

Abstract
BACKGROUND & AIMS:
Fecal microbiota transplantation (FMT) is an effective therapy for recurrent Clostridioides difficile infection (rCDI). However, the overall mechanisms underlying FMT success await comprehensive elucidation, and the safety of FMT has recently become a serious concern because of the occurrence of drug-resistant bacteremia transmitted by FMT. We investigated whether functional restoration of the bacteriomes and viromes by FMT could be an indicator of successful FMT.
METHODS:
The human intestinal bacteriomes and viromes from 9 patients with rCDI who had undergone successful FMT and their donors were analyzed. Prophage-based and CRISPR spacer-based host bacteria-phage associations in samples from recipients before and after FMT and in donor samples were examined. The gene functions of intestinal microorganisms affected by FMT were evaluated.
RESULTS:
Metagenomic sequencing of both the viromes and bacteriomes revealed that FMT does change the characteristics of intestinal bacteriomes and viromes in recipients after FMT compared with those before FMT. In particular, many Proteobacteria, the fecal abundance of which was high before FMT, were eliminated, and the proportion of Microviridae increased in recipients. Most temperate phages also behaved in parallel with the host bacteria that were altered by FMT. Furthermore, the identification of bacterial and viral gene functions before and after FMT revealed that some distinctive pathways, including fluorobenzoate degradation and secondary bile acid biosynthesis, were significantly represented.
CONCLUSIONS:
The coordinated action of phages and their host bacteria restored the recipients’ intestinal flora. These findings show that the restoration of intestinal microflora functions reflects the success of FMT.
Keywords: Bacteriophage, Bacteriome, Virome, Clostridioides difficile infection, Fecal Microbiota Transplantation
Graphical Abstract
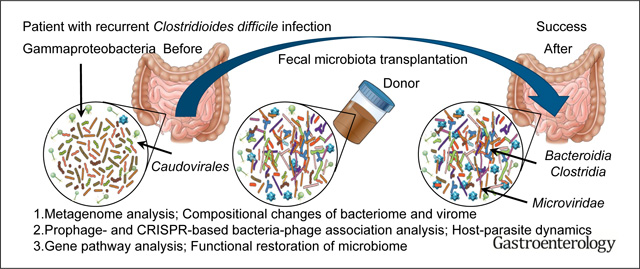
Clostridioides difficile, a Gram-positive, spore-forming anaerobic bacterium, is the representative cause of nosocomial diarrhea following antibiotic treatment. The treatment of C difficile infection (CDI) involves withdrawing the causative antibiotics and initiating antibiotic therapy (eg, vancomycin, metronidazole, or fidaxomicin). The treatment of recurrent CDI (rCDI) can be challenging, and fecal microbiota transplantation (FMT) is considered an effective therapy.1 However, 2 deaths caused by antibiotic-resistant bacterial infections after FMT have been reported,2,3 suggesting that a modification of FMT or alternative treatments are required to resolve safety concerns about FMT. Previous studies have revealed that the bacteriome can be altered by FMT.4–9 The dysbiotic state in patients with rCDI is reportedly characterized by a large expansion of Proteobacteria, and FMT is associated with an increased Bacteroidetes to Firmicutes ratio.10 In addition to the compositional changes occurring in the intestinal bacteriome, as assessed by fecal sampling before and after FMT, individual donor-recipient analysis of intestinal bacteria has been performed to examine the colonization and persistence of certain donor microbial species after FMT.6,7 Previous studies also examined specific strain transfer using intraspecies genetic variation5 or phylogenetic marker genes.8 It was possible to follow the transfer of some intestinal bacteria from donor feces to the recipient feces.5,8 However, many bacteria of unknown origin newly appeared after FMT because the gastrointestinal tract is an open system and colonization of microorganisms from the outer environment is always possible. Furthermore, the identified transfer of donor-derived bacteria in the recipient samples was clearly not associated with the success of FMT.5,7,8 Thus, although FMT certainly leads to donor-derived bacterial colonization in the intestinal tract of recipients and contributes to the improvement of rCDI, clarifying the origin of the bacteria detected in the recipient feces after FMT is extremely difficult, and this colonization is not considered a good indicator of the success of FMT.
In addition to alterations in the bacteriome, FMT leads to the transfer of phages in the feces. Previous studies have revealed decreased relative abundances of Caudovirales and increased relative abundances of Microviridae in patients with CDI after FMT.11,12 In particular, a high abundance of donor-derived Caudovirales is a key factor for the efficacy of FMT in patients with CDI.11–13 Furthermore, sterile fecal filtrates from donor feces were effective at treating rCDI,14 suggesting that bacteriophages have the potential to regulate the gastrointestinal microbiome in patients with CDI. However, multiple displacement amplification, which was used in most intestinal virome analyses in previous studies including the aforementioned study, was documented to be biased by the preferential amplification of circular single-stranded DNA (ssDNA), which likely explains the dominance of the Microviridae ssDNA phage in the human intestine. Thus, few studies have precisely and comprehensively demonstrated that FMT causes alterations in the virome. Recently, an adaptase plus linker amplification method that enables the composition of ssDNA viruses to be detected without bias was developed.15 To prevent bias in the quantitation of ssDNA viruses, including those of the Microviridae family, we obtained all data using the adaptase plus linker amplification method instead of multiple displacement amplification in this study. Importantly, because temperate phages can also exchange their genetic materials through infection, FMT might also contribute to the transfer of high-risk genetic information for the maintenance of host health from donor bacteriophages to recipients. In addition, host bacteria-phage dynamics and alterations in the gene function of the intestinal microbiota in FMT are unclear because whole-genome sequencing of the virome and bacteriome in the same fecal samples from patients with CDI who underwent FMT has not yet been performed. Therefore, it is clear that the safety of donor feces for FMT is not completely guaranteed. Thus, to ensure the efficacy of FMT in future, more detailed analyses of FMT, including the characteristics and function of its microbiota, are urgently needed.
