

Abstract
A functional approach to generate tumor-targeting human monoclonal antibodies is through selection of phage antibody display libraries directly on tumor cells. Although technically convenient, the use of cancer cell lines for selection has limitations as those cell lines often undergo genetic and epigenetic changes during prolonged in vitro culture and alter their cell surface antigen expression profile. The key is to develop a technology that allows selection of phage antibody display libraries on tumor cells in situ residing in their natural tissue microenvironment. Laser capture microdissection (LCM) permits the precise procurement of tumor cells from human cancer patient tissue sections. Here we describe a LCM-based method for selecting phage antibodies against tumor cells in situ using both fresh frozen and paraffin-embedded tissues. To restrict the selection to antibodies that bind internalizing epitopes, the method utilizes a polyclonal phage population pre-enriched for internalizing phage antibodies. The ability to recognize tumor cells in situ residing in their natural tissue microenvironment and to deliver payload intracellularly makes these LCM-selected antibodies attractive candidates for the development of targeted cancer therapeutics.
Keywords: Laser capture microdissection, phage antibody library, internalizing human monoclonal antibody, macropinocytosis, solid tumor, natural cell surface epitope, tissue microenvironment, human cancer specimen, targeted therapy, intracellular payload delivery
Introduction:
Tumor cell surface antigens are excellent targets for antibody-based therapy development. Identification of novel tumor-specific or tumor-associated cell surface antigens can result in significant improvements in detection and treatment of malignant tumors. The antigenic epitope space at the tumor cell surface is highly complex and consists of extensive post-translational modifications [1–5]. Monoclonal antibodies (mAbs) can recognize with high affinity and specificity a wide range of antigenic determinants and discern subtle differences in antigen structure and conformation, which can be used to effectively map the tumor cell surface epitope space independent of gene expression analysis [6–14]. Phage antibody display technology has been widely used to develop novel human monoclonal antibodies [15–26]. Phage antibody display libraries serve as a source of random shape repertoire that can be used to probe neoplastic alterations on cancer cell surface [6–8]. Selecting phage antibody libraries directly on cancer cell lines has enabled the identification of tumor-targeting antibodies without prior knowledge of target antigens [6, 7, 13, 27]. However, when tumor cells are removed from their natural environment, they undergo genetic and epigenetic changes yielding different surface antigens than those seen in actual cases of cancer. Laser capture microdissection (LCM) under direct microscopic visualization has allowed small clusters of tumor cells to be isolated and removed from heterogeneous human tissue sections [28–34]. This technology, when combined with either phage peptide display [35–37] or phage antibody display [7, 38], permits the selection of phage binding specifically to tumor cells in their native tissue environment. We have previously developed the LCM-based phage antibody display library selection technique and used it to select non-immune human antibody phage display libraries on cancer patient tissues to identify novel human antibodies targeting clinically relevant tumor epitopes [7]. To further identify internalizing antibodies for tumor-specific intracellular payload delivery, we generated sublibraries enriched for internalizing phage antibodies as input for LCM-based selection [7]. We hereby describe this LCM selection protocol for the identification and characterization of tumor-specific internalizing phage antibodies (Figure 1). Detailed methods are provided for phage sublibrary construction, antibody selection and identification on tissue specimen using LCM, validation of internalization with single-chain variable fragment antibodies (scFvs), and tumor-targeted intracellular payload delivery.
Figure 1.
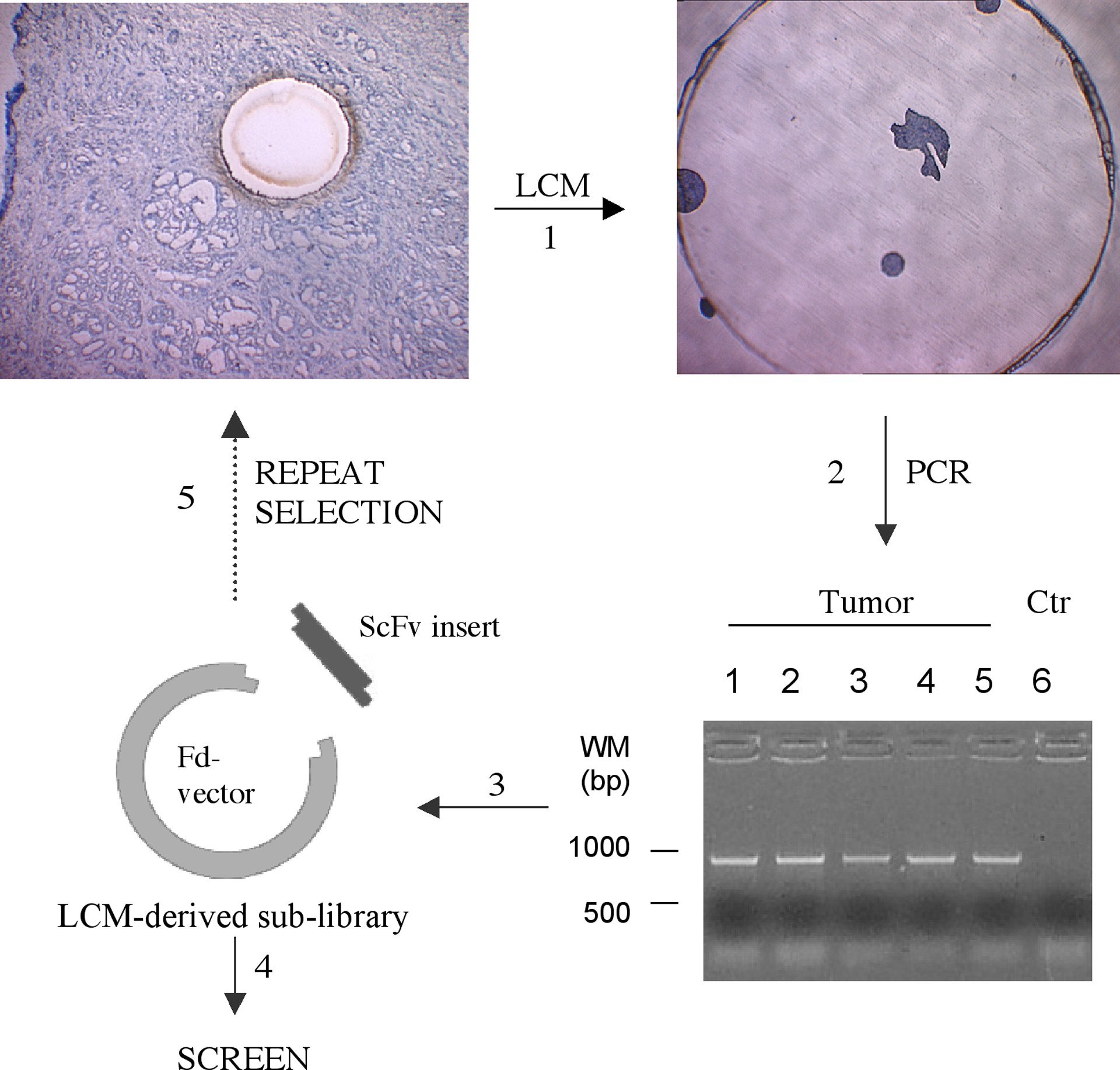
