

Abstract
Dilated cardiomyopathy (DCM) is a significant cause of pediatric heart failure. Mutations in proteins that regulate cardiac muscle contraction can cause DCM; however, the mechanisms by which molecular-level mutations contribute to cellular dysfunction are not well understood. Better understanding of these mechanisms might enable the development of targeted therapeutics that benefit patient subpopulations with mutations that cause common biophysical defects. We examined the molecular- and cellular-level impacts of a troponin T variant associated with pediatric-onset DCM, R134G. The R134G variant decreased calcium sensitivity in an in vitro motility assay. Using stopped-flow and steady-state fluorescence measurements, we determined the molecular mechanism of the altered calcium sensitivity: R134G decouples calcium binding by troponin from the closed-to-open transition of the thin filament and decreases the cooperativity of myosin binding to regulated thin filaments. Consistent with the prediction that these effects would cause reduced force per sarcomere, cardiomyocytes carrying the R134G mutation are hypocontractile. They also show hallmarks of DCM that lie downstream of the initial insult, including disorganized sarcomeres and cellular hypertrophy. These results reinforce the importance of multiscale studies to fully understand mechanisms underlying human disease and highlight the value of mechanism-based precision medicine approaches for DCM.
INTRODUCTION
Familial dilated cardiomyopathy (DCM) is a major cause of heart failure in both adult and pediatric patients and a significant cause of heart transplantation (McNally and Mestroni, 2017; Patel et al., 2017; Towbin et al., 2017). DCM is characterized by dilation of the left ventricular chamber and impaired cardiac contractility (Mestroni et al., 1999). Current treatments developed for adult heart failure have limited efficacy in the pediatric population, and the 5-y transplant-free survival rate is <50% in children with DCM (Shaddy et al., 2007; Kantor et al., 2010). Therefore, there is an outstanding clinical need to develop new therapeutics for pediatric DCM.
Familial DCM is often caused by point mutations in proteins involved in cardiac muscle contraction and mechanosensation, including troponin T (TnT) (McNally and Mestroni, 2017). Troponin T is part of the troponin complex, which, along with tropomyosin, regulates the calcium-dependent interactions between force-generating myosin heads and the thin filament. Several mutations in TnT are associated with severe, early-onset disease (Hershberger et al., 2009). However, the mechanisms by which mutation-induced changes in protein interactions at the molecular level affect cardiac contraction and drive the disease pathogenesis at the cellular level are not well understood. Furthermore, these disease-driving defects can vary between mutations, even within the same molecule (Lynn et al., 2018; Greenberg and Tardiff, 2021; Lavine and Greenberg, 2021). Understanding these mechanisms is a critical step in developing precision medicine therapeutics that target mutation-specific defects.
To better understand the molecular mechanisms that can drive DCM disease pathogenesis, we examined the molecular and cellular consequences of a variant in troponin T, R134G, which has been implicated in pediatric-onset DCM (Hershberger et al., 2008) (Figure 1). Several variants at the R134 position have been implicated in cardiomyopathy, with clinical significance of individual variants ranging from uncertain to likely pathogenic (Landrum et al., 2018). We chose to study the likely pathogenic R134G variant, which was shown to segregate with DCM in a patient cohort (Hershberger et al., 2009). The small number of patients known to carry this specific variant makes it difficult to definitively determine pathogenicity from the currently available clinical data (Hershberger et al., 2009; Landrum et al., 2018). Functional data supporting a plausible pathogenic mechanism would increase the confidence that this variant is indeed causative for DCM.
FIGURE 1:
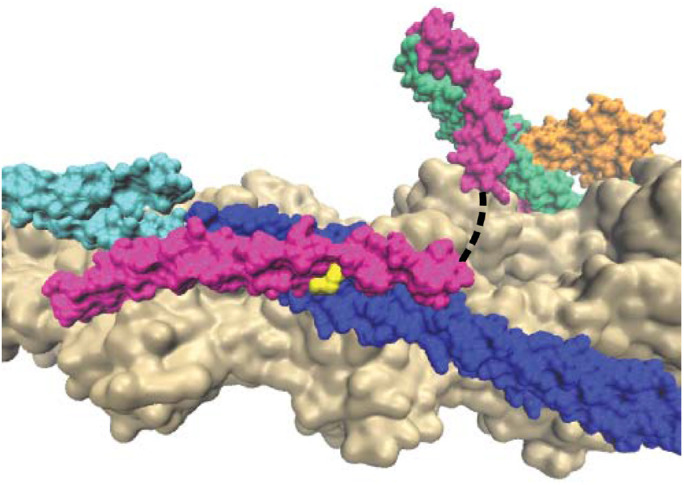
Structure of the RTF (PDB ID 6KN7) showing actin (tan), tropomyosin (blue/cyan), troponin I (green), troponin C (orange), troponin T (magenta), and R134 of troponin T (yellow). The dashed line represents an unstructured region of troponin T that is not resolved in the structure. Adjacent tropomyosin units are shown as cyan and blue to demonstrate the proximity of R134 to the overlap region.
Because troponin T plays a critical role in transmitting changes in calcium binding to troponin C to changes in the tropomyosin position on the thin filament, we tested whether R134G affects calcium-based regulation of interactions between myosin and the thin filament. Our results reveal that the R134G mutation in TnT depresses both calcium- and myosin-based activation of the thin filament. We find that R134G decouples calcium binding from the closed-to-open transition of the thin filament, resulting in reduced thin filament activation at saturating calcium concentrations. R134G also decreases the cooperativity of myosin binding to regulated thin filaments (RTFs). In gene-edited, stem cell–derived cardiomyocytes, we see reduced contractility that is consistent with the molecular-level defects, as well as downstream effects on sarcomeric organization and cell size that are consistent with features of DCM.
RESULTS
To study the effects of the R134G variant at the molecular scale, we purified recombinant human troponin complex and tropomyosin. We purified actin and myosin from porcine ventricles. Porcine cardiac actin is identical to human cardiac actin, and porcine cardiac myosin is 97% identical to human cardiac myosin, with indistinguishable biophysical properties (Deacon et al., 2012; Greenberg et al., 2014; Sung et al., 2015).
R134G decreases thin filament calcium sensitivity
We used an in vitro motility assay to assess the effect of the R134G mutation in TnT on thin filament regulation (Figure 2). The speed of fluorescent RTFs gliding across a myosin-functionalized substrate was measured as a function of calcium concentration. At low calcium, tropomyosin blocks myosin strong binding to actin and the RTFs are immobile (stuck). At saturating calcium, the RTFs are fully activated and the filaments glide at maximal speed. As the calcium concentration was increased from pCa 9 to pCa 4, the percent of stuck RTFs decreased from 97 ± 2% to 2 ± 1% for wild type (WT) and from 97 ± 1% to 9 ± 4% for R134G (Figure 2A). Thus, the stuck filament percent was similar for WT and R134G at pCa 9 (p = 0.91) and significantly higher for R134G relative to WT at pCa 4 (p = 0.003).
FIGURE 2:
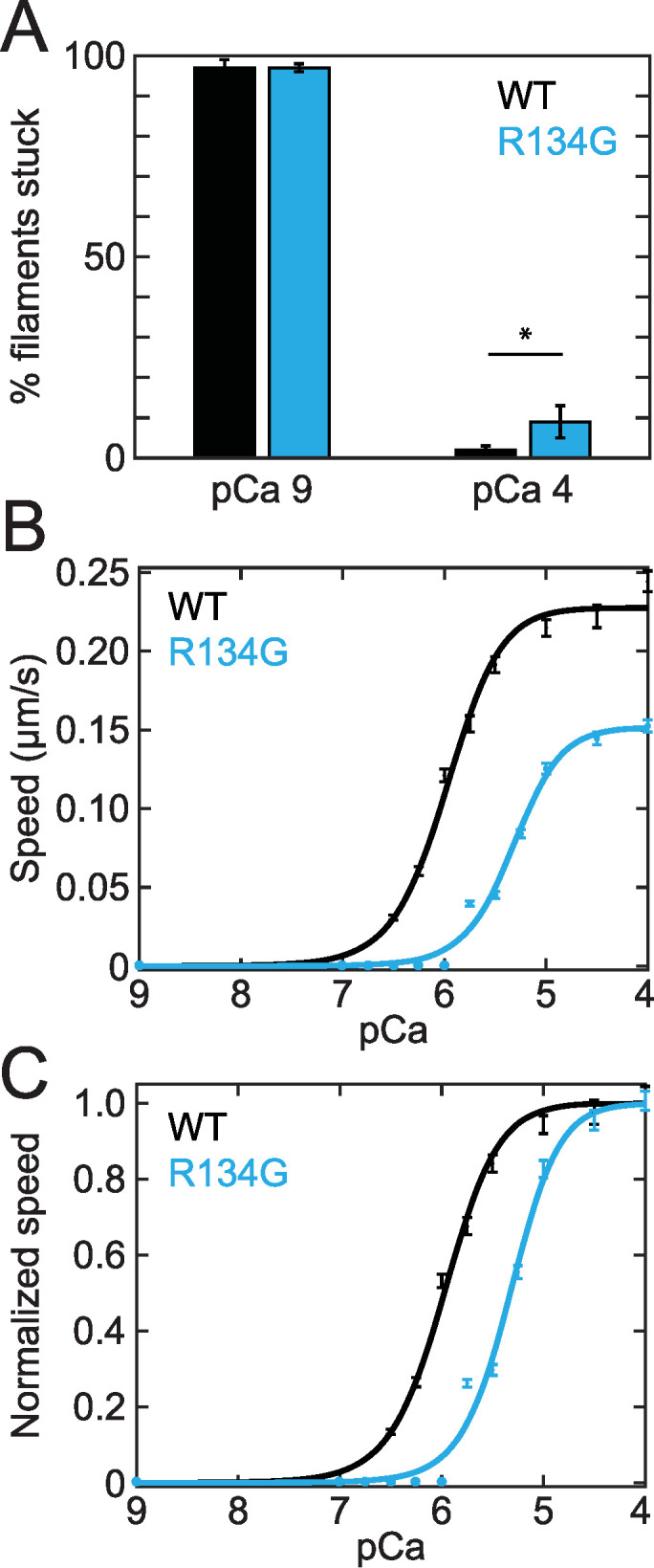
(A) Percent stuck filaments in an in vitro motility assay at pCa 9 (97 ± 2% for WT, 97 ± 1% for R134G; p = 0.91) and pCa 4 (2 ± 1% for WT, 9 ± 4% for R134G; p = 0.003). Asterisk indicates a statistically significant difference in the percent stuck filaments at pCa 4. Raw (B) and normalized (C) RTF gliding speed as a function of calcium concentration in the in vitro motility assay. Curves are fits to a Hill equation. Error bars represent SEM of 150 tracked RTFs from three independent experimental days. R134G (Vmax = 0.15 ± 0.1 μm/s) results in a decrease in the maximum gliding speed relative to WT (Vmax = 0.23 ± 0.1 μm/s; p < 0.001). Normalized RTF gliding speed is shown to more clearly illustrate the decrease in calcium sensitivity caused by the R134G variant (pCa50 [R134G] = 5.3 ± 0.1, pCa50 [WT] = 6.0 ± 0.1; p < 0.001). Note that data collected at pCa 6.75, 7, 8, and 9 are obscured by the fit line.
