

Abstract
Gap junctions mediate direct cell-to-cell communication by forming channels that physically couple cells, thereby linking their cytoplasm, permitting the exchange of molecules, ions, and electrical impulses. Gap junctions are assembled from connexin (Cx) proteins, with connexin 43 (Cx43) being the most ubiquitously expressed and best studied. While the molecular events that dictate the Cx43 life cycle have largely been characterized, the unusually short half-life of Cxs of only 1–5 h, resulting in constant endocytosis and biosynthetic replacement of gap junction channels, has remained puzzling. The Cx43 C-terminal (CT) domain serves as the regulatory hub of the protein affecting all aspects of gap junction function. Here, deletion within the Cx43 CT (amino acids 256–289), a region known to encode key residues regulating gap junction turnover, is employed to examine the effects of dysregulated Cx43 gap junction endocytosis using cultured cells (Cx43∆256-289) and a zebrafish model (cx43lh10). We report that this CT deletion causes defective gap junction endocytosis as well as increased gap junction intercellular communication. Increased Cx43 protein content in cx43lh10 zebrafish, specifically in the cardiac tissue, larger gap junction plaques, and longer Cx43 protein half-lives coincide with severely impaired development. Our findings demonstrate for the first time that continuous Cx43 gap junction endocytosis is an essential aspect of gap junction function and, when impaired, gives rise to significant physiological problems as revealed here for cardiovascular development and function.
INTRODUCTION
Direct cell-to-cell communication is pivotal for the development and function of multicellular organisms. It is achieved by gap junctions (GJs), arrays (termed plaques) consisting of hundreds to thousands of GJ channels that traverse the plasma membranes of neighboring cells to allow the exchange of electrical currents, and small signaling molecules to a size of approximately 1.5 kDa (Thévenin et al., 2013; Falk et al., 2014). GJ channels are composed of two hemichannels (termed connexons) that dock head-on in the extracellular space to form the complete, double-membrane traversing GJ channel. Connexons consist of six connexin (Cx) proteins that traverse the membrane four times, with N-terminal and C-terminal (CT) domains located in the cytoplasm. Twenty-one different Cxs are expressed in humans (Söhl and Willecke, 2004), with connexin 43 (Cx43) being the most abundant, most widely expressed, and best studied Cx protein. While the molecular events that dictate the Cx43 life cycle, including protein biosynthesis in the endoplasmic reticulum, oligomerization, trafficking via the Golgi apparatus along microtubules, insertion into the plasma membrane, connexon docking, and channel clustering, have largely been characterized (Thévenin et al., 2013; Falk et al., 2016), the physiological role of GJ endocytosis is still not well understood. Yet, this aspect of the GJ life cycle is of particular interest as GJ Cxs have an unusually short half-life of only 1–5 h, requiring GJ channels to be constantly endocytosed and replaced (Fallon and Goodenough, 1981; Chu and Doyle, 1985; Hare and Taylor, 1991; Lauf et al., 2002; Gaietta et al., 2002; Berthoud et al., 2004; Piehl et al., 2007; Falk et al., 2009). So far, we can only speculate on why such a rapid GJ channel replacement is necessary. Consequently, we also do not know whether aberrant GJ endocytosis, caused, for example, by mutations that occur in the Cx43 amino acid sequence, may cause disease, a question of seemingly utmost importance.
The Cx43 CT, spanning residues 232–382 (232–381 in zebrafish Cx43), is a regulatory hub serving as a substrate for key posttranslational modifications and accessory protein binding (Lampe and Lau, 2004; Thévenin et al., 2013, 2017; Falk et al., 2014; Leithe et al., 2018). A coordinated series of phosphorylation and dephosphorylation events on serine and tyrosine residues in the Cx43 CT control trafficking of oligomerized Cx complexes (termed connexons or hemichannels) to the plasma membrane, recruitment and docking of connexons to GJ plaques regulated by the scaffolding protein, zonula occludens-1 (ZO-1), and finally, ubiquitin-dependent clathrin-mediated endocytosis of GJ channels (Park et al., 2006, 2009; Piehl et al., 2007; Solan and Lampe, 2007; Gumpert et al., 2008; Falk et al., 2009, 2012; Rhett et al., 2011; Fong et al., 2013; Thévenin et al., 2013; Cone et al., 2014; Dunn and Lampe, 2014).
To test whether impaired GJ endocytosis results in physiological defects, we replaced the wild-type (WT) Cx43 coding sequence with a mutant in which key residues that were previously determined to be critical for Cx43 GJ endocytosis, including the conserved AP-2/clathrin binding site, S2, the S279/282 phosphorylation site, the binding site for the E3-ligase Nedd4 that ubiquitinates K264 and K303, and the ubiquitination site at K264 (Fong et al., 2012, 2013, 2014; Nimlamool et al., 2015; Thévenin et al., 2017; Kells-Andrews et al., 2018), were deleted (Cx43Δ256-289) using CRISPR/Cas9 gene-editing technology in the zebrafish, Danio rerio. Choosing zebrafish, and not mice, as a model system is feasible as Cx43 GJs have conserved roles in vertebrate function and development across species (Chatterjee et al., 2005), and Cx43-encoded endocytic signals are largely conserved in mammals and teleosts (see Figure 1). In addition, the transparency of zebrafish embryos readily allows the detection of developmental defects that are not easily accessible in developing mouse embryos. We found that zebrafish harboring a homozygous deletion of amino acid residues 256–289 in the Cx43 CT domain (designated cx43lh10) exhibit, besides other defects, a suite of cardiovascular phenotypes including heart malformation, bradycardia, and aberrant vasculature structure and function correlating well with the known high expression level of Cx43 in the cardiovascular system and the well-established role of Cx43 in cardiovascular development and function (Severs, 1999; Jongsma and Wilders, 2000; Michela et al., 2015) that partially were rescued by the GJ-specific blocker, Gap27. Cx43∆256-289-dependent cardiovascular phenotypes coincide with significantly increased Cx43 protein levels while Cx43 mRNA levels remained unchanged. Moreover, analyses of the Cx43∆256-289 mutation in a cell culture model revealed increases in GJ plaque size, prolonged Cx43 protein half-life, and increased dye transfer capability, indicative of increased GJ intercellular communication (GJIC). Our findings demonstrate for the first time that undisturbed GJ endocytosis and turnover is critical for proper GJ function as demonstrated here in the cardiovascular system.
FIGURE 1:

Generation of the cx43lh10 zebrafish mutant using the CRISPR/Cas9 gene-editing tool. (A) Schematic of the zebrafish WT Cx43 CT (top) and cx43lh10 CT (bottom). Two gRNAs were designed, one that targeted amino acid 256 and the other that targeted amino acid 289. After Cas9-mediated generation of double strand breaks, cells employ nonhomologous end joining to repair the breaks, omitting amino acids 256–289 from the Cx43 gene. The portion of Cx43 deleted in cx43lh10 (green) contains most of the key residues involved in GJ endocytosis including the conserved AP2/clathrin binding site, critical MAPK phosphorylation sites on serine residues 261 and 279/282, and one of the two polyubiquitination sites (blue), whereas residues known to be involved in plaque assembly, stabilization, and increased GJIC (red) are left intact. (B) Amino acid sequence alignment of the CT of human Cx43 (top) and zebrafish Cx43 (bottom). The region boxed in red encompasses the deleted amino acid residues and displays the critical endocytosis-related amino acid motifs. Note that except for the second AP-2/clathrin binding site (S3, underlined blue), endocytic motifs identified in mammalian Cx43 are conserved in zebrafish Cx43 (underlined green). All residues in A and B are labeled following zebrafish nomenclature, which is partially shifted one position to the left compared with mammalian Cx43 due to the deletion of one amino acid residue in the CT of zebrafish Cx43 at position 251.
RESULTS
cx43 lh10 zebrafish exhibit significantly increased Cx43 protein levels
To investigate the effects of impaired Cx43 GJ endocytosis in zebrafish, we generated a zebrafish transgenic line (designated cx43lh10) using the CRISPR/Cas9 gene-editing tool in which most of the conserved key regulatory amino acid residues in the Cx43 CT, involved in GJ endocytosis, are deleted (Figure 1A) (Thévenin et al., 2013). These residues include the critical MAPK phosphorylation sites S261, S279/S282 (involved in channel closure and decreased GJIC), and residues triggering clathrin-mediated endocytosis including K264 (ubiquitination), the Nedd4 E3-ubiquitin ligase binding site, and the conserved AP2/clathrin binding site (S2) (Falk et al., 2009, 2014; Girão et al., 2009; Fong et al., 2012, 2013; Thévenin et al., 2013; Martins-Marques et al., 2015b; Nimlamool et al., 2015; Kells-Andrews et al., 2018) (Figure 1, A and B). Note that amino acid numbering in the zebrafish Cx43 is partially shifted one position to the left compared with Mammalia due to the deletion of one amino acid residue on position 251 in the Cx43 zebrafish CT (see Figure 1). Guide RNAs (gRNAs) were designed using the ChopChop web program (Montague et al., 2014; Labun et al., 2016, 2019). ChopChop ranks potential gRNAs based on their “efficiency” score, which is based on each gRNA’s predicted on-target activity and potential off-target effects (as described in Doench et al., 2016). In addition, ChopChop describes the directionality of each gRNA, calculates the guanine and cytosine (GC) content for self-complementarity consideration, and lists all potential off targets for each gRNA, including off targets when 1, 2, or 3 mismatches per gRNA are allowed. gRNAs were chosen based on their proximity to the genomic regions of interest (amino acids 256 and 289), an optimal GC content (between 45 and 65%), and their lack of off-target binding sites at other locations of the zebrafish genome (Supplemental Figure 1, A and B). Potential Cas9 off-target effects on cx40.8, a cx43-like gene in zebrafish, are discussed below.
Zebrafish embryos were microinjected at the one-cell stage of development with the CRISPR components (gRNAs, Cas9 protein), and the efficiency of the CRISPR/Cas9 system was verified by PCR and sequencing at 24 hours postfertilization (hpf) (Figure 1B; Supplemental Figure 2A). When 20% of the injected embryos were analyzed at 24 hpf, on average about 10% of the embryos were mutated on one allele (indicated by a faster-migrating band of ∼450 nucleotides generated by PCR amplification of the mutated cx43 gene region compared with a band of ∼550 nucleotides of the WT cx43 gene) (Supplemental Figure 2, B and C). Note that the intensity of the faster-migrating fragment varies between embryos, which is indicative of mosaic mutants that were generated by Cas9 mutagenesis activity after initial cell divisions had occurred (Supplemental Figure 2B). To select for germline mutants, mutant fish were outcrossed with WT and raised to adulthood. To finally generate homozygous mutant zebrafish that carry the mutation on both alleles, mutants were intercrossed and homozygotes selected based on PCR analyses as described above. Mutant fish were sequenced to confirm identify of the desired cx43 deletion (Supplemental Figure 3). Interestingly, developing cx43lh10 zebrafish embryos containing the Cx43∆256-289 deletion exhibited no obvious decrease in viability or any evident morphological defects during early stages of development (Supplemental Figure 4). However, surviving fish developed a suite of developmental defects, most obviously significant defects in cardiovascular morphology and function that correlate well with the well-known important role of Cx43 for vasculature development and function (Pepper et al., 1992; Pepper and Meda, 1992; Christ et al., 1996; Inoguchi et al., 2001; Haefliger et al., 2004; Rummery and Hill, 2004; Figueroa et al., 2006; Figueroa and Duling, 2008; Schmidt et al., 2008).
