

Abstract
Viruses are pathogenic agents that can infect all varieties of organisms, including plants, animals, and humans. These microscopic particles are genetically simple as they encode a limited number of proteins that undertake a wide range of functions. While structurally distinct, viruses often share common characteristics that have evolved to aid in their infectious life cycles. A commonly underappreciated characteristic of many deadly viruses is a lipid envelope that surrounds their protein and genetic contents. Notably, the lipid envelope is formed from the host cell the virus infects. Lipid-enveloped viruses comprise a diverse range of pathogenic viruses, which often lead to high fatality rates and many lack effective therapeutics and/or vaccines. This perspective primarily focuses on the negative-sense RNA viruses from the order Mononegavirales, which obtain their lipid envelope from the host plasma membrane. Specifically, the perspective highlights the common themes of host cell lipid and membrane biology necessary for virus replication, assembly, and budding.
INTRODUCTION
Viruses have been on the world’s stage with the identification of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the causative agent of COVID-19. SARS-CoV-2 is a lipid-enveloped coronavirus that assembles and buds from the host cell ER-Golgi intermediate compartment (Boson et al., 2020; Plescia et al., 2020) and traffics out of the host cell through lysosomes (Ghosh et al., 2020). Little is known on how coronaviruses utilize host cell lipids for their assembly and budding, but based upon the presence of a transmembrane M protein, which is essential to the formation of sites of virus assembly, lipid composition undoubtedly plays a critical role. More detailed investigations will be needed into the mechanisms by which SARS-CoV-2 (and coronaviruses as a whole) interacts and modifies host cell lipid composition or lipid metabolism (Archambault et al., 2021; Pérez-Torres et al., 2021) to unveil how this family of viruses interacts with membranes. Many of the methods and strategies utilized for other lipid-enveloped viruses should be applicable to understanding and ultimately elucidating the host cell lipid characteristics critical to the spread of SARS-CoV-2. With the increase in global travel and the potential for more zoonotic transmissions of novel lipid-enveloped viruses, basic research geared toward an understanding of virus–host cell hijacking should help shed light on fundamental strategies of drug targeting and perhaps opportunities for panviral therapies.
Mononegavirales is a diverse order of RNA viruses established in 1991, initially comprised of three viral families grouped by their structure and morphology. As of 2018, the Mononegavirales has expanded to eight families encompassing some of the most infectious and fatal viruses known to cause disease in humans and animals (Amarasinghe et al., 2018). Mononegavirales include both well established and emerging viruses, such as Rhabdoviridae (includes rabies virus, which can lead to 95% fatality in untreated cases), Pneumoviridae (includes respiratory syncytial virus [RSV], one of the most common human viral infections), and the highly infectious and lethal Filoviridae (Ebola virus [EBOV] and Marburg virus [MARV]). This order also includes the diverse family Paramyxoviridae (e.g., measles morbillivirus [MeV], mumps, and Henipaviridae Hendra virus [HeV] and Nipah virus [NiV]; Stallcup et al., 1983; Liu, 2014; Cox and Plemper, 2017; Amarasinghe et al., 2018; Emanuel et al., 2018).
Members of the Mononegavirales order are lipid enveloped and constructed from a 10–20-kb-long single-stranded nonsegmented RNA genome (Liu, 2014), which encodes 5–10 proteins (Latorre et al., 2018). These proteins subvert host processes during infection, leading to a wide range of diseases. The structure and morphology of viruses arise from a complex network of protein–protein, protein–RNA, and protein–lipid interactions. These interactions are dependent on multimeric complexes of thousands of viral proteins. All members of the Mononegavirales order of viruses carry along a core set of proteins encoded by their RNA genome. There are four principal units of Mononegavirales viral structure: a linear (−) sense genome, helical nucleocapsid structure, extensive matrix layer, and the lipid envelope studded with surface glycoproteins. The genome is protected by a nucleocapsid complex, comprised of the nucleoprotein (NP) and accessory proteins. The genome−nucleocapsid structure is encapsulated by an extensive grid-like scaffold comprised by the self-assembly of the matrix protein. Lastly, the virus is bound by a lipid envelope derived from the host plasma membrane, which is studded with glycoproteins to aid in attachment and fusion (Latorre et al., 2018) to continue the viral life cycle.
Filoviruses and paramyxoviruses contain several proteins with conserved structure and function. The matrix protein in filoviruses and paramyxoviruses is highly abundant, viral protein 40 (VP40) and matrix protein (M), respectively. The matrix protein, initially made up of dimers (Adu-Gyamfi et al., 2012a; Bornholdt et al., 2013; Oda et al., 2016; Liu et al., 2018; Wan et al., 2020), oligomerizes into an extensive matrix interlaced between the nucleocapsid complex and lipid envelope (Adu-Gyamfi et al., 2012a, 2013, 2015; Bornholdt et al., 2013; Oda et al., 2015; Liu et al., 2018b) as an assembly of dimers (Wan et al., 2020). The matrix layer is paramount to the structural morphology and integrity observed in both viral families. Within the nucleocapsid lies the NP, considered the main structural component of the nucleocapsid complex, which interacts with both the RNA genome and other viral proteins. Additionally, filoviruses and paramyxoviruses encode an RNA-dependent RNA polymerase (RdRP), coined the L protein. The L protein of filoviruses and paramyxoviruses is quite large (more than 2000 amino acids) and is the only viral protein known to possess enzymatic activity. An essential polymerase cofactor can be found in both filoviruses (viral protein 35, VP35) and paramyxoviruses (phosphoprotein, P). Lastly, critical to viral attachment and fusion are the glycoproteins. EBOV and MARV contain one glycoprotein (GP; Emanuel et al., 2018), while two are found within paramyxoviruses such as MeV (fusion protein, F, and attachment protein, H) and NiV (fusion protein, F, and attachment protein, G; Thibault et al., 2017).
VIRUS REPLICATION
Filoviruses and paramyxoviruses have an ∼19-kb nonsegmented genome with conserved gene order, encoding seven and six structural proteins, respectively (Liu et al., 2014; Thibault et al., 2017). Additionally, these viral families utilize a RdRP for viral transcription and replication. Due to the “stop-start” model of gene transcription by the RdRP on the nonsegmented RNA genome, a gradient of gene products is observed with genes at the 3′ end of the genome transcribed more abundantly than those at the 5′ end. Transcription and replication of filoviruses and paramyxoviruses takes place within the cytosol in inclusion bodies (Thibault et al., 2017; Emanuel et al., 2018).