We fitted a Hill equation to the measured filament speed versus calcium data to obtain the maximal sliding speed (Vmax) and the calcium concentration at which RTFs glide at half-maximal speed (pCa50). We found that Vmax was reduced for R134G RTFs compared with WT (Vmax [WT] = 0.23 ± 0.01 μm/s, Vmax [R134G] = 0.15 ± 0.01 μm/s; p < 0.001) (Figure 2B). We did not detect a statistically significant change in the Hill coefficient (nH [WT] = 1.5 ± 0.5, nH [R134G] = 1.8 ± 0.6; p = 0.66). We plotted the normalized gliding speeds as a function of calcium concentration to illustrate the decrease in calcium sensitivity caused by the R134G variant (Figure 2C). The pCa50 for RTFs regulated by R134G troponin was significantly lower than that for WT troponin (pCa50 [WT] = 6.0 ± 0.1, pCa50 [R134G] = 5.3 ± 0.1; p < 0.001). This decrease in the calcium sensitivity of RTF motility indicates that more calcium is needed for the variant to achieve the same level of activation as the WT. These results suggest that cardiomyocytes expressing the R134G variant might demonstrate decreased force per sarcomere during a calcium transient, which would be consistent with the impaired systolic function observed in DCM patients carrying the R134G mutation (Hershberger et al., 2008).
Muscle activation requires both calcium- and myosin-dependent activation of the thin filament (Houmeida et al., 2010). The decreased calcium sensitivity observed in the in vitro motility assay could be caused by mutation-induced changes in 1) cross-bridge dissociation kinetics, 2) calcium binding affinity to troponin C, and/or 3) equilibrium positioning or cooperativity of tropomyosin movement along the thin filament. We measured each of these properties to determine which are responsible for the shift in calcium sensitivity caused by R134G.
R134G does not affect actomyosin dissociation kinetics
Thin filament activation depends on myosin binding to the thin filament, which locks tropomyosin in a position that allows cooperative binding of additional nearby myosin molecules. A mutation-induced increase in the rate of myosin cross-bridge detachment would decrease thin filament activation by decreasing the time myosin spends in the activating, strongly bound state. The rate of cross-bridge detachment in the absence of load is limited by the rate of ADP release from actomyosin (Bárány, 1967; Siemankowski et al., 1985). To test whether the decreased calcium sensitivity in the motility assay was due to changes in the cross-bridge detachment rate, we used a stopped-flow transient kinetic assay to measure the rate of ADP release from actomyosin S1 (Figure 3). We found that the R134G variant had no effect on the ADP release rate constant (k [WT] = 76.7 ± 0.2 s–1, k [R134G] = 78.8 ± 0.2 s–1; p = 0.35). This is consistent with previous measurements of ADP release from cardiac actomyosin S1 (Deacon et al., 2012; Greenberg et al., 2014; Clippinger et al., 2019, 2021). Thus, altered kinetics of actomyosin dissociation are not responsible for the decreased calcium sensitivity observed in the in vitro motility assay (Figure 2).
FIGURE 3:
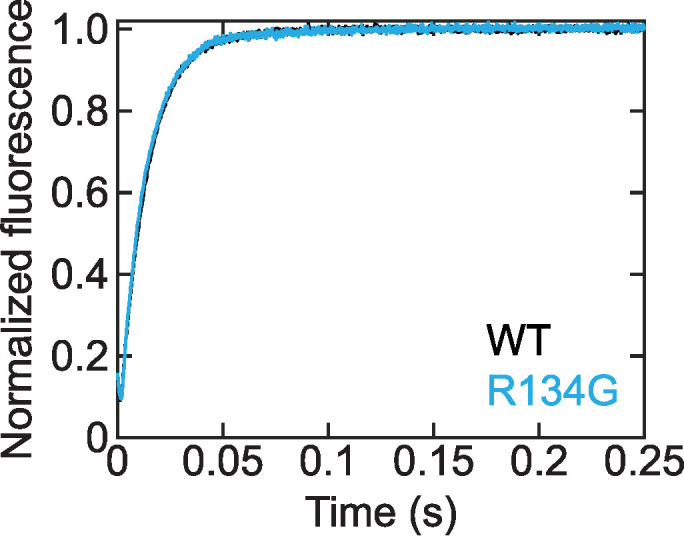
Stopped-flow kinetic traces measuring the ADP release rate from actomyosin after rapid mixing of ATP with pyrene-labeled RTFs bound to myosin and ADP. The pyrene fluorescence increases as myosin S1 dissociates from actin after release of ADP. Traces are the average of three (WT) or two (R134G) independent experiments, each the average of five technical replicates. The WT trace (k = 76.7 ± 0.2 s–1) is partially obscured by the R134G trace (k = 78.8 ± 0.2 s–1) due to the similar rates. R134G does not alter the rate of ADP release from actomyosin S1 (p = 0.35).
R134G increases the calcium sensitivity of troponin C
The decreased calcium sensitivity observed in the motility assay could also be caused by a reduction in the calcium binding affinity of troponin through an allosteric effect of the R134G mutation in TnT on troponin C (TnC). To test this possibility, we measured the calcium binding affinity of RTFs containing TnC site–specifically labeled with IAANS (2-(4′-(iodoacetamido)anilino)napthalene-6-sulfonic acid), an environmentally sensitive fluorophore. The response of IAANS fluorescence to calcium-dependent changes in TnC conformation provides a convenient method for monitoring calcium binding to TnC (Davis et al., 2007; Tikunova et al., 2010; Liu et al., 2012). Unexpectedly, we found that R134G TnT increased calcium binding to TnC within RTFs (pCa50 [WT] = 6.29 ± 0.07, pCa50 [R134G] = 6.6 ± 0.1; p < 0.001) (Figure 4). This increased calcium binding sensitivity for R134G troponin cannot explain the decrease in calcium sensitivity observed in the motility assay (Figure 2).
FIGURE 4:
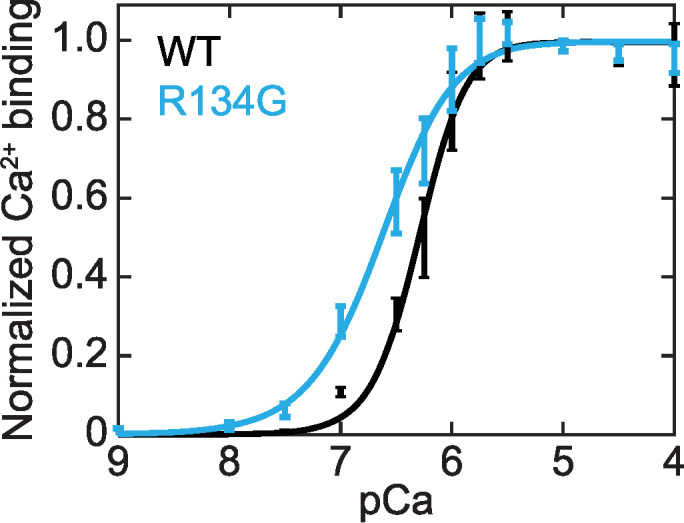
Calcium binding affinity of TnC within RTFs as a function of calcium concentration. Normalized calcium binding was determined from steady-state fluorescence of IAANS-labeled TnC. Data were fitted with the Hill equation. Error bars represent SD of five replicates. TnC within RTFs containing R134G TnT (pCa50 = 6.6 ± 0.1) showed increased calcium sensitivity compared with RTFs containing WT TnT (pCa50 = 6.29 ± 0.07; p < 0.001).
R134G decouples the closed-to-open transition from calcium binding and reduces the cooperativity of myosin binding to RTFs
Tropomyosin can lie in three distinct positions along the thin filament: blocked, closed, and open (McKillop and Geeves, 1993; Vibert et al., 1997). The distribution of tropomyosin among these three positions is affected by both calcium binding to troponin C and myosin binding to the thin filament (Figure 5A). The blocked state, in which tropomyosin blocks the myosin binding site on the thin filament, is significantly occupied only at low calcium concentrations. Myosin strong binding and subsequent force generation occur only in the open state of the thin filament. The fraction of force-generating thin filament regulatory units will thus depend on the equilibrium constants that define the transitions between these states. The process of thin filament activation is cooperative due to coupling between thin filament regulatory units via tropomyosin end-to-end bonds (Greene and Eisenberg, 1980; Moore et al., 2016). The R134G mutation could decrease the calcium sensitivity of RTFs by shifting the equilibrium positioning of tropomyosin and/or by decreasing the cooperativity of activation, resulting in the higher calcium concentrations required to activate the RTF and generate force. We examined the effect of the R134G variant on tropomyosin positioning using fluorescence-based assays of regulated actomyosin binding to determine the equilibrium constants for the blocked-to-closed (KB) and closed-to-open (KT) transitions of the thin filament (Figure 5).
FIGURE 5:

Effects of R134G on tropomyosin positioning along the thin filament. (A) Schematic of the three-state model of thin filament regulation. The equilibrium constants KB and KT describe the blocked-to-closed and closed-to-open transitions of the thin filament, respectively. KW and KS describe weak and strong myosin binding, respectively. (B, C) Measurement of KB. Stopped-flow kinetic traces of myosin S1 binding to RTFs containing pyrene-actin and WT (B) or R134G (C) troponin T. Traces are the average of 10 independent experiments, each the average of at least three technical replicates. S1 binding quenches the pyrene fluorescence at a higher rate at high calcium (pCa 4, green) than at low calcium (pCa 9, magenta) in each case. The value of KB was determined using Eq. 1 (Materials and Methods). (D, E) Measurement of KT and KW. Steady-state titrations of RTFs with S1 for WT (D) and R134G (E) at three calcium concentrations: saturating (pCa 4, green), intermediate (pCa 6.25, orange), and low (pCa 9, magenta). Curves are fits of Eq. 2 (Materials and Methods) to the data. Error bars represent the SD of six technical replicates collected on six independent experimental days. (F) Table of parameter values determined for WT and R134G. KB was determined from the stopped-flow measurements and is given as the average ± SD of 10 replicates. All other parameters were determined from the steady-state titrations and are given as best-fit values with 95% confidence intervals. Asterisks indicate statistical significance at the 95% confidence interval.