Methods
Human Subjects
We collected fecal samples from 9 patients with rCDI (defined as 3 or more confirmed episodes of CDI detected by either polymerase chain reaction or glutamate dehydrogenase reflexed to enzyme immunoassay toxin testing) who were referred for FMT as part of their clinical care. FMT was performed under the Food and Drug Administration policy of enforcement discretion. Samples were collected from donors and recipients 2 weeks before FMT and 8 weeks after FMT in the recipients. Anonymous rigorously screened stool donor samples were obtained from a stool bank (OpenBiome). The patients were assessed at week 8 after FMT and considered cured if there was an absence of diarrhea at assessment. Among the FMT recipients, 8 were women and 8 were White, with an average age at the time of FMT of 54.7 ± 15.6 years (Supplementary Table 1). Additionally, the average number of CDI episodes before FMT was 3.7 ± 0.8 (Supplementary Table 1). The antibiotic courses used for prior CDI episodes varied; however, 8 patients had received at least 1 vancomycin taper and 1 patient had been treated with fidaxomicin (Supplementary Table 1). All patients were on vancomycin before FMT. The study protocol was approved by the Ethics Committee of the Institute of Medical Science, The University of Tokyo (28-52-0210) and Brigham and Women’s Hospital (2014P001143), and signed informed consent was obtained from each participant.
Human Fecal Sample Fractionation
The human fecal samples were immediately stored at 4°C under anaerobic conditions after collection. One gram of each sample was stored at −80°C until use. Stocks of human fecal samples were homogenized in 3 mL of SM-plus buffer (100 mM NaCl, 50 mM Tris-HCl [pH 7.4], 8 mM MgSO4·7H2O, 5 mM CaCl·2H2O, and 0.01% [wt/vol] gelatin in distilled water) with vortex mixing and passage through a 100-μm cell strainer. SM-plus buffer was passed through a 0.22-μm syringe filter before use at each step. The debris on the cell strainer was washed twice with 3 mL of SM-plus buffer, and the filtrates were centrifuged at 6000g for 5 minutes. The supernatants were transferred to new tubes, and the pellets were suspended in 1 mL of SM-plus buffer. After centrifugation at 6000g for 5 minutes, the supernatants were combined with those obtained from the previous step. The combined supernatants were recentrifuged at 6000g for 15 minutes and carefully recovered as the “viral fraction.” The remaining pellets were suspended in 1 mL of SM-plus buffer, and 100 μL of the suspensions was used as the “bacterial fraction.”
Treatment of Viral Fraction
The supernatant fractions from the human feces were separately passed through unused 0.45-μm syringe filters to remove contaminated debris. Cell-free DNA in the samples was degraded by incubation with DNase mix (1 U/mL DNase I [Roche, Basel, Switzerland], 10 U/mL benzonase [Merck, Darmstadt, Germany], and 1 U/mL Baseline-Zero DNase [Epicentre, Madison, WI]) at 37°C for 1 hour. After stopping the DNase reaction with EDTA (final concentration, 20 mM), the DNA was extracted from each sample. To establish the absence or presence of negligible bacterial DNA contamination using our method for viral DNA purification from the fecal samples, we performed quality-control assays using 16S ribosomal DNA polymerase chain reaction.16
Treatment of the Bacterial Fraction
To extract DNA from each bacterial fraction, we used the protocol reported previously.17 The samples were incubated with 1 mL of SM-plus buffer containing 20 mM EDTA, 100 μg/mL recombinant human lysozyme (Sigma-Aldrich, St. Louis, MO), and 0.5 U/mL achromopeptidase at 37°C for 1 h, followed by DNA extraction.
DNA Extraction
Each supernatant sample from the viral and bacterial fractions was incubated with a 1/400 volume of 20 mg/mL proteinase K (Nacalai Tesque, Kyoto, Japan) and a 1/20 volume of 10% sodium dodecyl sulfate at 55°C for 1 hour. Thereafter, each sample was added to an equal volume of phenol/chloroform/isoamyl alcohol and mixed vigorously. After centrifugation at 16,000g for 5 minutes, the aqueous phase of each sample was transferred to a new tube, followed by chloroform extraction. Again, the aqueous phase of each sample was transferred to a new tube, and mixed with a 1/10 volume of 3 M sodium acetate and an equal volume of isopropanol. For each viral fraction, 40 mg of glycogen (Roche) was added as a co-precipitate. The samples were centrifuged at 16,000g for 15 minutes. After discarding each supernatant, each pellet was washed with 70% ethanol and centrifuged at 16,000g for 5 minutes. The supernatants were removed completely, and the pellets were air-dried for 5 minutes. Each DNA sample was resuspended in 10 mM Tris-HCl buffer (pH 8.0), and the concentration quantified using a Quantus double-stranded DNA (dsDNA) kit (Promega, Madison, WI).
Data Availability
All sequencing data supporting the findings of this study have been deposited in the National Center for Biotechnology Information Sequence Read Archive (PRJNA693652). They are available from the corresponding authors on reasonable request, with the approval of our ethics committee. Please contact the corresponding authors for additional information.
Quantification and Statistical Analysis
Statistical details relating to the sequencing data and experiments can be found in the figure legends and Methods. P values of <.05 were considered significant.
Results
Characterization of the Human Gastrointestinal Bacteriome in Patients Following FMT
We first analyzed the bacteriomes of the fecal samples from the donors and recipients before FMT, and the recipients after FMT. The relative abundance in each sample was calculated using sequencing reads (Figure 1A and Supplementary Figure 1A–D). In common with that reported previously,18 C difficile–associated sequence reads were barely detected in the recipients before FMT (Supplementary Figure 1E). Before FMT, the proportions of Negativicutes (Firmicutes), Gammaproteobacteria (Proteobacteria), and Fusobacteria (Fusobacteria) were significantly elevated in their relative bacterial abundances (Figure 1B). By contrast, the proportions of Clostridia (Firmicutes), Erysipelotrichia (Firmicutes), and Bacteroidia (Bacteroidetes) increased significantly after FMT (Figure 1B). In the multivariate analysis, hospitalization was not found to be significantly associated with FMT efficacy.19 In fact, although 5 of 9 patients required hospitalization for rCDI (Supplementary Table 1), no significant differences in the principal component (PC) scores (PC1, PC2, and PC3) obtained for the relative bacterial abundances were found between the hospitalization group (FMT2, FMT3, FMT6, FMT8, and FMT9) and the nonhospitalization group (FMT1, FMT4, FMT5, and FMT7) (Supplementary Figure 2). The richness and diversity of the bacterial species were significantly higher in recipients after FMT than before FMT, indicating that bacterial dysbiosis was ameliorated by FMT (Figure 1C and D). Furthermore, Bray–Curtis dissimilarity revealed that the bacteriome in the recipient feces after FMT was similar to that of the donor feces but not to that of the recipient feces before FMT (Figure 1E). Thus, FMT contributed to the amelioration of dysbiosis in the recipients, and the bacteriomes of the recipients tended to approach those of the donors after FMT.20 However, as observed previously,5,8 the recipient bacteriome did not completely resemble the donor bacteriome despite the disappearance of clinical symptoms.