Following uncoating of the viral matrix layer within the cytosol, the nucleocapsid complex proteins commence coordinating transcription of the (−) sense RNA genome. Transcription of the negative-sense genome must occur before replication, to produce the (+) sense RNA gene products required for protein translation and the production of new (−) sense genomes. Three viral proteins have emerged as required components for replication: NP, P or VP35, and L (Conzelmann, 2004; Whelan et al., 2004; Emanuel et al., 2018). NP is an RNA-binding protein that multimerizes into a helical assembly wrapped around the genome. Upon independent expression in mammalian cells, NP forms cytoplasmic inclusion bodies, where it has been shown to interact with host RNA (Noda et al., 2010; Emanuel et al., 2018). L is a multidomain enzymatic component of the system, responsible for transcription, replication, genome capping, and polyadenylation (Latorre et al., 2018). Lastly, P/VP35 serve as a cofactor by linking the L and NP proteins (P in paramyxoviruses and VP35 in filoviruses). Moreover, it has been proposed that P/VP35 carry out chaperone functions by obstructing the self-association of NP and nonproductive associations between NP and RNA, allowing replication to complete before NP begins its helical assembly around the viral genome (Latorre et al., 2018). When expressed together, NP, P/VP35, and L colocalize in the cell and fulfill each requirement for productive viral transcription and replication.
Cytoplasmic inclusion bodies form with Mononegavirale cell infections and are thought to be the sites of virus translation and transcription (Figure 1). While the origins of such inclusion bodies (reviewed in detail in Hume and Mühlberger, 2019) are unknown for filoviruses, they resemble liquid phase-separated structures or structures enriched in membrane components. These inclusions are formed in the cytoplasm of infected cells (Hoenen et al., 2012; Dolnik et al., 2015; Kolesnikova et al., 2017) and are sites of viral RNA synthesis as well as nucleocapsid formation. EBOV nucleocapsids are transported via an actin-dependent process to sites of virus assembly at the plasma membrane inner leaflet (Schudt et al., 2013). Despite lack of mechanistic knowledge on filovirus inclusion body formation, a number of studies have elucidated the origins of inclusion body formation for paramyxovirus replication.
FIGURE 1:
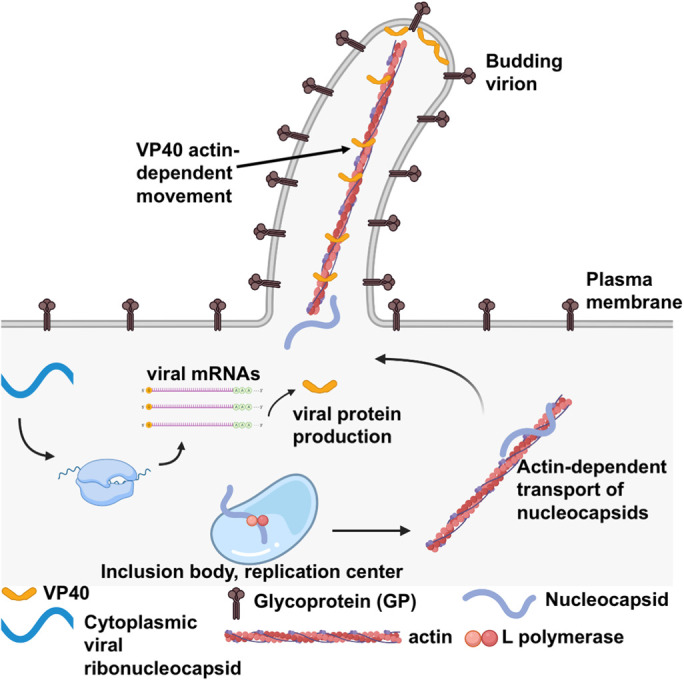
Schematic of Ebola virus replication and budding. Ebola virus (EBOV) enters the host cell via fusion at the lysosomal membrane. The viral ribonucleocapsid harbors the negative-sense RNA genome and becomes cytosolic postfusion of the virus and host membranes. The negative-sense RNA genome is used as a template to produce viral mRNAs for viral protein synthesis such as that of VP40. The negative-sense genome is also used to generate a complementary strand of RNA (positive strand), which is used to generate copies of the negative-sense RNA genome that can be packaged in virions. These replication processes occur via the EBOV L polymerase in the host cell cytoplasm. Inclusion bodies (virus replication centers) have been observed in the cytosol for both EBOV and MARV and are deemed replication centers for viral RNA synthesis and nucleocapsid (NC) assembly. The biophysical and biochemical nature of these viral inclusions is still unknown and may form via liquid–liquid phase separation as shown for other virus replication centers or through selective hijacking of host membrane components. Following NC assembly, NCs are trafficked in an actin-dependent manner to sites of virus assembly where the VP40 matrix layer underlies the plasma membrane. The host cell plasma membrane and budding virions are studded with transmembrane EBOV glycoproteins. VP40 dimers are peripheral proteins that interact with the plasma membrane inner leaflet (PS and PI(4,5)P2) and a VP40 matrix layer is formed via oligomerization of VP40 dimers in an end-to-end manner. VP40 has been shown to be transported on actin in filaments extending from the plasma membrane. Virus nucleocapsids recruited to VP40 assembly sites harbor the encapsulated negative-sense RNA genome and interactions between VP40 and NP most likely stabilize the NC in filaments emanating from the plasma membrane. Following the assembly of new virions at the plasma membrane, membrane scission occurs releasing the virion (or VLP) into the extracellular space giving rise to filamentous structures with a consistent diameter (∼80 nm) and variable lengths (1–14 μm). This figure was prepared in BioRender.
In the case of measles virus infection, N and P form a membraneless organelle (Guseva et al., 2020) at least in vitro. The proposed mechanism of this phase separation, which may come as a surprise, is attributed to intrinsically disordered regions in both the N and P proteins, which are sufficient to recruit RNA to the surface of the membraneless organelle (Guseva et al., 2020). The addition of RNA hexamers to N and P membraneless organelles triggered formation of nucleocapsids at a constant concentration of N. The rate at which RNA encapsulation occurred was significantly higher in phase-separated liquid droplets compared with the more dilute phase, strongly suggesting an important role for nucleocapsid assembly in the host cell cytoplasm. While the role of liquid–liquid phase separation is only beginning to unravel in Mononegavirale replication, it has become clear it likely plays a significant role in localization of viral machinery for genome replication and nucleocapsid formation. Whether or not lipids are involved in the localization and regulation of the viral replication machinery is still unknown, but warrants careful and critical further investigation into polymerase activity and localization and packaging of the viral nucleocapsid.
