line was drawn from the midpoint of the neck to the rear of the cell body or to the tip of the leader bleb. To measure width, a line was drawn perpendicular to the length measurement at the widest point of the cell body or the leader bleb. For each cell, the average length and width from at least three time points in the time series were calculated in Microsoft Excel. Phase-contrast images were used to confirm that beads were not obstructing bleb formation.
Determination of nuclear localization in cell body or leader bleb.
Nuclear position in cells under nonadhesive confinement was measured from spinning-disk confocal time-lapse images (10 min at 30 s or 1 min intervals) of FusionRed-F-Tractin, which allowed localization of the nucleus due to its exclusion of fluorescence. Only cells with leader bleb morphology were considered in the analysis. Using the Fiji plug-in, Cell counter https://imagej.nih.gov/ij/plugins/cell-counter.html, the total number of cells with the nucleus in the cell body, leader bleb, or between compartments in the neck in the frame were recorded in Microsoft Excel (Redmond, WA). In a time-lapse movie, the nuclear position for each cell was determined by where the nucleus resided for the duration of the movie. If it remained between compartments or transited between compartments during the movie it was scored as between compartments. All statistical analyses were performed in GraphPad Prism (La Jolla, CA).
Fluorescence intensity line scan analysis.
Average plots of intensity of mEmerald- or FusionRed-fusion protein along the cortex and the center of leader bleb cells under nonadhesive confinement were made from spinning-disk confocal images taken at the central cell plane. Using the multi-line tool from Metamorph software (Molecular Devices, Sunnyvale, CA), 7–10-pixel-wide lines were traced along the long axis of the middle of the cell or along the cortex starting from the neck to the distal tip of the cell body and from the neck to the distal tip of the leader bleb. On average n = 10 of cells were traced for each condition, with three-to-six time points, spaced 30 s to 1 min apart for 5 or 10 min, respectively, per cell. The intensity line profiles for 488 and 561 nm channels for both body and bleb for all time points were stored in an Excel spreadsheet (Microsoft, Seattle, WA) that was loaded into Matlab (Mathworks, Natick, MA) for analysis with a script that performed the following tasks: 1) Extracted the intensities from the spreadsheet and stored them in a structure array where each element corresponded to an imaged cell. Each cell element contained matrices for position and intensity along the body and bleb for all time points, separated by channel. The intensities were background subtracted using the lowest-intensity value and normalized by the maximum-intensity value along the profile. 2) Given that the length of the bleb and body fluctuate over time, the intensity data for each channel were binned into 50 bins (an arbitrary choice). The binning was performed by dividing each value in the position vector by the maximum length, multiplying by 50, and rounding off to the nearest digit. The average intensity for each bin was then calculated with the accumarray function using the positional bins and intensity values along the whole profile, applying the mean function. The position functions were correspondingly rescaled to a 50-element vector running from –1 to zero for the body and from zero to +1 for the leading bleb. This was performed for body and bleb for all channels and all time points independently. 3) Having thus appropriately normalized the length of each line profile, we averaged the intensity for body and bleb for each cell and each channel over all time points. Using these values we built two matrices (one for each channel) where each column represented the average normalized intensity for a specific cell. 4) Most of the graphs presented in this article are plots of the above-mentioned matrices, where the thick line in the middle represents the average and the shaded areas above and below represent the SD over all the cells imaged. In addition to these plots, using this script we also extracted the average and SD of the cell body and leader bleb’s longest axis.
Determination of the fraction of fluorescence in the cell body and cell bleb.
The fraction of mEmerald-fusion protein fluorescence in the cell body and the leader bleb was determined from spinning-disk confocal images taken in the central plane of the cell for cells in nonadhesive confinement in which the nucleus (visualized by FusionRed-F-Tractin exclusion) was in either compartment. The outline of the whole cell, the leader bleb, and the cell body was traced to define regions. Fluorescence intensity was calculated as the mean fluorescence intensity of the cytoplasm minus the background fluorescence calculated in Microsoft Excel. Statistical analysis and plots were generated using GraphPad Prism.