Figure 1.

Characterization of the human gastrointestinal bacteriome in recipients before and after FMT and in donors. (A) Relative abundances of bacterial classes in the fecal samples from 9 patients who underwent FMT. B: before FMT; A: after FMT; D: donor. (B) Comparison of the relative abundances of the bacterial classes between the samples obtained from recipients before and after FMT. *P < .05, **P < .01, ***P < .001, paired Student t test. (C) Comparison of bacterial richness between the samples obtained from recipients before and after FMT. ***P < .001, paired Student t test. (D) Alpha diversity of the bacterial species in the bacterial fraction of the samples obtained before and after FMT (n = 9). **P < .01, paired Student t test. (E) Bray–Curtis dissimilarity of the bacteriome communities at the contig level between pre-FMT samples and donor samples, between post-FMT samples and donor samples, and between pre- and post-FMT samples (n = 9). **P < .01, ***P < .001, paired Student t test. Boxplots were drawn using R with the default parameter setting.
Characterization of the Human Gut Virome in Patients After FMT
We next analyzed the virome in the donors’ fecal samples, the recipients before FMT, and the recipients after FMT. The analysis pipeline of the virome is shown in Supplementary Figure 1A and B, and Supplementary Figure 3A and B.21 Before FMT, the proportions of Caudovirales, especially unclassified Caudovirales (uc_Caudovirales) and Myoviridae, were high in terms of relative viral abundance (Figure 2A and B). In contrast to Caudovirales, the proportions of Microviridae and crAss-like phages, including the p-crAssphage, increased significantly after FMT compared with those before FMT (Figure 2A and B). Viral richness showed an increasing trend in patients after FMT compared with that before FMT (Figure 2C), but in general, the viral diversity after FMT was equivalent to that before FMT (Figure 2D). In detail, the diversity and richness of Caudovirales increased in patients after FMT compared with the findings before FMT, although the differences were not statistically significant (Figure 2E). In addition, Microviridae diversity and richness and crAss-like phage richness were significantly enhanced in patients after FMT compared with the findings before FMT (Figure 2F and G), indicating that FMT influences the virome, especially in terms of Microviridae and crAss-like phages. Bray-Curtis dissimilarity also showed that the fecal virome after FMT was similar to that of the donor feces (Figure 2H). Collectively, our findings suggest that FMT drastically changes the intestinal virome, a result similar to that for the bacteriome. The abundance of Microviridae and Proteobacteria in the fecal samples from the rCDI patients before and after FMT was negatively correlated (Supplementary Figure 4). Thus, although previous studies have revealed that donor-derived Caudovirales species were important for the efficacy of FMT,11,13 FMT also has the potential to increase the proportion of Microviridae in recipients.
Figure 2.

Characterization of the human gastrointestinal virome in recipients before and after FMT and in donors. (A) Relative abundances of viral taxa in the fecal samples from 9 patients who underwent FMT. B: before FMT; A: after FMT; D: donor. (B) Comparison of the relative abundance in viral taxa between samples obtained from recipients before and after FMT. *P < .05, ***P < .001, paired Student t test. (C) Richness of the viral contigs in the viral fraction in samples obtained from recipients before and after FMT (n = 9). (D) Alpha diversity of the viral contigs in the viral fraction from samples obtained from recipients before and after FMT (n = 9). (E) Alpha diversity and richness of Caudovirales viral contigs in samples obtained from recipients before and after FMT (n = 9). (F) Alpha diversity and richness of Microviridae viral contigs in samples obtained from recipients before and after FMT (n = 9). *P < .05, **P < .01, paired Student t test. (G) Alpha diversity and richness of crAss-like phage viral contigs in samples obtained from recipients before and after FMT (n = 9 patients). **P < .01, paired Student t test. (H) Bray–Curtis dissimilarity of the virome communities at the contig level between pre-FMT and donor samples, between post-FMT and donor samples, and between pre- and post-FMT samples (n = 9). **P < .01, paired Student t test. Boxplots were drawn using R with the default parameter setting.
Prophage-based Host Bacteria-Phage Associations in Patients After FMT
The recipient bacteriome and virome were altered by FMT, and therefore we analyzed prophage-based host bacteria-phage associations in samples from the recipients before and after FMT, and in the donor samples also. As shown in Supplementary Figure 5,21 prophage sequences were extracted from the bacterial (Supplementary Figure 1A) and viral contigs (Supplementary Figure 3A). Then, each detected prophage sequence was classified according to its host bacterium. With the expansion of bacterial richness and diversity (Figure 1C and D), the number of detected prophages increased (Figure 3A). Corresponding to the composition of the bacteriome (Figure 1A), the proportions of Proteobacteria with prophages in the recipients before FMT were elevated (Figure 3A). By contrast, the proportions of Firmicutes and Bacteroidetes with prophages dominated the recipients’ samples after FMT and in the donors also (Figure 3A). These data indicate that prophage numbers and compositions correspond to the dynamics of the bacteriome. We then classified the detected prophage sequences according to their bacterial phyla based on the viral classifications shown in Supplementary Figure 3B. Forty-six percent of the Proteobacteria-derived prophage sequences from the recipients before FMT were identified as Myoviridae (Figure 3B). Microviridae has been generally found to function as a lytic phage; however, fewer prophage sequences derived from Firmicutes and Bacteroidetes were identified as Microviridae (Figure 3B). No prophage sequences were identified as p-crAssphage or crAss-like phages (Figure 3B). We next examined whether the prophages we detected (Figure 3B) exist as viral particles in the recipients. To accurately demonstrate that viral particles were released from host commensal bacteria in the gastrointestinal tract, we analyzed the circular contigs obtained from each viral fraction.21 We obtained 25 prophage-based host bacteria-phage associations in the recipients’ fecal samples, and each prophage sequence on the bacterial contig corresponded to only one circular viral contig (Figure 3C). In addition, each bacterial contig was associated with a single bacterial class, and each circular viral contig was associated with a single viral order or family (Figure 3C). Among the 25 detected prophage-based host bacteria-phage associations, 13 were diminished by FMT, in parallel with the disappearance of host bacteria after FMT. Interestingly, the relative abundance of Proteobacteria decreased after FMT, and none of the 5 detected prophage-based Proteobacteria (Gammaproteobacteria and Betaproteobacteria)-phage associations remained after FMT. Thus, no lysogenic phages were detected when their host bacteria were not identified in the fecal samples from the rCDI patients after FMT.
