

Abstract
The Ca2+ hypothesis for Alzheimer’s disease (AD) conceives Ca2+ dyshomeostasis as a common mechanism of AD; the cause of Ca2+ dysregulation, however, is obscure. Meanwhile, hyperactivities of NMDA receptors (NMDARs), the primary mediator of Ca2+ influx, are reported in AD. GluN3A (NR3A) is a NMDAR inhibitory subunit. We hypothesize that GluN3A is critical for Ca2+ homeostasis, its deficiency is pathogenic for AD. Cellular, molecular and functional changes were examined in GluN3A knockout (KO) mice during aging. The GluN3A KO mouse brain displayed age-dependent moderate but persistent neuronal hyperactivity, elevated intracellular Ca2+, neuroinflammation, impaired synaptic integrity/plasticity, and neuronal loss. GluN3A KO mice developed olfactory dysfunction followed by psychological/cognitive deficits prior Aβ/tau pathology. Memantine at preclinical stage prevented/attenuated AD syndromes. AD patients’ brains show reduced GluN3A expression. We propose that chronic “degenerative excitotoxicity” leads to sporadic AD, while GluN3A represents a primary pathogenic factor, an early biomarker and an amyloid-independent therapeutic target.
Keywords: Alzheimer’s disease, N-methyl-D-aspartate receptor, NR3A, Dementia, Prevention, Degenerative excitotoxicity, Subhealth status
Graphical Abstract
PART 1: NARRATIVE
1.1. Objective and research goal
Alzheimer’s disease (AD) affects 1 in 10 people of age 65 and older, while the exact pathogenic factor remains ambiguous. Among all proposed hypotheses for AD, the “Ca2+ hypothesis” has gained increasing supports with the observations that Ca2+ dysregulation is a common and prevalent pathophysiology in AD. However, the initiating mechanism that causes lifelong alterations in Ca2+ homeostasis leading to neuronal dysfunction and cognition decline has not been identified. Here, we aim to disclose a root mechanism that can explain the origin of Ca2+ dyshomeostasis and its link to “degenerative excitotoxicity” responsible for age-dependent cognition decline and amyloid cascade pathology.
1.2. Background and rationale
Alzheimer’s disease (AD) is characterized by the development of age-dependent dementia accompanied by the formation of amyloid β (Aβ) plaques and neuronal fibrillary tangles (NFTs) [1–4]. Synaptic disruption and neuronal loss in the neocortex and hippocampus as well as brain atrophy are structural characteristics of AD [3]. In addition to cholinesterase inhibitors and the NMDA receptor (NMDAR) antagonist memantine (MEM) approved by FDA as symptomatic treatments for moderate to severe patients, there is no treatment that can cure or prevent AD. Extensive research has identified three typical genetic mutations including amyloid precursor protein (APP) [5], presenilin 1 (PS1) and PS2 [6], which are responsible for genetically inherited forms of AD (familial AD or FAD). FAD shows early onset and represents less than 1–2% of total AD cases [7]. Most AD patients (>95%) are sporadic type of AD (SAD) featuring late onset [8] with no clearly identified etiology. The genetic mutations in FAD support the “amyloid cascade hypothesis”, which suggests that AD pathogenesis is initiated by overproduction of Aβ and/or failure of its clearance, upstream of tau dysregulation and dementia [3, 9]. While the Aβ and tau pathology remains the current diagnostic standard of AD, they are poorly correlated with symptoms of AD such as progressive memory loss and cognitive decline; i.e. individuals may have significant Aβ plaques and tau tangles in the hippocampus and neocortex but show no sign of cognitive deficits [10–13]. Nevertheless, the majority of AD investigations have been performed on transgenic rodent models expressing FAD genes. The failure of clinical translation of experimental therapies based on the amyloid hypothesis and the lack of correlation between brain amyloid levels and cognitive status have led to the proposition to reevaluate or even reject the conventional, although very popular, amyloid cascade hypothesis so as to explore alternative pathogenic mechanisms [13–16]. Different hypotheses such as the “inflammatory hypothesis” and the “oligomer/soluble Aβ hypothesis” modified from original amyloid hypothesis have been proposed alongside others for a number of years [17]. However, so far few of these hypotheses have been able to account for all the hallmark pathologies and symptoms of AD or result in clinical therapies to show significant therapeutic effects against the disease progression.
Meanwhile, the “Ca2+ hypothesis” for AD/dementia has gained increasing supports from basic and clinical research [18–21]. The Ca2+ hypothesis was initially proposed by Khachaturian, predicted that a sustained dysregulation of cellular Ca2+ homeostasis could disrupt neuronal function and lead to neurodegenerative diseases such as AD [22]. This hypothesis has been endorsed by consistent observations that neuronal Ca2+ dysregulation is a common mechanism in the development of AD in both animal models and clinical studies [18, 19, 23–27]. Perturbations or increases in intracellular free Ca2+ ([Ca2+]i) were also demonstrated in cells and organoids from AD patients and in clinical imaging studies [28–30]. It is thus widely recognized that Ca2+ dysregulation is an important pathophysiology of familial and sporadic AD, although the causal mechanism that triggers the [Ca2+]i disturbance has not been clearly defined.