PERSPECTIVES ON VIRUS ASSEMBLY, BUDDING, AND EXIT
The matrix (or M) proteins direct the trafficking of viral components to the plasma membrane. Consequently, proper trafficking of the matrix proteins to the plasma membrane is a prerequisite for efficient viral assembly and budding. Early in infection, matrix proteins are cytosolically localized, and in the case of MARV, highly associated with intracellular membranes (Kolesnikova et al., 2004). Interestingly, VP40 has been observed to transiently translocate into the nucleus; however, the function of nuclear import and export is not well understood in filoviruses (Del Vecchio et al., 2018a). In the case of M proteins, ubiquitination and nuclear-cytoplasmic transport is required for efficient viral production of several members of the paramyxovirus family (Pohl et al., 2007; Harrison et al., 2010; Wang et al., 2010, 2012). Additionally, there is significant evidence of filoviral VP40 ubiquitination, suggested by pronounced interactions between VP40 and E3 ubiquitin ligases from the neural precursor cell expressed developmentally down-regulated protein 4 (NEDD4) family (Harty et al., 2000; Han et al., 2016, 2017; Urata and Yasuda, 2010; Zhang et al., 2021).
Matrix protein–mediated viral assembly requires the spatial and temporal alignment of each viral component. Following viral replication, the ribonucleoprotein (RNP) complexes in filoviruses and paramyxoviruses traverse to the matrix protein–enriched regions of the plasma membrane via the actin network (Schudt et al., 2013, 2015). In-depth live cell imaging experiments have highlighted that once at the cell periphery, filovirus RNPs are directed within filopodia by VP40 in an actin-dependent manner (Adu-Gyamfi et al., 2012b; Schudt et al., 2013; Takamatsu et al., 2018). Additionally, VP40 itself has been shown to move in an actin-dependent manner into sites of assembly at the plasma membrane (Adu-Gyamfi et al., 2012b) before RNP transport. However, the role of actin in M transport is not as clearly defined. Pharmacological inhibition of actin polymerization perturbs the assembly and release of MeV particles (Dietzel et al., 2013) and a recent proteomics analysis of NiV-infected cells suggested a role for actin in NiV trafficking and release (Johnston et al., 2019). Furthermore, the endocytic pathway and specifically Rab-11–positive recycling endosomes have also been implicated in MeV and NiV RNP transport (Nakatsu et al., 2013; Johnston et al., 2019) as well as in EBOV budding (Nanbo and Ohba, 2018; Nanbo et al., 2018).
In addition to the RNPs, the glycoproteins must convene with the matrix proteins at sites of viral assembly. During viral infection, GP travels independently of other viral proteins to the plasma membrane where they are randomly distributed. Filovirus GP hijacks the secretory pathway to arrive at the plasma membrane; however, detailed reports of this observation are lacking (Becker et al., 1996; Bavari et al., 2002). In the case of paramyxoviruses, the glycoproteins (F, G, H) travel to the basolateral membrane when expressed independently in polarized cells. However, upon coexpression with the M protein, the glycoproteins are redistributed to the apical side of the cell where budding occurs (Maisner et al., 1998; Lamp et al., 2013). This phenomenon is also observed in MARV budding within polarized cells; however, in the opposite direction. In MARV budding, GP will localize to the apical membrane, and upon coexpression with mVP40 will redistribute to the basolateral membrane where budding will occur (Kolesnikova et al., 2007).
A direct interaction between matrix proteins and glycoproteins has been established for HIV-1 (Cosson, 1996), influenza (Jin et al., 1997), as well as respiratory syncytial virus (Ghildyal et al., 2005). Biochemical and cryo-electron tomography (cryo-ET) studies have shown MeV and NiV M proteins directly interact with the cytoplasmic tails of F and H proteins, respectively (Rima and Duprex, 2006; Tahara et al., 2007; Johnston et al., 2017; Ke et al., 2018). However, the function of this interaction is not well understood. Specifically, recent cryo-ET and superresolution imaging of MeV and NiV proteins has provided conflicting evidence as to whether M redistributes the glycoproteins within the apical membrane to sites of viral assembly or if glycoprotein localization is stochastic (Ke et al., 2018; Liu et al., 2018). Unlike paramyxoviruses, a direct filovirus matrix protein VP40 and GP interaction has not been established. However, a VP40-GP relationship is suggested by the finding that viral budding is enhanced when both proteins are expressed compared with VP40 alone (Licata et al., 2004). Whether through a direct or indirect interaction, glycoproteins play an imperative role in successful viral budding.
Viral assembly is the penultimate step of the viral life cycle, followed by membrane scission and release (Figure 1). Upon congregation of the glycoproteins, matrix proteins, and RNPs within the plasma membrane inner leaflet, progeny virions mature that must be released to propagate the viral life cycle. A required step for viral assembly and scission is the generation of significant plasma membrane deformation, or negative curvature generation (Rossman et al., 2010; Rossman and Lamb, 2013; Soni and Stahelin, 2014). The movement of the plasma membrane (i.e., pushing out away from the cytosol) is believed to be attributed to several factors including accumulation of viral proteins at the plasma membrane, rearrangement of the actin network, and the hijacking of host machinery such as the ESCRT proteins (Rossman and Lamb, 2013; Lee et al., 2015).
Irrespective of viral morphology, filovirus and paramyxovirus virions pinch off from the plasma membrane, ready to disseminate throughout the host. The intimate role between plasma membrane lipids and Mononegavirale components has not been studied in great detail. This interface of viral assembly remains an underexplored platform for examining virus–host interactions as well as targets of drug development. An in-depth understanding of assembly processes between the plasma membrane, host proteins, and viral matrix proteins may provide the framework for a panviral therapy.
THE HOST PLASMA MEMBRANE AND APOPTOTIC MIMICRY
Extensive experimental evidence has confirmed that viral pathogens mimic apoptotic “eat-me” signals as a mechanism to enhance cellular entry of virus in a process known as apoptotic mimicry (Moller-Tank and Maury, 2014; Amara and Mercer, 2015; Nanbo and Kawoaka, 2019). The exploitation of apoptosis has been experimentally substantiated for a diverse set of lipid-enveloped viruses, including (+) sense RNA-enveloped viruses (alphaviridaes, flaviviridae) and (−) sense RNA-enveloped viruses (arenaviruses, and the Mononegavirales Filoviridae and rhabodiviridae). There are currently no reports of apoptotic mimicry strategies employed by members of the Paramyxoviridae family; however, it is important to note that during entry of paramyxoviruses, the viral envelope is thought to directly fuse with the host cell, therefore no true engulfment mechanism occurs.