Outline of the LCM-based method that allows selection of phage antibody display library on cancer patient specimen to identify novel antibodies targeting tumor cells in situ. Non-immune phage antibody display libraries were counter-selected on a panel of normal cells to remove binders to normal cell surface molecules, then selected on cancer cell lines and/or primary tumor cells to enrich for tumor specificity. When the selection is performed under internalizing conditions, a sublibrary enriched for tumor-specific internalizing antibodies is created and used as input for LCM-based selection on patient tissue specimen. Following incubation with slides containing sectioned tumor tissues, a small cluster of tumor cells along with tumor-bound phages were procured by LCM and collected on the cap of a PCR tube (step 1). Genes of scFv-coding regions were amplified by PCR (step 2) and spliced into a phage display vector to create secondary libraries (step 3) that were used for screening (step 4) or additional rounds of LCM-based selection (step 5). Ctr, control; MW, molecular weight (Adopted from a figure that was originally published in reference [7] in Mol. Cell Proteomics).
2. Materials
2.1. Sublibrary construction
Cell growth medium: DMEM supplemented with 10% fetal bovine serum and 100 μg/ml penicillin-streptomycin.
Phosphate-buffered saline (PBS), pH 7.4.
0.25% trypsin/EDTA.
Glycine wash buffer: 100 mM glycine, pH 2.8, 150 mM NaCl.
Phage elution buffer: 100 mM trimethylamine (TEA).
Neutralizing buffer: 1 M Tris-HCl, pH 6.8.
Escherichia coli TG1 (Lucigen, Middleton, WI, USA).
2xYT (1L): 16 g tryptone, 10 g yeast extract, 5 g NaCl.
Bacteria/phage growth media: 2xYT, 50ug/ml tetracycline.
Bacteria/phage plate: YT, 15 μg/ml tetracycline.
PEG8000/NaCl solution (5X): 20% (w/v) polyethylene glycol 8000, 2.5 M NaCl in PBS.
0.45 μm sterile syringe filter (Corning, New York, USA).
Phage or bacteria storage buffer: 25% glycerol (v/v) in PBS, pH 7.4.
2.2. Selection of phage antibodies targeting tumor cells in situ by LCM
Leica Membrane Slides (MicroDissect, Mittenaar, Germany).
Fixation buffer 1: acetone
Fixation buffer 2: 4% paraformaldehyde (PFA)
Blocking buffer: 3% H2O2 in PBS.
Hematoxylin (H-3401, Vector Laboratories, Burlingame, CA, USA)
Microcentrifuge PCR tubes (BioExpress, Kaysville, UT, USA)
Leica AS LMD (Leica Microsystems GmbH, Wetzlar, Germany)
2.3. PCR recovery of scFv genes from LCM-procured tissue pieces
PCR kit (Lucigen)
-
Primers for recovery and transfer of scFv gene to fd phage display vector:
Fd2 (TTTTTGGAGATTTTCAAC)
Fdseq (GAATTTTCTGTATGAGG)
QIAquick PCR purification kit (Qiagen, Germantown, MD, USA)
QIAquick gel extraction kit (Qiagen)
10x digest buffer (New England Biolabs, Ipswich, MA, USA)
SfiI restriction enzyme (New England Biolabs)
NotI restriction enzyme (New England Biolabs)
10x T4 DNA ligation buffer (New England Biolabs)
T4 DNA ligase (New England Biolabs)
Chemically competent TG1 (Lucigen)
BstNI restriction enzyme (New England Biolabs)
Spectrophotometer (NanoDrop from ThermoScientific, Waltham, MA, USA)
2.4. FACS analysis of selection output
96-well polystyrene round-bottom plates (Corning)
96-well polypropylene v-bottom plates (Corning)
Binding buffer: PBS, pH 7.4, 1% BSA
Biotinylated anti-M13 antibody (Sigma, St. Louis, MO, USA)
Streptavidin-phycoerythrin (Invitrogen, Carlsbad, CA, USA)
Flow cytometer (Accuri™ C6 from BD Biosciences, San Jose, CA, USA)
2.5. ScFv construction, production, and biotin labeling
pSyn1 scFv expression vector
NcoI restriction enzyme (New England Biolabs)
NotI restriction enzyme (New England Biolabs)
Bacteria/scFv-pSyn1 plate: YT, 50 μg/ml ampicillin
Overnight medium: 2xYT, 2% (w/v) glucose, 50 μg/ml ampicillin
Bacterial growth medium: 2xYT, 0.1% (w/v) glucose, 100 mM ampicillin
Induction solution: 1 mM IPTG (1:1000 diluted from 1 M stock)
Periplasmic prep buffer (PPB): 200 mg/ml sucrose, 1 mM EDTA, 30 mM Tris- HCl, pH 8.0, filter-sterilized
Osmotic shock buffer: 5 mM MgSO4
Ni-NTA agarose resin (Qiagen)
Wash buffer: PBS containing 20 mM imidazole
Elution buffer: PBS containing 250 mM imidazole
High-speed centrifuge tubes (Nalgene)
Zeba™ spin desalting columns (#87766, ThermoFisher)
EZ-Link Sulfo-NHS-LC-Biotin (#21327, ThermoFisher)
1 M Tris-HCl, pH 8.0
Ultra-15 concentrator tube (Millipore, Billerica, MA, USA)
2.6. Analysis of selection output by immunohistochemistry
Unmasking solution (Vector Laboratories)
Blocking buffer: 2% (v/v) goat serum in PBS, pH7.4
