

Abstract
Mitochondrial division is an important cellular process in both normal and pathological conditions. The dynamin GTPase Drp1 is a central mitochondrial division protein, driving constriction of the outer mitochondrial membrane (OMM). In mammals, the OMM protein mitochondrial fission factor (Mff) is a key receptor for recruiting Drp1 from the cytosol to the mitochondrion. Actin filaments are also important in Drp1 recruitment and activation. The manner in which Mff and actin work together in Drp1 activation is unknown. Here we show that Mff is an oligomer (most likely a trimer) that dynamically associates and disassociates through its C-terminal coiled coil, with a Kd in the range of 10 µM. Dynamic Mff oligomerization is required for Drp1 activation. While not binding Mff directly, actin filaments enhance Mff-mediated Drp1 activation by lowering the effective Mff concentration 10-fold. Total internal reflection microscopy assays using purified proteins show that Mff interacts with Drp1 on actin filaments in a manner dependent on Mff oligomerization. In U2OS cells, oligomerization-defective Mff does not effectively rescue three defects in Mff knockout cells: mitochondrial division, mitochondrial Drp1 recruitment, and peroxisome division. The ability of Mff to assemble into puncta on mitochondria depends on its oligomerization, as well as on actin filaments and Drp1.
INTRODUCTION
Mitochondrial division is important for mitochondrial stress response, correct mitochondrial distribution in polarized cells, and mitochondrial partitioning during cell division (Friedman and Nunnari, 2014; Mishra and Chan, 2014; Kraus and Ryan, 2017; Eisner et al., 2018; Ramachandran, 2018). Defects in mitochondrial division lead to a number of diseases, particularly in the nervous system (Nunnari and Suomalainen, 2012; Galloway and Yoon, 2013; Serasinghe and Chipuk, 2017). A key player in mitochondrial division is the dynamin family GTPase Drp1, which is recruited from the cytosol to the outer mitochondrial membrane (OMM), where it oligomerizes and hydrolyzes GTP to constrict the OMM en route to division (Bui and Shaw, 2013). Whole-body deletion of Drp1 is lethal in mice (Ishihara et al., 2009), with tissue-specific deletions causing major defects (Wakabayashi et al., 2009). Mutations in Drp1 are linked with severe neurological and other conditions in humans (Waterham et al., 2007; Fahrner et al., 2016; Longo et al., 2020).
Mitochondrial recruitment of Drp1 relies on Drp1 “receptor” proteins on the OMM (Hoppins et al., 2007; Kraus and Ryan, 2017). Budding yeast uses the OMM protein Fis1p, which binds the dimeric cytosolic protein Mdv1p to recruit Drp1 (Lackner et al., 2009; Koirala et al., 2010; Zhang et al., 2012). Mammals do not use this mechanism, but have several OMM proteins that can act as Drp1 receptors. One such receptor is the tail-anchored protein Mff (mitochondrial fission factor). Knockdown or knockout (KO) of Mff results in a dramatic decrease in mitochondrial division in multiple cell types and an almost complete absence of punctate Drp1 accumulation on mitochondria, indicative of Drp1 oligomerization (Gandre-Babbe and van der Bliek, 2008; Otera et al., 2010; Losón et al., 2013; Osellame et al., 2016; Ji et al., 2017). Patients with Mff deficiency display developmental and neurological abnormalities (Koch et al., 2016; Nasca et al., 2018). Two other proteins, MiD49 and MiD51 (also called MIEF2 and MIEF1, respectively), can also act as Drp1 receptors (Palmer et al., 2011; Zhao et al., 2011; Losón et al., 2014; Kalia et al., 2018). However, the effects of Mff depletion on mitochondrial division are generally greater (Otera et al., 2010; Losón et al., 2013). In addition, Mff is the sole receptor required for Drp1-mediated peroxisomal division, suggesting its central importance in Drp1 recruitment (Gandre-Babbe and van der Bliek, 2008; Ji et al., 2017).
The mechanisms by which Mff activates Drp1, however, are unclear. In biochemical assays, multiple studies have shown that the cytoplasmic region of Mff alone either stimulates Drp1 weakly or not at all (Otera and Mihara, 2011; Koirala et al., 2013; Ji et al., 2015; Clinton et al., 2016). Two studies show that Mff is insufficient to bind and activate Drp1 unless an inhibitory region of Drp1, the variable domain (VD), is removed (Liu and Chan, 2015; Clinton et al., 2016). How the inhibitory effects of the VD are overcome in vivo is unknown. In cells, Drp1 does not coimmunoprecipitate with Mff unless chemical cross-linking is employed prior to lysis, suggesting that the interaction is relatively low affinity (Gandre-Babbe and van der Bliek, 2008; Otera et al., 2010; Strack and Cribbs, 2012; Clinton et al., 2016).
Another area of uncertainty is Mff’s oligomeric state. While several studies show that Mff oligomerizes through its coiled-coil (CC) region, the nature of this oligomer is unclear, with one report suggesting a tetramer and another report suggesting a dimer (Koirala et al., 2013; Clinton et al., 2016). In addition, a cellular study shows evidence that mutation of the CC region has minimal effect on Mff’s cellular function (Otera et al., 2010).
These results suggest that other factors must be present to assist Mff, and there has been evidence for multiple such factors. Phosphorylation of both Drp1 and Mff modulate mitochondrial division, with Drp1 subjected to both activating and inhibitory phosphorylation while Mff phosphorylation is activating (Chang and Blackstone, 2007; Cribbs and Strack, 2007; Toyama et al., 2016). Drp1 is subject to a number of other post-translational modifications that modulate mitochondrial division (Harder et al., 2004; Nakamura et al., 2006; Braschi et al., 2009; Cho et al., 2009; Gawlowski et al., 2012). Phospholipids also influence Drp1 activity, with phosphatidic acid being inhibitory and cardiolipin activating (Bustillo-Zabalbeitia et al., 2014; Macdonald et al., 2014; Adachi et al., 2016). Finally, interaction between mitochondria and endoplasmic reticulum (ER) stimulates mitochondrial Drp1 recruitment (Friedman et al., 2011; Ji et al., 2017).
We have shown that the mechanism for ER-mediated stimulation may be through actin polymerization, through the ER-bound formin INF2-CAAX (Korobova et al., 2013). Inhibition of INF2, or of actin polymerization in general, inhibits Drp1 puncta assembly in cells (Korobova et al., 2013; Ji et al., 2015). INF2 activation increases mitochondrial Drp1 puncta accumulation (Ji et al., 2015), and this effect requires Mff (Ji et al., 2017). Drp1 binds directly to actin filaments, and actin binding increases Drp1 GTPase activity sixfold (Ji et al., 2015; Hatch et al., 2016). These results suggest that INF2-mediated actin polymerization on ER stimulates Drp1 oligomerization, and that actin polymerization works with Mff in this process. One possibility is that actin binding might enhance the relatively weak interaction between Mff and full-length Drp1 to drive Drp1 oligomerization forward.
We show here that a key element of Mff’s ability to activate Drp1 is its own oligomerization, which is of low affinity. Actin filaments increase Mff-mediated Drp1 activation by reducing the Mff concentration needed for activation. This effect of actin requires Mff oligomerization. Mff oligomerization is needed in cells for Drp1 recruitment and mitochondrial division. These results provide a mechanistic explanation for actin’s role in Drp1 activation.
RESULTS
Mff stimulates Drp1 activity and oligomerization in a concentration-dependent manner
Previous studies have shown that the cytosolic region of Mff has modest effects on Drp1 GTPase activity (Otera and Mihara, 2011; Koirala et al., 2013; Ji et al., 2015; Clinton et al., 2016). We re-examined these properties using the cytoplasmic region of human Mff isoform 4 (also called 0000) as a C-terminally 6His-tagged construct expressed in Escherichia coli and purified the protein to apparent homogeneity (Mff-∆TM) (Figure 1, A and B). We then tested the effect of Mff-∆TM on Drp1 GTPase activity using full-length Drp1 000 (containing none of the alternative splice inserts) expressed and purified as previously described (Hatch et al., 2016).
FIGURE 1:

Mff stimulates Drp1 activity and oligomerization in a concentration-dependent manner. (A) Bar diagrams of Mff constructs based on the isoform with no optional splice inserts (isoform 4, UniProt ID Q9GZY8-4). The 6xHis tag is the purple ball. (B) Coomassie-stained SDS–PAGE of Mff constructs (2 µg loaded). (C) GTPase assay for Drp1 (0.75 µM) in the absence and presence of 100 µM Mff-∆TM; data are from five independent experiments. GTPase activities: 1.55 ± 0.05 and 15.53 ± 1.04 µM GTP hydrolyzed/min/µM Drp1 without and with Mff, respectively. (D) GTPase activity for Drp1 (0.75 µM) as a function of Mff concentration added. (E) High-speed pelleting assay of Drp1 (1.3 µM) in the presence GTP (1 mM) and of varying Mff concentrations. Drp1 and Mff regions from the same SDS–PAGE gel are presented. Standards equivalent to the indicated concentrations of Drp1 or Mff in the assay are on the left of the gels. Pellet fractions are shown to the right. (F) Quantification of pelleting assay results from panel E.
At 100 µM, Mff-∆TM stimulates Drp1 GTPase activity 10-fold (Figure 1C) from a baseline of 1.55 ± 0.05 µM/min/µM to 15.53 ± 1.04 µM/min/µM. We then assessed the concentration dependence of this stimulation. Mff-∆TM stimulates Drp1 GTPase activity poorly at low concentrations similar to those tested previously and has an EC50 of 19.7 µM (Figure 1D).
We also examined the ability of Mff-∆TM to mediate Drp1 oligomerization, as assessed by high-speed pelleting assay. In the presence of 1 mM GTP, only a small fraction of Drp1 pellets at 1.3 µM. Increasing concentrations of Mff-∆TM result in increasing amounts of both Drp1 and Mff in the pellet, with Mff concentrations >10 µM necessary for observable effects (Figure 1, E and F).
These results show that Mff engages in low-avidity interaction with Drp1, resulting in activation of its GTPase activity and oligomerization. The low binding and activation of Drp1 observed in previous assays is likely due to the low concentrations of both proteins used in these assays.
Mff undergoes reversible oligomerization, dependent on the CC region
We next asked why such a high concentration of Mff-∆TM is needed to stimulate Drp1 activity. One possibility is that Mff oligomerization is necessary for Drp1 binding, and that this oligomerization is of relatively low affinity. Mff has been reported to be an oligomer (Koirala et al., 2013; Clinton et al., 2016) through a CC region at the C-terminus of the cytoplasmic segment. However, the nature of the oligomer is unclear, being reported as a dimer in one study (Koirala et al., 2013) and a tetramer in another study (Clinton et al., 2016). To resolve this discrepancy, we analyzed the CC in detail.