To assess whether the deleted sequence (Cx43∆256-289) in the cx43lh10 zebrafish affects Cx43 GJ endocytosis, immunofluorescence (IF) analyses of Cx43 levels was employed in cx43lh10 zebrafish and total Cx43 protein levels determined (Figure 2). Whole 24 hpf zebrafish embryos were fixed and stained for Cx43 protein, and levels were quantified using ImageJ (Schindelin et al., 2012). We found that cx43lh10 embryos exhibit significantly increased total Cx43 protein levels relative to WT zebrafish embryos (1.7-fold increase compared with WT embryos, n = 3) (Figure 2, A and B), consistent with our previously reported immunoblot analyses of Cx43 protein levels from regenerating fins of cx43lh10 zebrafish (Figure 2, C and D, and Bhattacharya et al., 2020). These results are consistent with the hypothesis that the Cx43∆256-289 deletion impairs GJ endocytosis, which is likely to lead to an increase in Cx43 protein levels, as endocytosis-impaired Cx43 GJs would be “trapped” in the plasma membrane. This hypothesis is explored and substantiated further below.
FIGURE 2:

Cx43 protein levels are up-regulated in cx43lh10 zebrafish. (A) Representative IF staining of Cx43 (magenta) and DAPI (blue) in 24 hpf WT (top) and cx43lh10 (bottom) zebrafish embryos. (B) Quantification of the IF staining indicates increased Cx43 protein levels in cx43lh10 embryos compared with WT (n = 5 embryos for WT and cx43lh10 each, p ≤ 0.05; error: SEM). (C, D) Representative Western blot of Cx43 protein levels in WT and cx43lh10 zebrafish embryos. Quantification of normalized Cx43 levels indicate that Cx43 protein levels in cx43lh10 mutant embryos are increased by ∼45% in comparison to WT (error: SD). (C and D are reproduced from Bhattacharya et al., 2020, with permission from The Company of Biologists.)
Cx43 ∆256-289 expressed in cultured cells exhibits significantly larger GJ plaques, longer protein half-life, and increased dye transfer capability
The Cx43 CT residues, 256–289, contain targets that regulate Cx43 endocytosis. To assess whether the observed increased protein levels in cx43lh10 zebrafish are due to increased accumulation of Cx43 GJs in the plasma membrane, constructs bearing the zebrafish coding sequence for the Cx43Δ256-289 allele were expressed in HeLa and Madin–Darby canine kidney (MDCK) cell models that do not contain endogenous Cx43 (Elfgang et al., 1995; Dukes et al., 2011). HeLa cells were transfected with either Cx43∆256-289 or WT constructs and fixed 24 h posttransfection. After fixation, HeLa cells were stained for Cx43 via IF. As expected, HeLa cells expressing Cx43∆256-289 exhibited significantly increased overall GJ plaque size and number compared with HeLa cells transfected with WT Cx43 (twofold increase compared with WT Cx43 plaques, n = 3) (Figure 3, A and B). To test whether increased GJ plaque size and number in Cx43∆256-289-expressing cells coincided with decreased Cx43 endocytosis, protein half-life analysis of Cx43∆256-289 and WT Cx43-expressing HeLa cells was employed. HeLa cells were transfected with either Cx43∆256-289 or WT Cx43 constructs, and 24 h posttransfection, the cells were treated with the ribosomal blocker cycloheximide to halt new protein biosynthesis. HeLa cells expressing WT zebrafish Cx43 exhibited a half-life of 5.0 ± 0.13 h, consistent with previous findings (Fallon and Goodenough, 1981; Beardslee et al., 1998; Falk et al., 2009, 2014). However, HeLa cells transfected with Cx43∆256-289 failed to exhibit an appreciable decrease in Cx43 protein levels within the 6-h incubation period (±0.26 h) in the presence of cycloheximide (Figure 3, C and D), indicating a significantly longer half-life of the Cx43∆256-289 protein. This is consistent with a similar mutant (Δ254–290) that we constructed and tested previously (Fong et al., 2013). To test whether Cx43∆256-289-expressing cells have altered GJIC, we performed scrape-loading Lucifer Yellow (LY) dye transfer assays in Cx43∆256-289- and WT Cx43-expressing MDCK cells. The distance of LY (a GJ permeable dye) transferring between cells, through GJs, away from the site of injury (scrape), was determined by quantitatively measuring LY fluorescence signals. LY transfer in cells expressing WT Cx43 is significantly increased compared with the untransfected control (no GJs) (19.7 ± 6.0 µm or 0 cell layers in untransfected cells, vs. 37.3 ± 9.5 µm or approximately one cell layer in WT Cx43-expressing cells). In addition, Cx43∆256-289-expressing MDCK cells exhibited significantly further increased dye transfer compared with MDCK cells expressing WT Cx43 (55.5 ± 11.7 µm or approximately two cell layers in Cx43∆256-289-expressing cells) (Figure 3, E and F). This indicates that cells expressing Cx43∆256-289 have more functional GJ channels in their plasma membranes, which results in increased GJIC. This result is consistent with our previous findings suggesting increased GJIC in regenerating fins in cx43lh10 zebrafish (Bhattacharya et al., 2020). Taken together, the increased half-life and the observed increase in the overall size of GJ plaques, combined with increased dye transfer in cells expressing Cx43∆256-289, is indicative of aberrant Cx43 GJ endocytosis, as well as increased GJIC in cx43lh10 zebrafish and is consistent with a role for residues 256–289 in the regulation of Cx43 endocytosis (Fong et al., 2013; Thévenin et al., 2017). We also measured Cx43 mRNA levels in cx43lh10 mutant embryos and found that they were unchanged (see below).
FIGURE 3:

zfCx43∆256-289 forms larger and more GJ plaques and exhibits longer protein half-lives and increased dye transfer in cultured cells. (A) Representative IF images of Cx43 (magenta) and DAPI (blue) showing GJ plaques in the membranes of HeLa cells 24 h posttransfection. (B) Plaque size analyses plotted as total fluorescence intensity indicate that Cx43∆256-289-expressing cells on average exhibit approximately two times the area (plaque number and size combined) of GJs in their plasma membranes compared with WT Cx43-expressing cells (n = 258 WT and 190 Cx43∆256-289 plaques, p ≤ 0.05). (C) Representative Western blot of Cx43 protein half-life at 0 and 6 h after cycloheximide treatment. (D) Quantification indicates that Cx43∆256-289 protein remains stable even after 6 h, while WT Cx43 protein is degraded with a typical half-life of approximately 4 h (n = 3 transfected cultures, p ≤ 0.05). (E) Representative images of LY scrape-loading dye transfer assays of untransfected MDCK cells (–), MDCK cells transfected with WT Cx43, and MDCK cells transfected with Cx43∆256-289. (F) Quantification of distances LY diffused away from the scrapes indicates that cells expressing Cx43∆256-289 exhibit significantly increased dye transfer compared with cells expressing WT Cx43 or cells without Cx43 protein (n = 3 transfected cultures, p ≤ 0.05; error: SD).
cx43 lh10 zebrafish have abnormal cardiac morphology and function
Having established that deletion of amino acid residues 256–289 results in impaired Cx43 GJ endocytosis, we wanted to investigate whether impaired turnover causes developmental and functional phenotypes. In fact, many cx43lh10 embryos developed, besides many other phenotypes (see Discussion), malformed, elongated hearts, and pericardial edema that were readily detectable in the transparent zebrafish embryos 3 days postfertilization (dpf) (Figure 4, A and C). Pericardial edema suggests cardiac insufficiency, whereas the malformed elongated heart might indicate an issue with cell migration, preventing further heart development into atrium and ventricle, valve formation, looping, ballooning, and chamber expansion (Bakkers, 2011).
FIGURE 4:
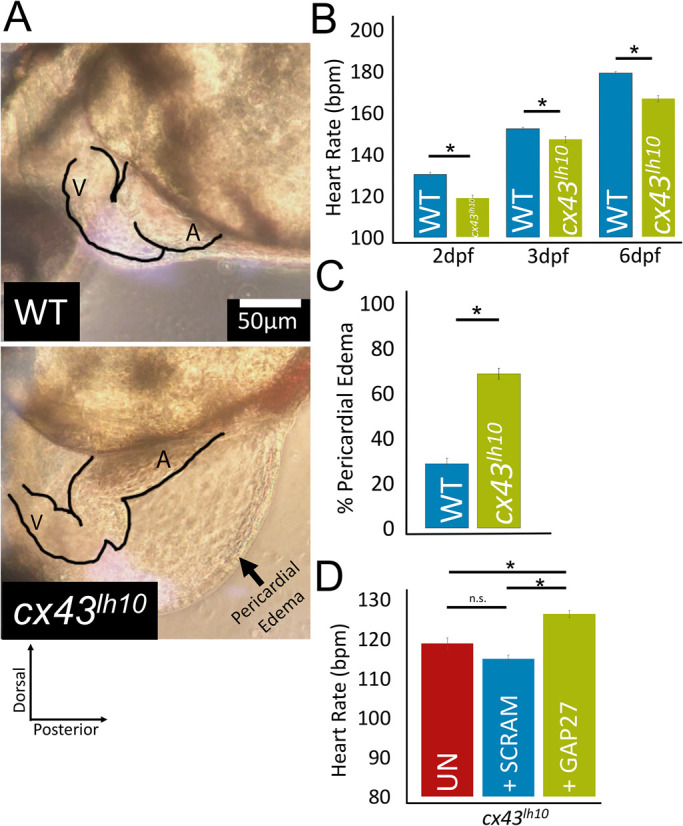
cx43lh10 zebrafish exhibit pronounced cardiac defects. (A) Representative images of the heart region of WT and cx43lh10 3 dpf embryos. cx43lh10 embryos exhibit malformed, underdeveloped, and elongated hearts, bradycardia, and pericardial edema. (B) Quantification of WT and cx43lh10 embryos at 2, 3, and 6 dpf indicates a significantly slower heart rate at all time points (n = 100 for WT and cx43lh10 embryos at 2, 3, and 6 dpf each, p ≤ 0.05). (C) Percentage of embryos exhibiting profound pericardial edema at 3 dpf (n = 496 for WT and 468 for cx43lh10 embryos, p ≤ 0.05). (D) Heart rates of uninjected 2 dpf cx43lh10 embryos (UN, red), 2 dpf cx43lh10 embryos injected with scrambled Gap27 peptide (SCRAM, blue), and 2 dpf cx43lh10 embryos injected with Gap27 peptide inhibitor (green). Note that the heart rate of embryos injected with Gap27 was restored close to WT levels (compare A with D) (n = 100 [uninfected], 21 [scrambled], 16 [Gap27 injected], p ≤ 0.05; error: SEM). (Also see Supplemental Movies 1 and 2.)