To determine KB, we measured the calcium-dependent binding of myosin subfragment-1 (myosin S1) to pyrene-labeled RTFs in a stopped-flow kinetic assay (McKillop and Geeves, 1993; Barrick et al., 2019). Myosin strong binding quenches the fluorescence of pyrene-labeled actin, resulting in a decrease in fluorescence. KB, the equilibrium constant for the blocked-to-closed transition, can be determined from the relative rates of S1 binding at pCa 9 (low calcium) and pCa 4 (saturating calcium) (see Materials and Methods for details). The calcium-dependent rates of binding to the RTF were similar for WT and R134G (Figure 5, B and C), and the calculated KB values were indistinguishable (KB [WT] = 0.3 ± 0.2, KB [R134G] = 0.3 ± 0.1; p = 0.76). Thus, R134G does not affect the equilibrium constant between the blocked and closed states.
To measure the equilibrium constant for the closed-to-open transition of the thin filament, KT, we performed fluorescence titrations of steady-state myosin S1 binding to pyrene-labeled RTFs (McKillop and Geeves, 1993; Barrick et al., 2019) (Figure 5, D and E). By fitting the titration data with an equation relating the fractional change in pyrene fluorescence to the extent of S1 binding (see Materials and Methods), we obtained estimates for the values of KT, as well as KW, the equilibrium constant for myosin weak binding to actin, and nH, the apparent size of the cooperative unit, as previously described in detail (Barrick et al., 2019). The values of these parameter estimates are summarized in Figure 5F.
For RTFs regulated by WT troponin, the value of KT increased with calcium concentration, as previously observed by us and others (McKillop and Geeves, 1993; Barrick et al., 2019; Clippinger et al., 2019). In contrast, the KT value did not depend on calcium for RTFs regulated by R134G troponin (Figure 5F). This can be seen qualitatively from the plots of fractional binding as a function of myosin concentration. In contrast to the WT (Figure 5D), the fractional bindings to RTFs containing R134G troponin (Figure 5E) are very similar at pCa 6.25 and 4. This suggests that the R134G mutation decouples the thin filament closed-to-open transition from calcium binding to TnC. Compared to WT, the KT value for R134G was higher at low calcium (pCa 9) and lower at saturating calcium (pCa 4). At pCa 9, the level of thin filament activation is low despite the increased value of KT because most thin filaments adopt the blocked state. At pCa 4, tropomyosin is less likely to adopt the open position for R134G compared with WT. This predicts a mutation-induced reduction in population of the myosin-bound states, consistent with the shift in calcium sensitivity and decrease in Vmax seen in the motility assay.
The R134G mutation resulted in a significant decrease in the Hill coefficient nH (Figure 5F), which can be interpreted as the apparent size of the cooperative unit (McKillop and Geeves, 1993; Mijailovich et al., 2012). Higher values of nH suggest longer-range cooperativity. For WT troponin, the best-fit value was nH = 6 (–1/+1), which is similar to previously reported values (Maytum et al., 1999; Clippinger et al., 2019). R134G thin filaments showed reduced cooperativity (nH = 3.2 [–0.2/+0.3]; p = 0.002), indicating that the R134G mutation limits the propagation of thin filament activation. This reduction in cooperativity would be expected to reduce activation, consistent with the shift in calcium sensitivity seen in the motility assay.
There was also a small but statistically significant increase in KW associated with the R134G variant (Figure 5F). This corresponds to a shift in the equilibrium toward weakly bound rather than dissociated myosin; however, it is unlikely that an increase of this magnitude would have biological significance. Regardless, an increase in KW predicts slightly increased myosin weak binding, so this change cannot explain the decreased calcium sensitivity caused by the R134G mutation in the in vitro motility assay (Figure 2).
Together, these molecular-level measurements demonstrate that the mutation-induced decrease in calcium sensitivity observed in the in vitro motility assay is primarily caused by reduced coupling of calcium binding to changes in tropomyosin positioning, resulting in reduced occupancy of the open state in the presence of calcium, and/or a decrease in the cooperativity of myosin binding to RTFs.
R134G results in increased cell area and sarcomeric disorganization in hiPSC-CMs
To examine the effect of the R134G mutation in a cellular context, we used CRISPR/Cas9 gene editing to generate human induced pluripotent stem cell–derived cardiomyocytes (hiPSC-CMs) homozygous for the R134G mutation in TnT. hiPSCs were differentiated to cardiomyocytes by temporal modulation of Wnt signaling and aged at least 30 d before any assays were performed, as previously described (Clippinger et al., 2019). hiPSC-CMs showed spontaneous beating starting at days 7–10 after the initiation of the differentiation.
R134G and WT hiPSC-CMs were each micropatterned onto glass coated with rectangular patterns of extracellular matrix, fixed, stained for troponin I and α-actinin to visualize the sarcomeres, and imaged using confocal microscopy (Figure 6, A and B). We observed larger cell areas for the R134G hiPSC-CMs (1800 ± 200 μm2) compared with WT hiPSC-CMs (1000 ± 50 μm2; p = 0.002) (Figure 6; Table 1), as we previously observed for a different DCM-causing mutation in TnT (Clippinger et al., 2019). We also observed a decreased aspect ratio for the R134G hiPSC-CMs relative to WT (Figure 6; Table 1). Moreover, the sarcomeres in the micropatterned WT hiPSC-CMs are well organized and tend to align along the long axis of the cell, as previously described (Clippinger et al., 2019). In contrast, the sarcomeres in the R134G hiPSC-CMs appear more disorganized and fail to align along a single axis. We frequently observe sarcomeres that appear more wavy than linear (see Figure 6B). The sarcomere lengths of fixed cells for the mutant were longer than those for the WT cells on glass, but this difference disappeared on 10 kPa hydrogels. Taken together, these results demonstrate mutation-induced changes in cell shape and sarcomeric organization.
FIGURE 6:
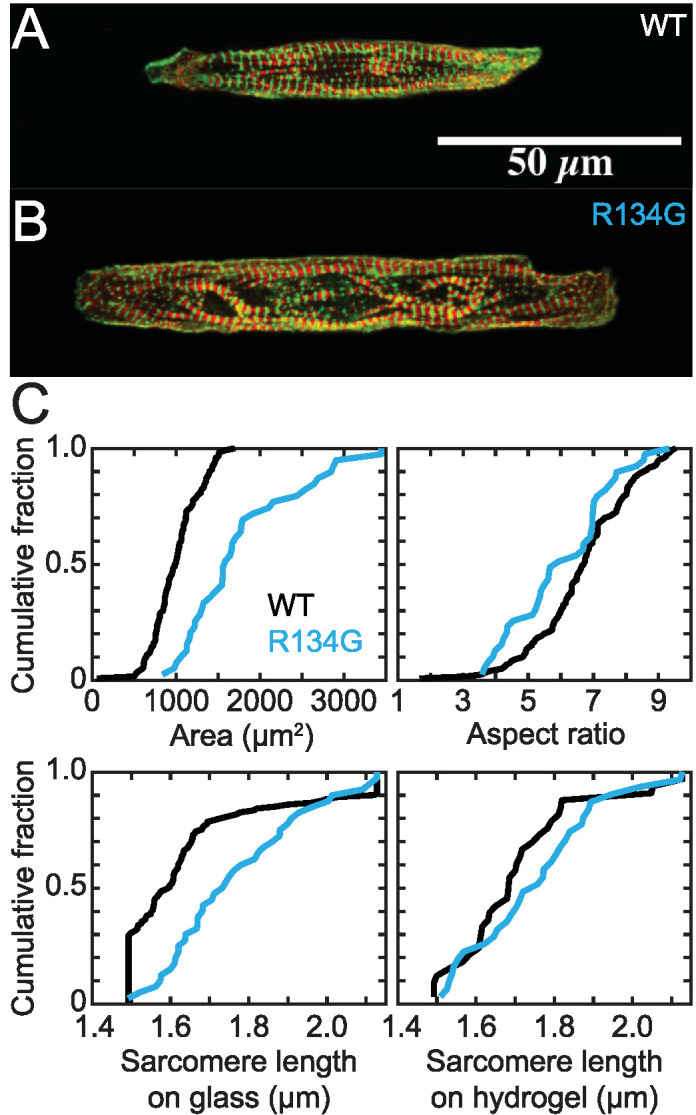
Representative immunofluorescence images of sarcomeres in hiPSC-CMs on a rectangular pattern on glass. Troponin I is red, and α-actinin is green. Images are z-projections. (A) WT hiPSC-CM. (B) R134G hiPSC-CM. R134G hiPSC-CMs exhibit sarcomeric disorganization and increased cell area. (C) Quantification of areas, aspect ratios, and sarcomere lengths from immunofluorescence images of WT (n = 120) and R134G (n = 39) hiPSC-CMs on glass and sarcomere lengths of WT (n = 33) and R134G (n = 31) hiPSC-CMs on hydrogel. Data are plotted as cumulative distributions, and parameter values are given in Table 1. R134G hiPSC-CMs had a larger cell area (p = 0.002) and a smaller aspect ratio (p = 0.028) than WT hiPSC-CMs. R134G hiPSC-CMs had longer sarcomere lengths than WT hiPSC-CMs on glass (p = 0.004) but not on hydrogel (p = 0.342).
TABLE 1:
Comparison of hiPSC-CMs expressing WT or R134G troponin T.
| Parameter | WT | R134G | p |
|---|---|---|---|
| Traction force (μN) | 0.27 (–0.03/+0.03) | 0.17 (–0.03/+0.04) | 0.002 |
| Power (pW) | 0.10 (–0.02/+0.02) | 0.06 (–0.02/+0.03) | 0.018 |
| Contraction velocity (μm/s) | 0.33 (–0.04/+0.04) | 0.22 (–0.04/+0.05) | 0.006 |
| Relaxation velocity (μm/s) | 0.24 (–0.03/+0.03) | 0.18 (–0.03/+0.04) | 0.012 |
| Cell area (μm2) | 1000 (–50/+50) | 1800 (–200/+200) | 0.002 |
| Aspect ratio | 6.7 (–0.3/+0.2) | 6.1 (–0.5/+0.5) | 0.028 |
| Sarcomere length (μm) | 1.65 (–0.03/+0.04) | 1.77 (–0.05/+0.06) | 0.004 |
| Sarcomere length on hydrogel (μm) | 1.71 (–0.06/+0.06) | 1.75 (–0.06/+0.06) | 0.342 |
Average traction force, contraction power, and contraction and relaxation velocities of WT (n = 158) and R134G (n = 97) hiPSC-CMs were measured using traction force microscopy (see Figure 7). Average cell areas, aspect ratios, and sarcomere lengths were determined from immunofluorescence images of WT (n = 120) and R134G (n = 39) hiPSC-CMs on rectangular patterns on glass (see Figure 6). Sarcomere lengths were also measured for WT (n = 33) and R134G (n = 31) hiPSC-CMs on rectangular patterns on hydrogel. Reported uncertainties are 95% confidence intervals determined by bootstrapping.
