

Abstract
Neurons must remove aggregated, damaged proteins in order to survive. Among the ways of facilitating this protein quality control is the ubiquitin-proteasomal system (UPS). Aggregated, damaged proteins are targeted for destruction by the UPS by acquiring a polymer of ubiquitin residues that serves as a signal for transport to the UPS. However, before this protein degradation can occur, the polyubiquitin chain must be removed, one residue at a time, a reaction facilitated by the enzyme, ubiquitin C-terminal hydrolase (UCH-L1). In Alzheimer disease brain, this normally abundant protein is both of lower levels and oxidatively and nitrosatively modified than in control brain. This causes diminished function of the pleiotropic UCH-L1 enzyme with consequent pathological alterations in AD brain, and the author asserts the oxidative and nitrosative alterations of UCH-L1 are major contributors to mechanisms of neuronal death in this devastating dementing disorder and its earlier stage, mild cognitive impairment (MCI). This review paper outlines these findings in AD and MCI brain.
Keywords: ubiquitin carboxyl-terminal hydrolase-L1, Ubiquitin-proteasomal system, Alzheimer disease and mild cognitive impairment, oxidative and nitrosative stress, loss of function, neuronal death
1.0. Introduction
Alzheimer disease (AD) is the most prevalent dementing disorder in aged individuals and one of several age-related neurodegenerative disorders associated with protein aggregation in brain [1]. AD is characterized histopathologically by the presence in brain of: a) extracellular senile plaques, which are composed of aggregated amyloid β-peptide42 (Aβ42) and Aβ40 and surrounded by dystrophic neurites; b) neurofibrillary tangles (NFT), which are composed of aggregated hyperphosphorylated tau protein that had fallen off axonal and dendritic microtubules; and c) loss of neuronal synapses [1]. Clinically, AD is characterized by progressive loss of higher executive functioning, among which are loss of reasoning, ability to draw conclusions from presented data, memory, and other aspects of cognition, ultimately leading to dementia. Since synapses are involved in these processes, synaptic dysfunction is highly prevalent in AD.
An earlier stage of AD is mild cognitive impairment (MCI), which exists in two forms, associated with memory loss (amnestic MCI, aMCI) and non-amnestic MCI [2]. Persons with aMCI are characterized by having significant levels of AD neuropathology, but such persons have normal activities of daily living [3].
The presence of protein aggregates in AD and MCI brains is in large part due to a failure of protein quality control processes, including, among others, cytoplasmic resident autophagy, removal of damaged mitochondria (mitophagy), intracellular unfolded protein response, and cytoplasmic ubiquitin proteasomal system (UPS) all involved in degradation of damaged and/or aggregated proteins [4–6]. Collectively, these quality control processes are often referred to as the proteostasis network [7].
The current review article emphasizes the role played by one aspect of the UPS, i.e., ubiquitin carboxyl-terminal hydrolase L-1 (UCH-L1), the dysfunction of which as a consequence of oxidative and/or nitrosative damage leads to at least three pathological aspects in AD brain. Our laboratory also hypothesized that UCH-L1 represents a promising pharmacological target for this dementing disorder.
2.0. Alzheimer disease and Oxidative/Nitrosative Stress
2.1. Oxidative and Nitrosative Stress.
Oxidative stress results when the production of free radicals exceeds the rate of cellular small-molecule or protein-based antioxidant scavenging of these free radicals [8–10]. Some examples of reactive oxygen species (ROS) or reactive nitrogen species (RNS) that are either free radicals or oxygen-containing molecules that can react to produce free radicals are superoxide free radical, hydroxyl free radical, hydrogen peroxide. A more extensive list of ROS and RNS and sulfur-containing free radicals is found in [9,10]. Indices of protein oxidation resulting from reaction of ROS and/or RNS are elevated levels of protein-resident carbonyls or 3-nitrotyrosine (3-NT) moieties, while the principal protein modification resulting from lipid peroxidation is protein-bound 4-hydroxy-2-nonenal (HNE).
2.1.1. Protein Carbonyls.
Protein carbonyls are formed by four main processes [8): a) Cleavage of the protein’s primary amino-acid chain by free radicals leading to aldehyde or ketone functionalities on the cleavage ends of each of the two strands; b) oxidation of side chain portions of the primary amino acid sequence of proteins; c) By the aldehyde functionality of lipid peroxidation-derived alkenals that can covalently bind by Michael addition to His, Cys, or Lys residues of proteins (see below); and d) By not well-understood processes of Amadori chemistry that are initiated by formation of an imine bond (aka Schiff base) between the aldehydic portion of a reducing sugar with a protein-resident Lys residue. Protein carbonyls are often detected immunochemically [11]. Following formation of the 2,4-dinitrophenylhydrazone on proteins by reaction of protein-resident carbonyls with 2,4-dintrophenylhydrazine, antibodies specific for the protein-resident hydrazone are employed (Figure 1). Antibodies are shown to be specific for the hydrazone by first treating the protein samples with the strong reducing agent, NaBH4 that converts the carbonyl functionality to an alcohol, to which the antibodies do not bind.
Figure 1.
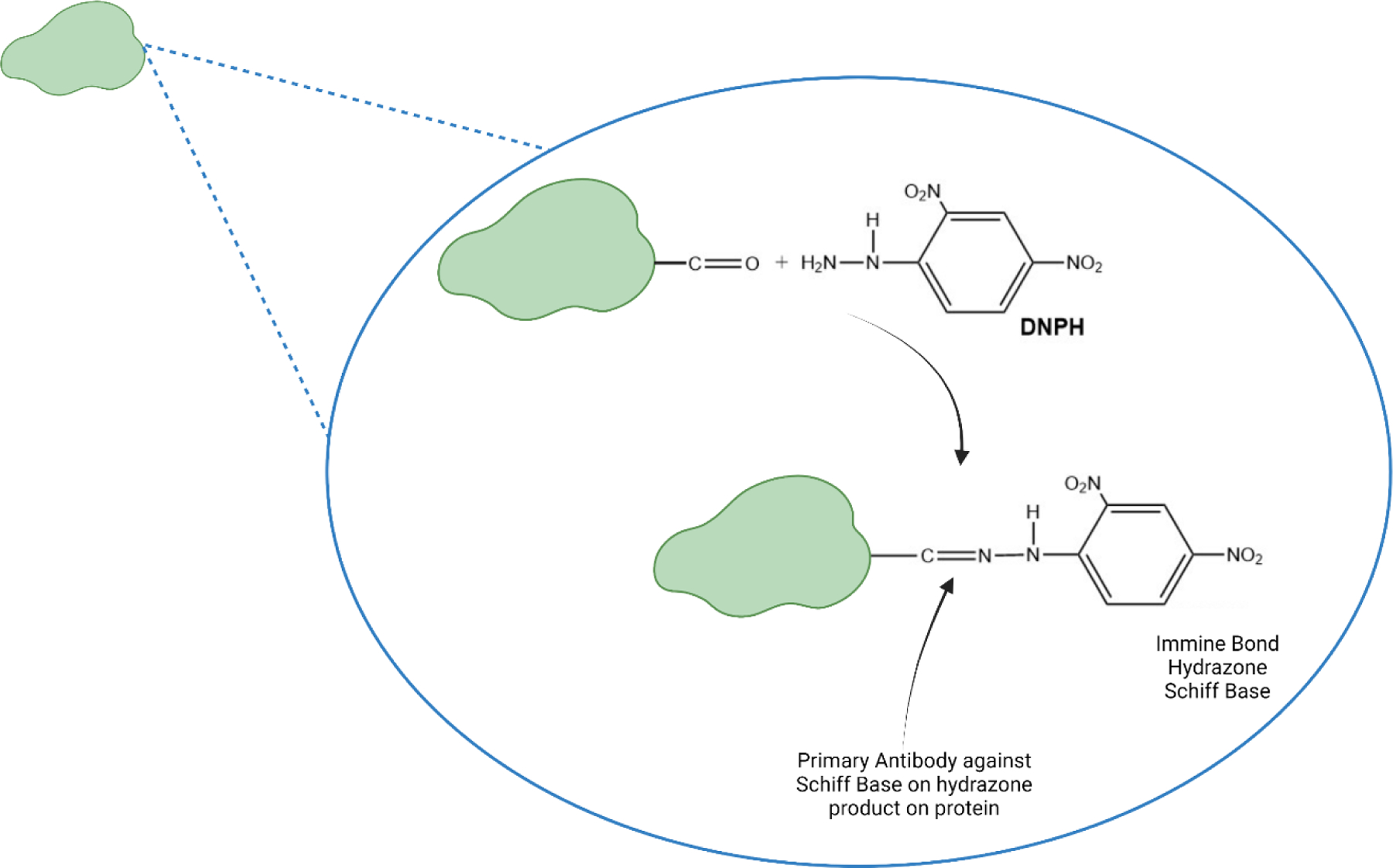
Formation of the Schiff base (aka protein hydrazone) by reaction of a protein carbonyl moiety that results from protein oxidation with 2,4,-dinitrohydrazine.