Filoviruses are one family of Mononegavirales known to exploit apoptotic mimicry to gain entry into host cells. During EBOV entry, host cell T-cell immunoglobulin and mucin domain protein 1 (TIM-1) receptors recognize phosphatidylserine (PS) residing in the outer viral envelope (Kondratowicz et al., 2011; Liu, 2014). During infection, eVP40 is the main viral component postulated to initiate exposure of PS to the outer leaflet during budding (Adu-Gyamfi et al., 2015). However, recent evidence reports that GP and eVP40 accomplish this mechanism through activation of Xkr8 activation (Nanbo et al., 2018). In contrast, another study performed with authentic EBOV found that TMEM16F and not Xkr8 was the main host scramblase required for PS incorporation and exposure in virions (Younan et al., 2018). Future investigations will need to be performed to distill a thorough understanding of PS movement across the plasma membrane in the viral budding process and if there are any cell type–specific differences.
LIPID-INDUCED MATRIX PROTEIN ASSEMBLY AND MEMBRANE REMODELING
The structural plasticity of matrix proteins was first documented in 1982 when it was observed that the matrix protein of Sendai virus (SeV-M) self-assembled into ordered tubes and sheets in vitro (Heggeness et al., 1982). The in vitro self-assembly of matrix proteins upon association with lipids has also been widely reported across the Mononegavirales order. In the presence of PC, the Pneumoviridae human metapneumovirus calcium-binding matrix protein assembles into flexible tubes (Leyrat et al., 2014). Further, atomic force microscopy was employed to investigate the assembly of the paramyxovirus Newcastle disease virus (NDV-M). Upon incubation with a negatively charged mica surface (representative of the negative charge of the plasma membrane inner leaflet), NDV-M assembled into an extensive scaffold (Shytkova et al., 2018).
Similar observations have been reported outside of the Mononegavirales order using high-resolution imaging analysis. The interaction of the influenza virus matrix protein (M1; Orthomyxoviridae family) with anionic lipids has been long established (Baudin et al., 2001). Recent investigations have aimed to delineate which lipids M1 interacts with and which M1 processes are influenced by lipid binding (Hilsch et al., 2014; Bobone et al., 2017). Fluorescence scanning microscopy experiments were performed utilizing fluorescently labeled M1 proteins and supported lipid bilayers containing PS, which confirmed that M1-membrane binding is mediated by PS (Hilsch et al., 2014). To understand the molecular implications of this interaction, the oligomerization of M1 was monitored in the presence of PS-containing supported lipid bilayers. M1 multimerized extensively upon incubation with supported lipid bilayers containing 40% PS (Hilsch et al., 2014), which is analogous to the observed self-assembly of other matrix proteins from the aforementioned EM and atomic force microscopy experiments.
During membrane remodeling, membrane shape and curvature are altered as the membrane yields new shapes, such as tubes and vesicles. This is observed throughout viral budding, as negative curvature occurs upon bending of the plasma membrane as a new virion is formed from the plasma membrane inner leaflet. Eventually, membrane scission occurs to release the new virions. Compelling evidence to support a role for matrix proteins in facilitating membrane remodeling during viral budding has been shown via the in vitro deformation of membranes in the absence of other viral components (Solon et al., 2005; Soni and Stahelin, 2014; Saletti et al., 2017; Dahmani et al., 2019).
Early evidence of membrane deformation induced by a matrix protein was from vesicular stomatitis virus (VSV-M), a member of the Rhabdoviridae family of Mononegavirales. VSV-M associates with membranes through basic patches on its N-terminal domain (Solon et al., 2005; Liu et al., 2014; Amarasinghe et al., 2018). Upon incubation of fluorescently labeled giant unilamellar vesicles (GUVs) with VSV-M, confocal microscopy revealed VSV-M induced significant membrane deformation when PS was incorporated into the GUVs. Moreover, VSV-M was shown to coalesce with fluorescently labeled PS on the surface of membranes, which resulted in invaginations of the membrane in PS-VSV-M–enriched regions (Solon et al., 2005).
Membrane deformations (e.g., vesiculation and tubulation) on GUVs have been observed for a number of matrix proteins of (−) sense RNA viruses. Both filovirus and paramyxovirus matrix proteins have demonstrated the capacity to remodel membranes. When incubated with fluorescently labeled GUVs, NDV-M transformed regions of the spherical GUVs into filamentous budding-like structures (Shnyrova et al., 2007). Notably, this observation was found using GUVs consisting of PC and PE (Shnyrova et al., 2007) and therefore no conclusions could be drawn on how an anionic lipid such as PS may contribute to NDV-M–mediated membrane deformation. Conversely, when incubated with fluorescently labeled GUVs, eVP40 selectively induced vesiculation from PS-containing membranes (Soni and Stahelin, 2014), which was corroborated by ultrastructural transmission electron microscopy (TEM) studies (Soni et al., 2013). Furthermore, eVP40 capacity to remodel membranes was abrogated when point mutations were made in a hydrophobic loop suggested to penetrate the plasma membrane and contribute to eVP40-induced membrane remodeling (Soni et al., 2013). The selectivity of PS in this process has been attributed to selective binding of PS to a cationic patch in the VP40 C-terminal domain (Del Vecchio et al., 2018). VP40 was also able to cluster PS in membranes, which promoted viral budding and could be dampened by treatment with an FDA-approved drug that lowered levels of PS in cells (Husby et al., 2021).
Membrane deformation was also observed in (−) sense RNA viruses outside of the Mononegavirales order. The ability of M1 of influenza virus A to deform membranes was investigated using GUVs and confocal microscopy, cryo-TEM, and fluorescence correlation spectroscopy (FCS). GUV studies highlighted that M1 binding to and deformation of GUVs was PS dependent. Moreover, FCS was used to show that M1 binding was insufficient to induce deformation, but that multimerization of M1 was responsible for deforming the membrane (Dahmani et al., 2019). A similar relationship between M1 and PS has also been shown through similar techniques for influenza C virus (Saletti et al., 2017).
How virus protein crowding/oligomerization on membranes induces membrane structural changes is still poorly understood; however, other related cell biology studies of the plasma membrane may give insights into some possibilities. For instance, Hirama et al. investigated the indirect effects of extracting cholesterol from the plasma membrane with methyl-β-cyclodextrin (MβCD; Hirama et al., 2017). Following treatment of cells with MβCD, the high-affinity PS-binding protein lactadherin C2 (LactC2) was significantly redistributed from the plasma membrane to intracellular membranes (Hirama et al., 2017). In vitro analysis of the affinity of LactC2 to liposomes confirmed that the presence of cholesterol did not alter the affinity of LactC2 to PS (Del Vecchio and Stahelin, 2018), ruling out that the displacement of LactC2 from the plasma membrane was a result of decreased LactC2 affinity to PS when cholesterol was depleted. Moreover, MβCD treatment resulted in an increase in the negative charge density within the inner leaflet of red blood cells (Hirama et al., 2017). An increase in anionic surface charge density has been reported to lead to the generation of positive curvature to relieve electrostatic repulsion between anionic lipids (Fuller et al., 2003; Kooijman et al., 2003). Taken together, Hirama et al. presented a model where cholesterol extraction results in an increase in the anionic surface charge density of the inner leaflet, which is relieved by the subsequent generation of spontaneous positive curvature.