Avidin/biotin blocking kit (Vector Laboratories)
R.T.U ABC REAGENT (Vector Laboratories)
DAB solution (ThermoFisher)
Acid rinse solution: 2% (v/v) glacial acetic acid in ddH2O
Bluing solution: 1.5 ml NH4OH (30% stock) with 98.5 ml of 70% ethanol
VectaMount™ Mounting Medium (H-5000, Vector Laboratories)
Keyence BZ-9000 digital microscope (Keyence-America, Itasca, IL, USA)
2.7. Microscopic detection of antibody internalization
Lab-Tek II glass chambers (ThermoFisher)
Permeabilization buffer: PBS, 1% BSA, 0.1% TritonX-100
Streptavidin-Cy3 (1 mg/ml) (Jackson immunoresearch, West Grove, PA, USA)
Fix/counterstain buffer: 4% PFA in PBS, 1:10,000 Hoechst
Confocal microscope (Fluoview FV10i from Olympus-America, San Jose, CA, USA)
2.8. Immunotoxin delivery (Functional internalization)
96-well tissue culture-treated flat-bottom plates (Corning)
Streptavidin-conjugated saporin (SA-ZAP, Advanced Targeting Systems, San Diego, CA, USA)
CCK-8 solution: (Dojindo, 10 μl CCK-8 solution mixed with 90 μl PBS just before use)
Plate reader (Synergy HT from Biotek, Winooski, VT, USA).
3. Methods
A summary of the experimental workflow of the protocols described below is as follows: First, a phage antibody display sublibrary enriched for antibodies that bind tumor cell-specific internalizing cell surface epitopes is generated (described in section 3.1). This sublibrary is created by first depleting a naive phage antibody display library against a mixed panel of normal cell lines, followed by selection for binding to a mixed panel of tumor cell lines under internalizing conditions. Next, this sublibrary is selected on cancer patient tissue sections (frozen or paraffin-embedded) and the tumor cells and associated binding phage antibodies are precisely excised by LCM (described in section 3.2). The tumor-binding scFv sequences are recovered from the LCM-procured tumor cells by PCR and re-cloned into the phage display vector (described in section 3.3). The LCM-selected phage antibodies are then screened for binding to tumor and normal cell lines by FACS and the tumor-specific binding clones are sequenced (described in section 3.4). The tumor-specific antibodies are then cloned into a scFv expression vector, produced as scFvs, and biotinylated (described in section 3.5). Finally, the biotinylated scFvs are used for immunohistochemistry analysis of tumor-specific binding to cancer patient tissue samples (described in section 3.6), microscopic analysis of tumor-specific internalization using tumor and normal cell lines (described in section 3.7), and tumor-specific intracellular delivery of immunotoxins (described in section 3.8).
3.1. Generation of phage antibody sublibrary enriched for internalizing antibody binding to tumor cell surface epitopes
To prepare cells for counter-selection, culture normal human fibroblasts, non-cancerous epithelial lines RWPE-1, BPH-1, MCF10A, and human mammary epithelial cells (HMEC) in three 10 cm diameter round cell culture-treated plates each in cell growth medium to approximately 80% confluence (see Note 1).
Remove the cell growth medium and wash cells once with 5 ml PBS. Add enough trypsin/EDTA solution to cover the cells and incubate at 37 °C, roll the flask gently to detach the cells from the flask. Add an equal volume of cell growth medium into the flask, pipette to mix and transfer the cells to centrifuge tubes.
Spin down and wash the cells three times with cell growth medium by centrifugation at 500g for 5 min.
Pool the cells (~108 total cells) by resuspending in 3 ml ice-cold cell growth medium, add 1 ml 1012 phage particles in PBS to the cells and incubate at 4°C for 4 h, with rotation.
Spin down the cells by centrifugation at 500g for 5 min. Carefully transfer the supernatant containing phage antibodies to a new tube. Spin again at 6,000g for 5 min and filter the supernatant with a 0.45 μm sterile syringe filter.
Prepare 106 target tumor cells (e.g., a panel of prostate cancer cell lines, PC3 and Du-145) in 1 ml cell growth medium as described in steps 1–4 (one 10 cm dish for each cell line will be sufficient). Add phage particles from step 5 to the target tumor cells and incubate in a humidified atmosphere of 95% air and 5% CO2 at 37 °C for 2 h (see Note 2).
Spin down and wash cells twice with 5 ml 100 mM glycine (pH 2.8) in the presence of 150 mM NaCl, followed with washing once with 10 ml PBS, pH 7.0 (see Note 3).