2.1.2. 3-Nitrotyrosine.
The free radical nitric oxide is formed from arginine by the action of nitric oxide synthase (NOS), of which there are three isoforms, the more important for brain being neuronal NOS (n-NOS) and inducible NOS (i-NOS). Superoxide free radicals are most commonly formed in Complex I on the matrix side of the inner membrane of mitochondria, but also by reactions involving NADPH oxidase or xanthine oxidase. Radical-radical recombination reactions of NO with superoxide free radicals are among the fastest reactions known and lead to the non-radical peroxynitrite, ONOO−. Through a series of other reactions, peroxynitrite leads to nitrogen dioxide, NO2, a free radical [12–14].
The OH moiety of tyrosine in the para-position of the aromatic ring of Tyr can be attacked by a free radical leading to abstraction of a hydrogen atom and a free radical on the oxygen atom. Because the OH group bound to an aromatic ring in the para-position is an ortho-para-directing substituent on an aromatic ring, the free electron on the 4-position of Tyr will delocalize to the ortho- or 3-position of the aromatic ring of Tyr. Then, in a rapid radical-radical recombination reaction, NO2 formed as from above binds to the free electron in the 3-position of Tyr to form 3-NT (Figure 2), which is often detected in samples immunochemically by reaction with specific antibodies to protein-resident 3-NT [14]. Specificity of the antibody used is shown by the absence of binding to samples that had been pretreated with the powerful reducing reagent, Na2S2O4 that converts 3-NT to 3-amino-Tyr that is no longer recognized by antibodies against 3-NT [15].
Figure 2.
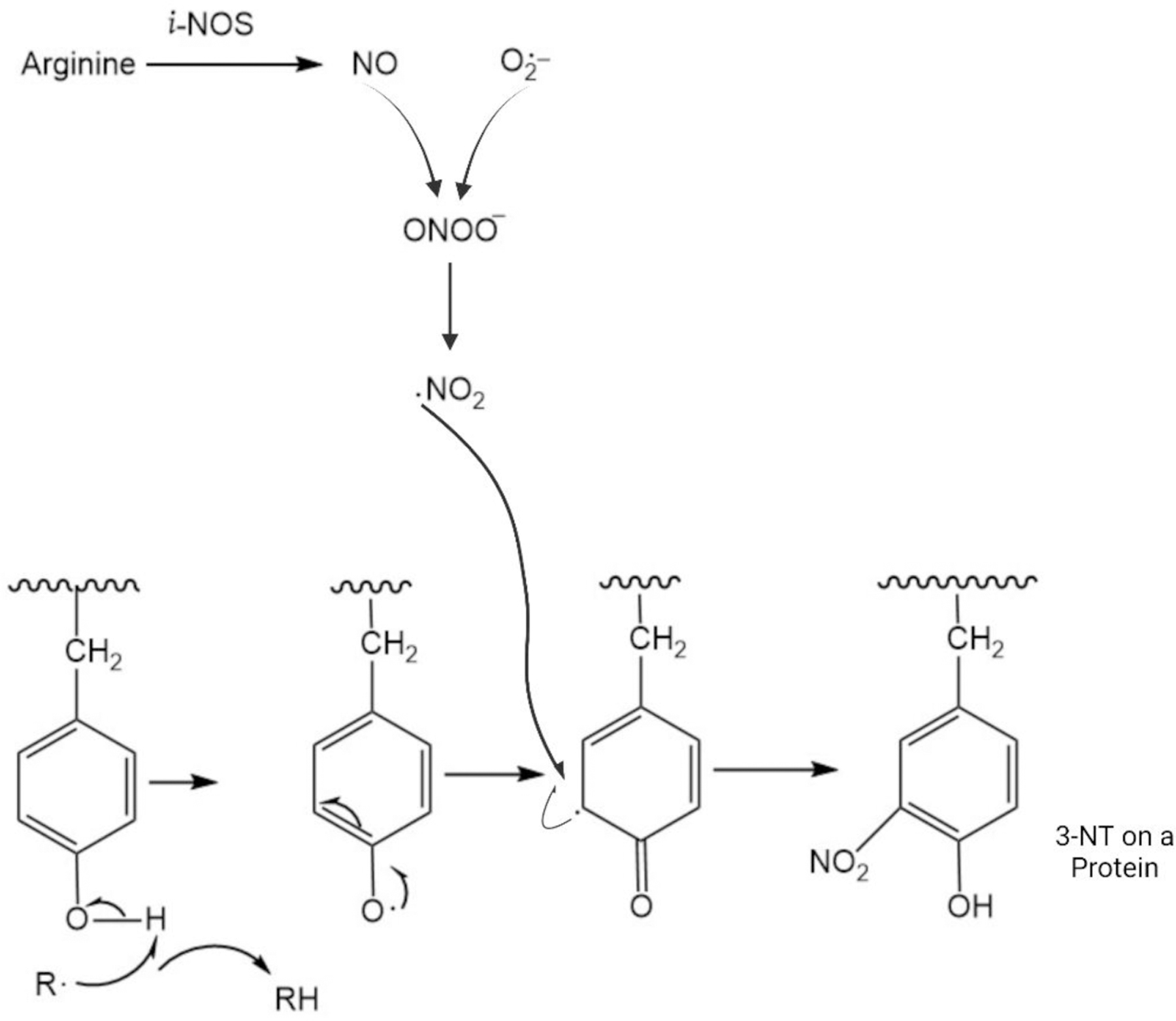
Schematic drawing of the formation of 3-nitrotyrosine (3-NT). Nitric oxide (NO), a free radical gaseous molecule formed by decomposition of arginine by the enzyme nitric oxide synthase (inducible NOS is shown), reacts with superoxide free radical anion by radical-radical recombination to form peroxynitrite, ONOO−. This, in turn, through a series of reactions, form nitrogen dioxide, NO2, also a free radical. Reaction of a radical R· with the OH group at the 4-position of the aromatic ring of Tyr leads to abstraction of H· and a resulting radical on the O-atom and RH. Because the OH functionality on aromatic 6-member rings is ortho- and para-directing of electron density, delocalization of the unpaired electron to the ortho 3-position of the aromatic ring of Tyr occurs (the para-position is already occupied as part of the Tyr structure). The NO2 free radical then reacts with this free electron on the 3-position of Tyr by radical-radical recombination to form 3-NT.
When present on proteins, steric hinderance of 3-NT of the OH group on the aromatic ring of Tyr interferes with cell-signaling processes involving receptor tyrosine kinases, i.e.,3-NT is highly detrimental to intracellular signaling [16].
2.1.3. Reactive Alkenals, Products of Lipid Peroxidation.
2.1.3.1. Lipid Peroxidation.
Lipid peroxidation leads to production of reactive alkenals that modify proteins (see below) [10,17–20]. The process of lipid peroxidation involves four steps:
Reaction of a free radical R· with a labile allylic H-atom on a beta-acyl chain of the phospholipid to form RH and a carbon-centered free radical C· on the C-atom from which the allylic H-atom was abstracted.
Molecular oxygen, which is a diradical with two unpaired electrons and is a molecule with zero dipole moment and therefore soluble in the low dielectric medium of the hydrophobic acyl chains of the phospholipid bilayer, reacts with the carbon-centered free radical via radical-radical recombination to form the lipid peroxyl free radical, COO·.
The lipid peroxyl free radical COO· reacts with another labile allylic H-atom on the beta-acyl chain of phospholipids to form the lipid hydroperoxide, COOH on the carbon atom from which the first H-atom was abstracted and another carbon-centered free radical, C· on the same or different acyl chain of phospholipids. Note that the carbon-centered free radical formed in this step can propagate these reactions by entering at reaction b above, that is, lipid peroxidation involves a chain reaction that will continue as long as molecular oxygen and a source of available labile allylic H-atoms are present. Moreover, the COOH moiety formed in reaction c, through a series of reaction steps, can from the highly reactive and neurotoxic alkenal, HNE, discussed below. An important point is that a small amount of reactive free radicals in the lipid bilayer can, via this chain reaction outlined above greatly amplify the amount of HNE formed.
As in essentially all chain reactions, recombination of free radical species, such as two carbon-centered free radicals in this case, will terminate the chain reaction.
2.1.3.2. 4-Hydroxy-2-nonenal (HNE).
HNE is a molecule with an alpha, beta double bond between carbon atoms 2 and 3 and adjacent to an aldehyde functional group on carbon-1 of this 9-carbon molecule. Due to resonance of the double bond between carbon atoms 2 and 3 and the aldehyde on carbon atom-1 of HNE, coupled to the inductive effect of the electronegative character of the O-atom of the OH group on carbon-4, the electron density surrounding carbon atom-3 is significantly lessened. Therefore, an electrophilic reaction of carbon atom-3 of HNE with the electron-rich S-atom of Cys, epsilon amino N-atom of Lys, or the reactive N-atom of His leads to a covalent bond formation in a process called Michael addition. The effects of HNE addition to proteins can be profound, ranging from major structural changes in the protein [20] to loss of activity of the protein to which HNE is bound, which can and does occur [9,17,20–29].