The CC region consists of three heptads (21 residues) N-terminal to the transmembrane (TM) domain (Figure 1A). The amino acid sequence conservation of the CC is impressive, with 100% sequence identity over a broad range of vertebrates (Supplemental Figure S1A), suggesting functional relevance. Analysis by LOGICOIL (Vincent et al., 2013) predicts that the CC is likely trimeric, followed by parallel dimeric, anti-parallel dimeric, and tetrameric in order of likelihood (Supplemental Figure S1B).
We produced two constructs postulated to disrupt Mff oligomerization (Figure 1A): 1) a construct lacking the CC and helical region (Mff-∆CC) and 2) a construct in which we mutated the d positions of two of the three predicted heptad repeats in the CC (Mff-L2P, Supplemental Figure S1C) (Chang et al., 1999). We then compared the hydrodynamic properties of these constructs with those of Mff-∆TM. By velocity analytical ultracentrifugation (vAUC), 100 µM of Mff-∆TM has a sedimentation coefficient of 2.7 (Figure 2A), with a resulting calculated mass of 65.6 kDa (Supplemental Figure S2A). In contrast, Mff-∆CC and Mff-L2P sediment at 1.2 and 1.4 S, respectively (Figure 2A), with calculated masses of 21.8 and 25.5 kDa respectively (Supplemental Figure S2A; Supplemental Table S1). The formula-calculated masses of these constructs are 23.6, 19.7, and 23.5 kDa, respectively. Conducting vAUC at a higher Mff-∆TM concentration (250 µM) produces similar results (3.0 S, 60.2 kDa, Figure 2A; Supplemental Figure S2A; Supplemental Table S1). These results suggest that Mff-∆TM is a trimer, whereas Mff-∆CC and Mff-L2P are monomers.
FIGURE 2:
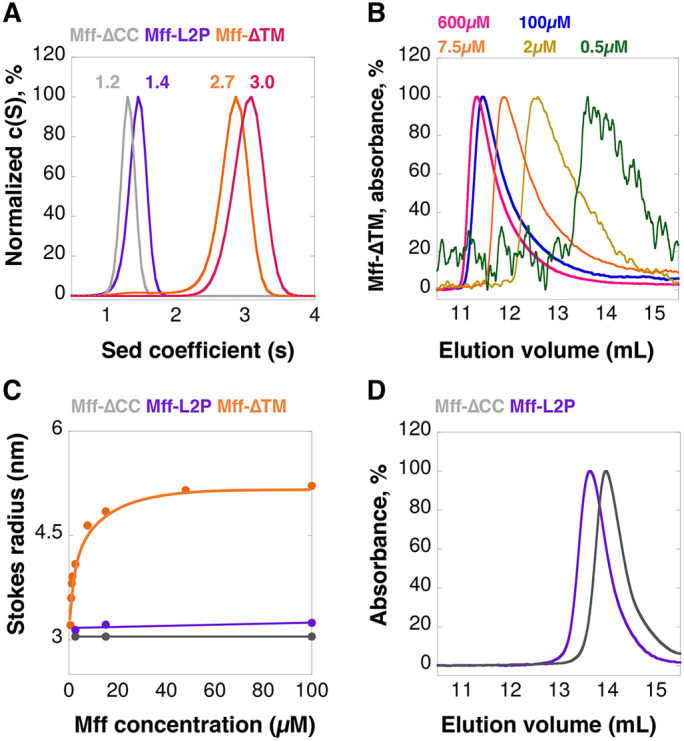
Mff undergoes reversible oligomerization, dependent on the CC region. (A) vAUC of Mff-∆TM at 100 µM (orange) or 250 µM (pink), and Mff-∆CC (gray) or Mff-L2P (purple) at 100 µM. The y-axis normalized to the peak c(S) for each sample; the maximum value of each curve is normalized as 100%, as for panels B–D. Peak sedimentation coefficients are listed on the graph. (B) SEC profiles for multiple concentrations of Mff-∆TM detected at 280 nm. The sample at 0.5 µM has low absorbance and thus gives a noisy trace. (C) Graph of calculated Stokes radius (from SEC results) versus Mff concentration for Mff-∆TM, Mff-∆CC, and Mff-L2P. (D) SEC for Mff-∆CC and Mff-L2P at 100 µM.
We examined the concentration dependence of Mff-∆TM oligomerization using size exclusion chromatography (SEC). At 100 and 600 µM, Mff-∆TM elutes in an early fraction, but lower concentrations elute progressively later (Figure 2B). Plotting the calculated Stokes radii versus concentration results in an apparent half-maximal concentration for the change in Stokes radius of 4.8 µM (Figure 2C). In contrast, 100 µM of both Mff-∆CC and Mff-L2P elute considerably later than Mff-∆TM (Figure 2D) and do not vary as a function of concentration (Figure 2C).
To determine whether the CC region is sufficient for oligomerization, we examined a fusion protein of the Mff CC on the C-terminus of GFP (GFP-CC), as well as the L2P mutant of this construct (GFP-L2P, Figure 3A). By vAUC, GFP-CC at 250 µM sediments primarily as a 4.9 S particle, corresponding to a mass of 100.3 kDa (Figure 3B; Supplemental Figure S2B; Supplemental Table S1), whereas the calculated monomer mass is 34.8 kDa. In contrast, GFP-L2P sediments at 2.7 S, corresponding to a mass of 29.3 kDa (Figure 3B; Supplemental Figure S2B; Supplemental Table S1). By SEC, a dilution series of GFP-CC results in steadily decreasing Stokes radii, with a half-maximal concentration of 15 µM, while GFP-L2P displays similar Stokes radii at low and high concentrations (Figure 3, C–E). GFP-CC itself has no ability to stimulate Drp1 GTPase activity (Supplemental Figure S2C) or to cause the pelleting of Drp1 (Supplemental Figure S2D), showing that the Mff-CC alone is not capable of Drp1 activation.
FIGURE 3:

Oligomerization of Mff CC region. (A) Bar diagrams of two GFP-fusion constructs: top, the CC region of Mff (GFP-CC) and bottom, the L2P mutant of the same construct (GFP-L2P). (B) vAUC of GFP-CC and GFP-L2P at 250 µM. Peak sedimentation coefficients are noted on the graph. (C) SEC profiles of GFP-CC at multiple concentrations detected at 490 nm. Absorbances normalized to peak absorbance at each group, the maximum value of each curve is normalized as 100%, the same for panel D. (D) SEC profiles of GFP-L2P at three concentrations. (E) Graph of calculated Stokes radius (from SEC results) versus GFP concentration for GFP-CC and GFP-L2P.
These results suggest that the cytoplasmic portion of Mff is oligomeric, and that its oligomerization is in dynamic equilibrium in the range of 5–15 µM in solution. The oligomer is most likely a trimer, although definitive resolution awaits structural analysis (see Discussion). Oligomerization is mediated by the CC region.
Mff oligomerization is necessary for productive Drp1 interaction
We next tested the effect of Mff oligomerization on its ability to influence Drp1, using Mff-∆CC or Mff-L2P as nonoligomeric constructs. In GTPase assays, neither Mff-∆CC nor Mff-L2P increases Drp1 GTPase activity at any concentration tested, in contrast to the effect of Mff-∆TM (Figure 1D). In Drp1 pelleting assays, these monomeric mutants do not cause an increase in Drp1 pelleting in the presence of GTP (Figure 1, E and F).
We also used total internal reflection fluorescence (TIRF) microscopy to assess the binding of Mff to Drp1 oligomers. In the presence of the nonhydrolyzable GTP analogue GMPPCP, Cy5-labeled Drp1 appears as heterogeneous punctae (Figure 4A), representing oligomeric rings (Koirala et al., 2013). Introduction of fluorescein-labeled Mff-∆TM causes fluorescein accumulation on Drp1 puncta (Figure 4, A and B). Significant fluorescein label remains on the puncta after washout, suggesting stable binding between Mff-∆TM and Drp1. In contrast, Mff-L2P displays fivefold less binding than Mff-∆TM and is removed after wash (Figure 4, A and B).
FIGURE 4:

Mff oligomerization is necessary for Drp1 interaction and activation. (A) TIRF microscopy of Drp1/Mff interaction. Cy5-labeled Drp1 (purple, 1 µM) was incubated with 0.5 mM GMPPCP and then flowed into the TIRF chamber. “Pre-flow” represents Drp1 puncta prior to Mff addition. Fluorescein-labeled Mff (2 µM, green, either Mff-∆TM or Mff-L2P) was then flowed in while imaging. “Flow Mff” represents an image taken after 30 s of Mff addition. After 1 min, the chamber was washed with buffer while imaging. The “After wash” image was taken at 30 s after buffer wash. “Enhanced” image is the After wash image with increased contrast to show background fluorescein channel signal. An example using Mff-∆TM is the top image set, and Mff-L2P is the bottom set. For each set, the fluorescein-Mff signal alone is shown on top, and the merged fluorescein-Mff/Cy5-Drp1 signal is shown on the bottom. Bar, 20 µm. (B) Quantification of 14 individual puncta for fluorescein-Mff:Cy5-Drp1 ratio for both Mff-∆TM and Mff-L2P.
These results suggest that Mff oligomerization is required for productive Drp1/Mff interaction, stimulating Drp1 GTP hydrolysis. The fact that oligomerization-competent Mff persists on Drp1 oligomers after washout of free Mff suggests that Drp1 binding stabilizes the Mff oligomerization interaction.
Actin filaments synergize with oligomeric Mff to stimulate Drp1 activity
We have previously shown that actin filaments stimulate Drp1 GTPase activity in a biphasic manner, with maximal stimulation occurring at 0.5 µM actin and then descending back to the level of Drp1 alone at higher actin concentrations (Hatch et al., 2016). Here we examined the combined effects of Mff-∆TM and actin on Drp1 GTPase activity. Either actin or Mff-∆TM alone stimulate Drp1, but a high concentration of Mff-∆TM is required for stimulation, with an EC50 of 17.4 µM (Figure 5A). However, the addition of 0.5 µM actin filaments greatly increases the potency of Mff’s effect on Drp1 by shifting the EC50 of Mff-∆TM to 2.2 µM (Figure 5A). Mff-∆TM does not affect the maximal effective concentration of actin (Figure 5B). The maximum combined effect of Mff-∆TM and actin is approximately 16 µM GTP hydrolyzed/min/µM Drp1. In contrast to its effect on Mff-∆TM, actin has no effect on the ability of Mff-L2P to activate Drp1, which is not able to enhance actin-induced Drp1 GTPase activity at any concentration tested (Figure 5A). Finally, the ability of Mff-∆TM and actin to stimulate Drp1 GTPase activity depends on Drp1’s ability to oligomerize, since an oligomerization-defective mutant (Drp1 401–404 AAAA) (Fröhlich et al., 2013; Hatch et al., 2016) is not activated by the Mff/actin combination (Supplemental Figure S3A).