Lyse the cells by adding 0.5 ml fresh 100 mM trimethylamine (TEA) to the cell pellet. Mix with pipette and rotate at room temperature for 5 min.
Add 250 μl Tris-HCl, pH 6.8 to neutralize the cell lysate. Mix gently.
Add total volume of neutralized cell lysate to 10 ml of exponentially growing TG1 (OD600 = 0.7), mix, and incubate without shaking at 37 °C for 30 min.
Titer the phage by making a 10-fold dilution of the culture in 2xYT/tet and plate 100 μl of each dilution (10 μl, 1μl, 0.1μl of original culture) on YT/tet plates.
Spin down the remaining bacterial culture at 4,000g for 15 min, resuspend in 0.5 ml of 2xYT/tet, plate on a 150 mm YT/tet plate, and incubate overnight at 37°C.
The next day, add 3–5 ml of 2xYT/tet to the plate, scrape the bacteria and mix with the 2xYT/tet. Add glycerol to a final concentration of 25% (v/v), aliquot, and store at −80 °C.
To prepare the phage sublibrary for the next selection step, inoculate 100 ml 2xYT with 0.1% of the panning output from step 13, culture with shaking (250 rpm) at 37 °C overnight (see Note 4).
Centrifuge the bacteria culture at 6,000g for 20 min at 4 °C.
Collect and transfer the supernatant to a new centrifuge tube, add 30 ml 20% PEG 8000/NaCl solution, mix and incubate on ice for 2 h. Phage precipitation should be visible as the supernatant should become cloudy.
Centrifuge the precipitated phage at 6,000g for 20 min at 4 °C. Remove as much supernatant as possible. Resuspend the phage sediment in 30 ml PBS and transfer to a new tube.
Add 10 ml 20% PEG 8000/NaCl solution to resupended phage, mix, and incubate on ice for 1 h.
Centrifuge the precipitated phage at 6,000g for 20 min at 4 °C. Remove as much supernatant as possible. Centrifuge at 6,000g again for 5 min at 4 °C and remove the residual supernatant.
Add 5 ml PBS to resuspend the phage. Transfer the supernatant to new tubes and centrifuge at 10,000g for 10 min at 4 °C.
Filter the supernatant with a 0.45 μm sterile syringe filter and store the phage for further selection (see Note 5).
3.2. Selection of antibodies targeting tumor cells in situ by LCM
Selections were performed on both frozen and paraffin-embedded prostate cancer tissues.
For selection on frozen tissue slides:
-
1
Cut cryostat sections of prostate cancer specimens at 5 μm and mount on Leica Membrane Slides. Fix with ice-cold acetone at −20°C for 20 min (see Note 6).
-
2
Air-dry the sections using a fan for 30 minutes at room temperature to prevent sections from falling off the slides during phage antibody incubations.
-
3
Add ice-cold 4% PFA to each tissue section, fix at 4 °C for 10 min.
-
4
Add one drop of Hematoxylin to the slide directly and counterstain for 3 min. Rinse slide with running tap water until rinse water is colorless.
-
5
Tap slides into PBS buffer, then incubate with the phage sublibrary (0.5 ml of 5 × 1011 colony forming unit (c.f.u.)/ml stock) from section 3.1, step 21 at room temperature for 1 h.
-
6
Proceed to step 12.
For the selection on paraffin-embedded tissue slides:
-
7
Deparaffinize paraffin tissue slides by immersing slides in xylene overnight.
-
8
Next day, immerse slide in a new xylene solution for 10 min.
-
9
Rehydrate the slides by sequential incubation in 100%, 95%, and 70% ethanol, followed by ddH2O for 5 min each. Wash the slides twice with PBS for 5 min each.
-
10
Incubate slides with blocking solution at room temperature for 1 h. Wash slides three times with PBS for 5 min each.
-
11
Incubate with the phage sublibrary (0.5 ml of 5 × 1011 cfu/ml stock) from section 3.1, step 21 at room temperature for 1 h.
-
12
Wash slides three times for 5 min each with PBS to remove unbound phage. Dehydrate the slides by sequential incubation in 70%, 95%, and 100% ethanol. Incubate for 5 min for each step (see Note 7).
-
13
Insert the dried slide into the specimen holder with section face down.
-
14
Open the cap of a 0.5 ml microcentrifuge PCR tube and place it in the collection area.
-
15
Focus on regions containing tumor cells of interest, draw a laser path around the target area.
-
16
Activate the laser, cut specimen along the pre-defined laser path, and drop the excised cells into the cap of the collection tube by electrostatic force and gravity (see Note 8).
-
17
Proceed immediately to PCR amplification or store the tissue pieces at −80 °C until analysis.
3.3. Recovery of phage antibody from LCM-procured tumor cells
- PCR amplify the genes encoding scFv fragments from LCM- collection tubes from section 3.2 step 16 using the following PCR cycling conditions:
Initial denature 95 °C 5 min Denature 94 °C 1 min Annealing 55 °C 45 sec Extending 68 °C 1 min Number of cycles 30 cycles Final extending 72 °C 10 min Purify the PCR products using a Qiagen PCR purification kit and digest the amplified fragments and fd phage display vector with SfiI and NotI at 37°C for 2–4 h.
Run the restriction digested products on a 1% agarose gel. Cut out target bands (approximately 800 bp for amplified scFv and 9 kb for fd phage vector) with a clean razor blade and isolate the PCR products using a Qiagen gel isolation kit. Elute in ddH2O and measure the concentration by a spectrophotometer.
Ligate precut PCR products into the fd phage display vector using T4 DNA ligase at room temperature for 15 min. Transform ligation products into chemically competent TG1. Culture at 37 °C at 225 rpm for 45 min and plate all the bacteria on YT/tet plate.