Importantly, once oxidized, protein structures are altered and their activities are significantly diminished and often completely inhibited [17,30–34].
2.2. Oxidative/Nitrosative Stress in Indices AD and aMCI Brains
2.2.1. AD and aMCI Brains
Relative to that in control brains, protein carbonyls were first reported to be elevated in AD brain in regions rich in Aβ42, such as hippocampus and inferior parietal, but not in Aβ42-poor cerebellum [35]. Another protein oxidation marker, 3-NT, was reported to be elevated in AD brains [33,36–39]. Lipid peroxidation, indexed by elevation of protein-bound HNE (among other indices), is found in AD brain [9,17,22–24,27]. Some recent reviews of oxidative/nitrosative stress in AD and MCI are cited here [24,25,27,40–42].
2.2.2. Redox Proteomics to Identify Oxidatively Modified Proteins in AD and aMCI Brains
Redox proteomics is that branch of the field of proteomics that identifies oxidatively or nitrosatively modified proteins [43,44]. Basically, current methods involve separation of subject and control proteins, with each group tagged with unique identifier probes for oxidative/nitrosative markers, and subjected to proteolysis and subsequent sequence analysis of resulting peptides using MS/MS methods by shotgun proteomics. Sequence analyses of individual peptides allows data-base facilitated identification of the oxidized proteins [45]. More detailed descriptions of methods of redox proteomics can be found in relatively recent reviews [45–49]. As mentioned elsewhere in this manuscript, our laboratory pioneered the use of redox proteomics to identify oxidatively or nitrosatively modified brain proteins in AD disease or MCI [23,24,27,31,32,36,47,50–57].
2.2.2.1. Major Pathways with Diminished Function and Constitutive Oxidatively Modified and Dysfunctional Proteins Identified by Redox Proteomics Following Oxidative/Nitrosative Modification of Constitutive Proteins.
Table 1 provides the major pathways that are affected in AD and MCI brains by Aβ42-associated oxidative and nitrosative stress [47]. Table 2 shows the specific proteins that are oxidatively modified and nearly always dysfunctional in common in the progression of AD from its earliest stages through late-stage AD indexed by protein carbonyls, protein-resident 3-NT, and protein-bound HNE [44]. In vivo mouse models of AD yield similar results in terms of pathways that are dysfunctional as Ab oligomers are formed [47].
TABLE1.
Major Dysfunctional Pathways in Brains of Late-Stage AD, Early-Stage AD, MCI, or Preclinical AD (as well as Down Syndrome with AD) [Ref. 47]
| Dysfunctional Pathway | Consequence(s) |
|---|---|
| Cellular redox homeostasis and detoxification defense | Build-up of oxidatively modified proteins and elevated lipid peroxidation |
| Glucose metabolism | Decreased ATP production, which can lead to loss of neuronal cell potential, opening of voltage-gated Ca2+ channels with resultant neuronal mitochondrial, ER, and cellular death |
| Glutamine shuttle | Repeated depolarization of post-synaptic neurons with result neuroal death (excitotoxicity) |
| Synaptic plasticity, neurotransmitter vesicle transport and pre-synaptic membrane docking mechanisms | Decreased neurotransmission capability; consequent decreased learning and memory ability |
| Brain inflammation | Release of pro-inflammatory cytokines, ROS, and i-NOS, all producing harmful effects in the brain parenchyma |
| Protein synthesis | Decreased production of proteins to replace oxidatively damaged proteins |
| Protein Chaperones, Protein Folding, Damaged protein degradation via proteostasis network, including the 26S proteasome | Oxidatively damaged, misfolded proteins not degraded efficiently with consequent build-up of such proteins in the neuronal cytosol with resultant neuronal death |
| Cellular Signal Transduction | Inefficient transduction of extracellular signals, for example, receptor binding by key ligands, would lead to disruption of regulation of key cellular processes necessary for proper functioning of brain cells. |
TABLE 2.
Oxidatively Modified Brain Proteins in Common Indexed by Oxidative Stress Marker [Ref. 44]
| Elevated Oxidative Stress Index | Stages of AD | Oxidatively Modified Proteins in Common |
|---|---|---|
| Protein Carbonyls | Late-AD; Early AD; MCI; PCAD | ATP synthase; UCH-L1 |
| Protein Carbonyls | Late AD; MCI; PCAD | ATP synthase; α-Enolase; UCH-L1; |
| Protein-Resident 3-NT | Late AD; Early AD; MCI | ATP synthase |
| Protein-Bound HNE | Late AD; Early AD; MCI | ATP synthase; α-Enolase |
As is evident from the Tables 1 and 2, glucose metabolism and proteostasis (protein quality control) mechanisms, the latter involving the UPS system and therefore UCH-L1, the subject of this review article, are found to be oxidatively and/or nitrosatively modified in brains from individuals with late-stage AD, early-stage AD, MCI and preclinical AD. Clearly, decreased glucose metabolism as a consequence of oxidative modification is a major contributor to brain dysfunction throughout the progression of AD, no matter which oxidative stress marker is elevated. In addition, in the case of brain proteins with elevated protein carbonyls, UCH-L1 also is oxidatively modified at each stage of AD investigated except the very earliest, preclinical AD (PCAD). Elevated protein carbonyls reflect damaged, often aggregated proteins, and oxidatively modified UCH-L1 is consistent with the notion that protein degradation via the proteasome is one of the major pathways that is dysfunctional throughout the progression of AD. This finding makes UCH-L1 a potentially promising therapeutic target to slow, or in the ideal case, retard the progression of this devastating dementing disorder. The remainder of this review article explores UCH-L1 in brains of AD and MCI individuals as well as in vivo and in vitro models of AD.
3.0. UCH-L1 in AD and MCI and Models thereof
3.1. UCH-L1 Background
UCH-L1 is found nearly exclusively in neurons, where it comprises 1–5% of total neuronal proteins [58–60]. That fact alone suggests that UCH-L1 is an important protein. The high abundance of UCH-L1 suggests one or more critical roles for this protein. Other locations include gonads and certain cancer cells.
As discussed further below, the primary function of UCH-L1 is to permit highly ubiquitinylated aggregated and/or damaged proteins to be degraded by the 26S proteasome following removal of the poly-ubiquitin tag, one ubiquitin residue at a time from the carboxyl end by UCH-L1. In addition, in neurons, UCH-L1 is known to be important for stability of axonal microtubules [58–60] and interactions with tau protein [61].
UCH-L1 is a relatively small protein of only 223 amino acids, with an active-site resident Cys residue. A characteristic of most de-ubiquintinylation enzymes is a highly knotted structure near the N-terminus. Indeed, this knotted structure is regarded as one of the most complicated protein structures known [62]. Structural analysis reveals two domains of multiple α-helices that overlap a tightly packed hydrophobic core of β-strands [63]. The knotted backbone serves to protect UCH-L1 from proteasomal degradation by blocking the active site [64].
Poly-ubiquitinylated proteins (see below) are targeted to the 26S proteasome, which consists of two 19S caps on either side of a 20S barrel-shaped structure, for destruction to recycle a fixed amount of ubiquitin in the brain and to recycle amino acids [65,66]. The 19S cap is triggered to open on which are proteases that cleave the poly-ubiquitinylated, damaged protein into small peptides with lysine-containing peptides capable of having a polymer of ubiquitin. These, in turn, are small enough to permit the carboxyl end of the ubiquitin chain to slip under the knot structure of UCH-L1 to be hydrolyzed and recycled. This process continues until all ubiquitin residues of all the small peptides are removed. The de-ubiquitinylated peptides then migrate to the 20S barrel of the proteasome for degradation of these small peptides into even smaller peptides that emerge from the bottom 19S cap to encounter non-specific proteases that degrade these very small peptides to single amino acids that can be recycled for protein synthesis.
UCH-L1 has binding affinities for two key proteins in neurons that are relevant to AD: amyloid precursor protein (APP) from which neurotoxic Aβ42 is derived, and tubulin, whose alpha- and beta-isoforms comprise microtubules that facilitate anterograde and retrograde axonal transport [58]. As elaborated further below, oxidative and/or nitrosative modification of UCH-L1 likely disrupts these interactions, conceivably leading to facilitated amyloidogenic processing of APP and disruption of axonal microtubules, with consequent decreased axonal transport of key cargo such as neurotransmitter vesicles and mitochondria [31,47,67–70]. In contrast, in the absence of oxidative/nitrosative modification of UCH-L1, APP conceivably undergoes non-amyloidogenic processing and microtubules are stabilized, consistent with the notion that UCH-L1 in its normal state demonstrates neuroprotective properties [58–60,71–73].