Figure 3.

Dynamics of prophage-based host bacteria-phage associations before and after FMT. (A) Distributions of host-related bacterial classifications for the prophages detected in pre-FMT, post-FMT, and donor samples. (B) Distributions of the viral taxa of the activated prophages from each bacterial phyla in the pre-FMT, post-FMT, and donor samples. (C) Detected circular prophage-based host bacteria-phage associations. The associations in red boxes are related to bacteria, which were observed in the pre-FMT samples but not in the post-FMT samples. Along with host bacteria, corresponding bacteriophages were not observed in the post-FMT samples.
CRISPR Spacer-based Host bacteria-phage Associations in Patients After FMT
We next examined clustered regularly interspaced short palindromic repeats (CRISPR) spacer-based bacteria-phage associations in recipient samples before and after FMT and in the donor samples also. The number of CRISPR spacers detected in the recipients’ fecal samples before and after FMT and in the donor samples were 2814, 6137, and 5863, respectively (Figure 4A and Supplementary Figure 6). A few spacers were detected in only 1 or 2 samples (Supplementary Figure 7A–C). The number of CRISPR spacers decreased in Gammaproteobacteria after FMT, whereas the number of spacers in Firmicutes and Bacteroidetes increased (Figure 4A). In particular, the numbers of CRISPR spacers in Clostridia and Bacteroidia drastically increased in the recipients’ feces after FMT (Figure 4A). Along with changes in bacterial composition following FMT, the number of CRISPR spacers in each bacterial class also changed. We next examined the distribution of the detected CRISPR spacers in the recipients’ samples before FMT, in the recipients’ samples after FMT, in the donor samples, and in their target phages. Figure 4B shows the number of common spacers and specific spacers in the recipients’ samples before and after FMT, and in the donors, according to 7 regions (B1–B7) on a Venn diagram. The bar plot shows the number of spacers against the associated viral taxa in each region. The spacers against Caudovirales were relatively abundant in the recipients’ samples before and after FMT and in the donor samples. Interestingly, the number of spacers against Microviridae was extremely large in the B6 donor samples and the post-FMT samples (χ2 test P = 2.45 × 10–6, Figure 4B). The number of spacers against Microviridae in the B1 region was relatively high in the post-FMT samples but low in the pre-FMT samples. Because Microviridae proportions increased in the virome after FMT (Figure 2A and B), bacteria with spacers against Microviridae might preferentially expand after FMT. Therefore, we next determined which bacteria carry the CRISPR spacer specific for Microviridae. The detected CRISPR spacers shown in Figure 4B are depicted as numbers in the Venn diagram (Figure 4C). The bar plot shows the number of spacers against Microviridae for the bacterial genera indicated in each region. Proteobacterial proportions, particularly Gammaproteobacteria, were abundant in the recipients’ feces before FMT (Figure 1A and B). However, although some Microviridae had proteobacterial hosts, almost no CRISPR spacers for Proteobacteria in the B1 region were detected (Figure 4C), implying that susceptibility to Microviridae may be one of the reasons for the extermination of Proteobacteria by FMT. In contrast with the samples obtained before FMT, CRISPR spacers against Microviridae were frequently detected for Bacteroidetes in the recipients’ feces after FMT (Figure 4C). In region B6, a common region between the donor and post-FMT samples, we found that Parabacteroides and Bacteroides possessed more than half of the CRISPR spacers against Microviridae (Figure 4C). The symbiotic relationship between Bacteroidetes and Microviridae may contribute to an improved intestinal environment for recipients after FMT.
Figure 4.

Dynamics of CRISPR-based host bacteria-phage associations before and after FMT. (A) The number of detected CRISPR spacers associated with bacterial classes in pre-FMT (left), post-FMT (middle), and donor (right) samples. (B) Venn diagram showing the overlapping CRISPR spacers in the bacteriomes pre-FMT and post-FMT, and the donor samples. Bar plots showing the number of viral families targeted by CRISPR spacers. (C) Venn diagram showing the CRISPR spacers targeting Microviridae in pre-FMT, post-FMT, and the donor samples. Bar plots showing the number of CRISPR spacers in the bacterial genera.
Alterations of Bacteriome and Virome Genes Associated With FMT
We identified characteristic changes in the bacteriome and virome by analyzing bacterial and viral compositions and bacteria-phage associations in the study groups. Previous works have tried to quantitatively identify the transfer of bacteria from donors to recipients.5,7,8 However, as they have previously demonstrated,5,7,8 the limitedly identified transfer of donor-derived bacteria in recipient samples was not correlated with the success of FMT. Microbial communities, including intestinal bacteria and bacteriophages, are thought to function as an “organ” that has immeasurable effects on human health and disease. Therefore, we considered that the functional restoration of the bacteriome and virome by FMT could be the best indicator of successful treatment. In fact, based on Kyoto Encyclopedia Genes and Genomes (KEGG) orthology (KO), we observed that the normalized abundance of recipient feces after FMT was similar to that of donor feces but not to that of recipient feces before FMT (Supplementary Figure 8).