Make the LCM-selected phage sublibrary as described in section 3.1, steps 14–21.
Amplify the scFv gene by colony PCR from phage-infected bacteria, digest the PCR products with BstNI, and analyze on a 1% agarose gel to estimate the diversity of recovered scFv sequences (see Note 9).
3.4. Analysis of selection output by FACS
Inoculate individual phage–infected bacteria in 96-well U-bottom plates by picking single colonies using sterile pipette tips or toothpicks and dipping into 120 ul of 2xYT/tet per well, leaving one or more mock well/plate without bacteria as a contamination control.
Culture the plates at 37 °C with shaking at 200 rpm for 18 h.
Transfer 50 μl of bacterial/phage culture per well into a new 96-well microtiter plate. Add 50 μl of 2xYT/tet containing 50% glycerol to each well, pipette up and down to mix, and store at −80 °C as master plates.
Centrifuge the remaining bacteria/phage in the U-bottom plate at 4,000g for 30 min for screening on cells by FACS (see below).
Prepare 105 cells/ml (10 ml for each 96-well plate) of prostate cancer (PC3 and Du-145) or non-tumorigenic control (BPH-1) cells as described in section 3.1, steps 1–3.
-
Resuspend cells in PBS with 1% BSA. Add 100 μl cells per well into 96-well V-bottom plates.
Transfer 30 μl of supernatant containing phage particles (5 × 1011 cfu/ml) in section 3.4, step 4 into the V-bottom 96-well plate containing tissue culture cells. Incubate at 4 °C for 1 h with rocking.
Centrifuge the cells at 500g for 5 min and remove the supernatant. Wash the cells three times with PBS containing 1% BSA.
Resuspend the cells in 100 μl of PBS with 1% BSA and biotinylated anti-M13 antibody (Sigma, diluted 1:1000). Incubate at 4 °C for 1 h with rocking.
Centrifuge the cells at 500g for 5 min, remove the supernatant. Wash cells three times with PBS containing 1% BSA.
Resuspend cells in 100 μl of PBS with 1% BSA and streptavidin-phycoerythrin (diluted 1:1000). Incubate at 4 °C for 1 h with rocking.
Centrifuge the cells at 500g for 5 min and remove the supernatant. Wash cells three times with PBS containing 1% BSA. Resuspend cells in 150 μl PBS.
Analyze cell fluorescence by FACS.
Sequence the phage antibodies that bind to tumor cells but not normal cells (see Note 10).
3.5. Construction, expression, purification, and biotinylation of scFv fragments
Analyze the sequences of phage antibodies and design primers for PCR amplification of scFv genes with NcoI and NotI restriction digest site (See Note 11).
PCR amplify the genes encoding scFv fragments using the cycling conditions described in section 3.3, step 1.
Purify the PCR products using a Qiagen PCR purification kit and digest the amplified fragments and pSyn-1 vector with NcoI and NotI at 37°C for 2–4 h.
Run the restriction-digested products on a 1% agarose gel. Cut out target bands with a clean razor blade and isolate the PCR products using a Qiagen gel isolation kit. Elute in ddH2O and measure the concentration by spectrophotometer.
Ligate precut PCR products into pSyn-1 vectors using T4 DNA ligase at room temperature for 15 min. Transform ligation products into chemically competent TG1. Culture at 37 °C at 225 rpm for 45 min and plate all the bacteria on YT/amp plate.
Miniprep and sequence to verify scFv-pSyn1 expression clones.
Pick and culture single-bacterial colony overnight at 37 °C in 5 ml of overnight medium (defined in section 2.5).
Add 1 ml of overnight cultured bacterial to 400 ml growth medium (defined in section 2.5). Culture bacteria to OD600 ~0.7 at 37 °C with shaking at 250 rpm.
Cool down the bacterial culture to room temperature. Add 1 M IPTG to culture to a final concentration of 1 mM. Continue culturing the bacteria at 30 °C for 16 h.
Collect and centrifuge the bacteria stock at 5,000g for 20 min at 4 °C. Remove all the supernatant.
Resuspend the bacterial pellet in 12.5 ml PPB. Keep the bacterial solution on ice for 20 min.
Centrifuge the bacteria at 5,000g for 15 min at 4 °C. Transfer the supernatant to a high-speed centrifuge tube.
Osmotically shock the cells by resuspending the pellet in 12.5 ml of 5 mM MgSO4 and incubate on ice for 20 min.
Combine osmotic shock prep with the periplasmic prep from step 7 and centrifuge at 10,000g for 15 min at 4 °C.
Transfer the supernatant to a new 50 ml Falcon tube. Add 25 ml PBS and 500 μl pre-washed Ni-NTA agarose resin beads to the tube. Incubate at 4 °C by rotating for 2 h (see Note 12).
Spin down the beads at 3000g for 15 min at 4 °C and carefully discard the liquid, and wash three times with 50 ml of PBS.
Transfer the beads to a new 2 ml Eppendorf tube. Spin down and wash the beads three times with wash buffer for 5 min each.
Add 1 ml of elution buffer to the beads and incubate at room temperature for 5 min. Spin down and transfer the liquid to a new Eppendorf tube.
Concentrate and buffer exchange the scFv antibody to PBS by using Millipore ultra-15 concentrator tube and Zeba™ spin desalting columns.
Measure the concentration of scFv antibody by using NanoDrop according to the manufacturer’s instruction (see Note 13).
To biotin-label antibody, prepare a 10 mM EZ-Link Sulfo-NHS-LC-Biotin solution using ultrapure water immediately before use. Add 27 μl of 10 mM biotin solution to 1 ml of 2 mg/ml purified scFv antibody in PBS at pH 7.4, rotate the mixture at room temperature for 45 min.