FIGURE 5:

Synergistic effects of actin and Mff on Drp1 activity require Mff oligomerization. (A) GTPase activity of Drp1 (0.75 µM) as a function of Mff-∆TM or Mff-L2P concentration added, in the absence or presence of 0.5 µM actin filaments. (B) GTPase activity of Drp1 (0.75 µM) as a function of actin filament concentration in the absence or presence of Mff-∆TM (20 µM). (C) TIRF microscopy of actin filaments and Mff-∆TM. Actin filaments (red) were polymerized at 4 µM, mixed with a 1.1 M excess of TRITC-phalloidin. Fluorescein-labeled Mff-∆TM of varying concentration was incubated with 0.1 µM TRITC-actin overnight and introduced into the TIRF chamber. Images of actin and Mff-∆TM were taken after 15 min of actin/Mff addition. After another 1 min, the chamber was washed with buffer while imaging. Bar, 20 µm. (D) Pyrene-actin polymerization assays of actin alone or in the presence of 200 nM INF2-FFC, a potent actin polymerization factor (Chhabra and Higgs, 2006). Also included was a varying concentration of Mff-∆TM.
The ability of actin to enhance the efficiency of the Drp1 GTPase activity stimulation by Mff-∆TM is not due to a direct affinity of Mff-∆TM for actin filaments. In high-speed pelleting assays, Mff-∆TM does not sediment appreciably with actin filaments (Supplemental Figure S3, B and C). In TIRF assays, fluorescein-Mff-∆TM does not accumulate on actin filaments at any concentration tested (Figure 5C). In pyrene-actin polymerization assays, Mff-∆TM does not influence the polymerization of actin alone or in the presence of the formin protein INF2 (Figure 5D). These results suggest that actin increases Mff’s ability to stimulate Drp1 activity through an effect on the Mff/Drp1 interaction.
We next examined the effect of Mff-∆TM on actin bundling by Drp1. In a previous study, we showed that Drp1 is a weak bundler of actin filaments, requiring at least 1 µM Drp1 for effective bundling (Hatch et al., 2016). At 0.2 µM Drp1, no actin bundles are observed (Figure 6A). Mff-∆TM addition results in the appearance of actin bundles, with increasing Mff resulting in increasing bundle thickness (Figure 6, A and B) and increasing Drp1 intensity in these bundles (Figure 6C). In contrast, Mff-L2P has minimal effect on Drp1-mediated bundling (Figure 6, A–C).
FIGURE 6:

Mff interacts with Drp1-bound actin filaments. (A) TIRF assay with 0.1 µM actin filaments (TRITC-phalloidin labeled), 0.2 µM Cy5-labeled Drp1, and varying concentrations of unlabeled Mff-∆TM or Mff-L2P. No GTP was present. Bar, 30 µm. (B) Quantification of actin filament number in individual bundles. (C) Quantification of Cy5-Drp1 intensity in actin bundles. (D) TIRF assay of Mff interaction with Drp1-bundled actin filaments. Actin bundles were preformed with 0.1 µM actin (unlabeled, phalloidin-stabilized) and 2.5 µM Cy5-Drp1. Fluorescein-labeled Mff (1 µM, either Mff-∆TM or Mff-L2P) was introduced to the chamber while imaging. After 1 min, the chamber was washed while imaging. Experiments ± 1 mM GTP throughout the whole process. Bar, 10 µm. (E) Quantification of fluorescein-Mff intensity on Cy5-Drp1-bundled actin filaments by taking the Mff:Drp1 ratio. (F) Comparison of fluorescein-Mff-∆TM binding to Drp1-bundled or fascin-bundled actin filaments. Conducted in same manner as in panel D, except actin filaments were labeled with TRITC-phalloidin only during the wash step to minimize bleed-through into the fluorescein channel. Bars, 10 µm.
We also evaluated Mff binding to Drp1-bundled actin filaments using TIRF assays in which fluorescein-labeled Mff (1 µM) was added to actin filaments (0.1 µM) prebundled by 2.5 µM Cy5-Drp1. We did not label actin filaments in this experiment because the slight bleed-through of the bright actin filaments into the fluorescein channel decreased detection sensitivity for fluorescein-Mff binding. In the absence of GTP, 1 µM Mff-∆TM is able to bind Drp1/actin bundles, and ∼50% of Mff-∆TM remains after wash (Figure 6, D and E). GTP significantly increases Mff-∆TM binding to the bundles and maintains Mff-∆TM on the bundles after wash (Figure 6, D and E). In contrast, 1 µM Mff-L2P displays no detectable binding to Drp1/actin bundles in the absence of GTP and low apparent binding to bundles in the presence of GTP. Increasing the Mff-L2P concentration causes detectable binding to Drp1/actin bundles in the absence of GTP, but Mff-L2P is removed by wash (Supplemental Figure S4). We also tested the possibility that Mff can directly bind to bundled actin in general by using fascin-assembled actin bundles. In contrast to Drp1-mediated bundles, Mff-∆TM displays no detectable binding to fascin/actin bundles (Figure 6F).
These results show that Mff and actin synergize in Drp1 activation in a manner dependent on Mff oligomerization. Mff does not interact with actin itself, but can interact with actin-bound Drp1. The presence of GTP strengthens this interaction.
Mff oligomerization is required for its role in mitochondrial division
To test the importance of Mff oligomerization in mitochondrial division, we expressed either full-length Mff-WT or Mff-L2P as GFP-fusions in Mff- KO U2OS cells. Expression levels of the GFP fusions were controlled by varying the concentration of transfected plasmid, to obtain expression levels close to those of endogenous Mff, with 25 and 50 ng being used in further experiments for GFP-Mff-WT and GFP-Mff-L2P, respectively (Supplemental Figure S5A).
We have previously reported that Mff-KO U2OS cells are strongly defective in mitochondrial division (Ji et al., 2017), similar to studies in other cell types (Otera et al., 2010, 2016; Losón et al., 2013; Shen et al., 2014; Osellame et al., 2016) This defect is characterized by increased mean mitochondrial size, and a decreased number of individual mitochondria (Figure 7, A–C). GFP-Mff-WT expression in Mff-KO U2OS cells largely rescues the KO phenotype, with an increase in mitochondrial number and a decrease in mitochondrial size (Figure 7, A–C). In contrast, expression of Mff-L2P at comparable levels causes significantly reduced recovery of these parameters (Figure 7, A–C).
FIGURE 7:

Mff oligomerization is required for mitochondrial and peroxisomal division. (A) Mff-KO U2OS cells were transfected with either GFP-Mff-WT (25 ng plasmid) or GFP-Mff-L2P (50 ng) for 24 hr, then fixed and stained with anti-Tom20 (red). GFP fluorescence is green. Controls are untransfected WT and Mff-KO U2OS cells. Images at right are merges or GFP signal sof boxed regions. (B and C) Quantification of mean mitochondrial number (B) and size (C) for the indicated conditions, from ROI like those in panel A. Cells analyzed: 147, 148, 142, and 147 for U2OS WT, Mff-KO U2OS, and transfected GFP-Mff-WT or GFP-Mff-L2P, respectively. *p ≤ 0.005. ***p ≤ 0.001. ****p ≤ 0.0001; n.s. = not significant, p > 0.05. (D) Mff-KO U2OS cells were transfected with either GFP-Mff-WT or GFP-Mff-L2P as in panel A, then fixed and stained with anti-Tom20 (yellow) and anti-PMP70 (red, peroxisomes). GFP fluorescence is green. Images at right are inverted PMP70 staining of boxed regions. (E) Quantification of mean peroxisome area for the indicated conditions, from ROIs of PMP70 micrographs like those in panel D. Cells analyzed: 119, 126, 108, and 108 for U2OS WT, Mff-KO U2OS, and the retransfected GFP-Mff-WT or GFP-Mff-L2P, respectively. Numbers represent mean values. *p ≤ 0.005. ***p ≤ 0.001. ****p ≤ 0.0001. (F) Contrast-enhanced images of GFP-Mff distribution (either GFP-Mff-WT or GFP-Mff-L2P) from panel D. Scale bar, 10 µm (3 µm in insets) in panels A and D; 5 µm in panel F.
We next examined the effect of Mff-WT and Mff-L2P on peroxisome size, since Mff is also required for peroxisome division (Gandre-Babbe and van der Bliek, 2008; Ji et al., 2017; Passmore et al., 2020). Mff-KO cells display elongated peroxisomes (Figure 7D), with mean peroxisomal area 8.4-fold larger than that of WT cells (Figure 7E). Expression of Mff-WT partially rescues the peroxisome size defect, with 1.6-fold larger area than WT (Figure 7, D and E). In contrast, Mff-L2P is much less effective at reducing peroxisomal size, with 5.9-fold larger area than WT (Figure 7, D and E). Interestingly, while Mff-WT clearly enriches on the majority of peroxisomes, Mff-L2P does not appear to display peroxisomal enrichment (Figure 7F).
We also examined the distribution of Mff and Drp1 under these conditions. The two Mff constructs display clear differences in distribution, with GFP-Mff-WT displaying a punctate pattern on mitochondria while Mff-L2P is evenly distributed on mitochondria (Figure 7A). In addition, some GFP-Mff-L2P appears to be present in the cytosol, similar to previous observations for an Mff construct with deletion of the CC (Otera et al., 2010). We quantified the distribution of Mff-WT and Mff-L2P through blinded classification into three categories: uniform distribution, few puncta within a background of uniform distribution, and puntate (Supplemental Figure S5B). Mff-WT displays punctate distribution in over 70% of the cells, whereas Mff-L2P is punctate in less than 10% of cells (Supplemental Figure S5C).
We also examined the effect of Mff on mitochondrial Drp1 puncta, which presumably reflects Drp1 oligomerization. Our previous work has shown that Mff-KO or knockdown U2OS cells decreases Drp1 puncta (Ji et al., 2017), similar to other studies (Otera et al., 2010; Losón et al., 2013). GFP-Mff-WT expression in the Mff-KO U2OS cells results in recovery of larger Drp1 puncta (Supplemental Figure S6A), as quantified by puncta area and mean intensity (Supplemental Figure S6, B and C). In contrast, GFP-Mff-L2P induces ∼30% smaller Drp1 puncta (Supplemental Figure S6, A–C), suggesting that Mff oligomerization increases cellular Drp1 oligomerization.