R134G decreases the force generated during cardiomyocyte contraction
To test whether R134G affects contractility, as predicted from the molecular mechanism, we performed traction force microscopy on hiPSC-CMs to measure cardiomyocyte contraction. Spontaneously beating hiPSC-CMs were seeded onto 10 kPa hydrogels micropatterned with extracellular matrix in rectangles with a 7:1 aspect ratio. This patterning onto hydrogels of physiological stiffness promotes sarcomeric alignment and cellular maturation (Ribeiro et al., 2015). The displacement of fluorescent microbeads embedded in the hydrogels was used to monitor cardiomyocyte beating (Figure 7A). The force of contraction for each cell was calculated using a MATLAB program developed by the Pruitt laboratory (Ribeiro et al., 2017). The contractile force exerted by R134G cardiomyocytes (0.17 [–0.03/+0.04] μN) was lower than the WT force (0.27 [–0.03/+0.03] μN; p = 0.002) (Figure 7; Table 1). The R134G variant also reduced the contractile power exerted by the cells, as well as the contraction and relaxation velocities, relative to WT (Figure 7; Table 1). These effects are consistent with our proposed molecular mechanism as well as the diminished force production observed for other known pathogenic DCM mutations (Sun et al., 2012; Clippinger et al., 2019).
FIGURE 7:
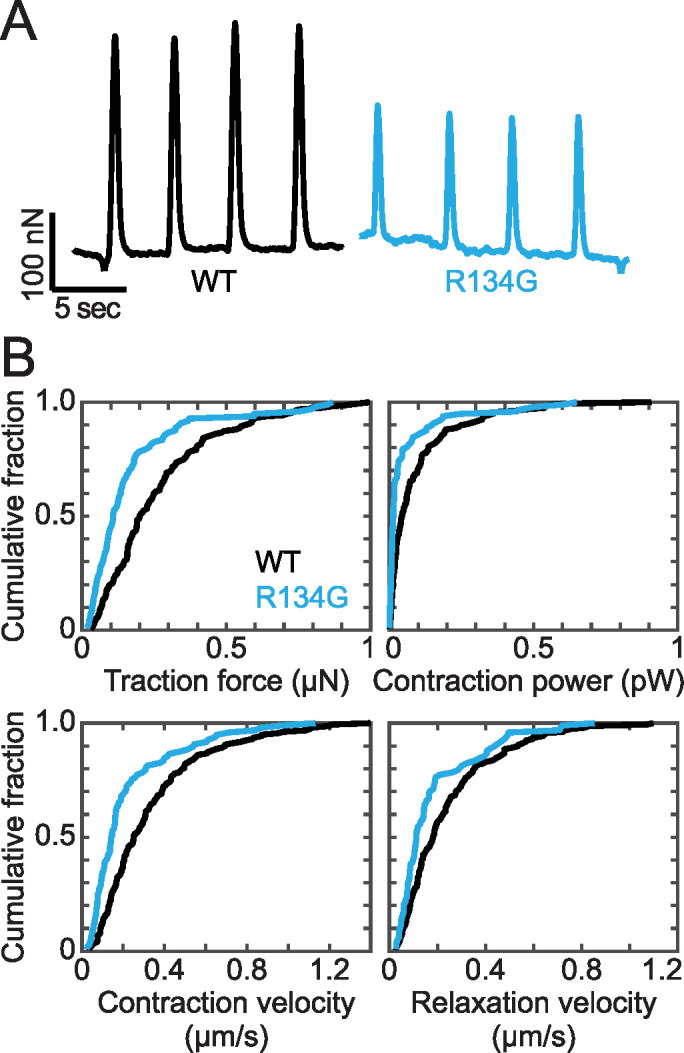
(A) Representative force transients of beating hiPSC-CMs measured using traction force microscopy. R134G hiPSC-CMs show reduced contractility compared with WT. (B) Quantification of force, power, and contraction and relaxation velocities measured using traction force microscopy. Data are plotted as cumulative distributions, and parameter values are given in Table 1. R134G (n = 97 cells) decreased the traction force (p = 0.002), power (p = 0.018), and contraction (p = 0.006) and relaxation (p = 0.012) velocities relative to WT (n = 158 cells).
DISCUSSION
Evidence for the pathogenicity of R134G
One challenge for the field has been connecting genotype and patient phenotype, in part because many variants occur in a small number of patients, with insufficient numbers to draw firm conclusions about the pathogenicity of a given variant. Mechanistic studies can inform whether a given variant is likely pathogenic or not; however, these mechanistic studies are currently not a substitute for clinical genetics studies.
Our molecular studies of the R134G variant revealed decreased calcium sensitivity for activation, similar to those observed for troponin T mutations known to be pathogenic for DCM (Hershberger et al., 2009; Gangadharan et al., 2017; Clippinger et al., 2019). Previous studies of R134G force development in porcine skinned cardiac muscle fibers or in vitro actomyosin ATPase activity each detected small decreases in calcium sensitivity that did not reach statistical significance (Hershberger et al., 2009; Gangadharan et al., 2017). Here, we report a statistically significant decrease in the calcium sensitivity of sliding speed in our in vitro motility measurements. Moreover, we generated a mutant stem cell line carrying the R134G variant that was isogenic to the parent WT line, except for the mutation. These cells showed reduced contractile function, sarcomeric disarray, and changes in cell size that are consistent with studies of known pathogenic DCM mutations in hiPSC-CMs (Sun et al., 2012; Hinson et al., 2015; Clippinger et al., 2019; Lee et al., 2019). Cellular hypocontractility is also consistent with the systolic dysfunction associated with DCM (Mestroni et al., 1999; Davis et al., 2016). Therefore, the data presented here are consistent with the classification of R134G as a pathogenic variant.
Uncoupling of calcium binding and tropomyosin positioning
The R134G variant decreased the calcium sensitivity of RTF motility (Figure 2) despite the increased affinity of TnC for calcium within RTFs containing R134G TnT, relative to WT (Figure 4). The effect of a mutation in TnT on calcium binding by TnC is consistent with previous work showing that two mutations at the R92 position of troponin T differentially altered calcium binding through a complex network of allosteric interactions (Williams et al., 2016). Our data suggest that the R134G mutation alters allosteric communication among thin filament proteins such that the coupling between tropomyosin positioning and calcium binding to troponin C is reduced. This is consistent with a study showing that the nearby hypertrophic cardiomyopathy mutation ΔE160 can affect the coupling between the troponin core complex and the tropomyosin overlap region (Abdullah et al., 2019).
The notion that R134G affects the coupling between calcium binding and tropomyosin movement is also supported by our measurements of tropomyosin positioning (Figure 5). The activation of thin filaments regulated by WT troponin contains two calcium-dependent transitions, namely between the blocked and closed states and between the closed and open states (McKillop and Geeves, 1993). R134G abolishes the calcium dependence of KT, the equilibrium constant between the closed and open states of the thin filament, without affecting KB (Figure 5). Thus, the R134G mutation uncouples the closed-to-open transition of the thin filament from calcium binding to troponin.
The uncoupling of tropomyosin positioning from calcium binding results in different effects of R134G on thin filament activation at different calcium concentrations. At low calcium (pCa 9), the larger value of KT for R134G signifies increased thin filament activation relative to WT; however, the overall level of activation for both WT and R134G RTFs is low because both KB and KT are small (≤0.3), indicating that most of the regulatory units are in the blocked state. Consistent with this observation, the fraction of stuck filaments in the in vitro motility assay at pCa 9 is the same for both the WT and mutant troponin. Therefore, the increase in KT at pCa 9 is not sufficient to increase the motility of RTFs containing R134G troponin (Figure 2). At pCa 4, the value of KT is small for R134G relative to WT, which demonstrates reduced activation at high calcium concentrations and could account for the decrease in Vmax in the in vitro motility assay. A shift in equilibrium tropomyosin positioning toward the closed state at saturating calcium levels is also consistent with reduced force generation, as has been described for a double mutation in troponin C thought to cause dilated cardiomyopathy (Dweck et al., 2010).
A mutation-induced reduction in thin filament activation at saturating calcium concentrations may also help explain the shift in calcium sensitivity of RTF motility (Figure 2). The motility rate in the absence of regulatory proteins is frequently assumed to be limited by the rate of actomyosin dissociation (Huxley, 1990); however, we do not observe a change in the rate of ADP release with the mutant protein (Figure 3). Therefore, it is likely that the mutant is affecting attachment kinetics, which is reasonable because the transition to strong binding is regulated by troponin and tropomyosin. Therefore, the reduced maximal speed in the motility assay is consistent with the reduced actin-activated ATPase rate measured with R134G-RTFs at fully activating calcium concentrations (Gangadharan et al., 2017).
Although pCa 9 and 4 are extreme values relative to the typical range of calcium concentrations in a physiological calcium transient, pCa 6–7 (Bers, 2002), we believe that measurements at pCa 9 and 4 provide meaningful insight into relaxed and fully activated conditions, respectively. We did not detect a significant difference in the value of KT at pCa 6.25, but we did observe decreased motility at intermediate calcium concentrations for R134G relative to WT. This discrepancy could be due to differences in the sensitivity of the respective assays and/or slight differences in the calcium-dependent behavior measured under different conditions (i.e., different buffers, protein concentrations, etc.). Alternatively, these results may suggest that the observed decrease in thin filament cooperativity is the primary physiological effect of the mutation.