Determination of MTOC polarization relative to the nucleus.
The position of the MTOC relative to the nucleus was determined from time-lapse series (30 s intervals for 10 min) of spinning-disk confocal images of mEmerald-gamma-tubulin and FusionRed-F-Tractin (which allowed localization of the nucleus due to its exclusion of fluorescence) in leader bleb cells under nonadhesive confinement. The line scan tool in Fiji was used to draw a line perpendicular to the long axis of the cell, dividing the nucleus roughly in half. The position of the MTOC (marked by mEmerald-gamma-tubulin) between the line and the rear of the cell body or tip of the leader bleb was recorded for cells in which the nucleus was in the cell body or in the nucleus.
siRNA knockdown and re-expression rescue
siRNAs and reagents were purchased from Dharmacon RNA Technologies: Accell Non-targeting Control siRNA (D-001910-01-05), filamin-A 3′UTR (A-012579-16-0005), fascin-1 3′UTR (A-019576-13-005). Briefly, cells were incubated with siRNA for 48–72 h before experiments were performed. For rescue, cells were incubated with siRNA for 48 h and then mEmerald-fascin-1 or mEmerald-filamin-A was transfected for 24 h before experiments were performed. Knockdown and rescue were confirmed by Western blotting of whole-cell lysates for the presence of fascin-1 or filamin-A and/or by immunofluorescence for the presence of fascin-1 or filamin-A in adhered or confined cells.
Western blotting
Western blotting was performed as follows. In brief, cells were lysed in Laemmli sample buffer, lysates were separated by SDS–PAGE, and proteins were electrotransferred overnight at 4°C to an Immobilon-p polyvinylidene fluoride (PVDF) membrane. Membranes were blocked for 1 h at RT with 5% nonfat dry milk (wt/vol) in TBS-T buffer (TBS + 0.1% Tween-20 [vol/vol]) and then incubated overnight at 4°C with the indicated primary antibodies. Subsequently, membranes were washed 3 × 5 min in TBS-T, incubated with appropriate horseradish peroxidase (HRP)-conjugated secondary antibodies (1:10,000) for 1 h at RT, and then washed 3 × 5 min in TBS-T. An ECL detection system (Millipore) was used to visualize protein bands. The following antibodies were used: rabbit anti–filamin-A (1:1000; Sigma-Aldrich; HPA002925), rabbit anti–fascin-1 (1:1000; Sigma-Aldrich; HPA005723), rabbit anti-paxillin (1:1000; Invitrogen; 10029-1-IG), rabbit anti-cortactin (1:1000; Millipore-Sigma; SAB4500766), mouse tropomyosin (1:1000; Millipore-Sigma; T2780), rabbit anti-GAPDH (1:2000; Clone 14C10; Cell Signaling; 2118S). The secondary antibodies (HRP-conjugated goat anti-rabbit [1:10,000; 111-035-003]; HRP-conjugated anti-mouse [1:10,000; 115-035-003]) were from Jackson ImmunoResearch Laboratories.
Statistics
All statistical analysis was performed in Prism (GraphPad, San Diego, CA). All sample sizes were empirically determined based on saturation. As noted in each figure legend, statistical significance was determined by a two-tailed (unpaired) Student’s t test, F test, Mann–Whitney (Figure 6K), or one-way analysis of variance (ANOVA). **p ≤ 0.01 , ***p ≤ 0.001, ****p ≤ 0.0001, NS not significant.
Data availability
The data that support the findings of this study are available from the corresponding author, C.M.W., upon reasonable request.