We then searched for the key functional pathways associated with the differences between samples from before and after FMT. In the post-FMT samples, 3 pathways, namely D-arginine and D-ornithine metabolism, non-homologous end-joining, and secondary bile acid biosynthesis, were detected as significant functional KEGG pathways (Figure 5A). In the pre-FMT samples, fluorobenzoate degradation was identified as a significant functional KEGG pathway (Figure 5A). The relative concentrations of bile acids are especially important in CDI.10,22 Primary bile acids such as taurocholic acid induce the germination of metabolically latent spores, whereas secondary bile acids inhibit both germination and vegetative growth in bacteria. A recent report found that FMT is associated with the functional restoration of secondary bile acid metabolism mediated by colon bacteria.10 Thus, we focused on secondary bile acid biosynthesis among the 3 significant functional KEGG pathways in the post-FMT samples. As shown in Figure 5B, FMT contributed to the acquisition of genes involved in the secondary bile acid biosynthesis pathway (eg, baiB and baiF) in the recipients’ bacteriomes. We next examined the proportions of bacteria known to be associated with the secondary bile acid biosynthesis pathway in pre- and post-FMT samples from recipients and donors. The high abundance of Clostridium scindens, a major bacterium in intestinal secondary bile acid biosynthesis, as well as Clostridium leptum, was found in recipients’ feces after FMT (Figure 5C). These findings confirmed that secondary bile acid biosynthesis was restored in the recipients after FMT. The fluorobenzoate degradation pathway was also highly represented in the recipients’ feces before FMT. Previous reports indicate that this pathway is linked with the severity of intestinal inflammation, such as that seen in Crohn’s disease.23 In fact, KO terms related to the fluorobenzoate degradation pathway were frequently observed in the pre-FMT samples (Figure 5D). We further determined which bacteria were involved in this pathway. Figure 5E is a Venn diagram showing the bacterial contigs possessing open-reading frames annotated with KO terms that are involved in the fluorobenzoate degradation pathway in the pre- and post-FMT samples and donor samples. The bar plot shows the number of contigs for each bacterial genera in each region. Most of the bacteria possessing such genes were identified in the B1 region. Interestingly, all of them were Proteobacteria (eg, Klebsiella, Escherichia, unclassified Enterobacteriaceae, and Salmonella), the counts of which diminished after FMT. Thus, the disappearance of Proteobacteria and the subsequent decreased level of fluorobenzoate degradation after FMT might contribute to an improvement in intestinal CDI. Finally, the KO terms detected in the viromes from the pre- and post-FMT samples are presented as a volcano plot (Figure 5F). Ten genes were highly abundant in the post-FMT samples. RNASAH1, dut, dnaQ, dnaG, polA, and DNMT1 are involved in viral replication; ABC.FEV.A and ABC.FEV.P are associated with iron transporters; and trxA is necessary for ssDNA phage assembly. The final gene, amiABC, is correlated with antimicrobial peptide resistance, which may be important for regulating intestinal dysbiosis. Therefore, we also determined which viral contigs possessed amiABC. Ninety-seven viral contigs included amiABC, almost all of which were classified as Caudovirales (Figure 5G). Among them, 7 viral contigs were integrated into bacterial contigs classified as Bacteroidetes, Firmicutes, Verrucomicrobia, and Fusobacteria (Figure 5H). Thus, pathway analysis of the bacteriome and virome is important for understanding the functional aspects of the microflora under pathological conditions, and these findings might provide insights into potential novel therapeutic targets.
Figure 5.

Bacterial and viral functional analyses in FMT recipients. (A) Volcano plot comparing the KEGG pathways in the bacteriomes of pre- and post-FMT samples (n = 9). Circles colored blue and red indicate significantly different pathways (>2-fold, q < 0.05 by paired Student t test with false discovery rate correction) between pre- and post-FMT samples. (B) The number of samples with bacteriomes that had KOs associated with the “secondary bile acid biosynthesis” pathway. (C) The number of contigs in bacterial species related to bile acid 7α-dehydroxylating activity. (D) The number of samples with bacteriomes that had KO terms associated with the “fluorobenzoate degradation” pathway. (E) Venn diagram showing the bacterial contigs overlapping with KO terms involved in the “fluorobenzoate degradation” pathway in pre-FMT, post-FMT, and donor samples. Bar plot showing the number of contigs in the bacterial genera. (F) Volcano plot comparing the KO terms in the virome between pre- and post-FMT samples (n = 9). Circles colored blue and red indicate significantly different KO terms (>2-fold, q < 0.1 by paired Student’s t test with false discovery rate correction) in the post-FMT samples. (G) Viral taxonomic distribution of viral contigs containing amiABC (K01448). (H) Viral taxonomic distribution of the viral contigs detected as prophages in Figure 5G (left) and the bacterial taxonomic distribution of the bacterial hosts’ contigs (right).
Discussion
We metagenomically analyzed the intestinal virome together with the bacteriome of fecal samples from patients with rCDI who underwent FMT, demonstrating bacteria-phage dynamics in the host and functional restoration of the microbiome in patients who underwent successful CDI treatment.
In patients with CDI before FMT, marked dysbiosis, including reductions in the prevalence of Bacteroidia and Clostridia and increased counts of Negativicutes and Gammaproteobacteria, was observed (Figure 1A, B, and D). After FMT, large numbers of Firmicutes and Bacteroidetes were detected in the recipients of FMT (Figure 1A and B). Along with these alterations, bacterial diversity and richness increased significantly, and the bacteriomes of the recipients approached those of the donors (Figure 1C and D). The sequence reads associated with C difficile were barely detectable in the CDI patients’ feces (Supplementary Figure 1E),18 making it difficult to correlate the relative abundance of fecal C difficile with the pathogenesis of rCDI. However, C difficile-specific depletion, such as that achieved by treatment with C difficile-associated phage-derived endolysins, is effective against CDI.21,24,25 Therefore, in addition to regulating Proteobacteria expansion, a characteristic of chronic intestinal inflammation during rCDI, the elimination of C difficile might be required.
In common with previous studies,11,12 the relative abundance of Microviridae in the viromes increased in the recipients after FMT, whereas that of Caudovirales decreased after FMT (Figure 2A and B). Bray-Curtis dissimilarity showed that the viromes from the recipients’ feces after FMT were similar to those from the donors (Figure 2H). However, it is noteworthy that the diversity and richness of Microviridae expanded significantly in patients after FMT (Figure 2F). In addition to the transfer of Caudovirales seen in previous studies,11–13 the increased Microviridae counts in the recipients after FMT reveal the potentially critical role played by this family in the therapeutic efficacy of FMT.
In this study and others,8,10,11,20,26–31 the relative abundance of Proteobacteria in recipients after FMT decreased significantly when compared with the relative abundance before FMT (Figure 1A and B, and Supplementary Figure 4). In addition to this FMT-associated alteration, the relative abundance of Microviridae in the recipients after FMT increased significantly when compared with the abundance before FMT (Figure 2A and B, and Supplementary Figure 4). By analyzing the bacteriome and virome from the same fecal samples, we also identified a negative correlation between the abundance of Microviridae and the abundance of Proteobacteria in the fecal samples from the rCDI patients before and after FMT (Supplementary Figure 3). This correlation might act as a prominent index for evaluating FMT efficacy.