Interestingly the malformed, elongated hearts were able to contract and pump and circulate blood, though much less efficiently compared to 3 dpf WT embryos (Figure 4; Supplemental Movies 1 and 2). To further test cardiac function, heart rates were measured in 2-, 3-, and 6-dpf embryos. WT embryos exhibited an average heart rate of about 130 ± 11 beats per minute (bpm) at 2 dpf, 152 ± 9 bpm at 3 dpf, and 179 ± 9 bpm at 6 dpf (n = 100 each), consistent with published data (Sampurna et al., 2018), whereas cx43lh10 embryos exhibited a significantly slower average heart rate of about 119 ± 16 bpm at 2 dpf, 147 ± 18 bpm at 3 dpf, and 167 ± 19 bpm at 6 dpf (n = 100 each) (Figure 4B; Supplemental Movies 1 and 2). These results show that impaired Cx43 endocytosis results in readily detectable morphological and functional defects in organs in which Cx43 is expressed at high levels and is required for their morphogenesis and function. To test whether increased GJIC resulting from impaired GJ turnover could be accountable for the bradycardia exhibited in the cx43lh10 zebrafish, we inhibited Cx43 GJIC using the well-established mimetic peptide inhibitor, Gap27 (Evans and Leybaert, 2007; Evans et al., 2012). Gap27 inhibits the formation of GJs and GJIC by binding to the second extracellular loop of Cx43 (Warner et al., 1995). We injected Gap27 or, as a negative control, a scrambled Gap27 (Scram) into the pericardium of 24 hpf cx43lh10 embryos. Heart rates were analyzed at 2 dpf, and we found that cx43lh10 embryos injected with Gap27 exhibited a significantly increased heart rate (126 ± 10 bpm, n = 16) compared with cx43lh10 embryos that were uninjected (119 ± 16 bpm, n = 100) or injected with scrambled Gap27 (115 ± 11 bpm, n = 21) (Figure 4D). In fact, cx43lh10 embryos injected with Gap27 exhibited a heart rate that was statistically like that of WT embryos at 2 dpf (130 ± 11 bpm, n = 100) (Figure 4B). These results suggest that increased levels of Cx43 GJIC, resulting from aberrant GJ endocytosis, contribute to the abnormal cardiac function seen in cx43lh10 zebrafish. This is similar to the finding described in Bhattacharya et al. (2020), where the abnormal bone growth phenotype exhibited in cx43lh10 zebrafish was rescued by treatment with Gap27, indicating an important role for Cx43 GJIC in bone growth and regeneration as well.
Movie S1.
Normal cardiac beat cycle and blood circulation in WT zebrafish at 3dpf.
Movie S2.
Bradycardia in cx43lh10 zebrafish at 3dpf also exhibiting pericardial edema and severely impaired blood circulation.
Malformed and dysfunctional hearts in cx43 lh10 zebrafish embryos correlate with increased Cx43 protein levels and larger GJ plaques in cardiac tissue due to impaired endocytosis
To test whether the observed morphological and functional heart phenotype correlates with an increased number of GJs in the cx43lh10 mutant embryos, we dissected hearts from 1-y-old WT and cx43lh10 zebrafish, performed Cx43 IF analyses, quantified total fluorescence intensity per square area, and measured and quantified GJ plaque size as described for cells shown in Figure 3. In WT zebrafish, Cx43 is heavily expressed in the ventricle, while also expressed in the atrium to a lesser extent. IF analysis of dissected cardiac tissue from cx43lh10 zebrafish confirmed this divergent pattern of Cx43 expression in the WT ventricle and the WT atrium (Figure 4, A and B). It also revealed a 1.3-fold increase in Cx43 levels in the cx43lh10 zebrafish ventricle relative to the WT ventricle and a 1.5-fold increase in Cx43 levels in the cx43lh10embryo atria, reaching levels as high as those present in normal WT ventricles (Figure 5, A and B). Moreover, increased Cx43 levels in cx43lh10 zebrafish hearts coincided with 3.4- and 2.7-fold increases in GJ plaque size in the cx43lh10 zebrafish ventricle and atria, respectively, relative to WT zebrafish (Figure 5, A and C). Taken together, these results indicate that the observed heart phenotypes in the cx43lh10 mutant fish described above, and the bone phenotype described in Bhattacharya et al. (2020), correlate with an increased number of GJs in these organs and an increased level of GJIC.
FIGURE 5:
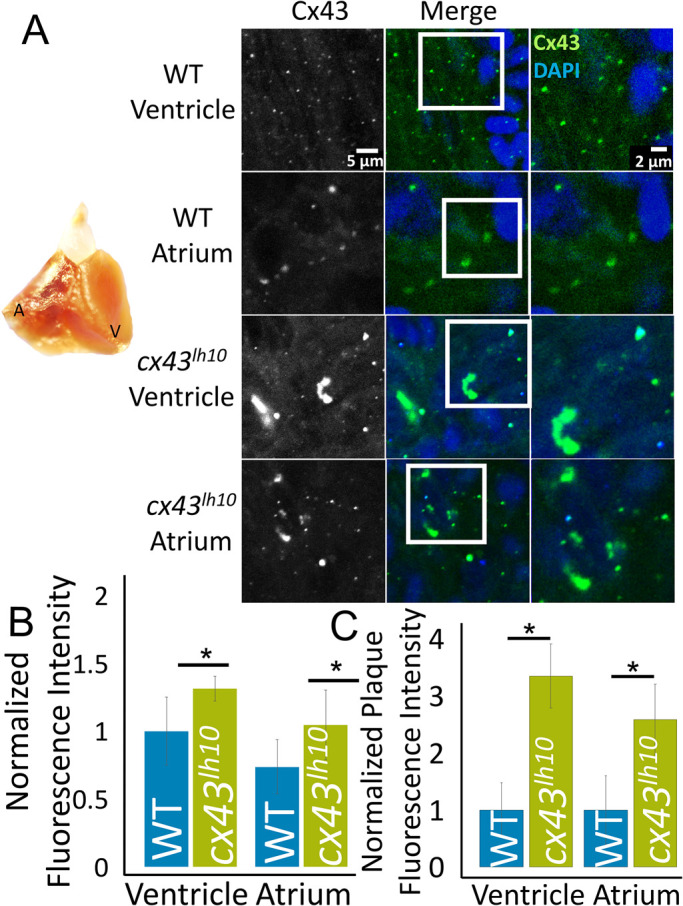
Cx43 protein levels and GJ size and number are increased in cx43lh10 zebrafish hearts. (A) A zebrafish heart (left) dissected from 1-y-old WT zebrafish indicating location of atrium and ventricle (also see Figure 4A). Representative IF images (right) of Cx43 (green) and DAPI (blue) in the atrium and ventricle of corresponding WT and cx43lh10 zebrafish hearts. (B) Quantification of total Cx43 protein levels per square area (plotted as total normalized fluorescence intensity) and (C) total plaque size and number analyses of outlined GJ plaques (plotted as normalized total plaque fluorescence intensity) reveal significantly increased levels of Cx43 protein as well as significantly increased sizes and numbers of GJ plaques in the plasma membranes of myocytes in both ventricle and atrium (n = 3 in both analyses for WT and cx43lh10 hearts, respectively, p ≤ 0.05; error: SEM).
cx43 lh10 zebrafish also exhibit a malformed and disorganized vasculature
Given the cardiac phenotypes exhibited in cx43lh10 embryos, we thought to determine whether cx43lh10 embryos and fish would also exhibit vascular abnormalities. Cx43 is primarily expressed in vasculature smooth muscle cells and vascular endothelial cells. In the vasculature, Cx43 coordinates synchronous vasomotor tone, vessel constriction, cell proliferation, and cell migration (Figueroa and Duling, 2008). To determine whether vasculature abnormalities are detectable, we first investigated the intersegmental vessels in developing embryos 5 dpf by outcrossing our cx43lh10 line with the TG(fli1:EGFP) transgenic line that drives green fluorescent protein (GFP) expression in all vascular cells throughout development (Lawson and Weinstein, 2002). The intersegmental vessels provide a unique view of vascular development, as these vessels are some of the first angiogenic vessels to assemble. During the early stages of vascular development, the individual intersegmental vessels are not defined, but as the vessels develop and blood flow begins (24 hpf), arterial–venous fate is established (Gore et al., 2012). Thus, this approach allowed us to investigate the developing vasculature before arteries and veins fully differentiated. We found that the organization of the developing vasculature in cx43lh10 embryos appears largely normal; however, vessel diameter in mutant embryos is significantly larger than in WT embryos (21.5 ± 5.03 μm in cx43lh10 embryos vs. 18.6 ± 5.18 μm in WT embryos) (Figure 6, A and B). This finding indicates that some early angiogenic developmental processes are affected in the GJ endocytosis-impaired cx43lh10 zebrafish.
FIGURE 6:

cx43lh10 zebrafish embryos exhibit defects of the vasculature. To evaluate blood vessel morphology in cx43lh10 embryos, cx43lh10 fish were crossed with TG(fli1:EGFP) fish in which all cells of the vasculature are GFP labeled. (A) Representative fluorescence images of GFP-tagged intersegmental vessels of 5 dpf WT/TG(fli1:EGFP) (left) and cx43lh10/TG(fli1:EGFP) mutant (right) embryos. Measurements of vessel diameters is indicated by a blue line in A. (B) Quantification of intersegmental vessels indicates significantly larger vessel diameters in 5 dpf cx43lh10 embryos (n = 5 [WT] and 3 [cx43lh10], p ≤ 0.05; error: SEM).
Next, to investigate later developmental processes, we analyzed the structure, organization, and function of the vasculature in caudal fins of cx43lh10 mutant zebrafish that developed to adulthood. Anesthetized 1-y-old fish were used to measure blood vessel diameter and record blood flow in the vasculature. In fish fins, veins are organized flanking each fin ray bone and an artery extends along the center of the ray bone between the two veins (Figure 7A). In our WT zebrafish, consistent with published data, veins are typically two times the diameter of arteries (Sugden et al., 2017) (Figure 7, A and B) and blood flow is significantly slower in veins than in arteries (Sugden et al., 2017) (Figure 7E; Supplemental Movie 3). In contrast, in cx43lh10 mutant fish, the arteries are significantly larger than arteries in WT fins (38.4 ± 15.4 μm in WT vs. 48.5 ± 17.5 μm in cx43lh10 fish) and veins are significantly smaller than veins in WT fins (86.4 ± 28.0 μm in WT vs. 57.2 ± 19.8 μm in cx43lh10 fish). Moreover, while WT veins normally are larger than WT arteries, cx43lh10 veins and arteries are of similar size (WT veins: 86.4 ± 28.0 μm, WT arteries: 38.4 ± 15.4 μm [p < 0.05] vs. cx43lh10 veins: 57.2 ± 19.8 μm, cx43lh10 arteries: 48.5 ± 17.5 μm) (Figure 7B). The vasculature malformation seen in the adult cx43lh10 fish is consistent with what is seen in the developing cx43lh10 embryo, indicating that the observed size aberration is likely caused by abnormal early developmental processes that are consistent throughout development. In addition, and even more strikingly, arteries and veins in the fins of adult cx43lh10 fish are profoundly disorganized and often largely or completely dysfunctional. Based on the direction of blood flow, in many cases, arteries are positioned flanking fin rays, where veins in WT fish are normally positioned. Also, veins are positioned along the center of the fin ray, where arteries in WT fish are normally positioned (Figure 7C; Supplemental Movies 3 and 4). Moreover, vasculature malformation in cx43lh10 fish regularly resulted in blood vessel dysfunction as determined by drastically reduced, or even completely halted, blood flow (Figure 7, D and E; Supplemental Movies 4 and 5). In contrast to the size aberration, the disorganization of the vasculature might be caused by later developmental aberrations such as improper arterial–venous differentiation caused by improper blood flow (Gore et al., 2009, 2012) or as a secondary effect caused by the described heart function defects. However, because proper GJ coupling is required for maintaining vasomotor tone (Christ et al., 1996; Schmidt et al., 2008; Figueroa and Duling, 2008), the described vasculature phenotypes in cx43lh10 fish are likely caused by aberrant GJIC between cells of the vasculature due to dysregulated Cx43 endocytosis.