3.2. Mechanisms of Protein Polyubiquitinylation
Ubiquitin is a small 76-amino acid protein that is critical in marking aggregated and/or damaged proteins for degradation via the UPS requires activation. This is accomplished by the enzyme, ubiquitin activating enzyme, designated at E1, which catalyzes the reactions:
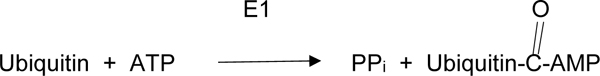 |
(1A) |
 |
(1B) |
In a second enzyme-catalyzed reaction, the enzyme, ubiquitin-carrier enzyme, E2, facilitates ubiquitin transfer to E2 from E1:
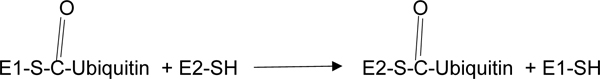 |
(2) |
In a final enzyme-catalyzed reaction, an enzyme, E3 ligase, which is unique for the protein being ubiquitinylated, binds the protein of interest to ubiquitin at lysine residue 63 or lysine residue 48, followed by a ligation of ubiquitin to the E3 ligase:protein adduct and separation of the E3 ligase:
| (3A) |
 |
(3B) |
These reactions repeat until a poly-ubiquitin chain exists on the protein, acting as a signal for the poly-ubiquitinylated protein to be subjected to the 26S proteasome for degradation as outlined above.
3.3. Oxidative and Nitrosative Modification of UCH-L1 in AD and MCI Brains and in Models Thereof.
Oxidative and/or nitrosative modifications of enzymes most often reduce or completely inhibit their activities [9]. Consequently, oxidative and/or nitrosative modification of UCH-L1 would lead to three predictions in the AD brain: a) there would be many poly-ubiquitinylated proteins present; b) because removal of ubiquitin residues from aggregated/damaged proteins is not efficiently facilitated by oxidized or nitrated UCH-L1, the 26S proteasome would develop lack of functionality; and c) because the 26S proteasomal activities would not be accessible as noted in b), there would be excess presence of aggregated, damaged proteins in AD brains. All three of these predictions are observed in AD brains [9,74–76]. The consequences of the inability of the 26S proteasome to degrade aggregated/damaged proteins in neurons and other brain cells are profound. Namely, the presence of such aggregates would slow or stop intracellular trafficking, inhibit intracellular movements of substrates, and potentially starve the cell for energy [65,66].
UCH-L1 was first identified to be oxidatively modified in autopsied brains (very short post-mortem interval, typically fewer than 4 h) from persons with sporadic AD compared to brains from control subjects [31] using redox proteomics, techniques pioneered in the author’s laboratory [44–47]. Later, Choi et al. using proteomics replicated our findings that UCH-L1 was oxidatively modified in AD brains [70]. Subsequently, our group identified UCH-L1 in brains of persons with inherited AD as oxidatively modified, suggesting that oxidative dysfunction of this protein, which normally is important in maintaining protein quality control in cells, is of fundamental importance with the pathogenesis of AD [69].
Elevated 3-NT is observed in AD and MCI brains [36,51,77,78]. Nitrosative modification of UCH-L1, indexed by elevated 3-NT, occurs in AD and MCI brains [45,56] as does another nitrosative modification, nitrosylation of key Cys residues in AD brains [68]. Lipton and co-workers demonstrated a tricomponent transnitrosylation network among UCH-L1 to cyclic-dependent kinase-5 to Drp1 in which each component could transnitrosylate the others in processes associated with aging and inflammation [67,68].
3.3.1. Model Systems and UCH-L1 and AD.
Recombinant UCH-L1 was treated with HNE with the result that decreased activity of the enzyme was found, presumably because components of the UCH-L1 catalytic triad of Cys, His, and asparagine were covalently by this highly reactive and neurotoxic alkenal [79]. Similarly, UCH-L1 treated with peroxynitrite led to tyrosine nitration of the hydrolase on specific residues, demonstrating UCH-L1 was subject to nitration reactions [80].
A study by Guglielmotto et al. [81] demonstrated that elevated Aβ42 led to increased levels of beta-site amyloid precursor protein cleaving enzyme 1 (BACE1), while levels of UCH-L1 were decreased. At the same time, NF-kB was activated, which would produce more inducible nitric oxide synthase. As noted above, Aβ oligomers are associated with increased oxidative stress, synaptic dysfunction, elevated intracellular Ca2+ and decreased glucose metabolism in AD brain [9,82,83], and following activation of NF-kB [84,85]. Consistent with the study by Guglielmotto at al. [81] but from the opposite direction, Wada and colleagues used stably expressed UCH-L1 in HEK cells to show that overexpression of UCH-L1 decreased BACE activity as demonstrated by decreased levels of C99 fragment of APP and, importantly, decreased Aβ levels [86]. These results are consistent with the notion that UCH-L1 leads to BACE degradation, and as such suggests upregulation of UCH-L1 potentially could be neuroprotective strategy in MCI and AD (see further discussion below).
Caenorhabditis elegans provide a relatively simple organism to study molecular biological aspects of aging and age-related disorders, including AD [87,88]. We showed that among the oxidized proteins identified by redox proteomics in C.elegans that expressed human Aβ42 was the worm equivalent of UCH-L1 [89]. Similarly, injection of Aβ42 in the forebrain of adult rats led to oxidative modification of hippocampal proteins, including UCH-L1 [90]. Moreover, addition of Aβ42 oligomers to gerbil synaptic membranes led to identification of UCH-L1 as oxidatively modified and dysfunctional [91]. These results are consistent with our laboratory’s overall hypothesis of AD that Aβ42 oligomer-induced oxidative damage, particularly HNE formation following lipid peroxidation, with subsequent modification to key molecular targets underlies synaptic loss and neuronal death in AD and MCI, including the UCH-L1-relevant proteostasis network, with consequent loss of cognitive function [9,25,82,91].
Wada and colleagues studied the gracile axonal dystrophic (gad) mice, which have sensory defects and increased motor ataxia with age associated with an in-frame deletion of exons on chromosome 5 comprising much of the active site of UCH-L1 [92]. gad mice have significantly elevated levels of APP and Aβ and deletion of the gracile tract [92]. Our group collaborated with Prof. Wada to perform redox proteomics on brains from these mice with inactive UCH-L1 [93]. Oxidized proteins were identified with functions ranging from antioxidant, glycolytic, cell signaling, and structural proteins, including neurofilament-L, recently identified as a biomarker for neuronal damage [94,95]. These results are consistent with the notion that in addition to its role in protein quality control mechanisms, UCH-L1 may influence or regulate many types of protein functions.
The results above showing AD-like elevations in APP and Aβ with the gad mouse are consistent with the idea that decreased levels of intact UCH-L1 are associated with damage to brain cells reminiscent of those in AD brain. Consonant with this idea, in AD brains there are found decreased levels of soluble UCH-L1 but elevated levels of UCH-L1 in NFT [96], which as mentioned at the beginning of this review are brain deposits composed of hyperphosphorylated tau protein and, along with amyloid plaques, a quintessential pathological hallmark of AD. Based on these results, Gong et al. [97] reasoned that elevation of soluble UCH-L1 might rescue cognitive loss in a mouse model of AD in which UCH-L1 were low as they are in AD subjects. The fusion product of UCH-L1 protein to the transduction domain of HIV-transactivator protein (TAT) was transduced to hippocampal slices treated with oligomeric Aβ42, a process that restored normal synaptic function. Similar studies with a human APP/PS-1 transgenic mouse model of AD demonstrated normal synaptic function and improved contextual memory in marked contrast to those characteristics in control APP/PS-1 mice [97].
Taken together, the results of these model studies suggest UCH-L1 levels in brain are inversely proportional to levels of enzymes involved in producing Aβ and NFT deposits, classic neuropathological hallmarks of AD. On the contrary, procedures that lead to elevation of UCH-L1 may be a promising therapeutic strategy for treatment of AD and MCI.
4.0. Conclusions and Future Studies
A protein such as UCH-L1 that comprises 2–5% of total brain protein, mostly localized to neurons, would be predicted to be critically important for efficient brain functions related to learning and memory. Indeed, this seems to be the case. Timely removal of aggregated, damaged proteins via the cytosolic-resident UPS system and autophagy, and the ER-resident unfolded protein response (UPR) is essential for maximum function of neurons [98]. In AD and MCI brains, evidence of dysfunction in autophagy and the UPR exist [16,99,100].
Figure 3 summarizes some of the key features of AD brain associated with oxidative modification of UCH-L1 and its decreased level of this soluble enzyme. For the UPS system, the 26S proteasome is known to be oxidized and dysfunctional in AD and MCI brains [6,47] as is UCH-L1, a key protein needed to facilitate proteolysis by the 20S proteasome components of originally aggregated and damaged proteins [31,47,67–70]. Neuronal survival depends in part on keeping the cell free of aggregated, oxidatively damaged proteins, and UCH-L1 is an important contributor to this function in normal brains but is oxidatively dysfunctional in AD and MCI brains.