Add 20% (v/v) of 1 M Tris-HCl (pH 8.0) to quench the reaction and mix gently by pipetting up and down.
Buffer-exchange to PBS and remove nonreacted biotin using Zebaspin desalting columns according to the manufacturer’s instruction. Biotin-labeled antibody can be stored at −20 °C for months or 4 °C for a couple of weeks until use in immunotoxin assays.
FACS analyze the biotinylated scFv antibody binding to prostate cancer lines as described in section 3.4 by using 10 μg/ml biotinylated scFv antibody followed detection with streptavidin-phycoerythrin.
3.6. Analysis of selection output by immunohistochemistry
For frozen tissue slides:
-
1
Prepare the frozen tissue sections as described in section 3.2 steps 2–3. Proceed to step 4.
For paraffin-embedded tissue slides:
-
2
Deparaffinize and rehydrate the paraffin-embedded tissue as described in section 3.2 steps 7–9.
-
3
Place the slide from step 2 into a glass jar filled with unmasking solution (diluted 1:100). Incubate the jar at 95–100 °C for 10 min in a pressure cooker. Remove the jar to room temperature and allow the slides to cool to room temperature (in about 20 min). Rinse the slide twice with PBS for 5 min each.
-
4
Use “liquid blocker” pen to demarcate the tissue.
-
5
Block endogenous peroxidase activity by incubating the slide in 3% H2O2 in PBS, 10 min. Rinse the slide 3 times in PBS for 5 min each.
-
6
Incubate the slide with 2% goat serum at room temperature for 30 min. Rinse briefly with PBS and incubate with avidin solution for 15 min. Rinse briefly with PBS followed by incubating with biotin solution 15 min. Rinse briefly with PBS.
-
7
Add 50 μg/ml of biotinylated scFv antibody in PBS with 2% goat serum, incubate for 1 h at room temperature, rinse 3 times in PBS for 5 min each.
-
8
Add R.T.U ABC REAGENT and incubate the slides for 30 min at room temperature. Rinse 3 times in PBS for 5 min each.
-
9
Add the DAB solution to the slide and check the reaction under a microscope within 5 min. Rinse in PBS to stop the reaction.
-
10
Add hematoxylin to the slide directly for counter staining for 3 min. Rinse slide with running tap water until rinse water is colorless.
-
11
Dip slides 10 times in acid rinse solution, followed by 10 dips in tap water.
-
12
Incubate slides in bluing solution for 1 min followed by 10 dips in tap water.
-
13
Dehydrate in 75% ethanol, 95% ethanol, 100% ethanol for 5 min each. Then clear the slides by incubating in xylene twice for a total of 15 min and allow the slide to air dry.
-
14
Add VectaMount™ mounting medium and apply coverslip. Analyze antibody staining under a microscope.
3.7. Microscopic analysis of antibody internalization
Grow prostate cancer (DU145) cells in Lab-Tek II glass chambers to 50–60% confluence.
Wash the cells once with pre-warmed fresh growth medium.
Add cell growth medium (defined in section 2.1) containing 15 μg/ml of biotinylated scFv and 50 μg/ml of ND70-TR to cells, incubate at 37 °C for 3 h.
Wash the cells three times with PBS for 5 min each.
Fix the cells with 4% PFA in PBS for 15 min at 4 °C.
Wash the cells three times with PBS for 5 min each.
Permeabilize the cells with permeabilization buffer for 20 min at room temperature.
Add 1:300 diluted streptavidin-Cy3 in 1:5 PBS-diluted permeabilization buffer, incubate at room temperature for 30 min.
Wash the cells three times with PBS for 5 min each, fix and counterstain cells with 4% PFA containing Hoechst in PBS for 10 min at room temperature.
Image and analyze the staining using a confocal microscope.
3.8. Intracellular delivery of immunotoxin
Seed 3,000 cells per well of prostate cancer (PC3 or Du-145) or non-tumorigenic control (BPH-1) in 96-well flat bottom plates with 50 μl cell growth medium and culture overnight at 37 °C with 5% CO2.
Prepare the immunotoxin by mixing biotinylated scFv with SA-ZAP at a molar ratio of 1:1, incubate on ice for 30 min.
Add 50 μl of serially diluted immunotoxin in PBS to each well and incubate for 96 h at 37 °C with 5% CO2 (see Note 14).
Carefully remove the cell growth medium from each well.
Add 100 μl of diluted CCK-8 to each well in the 96-well plates, incubate for 1 – 4 h at 37 °C in 5% CO2 (see Note 15).
Measure the absorbance at 450 nm using a microtiter plate reader and determine the EC50 value by curve fitting using appropriate software (e.g., GraphPad Prism).
4. Notes
The list of normal cells can be expanded to additional non-tumorigenic cell lines and normal primary cells when available.
Incubation at 37° in cell growth media allows for internalization of phage antibodies.
Wash with glycine for no longer than 5 min.
The inoculated media should look very slightly turbid (initial OD600 = 0.05~0.1).
The sublibrary contained 1 – 5 × 105 copies of about 106 independent clones at the concentration of 1 – 5 × 1011 cfu/ml.
The suggested cryostat temperature is between −15 and −23 °C. Slides can be stored unfixed for several months at −80 °C. Frozen tissue samples saved for later analysis should be stored intact.
All the slides should be reviewed by a board-certified pathologist and regions containing clusters of tumor cells should be confirmed and marked.
Laser microdissection microscope that uses a UV pulse laser to excise selected cells from surrounding tissues. Typically 20–50 tumor cells were procured at a time by generating a closed laser path around the group of cells of interest.
Each LCM selection library contained >105 independent clones. The number of unique phage antibodies was determined by patterns of BstNI digestion [6, 7, 26]. When restriction digestion patterns showed ambiguity, phage antibody genes were sequenced to determine their uniqueness.