Reduced coupling of myosin binding to thin filament activation
Our measurements also revealed a decrease in the cooperativity of myosin binding to thin filaments regulated by R134G TnT, relative to WT. This is important because full thin filament activation depends on both calcium and myosin binding (Houmeida et al., 2010). This finding is qualitatively consistent with the decreased Hill coefficient of actomyosin ATPase activity observed for R134G TnT relative to WT (Gangadharan et al., 2017). While we did not observe a statistically significant change in the Hill coefficient in the motility assay, this could be due to the large uncertainty in determining this value. Moreover, it should be noted that the Hill coefficients measured for motility, ATPase activity, and myosin binding to RTFs in equilibrium titrations are not necessarily equivalent. That being said, decreased cooperativity of thin filament activation would result in decreased accessibility of myosin binding sites along the thin filament. This contributes to the decreased calcium sensitivity in the in vitro motility assay because a decrease in myosin-induced activation should result in a requirement for additional calcium-based activation to achieve the same level of thin filament activation. Decreased accessibility of myosin binding sites along the thin filament is also predicted to reduce force generation in cardiomyocytes.
Potential structural mechanism of reduced cooperativity
We can gain insights into the potential structural mechanism of the reduced coupling of thin filament activation to myosin binding by examining recent crystal and cryogenic electron microscopy structures (Yamada et al., 2020; Risi et al., 2021). The core of the troponin complex consists of troponin C, most of troponin I, and the C-terminal region of troponin T (Takeda et al., 2003). The N-terminal 2/3 of troponin T stretches from this core complex in an elongated conformation that interacts with tropomyosin filaments on both sides of the thin filament. This extended structure thus couples the core complex to tropomyosin. R134 is located within the region of TnT that binds the tropomyosin overlap region (Jin and Chong, 2010; Gangadharan et al., 2017), near the end of the linker between the core complex and the tropomyosin on the other side of the thin filament (see Figure 1). In vitro measurements of tropomyosin-TnT binding affinity revealed that the R134G variant, along with several other DCM-causing mutations within the TnT N-terminal domain, increases the affinity of TnT for tropomyosin (Gangadharan et al., 2017). This result is consistent with the notion that altered interactions between TnT and tropomyosin can be important for cardiomyopathy pathogenesis (Palm et al., 2001; Manning et al., 2012; Gangadharan et al., 2017; McConnell et al., 2017; Madan et al., 2020). Additionally, it has been shown that hypertrophic cardiomyopathy-associated mutations impair the ability of a TnT fragment (TnT70-170) to increase the affinity of tropomyosin for actin and to stabilize the tropomyosin overlap region, where tropomyosin molecules interact in a head-to-tail manner (Palm et al., 2001). Taken together, these observations suggest that mutation-induced alterations in TnT-tropomyosin contacts could affect the head-to-tail interactions within the tropomyosin polymer as well as the actin-tropomyosin binding interface. We propose that these changes could contribute to the decrease in cooperativity of regulated actomyosin binding induced by the R134G mutation. Further structural and computational studies are needed to shed light on this potential mechanism.
Cellular consequences of the R134G variant
Our molecular studies suggest that the initial molecular insult driving disease pathogenesis is altered thin filament regulation that decreases thin filament activation, which is consistent with the decreased sarcomeric contractility observed in our stem cell–derived cardiomyocyte model of R134G (Figure 7; Table 1). This molecular and cellular hypocontractility has also been observed with other DCM-causing mutations (Sun et al., 2012; Davis et al., 2016; Yuan et al., 2018; Clippinger et al., 2019). We also observed downstream effects of the initial molecular insult in hiPSC-CMs engineered to express R134G TnT; namely, sarcomeric disorganization and increased cell area (Figure 6). These observations suggest that the initial molecular insult of altered mechanics can drive downstream alterations in cellular function beyond contractility. The exact mediators of these cellular changes are unknown; however, they likely involve the activation of mechanobiological signaling pathways.
Implications for precision medicine
Our study adds to the growing body of evidence that even within the same gene, not all mutations will have the same underlying molecular mechanism (Harada and Potter, 2004; Schmitt et al., 2006; Williams et al., 2016; Barrick et al., 2019; Clippinger et al., 2019, 2021; Ezekian et al., 2020). For example, a previous study of another DCM-causing mutation in troponin T, ΔK210, also showed decreased calcium sensitivity of RTF motility and increased cell size in hiPSC-CMs, in addition to hypocontractility and sarcomeric disorganization (Clippinger et al., 2019). However, the molecular-level effects of R134G are distinct from those of ΔK210, which uniformly decreased thin filament activation (KT) without altering cooperativity (nH) in steady-state measurements of regulated actomyosin binding.
Our observation of the increased calcium binding affinity of R134G troponin was unexpected because increased calcium binding to the troponin complex has been associated with mutations causing hypertrophic cardiomyopathy and molecular hypercontractility, not dilated cardiomyopathy (Robinson et al., 2007, 2018). However, as we demonstrate here, measurement of calcium binding affinity alone is insufficient to determine the consequences on thin filament regulation and contractility. The sarcomere is a complicated macromolecular complex, and mutation-induced changes in protein structure and function can lead to alterations in its interactions with multiple binding partners. As such, our results reiterate the complexity of disease pathogenesis and the value of multiscale characterization of disease-causing mutations. Although the molecular-level results do not uniformly predict molecular hypocontractility, the cellular phenotype conferred by the R134G variant is wholly consistent with cellular hypocontractility and DCM.
These results have important implications for the design of precision medicine therapeutics. Multiple molecular insults can lead to DCM. This heterogeneity has made it difficult to design therapeutics for DCM that work for all patients. We propose binning patient mutations based on the underlying molecular mechanism and designing precision therapeutics for these patient subpopulations based on reversing the underlying molecular mechanism (Greenberg and Tardiff, 2021; Lavine and Greenberg, 2021). Although the R134G variant increases calcium binding to the thin filament, a calcium-desensitizing drug that renormalizes this calcium binding would likely exacerbate the disease phenotype rather than repair it. Conversely, a drug designed to treat DCM by increasing the affinity of TnC for calcium would not be likely to improve the condition of a patient carrying the R134G mutation, but such a drug might be effective for DCM-causing mutations shown to decrease the calcium affinity of TnC within RTFs (Robinson et al., 2007; Liu et al., 2012). We speculate that omecamtiv mecarbil (Malik et al., 2011), which increases thin filament activation by increasing myosin binding (Woody et al., 2018), would be better tailored for repairing the molecular insult of R134G. In summary, our results emphasize the value of mechanism-based precision medicine in treating genetic cardiomyopathies.
Limitations of our study
It is important to note that our study has several limitations in the extrapolation to DCM more generally. Stem cell–derived cardiomyocytes are developmentally immature, and therefore, give windows into the early disease pathogenesis, rather than late-stage disease (Musunuru et al., 2018). Although our results are consistent between the molecular and cellular levels, the contractility in hiPSC-CMs can also be modified by factors beyond those considered here, including adaptive changes in calcium handling, protein isoform switching, and posttranslational modifications of proteins. These cellular changes will vary over the course of the disease progression. In addition, we used homozygous isogenic lines to facilitate the comparison between the molecular and cellular levels, whereas patients are usually heterozygous. That being said, homozygous cells carrying DCM mutations can recapitulate many disease features seen with heterozygous lines (Hinson et al., 2015). Finally, it should be noted that although we see changes consistent with a DCM phenotype with R134G at both the molecular and cellular levels, definitive determination of pathogenicity in patients requires further genetic and clinical studies.
Conclusions
In summary, we provide functional data supporting the classification of the R134G mutation in TnT as a likely pathogenic mutation. Specifically, R134G induces a statistically significant decrease in calcium sensitivity of regulated actomyosin motility, which results in decreased cardiomyocyte force generation. Our data suggest that decreased cooperativity of regulated actomyosin binding and uncoupling of calcium binding from tropomyosin movement are the primary drivers of R134G pathogenicity, which is, to our knowledge, a novel pathogenic mechanism. These results emphasize the value of a precision medicine approach to treating cardiomyopathies, wherein knowledge of mutation-specific effects on cardiac function are used to more effectively treat patients diagnosed with genetic DCM.
MATERIALS AND METHODS
Request a protocol through Bio-protocol.
Protein expression and purification
Cardiac ventricular myosin and actin were each prepared from cryoground porcine ventricles (Pelfreez) as previously described (Greenberg et al., 2014). Myosin S1 was isolated from full-length myosin by digestion with chymotrypsin (Margossian and Lowey, 1982; Eads et al., 1984), and its purity was assessed using SDS–PAGE. Cardiac actin was labeled with N-(1-pyrenyl)iodoacetamide (pyrene) as described in Pollard (1984). Protein concentrations were measured spectroscopically for actin (290 nm), pyrene-actin (290 and 344 nm), and myosin S1 (280 and 320 nm). The concentration of full-length myosin was measured using a Bradford assay.
Recombinant human tropomyosin (NP_001018005.1) was expressed in BL21-CodonPlus cells (Agilent) and purified using established protocols (Hitchcock-DeGregori and Heald, 1987), as we have done previously (Barrick et al., 2019; Clippinger et al., 2019). The concentration of tropomyosin was determined by measuring the absorbance at 280 nm. To remove aggregates before each experiment, tropomyosin was reduced by the addition of 50 mM dithiothreitol (DTT), heated at 56°C for 5 min, and allowed to cool to room temperature before centrifuging for 30 min at 436,000 × g at 4°C in an Optima MAX-TL Ultracentrifuge (Beckman Coulter) equipped with a TLA 120.2 rotor (Beckman Coulter).
Human recombinant troponins I (NP_000354.4), C (NP_003271.1), and T (NP_001001430.1) were expressed in BL21-CodonPlus cells, complexed, and purified using established protocols (Kozaili et al., 2010), with the modification that the complexed protein was purified using a MonoQ column (GE Healthcare). The concentration of the troponin complex was measured spectroscopically using a Bradford assay (Coomassie Plus Assay kit; Thermo Scientific). The R134G mutation was introduced into troponin T using the QuikChange Site-Directed Mutagenesis Kit (Agilent) and verified by sequencing (GENEWIZ). R134G troponin T was expressed, complexed with troponins I and C, and purified using the same procedure as the WT protein. All assays were conducted using two independent preparations of the R134G troponin T variant.
In vitro motility assay
Experiments were conducted as previously described (Clippinger et al., 2019). Unless otherwise specified, reagents were diluted in KMg25 buffer (25 mM KCl, 2 mM ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), 60 mM MOPS, pH 7.0, 4 mM free MgCl2, 1 mM DTT). Phalloidin-stabilized F-actin (unlabeled or rhodamine labeled) was prepared by incubating G-actin (23.8 or 2 µM) with 26.2 µM phalloidin or 3.75 µM rhodamine-phalloidin for at least 30 min at room temperature. Before measurements, nonfunctional myosin “dead heads” were removed by centrifuging myosin solution (2 µM myosin, 4 µM phalloidin-stabilized F-actin, 1 mM ATP in high-salt buffer [KMg25 with 300 mM KCl]) for 30 min at 436,000 × g at 4°C in an Optima MAX-TL Ultracentrifuge (Beckman Coulter) equipped with a TLA 120.2 rotor (Beckman Coulter). The concentration of myosin in the supernatant was measured using a Bradford reagent (Coomassie Plus Assay kit; Thermo Scientific) and diluted to the desired concentration in high-salt buffer. Coverslips (VWR) were cleaned with compressed air and coated with 1% nitrocellulose in amyl acetates (both from Electron Microscopy Sciences) using the side of a pipette tip to spread the nitrocellulose across the coverslip. Nitrocellulose-coated coverslips were allowed to dry in a covered container for at least 30 min before the flow cells were assembled.