FIGURE 7:

Adult cx43lh10 zebrafish exhibit misdeveloped, unorganized, and functionally impaired vasculature. (A) Representative images of vasculature organization and morphology in the caudal fins of 1-y-old WT and cx43lh10 fish. Typical organization of veins (located along the periphery of fin rays) and arteries (located at the center of fin rays), direction of blood flow (indicated by blue and red arrows), and typical diameter of veins and arteries in WT (left) and vessel disorganization typically seen in cx43lh10 fish (right). (B) Quantification of vessels indicates a significant diameter disproportion in cx43lh10 compared with WT fins (n = 14 for WT, 17 for cx43lh10 fish, p ≤ 0.05). (C) Mislocalization of arteries and veins (based on the direction of blood flow) in WT and cx43lh10 mutant fish (n = 14 for WT, 17 for cx43lh10 fish). (D) Representative still images at 5 s intervals taken from movie sequences indicating the distance blood flowed in vessels of WT and cx43lh10 fins. (E) Quantification indicates a significantly reduced blood flow in veins and arteries in cx43lh10 vs. WT fins (n = 10 for WT and cx43lh10 each, p ≤ 0.05; error: SEM). (Also see Supplemental Movies 3 and 4.)
Movie S3.
Normal vasculature organization and blood flow in WT adult 1 year old zebrafish caudal fins (Distal, top and Proximal, bottom). Arteries are located in the center of the bony fin rays, while veins flank the fin ray on either side.
Movie S4.
Abnormal vasculature organization in cx43lh10 adult (12-month-old) zebrafish caudal fins (Distal, top and Proximal, bottom). In addition to disorganized vasculature (mis-localization of arteries and veins, branched morphology, untypical vessel diameters), it is also apparent that some vessels have dramatically reduced and even halted blood flow.
vegf gene expression is down-regulated in cx43 lh10 fish while Cx43 mRNA levels remain unchanged
In addition to morphological and functional defects in the heart and vasculature, the cx43lh10 mutation may promote compensatory molecular responses in zebrafish cells expressing the cx43lh10 allele to cope with the physiological stresses of this deletion. Based on our data, the cx43lh10 mutation results in decreased Cx43 endocytosis and ensuing dysregulated GJIC; therefore, cells may respond through transcriptional repression of Cx43 mRNA levels to recover GJIC regulation. To test this hypothesis, real-time quantitative reverse transcription (qRT)-PCR was employed to analyze Cx43 mRNA levels in cx43lh10 zebrafish relative to WT zebrafish. We found cx43 mRNA levels in the cx43lh10 line to be similar to that of WT zebrafish (1.05 ± 0.25, n = 3), suggesting that transcriptional up-regulation of cx43 mRNA does not occur in the mutant fish and therefore is not responsible for the observed increased Cx43 protein and GJ level (Figure 8; Supplemental Table 1).
FIGURE 8:
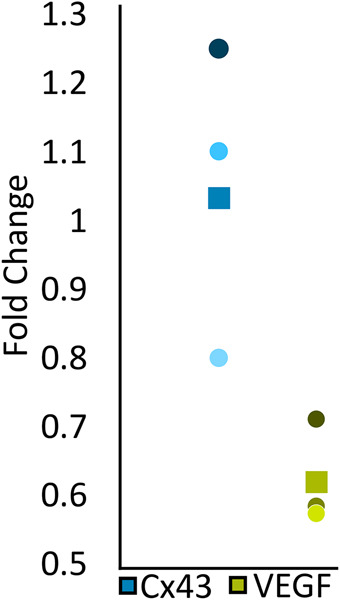
cx43lh10 fish exhibit down-regulated vegf gene expression. Fifteen 2 dpf WT and cx43lh10 zf embryos each were pooled, total mRNA prepared and retranscribed and mRNA levels of cx43 and vegf quantitatively amplified by qRT-PCR using specific primer pairs. Expression fold changes of three independent biological replicates were quantified (cx43, blue; and vegf, green). The box in the plot indicates the average fold change, and circles represent the fold change of each replicate. Note unchanged cx43 mRNA expression in cx43lh10 compared with WT fish, while vegf mRNA expression is reduced significantly.
Earlier work in mouse tumor models demonstrated that Cx43 levels are inversely related to mRNA expression of the angiogenesis signaling molecule, vascular endothelial growth factor (VEGF) (Suarez and Ballmer-Hofer, 2001; Nimlamool et al., 2015). Given abnormal blood vessel structuring in cx43lh10 zebrafish, we tested expression changes in vegf mRNA in response to the Cx43∆256-289 allele. Interestingly, vegf mRNA levels were found to be decreased (0.62 ± 0.09) in cx43lh10 zebrafish relative to WT zebrafish, which is consistent with earlier findings in mouse tumor models that overexpression of Cx43 is associated with down-regulated vegf mRNA expression (McLachlan et al., 2006; Wang et al., 2014) (Figure 8). These results may point to compensatory transcriptional changes that occur in the cx43lh10 mutant fish to modulate VEGF (and potentially other signaling molecules) expression to counteract the arterial hypertrophy and venous hypotrophy in the cx43lh10 zebrafish described above.
DISCUSSION
Cell-to-cell communication, mediated by GJ channels, is crucial for multicellular organisms. When dysregulated, a variety of damaging consequences may be encountered. Regulated turnover (biosynthesis and endocytosis) is of particular interest as GJs and Cxs exhibit an unusual, not well understood, rapid turnover of only a few hours (Fallon and Goodenough, 1981; Chu and Doyle, 1985; Hare and Taylor, 1991; Lauf et al., 2002; Gaietta et al., 2002; Berthoud et al., 2004; Piehl et al., 2007; Falk et al., 2009). Furthermore, whether regulated GJ turnover is crucial for physiological GJ function, and whether impaired turnover could cause disease, has not been known. Research has shown that the Cx43 CT harbors key regulatory residues that control GJ endocytosis (Berthoud et al., 2004; Thévenin et al., 2013; Falk et al., 2016). Binding and release of a scaffolding protein (ZO-1), and a series of posttranslational modifications that include phosphorylation/dephosphorylation, ubiquitination, and adaptor protein (AP-2, Eps15) and clathrin binding on key regulatory amino acid residues located in the Cx43 CT, transition functional GJ channels for endocytosis (Park et al., 2006, 2009; Piehl et al., 2007; Solan et al., 2007; Gumpert et al., 2008; Falk et al., 2009. 2012; Girão et al., 2009; Rhett et al., 2011; Fong et al., 2013; Thévenin et al., 2013; Cone et al., 2014; Dunn and Lampe, 2014). Here, we used the CRISPR/Cas9 gene-editing tool to generate a zebrafish line in which amino acids 256–289 (Cx43∆256-289, termed cx43lh10) that encompasses most of the above-described amino acid residues found critical for Cx43 GJ endocytosis were deleted. These fish developed a suite of developmental defects that include cardiovascular morphology and function (reported here), fin ray morphology (Bhattacharya et al., 2020), likely higher early embryo misdevelopment and mortality, problems with proper oocyte and sperm overproduction, pigmentation (see Figure 7 and Supplemental Movies 3–5), and others (unpublished data).
Movie S5.
Dramatically reduced blood flow and blood clotting in cx43lh10 adult zebrafish caudal fins. Distal (top) and Proximal (bottom).
To minimize off-target effects of Cas9, we designed gRNAs that are predicted to bind only to the cx43 gene and not to any other sites, even when three mismatches are allowed, and used the improved VP12 Cas9 enzyme that minimizes off-target activity (Kleinstiver et al., 2016). In addition, backcrossing to eliminate mosaic mutagenesis and to generate homozygotes should have eliminated any potential off-target effects. Most importantly, we obtained the “expected” effects (vasculature and other organs where Cx43 protein function is known to be important). Of note, zebrafish have a cx43-like gene, cx40.8, that results from the whole genome duplication in the teleost lineage that is not present in Mammalia (Watanabe, 2017; Eastman et al., 2006; Gerhart et al., 2009). Although, cx40.8 shares about 80% nucleotide and amino acid sequence identity with cx43, our gRNAs are cx43 specific, exhibiting eight and four mismatches with cx40.8 and thus are not likely to bind to the cx40.8 gene (Supplemental Figure 1C). Thus, it is highly unlikely that potential off-target activity by Cas9 could have led to any confounding or misleading results.
While Cx43 mRNA levels remained unchanged, the mutation correlated with increased Cx43 protein levels, which coincided with significantly more and larger GJ plaques, a longer mutant Cx43 half-life, and an increase in dye transfer. Our findings correlate with an earlier study that we conducted in cells in culture where mutating/removal of specific tyrosine-based sorting signals (the AP-2/clathrin binding sites S2 and S3) and other key endocytic residues located in the Cx43 CT between amino acids 254 and 290 abolished clathrin to associate with Cx43 GJs, resulting in larger GJ plaques with decreased endocytic kinetics and dramatically extended Cx43 half-life (Fong et al., 2013), and a very recent study in zebrafish where the cx43lh10 mutant exhibited increased Cx43 protein levels in the fins, as well as increased fin regenerate and segment length compared with WT fins (Bhattacharya et al., 2020). Taken together, our findings indicate that removing amino acids 256–289, similarly to previous reports, causes impaired GJ endocytosis and an increase in GJIC.
Interestingly, hundreds of mutations are known in Cx genes that associate with human disease; however, with a few exceptions all these disease-associated Cx mutations occur in the N-terminal portion of the Cxs (N-terminus to end of transmembrane domain 4) and only a very few are present in the CT portion (11 in Cx32 in X-linked Charcot-Marie-Tooth disease [CMTX] patients, none in Cx26 hearing impaired patients, and two in Cx43 in Oculodentodigital dysplasia [ODDD] patients) (Laird et al., 2017), suggesting that mutations that occur in the CT and may dysregulate GJ turnover are not tolerated. This is further supported by the finding that more than 60,000 healthy, unrelated humans whose exome sequences are compiled in the Exome Aggregation Consortium (ExAC) database (Lek et al., 2016; Karczewski et al., 2020) do not have mutations—not even on one allele—on sites that we, and others, have identified as critical for GJ internalization, including the ZO-1 binding site (D379LIE382); the phosphorylation sites on serine residues 372/373, 368, and 279/282; the ubiquitination site on K303; and the AP-2/clathrin binding sites S2 (Y265AYF268), and S3 (Y286KLV290), while mutations on other, unrelated, amino acid residues in the Cx43 CT do occur regularly (1117 missense mutations of CT Cx43 amino acid residues 245–382), further reiterating the importance of these amino acid residues for GJ function.