More than 600 clones from various LCM-derived sublibraries were screened. Only those clones that bound to both PC3 and Du-145 cells but not BPH-1cells were chosen for further analysis because they were more likely to recognize tumor cell surface antigens as opposed to artifacts associated with a particular tissue slide.
Two forms of soluble antibody fragments, scFv and (scFv′)2, can be produced [6, 7, 39, 40]. The scFv gene was subcloned into the secretion vector pUC119mycHis, adding a c-Myc epitope tag and hexahistidine tag at the c-terminus of the scFv. To create the (scFv′)2 dimer for immunoliposome studies, the c-Myc epitope tag was removed, and a free cysteine was introduced at the c-terminus of the scFv preceding the hexahistidine tag.
Purification can be done with Ni-NTA agarose beads/gravity method or GE HisTrap column/FPLC method.
The purified scFv should also be analyzed by SDS-PAGE. The molecular weight of monomeric scFv is about 27 kD.
For initial assessment, 1:10 serial dilutions are often used to find the linear range of activity. Once the linear range has been determined, 1:3 serial dilutions can be used to improve the accuracy of the EC50 measurement.
Remove any air bubbles in the well, as they interfere with the absorbance measurement.
Acknowledgment
Work in our laboratory is supported by grants from the National Institutes of Health/National Cancer Institute (R01 CA171315, R01 CA118919, and R01 CA129491). NKL received fellowship support from Basic Science Research Program of the National Research Foundation of Korea (NRF) that is funded by the Ministry of Education, Science and Technology (2013R1A6A3A03060495).
Reference:
- 1.Kobata A and Amano J (2005) Altered glycosylation of proteins produced by malignant cells, and application for the diagnosis and immunotherapy of tumours. Immunol Cell Biol. 83, 429–39. [DOI] [PubMed] [Google Scholar]
- 2.Birkle S, Zeng G, Gao L et al. (2003) Role of tumor-associated gangliosides in cancer progression. Biochimie. 85, 455–63. [DOI] [PubMed] [Google Scholar]
- 3.Hakomori S (2001) Tumor-associated carbohydrate antigens defining tumor malignancy: basis for development of anti-cancer vaccines. Adv Exp Med Biol. 491, 369–402. [DOI] [PubMed] [Google Scholar]
- 4.Hanisch FG (2001) O-glycosylation of the mucin type. Biol Chem. 382, 143–9. [DOI] [PubMed] [Google Scholar]
- 5.Ugorski M and Laskowska A (2002) Sialyl Lewis(a): a tumor-associated carbohydrate antigen involved in adhesion and metastatic potential of cancer cells. Acta Biochim Pol. 49, 303–11. [PubMed] [Google Scholar]
- 6.Liu B, Conrad F, Cooperberg MR et al. (2004) Mapping tumor epitope space by direct selection of single-chain Fv antibody libraries on prostate cancer cells. Cancer Res. 64, 704–10. [DOI] [PubMed] [Google Scholar]
- 7.Ruan W, Sassoon A, An F et al. (2006) Identification of clinically significant tumor antigens by selecting phage antibody library on tumor cells in situ using laser capture microdissection. Mol Cell Proteomics. 5, 2364–73. doi: 10.1074/mcp.M600246-MCP200 [DOI] [PubMed] [Google Scholar]
- 8.An F, Drummond DC, Wilson S et al. (2008) Targeted drug delivery to mesothelioma cells using functionally selected internalizing human single-chain antibodies. Mol Cancer Ther. 7, 569–78. doi: 10.1158/1535-7163.MCT-07-2132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bidlingmaier S, He J, Wang Y et al. (2009) Identification of MCAM/CD146 as the target antigen of a human monoclonal antibody that recognizes both epithelioid and sarcomatoid types of mesothelioma. Cancer Res. 69, 1570–7. doi: 10.1158/0008-5472.CAN-08-1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bidlingmaier S, Su Y, Liu B (2015) Combining Phage and Yeast Cell Surface Antibody Display to Identify Novel Cell Type-Selective Internalizing Human Monoclonal Antibodies. Methods Mol Biol. 1319, 51–63. doi: 10.1007/978-1-4939-2748-7_3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ha KD, Bidlingmaier SM, Zhang Y et al. (2014) High-content analysis of antibody phage-display library selection outputs identifies tumor selective macropinocytosis-dependent rapidly internalizing antibodies. Mol Cell Proteomics. doi: 10.1074/mcp.M114.039768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu X, Bidlingmaier S, Hashizume R et al. (2010) Identification of internalizing human single-chain antibodies targeting brain tumor sphere cells. Mol Cancer Ther. 9, 2131–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sherbenou DW, Aftab BT, Su Y et al. (2016) Antibody-drug conjugate targeting CD46 eliminates multiple myeloma cells. J Clin Invest. 126, 4640–4653. doi: 10.1172/JCI85856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bidlingmaier S, Zhu X, Liu B (2008) The utility and limitations of glycosylated human CD133 epitopes in defining cancer stem cells. J Mol Med (Berl). 86, 1025–32. doi: 10.1007/s00109-008-0357-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marks J, Hoogenboom H, Bonnert T et al. (1991) By-passing immunization. Human antibodies from V-gene libraries displayed on phage. J Mol Biol. 222, 581–97. [DOI] [PubMed] [Google Scholar]
- 16.McCafferty J, Griffiths A, Winter G et al. (1990) Phage antibodies: filamentous phage displaying antibody variable domains. Nature. 348, 552–4. [DOI] [PubMed] [Google Scholar]