Flow cells were created by fixing a nitrocellulose-coated coverslip to an uncoated coverslip using double-sided tape and vacuum grease. For each measurement, a flow cell was loaded by pipetting 50 µl (∼1 volume) of each of the following reagents into the channel: myosin (0.2–0.4 µM, 1 min); bovine serum albumin (1 mg/ml, 2 × 1 min); phalloidin-stabilized F-actin (1 µM, 2 × 1 min); Mg·ATP (5 mM each ATP and MgCl2, 100 µl wash); KMg25 (200 µl wash); rhodamine-labeled phalloidin-stabilized F-actin (40 nM, 1 min); tropomyosin and troponin (0.2 µM each, 1 min); activation buffer (KMg25 supplemented with 5 mM MgCl2, 10 mM DTT, 5 mM ATP, 0.5% methylcellulose, 0.2 µM each tropomyosin and troponin, 2.5 mg/ml glucose, 5 U/ml glucose oxidase [Sigma], 11 µg/ml catalase [Sigma], and CaCl2 to the desired calcium concentration). The added calcium required to achieve each desired final free calcium concentration was determined using MaxChelator (Bers et al., 2010).
Flow cells were imaged using an IX70 fluorescence microscope (Olympus) controlled by Ocular software (QImaging). Rhodamine fluorescence was excited through a 532 nm filter, and movies were captured by a Retiga R1 charge-coupled device camera (QImaging) with an exposure time of 400 ms. The frame rate and total number of frames were empirically set for each calcium concentration: typically 20 frames at 3-s intervals for high calcium and 10 frames at 10-s intervals for low calcium. Pixels were binned 2 × 2 to improve the signal-to-noise ratio. The percent stuck filaments was estimated by manually counting the gliding filaments within a randomly selected area of each video. Filament speeds were determined by manually tracking gliding filaments using the MTrackJ plug-in in ImageJ (National Institutes of Health [NIH]). The speed at each calcium concentration was determined as the average of the speeds of n = 150 filaments from three independent experimental days (with 2–3 technical replicates per experimental day). For calcium concentrations that did not support continuous, directional filament gliding (pCa > 6.5 for WT, pCa > 5.75 for R134G), the speed was recorded as 0. The pCa50, the calcium concentration that supported half-maximal filament gliding speed, was determined by fitting a Hill equation to the mean filament speed as a function of pCa. Statistical significance was assessed from the 95% confidence intervals of the fit parameters (Altman and Bland, 2011).
Stopped-flow kinetic measurements of ADP release
Stopped-flow transient kinetic measurements were carried out in buffer containing 60 mM MOPS, 200 mM KCl, 5 mM MgCl2, 1 mM DTT, 2 mM EGTA, and 2.15 mM CaCl2 (pCa 4). Total concentrations of calcium, magnesium, ATP, and EGTA added were calculated using MaxChelator (Bers et al., 2010). Phalloidin-stabilized, pyrene-labeled actin filaments were prepared by incubating 50 µM pyrene-actin with 55 µM phalloidin for at least 10 min at room temperature.
Fluorescent thin filaments (0.5 µM phalloidin-stabilized pyrene-actin, 1 µM each troponin and tropomyosin) were preincubated with myosin S1 (0.5 µM) and Mg·ADP (50 µM) and Mg·ATP (2.5 mM), as previously described (De La Cruz and Ostap, 2009; Clippinger et al., 2019). All concentrations refer to the final concentrations after mixing. Release of ADP from the myosin active site (the rate-limiting step) is followed by rapid binding of ATP, which results in myosin dissociation from the thin filament and an exponential increase in pyrene fluorescence. A single experiment consisted of recording at least three kinetic time traces followed by fitting of a single exponential function to the averaged traces. The reported rate constant (k) is the average of three (WT) or two (R134G) independent experiments, and reported errors are the propagated SEs from the fits. Statistical significance was assessed using a Student’s two-tailed t test executed in Excel (Microsoft).
Stopped-flow kinetic measurements of regulated actomyosin binding
Stopped-flow transient kinetic measurements were carried out in buffer containing 60 mM MOPS, pH 7.0, 200 mM KCl, 5 mM MgCl2, 1 mM DTT, 2 mM EGTA, and either 5.2 µM CaCl2 (pCa 9) or 2.15 mM CaCl2 (pCa 4). Total concentrations of calcium, magnesium, ATP, and EGTA added were calculated using MaxChelator (Bers et al., 2010). Phalloidin-stabilized, pyrene-labeled actin filaments were prepared by incubating 50 µM pyrene-actin with 55 µM phalloidin in pCa 9 buffer at least 10 min at room temperature.
Fluorescent RTFs (2.5 µM phalloidin-stabilized pyrene-actin, 1 µM each troponin and tropomyosin) were rapidly mixed with 0.25 µM myosin S1 using an SX20 stopped-flow mixer (Applied Photophysics). Apyrase VII (0.02 U/ml; Sigma) was included in each protein solution to ensure the absence of nucleotide (i.e., ATP and ADP). All concentrations are given as final concentrations after mixing. Pyrene was excited at 365 nm, and the fluorescence emission was detected using a 395 nm cutoff filter. The fluorescence of the pyrene-actin in the thin filaments was quenched by myosin strong binding, yielding an exponential decrease in fluorescence. KB, the equilibrium constant between the blocked and closed states of the thin filament, was calculated by taking the ratio of the rate constants of myosin binding at low (k [–Ca2+]; pCa 9) and high (k [+Ca2+]; pCa 4) calcium, as described by McKillop and Geeves (1993):
| (1) |
A single experiment consisted of recording at least three kinetic time traces at each calcium concentration followed by fitting of a single exponential function to the averaged traces. The reported KB values are the average of 10 independent experiments, and reported errors are the SDs of these values. The statistical significance between KB values was evaluated using a Student’s two-tailed t test executed in Excel (Microsoft).
Fluorescence titrations of myosin binding to RTFs
Fluorescence titrations were performed at 20°C in buffer containing 60 mM MOPS, pH 7.0, 200 mM KCl, 5 mM MgCl2, 1 mM DTT, 2 mM EGTA, and either 5.2 µM CaCl2 (pCa 9), 1.19 mM CaCl2 (pCa 6.25), or 2.15 mM CaCl2 (pCa 4). Phalloidin-stabilized pyrene-actin was prepared as described above. Measurements were performed using a Fluoromax spectrophotometer (Horiba Scientific) equipped with a TC 1 temperature controller (Quantum Northwest) and a Microlab 600 autotitrator (Hamilton). Myosin S1 was added to continuously stirred RTFs (0.5 µM phalloidin-stabilized pyrene-actin and 0.27 µM each troponin and tropomyosin) in the presence of 2 mM ADP, as well as 50 µM P1,P5-di(adenosine-5′) pentaphosphate (Ap5a), 1 µM hexokinase, and 2 mM glucose to maintain the nucleotide in the ADP state, as described previously (McKillop and Geeves, 1993; Barrick et al., 2019; Clippinger et al., 2019). S1 was added at 1-min intervals in increments of 0.05 µM up to 0.2 µM, then at 0.2 µM increments up to 2 µM, and finally at 1 µM increments up to 10 µM. The steady-state pyrene fluorescence was measured at 1-min intervals. Six technical replicates per condition were collected.
The fraction of myosin-bound subunits, f ([m]), is given by the fractional change in pyrene fluorescence upon myosin binding, which is related to parameters describing thin filament activation by the following equation (McKillop and Geeves, 1993):
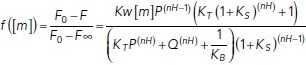 |
(2) |
where F is the measured pyrene fluorescence; F0 and F∞ are the pyrene fluorescence in the absence of myosin and at saturating myosin, respectively; [m] is the concentration of myosin; KW and KS are the equilibrium constants for weak and strong myosin binding, respectively; KB and KT are the equilibrium constants for the described blocked-to-closed and closed-to-open transitions of the thin filament, respectively; nH is the Hill coefficient for myosin binding to RTFs; P = 1 + [m]*KW(1 + KS); and Q = 1 + [m]*KW. KB at pCa 9 was set to the value determined by stopped-flow kinetic measurements (see above). KB at high calcium and KS were set to 20 and 18, respectively, based on McKillop and Geeves (1993). The best-fit values and 95% confidence intervals for nH, KW, and KT were determined by fitting Eq. 2 to the data using a MATLAB-based computational tool (Barrick et al., 2019). The statistical significance of differences in these parameter values was determined by bootstrapping, as previously described (Barrick et al., 2019).
IAANS titrations
TnC carrying three mutations (C35S, T53C, C84S) was expressed, purified, and labeled with 2-(4′-(iodoacetamido)anilino)napthalene-6-sulfonic acid (IAANS; Toronto Research Chemicals) following published protocols (Davis et al., 2007; Clippinger et al., 2021). This TnCT53C variant is fluorescently labeled by IAANS at its single cysteine residue. IAANS fluorescence is sensitive to the fluorophore environment, allowing IAANS-TnCT53C to report on the conformational state of TnC. IAANS-TnCT53C was complexed with troponin I and either WT or R134G troponin T as described above.
IAANS titrations were carried out in buffer containing 10 mM MOPS, 150 mM KCl, 3 mM MgCl2, and 1 mM DTT. Phalloidin-stabilized actin filaments were prepared by incubating 50 µM G-actin with 55 µM phalloidin for at least 30 min at room temperature. Measurements were performed using the Fluoromax system described above. Ca2+ was added to continuously stirred RTFs (2 µM phalloidin-stabilized actin, 0.45 µM tropomyosin, 0.15 µM troponin complex containing IAANS-TnCT53C) in the presence of 2 mM EGTA and 4.1 µM CaCl2 (pCa 9). Volumes of added calcium required to obtain the desired final calcium concentrations were calculated using MaxChelator (Bers et al., 2010) and automatically added by the autotitrator. IAANS fluorescence was excited at 330 nm, and the emission spectrum (370 to 550 nm) was recorded at 1-min intervals. Five technical replicates per condition were collected. The IAANS fluorescence was plotted against calcium concentration and fitted by a Hill equation to determine the pCa50 as previously described (Davis et al., 2007), with the modification that we analyzed the integrated fluorescence from 530 to 550 nm. The statistical significance of the difference in pCa50 for WT versus R134G was assessed by calculating a p value from the 95% confidence intervals of the fits (Altman and Bland, 2011). Plots and fits were generated in MATLAB (MathWorks), and all other analysis was performed in Excel (Microsoft).