Supplementary Material
Acknowledgments
We thank Michael Schell (USHS) and the late Michael Davidson for reagents, Bob Fischer and Ankita Jha for insightful comments on the manuscript, and the National Heart, Lung and Blood Institute (NHLBI) Light Microscopy Core Facility. The work was supported by the intramural research programs of the NHLBI (G.A., Jr., M.P.L., and C.M.W.), the National Institute of Biomedical Imaging and Bioengineering, and the Distinguished Scholars Program, Office of the Director at the National Institutes of Health (A.X.C-R.) and by a Rubicon Grant from the Dutch Research Council (NWO; project number 82515005) (M.P.L.)
Abbreviations used:
- BSA
Bovine Serum-Albumin
- CAAX
Farnesylated
- DAPI
4’,6-diamidino-2-phenylindole
- ECM
extracellular matrix
- EMT
epithelial-to-mesenchymal transition
- ER
endoplasmic reticulum
- FA
focal adhesion
- FN
fibronectin
- GFP
green fluorescence protein
- GFP
3X-EMTB- ensconsin
- LBBM
leader bleb based migration
- MAT
mesenchymal-to-ameboid transition
- gtubulin
mEmerald-gT-gamma-Tubulin
- MSD
mean squared displacement
- MT
microtubules
- MTOC
mitotic organization center
- PDHA
pyruvate dehydrogenase E1 component subunit alpha
- PDMS
polydimethylsiloxane
- PVDF
polyvinylidene difluoride (PVDF) membrane
- PXMP2
peroxisomal membrane protein 2
- siRNA
small interfering RNA
- SiT
sialyltransferase
- TBST
Tris-buffered saline (TBS) and Tween 20
- TIRF
Total Internal Reflection Fluorescence
- VIF
vimentin intermediate filaments.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E21-04-0174) on July 14, 2021.
REFERENCES
- Aoki K, Harada S, Kawaji K, Matsuzawa K, Uchida S, Ikenouchi J (2021). STIM-Orai1 signaling regulates fluidity of cytoplasm during membrane blebbing. Nat Commun 12, 480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beach JR, Bruun KS, Shao L, Li D, Swider Z, Remmert K, Zhang Y, Conti MA, Adelstein RS, Rusan NM, et al. (2017). Actin dynamics and competition for myosin monomer govern the sequential amplification of myosin filaments. Nat Cell Biol 19, 85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergert M, Chandradoss SD, Desai RA, Paluch E (2012). Cell mechanics control rapid transitions between blebs and lamellipodia during migration. Proc Natl Acad Sci USA 109, 14434–14439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergert M, Erzberger A, Desai RA, Aspalter IM, Oates AC, Charras G, Salbreux G, Paluch EK (2015). Force transmission during adhesion-independent migration. Nat Cell Biol 17, 524–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaser H, Reichman-Fried M, Castanon I, Dumstrei K, Marlow FLL, Kawakami K, Solnica-Krezel L, Heisenberg CP, Raz E (2006). Migration of zebrafish primordial germ cells: a role for myosin contraction and cytoplasmic flow. Dev Cell 11, 613–627. [DOI] [PubMed] [Google Scholar]
- Brayford S, Bryce NS, Schevzov G, Haynes EM, Bear JE, Hardeman EC, Gunning PW (2016). Tropomyosin promotes lamellipodial persistence by collaborating with Arp2/3 at the leading edge. Curr Biol 26, 1312–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitsprecher D, Koestler SA, Chizhov I, Nemethova M, Mueller J, Goode BL, Small JV, Rottner K, Faix J (2011). Cofilin cooperates with fascin to disassemble filopodial actin filaments. J Cell Sci 124, 3305–3318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugyi B, Didry D, Carlier MF (2010). How tropomyosin regulates lamellipodial actin-based motility: a combined biochemical and reconstituted motility approach. EMBO J 28, 14–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartagena-Rivera AX, Logue JS, Waterman CM, Chadwick RS (2016). Actomyosin cortical mechanical properties in nonadherent cells determined by atomic force microscopy. Biophys J 110, 2528–2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charras G, Paluch E (2008). Blebs lead the way: how to migrate without lamellipodia. Nat Rev Mol Cell Biol 9, 730–736. [DOI] [PubMed] [Google Scholar]