The cx43lh10 zebrafish exhibit severe defects of the cardiovascular system including malformed, elongated hearts, decreased heart rate, disorganized and malformed vasculature, and impaired blood flow, indicating that undisturbed GJ endocytosis is crucial for normal organ development. Cardiovascular phenotypes in the cx43lh10 mutant fish correlate with the well-established and documented role GJs play in heart development and function (Reaume et al., 1995; Ya et al., 1998; Severs, 1999; Severs et al., 2004; Jongsma and Wilders, 2000; Gutstein et al., 2001; Inoguchi et al., 2001; Winterhager et al., 2007; Bruce et al., 2008; Maass et al., 2009; Bakkers, 2011; Martins-Marques et al., 2015a–d; Michela et al., 2015). In addition, altered Cx43 function and GJIC is known to be associated with cardiomyopathies that include heart failure, myocardial ischemia, and cardiac arrhythmias (Beardslee et al., 2000; Bruce et al., 2008; Duffy, 2012; Martins-Marques et al., 2015d). Previous findings have shown that deletion of Cx43 in mice causes defects in the interface between the right ventricle and outflow tract, which results in blood flow obstruction and ultimately neonatal lethality due to delayed cardiac looping and malformation (Reaume et al., 1995; Ya et al., 1998). A premature stop codon, preventing the translation of most of the Cx43 CT (K258Stop), has been characterized to generate functional channels in mice, with larger plaques at the intercalated disk, and no abnormalities in heart morphology, yet increased infarct size and susceptibility to arrhythmias were reported (Maass et al., 2007, 2009). Furthermore, mice lacking the last five CT amino acid residues (Cx43Δ378stop), abolishing ZO-1 binding, displayed aberrant cardiac electrical activation properties that ultimately led to lethal ventricular arrhythmias, yet channel function was not impaired, detailing Cx43-dependent cardiac defects that are independent of channel function (Lübkemeier et al., 2013). It has also been shown that changes in Cx43 expression, phosphorylation, and distribution lead to cardiac arrhythmias, such as atrial fibrillation, and diseases of the myocardium such as hypertrophic cardiomyopathy and ischemic cardiomyopathy (Michela et al., 2015). Furthermore, defective Cx43 plays a role in diseases of the vasculature, such as hypertension and diabetes (Inoguchi et al., 2001; Rummery and Hill, 2004; Zhang and Hill, 2005; Figueroa et al., 2006; Figueroa and Duling, 2008). However, there has been very little evidence to support the mechanism/s behind the defective GJ function that leads to these disorders.
The cx43lh10 zebrafish exhibit bradycardia, malformed hearts, and pericardial edema. The bradycardia exhibited in cx43lh10 fish was rescued by treatment with the GJ blocker, Gap27, an observation that correlates with our recent finding in zebrafish caudal fins where Gap27 rescued the segment length defect of the cx43lh10phenotype (Bhattacharya et al., 2020). The cx43lh10 fish also exhibit severe defects of the vasculature including impaired blood flow, as well as disorganized and malformed vasculature. It has been previously reported that increased Cx43 expression and GJIC signal endothelial cells of the vasculature to migrate due to the necessity of establishing a continuous covering during formation and renewal of blood vessels and wound repair (Pepper and Meda, 1992; Pepper et al., 1992; Kwak et al., 2001; Haefliger et al., 2004). In addition, hypertension mouse models reveal increased Cx43 in the plasma membranes of vascular smooth muscle cells and cardiovascular hypertrophy (Haefliger et al., 2004). Together, these data indicate that increased levels of GJIC are likely to contribute to the developmental and functional phenotypes observed in the cx43lh10 mutant. However, GJs are also well known to provide a role in cell guidance and physical cell–cell adhesion (Huang et al., 1998; Xu et al., 2006; Elias et al., 2007; Francis et al., 2011; Liu et al., 2012; Matsuuchi and Naus, 2013; Zhou and Jiang, 2014; Karpinich and Caron, 2015; Naus et al., 2016; Polusani et al., 2016). Thus, GJs that cannot be removed from the plasma membrane may also severely impact cell migration, which is particularly important during development. Indeed, while many of the cx43lh10 mutant embryos survive and early heart development (at least the first 30 h) appears to occur mostly undisturbed (although the heart beats slower, the elongated heart tube forms), further development (differentiation into mature atrium, ventricle, valve, looping, ballooning, and expansion) (Bakkers, 2011) appears to be halted, which potentially suggests issues with cell migration and guiding in the cx43lh10 mutant fish. Previous work has shown that mechanical and electrical forces are required for proper chamber formation. For example, in zebrafish with deficiencies in ventricular or atrial contraction, the resultant phenotype includes larger, elongated chambers (Chi et al., 2010), similar to what we see in the cx43lh10 zebrafish.
While the data presented here provide evidence for Cx43 regulatory reactions in cardiovascular development, our findings also reveal a complex interplay between Cx43 and other molecules implicated in GJIC. For instance, we found that transcription of vegf mRNA is repressed in the cx43lh10 mutant fish. This suggests that VEGF may be an important player in cardiovascular development, functioning downstream of Cx43. However, the putative role of altered vegf transcription in arterial hypertrophy and venous hypotrophy in relation to the cx43lh10 mutation remains to be explored further. One possibility is that dysregulated Cx43 endocytosis directly leads to vasculature malformations, which induce transcriptional modulation of vegf to compensate for aberrant vasculature patterning. This mechanism may be initiated in response to increased artery growth but also affects the structuring of veins, leading to drastic reductions in venous lumen size. Another possibility is that Cx43-dependent GJIC directly influences transcriptional regulators of vegf, but this response bears different consequences in arteries compared with veins.
Another interesting revelation of this study is that impaired GJ turnover exhibited in the cx43lh10 embryos allows development into adulthood although, generally, with severe developmental defects, which is seemingly different from that of humans based on the ExAC database analysis described above. In the zebrafish embryo, Cx43 is expressed as early as 1.25 hpf (eight-cell stage) (Hardy et al., 1996; Chatterjee et al., 2005; Cofre and Abdelhay, 2007), but cardiovascular morphological abnormalities in the cx43lh10 embryos are apparent only beginning at ∼24 hpf. Also, the severity of phenotypes was observed to be quite variable in the cx43lh10 mutant fish. One possible explanation for this delayed onset of visible abnormalities and variability in phenotype severity is that other Cxs (not present or functional in humans, such as Cx40.8) compensate for aberrant Cx43 function. Although zf Cx40.8 is expressed in the same tissues as Cx43, colocalizes with Cx43 in the plasma membrane, and exhibits similar channel properties as Cx43, it only inefficiently forms GJ channels, for example, in the absence of Cx43 (Eastman et al., 2006; Gerhart et al., 2009, 2012) and thus is not likely to play a major role in compensation. However, it was reported that several Cx43 knockout phenotypes in mice can be rescued through replacement with other Cx types, such as Cx26, Cx32, Cx40, and Cx31 (Reaume et al., 1995; Plum et al., 2000; Gutstein et al., 2001; Brehm et al., 2007; Sridharan et al., 2007; Winterhager et al., 2007; Bedner et al., 2012). Hence, it is possible that one of these Cxs partially compensates the developmental and functional phenotypes seen in the cx43lh10 fish. Finally, it is questionable whether a developing human embryo, due to its much larger size, could tolerate even the mildest cardiovascular developmental defects seen in the cx43lh10 embryos, particularly. the associated insufficient tissue oxygenation defects.
Taken together, the data presented here highlight the importance of undisturbed Cx43 GJ endocytosis as a critical aspect of normal GJ function that should be considered as a possible mechanism for causing GJ-related disease. Further analyses will generate a deeper mechanistic understanding of the role that regulated Cx43 endocytosis plays for development, morphogenesis, and tissue function.
MATERIALS AND METHODS
Request a protocol through Bio-protocol.
Fish maintenance
Zebrafish (D. rerio) were maintained in the water system built by Aquatic Habitats (now Pentair) at 27°C–28°C temperature in a 14:10 light:dark period. Water quality was monitored and dosed to maintain conductivity (400–600 ms) and pH (6.95—8/27/2019 approval). Research was performed per the IACUC for Lehigh University (protocol #231). Food was provided three times a day. Brine shrimp (hatched from INVE artemia cysts) was fed once, and flake food (Aquatox AX5) supplemented with 7.5% micropellets (Hikari), 7.5% Golden Pearl (300–500 micron; Brine Shrimp direct), and 5% Cyclo-Peeze (Argent) twice per day.
Generation of cx43 lh10 transgenic fish
A transgenic zebrafish line was generated that deletes amino acids 256–289 in the CT of Cx43 using the CRISPR/Cas9 genomeediting tool. To generate the deletion, two gRNAs were designed using the ChopChop web tool (Montague et al., 2014; Labun et al., 2016, 2019). One gRNA was designed to target the CT T256 region, and the other gRNA was designed to target the CT A289 region of the gene. Designed gRNAs were purchased from Genscript’s synthetic gRNA service (gRNA T256: UAGACAGUUCCUUCGGCGUG; gRNA A289: CCAGGCUACAAACUGGCCAC) (Cat. No. SC1838, SC1933). Improved VP12 Cas9 plasmid was a gift from Keith Joung, Massachusetts General Hospital, Charlestown, MA (Addgene plasmid #72247; Kleinstiver et al., 2016) and transcribed using the mMessage mMachine T7 Transcription Kit according to the manufacturer’s instructions (Invitrogen; Cat. No. AM1344). Cas9 mRNA (500 ng) and each gRNA (1 µg) were dissolved in water and coinjected into one-cell-stage WT zebrafish embryos. At 24 hpf, 20% of the embryos were genotyped using PCR (forward primer [IDT]: 5′-CCCTCATAGGGTGGACTGTTTCCTTTCTCGGCCCACCGAG-3′, reverse primer [IDT]: 5′-GACGTCCAGGTCATCAGGCCTCGCTCGACTGCTCATGCGGCTGC-3′). The remaining embryos were raised to adulthood and outcrossed to WT fish for detection of germline transmission. Heterozygous embryos were then raised to adulthood and intercrossed to generate homozygous ∆256–289 mutant zebrafish (designated cx43lh10) and the progeny sequenced.
Generation of zebrafish WT Cx43 and Cx43 ∆256-289 mutant mammalian cell culture expression plasmids
Untagged zebrafish WT Cx43 and Cx43∆256-289 plasmids were generated using the pEGFP-N1 vector including the full-length rat Cx43 cDNA (described in Fong et al., 2013) as the template. The entire rat Cx43 sequence was removed using EcoRI (Cat. No. R3101S; NEB) and BamHI (Cat. No. R3136S; NEB) restriction enzymes. The linearized and empty pEGFP-N1 vector was extracted and purified using Qiagen’s QIAquick gel extraction kit (Cat. No. 28704) for use as the backbone for the constructs. Gibson assembly (Cat. No. E5510S; NEB) was used to replace the full-length rat Cx43 in the pEGFP-N1 vector with either full-length zebrafish Cx43 (including its authentic stop codon) obtained via PCR amplification from WT zebrafish or Cx43∆256-289 (including its authentic stop codon) obtained via PCR amplification from cx43lh10 zebrafish. Constructs were transformed into DH5α competent Escherichia coli cells. Plasmids were purified using a Midiprep plasmid DNA purification kit (Cat. No. 12143; Qiagen). All constructs were verified via DNA sequence analyses.
Whole-mount IF of zebrafish embryos/hearts and confocal microscopy
Zebrafish embryos were fixed at 24 hpf using 4% paraformaldehyde overnight at 4°C. Adult zebrafish were killed in an excess of 0.1% tricaine solution and fixed in 4% paraformaldehyde overnight and hearts dissected according to Singleman and Holtzman (2011). To improve staining, antigen retrieval was implemented using 1% SDS/phosphate-buffered saline (PBS) for 5 min while rocking at room temperature immediately before immunostaining. Embryos/hearts were washed in PBST (PBS + 0.1% Tween) and, for 1 h before blocking, PBSTX (PBS + 0.1% Tween + 0.1% Triton X-100). Embryos/hearts were blocked for 1 h using 10% bovine serum albumin (BSA) (Cat. No. A7906; Sigma)/PBSTX and incubated for 48 h at 4°C in rabbit anti-Cx43 primary antibody (Cat. No. 3512; Cell Signaling) diluted 1:100 in 1% BSA/PBSTX. Embryos/hearts were then washed with 1% BSA/PBSTX and incubated for 48 h at 4°C in goat anti-rabbit Alexa Fluor 568 (embryos) (Cat. No. A-11036; Invitrogen) or goat anti-rabbit Alexa Fluor 488 (hearts) (Cat. No. A-32731; Invitrogen) diluted 1:100 in 1% BSA/PBSTX. Embryos/hearts were also stained with 1 µg/mg 4′,6-diamidino-2-phenylindole (Cat. No. D1306; Molecular Probes). Embryos/hearts were washed with PBST and mounted onto glass slides with 3% methylcellulose, and z-stacks were generated with confocal microscopy (Zeiss LSM 880) at 25× (embryos) or 63× (hearts) using argon laser bands at 568, 488, and 405 nm. To quantify total Cx43 protein levels, fluorescence intensity/outlined region was measured in 50 separate regions in each image of the z-stack via ImageJ (National Institutes of Health, Bethesda, MD) using the rectangle tool and averaged. Five biological replicates were performed in embryos, and three biological replicates were performed in hearts.