Preparation of human induced pluripotent stem cell–derived cardiomyocytes
Parent and homozygous mutant induced pluripotent stem cells were generated from the human BJ fibroblast line (CRL-2522; American Type Culture Collection) by the Genome Engineering and iPSC Center (GEiC, Washington University in St. Louis) with guide RNA targeting TTCTCCTTAGGAGAGACGTCGGG and validated as previously described (Clippinger et al., 2019). We previously showed by whole exome sequencing that the parent line has no known mutations associated with cardiomyopathy (Clippinger et al., 2019). Cell identity was confirmed by the GEiC using short tandem repeat (STR) sequencing. These cells had normal karyotypes (Cell Line Genetics; Figure 8) and were tested for mycoplasma by the GEiC. Gene-edited cells were pluripotent, as determined by immunofluorescence staining for SSEA4, OCT4, SOX2, and TRA-1-61 using the Pluripotent Stem Cell 4-Marker Immunocytochemistry kit (A25526; Thermo Fisher) (Figure 8). Cells were cultured in feeder-free conditions as previously described (Clippinger et al., 2019). These hiPSCs were differentiated into cardiomyocytes (hiPSC-CMs) by temporal modulation of Wnt signaling as previously described (Lian et al., 2013; Clippinger et al., 2019). hiPSC-CMs started spontaneously beating 7–10 d after the differentiation was initiated, and after metabolic selection, >90% hiPSC-CMs were obtained, as previously described (Clippinger et al., 2019). hiPSC-CMs were aged at least 30 d in culture before being used, and at least three differentiations were used for the collection of all data sets.
FIGURE 8:

Validation of gene-edited hiPSCs carrying the R134G mutation in troponin T. (A) Immunofluorescence staining for pluripotency markers SSEA4, OCT4, SOX2, and TRA-1-61 in R134G hiPSCs. Cell nuclei are visualized using 4’,6-diamidino-2-phenylindole (DAPI) stain. (B) Karyotypes of R134G hiPSCs are normal.
Immunofluorescence imaging
(hiPSC-CMs were immunostained for α-actinin and troponin I (1:500 anti–α-actinin [A7811; Sigma]; 1:300 Cy2 anti-mouse [715225150; Jackson ImmunoResearch]; 1:300 anti–troponin I [SC-15368; Santa Cruz Biotechnology]; 1:300 Cy3 anti-rabbit [711165152; Jackson ImmunoResearch]) and imaged using confocal microscopy, as previously described (Clippinger et al., 2019). Cell areas were calculated from thresholded images in ImageJ (NIH). The sarcomere lengths were calculated from the Fourier transform of the sarcomeric staining, as described in Pasqualini et al. (2015). Some data for WT hiPSC-CMs were previously published in (Clippinger et al., 2019). The statistical significance of differences was assessed using a MATLAB-based computational tool (Barrick et al., 2019).
Traction force microscopy
Traction force microscopy was performed as previously described (Clippinger et al., 2019). Briefly, singularized hiPSC-CMs were seeded onto rectangular Geltrex (Thermo Fisher) patterns stamped onto 10 kPa hydrogels containing fluorescent marker beads. The spontaneous beating of hiPSC-CMs was monitored using confocal microscopy. Traction force videos were analyzed using a MATLAB program developed by the Pruitt lab (Ribeiro et al., 2017). Some data for WT hiPSC-CMs were previously published in (Clippinger et al., 2019). The statistical significance of differences in force generation was assessed using a MATLAB-based computational tool (Barrick et al., 2019).
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (R01 HL141086 to M.J.G.), the March of Dimes Foundation (FY18-BOC-430198 to M.J.G.), the Children’s Discovery Institute of Washington University and St. Louis Children’s Hospital (PM-LI-2019-829 to M.J.G.), and the Washington University Center for Cellular Imaging (WUCCI) (CDI-CORE-2015-505 to M.J.G.).
Abbreviations used:
- DCM
dilated cardiomyopathy
- hiPSC-CM
human induced pluripotent stem cell–derived cardiomyocyte
- myosin S1
myosin subfragment-1
- RTF
regulated thin filament
- TnC
troponin C
- TnT
troponin T.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E21-02-0082) on June 23, 2021.
REFERENCES
- Abdullah S, Lynn ML, McConnell MT, Klass MM, Baldo AP, Schwartz SD, Tardiff JC (2019). FRET-based analysis of the cardiac troponin T linker region reveals the structural basis of the hypertrophic cardiomyopathy-causing Δ160E mutation. J Biol Chem 294, 14634–14647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altman DG, Bland JM (2011). How to obtain the P value from a confidence interval. BMJ 343, d2304. [DOI] [PubMed] [Google Scholar]
- Bárány M (1967). ATPase activity of myosin correlated with speed of muscle shortening. J Gen Physiol 50, 197–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrick SK, Clippinger SR, Greenberg L, Greenberg MJ (2019). Computational tool to study perturbations in muscle regulation and its application to heart disease. Biophys J 116, 2246–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers DM (2002). Cardiac excitation-contraction coupling. Nature 415, 198–205. [DOI] [PubMed] [Google Scholar]
- Bers DM, Patton CW, Nuccitelli R (2010). A practical guide to the preparation of Ca2+ buffers. Methods Cell Biol 99, 1–26. [DOI] [PubMed] [Google Scholar]
- Clippinger SR, Cloonan PE, Greenberg L, Ernst M, Stump WT, Greenberg MJ (2019). Disrupted mechanobiology links the molecular and cellular phenotypes in familial dilated cardiomyopathy. Proc Natl Acad Sci USA 116, 17831–17840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clippinger SR, Cloonan PE, Wang W, Greenberg L, Stump WT, Angsutararux P, Nerbonne JM, Greenberg MJ (2021). Mechanical dysfunction of the sarcomere induced by a pathogenic mutation in troponin T drives cellular adaptation. J Gen Physiol 153, e202012787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis J, Davis LC, Correll RN, Makarewich CA, Schwanekamp JA, Moussavi-Harami F, Wang D, York AJ, Wu H, Houser SR, et al. (2016). A tension-based model distinguishes hypertrophic versus dilated cardiomyopathy. Cell 165, 1147–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JP, Norman C, Kobayashi T, Solaro RJ, Swartz DR, Tikunova SB (2007). Effects of thin and thick filament proteins on calcium binding and exchange with cardiac troponin C. Biophys J 92, 3195–3206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deacon JC, Bloemink MJ, Rezavandi H, Geeves MA, Leinwand LA (2012). Identification of functional differences between recombinant human α and β cardiac myosin motors. Cell Mol Life Sci 69, 2261–2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De La Cruz EM, Michael Ostap E (2009). Kinetic and equilibrium analysis of the myosin ATPase. Methods Enzymol 455, 157–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dweck D, Reynaldo DP, Pinto JR, Potter JD (2010). A dilated cardiomyopathy troponin C mutation lowers contractile force by reducing strong myosin-actin binding. J Biol Chem 285, 17371–17379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eads TM, Thomas DD, Austin RH (1984). Microsecond rotational motions of eosin-labeled myosin measured by time-resolved anisotropy of absorption and phosphorescence. J Mol Biol 179, 55–81. [DOI] [PubMed] [Google Scholar]
- Ezekian JE, Clippinger SR, Garcia JM, Yang Q, Denfield S, Jeewa A, Dreyer WJ, Zou W, Fan Y, Allen HD, et al. (2020). Variant R94C in TNNT2-encoded troponin T predisposes to pediatric restrictive cardiomyopathy and sudden death through impaired thin filament relaxation resulting in myocardial diastolic dysfunction. J Am Heart Assoc 9, e015111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangadharan B, Sunitha MS, Mukherjee S, Chowdhury RR, Haque F, Sekar N, Sowdhamini R, Spudich JA, Mercer JA (2017). Molecular mechanisms and structural features of cardiomyopathy-causing troponin T mutants in the tropomyosin overlap region. Proc Natl Acad Sci USA 114, 11115–11120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg MJ, Shuman H, Ostap EM (2014). Inherent force-dependent properties of β-cardiac myosin contribute to the force-velocity relationship of cardiac muscle. Biophys J 107, L41–L44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg MJ, Tardiff JC (2021). Complexity in genetic cardiomyopathies and new approaches for mechanism-based precision medicine. J Gen Physiol 153, e202012662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene LE, Eisenberg E (1980). Cooperative binding of myosin subfragment-1 to the actin-troponin-tropomyosin complex. Proc Natl Acad Sci USA 77, 2616–2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada K, Potter JD (2004). Familial hypertrophic cardiomyopathy mutations from different functional regions of troponin T result in different effects on the pH and Ca2+ sensitivity of cardiac muscle contraction. J Biol Chem 279, 14488–14495. [DOI] [PubMed] [Google Scholar]
- Hershberger RE, Parks SB, Kushner JD, Li D, Ludwigsen S, Jakobs P, Nauman D, Burgess D, Partain J, Litt M (2008). Coding sequence mutations identified in MYH7, TNNT2, SCN5A, CSRP3, LBD3, and TCAP from 313 patients with familial or idiopathic dilated cardiomyopathy. Clin Transl Sci 1, 21–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershberger RE, Pinto JR, Parks SB, Kushner JD, Li D, Ludwigsen S, Cowan J, Morales A, Parvatiyar MS, Potter JD (2009). Clinical and functional characterization of TNNT2 mutations identified in patients with dilated cardiomyopathy. Circ Cardiovasc Genet 2, 306–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinson JT, Chopra A, Nafissi N, Polacheck WJ, Benson CC, Swist S, Gorham J, Yang L, Schafer S, Sheng CC, et al. (2015). Titin mutations in iPS cells define sarcomere insufficiency as a cause of dilated cardiomyopathy. Science 349, 982–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitchcock-DeGregori SE, Heald RW (1987). Altered actin and troponin binding of amino-terminal variants of chicken striated muscle alpha-tropomyosin expressed in Escherichia coli. J Biol Chem 262, 9730–9735. [PubMed] [Google Scholar]
- Houmeida A, Heeley DH, Belknap B, White HD (2010). Mechanism of regulation of native cardiac muscle thin filaments by rigor cardiac myosin-S1 and calcium. J Biol Chem 285, 32760–32769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huxley HE (1990). Sliding filaments and molecular motile systems. J Biol Chem 265, 8347–8350. [PubMed] [Google Scholar]