We have previously shown that the punctate pattern of Mff is dependent on Drp1, similar to another study (Otera et al., 2016; Ji et al., 2017). We find the same result here, with Drp1 knockdown resulting in a uniform distribution of GFP-Mff-WT on mitochondria (Supplemental Figure S7A). We also tested whether actin filaments are required for the punctate pattern of GFP-Mff-WT. WT U2OS cells were treated with the actin monomer sequestering drug latrunculin A (LatA) for 15 min, then stained with anti-Mff to detect the endogenous protein. As observed previously (Korobova et al., 2013), LatA treatment results in mitochondrial elongation (Supplemental Figure S7B). LatA treatment also results in a change in mitochondrial Mff staining pattern from punctate to more uniform (Supplemental Figure S7B). We quantified this effect by measuring the percentage of mitochondrial area covered by Mff, with an increase in coverage denoting a less punctate Mff pattern. The Mff area increases twofold in LatA-treated cells (Supplemental Figure S7C). These results suggest that Drp1 and Mff reciprocally enhance each other’s oligomerization in cells, and that actin filaments are important for this effect.
DISCUSSION
We show here that oligomerization of Mff plays an important role in its ability to recruit and activate Drp1. Without the capacity to oligomerize, Mff does not efficiently affect Drp1 oligomeric recruitment or mitochondrial division in cells. Furthermore, Mff and actin filaments act synergistically in Drp1 activation, and this synergy also depends on Mff oligomerization. Actin filaments lower the concentration of Mff needed to activate Drp1.
Our results suggest a fundamentally similar requirement for Drp1 receptor oligomerization in mammalian and yeast mitochondrial division, although the Drp1 receptors are quite different. In yeast, Fis1p is the relevant OMM receptor, but does not bind Drp1 directly. Instead, Fis1p recruits the cytoplasmic adaptor protein Mdv1 to the OMM, with Mdv1 in turn recruiting Drp1 (Lackner et al., 2009; Koirala et al., 2010; Zhang et al., 2012). Mdv1 forms a dimer through a CC interaction, and this dimerization is essential for both Drp1 recruitment and mitochondrial division (Lackner et al., 2009; Koirala et al., 2010; Zhang et al., 2012). Yeast has a second protein, Caf4, with similar properties to Mdv1 (Griffin et al., 2005; Guo et al., 2012). Although mammals have a Fis1p homologue, they do not have a Caf4/Mdv1 homologue, and it is becoming increasingly clear that Fis1p has distinct functions from mitochondrial division (Otera et al., 2010; Yu et al., 2019).
We show that Mff oligomerization is important for three cellular effects: Drp1 puncta assembly on mitochondria, mitochondrial division, and peroxisomal division. In addition, oligomerization-deficient Mff itself does not display a punctate appearance on mitochondria, unlike wild-type Mff. We suspect that these cellular Mff puncta are structures that we have observed previously to colocalize with Drp1 puncta (Ji et al., 2017). It is not clear whether they represent solely the CC-mediated oligomerization or a larger oligomer. The fact that puncta of Mff-WT are reduced by either actin depolymerization or Drp1 KD suggests that the three entities (Mff, Drp1, and actin filaments) are all required to promote oligomeric Drp1-containing assemblies capable of driving mitochondrial division.
Previous results suggested that oligomerization was not required for Mff’s cellular function, because an Mff construct in which the CC is deleted rescues the mitochondrial phenotype of Mff-depleted cells nearly to the same degree as wild-type Mff (Otera et al., 2010). We found similar effects when our nonoligomerizable Mff mutant was expressed at levels much higher than endogenous Mff, but at levels closer to that of endogenous Mff, the functional difference between WT Mff and nonoligomerizable Mff is clear. High Mff expression levels likely can overcome defects in Mff oligomerization through crowding on the OMM. This effect has been suggested by biochemical results showing that monomeric Mff alone has no ability to stimulate Drp1, but can stimulate Drp1 when attached to liposomes (Clinton et al., 2016; Osellame et al., 2016), although the relative surface concentrations of monomeric and oligomerization-competent Mff necessary for this effect were not assessed. It is also possible that additional factors such as phosphorylation of Drp1 or Mff (Chang and Blackstone, 2007; Cribbs and Strack, 2007), cardiolipin levels (Bustillo-Zabalbeitia et al., 2014; Macdonald et al., 2014), or the presence of MiD proteins (Palmer et al., 2011; Zhao et al., 2011) might allow some Drp1 recruitment by oligomerization-defective Mff.
It has been unclear as to whether the many factors that can activate Drp1 act together in the same pathway, or as independent mechanisms for inducing mitochondrial division (Hatch et al., 2014). Our results strongly suggest that Mff and actin filaments operate in the same pathway. Previously, we have shown that increasing cytosolic calcium causes a fourfold increase in mitochondrial division and an increase in mitochondrial Drp1 oligomerization, and that these responses require actin polymerization through the formin protein INF2 (Korobova et al., 2013; Ji et al., 2015) as well as Mff (Ji et al., 2017). Drp1 binds directly to actin filaments, and this binding increases Drp1 GTPase activity (Ji et al., 2015; Hatch et al., 2016). How actin and Mff work together, however, has been unclear.
In this paper, we show a mechanism for this actin/Mff synergy. Mff is a poor Drp1 activator unless Drp1’s VD is removed (Liu and Chan, 2015; Clinton et al., 2016). The VD acts to inhibit aberrant interaction between Drp1 dimers (Lu et al., 2018), and oligomerization-deficient Drp1 mutants cannot be activated by Mff (Liu and Chan, 2015; Clinton et al., 2016). We show here that high concentrations of Mff alone can activate Drp1 even with the VD present. Actin filaments synergize by lowering the effective concentration of Mff for Drp1 activation. The inhibitory effects of the VD on Drp1 oligomerization might be overcome by Drp1 binding to actin filaments, allowing preassembly of Drp1 oligomers which can bind Mff more effectively. Another possibility, not mutually exclusive with the first, is that Drp1 oligomer assembly on the actin filament enhances Mff oligomerization by providing a multi-valent binding site that stabilizes Mff oligomers, allowing Mff to exert its full stimulation of Drp1 at lower concentration. Others have shown that Drp1 must be oligomerization-competent for activation by Mff (Liu and Chan, 2015; Clinton et al., 2016), and we show similar results here for Drp1 activation by Mff and actin. In either case, it is clear that Mff’s effect on Drp1 is greater than that of actin, suggesting that Mff allows a more productive Drp1 assembly for GTP hydrolysis.
The CC region, consisting of three heptad repeats, is required for Mff oligomerization. Mutation of two of the three heptad repeats is sufficient to disrupt this oligomerization. Given that Mff is a tail-anchored protein on the OMM, with the CC only eight residues from the transmembrane domain, it is highly likely that the oligomerization is parallel.
From the ensemble of the techniques used here, we conclude that Mff is most likely a trimer. Our rationale are: 1) that both the full cytoplasmic region of Mff and the CC alone as a GFP fusion sediment as trimers by vAUC, and 2) that prediction software gives the highest probability for a trimer. Our results differ to published results suggesting two different oligomeric states for Mff: a dimer (Koirala et al., 2013; Lu et al., 2018) or a tetramer (Clinton et al., 2016). The dimerization prediction made from an eAUC study (Koirala et al., 2013) may have been influenced by the slow speeds used, which did not provide sufficiently broad curves for high-confidence analysis. A second prediction of dimerization (Lu et al., 2018) as well as a prediction of tetramerization (Clinton et al., 2016) were based on SEC-MALS measurements. These discrepancies are likely due to the low affinity nature of Mff oligomerization, and the fact that most of the Mff polypeptide is not predicted to adopt a stable fold (Liu and Chan, 2015). Definitive determination of the oligomerization stoichiometry for Mff awaits more detailed structural analysis.
Regardless of the stoichiometry, our data clearly show that Mff oligomerization is relatively low affinity in solution, with dissociation constants in the range of 10 µM. On membranes, oligomerization would be significantly more favored by the reduction in dimensionality and generally confined membrane area of the OMM. Still, we find that the oligomerization-deficient mutant has a low tendency to display a punctate mitochondrial appearance when expressed at concentrations similar to endogenous, suggesting that assembly of these mitochondrial aggregates of Mff require oligomerization.
At present, it is not possible to determine if these puncta themselves represent the Mff oligomers determined biochemically, but we believe that the puncta are considerably bigger than trimers of Mff. Our model is that Mff oligomerization is a necessary step in the assembly of higher-order structures consisting of Drp1 oligomers and multiple Mff trimers. An additional required step is nucleation of Drp1 oligomers, which we postulate occurs here through actin binding. We and others have shown that knockdown of Drp1 disrupts Mff puncta (Otera et al., 2010, 2016; Ji et al., 2017), suggesting that Drp1 oligomerization is necessary for Mff puncta. In addition, others have shown that Mff binds preferentially to Drp1 oligomers, suggesting that this actin-mediated nucleation is necessary to increase the affinity of Mff for Drp1 (Liu and Chan, 2015). It is possible that other Drp1 stimuli, such as cardiolipin and MiD49/51, serve the same purpose as actin in different contexts.
Overall, our understanding of actin’s role in mitochondrial division is evolving as we learn more about its functional interactions with other division factors. Previously, we postulated that actin and myosin II mediated a “mitokinetic ring” to cause OMM preconstriction prior to Drp1 action (Korobova et al., 2013; Hatch et al., 2014). Subsequent studies from our lab and others revealed that this preconstriction is due to calcium-induced constriction of the inner mitochondrial membrane, and that actin/myosin II are necessary for the increase in mitochondrial calcium (Cho et al., 2017; Chakrabarti et al., 2018; Lee et al., 2018). In parallel, however, actin plays a second role in mitochondrial division: direct binding to Drp1, which initiates Drp1 assembly. Mff is then able to recruit these actin-nucleated Drp1 oligomers to assemble the functional constrictive ring (Ji et al., 2015, 2017; Hatch et al., 2016). This paper provides biochemical evidence for the model. We still do not know how myosin II or other proteins might enhance this process but, since myosin II is necessary for both the calcium effect (Chakrabarti et al., 2018) and Drp1 recruitment (Korobova et al., 2014), we postulate that its ability to organize/constrict the actin filaments assembled by INF2 create a higher affinity interface for Drp1 nucleation.
MATERIALS AND METHODS
Request a protocol through Bio-protocol.