The observed positive curvature facilities endocytic events and the concomitant loss of the abundant anionic lipid, PS, from within the plasma membrane (Hirama et al., 2017). One could envision a similar mechanism whereby PS exposure on the outer leaflet of the plasma membrane during viral budding leads to an abrupt change in the lipid and electrostatics per surface area of the plasma membrane inner and outer leaflets with more favorable spontaneous positive curvature generation on the outer plasma membrane. More detailed mechanistic experiments are necessary on the process of viral budding to envision how and when PS, cholesterol, and other plasma membrane lipid abundancies change to fully understand the host plasma membrane contribution to the viral budding process. One distinct possibility is the formation of PS and cholesterol-enriched nanodomain formation at sites of VP40 or M assembly where transbilayer interactions can form between saturated PS with long acyl chains and cholesterol (Raghupathy et al., 2015). Transbilayer interactions can be attributed to cholesterol stabilization and ordered phase formation with PS with long acyl chains on the opposite leaflet (Raghupathy et al., 2015). These types of transbilayer interactions may lead to enhanced liquid-ordered phase on the plasma membrane inner leaflet as PS becomes more significantly enriched on the outer plasma membrane during viral budding.
CONCLUSIONS AND FUTURE PERSPECTIVES
Viral budding by lipid-enveloped viruses is an exemplary illustration of how viruses hijack their host to support their own replication. Budding is predominately facilitated by the multifunctional properties of their matrix proteins, some of which are sufficient to form virus-like particles in the absence of other viral proteins. Moreover, the pleomorphic structure of lipid-enveloped viruses highlights the high conformational plasticity and multifunctionality of matrix proteins.
Matrix proteins transform into different higher ordered structures to execute necessary tasks. As the central organizer of viral budding, matrix proteins assemble into an extensive scaffold underneath the plasma membrane. This scaffold serves as a bridge between the viral envelope (derived from the plasma membrane) and the internal nucleocapsid-containing genome. Moreover, matrix proteins are responsible for actively recruiting viral components to viral budding sites (Baudin et al., 2001; Kolesnikova et al., 2007, 2012; Nanbo et al., 2013). Matrix proteins may also cluster or restrict certain lipid formations to the sites of assembly and budding. For instance, EBOV VP40 clustered PI(4,5)P2 beneath oligomers due to the abundancy of multiple cationic basic patches (Gc et al., 2016) and HIV-1 gag restricted cholesterol and PI(4,5)P2 (Favard et al., 2019). Although these lipid restrictions seem crucial to the viral assembly and budding process, the molecular details as to how these processes contribute to effective viral egress and infectivity is still not well understood. There are a plethora of critical questions regarding how these viruses, and especially the matrix protein, utilize the host cell lipid network for their infection and replication scheme. For instance:
How do matrix proteins utilize host cell lipids and/or vesicular pathways for plasma membrane trafficking?
Do these viruses, or their matrix proteins alone, induce lipid metabolic changes to favor lipid synthesis necessary for building new virions from the plasma membrane?
What cues of matrix protein assembly prompt changes in PS distribution across the plasma membrane?
What cofactors play a role in the initiation of membrane curvature changes in the plasma membrane at sites of assembly?
With the increasingly available technologies to image at high resolution and manipulate host cell lipid composition and trafficking pathways, many of these questions should be answered in the coming decade. While it seems likely that the cellular and biophysical details of how one virus family commandeers the host cell for virus assembly and spread will be elucidated, there are thousands of viruses in nature still unknown to humans. Thus, maintaining a keen understanding of virus–host cell targets and technologies that can be used to elucidate virus–host interactions will help prepare scientists to tackle new viruses if and when they emerge.
Acknowledgments
Virus research in the Stahelin lab is funded by the National Institutes of Health (AI-081077, AI-130609, AI-139950, and AI-142651 to R.V.S). M.L.H. was partially supported by a National Institute of General Medical Sciences T32 fellowship (GM075762).
Abbreviations used:
- EBOV
Ebola virus
- MARV
Marburg virus
- MeV
measles morbillivirus
- NiV
Nipah virus
- RdRp
RNA-dependent RNA polymerase
- VP40
viral protein 40 kDa
- VSV
vesicular stomatitis virus.
Footnotes
REFERENCES
- Adu-Gyamfi E, Digman MA, Gratton E, Stahelin RV (2012a). Investigation of Ebola VP40 assembly and oligomerization in live cells using number and brightness analysis. Biophys J 102, 2517–2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adu-Gyamfi E, Digman MA, Gratton E, Stahelin RV (2012b). Single-particle tracking demonstrates that actin coordinates the movement of the Ebola virus matrix protein. Biophys J 103, L41–L43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adu-Gyamfi E, Johnson KA, Fraser ME, Scott JL, Soni SP, Jones KR, Digman MA, Gratton E, Tessier CR, Stahelin RV (2015). Host cell plasma membrane phosphatidylserine regulates the assembly and budding of Ebola virus. J Virol 89, 9440–9453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adu-Gyamfi E, Soni SP, Xue Y, Digman MA, Gratton E, Stahelin RV (2013). The Ebola virus matrix protein penetrates into the plasma membrane: a key step in viral protein 40 (VP40) oligomerization and viral egress. J Biol Chem 288, 5779–5789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali A, Nayak DP (2000). Assembly of Sendal virus: M protein interacts with F and HN proteins and with the cytoplasmic tail and transmembrane domain of F protein. Virology 276, 289–303. [DOI] [PubMed] [Google Scholar]
- Amara A, Mercer J (2015). Viral apoptotic mimicry. Nat Rev Microbiol 13, 461–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amarasinghe GK, Arechiga Ceballos NG, Banyard AC, Basler CF, Bavari S, Bennett AJ, Blasdell KR, Briese T, Bukreyev A, Cai Y, et al. (2018). Taxonomy of the order Mononegavirales: update. Arch Virol 163, 2283–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archambault AS, Zaid Y, Rakotoarivelo V, Turcotte C, Doré É, Dubuc I, Martin C, Flamand O, Amar Y, Cheikh A, et al. (2021). High levels of eicosanoids and docosanoids in the lungs of intubated COVID-19 patients. FASEB J 35, e21666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudin F, Petit I, Weissenhorn W, Ruigrok RWH (2001). In vitro dissection of the membrane and RNP binding activities of influenza virus M1 protein. Virology 281, 102–108. [DOI] [PubMed] [Google Scholar]