In the present study, FMT influenced the bacteria-phage association between bacteria and temperate phages in the host. Most temperate phages behaved similarly to host bacteria in the FMTs (Figure 3C). Compared with the recipients’ samples before FMT, many CRISPR spacers against Microviridae were detected in the recipients’ samples after FMT (Figure 4B). Interestingly, Bacteroidetes (eg, Parabacteroides and Bacteroides) preferentially possessed spacers against Microviridae. Although Bullavirinae (a Microviridae sub-family member) infect Enterobacteriaceae such as Escherichia, Klebsiella, and Salmonella,32 we identified few spacers against Microviridae on the contigs identified as Proteobacteria in the pre-FMT samples (Figure 4C). Thus, the expansion of Bacteroidetes and Firmicutes based on symbiosis with temperate and virulent phages is another important step toward the amelioration of dysbiosis.
In addition to the altered intestinal bacteriome and virome by FMT, we showed that the series of microbiome functions in the recipients’ feces after FMT resembled those of the donor feces but not those of the recipients’ feces before FMT (Supplementary Figure 8). These results clearly demonstrate that the restoration of intestinal microflora functions reflected the success of FMT. As shown in Figure 5A, fluorobenzoate degradation, a process closely related to the severity of intestinal inflammation such as that linked with Crohn’s disease, was identified as a significant functional KEGG pathway in the samples obtained before FMT.23 Proteobacteria (eg, Klebsiella, Escherichia, unclassified Enterobacteriaceae, and Salmonella), which were only present in samples from donors before FMT, were responsible for this pathway (Figure 5E). Thus, the disappearance of Proteobacteria is possibly an important step in the treatment of FMT. After FMT, D-arginine and D-ornithine metabolism, non-homologous end-joining, and secondary bile acid biosynthesis were detected as significant functional KEGG pathways (Figure 5A). According to previous reports, arginine deficiency causes poor growth and enhanced toxin production in some strains of C difficile, suggesting that arginine synthesis activation may contribute to the inhibition of toxin production by C difficile.33 Secondary bile acids also inhibit both germination and vegetative growth in C difficile. Interestingly, we detected an increased number of genes, and an expansion of C scindens and C leptum, involved in secondary bile acid biosynthesis after FMT (Figure 5B and C). Thus, the colonization of Clostridia species in FMT recipients may inhibit C difficile reactivation. Furthermore, after FMT, amiABC, a gene associated with antimicrobial peptide resistance, was harbored by Caudovirales and overrepresented in the recipients’ viromes (Figure 5F and G). It is interesting that Bacteroidetes, Firmicutes, Verrucomicrobia, and Fusobacteria acquired the amiABC gene by inserting these lysogenic phage genomes into their own.
This study had several limitations. Among the FMT recipients, 8 were women (Supplementary Table 1); however, there were no sex differences with regard to FMT efficacy.19,34,35 Future studies will be required to address whether biological sex influences bacteriome and virome alterations in patients with rCDI who receive FMT. Furthermore, we only analyzed the samples collected from donors and recipients 2 weeks before FMT and 8 weeks after FMT from the recipients, and all recipients were FMT-responders. Our comprehensive analyses of intestinal bacteriomes and viromes revealed that FMT changed the functions of intestinal bacteriomes and viromes in the recipients after FMT compared with those before FMT. What is uncertain, however, is whether these functional changes are durable over time in patients after successful FMT. It would be an interesting future direction, therefore, to address this question by analyzing the bacteriomes and viromes from recipients with successful and unsuccessful FMTs longitudinally at different time points.
Our data show that FMT changes the intestinal microflora to resemble more closely that of the physiological healthy state by systemically affecting both the bacteriome and virome of the recipients. Our results on the host bacteria-phage associations between intestinal bacteria and phages provide fundamental information on the effects of FMT. When CDI develops after initial antibiotic administration, it is desirable to discontinue the agent immediately and eliminate C difficile as specifically as possible without inducing dysbiosis. However, in rCDI, proteobacterial expansion might induce severe secondary enterocolitis,36,37 thereby preventing the intestinal microbiota from recovering to their normal state. Thus, eliminating such “pathobiont” Proteobacteria in addition to C difficile before FMT could potentially promote the reconstitution of intestinal microbiota. However, 2 independent cases of extended-spectrum beta-lactamase-producing Escherichia coli bacteremia were recently reported after FMT.2 Considering that FMT can alter host bacteria-phage dynamics, in addition to the clinical survey about short-term and long-term safety of FMT for rCDI,38 more detailed analyses on intestinal viromes and bacteriomes are required to determine safe methods to specifically exclude pathogenic Proteobacteria and efficiently promote colonization by beneficial Bacteroidetes and Firmicutes in recipients to improve the therapeutic efficacy of FMT.
Supplementary Material
WHAT YOU NEED TO KNOW.
BACKGROUND AND CONTEXT
The overall effects of fecal microbiota transplantation (FMT) are not fully understood. We comprehensively investigated the human intestinal bacteriomes and viromes from patients with recurrent Clostridioides difficile infections (rCDIs) after successful FMT and their donors.
NEW FINDINGS
Microviridae and Proteobacteria abundances in fecal samples from rCDI patients pre- and post-FMT are negatively correlated. Host bacteria-phage associations, and a functionally restored bacteriome and virome in the intestine, provide fundamental information about the effects of FMT.
LIMITATIONS
This study was performed in patients who underwent successful FMT. Studies in patients who undergo unsuccessful FMT are needed.
IMPACT
Restoration of intestinal bacterial and viral functions in patients after FMT evidences the success of FMT as a treatment for rCDI.
Acknowledgments
We thank S. Yin, B. Baigalmaa, H. Ohmiya, and S. Hatakeyama for technical assistance and K. Ogawa, M. Maeda, and K. Suetsugu for secretarial assistance. We also thank Colin Hill (APC Microbiome Ireland & School of Microbiology, University College Cork, Cork T12 YT20, Ireland) and his colleagues for their scientific insights. The super-computing resource was provided by the Human Genome Center, Institute of Medical Science, The University of Tokyo (http://sc.hgc.jp/shirokane.html). We thank Edanz Group (https://en-author-services.edanz.com/ac) for editing a draft of this manuscript.