Plasmids
For bacterial expression, the full-length of human Drp1 isoform 3 (NP_055681.2, UniProt ID O00429-4) and truncated human Mff isoform 4 (UniProt ID Q9GZY8-4) (Mff-∆TM) have been described previously (Hatch et al., 2016; Osellame et al., 2016). Mff-L2P double mutant (L179P and L186P) was created in Mff-∆TM construct by Quick Change mutagenesis (Stratagene, Santa Clara, CA). For Mff-ΔCC, hMff-isoform 4 lacking CC domain and TM segment was cloned into a modified pET28a vector using Nco1 and Xho1 sites. Mff-∆CC is followed by a C-terminal HRV3C protease site, an additional cystine, and a 6-His affinity tag; a cystine was inserted between the Mff-∆CC and HRV3C sites for labeling purpose. For GFP-CC, DNA coding for the CC domain of hMff-isoform 4 (amino acids 149–197) flanked by linkers (N-terminal SGGG, C-terminal GGGS) was synthesized by Integrated DNA Technologies and inserted into a modified GFP-fusion pGEX-KT vector as previously described (Gurel et al., 2014). The GFP contains the A206K mutation, which reduces the ability to dimerize (Zacharias et al., 2002). Quick Change mutagenesis was performed to make GFP-L2P double mutant. For cellular assays, full-length hMff-isoform 4 was inserted into GFP-C1 vector as previously described (Strack and Cribbs, 2012).
Protein expression, purification
Drp1 was expressed and purified as previously described with modifications (Hatch et al., 2016). Briefly, Drp1 construct was expressed in One Shot BL21 Star (DE3) E. coli (C6010-03; Life Technologies, Carlsbad, CA) in LB broth, induced by isopropyl-β-d-thiogalactoside (IPTG) at 16°C for 16 h when OD reached 1.5. Cell pellets were resuspended in lysis buffer (100 mM Tris-Cl, pH 8.0; 500 mM NaCl, 1 mM dithiothreitol [DTT], 1 mM EDTA, 2 µg/ml leupeptin, 10 µg/ml aprotinin, 2 µg/ml pepstatin A, 2 mM benzamidine, 1 µg/ml calpain inhibitor I [ALLN], and 1 µg/ml calpeptin) and lysed using a high-pressure homogenizer (M-110L Microfluidizer Processor; Microfluidics, Newton, MA). The lysate was cleared by centrifugation at 40,000 rpm (type 45 Ti rotor; Beckman, Brea, CA) for 1 h at 4°C. Avidin (20 µg/ml; PI-21128; Thermo Fisher Scientific, Waltham, MA) was added to the supernatant and then was loaded onto Strep-Tactin Superflow resin (2-1206-025; IBA, Göttingen, Germany) by gravity flow. The column was washed with 20 column volumes (CV) of lysis buffer without protease inhibitors. To elute Drp1, 0.01 mg/ml HRV3C protease in lysis buffer without protease inhibitors was added for 16 h at 4°C. The Strep-Tactin Superflow eluate was further purified by SEC on Superdex200 (GE Biosciences, Piscataway, NJ) with Drp1-S200 buffer (20 mM HEPES- KOH, pH 7.5; 150 mM KCl, 2 mM MgCl2, 1 mM DTT, and 0.5 mM ethylene glycol tetraacetic acid [EGTA]), spin concentrated, frozen in liquid nitrogen, and stored at −80°C.
Mff-∆TM and Mff-L2P were expressed in Rosetta 2 BL21-(DE3) E. coli (71400; EMD Millipore Corporation, Burlington, MA) in LB broth, induced by 1 M IPTG at 30°C for 4 h when OD reached to 1.5. Cell pellets were resuspended in lysis buffer (50 mM Tris-HCl, pH 7.5; 500 mM NaCl, 20 mM imidazole, pH 7.5; 1 mM DTT, 1 mM EDTA, 2 µg/ml leupeptin, 10 µg/ml aprotinin, 2 µg/ml pepstatin A, 2 mM benzamidine, 1 µg/ml calpain inhibitor I [ALLN], and 1 µg/ml calpeptin) and lysed using M-110 microfluidizer processor. The lysate was cleared by centrifugation at 40,000 rpm (type 45 Ti rotor; Beckman, Brea, CA) for 40 min at 4°C, the supernatant was saved. Affinity capture was performed using FPLC and a HiTrap IMAC column (17-5248-01, GE Healthcare, Chicago, IL). Prepacked HiTrap IMAC column was equilibrated with IMAC-A buffer (50 mM Tris-HCl, pH 7.5; 0.1 M NaCl, and 20 mM imidazole). Cleared lysate was loaded onto the column with a rate of 3 ml/min and washed to baseline with IMAC-A. Mff was eluted from the column with gradient-step washes by IMAC-B buffer (50 mM Tris-HCl, pH 7.5; 0.1 M NaCl, 500 mM imidazole): step1, 10% IMAC-B for 5 CV; step2, 20% IMAC-B for 5 CV; step3, 100% for 5 CV. Fractions from step3 were pooled and diluted 10-fold in ion exchange (IEX)-A buffer (50 mM Tris-HCl, pH 7.5; 1 mM DTT). Diluted fractions were loaded onto a HiTrap Q anion exchange column (54816, EMD Millipore Corporation, Burlington, MA). The column was washed to baseline with IEX-A and Mff was eluted by IEX-B buffer (50 mM Tris-HCl, pH 7.5; 1 M NaCl, and 1 mM DTT) with a step gradient: step1, 10% 5 CV, linear 10–50% 30 CV followed by linear 50–100% 5 CV. Peak Mff fractions were concentrated by reloading onto the HiTrap IMAC column and eluted with 100% IMAC-B step wash. Mff fractions were pooled and further purified by SEC on Superdex200 with S200 buffer (20 mM HEPES, pH 7.4; 2 mM MgCl2, 0.5 mM EGTA, 65 mM KCl, and 1 mM DTT) and spin concentrated with a 30,000 MWCO centrifugal concentrator (UFC903024, EMD Millipore Corporation, Burlington, MA), and aliquots were frozen in liquid nitrogen and stored at −80°C.
Mff-∆CC was expressed and cleared as described above. Mff-∆CC supernatant was loaded onto HisPur Ni-NTA resin (88221, Thermo Fisher Scientific, Rockford, IL) by gravity flow. The column was washed with 10 CV IMAC-A buffer and 30 CV ATP wash buffer (200 mM Tris-HCl, pH 7.5; 50 mM KCl, 20 mM MgCl2, and 5 mM ATP) followed by 10 CV IMAC-A buffer. To elute Mff-∆CC, 0.16 mg/ml HRV3C protease was added for 16 h at 4°C. The Ni-NTA eluate was further purified by SEC on Superdex200 with S200 buffer and spin concentrated (UFC901024, EMD Millipore Corporation, Burlington, MA), and aliquots were frozen in liquid nitrogen and stored at −80°C.
GFP-CC and GFP-L2P were expressed in Rosetta 2 BL21-(DE3) E. coli in LB broth, induced by 1M IPTG at 16°C for 16 h when OD reached to 1.0. Cell pellets were resuspended in lysis buffer and cleared as described above; 1% Triton was added to clarified supernatant and incubated for 30 min. The supernatant was loaded onto a HiTrap IMAC column with a rate of 3 ml/min and washed to baseline with IMAC-A. Mff was eluted from the column by gradient-step washes with IMAC-B buffer (50 mM Tris-HCl, pH 7.5; 0.1 M NaCl, and 500 mM imidazole): step1, 10% IMAC-B for 5 CV; step2, 20% IMAC-B for 5 CV; step3, 100% for 5 CV. Fractions from step3 were pooled and diluted fivefold in GST buffer (50 mM HEPES, pH 7.4; 500 mM NaCl, 1 mM EDTA, 1 mM DTT). Diluted fractions were loaded onto Glutathione Sepharose 4B resin (17-0756-05, GE Healthcare, Chicago, IL) by gravity flow followed by 5 CV phosphate-buffered saline (PBS) wash. To elute protein, 0.32 mg/ml TEV protease was added to the column and incubated for 16 h at 4°C. The eluate was further purified by SEC on Superdex200 with S200 buffer and spin concentrated (UFC903024, EMD Millipore Corporation, Burlington, MA), and aliquots were frozen in liquid nitrogen and stored at −80°C.
Rabbit skeletal muscle actin was extracted from acetone powder as previously described (Spudich and Watt, 1971) and further gel-filtered on Superdex 75 16/60 columns (GE Healthcare). Actin was stored in G buffer (2 mM Tris, pH 8.0; 0.5 mM DTT, 0.2 mM ATP, 0.1 mM CaCl2, and 0.01% NaN3) at 4°C.
Actin and Drp1 preparation for biochemical assays
For high-speed pelleting assay, actin filaments were polymerized from 30 µM monomers for 3 h at 23°C by the addition of a 10× stock of polymerization buffer (200 mM HEPES, pH 7.4; 650 mM KCl, 10 mM MgCl2, 10 mM EGTA) to a final 1× concentration. For GTPase assay, actin monomers in G-buffer were incubated with AG1-X2 100–200 mesh anion exchange resin (Dowex; 1401241; Bio-Rad) at 4°C for 5 min to remove ATP, followed by low-speed centrifugation; 20 µM actin filaments were polymerized as described before. To maintain ionic strength across all samples, an actin blank was prepared in parallel using G-buffer in place of actin monomers and used to dilute actin filaments as needed for each sample. Drp1 was diluted in MEHD buffer (20 mM HEPES, pH 7.4; 2 mM MgCl2, 0.5 mM EGTA, 1 mM DTT) to adjust the ionic strength to the same as S200 buffer before biochemical assays.
Size exclusion chromatography assays
Mff-∆TM, Mff-L2P, Mff-∆CC, GFP-CC, and GFP-L2P oligomeric distribution was determined by Superdex200 increase 10/300 GL SEC column in S200 buffer (20 mM HEPES, pH 7.4; 65 mM KCl, 2 mM MgCl2, 0.5 mM EGTA, and 1 mM DTT). Protein at varying concentration was loaded onto the column in a total volume of 500 µl and gel-filtered with a flow rate of 0.7 ml/min.