Figure 3.
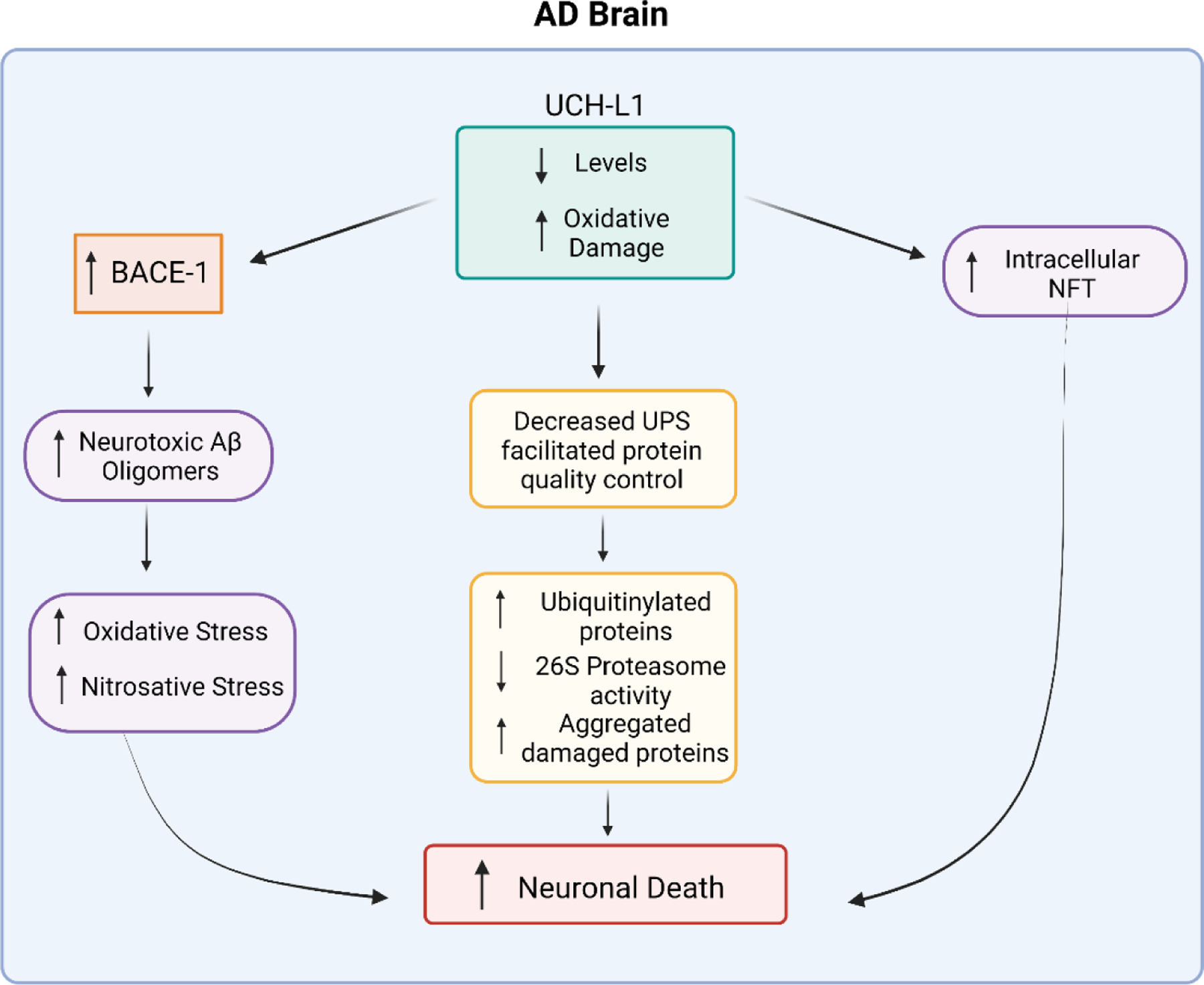
UCH-L1 is oxidatively modified and nitrosylated and of lower levels in AD brain compared to control brain. These characteristics of UCH-L1 in AD brain have the consequent effect of decreasing function of the Ubiquitin-Proteasomal System (UPS), which leads to accumulation of aggregated damaged proteins in the cell that are neurotoxic. Moreover, in AD brain, decreased levels of UCH-L1 are associated with elevated levels of BACE1, an enzyme that is involved in the processing of APP to eventually lead to neurotoxic Aβ peptide and subsequent oligomers. Aβ oligomers are associated with excess oxidative and nitrosative stress, causing neuronal death. In addition, in AD brain, UCH-L1 is associated with intracellular neurofibrillary tangles (NFT, composed of aggregates of hyperphosphorylated tau protein), possibly because of UCH-L1 normally associates with microtubules, the foundation of axonal microtubules, to stabilize them as does tau. NFT cause neuronal death. See text for more details.
As noted above, UCH-L1 has other functions in addition to facilitation of the UPS system. Namely, UCH-L1 is important for stabilization of axonal and dendritic microtubules that permits organelle (i.e., mitochondria; neurotransmitter vesicles, etc.) trafficking to and from the synapse from the neuronal cell body, and in synaptic protein remodeling, which is a key process associated with learning and memory [97]. Also, UCH-L1 is intimately associated with tau protein [61], the hyperphosphorylation of which leads to NFT. As also noted above, UCH-L1 normally plays a protective role in mitigating processing of APP by BACE1 [81]. In the absence of this UCH-L1-mediated protection, BACE1 is activated, which in AD brain is part of the mechanisms involved in producing highly neurotoxic Aβ. Small oligomeric Aβ peptide aggregates interact with synapses, leading to damage that results in loss of learning and memory [101–103]. Moreover, Aβ oligomer-associated lipid peroxidation and protein oxidation in neurons lead to glucose dysmetabolism with consequent loss of ATP that is needed for numerous neuronal functions, some of which are maintenance of neuronal membrane cell potential, prevention of Ca2+ overload, proper mitochondrial function, and, of course, execution of neurotransmission processes [9,16,17,24,25,100], all characteristics of AD. As indicated previously, in AD and MCI brains and models thereof, UCH-L1 levels are decreased while BACE1 levels are increased [61]. That is, lower levels of UCH-L1 reportedly lead to processes that themselves are involved in producing the two major pathological hallmarks of AD and MCI brains.
Consequently, UCH-L1 may be an attractive target in the arsenal of therapeutic strategies to slow progression of AD. Future research will determine if this prediction is confirmed.