- Bavari S, Bosio CM, Wiegand E, Ruthel G, Will AB, Geisbert TW, Hevey M, Schmalijohn C, Schmalijohn A, Aman MJ (2002). Lipid raft microdomains: a gateway for compartmentalized trafficking of Ebola and Marburg viruses. J Exp Med 195, 593–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker S, Klenk HD, Mühlberger E (1996). Intracellular transport and processing of the Marburg virus surface protein in vertebrate and insect cells. Virology 225, 145–55. [DOI] [PubMed] [Google Scholar]
- Bobone S, Hilsch M, Storm J, Dunsing V, Herrmann A, Chiantia S (2017). Phosphatidylserine lateral organization influences the interaction of influenza virus matrix protein 1 with lipid membranes. J Virol 91, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornholdt ZA, Noda T, Abelson DM, Halfmann P, Wood MR, Kawoaka Y, Saphire EO (2013). Structural rearrangement of Ebola virus VP40 begets multiple functions in the virus life cycle. Cell 154, 763–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boson B, Legros V, Zhou B, Siret E, Mathieu C, Cosset FL, Lavillette D, Denolly S (2020). The SARS-CoV-2 envelope and membrane proteins modulate maturation and retention of the spike protein, allowing assembly of virus-like particles. J Biol Chem 296, 100111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conzelmann KK (2004). Reverse genetics of Mononegavirales. Curr Top Microbiol Immunol 283, 1–41. [DOI] [PubMed] [Google Scholar]
- Cosson P (1996). Direct interaction between the envelope and matrix proteins of HIV-1. EMBO J 15, 5783–5788. [PMC free article] [PubMed] [Google Scholar]
- Cox RM, Plemper RK (2017). Structure and organization of paramyxovirus particles. Curr Opin Virol 24, 105–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahmani I, Ludwig K, Chiantia S (2019). Influenza A matrix protein M1 induces lipid membrane deformation via protein multimerization. Biosci Rep 39, BSR20191024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietzel E, Kolesnikova L, Maisner A (2013). Actin filaments disruption and stabilization affect measles virus maturation by different mechanisms. Virol J 10, 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Vecchio K, Frick CT, Gc JB, Oda SI, Gerstman BS, Saphire EO, Chapagain PP, Stahelin RV (2018). A cationic C-terminal patch and structural rearrangements in Ebola virus matrix VP40 protein control its interactions with phosphatidylserine. J Biol Chem 293, 3335–3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Vecchio K, Stahelin RV (2018). Investigation of the phosphatidylserine binding properties of the lipid biosensor, Lactadherin (Lact C2), in different membrane environments. J. Bioenerg Biomembr 50, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolnik O, Stevermann L, Kolesnikova L, Becker S (2015). Marburg virus inclusions: a virus-induced microcompartment and interface to multivesicular bodies and the late endosomal compartment. Eur J Cell Biol 94, 323–331. [DOI] [PubMed] [Google Scholar]
- Emanuel J, Marzi A, Feldmann H (2018). Filoviruses: ecology, molecular biology, and evolution. Adv Virus Res 100, 189–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favard C, Chojnacki J, Merida P, Yandrapalli N, Mak J, Eggeling C, Muriaux D (2019). HIV-1 Gag specifically restricts PI(4,5)P2 and mobility in living cells creating a nanodomain platform for virus assembly. Sci Adv 5, eaaw8651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller N, Benatti CR, Rand RP (2003). Curvature and bending constants for phosphatidylserine-containing membranes. Biophys J 85, 1667–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gc JB, Gerstman BS, Stahelin RV, Chapagain PP (2016). The Ebola virus VP40 hexamer enhances clustering of PI(4,5)P2 lipids in the plasma membrane. Phys Chem Chem Phys 18, 28409–28417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghildyal R, Li D, Peroulis I, Shields B, Bardin PG, Teng MN, Collins PL, Meanger J, Mills J (2005). Interaction between the respiratory syncytial virus G glycoprotein cytoplasmic domain and the matrix protein. J Gen Virol 86, 187–1884. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Dellibovi-Ragheb TA, Kerviel A, Pak E, Qiu Q, Fisher M, Takvorian PM, Bleck C, Hsu VW, Fehr AR, et al. (2020). β-coronaviruses use lysosomes for egress instead of the biosynthetic secretory pathway. Cell 183, 152–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guseva S, Milles S, Ringkjøbing Jensen M, Schoehn G, Ruigrok RWH, Blackledge M (2020). Structure, dynamics, and phase separation of measles virus RNA replication machinery. Curr Opin Virol 41, 59–67. [DOI] [PubMed] [Google Scholar]
- Han Z, Sagum CA, Bedford MT, Sidhu SS, Sudol M, Harty RN (2016). ITCH E3 ubiquitin ligase interacts with Ebola virus VP40 to regulate budding. J Virol 90, 9163–9171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Z, Sagum CA, Takizawa F, Ruthel G, Berry CT, Kong J, Sunyer JO, Freedman BD, Bedford MT, Sidhu SS, et al. (2017). Ubiquitin ligase WWP1 interacts with Ebola virus VP40 to regulate egress. J Virol 91, e00812–e00817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison MS, Sakaguchi T, Schmitt AP (2010). Paramyxovirus assembly and budding: building particles that transmit infections. Intl J Biochem Cell Biol 42, 1416–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harty RN, Brown ME, Wang G, Huibregtse J, Hayes FP (2000). A PPxY motif within the VP40 protein of Ebola virus interacts physically and functionally with a ubiquitin ligase: implications for filovirus budding. Proc Natl Acad Sci USA 97, 13871–13876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heggeness MH, Smith PR, Choppin PW (1982) In vitro assembly of the nonglycosylated membrane protein (M) or Sendai virus. Proc Natl Acad Sci USA 79, 6232–6236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilsch M, Goldenbogen B, Sieben C, Hofer CT, Rabe JP, Klipp E, Herrmann A, Chiantia S (2014). Influenza A matrix protein m1 multimerizes upon binding to lipid membranes. Biophys J 107, 912–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirama T, Lu SM, Kay JG, Maekawa M, Kozlov MM, Grinstein S, Fairn GD (2017). Membrane curvature induced by proximity of anionic phospholipids can initiate endocytosis. Nat