Funding
This study was supported by the Ministry of Education, Culture, Sports, Science, and Technology of Japan (Grant-in-Aid for Challenging Exploratory Research [to Satoshi Uematsu]), the Japan Agency for Medical Research and Development (AMED) (to Satoshi Uematsu; JP18ak0101069), the Takeda Science Foundation (to Satoshi Uematsu), the Canon Foundation (to Satoshi Uematsu), Grants-in-Aid for Young Scientists (to Yasumasa Kimura) from the JSPS, Priority Issue on Post-K computer (hp150265, hp160219, hp170227), Strategic Programs for Innovative Research Field 1 (hp150232) from MEXT (to Satoru Miyano), and the Center of Innovation Program from Japan Science and Technology Agency (JST) (to Satoru Miyano and Seiya Imoto).
Abbreviations used in this paper:
- CDI
Clostridioides difficile infection
- CRISPR
clustered regularly interspaced short palindromic repeats
- FMT
fecal microbiota transplantation
- KEGG
Kyoto Encyclopedia Genes and Genomes
- KO
KEGG orthology
- rCDI
recurrent CDI
- ssDNA
single-stranded DNA
- uc_Caudovirales
unclassified Caudovirales
Footnotes
Supplementary Material
Note: To access the supplementary material accompanying this article, visit the online version of" Gastroenterology at www.gastrojournal.org, and at https://doi.org/10.1053/j.gastro.2021.02.013.
CRediT Authorship Contributions
Kosuke Fujimoto, MD, PhD (Conceptualization: Equal; Data curation: Equal; Formal analysis: Equal; Methodology: Equal; Project administration: Equal; Resources: Supporting; Writing - original draft: Equal; Writing - review & editing: Equal).
Yasumasa Kimura, PhD (Data curation: Equal; Formal analysis: Equal; Methodology: Equal).
Jessica R. Allegretti, MD, MPH (Data curation: Supporting; Resources: Lead; Writing - original draft: Supporting; Writing - review & editing: Supporting).
Mako Yamamoto, MS (Data curation: Supporting); Yao-zhong Zhang, PhD (Data curation: Supporting).
Kotoe Katayama, PhD (Data curation: Supporting).
Georg Tremmel, PhD (Data curation: Supporting).
Yunosuke Kawaguchi, MD (Data curation: Supporting).
Masaki Shimohigoshi, MD (Data curation: Supporting).
Tetsuya Hayashi, MD (Data curation: Supporting).
Miho Uematsu, MD, PhD (Data curation: Supporting).
Kiyoshi Yamaguchi, PhD (Data curation: Supporting).
Yoichi Furukawa, MD, PhD (Data curation: Supporting).
Yutaka Akiyama, PhD (Data curation: Supporting; Software: Equal).
Rui Yamaguchi, PhD (Data curation: Supporting).
Sheila E. Crowe, MD, PhD (Project administration: Supporting; Supervision: Supporting; Writing - review & editing: Supporting).
Peter B. Ernst, MD, PhD (Project administration: Supporting; Supervision: Supporting; Writing - review & editing: Supporting).
Satoru Miyano, PhD (Funding acquisition: Supporting; Software: Equal).
Hiroshi Kiyono, DDS, PhD (Funding acquisition: Supporting; Supervision: Supporting).
Seiya Imoto, PhD (Conceptualization: Equal; Data curation: Equal; Formal analysis: Equal; Funding acquisition: Equal; Methodology: Equal; Project administration: Equal; Software: Equal; Supervision: Lead; Writing - original draft: Equal; Writing - review & editing: Equal).
Satoshi Uematsu, M.D., Ph.D. (Conceptualization: Lead; Funding acquisition: Lead; Methodology: Equal; Project administration: Lead; Supervision: Lead; Writing - original draft: Lead; Writing - review & editing: Lead).
Conflicts of interest
The authors disclose no conflicts.
References
- 1.McDonald LC, Gerding DN, Johnson S, et al. Clinical practice guidelines for Clostridium difficile infection in adults and children: 2017 Update by the Infectious Diseases Society of America (IDSA) and Society for Healthcare Epidemiology of America (SHEA). Clin Infect Dis 2018;66:987–994. [DOI] [PubMed] [Google Scholar]
- 2.DeFilipp Z, Bloom PP, Torres Soto M, et al. Drug-resistant E. coli bacteremia transmitted by fecal microbiota transplant. N Engl J Med 2019;381:2043–2050. [DOI] [PubMed] [Google Scholar]
- 3.Grosen AK, Povlsen JV, Lemming LE, et al. Faecal microbiota transplantation eradicated extended-spectrum beta-lactamase-producing Klebsiella pneumoniae from a renal transplant recipient with recurrent urinary tract infections. Case Rep Nephrol Dial 2019; 9:102–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Nood E, Vrieze A, Nieuwdorp M, et al. Duodenal infusion of donor feces for recurrent Clostridium difficile. N Engl J Med 2013;368:407–415. [DOI] [PubMed] [Google Scholar]
- 5.Li SS, Zhu A, Benes V, et al. Durable coexistence of donor and recipient strains after fecal microbiota transplantation. Science 2016;352:586–589. [DOI] [PubMed] [Google Scholar]
- 6.Moss EL, Falconer SB, Tkachenko E, et al. Long-term taxonomic and functional divergence from donor bacterial strains following fecal microbiota transplantation in immunocompromised patients. PLoS One 2017;12: e0182585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kumar R, Yi N, Zhi D, et al. Identification of donor microbe species that colonize and persist long term in the recipient after fecal transplant for recurrent Clostridium difficile. NPJ Biofilms Microbiomes 2017;3:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smillie CS, Sauk J, Gevers D, et al. Strain tracking reveals the determinants of bacterial engraftment in the human gut following fecal microbiota transplantation. Cell Host Microbe 2018;23:229–240.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Siranosian BA, Tamburini FB, Sherlock G, et al. Acquisition, transmission and strain diversity of human gut-colonizing crAss-like phages. Nat Commun 2020;11:280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weingarden AR, Chen C, Bobr A, et al. Microbiota transplantation restores normal fecal bile acid composition in recurrent Clostridium difficile infection. Am J Physiol Gastrointest Liver Physiol 2014;306:G310–G319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zuo T, Wong SH, Lam K, et al. Bacteriophage transfer during faecal microbiota transplantation in Clostridium difficile infection is associated with treatment outcome. Gut 2018;67:634–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Draper LA, Ryan FJ, Smith MK, et al. Long-term colonisation with donor bacteriophages following successful faecal microbial transplantation. Microbiome 2018;6:220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baktash A, Terveer EM, Zwittink RD, et al. Mechanistic insights in the success of fecal microbiota transplants for the treatment of Clostridium difficile infections. Front Microbiol 2018;9:1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ott SJ, Waetzig GH, RehmanA, et al. Efficacy of sterile fecal filtrate transfer for treating patients with Clostridium difficile infection. Gastroenterology 2017;152:799–811. e7. [DOI] [PubMed] [Google Scholar]