Acknowledgements
This work was supported in part by a grant from National Institutes of Health [AG060056]. The author thanks his graduate student, Ms. Nicole Rummel, for assistance in preparation of the figures used in this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Markesbery WR Neuropathological criteria for the diagnosis of Alzheimer’s disease. Neurobiol Aging 1997. 18:S13–19. [DOI] [PubMed] [Google Scholar]
- [2].Markesbery WR Neuropathologic alterations in mild cognitive impairment: a review. J Alzheimers Dis 2010. 19: 221–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Petersen RC, Parisi JE, Dickson DW, Johnson KA, Knopman DS, Boeve BF, Jicha GA, Ivnik RJ, Smith GE, Tangalos EG, Braak H, Kokmen E Neuropathologic features of amnestic mild cognitive impairment. Arch Neurol 2006. 63:665–672. [DOI] [PubMed] [Google Scholar]
- [4].Uddin MS, Tewari D, Sharma G, Kabir MT, Barreto GE, Bin-Jumah MN, Perveen A, Abdel-Daim MM, Ashraf GM Molecular Mechanisms of ER Stress and UPR in the Pathogenesis of Alzheimer’s Disease. Mol Neurobiol 2020.57:2902–2919. [DOI] [PubMed] [Google Scholar]
- [5].Colacurcio DJ, Pensalfini A, Jiang Y, Nixon RA Dysfunction of autophagy and endosomal-lysosomal pathways: Roles in pathogenesis of Down syndrome and Alzheimer’s disease. Free Radic Biol Med 2018.114:40–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Keller JN, Hanni KB, Markesbery WR Impaired proteasome function in Alzheimer’s disease. J Neurochem 2000. 75:436–439. [DOI] [PubMed] [Google Scholar]
- [7].Di Domenico F, Tramutola A, Foppoli C, Head E, Perluigi M, Butterfield DA mTOR in Down syndrome: Role in Aß and tau neuropathology and transition to Alzheimer disease-like dementia. Free Radic Biol Med 2018.114:94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Butterfield DA, Stadtman ER Protein oxidation processes in aging brain, Adv. Cell Aging Gerontol 1997. 2:161–191. [Google Scholar]
- [9].Butterfield DA, Halliwell B Oxidative stress, glucose dysmetabolism and Alzheimer disease. Nature Rev Neurosci 2019. 20:148–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Halliwell B, Gutteridge JM Free Radicals in Biology & Medicine, 2015. Oxford University Press, 5th Ed., New York. [Google Scholar]
- [11].Butterfield DA, Yatin SM, Varadarajan S, Koppal T Amyloid β-peptide-induced oxidative pathways of cell death. Meth Enzymol 1999. 309:746–768. [DOI] [PubMed] [Google Scholar]
- [12].Ye YZ, Strong M, Huang ZQ, Beckman JS. Antibodies that recognize nitrotyrosine. Methods Enzymol 1996. 269:201–209. [DOI] [PubMed] [Google Scholar]
- [13].Calabrese V, Mancuso C, Calvani M, Rizzarelli E, Butterfield DA Giuffrida Stella AM. Nitric oxide in the nervous system: neuroprotection vs. neurotoxicity. Nature Rev Neurosci 2007. 8:766–775. [DOI] [PubMed] [Google Scholar]
- [14].Sultana R, Butterfield DA Slot blot analysis of 3-nitrotyrosine-modified brain proteins. Meth Enzymol 2008. 440:309–316. [DOI] [PubMed] [Google Scholar]
- [15].Keeney JT, Forster S, Sultana R, Brewer LD, Latimer CS, Cai J Klein JB, Porter NM, Butterfield DA Dietary vitamin D deficiency in rats from middle- to old-age leads to elevated tyrosine nitration and proteomics changes in levels of key proteins in brain: Implications for low vitamin D-dependent age-related cognitive decline. Free Radic Biol Med 2013. 65:324–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Perluigi M, Di Domenico F, Barone E, Butterfield DA mTOR in Alzheimer disease and its earlier stages: Links to oxidative damage in the progression of this dementing disorder. Free Radic Biol Med 2021. 169:382–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Butterfield DA, Lauderback CM Lipid peroxidation and protein oxidation in Alzheimer’s disease brain: potential causes and consequences involving amyloid β-peptide-associated free radical oxidative stress. Free Radical Biol Med 2002. 32:1050–1060. [DOI] [PubMed] [Google Scholar]
- [18].Higdon AN, Landar A, Barnes S, Darley-Usmar VM The electrophile responsive proteome: integrating proteomics and lipidomics with cellular function. Antioxid Redox Signal 2012. 17:1580–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Moreira PI, Sayre LM, Zhu X, Nunomura A, Smith MA, Perry G Detection and localization of markers of oxidative stress by in situ methods: application in the study of Alzheimer disease. Meth Mol Biol 2010. 610:419–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Mark RJ, Pang Z, Geddes JW, Uchida K, Mattson MP Amyloid beta-peptide impairs glucose transport in hippocampal and cortical neurons: involvement of membrane lipid peroxidation. J Neurosci 1997.17:1046–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Subramaniam R, Roediger F, Jordan B, Mattson MP, Keller JN, Waeg G, Butterfield DA The lipid peroxidation product, 4-hydroxy-2-trans-nonenal, alters the conformation of cortical synaptosomal membrane proteins. J Neurochem 1997. 69:1161–1169. [DOI] [PubMed] [Google Scholar]
- [22].Lauderback CM, Hackett JM, Huang FF, Keller JN, Szweda LI, Markesbery WR, Butterfield DA The glial glutamate transporter, GLT-1, is oxidatively modified by 4-hydroxy-2-nonenal in the Alzheimer’s disease brain: the role of Abeta1–42. J Neurochem 2001. 78:413–416. [DOI] [PubMed] [Google Scholar]
- [23].Sultana R, Perluigi M, Butterfield DA Lipid peroxidation triggers neurodegeneration: A redox proteomics view into the Alzheimer disease brain. Free Radic Biol Med 2013. 62:157–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Butterfield DA Brain lipid peroxidation and Alzheimer disease: Synergy between the Butterfield and Mattson laboratories. Ageing Res Rev 2020. 64:101049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Barone E, Di Domenico F, Perluigi M, Butterfield DA The interplay among oxidative stress, brain insulin resistance and AMPK dysfunction contribute to neurodegeneration in type 2 diabetes and Alzheimer disease. Free Radic Biol Med 2021. 176:16–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Jaganjac M, Milkovic L, Gegotek A, Cindric M, Zarkovic K, Skrzydlewska E, Zarkovic N The relevance of pathophysiological alterations in redox signaling of 4-hydroxynonenal for pharmacological therapies of major stress-associated diseases. Free Radic Biol Med 2020.157:128–153. [DOI] [PubMed] [Google Scholar]
- [27].Di Domenico F, Tramutola A, Butterfield DA Role of 4-hydroxy-2-nonenal (HNE) in the pathogenesis of Alzheimer disease and other selected age-related neurodegenerative disorders. Free Radic Biol Med 2017.111:253–261. [DOI] [PubMed] [Google Scholar]
- [28].Cioffi F, Ibrahim Adam RH, Bansal R, Broersen K A Review of Oxidative Stress Products and Related Genes in Early Alzheimer’s Disease. J Alzheimers Dis 2021. in press. doi: 10.3233/JAD-210497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Singh A, Kukreti R, Saso L, Kukreti S Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019. 24:1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Calabrese V, Mancuso C, Calvani M Rizzarelli E, Butterfield DA, Stella AM Nitric oxide in the central nervous system: neuroprotection versus neurotoxicity. Nat Rev Neurosci 2007. 8:766–775. [DOI] [PubMed] [Google Scholar]
- [31].Castegna A, Aksenov M, Aksenova M, Thongboonkerd V, Klein JB, Pierce WM, Booze R, Markesbery WR, Butterfield DA Proteomic identification of oxidatively modified proteins in Alzheimer’s disease brain. Part I: creatine kinase BB, glutamine synthase, and ubiquitin carboxy-terminal hydrolase L-1. Free Radic Biol Med 2002. 33:562–571. [DOI] [PubMed] [Google Scholar]
- [32].Castegna A, Aksenov M, Thongboonkerd V, Klein JB, Pierce WM, Booze R, Markesbery WR, Butterfield DA Proteomic identification of oxidatively modified proteins in Alzheimer’s disease brain. Part II: dihydropyrimidinase-related protein 2, alpha-enolase and heat shock cognate 71. J Neurochem 2002. 82:1524–1532. [DOI] [PubMed] [Google Scholar]
- [33].Beal MF Oxidatively modified proteins in aging and disease. Free Radic Biol Med 2002. 32:797–803. [DOI] [PubMed] [Google Scholar]
- [34].John A, Reddy PH Synaptic basis of Alzheimer’s disease: Focus on synaptic amyloid beta, P-tau and mitochondria. Ageing Res Rev 2021. 65:101208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Hensley K, Hall N, Subramaniam R, Cole P, Harris M, Aksenov M, Aksenova M, Gabbita SP, Wu JF, Carney JM, Lovell M, Markesbery WR, Butterfield DA Brain regional correspondence between Alzheimer’s disease histopathology and biomarkers of protein oxidation. J Neurochem 65:2146–2156. [DOI] [PubMed] [Google Scholar]