- Charras G, Sahai E (2014). Physical influences of the extracellular environment on cell migration. Nat Rev Mol Cell Biol 15, 813–824. [DOI] [PubMed] [Google Scholar]
- Charras GT, Yarrow JC, Horton MA, Mahadevan L, Mitchison TJ (2005). Non-equilibration of hydrostatic pressure in blebbing cells. Nature 435, 365–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AG, Dierkes K, Paluch EK (2013). Monitoring actin cortex thickness in live cells. Biophys J 105, 570–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedl P, Gilmour D (2009). Collective cell migration in morphogenesis, regeneration and cancer. Nat Rev Mol Cell Biol 10, 445–457. [DOI] [PubMed] [Google Scholar]
- Friedl P, Wolf K (2010). Plasticity of cell migration: a multiscale tuning model. J Cell Biol 188, 11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadea G, De Toledo M, Anguille C, Roux P (2007). Loss of p53 promotes RhoA-ROCK-dependent cell migration and invasion in 3D matrices. J Cell Biol 178, 23–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghisleni A, Galli C, Monzo P, Ascione F, Fardin MA, Scita G, Li Q, Maiuri P, Gauthier NC (2020). Complementary mesoscale dynamics of spectrin and acto-myosin shape membrane territories during mechanoresponse. Nat Commun 11, 5108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorelik R, Gautreau A (2014). Quantitative and unbiased analysis of directional persistence in cell migration. Nat Protoc 9, 1931–1943. [DOI] [PubMed] [Google Scholar]
- Goudarzi M, Banisch TU, Mobin MB, Maghelli N, Tarbashevich K, Strate I, van den Berg J, Blaser H, Bandemer S, Paluch E, et al. (2012). Identification and regulation of a molecular module for bleb-based cell motility. Dev Cell 23, 210–218. [DOI] [PubMed] [Google Scholar]
- Graham DM, Andersen T, Sharek L, Uzer G, Rothenberg K, Hoffman BD, Rubin J, Balland M, Bear JE, Burridge K (2018). Enucleated cells reveal differential roles of the nucleus in cell migration, polarity, and mechanotransduction. J Cell Biol 217, 895–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamidi H, Ivaska J (2018). Every step of the way: integrins in cancer progression and metastasis. Nat Rev Cancer 18, 533–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuzé ML, Vargas P, Chabaud M, Le Berre M, Liu YJ, Collin O, Solanes P, Voituriez R, Piel M, Lennon-Duménil AM (2013). Migration of dendritic cells: physical principles, molecular mechanisms, and functional implications. Immunol Rev 256, 240–254. [DOI] [PubMed] [Google Scholar]
- Hsiao JY, Goins LM, Petek NA, Mullins RD (2015). Arp2/3 complex and cofilin modulate binding of tropomyosin to branched actin networks. Curr Biol 25, 1573–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YJ, Samorajski J, Kreimer R, Searson PC (2013). The influence of electric field and confinement on cell motility. PLoS One 8, e59447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapushesky M, Emam I, Holloway E, Kurnosov P, Zorin A, Malone J, Rustici G, Williams E, Parkinson H, Brazma A (2010). Gene Expression Atlas at the European Bioinformatics Institute. Nucleic Acids Res 38, D690–D698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley CA, Triplett O, Mallick S, Burkewitz K, Mair WB, Cram EJ (2020). FLN-1/filamin is required to anchor the actomyosin cytoskeleton and for global organization of sub-cellular organelles in a contractile tissue. Cytoskeleton 77, 379–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenderink GH, Paluch EK (2018). Architecture shapes contractility in actomyosin networks. Curr Opin Cell Biol 50, 79–85. [DOI] [PubMed] [Google Scholar]