- 15.Roux S, Solonenko NE, Dang VT, et al. Towards quantitative viromics for both double-stranded and single-stranded DNA viruses. PeerJ 2016;4:e2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reyes A, Haynes M, Hanson N, et al. Viruses in the faecal microbiota of monozygotic twins and their mothers. Nature 2010;466:334–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fujimoto K, Kawaguchi Y, Shimohigoshi M, et al. Antigen-specific mucosal immunity regulates development of intestinal bacteria-mediated diseases. Gastroenterology 2019;157:1530–1543.e4. [DOI] [PubMed] [Google Scholar]
- 18.Vincent C, Miller MA, Edens TJ, et al. Bloom and bust: intestinal microbiota dynamics in response to hospital exposures and Clostridium difficile colonization or infection. Microbiome 2016;4:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ianiro G, Valerio L, Masucci L, et al. Predictors of failure after single faecal microbiota transplantation in patients with recurrent Clostridium difficile infection: results from a 3-year, single-centre cohort study. Clin Microbiol Infect 2017;23:337.e1–337.e3. [DOI] [PubMed] [Google Scholar]
- 20.Shahinas D, Silverman M, Sittler T, et al. Toward an understanding of changes in diversity associated with fecal microbiome transplantation based on 16S rRNA gene deep sequencing. MBio 2012;3. e00338–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fujimoto K, Kimura Y, Shimohigoshi M, et al. Metagenome data on intestinal phage-bacteria associations aids the development of phage therapy against pathobionts. Cell Host Microbe 2020;28:380–389.e9. [DOI] [PubMed] [Google Scholar]
- 22.Khoruts A, Sadowsky MJ. Understanding the mechanisms of faecal microbiota transplantation. Nat Rev Gastroenterol Hepatol 2016;13:508–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Montassier E, Gastinne T, Vangay P, et al. Chemotherapy-driven dysbiosis in the intestinal microbiome. Aliment Pharmacol Ther 2015;42:515–528. [DOI] [PubMed] [Google Scholar]
- 24.Mehner-Breitfeld D, Rathmann C, Riedel T, et al. Evidence for an adaptation of a phage-derived holin/endolysin system to toxin transport in Clostridioides difficile. Front Microbiol 2018;9:2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Q, Euler CW, Delaune A, et al. Using a novel lysin to help control Clostridium difficile infections. Antimicrob Agents Chemother 2015;59:7447–7457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dutta SK, Girotra M, Garg S, et al. Efficacy of combined jejunal and colonic fecal microbiota transplantation for recurrent Clostridium difficile Infection. Clin Gastroenterol Hepatol 2014;12:1572–1576. [DOI] [PubMed] [Google Scholar]
- 27.Hamilton MJ, Weingarden AR, Unno T, et al. High-throughput DNA sequence analysis reveals stable engraftment of gut microbiota following transplantation of previously frozen fecal bacteria. Gut Microbes 2013;4:125–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seekatz AM, Aas J, Gessert CE, et al. Recovery of the gut microbiome following fecal microbiota transplantation. mBio 2014;5:e00893–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shankar V, Hamilton MJ, Khoruts A, et al. Species and genus level resolution analysis of gut microbiota in Clostridium difficile patients following fecal microbiota transplantation. Microbiome 2014;2:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Song Y, Garg S, Girotra M, et al. Microbiota dynamics in patients treated with fecal microbiota transplantation for recurrent Clostridium difficile infection. PLoS One 2013; 8:e81330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jalanka J, Mattila E, Jouhten H, et al. Long-term effects on luminal and mucosal microbiota and commonly acquired taxa in faecal microbiota transplantation for recurrent Clostridium difficile infection. BMC Med 2016;14:155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Szekely AJ, Breitbart M. Single-stranded DNA phages: from early molecular biology tools to recent revolutions in environmental microbiology. FEMS Microbiol Lett 2016;363. [DOI] [PubMed] [Google Scholar]
- 33.Karasawa T, Maegawa T, Nojiri T, et al. Effect of arginine on toxin production by Clostridium difficile in defined medium. Microbiol Immunol 1997;41:581–585. [DOI] [PubMed] [Google Scholar]
- 34.Tariq R, Saha S, Solanky D, et al. Predictors and management of failed fecal microbiota transplantation for recurrent Clostridioides difficile infection [published online ahead of print July 21, 2020]. J Clin Gastroenterol. 10.1097/MCG.0000000000001398. [DOI] [PubMed] [Google Scholar]
- 35.Peri R, Aguilar RC, Tuffers K, et al. The impact of technical and clinical factors on fecal microbiota transfer outcomes for the treatment of recurrent Clostridioides difficile infections in Germany. United European Gastroenterol J 2019;7:716–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Seekatz AM, Young VB. Clostridium difficile and the microbiota. J Clin Invest 2014;124:4182–4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shin NR, Whon TW, Bae JW. Proteobacteria: microbial signature of dysbiosis in gut microbiota. Trends Biotechnol 2015;33:496–503. [DOI] [PubMed] [Google Scholar]
- 38.Saha S, Mara K, Pardi DS, et al. Long-term safety of fecal microbiota transplantation for recurrent Clostridioides difficile infection [published online ahead of print January 11, 2021]. Gastroenterology. 10.1053/j.gastro.2021.01.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data supporting the findings of this study have been deposited in the National Center for Biotechnology Information Sequence Read Archive (PRJNA693652). They are available from the corresponding authors on reasonable request, with the approval of our ethics committee. Please contact the corresponding authors for additional information.