- [36].Castegna A, Thongboonkerd V, Klein JB, Lynn B, Markesbery WR, Butterfield DA Proteomic identification of nitrated proteins in Alzheimer’s disease brain. J Neurochem 2003. 85:1394–1401. [DOI] [PubMed] [Google Scholar]
- [37].Smith MA, Richey Harris PL, Sayre LM, Beckman JS, Perry G Widespread peroxynitrite-mediated damage in Alzheimer’s disease. J Neurosci 1997. 17:2653–2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Su JH, Deng G, Cotman CW Neuronal DNA damage precedes tangle formation and is associated with up-regulation of nitrotyrosine in Alzheimer’s disease brain. Brain Res 1997. 774:193–199. [DOI] [PubMed] [Google Scholar]
- [39].Numomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, Jones PK, Ghanbari H, Wataya T, Shimohama S, Chiba S, Atwood CS, Petersen RB, Smith MA Oxidative damage is the earliest event in Alzheimer disease. N Neuropathol Exp Neurol 2001. 60:759–767. [DOI] [PubMed] [Google Scholar]
- [40].Angelova Plamena R, P.R., Esteras N, Abramov AY. Mitochondria and lipid peroxidation in the mechanism of neurodegeneration: Finding ways for prevention. Med Res Rev 2021. 41:770–784. [DOI] [PubMed] [Google Scholar]
- [41].Zarkovic N 4-hydroxynonenal as a bioactive marker of pathophysiological processes. Mol Aspects Med 2003. 24: 281–291. [DOI] [PubMed] [Google Scholar]
- [42].Reddy PH, Oliver DM Amyloid beta and phosphorylated tau-induced defective autophagy and mitophagy in Alzheimer’s disease. Cells 2019. 8:488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Dalle-Donne I Scaloni A, Butterfield DA, Eds., Redox Proteomics: From Protein Modifications to Cellular Dysfunction and Diseases, 2006. Wiley Press, New York. [DOI] [PubMed] [Google Scholar]
- [44].Butterfield DA, Perluigi M, Reed T, Hughes MT, Hughes CP, Robinson RA, Sultana R Redox proteomics in selected neurodegenerative disorders: from its infancy to future applications. Antioxid Redox Signal 2012. 17:1610–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Sultana R, Boyd-Kimball D, Poon HF, Cai J, Pierce WM, Klein JB, Merchant M, Markesbery WR, Butterfield DA Redox proteomics identification of oxidized proteins in Alzheimer’s disease hippocampus and cerebellum: an approach to understand pathological and biochemical alterations in AD. Neurobiol Aging 2006. 27:1564–1576. [DOI] [PubMed] [Google Scholar]
- [46].Butterfield DA, Gu L, Di Domenico F, Robinson RA Mass spectrometry and redox proteomics: applications in disease. Mass Spectrom Rev 2014. 33:277–301. [DOI] [PubMed] [Google Scholar]
- [47].Butterfield DA, Boyd-Kimball D Redox proteomics and amyloid β-peptide: insights into Alzheimer disease. J Neurochem 2019. 151:459–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Dyer RR, Ford KI, Robinson RAS The roles of S-nitrosylation and S-glutathionylation in Alzheimer’s disease. Methods Enzymol 2019. 626:499–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Ren RJ, Dammer EB, Wang G, Seyfried NT, Levey AI Proteomics of protein post-translational modifications implicated in neurodegeneration. Transl Neurodegener 2014. 3:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Butterfield DA, Poon HF, St Clair D, Keller JN, Pierce WM, Klein JB, Markesbery WR Redox proteomics identification of oxidatively modified hippocampal proteins in mild cognitive impairment: insights into the development of Alzheimer’s disease. Neurobiol Dis 2006. 22:223–232. [DOI] [PubMed] [Google Scholar]
- [51].Butterfield DA, Reed T, Sultana R Roles of 3-nitrotyrosine- and 4-hydroxynonenal-modified brain proteins in the progression and pathogenesis of Alzheimer’s disease. Free Radic Res 2011. 45:59–72. [DOI] [PubMed] [Google Scholar]
- [52].Reed TT, Pierce WM, Markesbery WR, Butterfield DA Proteomic identification of HNE-bound proteins in early Alzheimer disease: Insights into the role of lipid peroxidation in the progression of AD. Brain Res 2009. 1274:66–76. [DOI] [PubMed] [Google Scholar]
- [53].Swomley AM, Förster S, Keeney JT, Triplett J, Zhang Z, Sultana R, Butterfield DA Abeta, oxidative stress in Alzheimer disease: evidence based on proteomics studies. Biochim Biophys Acta 2014. 1842:1248–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Reed T, Perluigi M, Sultana R, Pierce WM, Klein JB, Turner DM, Coccia R, Markesbery WR, Butterfield DA Redox proteomic identification of 4-hydroxy-2-nonenal-modified brain proteins in amnestic mild cognitive impairment: insight into the role of lipid peroxidation in the progression and pathogenesis of Alzheimer’s disease. Neurobiol Dis 2008. 30:107–120. [DOI] [PubMed] [Google Scholar]
- [55].Butterfield DA, Reed T, Newman SF, Sultana R Roles of amyloid beta-peptide-associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer’s disease and mild cognitive impairment. Free Radic Biol Med 2007. 43:658–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Sultana R, Reed T, Perluigi M, Coccia R, Pierce WM, Butterfield DA Proteomic identification of nitrated brain proteins in amnestic mild cognitive impairment: a regional study. J Cell Mol Med 2007. 11:839–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Swomley AM, Butterfield DA Oxidative stress in Alzheimer disease and mild cognitive impairment: evidence from human data provided by redox proteomics. Arch Toxicol 2015. 89:1669–1680. [DOI] [PubMed] [Google Scholar]
- [58].Bishop P, Rocca D, Henley JM Ubiquitin C-terminal hydrolase L1 (UCH-L1): structure, distribution, and roles in brain function and dysfunction. Biochem J 2016. 473:2453–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Setsuie R, Wada K The functions of UCH-L1 and its relation to neurodegenerative diseases. Neurochem Int 2007. 51:105–111. [DOI] [PubMed] [Google Scholar]
- [60].Gong B, Leznik E The role of ubiquitin C-terminal hydrolase L1 in neurodegenerative disorders. Drug News Perspect 2007. 20:365–370. [DOI] [PubMed] [Google Scholar]
- [61].Zhao Z-B, Wu L, Xiong R, Wang L-L, Zhang B, Wang C, Li H, Liang L, Chen S-D MicroRNA-922 promotes tau phosphorylation by downregulating ubiquitin carboxy-terminal hydrolase L1 (UCHL1) expression in the pathogenesis of Alzheimer’s disease. Neuroscience 2014. 275:232–237. [DOI] [PubMed] [Google Scholar]
- [62].Sulkowska JI, Rawdon EJ, Millett KC, Onuchic JN and Stasiak A Conservation of complex knotting and slipknotting patterns in proteins. Proc. Natl. Acad. Sci. U.S.A 2012. 109:E1715–E1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Das C, Hoang QQ, Kreinbring CA, Luchansky SJ, Meray RK, Ray SS, Lansbury PT, Ringe D and Petsko GA Structural basis for conformational plasticity of the Parkinson’s disease-associated ubiquitin hydrolase UCH-L1. Proc. Natl. Acad. Sci. U.S.A 2006. 103:4675–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Virnau P, Mirny LA and Kardar M Intricate knots in proteins: Function and evolution. PLoS Comput. Biol 2006. 2:e122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Poppek D, Grune T Proteasomal defense of oxidative protein modifications. Antioxid Redox Signal 2006. 8:173–184. [DOI] [PubMed] [Google Scholar]
- [66].Ding Q, Dimayuga E, Keller JN Proteasome regulation of oxidative stress in aging and age-related diseases of the CNS. Antioxid Redox Signal 2006. 8:173–184. [DOI] [PubMed] [Google Scholar]
- [67].Nakamura T, Oh C-K, Zhang X, Lipton SA Protein S-nitrosylation and oxidation contribute to protein misfolding in neurodegeneration. Free Radic Biol Med 2021. 172:562–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Nakamura T, Oh C-K, Zhang X, Tannenbaum SR, Lipton SA Protein Transnitrosylation Signaling Networks Contribute to Inflammaging and Neurodegenerative Disorders. Antioxid Redox Signal 2021. 35:531–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Butterfield DA, Gnjec A, Poon HF, Castegna A, Pierce WM, Klein JB, Martins RN Redox proteomics identification of oxidatively modified brain proteins in inherited Alzheimer’s disease: an initial assessment. J Alzheimers Dis 2006. 10:391–397. [DOI] [PubMed] [Google Scholar]
- [70].Choi J, Levey AI, Weintraub ST, Rees HD, Gearing M, Chin LS, Li L Oxidative modifications and down-regulation of ubiquitin carboxyl-terminal hydrolase L1 associated with idiopathic Parkinson’s and Alzheimer’s diseases. J Biol Chem 2004. 279:13256–13264. [DOI] [PubMed] [Google Scholar]
- [71].Zhang M, Cai F, Zhang S, Zhang S, Song W Overexpression of ubiquitin carboxyl-terminal hydrolase L1 (UCHL1) delays Alzheimer’s progression in vivo. Sci Rep 2014. 4:7298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Xie M, Han Y, Yu Q, Wang X, Wang S, Liao X UCH-L1 inhibition decreases the microtubule binding function of Tau protein. J Alzheimers Dis 2016. 49:353–363. [DOI] [PubMed] [Google Scholar]