- Krueger D, Pallares Cartes C, Makaske T, De Renzis S (2020). βH-spectrin is required for ratcheting apical pulsatile constrictions during tissue invagination. EMBO Rep 5, e49858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lämmermann T, Bader BL, Monkley SJ, Worbs T, Wedlich-Söldner R, Hirsch K, Keller M, Förster R, Critchley DR, Fässler R, Sixt M (2008). Rapid leukocyte migration by integrin-independent flowing and squeezing. Nature 453, 51–55. [DOI] [PubMed] [Google Scholar]
- Lauffenburger DA, Horwitz AF (1996). Cell migration: a physically integrated molecular process. Cell 84, 359–369. [DOI] [PubMed] [Google Scholar]
- Lavenus SB, Tudor SM, Ullo MF, Vosatka KW, Logue JS (2020). A flexible network of vimentin intermediate filaments promotes migration of amoeboid cancer cells through confined environments. J Biol Chem 295, 6700–6709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YJ, Le Berre M, Lautenschlaeger F, Maiuri P, Callan-Jones A, Heuzé M, Takaki T, Voituriez R, Piel M (2015). Confinement and low adhesion induce fast amoeboid migration of slow mesenchymal cells. Cell 160, 659–672. [DOI] [PubMed] [Google Scholar]
- Logue JS, Cartagena-Rivera AX, Baird MA, Davidson MW, Chadwick RS, Waterman CM (2015). Erk regulation of actin capping and bundling by Eps8 promotes cortex tension and leader bleb-based migration. eLife 4, e08314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logue JS, Cartagena-Rivera AX, Chadwick RS (2018). C-Src activity is differentially required by cancer self-motility modes. Oncogene 37, 2104–2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomakin A, Cattin C, Cuvelier D, Alraies Z, Molina M, Nader G, Srivastava N, Sáez P, Garcia-Arcos J, Zhitnyak I, et al. (2020). The nucleus acts as a ruler tailoring cell responses to spatial constraints. Science 370, eaba2894.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin T, Ye L, Sanders A, Lane J, Jiang W (2013). Cancer invasion and metastasis: molecular and cellular perspective. In: Metastatic Cancer: Clinical and Biological Perspectives, ed. Jandial R, Austin TX: Landes Bioscience, 135–168. [Google Scholar]
- Maugis B, Brugués J, Nassoy P, Guillen N, Sens P, Amblard F (2010). Dynamic instability of the intracellular pressure drives bleb-based motility. J Cell Sci 123, 3884–3892. [DOI] [PubMed] [Google Scholar]
- Meijering E, Dzyubachyk O, Smal I (2012). Methods for cell and particle tracking. Methods Enzymol 504, 183–200. [DOI] [PubMed] [Google Scholar]
- Nabi IR (1999). The polarization of the motile cell. JCell Sci 112, 1803–1811. [DOI] [PubMed] [Google Scholar]
- Paluch EK, Raz E (2013). The role and regulation of blebs in cell migration. Curr Opin Cell Biol 25, 582–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paluch E, Sykes C, Prost J, Bornens M (2006). Dynamic modes of the cortical actomyosin gel during cell locomotion and division. Trends Cell Biol 16, 5–10. [DOI] [PubMed] [Google Scholar]
- Parsons JT, Horwitz AR, Schwartz MA (2010). Cell adhesion: integrating cytoskeletal dynamics and cellular tension. Nat Rev Mol Cell Biol 11, 633–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renkawitz J, Schumann K, Weber M, Lämmermann T, Pflicke H, Piel M, Polleux J, Spatz JP, Sixt M (2009). Adaptive force transmission in amoeboid cell migration. Nat Cell Biol 11, 1438–1443. [DOI] [PubMed] [Google Scholar]
- Ridley A (2011). Blebs on the move. Dev Cell 20, e1. [DOI] [PubMed] [Google Scholar]
- Rodriguez OC, Schaefer AW, Mandato CA, Forscher P, Bement WM, Waterman-Storer CM (2003). Conserved microtubule-actin interactions in cell movement and morphogenesis. Nat Cell Biol 5, 599–609. [DOI] [PubMed] [Google Scholar]