- Jin JP, Chong SM (2010). Localization of the two tropomyosin-binding sites of troponin T. Arch Biochem Biophys 500, 144–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantor PF, Abraham JR, Dipchand AI, Benson LN, Redington AN (2010). The impact of changing medical therapy on transplantation-free survival in pediatric dilated cardiomyopathy. J Am Coll Cardiol 55, 1377–1384. [DOI] [PubMed] [Google Scholar]
- Kozaili JM, Leek D, Tobacman LS (2010). Dual regulatory functions of the thin filament revealed by replacement of the troponin I inhibitory peptide with a linker. J Biol Chem 285, 38034–38041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Jang W, et al. (2018). ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res 46, D1062–D1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavine KJ, Greenberg MJ (2021). Beyond genomics—technological advances improving the molecular characterization and precision treatment of heart failure. Heart Fail Rev 26, 405–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Termglinchan V, Diecke S, Itzhaki I, Lam CK, Garg P, Lau E, Greenhaw M, Seeger T, Wu H, et al. (2019). Activation of PDGF pathway links LMNA mutation to dilated cardiomyopathy. Nature 572, 335–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian X, Zhang J, Azarin SM, Zhu K, Hazeltine LB, Bao X, Hsiao C, Kamp TJ, Palecek SP (2013). Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/β-catenin signaling under fully defined conditions. Nat Protoc 8, 162–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Lee RS, Biesiadecki BJ, Tikunova SB, Davis JP (2012). Engineered troponin C constructs correct disease-related cardiac myofilament calcium sensitivity. J Biol Chem 287, 20027–20036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynn ML, Lehman SJ, Tardiff JC (2018). Biophysical derangements in genetic cardiomyopathies. Heart Fail Clin 14, 147–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madan A, Viswanathan MC, Woulfe KC, Schmidt W, Sidor A, Liu T, Nguyen TH, Trinh B, Wilson C, Madathil S, et al. (2020). TNNT2 mutations in the tropomyosin binding region of TNT1 disrupt its role in contractile inhibition and stimulate cardiac dysfunction. Proc Natl Acad Sci USA 117, 18822–18831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik FI, Hartman JJ, Elias KA, Morgan BP, Rodriguez H, Brejc K, Anderson RL, Sueoka SH, Lee KH, Finer JT, et al. (2011). Cardiac myosin activation: a potential therapeutic approach for systolic heart failure. Science 331, 1439–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning EP, Guinto PJ, Tardiff JC (2012). Correlation of molecular and functional effects of mutations in cardiac troponin T linked to familial hypertrophic cardiomyopathy: an integrative in silico/in vitro approach. J Biol Chem 287, 14515–14523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margossian SS, Lowey S (1982). Preparation of myosin and its subfragments from rabbit skeletal muscle. Methods Enzymol 85, 55–71. [DOI] [PubMed] [Google Scholar]
- Maytum R, Lehrer SS, Geeves MA (1999). Cooperativity and switching within the three-state model of muscle regulation. Biochemistry 38, 1102–1110. [DOI] [PubMed] [Google Scholar]
- McConnell M, Tal Grinspan L, Williams MR, Lynn ML, Schwartz BA, Fass OZ, Schwartz SD, Tardiff JC (2017). Clinically divergent mutation effects on the structure and function of the human cardiac tropomyosin overlap. Biochemistry 56, 3403–3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKillop DF, Geeves MA (1993). Regulation of the interaction between actin and myosin subfragment 1: evidence for three states of the thin filament. Biophys J 65, 693–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNally EM, Mestroni L (2017). Dilated cardiomyopathy: genetic determinants and mechanisms. Circ Res 121, 731–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mestroni L, Maisch B, McKenna WJ, Schwartz K, Charron P, Rocco C, Tesson F, Richter A, Wilke A, Komajda M (1999). Guidelines for the study of familial dilated cardiomyopathies. Eur Heart J 20, 93–102. [DOI] [PubMed] [Google Scholar]
- Mijailovich SM, Li X, Griffiths RH, Geeves MA (2012). The Hill model for binding myosin S1 to regulated actin is not equivalent to the McKillop-Geeves model. J Mol Biol 417, 112–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore JR, Campbell SG, Lehman W (2016). Structural determinants of muscle thin filament cooperativity. Arch Biochem Biophys 594, 8–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musunuru K, Sheikh F, Gupta RM, Houser SR, Maher KO, Milan DJ, Terzic A, Wu JC (2018). Induced pluripotent stem cells for cardiovascular disease modeling and precision medicine: a scientific statement from the American Heart Association . Circ Genomic Precis Med 11, e000043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm T, Graboski S, Hitchcock-DeGregori SE, Greenfield NJ (2001). Disease-causing mutations in cardiac troponin T: identification of a critical tropomyosin-binding region. Biophys J 81, 2827–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasqualini FS, Sheehy SP, Agarwal A, Aratyn-Schaus Y, Parker KK (2015). Structural phenotyping of stem cell-derived cardiomyocytes. Stem Cell Rep 4, 340–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel MD, Mohan J, Schneider C, Bajpai G, Purevjav E, Canter CE, Towbin J, Bredemeyer A, Lavine KJ (2017). Pediatric and adult dilated cardiomyopathy represent distinct pathological entities. JCI Insight 2, e94382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard TD (1984). Purification of a high molecular weight actin filament gelatin protein from Acanthamoeba that shares antigenic determinants with vertebrate spectrins. J Cell Biol 99, 1970–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro AJS, Ang Y-S, Fu J-D, Rivas RN, Mohamed TMA, Higgs GC, Srivastava D, Pruitt BL (2015). Contractility of single cardiomyocytes differentiated from pluripotent stem cells depends on physiological shape and substrate stiffness. Proc Natl Acad Sci USA 112, 12705–12710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro AJS, Schwab O, Mandegar MA, Ang YS, Conklin BR, Srivastava D, Pruitt BL (2017). Multi-imaging method to assay the contractile mechanical output of micropatterned human iPSC-derived cardiac myocytes. Circ Res 120, 1572–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risi CM, Pepper I, Belknap B, Landim-Vieira M, White HD, Dryden K, Pinto JR, Chase PB, Galkin VE (2021). The structure of the native cardiac thin filament at systolic Ca2+ levels. Proc Natl Acad Sci USA 118, 2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson P, Griffiths PJ, Watkins H, Redwood CS (2007). Dilated and hypertrophic cardiomyopathy mutations in troponin and α-tropomyosin have opposing effects on the calcium affinity of cardiac thin filaments. Circ Res 101, 1266–1273. [DOI] [PubMed] [Google Scholar]
- Robinson P, Liu X, Sparrow A, Patel S, Zhang YH, Casadei B, Watkins H, Redwood C (2018). Hypertrophic cardiomyopathy mutations increase myofilament Ca2+ buffering, alter intracellular Ca2+ handling, and stimulate Ca2+-dependent signaling. J Biol Chem 293, 10487–10499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt JP, Debold EP, Ahmad F, Armstrong A, Frederico A, Conner DA, Mende U, Lohse MJ, Warshaw D, Seidman CE, et al. (2006). Cardiac myosin missense mutations cause dilated cardiomyopathy in mouse models and depress molecular motor function. Proc Natl Acad Sci USA 103, 14525–14530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaddy RE, Boucek MM, Hsu DT, Boucek RJ, Canter CE, Mahony L, Ross RD, Pahl E, Blume ED, Dodd DA, et al. (2007). Carvedilol for children and adolescents with heart failure. J Am Med Assoc 298, 1171. [DOI] [PubMed] [Google Scholar]
- Siemankowski RF, Wiseman MO, White HD (1985). ADP dissociation from actomyosin subfragment 1 is sufficiently slow to limit the unloaded shortening velocity in vertebrate muscle. Proc Natl Acad Sci USA 82, 658–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun N, Yazawa M, Liu J, Han L, Sanchez-Freire V, Abilez OJ, Navarrete EG, Hu S, Wang L, Lee A, et al. (2012). Patient-specific induced pluripotent stem cells as a model for familial dilated cardiomyopathy. Sci Transl Med 4, 130ra47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung J, Nag S, Mortensen KI, Vestergaard CL, Sutton S, Ruppel K, Flyvbjerg H, Spudich JA (2015). Harmonic force spectroscopy measures load-dependent kinetics of individual human β-cardiac myosin molecules. Nat Commun 6, 7931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda S, Yamashita A, Maeda K, Maéda Y (2003). Structure of the core domain of human cardiac troponin in the Ca2+-saturated form. Nature 424, 35–41. [DOI] [PubMed] [Google Scholar]
- Tikunova SB, Liu B, Swindle N, Little SC, Gomes AV, Swartz DR, Davis JP (2010). Effect of calcium-sensitizing mutations on calcium binding and exchange with troponin C in increasingly complex biochemical systems. Biochemistry 49, 1975–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towbin JA, Lowe AM, Colan SD, Sleeper LA, Orav EJ, Clunie S, Messere J, Cox GF, Lurie PR, Hsu D, et al. (2017). Incidence, causes, and outcomes of dilated cardiomyopathy in children. J Am Med Assoc 296, 1867–1876. [DOI] [PubMed] [Google Scholar]
- Vibert P, Craig R, Lehman W (1997). Steric-model for activation of muscle thin filaments. J Mol Biol 266, 8–14. [DOI] [PubMed] [Google Scholar]
- Williams MR, Lehman SJ, Tardiff JC, Schwartz SD (2016). Atomic resolution probe for allostery in the regulatory thin filament. Proc Natl Acad Sci USA 113, 3257–3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woody MS, Greenberg MJ, Barua B, Winkelmann DA, Goldman YE, Ostap EM (2018). Positive cardiac inotrope omecamtiv mecarbil activates muscle despite suppressing the myosin working stroke. Nat Commun 9, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada Y, Namba K, Fujii T (2020). Cardiac muscle thin filament structures reveal calcium regulatory mechanism. Nat Commun 11, 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan CC, Kazmierczak K, Liang J, Zhou Z, Yadav S, Gomes AV, Irving TC, Szczesna-Cordary D (2018). Sarcomeric perturbations of myosin motors lead to dilated cardiomyopathy in genetically modified MYL2 mice. Proc Natl Acad Sci USA 115, E2338–E2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.