- 17.O’Connell D, Becerril B, Roy-Burman A et al. (2002) Phage versus phagemid libraries for generation of human monoclonal antibodies. J Mol Biol. 321, 49–56. [DOI] [PubMed] [Google Scholar]
- 18.Hoogenboom HR and Winter G (1992) By-passing immunisation. Human antibodies from synthetic repertoires of germline VH gene segments rearranged in vitro. J Mol Biol. 227, 381–8. [DOI] [PubMed] [Google Scholar]
- 19.de Haard HJ vNN, Reurs A, Hufton SE, Roovers RC, Henderikx P, de Bruine, Arends JW, Hoogenboom HR (1999) A large non-immunized human Fab fragment phage library that permits rapid isolation and kinetic analysis of high affinity antibodies. J. Biol. Chem 274, 18218–18230. [DOI] [PubMed] [Google Scholar]
- 20.Clackson T, Hoogenboom HR, Griffiths AD et al. (1991) Making antibody fragments using phage display libraries. Nature. 352, 624–8. doi: 10.1038/352624a0 [DOI] [PubMed] [Google Scholar]
- 21.Winter G, Griffiths A, Hawkins R et al. (1994) Making antibodies by phage display technology. Annu Rev Immunol. 12, 433–455. [DOI] [PubMed] [Google Scholar]
- 22.Barbas CF 3rd, Kang AS, Lerner RA et al. (1991) Assembly of combinatorial antibody libraries on phage surfaces: the gene III site. Proc Natl Acad Sci U S A. 88, 7978–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huston JS and George AJ (2001) Engineered antibodies take center stage. Hum Antibodies. 10, 127–42. [PubMed] [Google Scholar]
- 24.Janda KD, Lo CH, Li T et al. (1994) Direct selection for a catalytic mechanism from combinatorial antibody libraries. Proc Natl Acad Sci U S A. 91, 2532–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sheets MD, Amersdorfer P, Finnern R et al. (1998) Efficient construction of a large nonimmune phage antibody library: the production of high-affinity human single-chain antibodies to protein antigens. Proc Natl Acad Sci U S A. 95, 6157–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu B and Marks JD (2000) Applying phage antibodies to proteomics: selecting single chain Fv antibodies to antigens blotted on nitrocellulose. Anal Biochem. 286, 119–28. doi: 10.1006/abio.2000.4788 [DOI] [PubMed] [Google Scholar]
- 27.Liu B, Huang L, Sihlbom C et al. (2002) Towards proteome-wide production of monoclonal antibody by phage display. J Mol Biol. 315, 1063–73. [DOI] [PubMed] [Google Scholar]
- 28.Bonner RF, Emmert-Buck M, Cole K et al. (1997) Laser capture microdissection: molecular analysis of tissue. Science. 278, 1481,1483. [DOI] [PubMed] [Google Scholar]
- 29.Emmert-Buck MR, Bonner RF, Smith PD et al. (1996) Laser capture microdissection. Science. 274, 998–1001. [DOI] [PubMed] [Google Scholar]
- 30.Best CJ and Emmert-Buck MR (2001) Molecular profiling of tissue samples using laser capture microdissection. Expert Rev Mol Diagn. 1, 53–60. doi: 10.1586/14737159.1.1.53 [DOI] [PubMed] [Google Scholar]
- 31.Mukherjee S, Rodriguez-Canales J, Hanson J et al. (2013) Proteomic analysis of frozen tissue samples using laser capture microdissection. Methods Mol Biol. 1002, 71–83. doi: 10.1007/978-1-62703-360-2_6 [DOI] [PubMed] [Google Scholar]
- 32.Espina V, Wulfkuhle JD, Calvert VS et al. (2006) Laser-capture microdissection. Nat Protoc. 1, 586–603. doi: 10.1038/nprot.2006.85 [DOI] [PubMed] [Google Scholar]
- 33.Espina V, Milia J, Wu G et al. (2006) Laser capture microdissection. Methods Mol Biol. 319, 213–29. doi: 10.1007/978-1-59259-993-6_10 [DOI] [PubMed] [Google Scholar]
- 34.Johann DJ, Rodriguez-Canales J, Mukherjee S et al. (2009) Approaching solid tumor heterogeneity on a cellular basis by tissue proteomics using laser capture microdissection and biological mass spectrometry. J Proteome Res. 8, 2310–8. doi: 10.1021/pr8009403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lu H, Jin D, Kapila YL (2004) Application of laser capture microdissection to phage display peptide library screening. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 98, 692–7. doi: 10.1016/S1079210404006134 [DOI] [PubMed] [Google Scholar]
- 36.Kubo N, Akita N, Shimizu A et al. (2008) Identification of oligopeptide binding to colon cancer cells separated from patients using laser capture microdissection. J Drug Target. 16, 396–404. doi: 10.1080/10611860802088796 [DOI] [PubMed] [Google Scholar]
- 37.Sun Y, Shukla GS, Kennedy GG et al. (2009) Biopanning Phage-Display Libraries on Small Tissue Sections Captured by Laser Capture Microdissection. J Biotech Res. 1, 55–63. [PMC free article] [PubMed] [Google Scholar]
- 38.Sun Y, Shukla GS, Weaver D et al. (2009) Phage-display selection on tumor histological specimens with laser capture microdissection. J Immunol Methods. 347, 46–53. doi: 10.1016/j.jim.2009.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roth A, Drummond DC, Conrad F et al. (2007) Anti-CD166 single chain antibody-mediated intracellular delivery of liposomal drugs to prostate cancer cells. Mol Cancer Ther. 6, 2737–46. [DOI] [PubMed] [Google Scholar]
- 40.Iyer AK, Su Y, Feng J et al. (2011) The effect of internalizing human single chain antibody fragment on liposome targeting to epithelioid and sarcomatoid mesothelioma. Biomaterials. 32, 2605–13. doi: 10.1016/j.biomaterials.2010.11.073 [DOI] [PMC free article] [PubMed] [Google Scholar]