High-speed pelleting assay
Interactions between Drp1 and Mff (Mff-∆TM, Mff-∆CC, Mff-L2P) were tested in the S200 buffer plus 1 mM GTP; 1.3 µM Drp1 was incubated with varying concentrations of Mff (0–50 µM) for 10 min at 4°C in a 200 µl volume. After incubation, samples were centrifuged at 80,000 rpm for 20 min at 4°C in a TLA-100.1 rotor (Beckman). The supernatant was carefully removed. Pellets were washed three times with S200 buffer plus 1 mM GTP and then resuspended in 100 µl of SDS–PAGE sample buffer and resolved by SDS–PAGE (LC6025; Invitrogen, Carlsbad, CA). To test the interaction between Mff and actin, 5 µM Mff-∆TM was incubated with varying concentration of actin filaments (0–20 µM) for 1 h at 23°C in a 200 µl volume. Samples were centrifuged and the pellets were resolved by SDS–PAGE. Gels were stained with Coomassie Brilliant Blue R-250 staining (1610400, Bio-Rad, Hercules, CA), and band intensity was analyzed using ImageJ software.
GTPase assay
Drp1 (0.75 µM) was mixed with the indicated concentration of Mff or actin filaments. Sample were incubated at 37°C for 5 min. At this point, GTP was added to a final concentration of 250 µM to start the reaction at 37°C. Reactions were quenched at designated time points by mixing 15 µl of sample with 5 µl of 125 mM EDTA in a clear, flat-bottomed, 96-well plate (Greiner, Monroe, NC). Six time points were acquired for all conditions, and high-speed reactions were monitored in a 12 min time range, while low-speed reactions were monitored in a 45 min time range. Released phosphate was determined by the addition of 150 µl of malachite green solution as previously described (Hatch et al., 2016) Absorbance at 650 nm was measured 15 min after malachite green solution incubation. GTP hydrolysis rates were determined by plotting phosphate concentration as a function of time.
Pyrene actin polymerization assay
Pyrene actin polymerization assay has been described previously (Gurel et al., 2015). Briefly, rabbit skeletal muscle actin (2 µM actin, 10% pyrene) was polymerized with varying concentrations of Mff-∆TM in polymerization buffer (50 mM KCl, 1 mM MgCl2, 1 mM EGTA, 10 mM HEPES, pH 7.4; 1 mM DTT, 2 mM Tris-HCl, 0.2 mM ATP, 2 mM Tris-HCl, 0.2 mM ATP, 0.1 mM CaCl2, and 0.01% wt/vol NaN3). Rabbit muscle actin and 200 nM INF2-FFC with varying concentrations of Mff-∆TM were used to test the effects of Mff on formin-induced actin polymerization (Chhabra and Higgs, 2006). Pyrene fluorescence (365/410 nm) was monitored in a 96-well fluorescence plate reader (Infinite M1000; Tecan, Mannedorf, Switzerland) after 1 min of inducing polymerization.
Protein labeling
Mff-∆TM and Mff-L2P N-term were labeled with fivefold molar excess of 5/6-carboxyfluorescein succinimidyl ester (46410, Thermo Scientific) in 20 mM HEPES, pH 7.4, at 4°C; 2 mM MgCl2, 0.5 mM EGTA at 4°C for 1 h. Reactions were quenched by 100-fold molar excess of Tris-HCl, pH 7.4, at 4°C. Drp1 N-term was labeled with 3.3-fold molar excess of Cy5 succinimidyl ester (1076-1, Click Chemistry Tools, Scottsdale, AZ) in 100 mM MES, pH 6.1, at 4°C, 2 mM MgCl2, 0.5 mM EGTA for 10 min. Reactions were quenched by Tris-HCl as described above. Free dye was removed by gel filtration using Superdex200 increase 10/30 (GE Biosciences) in S200 buffer. For ∆TM and L2P, final protein concentration was determined by Bradford (Bio-Rad) and fluorescein concentration using extinction coefficient 72,000 M–1 cm–1 at 494 nm. Calculated ratio of fluorescein-Mff-∆TM is 0.49 and fluorescein-Mff-L2P is 0.44. For Drp1, Cy5 concentration was determined by extinction coefficient 255,000 M–1 cm–1. Calculated ratio of Cy5-Drp1 is 0.75. Actin filaments (4 µM) were assembled from monomers for 3 h as described and then stabilized with either tetramethylrhodamine (TRITC)-phalloidin (4 µM P1951; Sigma-Aldrich) or unlabeled 4 µM phalloidin (17466-45-4, Sigma-Aldrich) for 5 min.
TIRF MICROSCOPY IMAGING
Mff GMPPCP-Drp1 interaction experiment
To measure Mff and GMPPCP-Drp1 interaction, 1 µM Cy5-labeled Drp1 was incubated in S200 buffer plus 500 µM GMPPCP at room temperature (RT) overnight. Drp1 was manually added to the flow cell in a 10 µl volume while the current flow cell solution was removed with Whatman paper. Once Drp1 attached to the cell (15 min incubation), 2 µM fluorescein-labeled Mff (Mff-∆TM and Mff-L2P) was flowed into the chamber. After 1 min incubation, Mff was washed by S200 buffer plus 500 µM GMPPCP. Two-color images were acquired at three time points: before, during Mff flow, and after buffer wash. ImageJ software was used to measure fluorescence intensity. Briefly, background-subtracted intensities of Drp1 and Mff on a punctum were measured at the time points mentioned above; the intensity ratio of Mff/Drp1 was calculated at each time point.
Mff-Drp1-mediated actin bundling experiment
TRITC-phalloidin-stabilized actin filaments (100 nM) were incubated with 200 nM Drp1 and varying concentrations of unlabeled Mff overnight at RT. Samples were manually added to the flow cell in a 10 µl volume as mentioned above. Two-color images were acquired after 15 min sample incubation, and the same exposure times and laser intensities were used for all conditions. Interestingly, due to the strong TRITC signal, actin filaments can be acquired by the 488-nm laser. Under this condition, TRITC signal kept in a linear range without saturation.
To determine the number of actin filaments in a bundle, maximum intensity of 50 background-subtracted single actin filaments from actin alone group was measured by line scan. An average intensity of the filaments was used as the value of single actin filament. The number of actin filaments in a bundle was calculated by the ratio of the intensity of actin bundle to single actin filament; 50 actin bundles were measured.
To determine the intensity of actin-colocalized Drp1, actin was thresholded using an ImageJ plugin, colocalization, with the following parameters: ratio, 30% (0–100%); threshold channel, 1: 50 (0–255); threshold channel, 2: 50 (0–255); display value, 255 (100–255). Colocalized Drp1 puncta intensities were measured with ImageJ using line scans.
Mff binding to Drp1-actin bundles experiment
To measure the interaction between Mff and Drp1-preassembled actin bundles, 2.5 µM Cy5-labeled Drp1 was incubated with 100 nM unlabeled actin filaments overnight at RT. Drp1 and actin mixture was manually added to the flow cell as described before. The mixture was incubated in the chamber for 15 min to let actin attach to the cell. Fluorescein-labeled Mff was flowed into the chamber. After 1 min incubation, Mff was washed by S200 buffer. To test the effects of GTP on the interaction, 1 mM GTP was added to the Drp1 and actin mixture right before adding it into the chamber. Mff flow and buffer wash steps also included 1 mM GTP throughout the experiment. Two-color images were acquired at three time points: before, during Mff flow, and after buffer wash. ImageJ software was used to measure fluorescence intensity. Briefly, background-subtracted intensities of Drp1 and Mff on a given punctum were measured at the time points mentioned above, and the intensity ratio of Mff/Drp1 was calculated at each time point.
Mff actin interaction experiment
To test the direct interaction between Mff and actin filaments, varying concentration of Mff were incubated with 100 nM TRITC-actin filaments overnight at RT. Samples were manually added to the flow cell in a 10 µl volume as mentioned above. Two-color images were acquired after 15 min sample incubation, and the same exposure times and laser intensities were used throughout all conditions tested.
Cell culture, transfection, and drug treatments
Human osteosarcoma U2OS cells (HTB96; American Type Culture Collection) were grown in DMEM (Invitrogen) supplemented with 10% calf serum (Atlanta Biologicals). All cells were cultivated at 37°C and in 5% CO2 atmosphere.
For the generation of the Mff KO U2OS cell line, an appropriate guide sequence (5′-CAC CGT GAT AAT GCA AGT TCC GGA G-3′) was cloned into LentiCRISPRv2 vector according to the protocol from http://genome-engineering.org/gecko/. The resulting guide plasmid, along with helper plasmids psPAX2 and pMD2.G were transfected into HEK293 cells using Lipofectamine LTX (Invitrogen). Supernatant containing lentivirus was collected 48 h post-transfection and filtered through a 0.45-μm filter. Fresh virus was used to infect U2OS cells. After 48 h, media containing virus was removed and replaced with fresh media containing 2 μg/ml puromycin. Cells clones were selected through puromycin resistance, and single cell clones were expanded and tested for Mff depletion by Western blotting.
For transfections, cells were seeded at 5 × 105 cells per well in 3-cm culture dishes ∼16 h before transfection. Plasmid transfections were performed in OPTI-MEM media (Invitrogen) with 2 µl Lipofectamine 2000 (Invitrogen) per plate for 6 h. For all experiments, the following amounts of DNA were transfected per plate: 25 ng for GFP-Mff WT and 50 ng for GFP-Mff-L2P. After ∼6 h incubation, cells were trypsinized and replated onto fibronectin (Sigma-Aldrich, 1:100 in PBS)-coated glass coverslips (18 mm diameter, Electron Microscopy Sciences) at a density of ∼3.5 × 105 cells per dish.
LatA (428021; Calbiochem) treatment (Supplemental Figure S7B) was performed before fixation of the cells; 2 µM LatA (from a 2 mM stock in DMSO) was dissolved in cell culture media and added to the cells for 15 min at 37°C. An equal volume of DMSO was used as control treatment.
Drp1 RNA interference
For siRNA transfections, Mff-KO cells were plated on 3-cm dishes, 1 × 105 cells were plated for control siRNA transfection, and 2 × 105 cells were prepared for Drp1 siRNA as the knockdown affected cell growth. Sequence for scrambled control siRNA was 5′-CGTTAATCGCGTATAATACGCGTAT-3′ and siRNA sequence for Drp1 silencing was 5′-GCCAGCTAGATATTAACAACAAGAA-3′. Transfections were carried out using 2 µl RNAi max (Invitrogen) and 63 pg siRNA per dish. siRNA transfections were repeated after 48 h, and cells were transfected with GFP-Mff constructs after 72 h and finally analyzed 96 h post-transfection. Knockdown cells were plated onto fibronectin-coated coverslips, fixed, and stained for Drp1 (Supplemental Figure S7A).