Cell culture and transient transfections
HeLa (GJ deficient; Cat. No. CCL2; American Type Culture Collection [ATCC]) (Elfgang et al., 1995) and MDCK cells (GJ deficient; Cat. No. NBL-2; ATCC) (Dukes et al.., 2011) were maintained in low-glucose DMEM (Cat. No. SH30021.01; Hyclone) supplemented with 50 I.U./ml penicillin and 50 µg/ml streptomycin (Cat. No. 30-001-C1; Corning), 2 mM ʟ-glutamine (Cat. No. 25-005-C1; Mediatech), and 10% fetal bovine serum (FBS) (Cat. No. S11150; Atlanta Biologicals) at 37°C, 5% CO2, and 100% humidity. Cells were washed with 1× PBS and treated with 0.25% trypsin/0.1% EDTA (Cat. No. 25-053-CI; Corning) for passaging. Twenty-four to forty-eight hours after passaging, 60–80% confluent HeLa cells were transiently transfected with WT Cx43 or Cx43∆256-289 constructs using Lipofectamine2000 (Cat. No. 11668019; Invitrogen) according to the manufacturer’s recommendations.
Cx43 plaque size analyses in HeLa cells
Cx43 GJ plaques in HeLa cells (Cat. No. CCL2; ATCC) were analyzed via IF and plaque size quantification. HeLa cells were grown on pretreated poly-l-lysine (Cat. No. P8920; Sigma)-coated coverslips in low-glucose DMEM at 37°C, 5% CO2, and 100% humidity. Cells were transiently transfected with either WT Cx43 or Cx43∆256-289 constructs. Cells were fixed and permeabilized in ice-cold methanol. Cells were blocked in 10 FBS/PBS for 30 min at room temperature and incubated with primary rabbit anti-Cx43 antibodies diluted 1:200 in 10% FBS/PBS and incubated overnight at 4°C. Cells were incubated in goat anti-rabbit Alexa Fluor 568 diluted 1:200 in 10% FBS/PBS and incubated for 1 h at room temperature. Cells were also stained with 1 µg/mg DAPI. Coverslips were mounted using Fluoromount G (Cat. No. 0100-01; Southern Biotechnology) and imaged using a 60× objective on a Nikon Eclipse TE2000 inverted fluorescence microscope. To quantify, GJ plaques clearly recognizable as such by their size (larger than 0.5 μm in diameter), bright fluorescence intensity, and string-like arrangement indicative of plasma membrane location (258 in WT-expressing cells and 190 in Cx43∆256-289-expressing cells) in 10 cell pairs/transient transfection were outlined using the freeform tool in ImageJ, and the fluorescence intensity/outlined area was quantified for cell pairs expressing Cx43. Three biological replicates were performed of each construct.
Cx43 protein half-life analyses and immunoblotting
Twenty-four hours posttransfection with either the WT Cx43 or Cx43∆256-289 constructs, HeLa cells were treated with 50 µg/ml cycloheximide (Cat. No. C655; Sigma) for up to 6 h at 37°C, 5% CO2, and 100% humidity. Cells were lysed in SDS–PAGE sample buffer at 0 and 6 h and boiled for 5 min. Samples were separated on 12% SDS–PAGE mini gels (BioRad). Proteins were transferred to nitrocellulose membranes and blocked overnight at 4°C or for 1 h at room temperature in 5% nonfat dry milk/TBS (Tris-buffered saline). Antibodies were diluted in 5% BSA/TBS as follows: rabbit anti-Cx43 (Cat. No. 3512; Cell Signaling) at 1:2000 and mouse anti-vinculin (Cat. No. V9131; Sigma) as a loading control at 1:5000. Blots were incubated with primary antibodies overnight at 4°C or for 2 h at room temperature and then washed with TBS-Tween (TBST). Secondary horseradish peroxidase–conjugated anti-rabbit or anti-mouse antibodies were diluted at 1:5000 in TBST and incubated overnight at 4°C or for 2 h at room temperature. Blots were incubated in ECL buffer (100 mM Tris, pH 8.8, 2.5 mM luminol, 0.4 mM p-coumaric acid, 0.02% hydrogen peroxide) before exposure to x-ray film. ImageJ software was used to measure relative densities normalized to vinculin. Cx43 protein intensities were quantified with ImageJ using the gel analyzer tool and normalized to corresponding vinculin intensities for three separately transfected cultures expressing each transfected construct.
Scrape loading dye transfer assay
MDCK cells were seeded into 3.5 cm dishes and grown to ∼70% confluency. Dishes were transfected with Cx43 WT and Cx43∆256-289 constructs as described above. One set of dishes was left as untransfected controls. Transfection efficiency was estimated by transfecting a Cx43-GFP–expressing construct (as described in Falk, 2000) concurrently. All transfection efficiencies were 80% or higher. At ∼24 h posttransfection, cells (∼100% confluent) were washed once with 1× PBS, and 1 ml of 0.05 weight % LY (Cat. No L682; Invitrogen) in 1× PBS was added to the culture medium of each dish. Monolayers were scraped with a sharp razor blade and incubated at 37°C, under 5% CO2, and at 100% humidity for 10 min. Cells were rinsed with 1× PBS and fixed in 3.7% formaldehyde for 10 min at room temperature. Cells were rinsed two times in 1× PBS and imaged with a 10× objective on a Nikon Eclipse TE2000 inverted fluorescence microscope. Images were analyzed by measuring the distance the dye transferred away from the scrape (10 measurements/scrape, three scrapes/ transfected dish, three separate transfected cultures expressing each construct) in ImageJ.
RNA extraction and qRT-PCR analyses
Total RNA was extracted and purified from killed, unfixed, 2 dpf zebrafish embryos (15 embryos/biological replicate) using the Trizol reagent and the standard protocol. The resulting RNA pellet was resuspended in a solution of DEPC water and RNase Inhibitor and the concentration recorded using the Thermo Scientific Nanodrop 2000. To generate cDNA, 1 µg of total RNA was reverse transcribed with SuperScript III reverse transcriptase (Invitrogen) using oligo (dT) primers. The resulting cDNA was diluted 1:10 for qRT-PCR. CT values were measured using a Rotor-Gene (Corbett) analyzer. Derived CT values using the following primer pairs for Cx43 (forward: TCGCGTACTTGGATTTGGTGA, reverse: CCTTGTCAAGAAGCCTTCCCA), VEGF (forward: TGCTCCTGCAAATTCACACAA, reverse: ATCTTGGCTTTTCACATCTGCAA), and the internal control, keratin (forward: TCATCGACAAAGTGCGCTTC, reverse: TCGATGTTGGAACGTGTGGT) were averaged. The ∆CT values represent the expression levels of the gene of interest normalized to the internal keratin control. ∆∆CT values represent the level of gene expression. The fold change differences were determined using the ∆∆CT method as previously described (Livak and Scmittgen, 2001, 2008). Three technical replicates/gene were performed for each of the five biological replicates.
Heart rate analyses
At 3 and 6 dpf, zebrafish embryos were collected for heart rate analyses. Individual embryos were placed on a glass coverslip in a drop of water. Heart rates were manually determined by counting individual beats visualized under a light microscope for a total of 15 s each, using a cell counter. This was performed three times for each embryo (100 for each genotype). Beats/15 s were averaged for each embryo and multiplied to obtain the average bpm for each individual embryo. Images and movies of the heart were obtained with an iPhone X camera mounted to an Olympus inverted light microscope.
Cardiac morphology analyses
Heart morphology was analyzed visually and by tracing the atrium and ventricle of the heart on still frames from the heartbeat movies using ImageJ. Pericardial edema was determined by visually assessing the amount of space between the pericardium and the ventricle and atrium. In WT embryos, the pericardium is flush with the atrium and ventricle, whereas embryos with pericardial edema exhibit a distended pericardium (Henry et al., 1997).
Peptide injections
The well-established GJ-specific mimetic peptide inhibitor, Gap27 (Evans and Leybaert, 2007; Evans et al., 2012) (Cat. No. 1476, Tocris Bio-Techne Corporation) and a scrambled Gap27 peptide were used. cx43lh10 embryos were injected with 0.5 mM peptide in phenol red into the pericardium at 24 hpf. At 2 dpf, injected cx43lh10 embryos were collected for heart rate analysis as described previously.
Vasculature analyses
Adult fin vasculature was analyzed in zebrafish that were anesthetized with 0.1% tricaine solution and imaged using a Nikon Eclipse 80i Microscope. Movies of fish were taken using a Levenhuk M300 BASE Camera and Levenhuk software. In addition to movies, three images/fin were obtained, and three diameter measurements were acquired/vessel for each image in ImageJ. In addition, blood flow distance/second was measured in ImageJ by tracing the paths of single red blood cells moving through the vasculature for 1 s and measuring the distance traveled. One measurement was taken/vessel for each movie. Juvenile vasculature was analyzed by crossing cx43lh10 with the Tg(fli1:egfp) transgenic line that drives GFP expression in all vasculature cells throughout development (Lawson and Weinstein, 2002) so the resultant progeny are cx43lh10 zebrafish with GFP-expressing vasculature. Tg(fli1:egfp)/cx43lh10 zebrafish were fixed in 4% formaldehyde at 5 dpf, and z-stacks were generated with confocal microscopy. Vasculature diameters for all intersegmental vessels were measured using ImageJ in five (WT) or three (cx43lh10) biological replicates.
Statistical analyses
Statistical significance was determined by Student’s t test (two tailed, unpaired). Statistical significance was defined by a p value of ≤0.05.
Supplementary Material
Acknowledgments
We thank current and previous Falk and Iovine lab members for constructive discussions. This work was supported by National Institutes of Health Grants RO1-GM55725 to M.M.F. and R15-HD080507 to M.K.I. and a 2018 Lehigh University’s Faculty Innovation Grant to M.M.F. and M.K.I.
Abbreviations used:
- CT
C-terminal
- Cx
connexin
- Cx43
connexin43
- GJIC
gap junction intercellular communication
- WT
wild type.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www .molbiolcell.org/cgi/doi/10.1091/mbc.E20-12-0797) on August 11, 2021.