Commun 8, 1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoenen T, Shabman RS, Groseth A, Herwig A, Weber M, Schudt G, Dolnik O, Basler CF, Becker S, Feldmann H (2012). Inclusion bodies are a site of ebolavirus replication. J Virol 86, 11779–11788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hume AJ, Mühlberger E (2019). Distinct genome replication and transcription strategies within the growing filovirus family. J Mol Biol 431, 4290–4320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husby ML, Amiar S, Prugar LI, David EA, Plescia CB, Huie KE, Brannan JM, Dye JM, Pienaar E, Stahelin RV (2021). The Ebola virus matrix protein clusters phosphatidylserine, a critical step in viral budding. BioRxiv 06.08.44755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin H, Leser GP, Zhang J, Lamb RA (1997). Influenza virus hemagglutinin and neuraminidase cytoplasmic tails control particle shape. EMBO J 16, 1236–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston GP, Bradel-Tretheway B, Piehowski PD, Brewer HM, Nae Rin Lee B, Usher NT, Reyes Zamora JL, Ortega V, Contreras EM, Teuton JR, et al. (2019). Nipah virus-like particle egress is modulated by cytoskeletal and vesicular trafficking Ppathways: a validated particle proteomics analysis. mSystems 4, e00194-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston GP, Contreras EM, Dabundo J, Henderson BA, Matz KM, Ortega V, Ramirez A, Park A, Aguilar HC (2017). Cytoplasmic motifs in the Nipah virus fusion protein modulate virus particle assembly and egress. J Virol 91, e02150-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke Z, Strauss JD, Hampton CM, Brindley MA, Dillard RS, Leon F, Lamb KM, Plemper RK, Wright ER (2018). Promotion of virus assembly and organization by the measles virus matrix protein. Nat Commun 9, 1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolesnikova L, Bamberg S, Berghofer B, Becker S (2004). The matrix protein of Marburg virus is transported to the plasma membrane along cellular membranes: exploiting the retrograde late endosomal pathway. J Virol 78, 2382–2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolesnikova L, Nanbo A, Becker S, Kawoaka Y (2017). Inside the cell: assembly of filoviruses. Curr Top Microbiol Immunol 411, 353–380. [DOI] [PubMed] [Google Scholar]
- Kondratowicz AS, Lennemann NJ, Sinn PL, Davey RA, Hunt CL, Moller-Tank S, Meyerholz DK, Rennert P, Mullins RF, Brindley M, et al. (2011). T-cell immunoglobulin and mucin domain 1 (TIM-1) is a receptor for Zaire Ebolavirus and Lake Victoria Marburgvirus. Proc Natl Acad Sci USA 108, 8426–8431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooijman EE, Chupin V, de Kruijff B, Burger KNJ (2003). Modulation of membrane curvature by phosphatidic acid and lysophosphatidic acid. Traffic 4, 162–174. [DOI] [PubMed] [Google Scholar]
- Kolesnikova L, Mittler E, Schudt G, Shams-Eldin H, Becker S (2012). Phosphorylation of Marburg virus matrix protein VP40 triggers assembly of nucleocapsids with the viral envelope at the plasma membrane. Cell Microbiol 14, 182–197. [DOI] [PubMed] [Google Scholar]
- Kolesnikova L, Ryabchikova E, Shestopalov A, Becker S (2007). Basolateral budding of Marburg virus: VP40 retargets viral glycoprotein GP to the basolateral surface. J Infect Dis 196, S232–S236. [DOI] [PubMed] [Google Scholar]
- Lamp B, Dietzel E, Kolesnikova L, Sauerhering L, Erbar S, Weingartl H, Maisner A (2013). Nipah virus entry and egress from polarized epithelial cells. J Virol 87, 3143–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latorre V, Mattenberger F, Geller R (2018). Chaperoning the mononegavirales: current knowledge and future directions. Viruses 10, 699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I-H, Kai H, Carlson L-A, Groves JT, Hurley JH (2015). Negative membrane curvature catalyzes nucleation of endosomal sorting complex required for transport (ESCRT)-III assembly. Proc Natl Acad Sci USA 112, 15892–15897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyrat C, Renner M, Harlos K, Huiskonen JT, Grimes JM (2014). Structure and self-assembly of the calcium binding matrix protein of human metapneumovirus. Structure 22, 136–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licata JM, Johnson RF, Han Z, Harty RN (2004). Contribution of Ebola virus glycoprotein, nucleoprotein, and VP24 to budding of VP40 virus-like particles. J Virol 78, 7344–7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L (2014). Fields virology, 6th edition. Clin Infect Dis 59, 613–613. [Google Scholar]
- Liu Q, Chen L, Aguilar HC, Chou KC (2018a). A stochastic assembly model for Nipah virus revealed by super-resolution microscopy. Nat Commun 9, 3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YC, Grusovin J, Adams TE (2018b). Electrostatic interactions between Hendra virus matrix proteins are required for efficient virus-like-particle assembly. J Virol 92, e00143-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maisner A, Klenk H, Herrler G (1998). Polarized budding of measles virus is not determined by viral surface glycoproteins. J Virol 72, 5276–5278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moller-Tank S, Maury W (2014). Phosphatidylserine receptors: enhancers of enveloped virus entry and infection. Virology 468-470, 565–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsu Y, Ma X, Seki F, Suzuki T, Iwasaki M, Yanagi Y, Komase K, Takeda M (2013). Intracellular transport of the measles virus ribonucleoprotein complex is mediated by Rab11A-positive recycling endosomes and drives virus release from the apical membrane of polarized epithelial cells. J Virol 87, 4683–4693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanbo A, Kawoaka Y (2019). Molecular mechanism of externalization of phosphatidylserine on the surface of Ebola virus particles. DNA Cell Biol 38, 115–120. [DOI] [PubMed] [Google Scholar]