Cell lysates and Western blotting
For generation of cell lysates (Supplemental Figure S5A), cells were grown and transfected on 3-cm dishes as described before. On the following day, cells were washed three times with PBS and lysed using 1× DB (125 mM Tris-HCl, pH 6.8; 1 mM EDTA, 10% glycerol, 0.4% SDS, 0.01% bromophenol blue, 500 mM NaCl, and 2 M urea). Cell lysates were collected from plates, boiled 5 min at 95°C, and sheared using a 27× G needle. Proteins were separated by standard SDS–PAGE and transferred to a PVDF membrane (polyvinylidine difluoride membrane; Millipore). The membrane was blocked with TBS-T (20 mM Tris-HCl, pH 7.6; 136 mM NaCl, and 0.1% Tween-20) containing 3% bovine serum albumin (Research Organics) for 1 h, then incubated with the primary antibody solution at 4°C overnight. After washing with TBS-T, the membrane was incubated with IRDye-680RD goat anti-mouse or IRDye-800CW goat anti-rabbit secondary antibodies (#926-68070, #926-32211, LI-COR) for 1 h at RT. Membrane was washed with TBS-T and dried before exposure. Signals were detected using the Odyssey CLx imaging system (LI-COR). Primary antibodies applied were as follows: GFP (rabbit, self-made, 1:1000), GAPDH (mouse, Santa Cruz (G-9) sc-365062, 1:1000), and Mff (rabbit, Proteintech 17090-1-AP, 1:1000).
Immunofluorescence staining
Cells were plated subconfluently onto fibronectin-coated coverslips and allowed to adhere overnight. The following day, cell culture media was removed, and cells were fixed in prewarmed 4% paraformaldehyde/PBS for 20 min at RT, followed by three PBS washes and permeabilization with 0.1% TritonX-100 in PBS for 1 min. Coverslips were blocked with 10% calf serum for 30 min, followed by primary antibody incubation for 1 h. After three PBS washes, coverslips were incubated with respective secondary antibodies (fluorescein anti-rabbit or -mouse, 1:300, FI-1000/FI-2000, Vector Laboratories and Texas-Red anti-rabbit or -mouse, 1:300, TI-1000/TI-2000, Vector Laboratories) for 45 min. Alexa Fluor 405-phalloidin (A30104, Invitrogen) was added with secondary antibodies. After three PBS washes, coverslips were mounted on glass slides using ProLong Gold Antifade mountant (P36930, Invitrogen). Primary antibodies were as follows: Mff (Proteintech 17090-1-AP, rabbit, 1:200), Tom20 (Santa Cruz [F10]: sc-17764, mouse, 1:50), Tom20 (Abcam ab78547, rabbit, 1:200), Drp1 (BD Transduction Laboratories 611112, mouse, 1:50), and PMP70 (Abcam ab3421, rabbit, 1:100).
Confocal microscopy
Imaging was performed at a Dragonfly 302 spinning disk confocal (Andor Technology, Inc.) on a Nikon Ti-E base and equipped with an iXon Ultra 888 EMCCD camera, and a Zyla 4.2 Mpixel sCMOS camera. Solid-state 405 smart diode 100-mW laser, solid-state 488 OPSL smart laser 50-mW laser, and solid-state 560 OPSL smart laser 50-mW laser were used (objective: 10× 1.4 NA CFI Plan Apo; Nikon). Images were acquired using Fusion software (Andor Technology).
Mitochondrial/peroxisomal length analysis
Mff-KO cells with restored expression of either GFP-Mff WT or GFP-Mff-L2P were analyzed for their mitochondrial and peroxisomal morphology (Figure 7). Untransfected U2OS WT and Mff-KO cells served as controls for normal and abnormal phenotypes, respectively. Regions of interest (ROIs) were selected in the flat cell periphery with well-resolvable, individual organelles. ROIs (400 µm2 area) of mitochondria/peroxisomes were converted to 8-bit binary masks and analyzed using the “analyze particles” plugin in Fiji to obtain the mean number of mitochondrial/peroxisomal fragments and the mean size of these organelles per ROI. Settings for analyzing particles were as follows: size (pixel2), 0.05—infinity, circularity 0.00–1.00 for mitochondria; and size (pixel2), 0.02—infinity, circularity 0.00–1.00 for peroxisomes, respectively. Data were plotted as bar graphs or box and whiskers plots.
Mff localization analysis
The 100× images of transfected cells were randomly taken and afterward analyzed in a blinded manner. The localization of GFP-Mff WT or GFP-Mff-L2P was categorized into three groups: uniform distribution of the GFP signal on mitochondria, mainly uniform distribution with few puncta visible, or a clear punctate localization on mitochondria. The percentage of cells falling into these three classes was represented in stacked bar graphs with SEM from three independent experiments (Supplemental Figure S5, B and C).
Analysis of Mff/Drp1:mitochondrial area ratio
ROIs containing mitochondria in spread, peripheral cell areas (Supplemental Figures S6 and S7, B and C) were thresholded using the same contrast settings for Tom20 staining. Anti-Mff stainings were first processed by background subtraction using Fiji (rolling ball radius 5 pixels), then further thresholded applying equal setting parameters. Mff or Drp1 with respective Tom20 stainings were analyzed using the Fiji “Colocalization” plugin with the following parameters: ratio 30% (0–100%), threshold channel 1: 50 (0–255), threshold channel 2: 50 (0–255), display value (0–255): 255. Colocalized pixels were then converted to a binary mask and quantified using the “analyze particles” tool with settings as follows: size (pixel2) 0.01—infinity, circularity 0.00–1.00.
Data processing and statistical analyses
Figures were processed and assembled with Photoshop CS4. Data analyses were carried out in ImageJ and Excel 2010. Statistical comparisons were performed with GraphPad Prism using unpaired t test. A probability of error of 5% (p ≤ 0.05; * in figure panels) was considered to indicate statistical significance; **, ***, **** indicated p values ≤ 0.01, 0.001, and 0.0001, respectively.
Supplementary Material
Acknowledgments
We thank Zdenek Svindrych for help with TIRF microscopy, Andreia Isabel Ferreira Verissimo for advice on protein expression and purification, Emilie Shipman for cell culturing support, Michael Ryan for Mff-∆TM construct, Stefan Strack for GFP-Mff construct, Roberto Dominguez for fascin, Ryn Geys for working with us so well, Dek Woolfson and Amy Keating for advice on coiled coil prediction, Peter Schuck for help with analytical ultracentrifugation analysis, and James Moseley and Michael Ragusa for advice on the project. This work was supported by National Institutes of Health Grants NIH GM122545 and DK088826 to H.N.H. and P20 GM113132 to the Institute for Biomolecular Targeting Centers of Biomedical Research Excellence (COBRE). This work was also supported by the German Research Foundation grant KA 5106/1-1 to F.K.
Abbreviations used:
- CC
coiled coil
- CV
column volume
- DTT
dithiothreitol
- EGTA
ethylene glycol tetraacetic acid
- ER
endoplasmic reticulum
- IEX
ion exchange
- IPTG
isopropyl-β- d-thiogalactoside
- KO
knockout
- LatA
latrunculin A
- Mff
mitochondrial fission factor
- OMM
outer mitochondrial membrane
- PBS
phosphate-buffered saline
- ROI
region of interest
- RT
room temperature
- SEC
size exclusion chromatography
- TIRF
total internal reflection fluorescence
- TM
transmembrane
- TRITC
tetramethylrhodamine
- vAUC
velocity analytical ultracentrifugation
- VD
variable domain.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E21-04-0224).
REFERENCES
- Adachi Y, Itoh K, Yamada T, Cerveny KL, Suzuki TL, Macdonald P, Frohman MA, Ramachandran R, Iijima M, Sesaki H (2016). Coincident phosphatidic acid interaction restrains Drp1 in mitochondrial division. Mol Cell 63, 1034–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braschi E, Zunino R, McBride HM (2009). MAPL is a new mitochondrial SUMO E3 ligase that regulates mitochondrial fission. EMBO Rep 10, 748–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui HT, Shaw JM (2013). Dynamin assembly strategies and adaptor proteins in mitochondrial fission. Curr Biol 23, 891–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustillo-Zabalbeitia I, Montessuit S, Raemy E, Basañez G, Terrones O, Martinou JC (2014). Specific interaction with cardiolipin triggers functional activation of dynamin-related protein 1. PLoS One 9, e102738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti R, Ji WK, Stan RV, Sanz JJ, Ryan TA, Higgs HN (2018). INF2-mediated actin polymerization at the ER stimulates mitochondrial calcium uptake, inner membrane constriction, and division. J Cell Biol 217, 251–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CR, Blackstone C (2007). Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J Biol Chem 282, 21583–21587. [DOI] [PubMed] [Google Scholar]
- Chang DK, Cheng SF, Trivedi VD, Lin KL (1999). Proline affects oligomerization of a coiled coil by inducing a kink in a long helix. J Struct Biol 128, 270–279. [DOI] [PubMed] [Google Scholar]
- Chhabra ES, Higgs HN (2006). INF2 is a WASP homology 2 motif-containing formin that severs actin filaments and accelerates both polymerization and depolymerization. J Biol Chem 281, 26754–26767. [DOI] [PubMed] [Google Scholar]
- Cho B, Cho HM, Jo Y, Kim HD, Song M, Moon C, Kim H, Kim K, Sesaki H, Rhyu IJ, et al. (2017). Constriction of the mitochondrial inner compartment is a priming event for mitochondrial division. Nat Commun 8, 15754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho D-H, Nakamura T, Fang J, Cieplak P, Godzik A, Gu Z, Lipton SA (2009). S-nitrosylation of Drp1 mediates β-amyloid-related mitochondrial fission and neuronal injury. Science (80) 324, 102–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clinton RW, Francy CA, Ramachandran R, Qi X, Mears JA (2016). Dynamin-related protein 1 oligomerization in solution impairs functional interactions with membrane-anchored mitochondrial fission factor. J Biol Chem 291, 478–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cribbs JT, Strack S (2007). Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep 8, 939–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisner V, Picard M, Hajnóczky G (2018). Mitochondrial dynamics in adaptive and maladaptive cellular stress responses. Nat Cell Biol 20, 755–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahrner JA, Liu R, Perry MS, Klein J, Chan DC (2016). A novel de novo dominant negative mutation in DNM1L impairs mitochondrial fission and presents as childhood epileptic encephalopathy. Am J Med Genet Part A 170, 2002–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK (2011). ER tubules mark sites of mitochondrial division. Science (80) 334, 358–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JR, Nunnari J (2014). Mitochondrial form and function. Nature 505, 335–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fröhlich C, Grabiger S, Schwefel D, Faelber K, Rosenbaum E, Mears J, Rocks O, Daumke O (2013). Structural insights into oligomerization and mitochondrial remodelling of dynamin 1-like protein. EMBO J 32, 1280–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galloway CA, Yoon Y (2013). Mitochondrial morphology in metabolic diseases. Antioxidants Redox Signal 19, 415–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandre-Babbe S, van der Bliek AM (2008). The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol Biol Cell 19, 2402–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gawlowski T, Suarez J, Scott B, Torres-Gonzalez M, Wang H, Schwappacher R, Han X, Yates JR, Hoshijima M, Dillmann W (2012). Modulation of dynamin-related protein 1 (DRP1) function by increased O-linked-β-N-acetylglucosamine modification (O-GlcNAc) in cardiac myocytes. J Biol Chem 287, 30024–30034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin EE, Graumann J, Chan DC (2005). The WD40 protein Caf4p is a component of the mitochondrial fission machinery and recruits Dnm1p to mitochondria. J Cell Biol 170, 237–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Q, Koirala S, Perkins EM, McCaffery JM, Shaw JM (2012). The mitochondrial fission adaptors Caf4 and Mdv1 are not functionally equivalent. PLoS One 7, e53523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurel PS, Ge P, Grintsevich EE, Shu R, Blanchoin L, Zhou ZH, Reisler E, Higgs HN (2014). INF2-mediated severing through actin filament encirclement and disruption. Curr Biol 24, 156–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurel PS, Mu A, Guo B, Shu R, Mierke DF, Higgs HN (2015). Assembly and turnover of short actin filaments by the formin INF2 and profilin. J Biol Chem 290, 22494–22506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder Z, Zunino R, McBride H (2004). Sumo1 conjugates mitochondrial substrates and participates in mitochondrial fission. Curr Biol 14, 340–345. [DOI] [PubMed] [Google Scholar]