REFERENCES
- Bakkers J (2011). Zebrafish as a model to study cardiac development and human cardiac disease. Cardiovasc Res 91, 279–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beardslee MA, Laing JG, Beyer EC, Saffitz JE (1998). Rapid turnover of connexin43 in the adult rat heart. Circ Res 83, 629–635. [DOI] [PubMed] [Google Scholar]
- Beardslee MA, Lerner DL, Tadros PN, Laing JG, Beyer EC, Yamada KA, Kléber AG, Schuessler RB, Saffitz JE (2000). Dephosphorylation and intracellular redistribution of ventricular connexin43 during electrical uncoupling induced by ischemia. Circ Res 87, 656–662. [DOI] [PubMed] [Google Scholar]
- Bedner P, Steinhäuser C, Theis M (2012). Functional redundancy and compensation among members of gap junction protein families? Biochim Biophys Acta 1818, 1971–1984. [DOI] [PubMed] [Google Scholar]
- Berthoud VM, Minogue PJ, Laing JG, Beyer EC (2004). Pathways for degradation of connexins and gap junctions. Cardiovasc Res 62, 256–267. [DOI] [PubMed] [Google Scholar]
- Bhattacharya S, Hyland C, Falk, MM, Iovine MK (2020). Connexin 43 gap junctional intercellular communication inhibits Evx1 expression and joint formation in regenerating fins. Development 147, dev190512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brehm R, Zeiler M, Rüttinger C, Herde K, Kibschull M, Winterhager E, Willecke K, Guillou F, Lécureuil C, Steger K, et al. (2007). A Sertoli cell-specific knockout of connexin43 prevents initiation of spermatogenesis. Am J Pathol 171, 19–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce AF, Rothery S, Dupont E, Severs NJ (2008). Gap junction remodelling in human heart failure is associated with increased interaction of connexin43 with ZO-1. Cardiovasc Res 77, 757–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee B, Chin AJ, Valdimarsson G, Finis C, Sonntag JM, Choi BY, Tao L, Balasubramanian K, Bell C, Krufka A, et al. (2005). Developmental regulation and expression of the zebrafish connexin43 gene. Dev Dyn 233, 890–906. [DOI] [PubMed] [Google Scholar]
- Chi NC, Bussen M, Brand-Arzamendi K, Ding C, Olgin JE, Shaw RM, Martin GR, Stainier DYR (2010). Cardiac conduction is required to preserve cardiac chamber morphology. Proc Natl Acad Sci USA 107, 14662–14667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christ GJ, Spray DC, el-Sabban M, Moore LK, Brink PR (1996) Gap junctions in vascular tissues. Evaluating the role of intercellular communication in the modulation of vasomotor tone. Circ Res 79, 631–646. [DOI] [PubMed] [Google Scholar]
- Chu FF, Doyle D (1985) Turnover of plasma membrane proteins in rat hepatoma cells and primary cultures of rat hepatocytes. J Biol Chem 260, 3097–3107. [PubMed] [Google Scholar]
- Cofre J, Abdelhay E (2007). Connexins in the early development of the African clawed frog Xenopus laevis (Amphibia): the role of the connexin43 carboxyl terminal tail in the establishment of the dorso-ventral axis. Genet Mol Biol 30, 483–493. [Google Scholar]
- Cone AC, Cavin G, Ambrosi C, Hakozaki H, Wu-Zhang AX, Kunkel MT, Newton AC, Sosinsky GE (2014). Protein kinase Cδ-mediated phosphorylation of connexin43 gap junction channels causes movement within gap junctions followed by vesicle internalization and protein degradation. J Biol Chem 289, 8781–8798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, et al. (2016). Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol 34, 184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy HS (2012). The molecular mechanisms of gap junction remodeling. Heart Rhythm 9, 1331–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dukes JD, Whitley P, Chalmers AD (2011). The MDCK variety pack: choosing the right strain. BMC Cell Biol 12, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn CA, Lampe PD (2014). Injury-triggered Akt phosphorylation of Cx43: a ZO-1-driven molecular switch that regulates gap junction size. J Cell Sci 127, 455–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eastman SD, Chen TH, Falk MM, Mendelson TC, Iovine MK (2006). Phylogenetic analysis of three complete gap junction gene families reveals lineage-specific duplications and highly supported gene classes. Genomics 87, 265–274. [DOI] [PubMed] [Google Scholar]
- Elfgang C, Eckert R, Lichtenberg-Fraté H, Butterweck A, Traub O, Klein RA, Hülser DF, Willecke K (1995). Specific permeability and selective formulation of gap junction channels in connexin-transfected HeLa cells. J Cell Biol 129, 805–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias LA, Wang DD, Kriegstein AR (2007). Gap junction adhesion is necessary for radial migration in the neocortex. Nature 448, 901–907. [DOI] [PubMed] [Google Scholar]
- Evans WH, Bultynck G, Leybaert L (2012). Manipulating connexin communication channels: use of peptidomimetics and the translational outputs. J Membr Biol 245, 437–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans WH, Leybaert L (2007). Mimetic peptides as blockers of connexin channel-facilitated intercellular communication. Cell Commun Adhes 14, 265–273. [DOI] [PubMed] [Google Scholar]
- Falk MM (2000). Connexin-specific distribution within gap junctions revealed in living cells. J Cell Sci 113, 4109–4120. [DOI] [PubMed] [Google Scholar]
- Falk MM, Baker SM, Gumpert AM, Segretain D, Buckheit RW (2009). Gap junction turnover is achieved by the internalization of small endocytic double-membrane vesicles. Mol Biol Cell 20, 3342–3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk MM, Bell CL, Kells-Andrews RM, Murray SA (2016). Molecular mechanisms regulating formation, trafficking, and processing of annular gap junctions. BMC Cell Biol 17, S22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk MM, Fong J, Kells R, O’Laughlin M, Kowal T, Thévenin A (2012). Degradation of endocytosed gap junctions by autophagosomal and endo-/lysosomal pathways: a perspective. J Membr Biol 245, 465–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk MM, Kells RM, Berthoud VM (2014). Degradation of connexins and gap junctions. FEBS Lett 588, 1221–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallon RF, Goodenough DA (1981). Five-hour half-life of mouse liver gap-junction protein. J Cell Biol 90, 521–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa XF, Duling BR (2008). Gap junctions in the control of vascular function. Antioxid Redox Signal 11, 251–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa XF, Isakson BE, Duling BR (2006). Vascular gap junctions in hypertension. Hypertension 48, 804–811. [DOI] [PubMed] [Google Scholar]
- Fong JT, Kells RM, Falk MM (2013). Two tyrosine-based sorting signals in the Cx43 C-terminus cooperate to mediate gap junction endocytosis. Mol Biol Cell 24, 2834–2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong JT, Kells RM, Gumpert AM, Marzillier JY, Davidson MW, Falk MM (2012). Internalized gap junctions are degraded by autophagy. Autophagy 8, 794–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong JT, Nimlamool W, Falk MM (2014). EGF induces efficient Cx43 gap junction endocytosis in mouse embryonic stem cell colonies via phosphorylation of Ser262, Ser279/282, and Ser368. FEBS Lett 588, 836–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis R, Xu X, Park H, Wei CJ, Chang S, Chatterjee B, Lo C (2011). Connexin43 modulates cell polarity and directional cell migration by regulating microtubule dynamics. PLoS One 6, e26379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaietta G, Deerinck TJ, Adams SR, Bouwer J, Tour O, Laird DW, Sosinsky GE, Tsien RY, Ellisman MH (2002). Multicolor and electron microscopic imaging of connexin trafficking. Science 296, 503–507. [DOI] [PubMed] [Google Scholar]
- Gerhart SV, Eble DM, Burger RM, Oline SN, Vacaru A, Sadler KC, Jefferis R, Iovine MK (2012). The Cx43-like connexin protein Cx40.8 is differentially localized during fin ontogeny and fin regeneration. PLoS One 7, e31364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhart SV, Jefferis R, Iovine MK (2009). Cx40.8, a Cx43-like protein, forms gap junction channels inefficiently and may require Cx43 for its association at the plasma membrane. FEBS Lett 583, 3419–3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gore AV, Monzo K, Cha YR, Pan W, Weinstein BM (2012). Vascular development in the zebrafish. Cold Spring Harb Perspect Med 2, a006684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gore AV, Monzo K, Cha YR, Pan W, Weinstein BM (2009). Gap junctions in the control of vascular function. Antioxid Redox Signal 11, 251–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girão H, Catarino S, Pereira P (2009). Eps15 interacts with ubiquitinated Cx43 and mediates its internalization. Exp Cell Res 315, 3587–3597. [DOI] [PubMed] [Google Scholar]
- Gumpert AM, Varco JS, Baker SM, Piehl M, Falk MM (2008). Double-membrane gap junction internalization requires the clathrin-mediated endocytic machinery. FEBS Lett 582, 2887–2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutstein DE, Morley GE, Tamaddon H, Vaidya D, Schneider MD, Chen J, Chien KR, Stuhlmann H, Fishman GI (2001). Conduction slowing and sudden arrhythmic death in mice with cardiac-restricted inactivation of connexin43. Circ Res 88, 333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haefliger J, Nicod P, Meda P (2004). Contribution of connexins to the function of the vascular wall. Cardiovasc Res 62, 345–356. [DOI] [PubMed] [Google Scholar]
- Hardy K, Warner A, Winston RML, Becker DL (1996). Expression of intercellular junctions during preimplantation development of the human embryo. Mol Hum Reprod 2, 621–632. [DOI] [PubMed] [Google Scholar]
- Hare JF, Taylor K (1991). Mechanisms of plasma membrane protein degradation: recycling proteins are degraded more rapidly than those confined to the cell surface. Proc Natl Acad Sci USA 88, 5902–5906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry TR, Spitsbergen JM, Hornung MW, Abnet CC, Peterson RE (1997). Early life stage toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin in zebrafish (Danio rerio). Toxicol Appl Pharmacol 142, 56–68. [DOI] [PubMed] [Google Scholar]
- Huang GY, Cooper ES, Waldo K, Kirby ML, Gilula NB, Lo CW (1998). Gap junction-mediated cell-cell communication modulates mouse neural crest migration. J Cell Biol 143, 1725–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoguchi T, Yu HY, Imamura M, Kakimoto M, Kuroki T, Maruyama T, Nawata H (2001). Altered gap junction activity in cardiovascular tissues of diabetes. Med Electron Microsc 34, 86–91. [DOI] [PubMed] [Google Scholar]
- Jongsma, HJ, Wilders, R (2000). Gap junctions in cardiovascular disease. Circ Res 86, 1193–1197. [DOI] [PubMed] [Google Scholar]
- Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, et al. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpinich NO, Caron KM (2015). Gap junction coupling is required for tumor cell migration through lymphatic endothelium. Arterioscler Thromb Vasc Biol 35, 1147–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kells-Andrews RM, Margraf RA, Fisher CG, Falk MM (2018). Connexin-43 K63-polyubiquitylation on lysines 264 and 303 regulates gap junction internalization. J Cell Sci 131, jcs204321. [DOI] [PMC free article] [PubMed]