- Nanbo A, Maruyama J, Imai M, Uije M, Fuijoka Y, Nishide S, Takada A, Ohba Y, Kawaoka Y (2018). Ebola virus requires a host scramblase for externalization of phosphatidylserine on the surface of viral particles. PLOS Pathog 14, e1006848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanbo A, Ohba Y (2018). Budding of Ebola virus particles requires the Rab11-dependent endocytic recycling pathway. J Inf Dis 218 (Suppl 5), S388–S396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanbo A, Watanabe S, Halfmann P, Kawaoka Y (2013). The spatio-temporal distribution dynamics of Ebola virus proteins and RNA in infected cells. Sci Rep 3, 1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda T, Hagiwara K, Sagara H, Kawaoka Y (2010). Characterization of the Ebola virus nucleoprotein-RNA complex. J Gen Virol 91, 1478–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda S, Noda T, Wijesinghe KJ, Halfmann P, Bornholdt ZA, Abelson DM, Armbrust T, Stahelin RV, Kawoaka Y, Saphire EO (2015). Crystal structure of Marburg virus VP40 reveals a broad, basic patch for matrix assembly and a requirement of the N-terminal domain for immunosupression. J Virol 90, 1839–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Torres I, Guarner-Lans V, Soria-Castro E, Manzano-Pech L, Palacios-Chavarría A, Valdez-Vázquez RR, Domínguez-Cherit JG, Herrera-Bello H, Castillejos-Suastegui H, Moreno-Castañeda L, et al. (2021). Alteration in the lipid profile and desaturase activity in patients with severe pneumonia by SARS-CoV-2. Front Physiol 12, 667024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plescia CB, David EA, Patra D, Sengupta R, Amiar S, Su Y, Stahelin RV (2020). SARS-CoV-2 viral budding and entry can be modeled using BSL-2 level virus-like particles. J Biol Chem 296, 100103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohl C, Duprex WP, Krohne G, Rima BK, Schneider-Schaulies S (2007). Measles virus M and F proteins associate with detergent-resistant membrane fractions and promote formation of virus-like particles. J Gen Virol 88, 1243–1250. [DOI] [PubMed] [Google Scholar]
- Raghupathy R, Ambika Anilkumar A, Polley A, Pal Singh P, Yadav M, Johnson C, Suryawanshi S, Saikam V, Sawant SD, Panda A, et al. (2015). Transbilayer lipid interactions mediate nanoclustering of lipid-anchored proteins. Cell 161, 581–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rima BK, Duprex WP (2006). Morbilliviruses and human disease. J Pathol 208, 199–214. [DOI] [PubMed] [Google Scholar]
- Rossman JS, Jing X, Leser GP, Lamb RA (2010). Influenza virus M2 protein mediates ESCRT-independent membrane scission. Cell 142, 902–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossman JS, Lamb RA (2013). Viral membrane Sscission. Annu Rev Cell Dev Biol 29, 551–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saletti D, Radzimanowski J, Effantin G, Midtvedt D, Mangenot S, Weissenhorn W, Bassereau P, Bally M (2017). The matrix protein M1 from influenza C virus induces tubular membrane invaginations in an in vitro cell membrane model. Sci Rep 7, 40801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schudt G, Dolnik O, Kolesnikova L, Biedenkopf N, Herwig A, Becker S (2015). Transport of ebolavirus nucleocapsids is dependent on actin polymerization: live-cell imaging analysis of Ebolavirus-infected cells. J Infect Dis 212 (Suppl 2), S160–S166. [DOI] [PubMed] [Google Scholar]
- Schudt G, Kolesnikova L, Dolnik O, Sodeik B, Becker S (2013). Live-cell imaging of Marburg virus-infected cells uncovers actin-dependent transport of nucleocapsids over long distances. Proc Natl Acad Sci USA 110, 14402–14407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shnyrova AV, Ayllon J, Mikhalyov II, Villar E, Zimmerberg J, Frolov VA (2007). Vesicle formation by self-assembly of membrane-bound matrix proteins into a fluidlike budding domain. J Cell Biol 179, 627–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solon J, Gareil O, Bassereau P, Gaudin Y (2005). Membrane deformations induced by the matrix protein of vesicular stomatitis virus in a minimal system. J Gen Virol 86, 3357–3363. [DOI] [PubMed] [Google Scholar]
- Soni SP, Adu-Gyamfi E, Yong SS, Jee CS, Stahelin RV (2013). The Ebola virus matrix protein deeply penetrates the plasma membrane: an important step in viral egress. Biophys J 104, 1940–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soni SP, Stahelin RV (2014). The Ebola virus matrix protein VP40 selectively induces vesiculation from phosphatidylserine-enriched membranes. J Biol Chem 289, 33590–33597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahelin RV (2014). Membrane binding and bending in Ebola VP40 assembly and egress. Frontiers Microbiol 5, 300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stallcup KC, Raine CS, Fields BN (1983). Cytochalasin B inhibits the maturation of measles virus. Virology 124, 59–74. [DOI] [PubMed] [Google Scholar]
- Tahara M, Takeda M, Yanagi Y (2007). Altered interaction of the matrix protein with the cytoplasmic tail of hemagglutinin modulates measles virus growth by affecting virus assembly and cell-cell fusion. J Virol 81, 6827–6836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamatsu Y, Kolesnikova L, Becker S (2018). Ebola virus proteins NP, VP35, and VP24 are essential and sufficient to mediate nucleocapsid transport. Proc Natl Acad Sci USA 115, 1075–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibault PA, Watkinson RE, Moreira-Soto A, Drexler JF, Lee B (2017). Zoonotic potential of emerging paramyxoviruses: knowns and unknowns. Adv Virus Res 98, 1–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urata S, Yasuda J (2010). Regulation of Marburg virus (MARV) budding by Nedd4.1: a different WW domain of Nedd4.1 is critical for binding to MARV and Ebola virus VP40. J Gen Virol 91, 228–234. [DOI] [PubMed] [Google Scholar]
- Wan W, Clarke M, Norris MJ, Kolesnikova L, Koehler A, Bornholdt ZA, Becker S, Saphire EO, Briggs JA (2020). Ebola and Marburg virus matrix layers are locally ordered assemblies of VP40 dimers. Elife 9, e59225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YE, Park A, Lake M, Pentecost M, Torres B, Yun TE, Wolf MC, Holbrook MR, Freiberg MR, Lee B (2010). Ubiquitin-regulated nuclear-cytoplasmic trafficking of the Nipah virus matrix protein is important for viral budding. PLoS Pathog 6, e1001186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YE, Pernet O, Lee B (2012). Regulation of the nucleocytoplasmic trafficking of viral and cellular proteins by ubiquitin and small ubiquitin-related modifiers. Biol Cell 104, 121–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelan SPJ, Barr JN, Wertz GW (2004). Transcription and replication of nonsegmented negative-strand RNA viruses. Curr Top Microbiol Immunol 283, 61–119. [DOI] [PubMed] [Google Scholar]
- Younan P, Iampietro M, Santos RI, Ramanathan P, Popov VL, Bukreyev A (2018). Role of transmembrane protein 16F in the incorporation of phosphatidylserine into budding Ebola virus virions. J Infect Dis 218 (Suppl 5), S335–S345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Zhou S, Chen M, Yan J, Yang Y, Wu L, Jin D, Yin L, Chen M, Qin Y (2021). P300-mediated NEDD4 acetylation drives ebolavirus VP40 egress by enhancing NEDD4 ligase activity. PLoS Pathogens 17, e1009616. [DOI] [PMC free article] [PubMed] [Google Scholar]