- Hatch AL, Gurel PS, Higgs HN (2014). Novel roles for actin in mitochondrial fission. J Cell Sci 127, 4549–4560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatch AL, Ji WK, Merrill RA, Strack S, Higgs HN (2016). Actin flaments as dynamic reservoirs for Drp1 recruitment. Mol Biol Cell 27, 3109–3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppins S, Lackner L, Nunnari J (2007). The machines that divide and fuse mitochondria. Annu Rev Biochem 76, 751–780. [DOI] [PubMed] [Google Scholar]
- Ishihara N, Nomura M, Jofuku A, Kato H, Suzuki SO, Masuda K, Otera H, Nakanishi Y, Nonaka I, Goto Y, et al. (2009). Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat Cell Biol 11, 958–966. [DOI] [PubMed] [Google Scholar]
- Ji WK, Chakrabarti R, Fan X, Schoenfeld L, Strack S, Higgs HN (2017). Receptor-mediated Drp1 oligomerization on endoplasmic reticulum. J Cell Biol 216, 4123–4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji WK, Hatch AL, Merrill RA, Strack S, Higgs HN (2015). Actin filaments target the oligomeric maturation of the dynamin GTPase Drp1 to mitochondrial fission sites. Elife 4, e11553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalia R, Wang RYR, Yusuf A, Thomas PV, Agard DA, Shaw JM, Frost A (2018). Structural basis of mitochondrial receptor binding and constriction by DRP1. Nature 558, 401–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch J, Feichtinger RG, Freisinger P, Pies M, Schrödl F, Iuso A, Sperl W, Mayr JA, Prokisch H, Haack TB (2016). Disturbed mitochondrial and peroxisomal dynamics due to loss of MFF causes Leigh-like encephalopathy, optic atrophy and peripheral neuropathy. J Med Genet 53, 270–278. [DOI] [PubMed] [Google Scholar]
- Koirala S, Bui HT, Schubert HL, Eckert DM, Hill CP, Kay MS, Shaw JM (2010). Molecular architecture of a dynamin adaptor: Implications for assembly of mitochondrial fission complexes. J Cell Biol 191, 1127–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koirala S, Guo Q, Kalia R, Bui HT, Eckert DM, Frost A, Shaw JM (2013). Interchangeable adaptors regulate mitochondrial dynamin assembly for membrane scission. Proc Natl Acad Sci USA 110, 1342–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korobova F, Gauvin TJ, Higgs HN (2014). A role for myosin II in mammalian mitochondrial fission. Curr Biol 24, 409–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korobova F, Ramabhadran V, Higgs HN (2013). An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2. Science 339, 464–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus F, Ryan MT (2017). The constriction and scission machineries involved in mitochondrial fission. J Cell Sci 130, 2953–2960. [DOI] [PubMed] [Google Scholar]
- Lackner LL, Horner JS, Nunnari J (2009). Mechanistic analysis of a dynamin effector. Science 325, 874–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KS, Huh S, Lee S, Wu Z, Kim AK, Kang HY, Lu B (2018). Altered ER-mitochondria contact impacts mitochondria calcium homeostasis and contributes to neurodegeneration in vivo in disease models. Proc Natl Acad Sci USA 115, E8844–E8853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Chan DC (2015). The mitochondrial fission receptor Mff selectively recruits oligomerized Drp1. Mol Biol Cell 26, 4466–4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo F, Benedetti S, Zambon AA, Sora MGN, Di Resta C, De Ritis D, Quattrini A, Maltecca F, Ferrari M, Previtali SC (2020). Impaired turnover of hyperfused mitochondria in severe axonal neuropathy due to a novel DRP1 mutation. Hum Mol Genet 29, 177–188. [DOI] [PubMed] [Google Scholar]
- Losón OC, Liu R, Rome ME, Meng S, Kaiser JT, Shan SO, Chan DC (2014). The mitochondrial fission receptor MiD51 requires ADP as a cofactor. Structure 22, 367–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losón OC, Song Z, Chen H, Chan DC (2013). Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell 24, 659–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B, Kennedy B, Clinton RW, Wang EJ, McHugh D, Stepanyants N, MacDonald PJ, Mears JA, Qi X, Ramachandran R (2018). Steric interference from intrinsically disordered regions controls dynamin-related protein 1 self-assembly during mitochondrial fission. Sci Rep 8, 10879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald PJ, Stepanyants N, Mehrotra N, Mears JA, Qi X, Sesaki H, Ramachandran R (2014). A dimeric equilibrium intermediate nucleates Drp1 reassembly on mitochondrial membranes for fission. Mol Biol Cell 25, 1905–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra P, Chan DC (2014). Mitochondrial dynamics and inheritance during cell division, development and disease. Nat Rev Mol Cell Biol 15, 634–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura N, Kimura Y, Tokuda M, Honda S, Hirose S (2006). MARCH-V is a novel mitofusin 2- and Drp1-binding protein able to change mitochondrial morphology. EMBO Rep 7, 1019–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasca A, Nardecchia F, Commone A, Semeraro M, Legati A, Garavaglia B, Ghezzi D, Leuzzi V (2018). Clinical and biochemical features in a patient with mitochondrial fission factor gene alteration. Front Genet 9, 625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunnari J, Suomalainen A (2012). Mitochondria: In sickness and in health. Cell 148, 1145–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osellame LD, Singh AP, Stroud DA, Palmer CS, Stojanovski D, Ramachandran R, Ryan MT (2016). Cooperative and independent roles of the Drp1 adaptors Mff, MiD49 and MiD51 in mitochondrial fission. J Cell Sci 129, 2170–2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otera H, Mihara K (2011). Discovery of the membrane receptor for mitochondrial fission GTPase Drp1. Small GTPases 2, 167–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otera H, Miyata N, Kuge O, Mihara K (2016). Drp1-dependent mitochondrial fission via MiD49/51 is essential for apoptotic cristae remodeling. J Cell Biol 212, 531–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, Mihara K (2010). Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol 191, 1141–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer CS, Osellame LD, Laine D, Koutsopoulos OS, Frazier AE, Ryan MT (2011). MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep 12, 565–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passmore JB, Carmichael RE, Schrader TA, Godinho LF, Ferdinandusse S, Lismont C, Wang Y, Hacker C, Islinger M, Fransen M, et al. (2020). Mitochondrial fission factor (MFF) is a critical regulator of peroxisome maturation. Biochim Biophys Acta - Mol Cell Res 1867, 118709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran R (2018). Mitochondrial dynamics: The dynamin superfamily and execution by collusion. Semin Cell Dev Biol 76, 201–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serasinghe MN, Chipuk JE (2017). Mitochondrial fission in human diseases. In: Handbook of Experimental Pharmacology, 159–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Q, Yamano K, Head BP, Kawajiri S, Cheung JTM, Wang C, Cho J-H, Hattori N, Youle RJ, van der Bliek AM (2014). Mutations in Fis1 disrupt orderly disposal of defective mitochondria. Mol Biol Cell 25, 145–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spudich JA, Watt S (1971). The regulation of rabbit skeletal muscle contraction. I. Biochemical studies of the interaction of the tropomyosin-troponin complex with actin and the proteolytic fragments of myosin. J Biol Chem 246, 4866–4871. [PubMed] [Google Scholar]
- Strack S, Cribbs JT (2012). Allosteric modulation of Drp1 mechanoenzyme assembly and mitochondrial fission by the variable domain. J Biol Chem 287, 10990–11001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyama EQ, Herzig S, Courchet J, Lewis TL Jr, Losón OC, Hellberg K, Young NP, Chen H, Polleux F, Chan D, et al. (2016). Metabolism: AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 351, 275–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent TL, Green PJ, Woolfson DN (2013). LOGICOIL—Multi-state prediction of coiled-coil oligomeric state. Bioinformatics 29, 69–76. [DOI] [PubMed] [Google Scholar]
- Wakabayashi J, Zhang Z, Wakabayashi N, Tamura Y, Fukaya M, Kensler TW, Iijima M, Sesaki H (2009). The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J Cell Biol 186, 805–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterham HR, Koster J, van Roermund CWT, Mooyer PAW, Wanders RJA, Leonard JV (2007). A lethal defect of mitochondrial and peroxisomal fission. N Engl J Med 356, 1736–1741. [DOI] [PubMed] [Google Scholar]
- Yu R, Jin S, Lendahl U, Nistér M, Zhao J (2019). Human Fis1 regulates mitochondrial dynamics through inhibition of the fusion machinery. EMBO J 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacharias DA, Violin JD, Newton AC, Tsien RY (2002). Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science 296, 913–916. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Chan NC, Ngo HB, Gristick H, Chan DC (2012). Crystal structure of mitochondrial fission complex reveals scaffolding function for Mitochondrial division 1 (Mdv1) coiled coil. J Biol Chem 287, 9855–9861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Liu T, Jin S, Wang X, Qu M, Uhlén P, Tomilin N, Shupliakov O, Lendahl U, Nistér M (2011). Human MIEF1 recruits Drp1 to mitochondrial outer membranes and promotes mitochondrial fusion rather than fission. EMBO J 30, 2762–2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.