- Kleinstiver BP, Pattanayak V, Prew MS, Tsai SQ, Nguyen NT, Zheng Z, Joung KJ (2016). High-fidelity CRISPR/Cas9 nucleases with no detectable genome-wide off-target effects. Nature 529, 490–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak BR, Pepper MS, Gros DB, Meda P (2001). Inhibition of endothelial wound repair by dominant negative connexin inhibitors. Mol Biol Cell 12, 831–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labun K, Montague TG, Gagnon JA, Thyme SB, Valen E (2016). CHOPCHOP V2: a web tool for the next generation of CRISPR genome engineering. Nucleic Acids Res 44, W272–W276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labun K, Montague TG, Krause M, Torres Cleuren YN, Tjeldnes H, Valen E (2019). CHOPCHOP V3: expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res 47, W171–W174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird DW, Naus CC, Lampe PD (2017). SnapShot: connexins and disease. Cell 170, 1260. [DOI] [PubMed] [Google Scholar]
- Lampe PD, Lau AF (2004). The effects of connexin phosphorylation on gap junctional communication. Int J Biochem Cell Biol 36, 1171–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauf U, Giepmans BN, Lopez P, Braconnot S, Chen SC, Falk MM (2002). Dynamic trafficking and delivery of connexons to the plasma membrane and accretion to gap junctions in living cells. Proc Natl Acad Sci USA 99, 10446–10451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson ND, Weinstein BM (2002). In vivo imaging of embryonic vascular development using transgenic zebrafish. Dev Biol 248, 307–318. [DOI] [PubMed] [Google Scholar]
- Leithe E, Mesnil M, Aasen T (2018). The connexin 43 C-terminus: a tail of many tales. Biochim Biophys Acta 1860, 48–64. [DOI] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. (2016). Exome aggregation C. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Sun L, Torii M, Rakic P (2012). Connexin 43 controls the multipolar phase of neuronal migration to the cerebral cortex. Proc Natl Acad Sci USA 109, 8280–8285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods 25, 402–408. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD (2008). Analyzing real-time PCR data by the comparative CT method. Nat Protoc 3, 1101–1108. [DOI] [PubMed] [Google Scholar]
- Lübkemeier I, Requardt R, Lin X, Sasse P, Andrié R, Schrickel J, Chkourko H, Bukauskas F, Kim J, Frank M, et al. (2013). Deletion of the last five C-terminal amino acid residues of connexin43 leads to lethal ventricular arrhythmias in mice without affecting coupling via gap junction channels. Basic Res Cardiol 108, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maass K, Chase SE, Lin X, Delmar M (2009). Cx43 CT domain influences infarct size and susceptibility to ventricular tachyarrhythmias in acute myocardial infarction. Cardiovasc Res 84, 361–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maass K, Shibayama J, Chase SE, Willecke K, Delmar M (2007). C-terminal truncation of connexin43 changes number, size, and localization of cardiac gap junction plaques. Circ Res 101, 1283–1291. [DOI] [PubMed] [Google Scholar]
- Martins-Marques T, Anjo SI, Pereira P, Manadas B, Girão H (2015a). Interacting network of the gap junction (GJ) protein connexin43 (Cx43) is modulated by ischemia and reperfusion in the heart. Mol Cell Proteomics 14, 3040–3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins-Marques T, Catarino S, Marques C, Matafome P, Ribeiro-Rodrigues T, Baptista R, Pereira P, Girão H (2015b). Heart ischemia results in connexin43 ubiquitination localized at the intercalated discs. Biochimie 112, 196–201. [DOI] [PubMed] [Google Scholar]
- Martins-Marques T, Catarino S, Marques C, Pereira P, Girão H (2015c). To beat or not to beat: degradation of Cx43 imposes the heart rhythm. Biochem Soc Trans 43, 476–481. [DOI] [PubMed] [Google Scholar]
- Martins-Marques T, Catarino S, Zuzarte M, Marques C, Matafome P, Pereira P, Girão H (2015d). Ischaemia-induced autophagy leads to degradation of gap junction protein connexin43 in cardiomyocytes. Biochem J 467, 231–245. [DOI] [PubMed] [Google Scholar]
- Matsuuchi L, Naus CC (2013). Gap junction proteins on the move: connexins, the cytoskeleton and migration. Biochim Biophys Acta 1828, 94–108. [DOI] [PubMed] [Google Scholar]
- McLachlan E, Shao Q, Wang H, Langlois S, Laird DW (2006). Connexins act as tumor suppressors in three-dimensional mammary cell organoids by regulating differentiation and angiogenesis. Cancer Res 66, 9886–9894. [DOI] [PubMed] [Google Scholar]
- Michela P, Velia V, Aldo P, Ada P (2015). Role of connexin 43 in cardiovascular diseases. Eur J Pharmacol 768, 71–76. [DOI] [PubMed] [Google Scholar]
- Montague TG, Cruz JM, Gagnon JA, Church GM, Valen E (2014). CHOPCHOP: a CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Res 42, W401–W407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naus CC, Aftab Q, Sin WC (2016). Common mechanisms linking connexin43 to neural progenitor cell migration and glioma invasion. Semin Cell Dev Biol 50, 59–66. [DOI] [PubMed] [Google Scholar]
- Nimlamool W, Andrews RMK, Falk MM (2015). Connexin43 phosphorylation by PKC and MAPK signals VEGF-mediated gap junction internalization. Mol Biol Cell 26, 2755–2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park DJ, Freitas TA, Wallick CJ, Guyette CV, Warn-Cramer BJ (2006). Molecular dynamics and in vitro analysis of connexin43: a new 14-3-3 mode-1 interacting protein. Protein Sci 15, 2344–2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park DJ, Wallick CJ, Martyn KD, Lau AF, Jin C, Warn-Cramer BJ (2009). Akt phosphorylates connexin43 on Ser373, a “mode-1” binding site for 14-3-3. Cell Commun Adhes 14, 211–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepper MS, Meda P (1992). Basic fibroblast growth factor increases junctional communication and connexin 43 expression in microvascular endothelial cells. J Cell Physiol 153, 196–205. [DOI] [PubMed] [Google Scholar]
- Pepper MS, Montesano R, el Aoumari A, Gros D, Orci L, Meda P (1992). Coupling and connexin 43 expression in microvascular and large vessel endothelial cells. Am J Physiol 262, 1246–1257. [DOI] [PubMed] [Google Scholar]
- Piehl M, Lehmann C, Gumpert A, Denizot J, Segretain D, Falk MM (2007). Internalization of large double-membrane intercellular vesicles by a clathrin-dependent endocytic process. Mol Biol Cell 18, 337–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plum A, Hallas G, Magin T, Dombrowski F, Hagendorff A, Schumacher B, Wolpert C, Kim J, Lamers WH, Evert M, et al. (2000). Unique and shared functions of different connexins in mice. Curr Biol 10, 1083–1091. [DOI] [PubMed] [Google Scholar]
- Polusani SR, Kalmykov EA, Chandrasekhar A, Zucker SN, Nicholson BJ (2016). Cell coupling mediated by connexin 26 selectively contributes to reduced adhesivity and increased migration. J Cell Sci 129, 4399–4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reaume AG, de Sousa PA, Kulkarni S, Langille BL, Zhu D, Davies TC, Juneja SC, Kidder GM, Rossant J (1995). Cardiac malformation in neonatal mice lacking connexin43. Science 267, 1831–1834. [DOI] [PubMed] [Google Scholar]
- Rhett JM, Jourdan J, Gourdie RG (2011). Connexin 43 connexon to gap junction transition is regulated by zonula occludens-1. Mol Biol Cell 22, 1516–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rummery NM, Hill CE (2004). Vascular gap junctions and implications for hypertension. Clin Exp Pharmacol Physiol 31, 659–667. [DOI] [PubMed] [Google Scholar]
- Sampurna BP, Audira G, Juniardi S, Lai Y-H, Hsiao C-D (2018). A simple ImageJ-based method to measure cardiac rhythm in zebrafish embryos. Inventions 3, 21. [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt VJ, Wölfle SE, Boettcher M, de Wit C (2008). Gap junctions synchronize vascular tone within the microcirculation. Pharmacol Rep 60, 68–74. [PubMed] [Google Scholar]
- Severs NJ (1999). Cardiovascular disease. Novartis Found Symp 219, 188–206. [DOI] [PubMed] [Google Scholar]
- Severs NJ, Coppen SR, Dupont E, Yeh H, Ko Y, Matsushita T (2004). Gap junction alterations in human cardiac disease. Cardiovasc Res 62, 368–377. [DOI] [PubMed] [Google Scholar]
- Singleman C, Holtzman NG (2011). Heart dissection in larval, juvenile and adult zebrafish, Danio rerio. J Vis Exp 55, 3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söhl G, Willecke K (2004). Gap junctions and the connexin protein family. Cardiovasc Res 62, 228–232. [DOI] [PubMed] [Google Scholar]
- Solan JL, Lampe PD (2007). Key connexin 43 phosphorylation events regulate the gap junction life cycle. J Membr Biol 217, 35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solan JL, Marquez-Rosado L, Sorgen PL, Thornton PJ, Gafken PR, Lampe PD (2007). Phosphorylation at S365 is a gatekeeper event that changes the structure of Cx43 and prevents down-regulation by PKC. J Cell Biol 179, 1301–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sridharan S, Simon L, Meling DD, Cyr DG, Gutstein DE, Fishman GI, Guillou F, Cooke PS (2007). Proliferation of adult Sertoli cells following conditional knockout of the gap junctional protein GJA1 (connexin 43) in mice. Biol Reprod 76, 804–812. [DOI] [PubMed] [Google Scholar]
- Suarez S, Ballmer-Hofer K (2001). VEGF transiently disrupts gap junctional communication in endothelial cells. J Cell Sci 114, 1229–1235. [DOI] [PubMed] [Google Scholar]
- Sugden WW, Meissner R, Aegerter-Wilmsen T, Tsaryk R, Leonard EV, Bussmann J, Hamm MJ, Herzog W, Jin Y, Jakobsson L, et al. (2017). Endoglin controls blood vessel diameter through endothelial cell shape changes in response to haemodynamic cues. Nat Cell Biol 19, 653–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thévenin AF, Kowal TJ, Fong JT, Kells RM, Fisher CG, Falk MM (2013). Proteins and mechanisms regulating gap-junction assembly, internalization, and degradation. Physiology (Bethesda) 28, 93–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thévenin AF, Margraf RA, Fisher CG, Kells-Andrews RM, Falk MM (2017). Phosphorylation regulates connexin43/ZO-1 binding and release, an important step in gap junction turnover. Mol Biol Cell 28, 3595–3608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Chen M, Leong H, Kuo Y, Kuo C, Lee C (2014). Connexin 43 suppresses tumor angiogenesis by down-regulation of vascular endothelial growth factor via hypoxic-induced factor-1α. Int J Mol Sci 16, 439–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner A, Clements DK, Parikh S, Evans WH, DeHaan RL (1995). Specific motifs in the external loops of connexin proteins can determine gap junction formation between chick heart myocytes. J Physiol 488, 721–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M (2017). Gap junction in the teleost fish lineage: duplicated connexins may contribute to skin pattern formation and body shape determination. Front Cell Dev Biol 5, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winterhager E, Pielensticker N, Freyer J, Ghanem A, Schrickel JW, Kim J, Behr R, Grümmer R, Maass K, Urschel S, et al. (2007). Replacement of connexin43 by connexin26 in transgenic mice leads to dysfunctional reproductive organs and slowed ventricular conduction in the heart. BMC Dev Biol 7, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Francis R, Wei CJ, Linask KL, Lo CW (2006). Connexin 43-mediated modulation of polarized cell movement and the directional migration of cardiac neural crest cells. Development 133, 3629–3639. [DOI] [PubMed] [Google Scholar]
- Ya J, Erdtsieck-Ernste EB, de Boer PA, van Kempen MJ, Jongsma H, Gros D, Moorman AF, Lamers WH (1998). Heart defects in connexin43-deficient mice. Circ Res 82, 360–366. [DOI] [PubMed] [Google Scholar]
- Zhang J, Hill CE (2005). Differential connexin expression in preglomerular and postglomerular vasculature: accentuation during diabetes. Kidney Int 68, 1171–1185. [DOI] [PubMed] [Google Scholar]
- Zhou JZ, Jiang JX (2014). Gap junction and hemichannel-independent actions of connexins on cell and tissue functions—an update. FEBS Lett 588, 1186–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.