- Ruprecht V, Wieser S, Callan-Jones A, Smutny M, Morita H, Sako K, Barone V, Ritsch-Marte M, Sixt M, Voituriez R, Heisenberg CP (2015). Cortical contractility triggers a stochastic switch to fast amoeboid cell motility. Cell 160, 673–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabeh F, Shimizu-Hirota R, Weiss SJ (2009). Protease-dependent versus-independent cancer cell invasion programs: three-dimensional amoeboid movement revisited.J Cell Biol 185, 11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahai E, Marshall CJ (2003). Differing modes for tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat Cell Biol 5, 711–719. [DOI] [PubMed] [Google Scholar]
- Salbreux G, Charras G, Paluch E (2012). Actin cortex mechanics and cellular morphogenesis. Trends Cell Biol 22, 536–545. [DOI] [PubMed] [Google Scholar]
- Schell MJ, Erneux C, Irvine RF (2001). Inositol 1,4,5-trisphosphate 3-kinase A associates with F-actin and dendritic spines via its N terminus. J Biol Chem 276, 37537–37546. [DOI] [PubMed] [Google Scholar]
- Shin WD, Fischer RS, Kanchawong P, Kim Y, Lim J, Myers KA, Nishimura Y, Plotnikov SV, Thievessen I, Yarar D, et al. (2010). A versatile, multicolor total internal reflection fluorescence and spinning-disk confocal microscope system for high-resolution live cell imaging . In: Live Cell Imaging: A Laboratory Manual, 2nd, ed. Goldman RD, Swedlow JR, Spector DL, Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press. [Google Scholar]
- Svitkina T (2018). The actin cytoskeleton and actin-based motility. Cold Spring Harb Perspect Biol 10, a018267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tozluogˇlu M, Tournier AL, Jenkins RP, Hooper S, Bates PA, Sahai E (2013). Matrix geometry determines optimal cancer cell migration strategy and modulates response to interventions. Nat Cell Biol 15, 751–762. [DOI] [PubMed] [Google Scholar]
- Trepat X, Fredberg JJ (2011). Plithotaxis and emergent dynamics in collective cellular migration. Trends Cell Biol 21, 638–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullo MF, Logue JS (2018). Re-thinking preclinical models of cancer metastasis. Oncoscience 5, 252–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venturini V, Pezzano FFCC, Häkkinen H, Jiménez-Delgado S, Colomer-Rosell M, Marro M, Tolosa-Ramon Q, Paz-López S, Valverde M, Weghuber J, et al. (2020). The nucleus measures shape changes for cellular proprioception to control dynamic cell behavior. Science 370, eaba2644.. [DOI] [PubMed] [Google Scholar]
- Vicente-Manzanares M, Ma X, Adelstein RS, Horwitz AR (2009). Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat Rev Mol Cell Biol 10, 778–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkelman JD, Suarez C, Hocky GM, Harker AJ, Morganthaler AN, Christensen JR, Voth GA, Bartles JR, Kovar DR (2016). Fascin- and α-actinin-bundled networks contain intrinsic structural features that drive protein sorting. Curr Biol 26, 2697–2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wioland H, Jegou A, Romet-Lemonne G (2019). Torsional stress generated by ADF/cofilin on cross-linked actin filaments boosts their severing. Proc Natl Acad Sci USA 116, 2595–2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong SY, Hynes RO (2006). Lymphatic or hematogenous dissemination: how does a metastatic tumor cell decide? Cell Cycle 5, 812–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada KM, Sixt M (2019). Mechanisms of 3D cell migration. Nat Rev Mol Cell Biol 20, 738–752. [DOI] [PubMed] [Google Scholar]
- Yang Y, Zheng H, Zhan Y, Fan S (2019). An emerging tumor invasion mechanism about the collective cell migration. Am J Transl Res 11, 5301–5312. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author, C.M.W., upon reasonable request.