- [73].Corsetti V, Florenzano F, Atlante A, Bobba A, Ciotti MT, Natale F, Della Valle F, Borreca A, Manca A, Meli G, Ferraina C, Feligioni M, D’Aguanno S, Bussani R, Ammassari-Teule M, M., Nicolin V, Calissano P, Amadoro G NH2-truncated human tau induces deregulated mitophagy in neurons by aberrant recruitment of Parkin and UCHL-1: implications in Alzheimer’s disease. Hum Mol Genet 2015. 24:3058–3081. [DOI] [PubMed] [Google Scholar]
- [74].Tramutola A, Di Domenico F, Barone E, Arena A, Giorgi A, di Francesco L, Schininà ME, Coccia R, Head E, Butterfield DA, Perluigi M Polyubiquitinylation Profile in Down Syndrome Brain Before and After the Development of Alzheimer Neuropathology. Antioxid Redox Signal 2017. 26:280–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Tramutola A, Triani F, Di Domenico F, Barone E, Cai J, Klein JB, Perluigi M, Butterfield DA Poly-ubiquitin profile in Alzheimer disease brain. Neurobiol Dis 2018. 118:129–141. [DOI] [PubMed] [Google Scholar]
- [76].Tramutola A, Perluigi M Polyubiquitin profile in Down syndrome and Alzheimer’s disease brain. Methods Mol Biol 2021. 2261:79–91. [DOI] [PubMed] [Google Scholar]
- [77].Keller JN, Schmitt FA, Scheff SW, Ding Q, Chen Q, Butterfield DA, Markesbery WR Evidence of increased oxidative damage in subjects with mild cognitive impairment. Neurology 2005. 64:1152–1156. [DOI] [PubMed] [Google Scholar]
- [78].Butterfield DA, Reed T, Perluigi M, De Marco C, Coccia R, Keller JN, Markesbery WR, Sultana R Elevated levels of 3-nitrotyrosine in brain from subjects with amnestic mild cognitive impairment: implications for the role of nitration in the progression of Alzheimer’s disease. Brain Res 2007. 114:243–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Nishikawa K, Li H, Kawamura R, Osaka H, Wang YL, Hara Y, Hirokawa T, Manago Y, Amano T, Noda M, Aoki S, Wada K Alterations of structure and hydrolase activity of parkinsonism-associated human ubiquitin carboxyl-terminal hydrolase L1 variants. Biochem. Biophys. Res. Commun 2003. 304, 176–183. [DOI] [PubMed] [Google Scholar]
- [80].Guingab-Cagmat J, Stevens SM Jr., Ratliff MV, Zhang Z, Gold MS, Anagli J, Wang KKW Wang, Kobeissy, F.H. Identification of tyrosine nitration in UCH-L1 and GAPDH. Electrophoresis 2011. 32:1692–1705. [DOI] [PubMed] [Google Scholar]
- [81].Guglielmotto M, Monteleone D, Boido M, Piras A, Giliberto L, Borghi R, Vercelli A, Fornaro M, Tabaton M, Tamagno E Aβ1–42-mediated down-regulation of Uch-L1 is dependent on NF-κB activation and impaired BACE1 lysosomal degradation. Aging Cell 2012. 11:834–844. [DOI] [PubMed] [Google Scholar]
- [82].Butterfield DA, Drake J, Pocernich C, Castegna A Evidence of oxidative damage in Alzheimer’s disease brain: central role of amyloid β-peptide. Trends Mol Med 2001. 7:548–554. [DOI] [PubMed] [Google Scholar]
- [83].Bezprozvanny I, Mattson MP Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci 2008. 31:454–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Keeney JT, Butterfield DA Vitamin D deficiency and Alzheimer disease: common links. Neurobiol Dis 2015. 84:84–98. [DOI] [PubMed] [Google Scholar]
- [85].Keeney JTR, Förster S, Sultana R, Brewer LD, Latimer CS, Cai J, Klein JB, Porter NM, Butterfield DA Dietary vitamin D deficiency in rats from middle to old age leads to elevated tyrosine nitration and proteomics changes in levels of key proteins in brain: implications for low vitamin D-dependent age-related cognitive decline. Free Radic Biol Med 2013. 65:324–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Zhang M, Deng Y, Zhang S, Zou H, Cai F, Wada K Control of BACE1 degradation and APP processing by ubiquitin carboxyl-terminal hydrolase L1. J Neurochem 2012. 120:1129–1138. [DOI] [PubMed] [Google Scholar]
- [87].Yatin SM, Link CD, Butterfield DA In vitro and in vivo oxidative stress associated with Alzheimer’s amyloid β-peptide (1–42). Neurobiol Aging 1999. 20:325–330. [DOI] [PubMed] [Google Scholar]
- [88].Drake J, Link CD, Butterfield DA Oxidative stress precedes fibrillar deposition of Alzheimer’s disease amyloid β-peptide (1–42) in a transgenic Caenorhabditis elegans model. Neurobiol Aging 2003. 2:415–420. [DOI] [PubMed] [Google Scholar]
- [89].Boyd-Kimball D, Poon HF, Lynn BC, Cai J, Pierce WM Jr, Klein JB, Ferguson J, Link CD, Butterfield DA Proteomic identification of proteins specifically oxidized in Caenorhabditis elegans expressing human Abeta(1–42): implications for Alzheimer’s disease. Neurobiol Aging 2006. 27:1239–1249. [DOI] [PubMed] [Google Scholar]
- [90].Boyd-Kimball D, Sultana R, Poon HF, Lynn BC, Casamenti F, Pepeu G, Klein JB, Butterfield DA Proteomic identification of proteins specifically oxidized by intracerebral injection of amyloid beta-peptide (1–42) into rat brain: implications for Alzheimer’s disease. Neuroscience 2005. 132:313–324. [DOI] [PubMed] [Google Scholar]
- [91].Rummel NG, Butterfield DA Altered Metabolism in Alzheimer Disease Brain: Role of Oxidative Stress. Antioxid Redox Signal 2021. in press. doi: 10.1089/ars.2021.0177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Saigoh K, Wang YL, Suh JG, Yamanishi T, Sakai Y, Kiyosawa H, Harada T, Ichihara N, Wakana S, Kikuchi T, Wada K Intragenic deletion in the gene encoding ubiquitin carboxy-terminal hydrolase in gad mice. Nature Genet 1999. 23:47–51. [DOI] [PubMed] [Google Scholar]
- [93].Castegna A, Thongboonkerd V, Klein J, Lynn BC, Wang YL, Osaka H, Wada K, Butterfield DA Proteomic analysis of brain proteins in the gracile axonal dystrophy (gad) mouse, a syndrome that emanates from dysfunctional ubiquitin carboxyl-terminal hydrolase L-1, reveals oxidation of key proteins. J Neurochem 2004. 88:1540–1546. [DOI] [PubMed] [Google Scholar]
- [94].Gaetani L, Blennow K, Calabresi P, Di Filippo M, Lucilla Parnetti L, Zetterberg H Neurofilament light chain as a biomarker in neurological disorders. J Neurol Neurosurg Psychiatry 2019. 90:870–881. [DOI] [PubMed] [Google Scholar]
- [95].Ashton NJ, Janelidze S, Al Khleifat A, Leuzy A, van der Ende EL, Karikari TK, Benedet AL, Pascoal TA, Lleó A, Parnetti L, Galimberti D, Bonanni L, Pilotto A, Padovani A, Lycke J, Novakova L, Axelsson M, Velayudhan L, Rabinovici GD, Miller B, Pariante C, Nikkheslat N, Resnick SM, Thambisetty M, Schöll M, Fernández-Eulate G, Gil-Bea FJ, López de Munain A, Al-Chalabi A, Rosa-Neto P, Strydom A, Svenningsson P, Stomrud E, Santillo A, Aarsland D, van Swieten JC, Palmqvist S, Zetterberg H, Blennow K, Hye A, Hansson O A multicentre validation study of the diagnostic value of plasma neurofilament light. Nature Commun 2021. 12:3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Lowe J, McDermott H, Landon M, Mayer RJ, Wilkinson KD Ubiquitin carboxyl-terminal hydrolase (PGP9.5) is selectively present in ubiquitinated inclusion bodies characteristic of human neurodegenerative diseases. J. Pathol 1990. 161:153–160. [DOI] [PubMed] [Google Scholar]
- [97].Gong B, Cao Z, Zheng P, Vitolo OV, Liu S, Staniszewski A, Moolman D, Zhang H, Shelanski M, Arancio O 2006. Ubiquitin hydrolase Uch-L1 rescues beta-amyloid-induced decreases in synaptic function and contextual memory. Cell 126, 775–788. [DOI] [PubMed] [Google Scholar]
- [98].Pomatto LCD, Sun PY, Davies KJA To adapt or not to adapt: Consequences of declining Adaptive Homeostasis and Proteostasis with age. Mech Ageing Dev 2019. 177:80–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Colacurcio DJ, Pensalfini A, Jiang Y, Nixon RA Dysfunction of autophagy and endosomal-lysosomal pathways: Roles in pathogenesis of Down syndrome and Alzheimer’s Disease. Free Radic Biol Med 2018. 114:40–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Di Domenico F, Barone E, Perluigi M, Butterfield DA The triangle of death in Alzheimer’s disease brain: the aberrant cross-talk among energy metabolism, mammalian target of rapamycin signaling, and protein homeostasis revealed by redox proteomics. Antioxid Redox Signal 2017. 26:364–387. [DOI] [PubMed] [Google Scholar]
- [101].Selkoe DJ Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav Brain Res 2008. 192:106–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Yang T, Li S, Xu H, Walsh DM, Selkoe DJ Large soluble oligomers of amyloid beta-protein from Alzheimer brain are far less neuroactive than the smaller oligomers to which they dissociate. J Neurosci 2017. 37:152–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Li S, Selkoe, D.J. A mechanistic hypothesis for the impairment of synaptic plasticity by soluble Abeta oligomers from Alzheimer’s brain. J Neurochem 2020. 154:583–597. [DOI] [PMC free article] [PubMed] [Google Scholar]