Ca2+ is a prevalent second messenger regulating numerous cellular functions, including, but not limited to, neuronal excitability, synapse integrity and plasticity associated with learning and memory [31]. The Ca2+ level in the extracellular space is typically maintained between 1 and 2 mM, whereas the resting cytosolic concentration of free Ca2+ in neurons is tightly regulated around the 100 nM range [32]. This substantial Ca2+ gradient is critical for normal neuronal activity and cell viability [33, 34]. Upon excitation, Ca2+ enters neurons via Ca2+ permeable channels and receptors on the plasma membrane and is quickly sequestered by intracellular buffering systems including Ca2+ binding proteins and internal organelles, such as the endoplasmic reticulum (ER), mitochondria, and lysosomes [35, 36]. Many studies have demonstrated their interactions with amyloid cascades, thereafter, resulting in Ca2+ dysregulation.
In the validation of the Ca2+ hypothesis of AD, we have focused on the regulation of Ca2+ influx via the NMDA receptor that plays primary roles in neuronal activity, synaptic transmission/plasticity, cognitive function as well as excitotoxicity of glutamatergic neurons [37, 38]. Under pathological conditions, overactivation of NMDARs and increased [Ca2+]i are biological signals that trigger a chain of molecular and cellular events leading to acute and delayed cell death and pathological consequences such as synapse loss, neurovascular damage, mitochondrial dysfunction, and failure of neural network communications [38–40]. Compelling evidence has shown hyperactivity of NMDARs in AD [28–30], but the precise mechanism that causes persistent NMDAR hyperactivity and whether this NMDAR misbehavior has a causal relationship to AD development remain unidentified.
1.3. Historical evolution and updated hypothesis
Excitotoxicity was initially described in neuronal cultures and animal models of stroke 30 years ago in investigations of glutamate-induced toxicity to neurons [34, 41, 42]. It is defined as excessive exposure to the neurotransmitter glutamate or overstimulation of its membrane receptors, leading to massive Ca2+ influx and intracellular Ca2+ overload [43, 44], resulting in acute neuronal injury i.e. necrotic cell death within several hours to a few days after the insult [34, 40, 45]. The Ca2+ hypothesis for AD resembled the similar concept of Ca2+ dysregulation, predicting that excitotoxicity is responsible for neurodegenration developed in the AD brain [18, 21, 24]. However, distinctions between the acute and chronic forms of excitotoxicity have not be explicitly defined.
We propose the concept of “degenerative excitotoxicity” whereby some ulterior cause of Ca2+ disregulation is the root of a slight but sustained increase in [Ca2+]i. The resultant “calciumopathy” yields brain region specific impairment that is not immediately injurous but induces enduring perturbations of multiple Ca2+-dependent cascades and generates a chronic pro-AD state prone to programmed cell degeneration involving synaptic disruption, recurring inflammation, metabolic interruption, aberrant autophagy, apoptosis, and other cytotoxic consequences including the activation of amyloid cascades [46–49]. According to this modification to the Ca2+ hypothesis, there must be a pathogenic mechanism that exists during presymtomatic stage and persists to advance to age-dependent abnormal consequenses. It is also hypothesized that for a proloned period of time (months in rodents and years in humans), the AD susceptible brain can still be functional under a “suboptimal health or subhealth” status [50]. This status could exist prior to the development of any brain pathology yet exert significant stress to neuronal infrastructure, brain matablism, and neural network functions. Aβ cascades have been proposed as triggers for excitotoxity in AD [51–53]. The proposition, however, cannot account for how neurodegeneration could develop before Aβ related pathologies in many AD cases. Our investigation is an initial effort to reveal the primary pathogenic mechanism mediated by continual hyperactivities of NMDAR in the deficiency of its unique inhibitory subunit GluN3A.
Ionotropic glutamate receptors, includind NMDA, AMPA, and kainate receptors, serve as important mediators of excitatory synaptic transmission in the brain [37, 54]. Of these, NMDAR plays a major role in Ca2+ influx of cellular physiology and pathophysiology [38, 39]. NMDARs comprise the GluN1 (NR1) subunit, GluN2 (NR2) subunits (GluN2A-2D), and a pair of GluN3 (NR3) subunits (GluN3A and GluN3B) [55]. Functional NMDARs are heterotetramers composed of two glycine/d-serine-binding GluN1 subunits paired with two glutamate-binding GluN2 subunits, or often with one GluN2 and one inhibitory GluN3 subunit [56]. GluN3A and 3B act in a dominant-negative manner to restrain receptor activity. Heterogeneous expression of NMDARs with a GluN3 subunit induces smaller NMDA currents with reduced Ca2+ permeability [57–60]. The GluN3A expression in dendritic spines of glutamatergic neurons implies that GluN3A can influence synaptic stability, receptor assembly, and functional activity of these neurons [61]. Different from GluN3A, GluN3B mainly expresses in motor neurons. GluN3A knockout (KO) cortical neurons exhibit increased NMDA currents and dendritic spine density, supporting that GluN3A plays a significant role in synaptic modulation/plasticity [57]. There is a high level of sequence homology (93%) between human and rodent GluN3A/3B, implying that their function is likely similar between mammalian species [62]. The lack of gene equivalents in D. melanogaster and C. elegans suggests that GluN3 emerged during evolution of vertebrates in the need for a more complicated and more precisely controlled nervous system. We propose that GluN3 genes are essential and critical regulators for optimal NMDAR function and sustained Ca2+ homeostasis in the mammal’s brain.
The control of NMDAR activity is imparitive for normal brain function. Repeated NMDAR activation enhances postsynaptic Ca2+ entry, which is a pre-requisite for AMPA receptor (AMPAR) activation and subsequent synaptic long-term potentiation (LTP) [38]. This essential role of NMDAR makes it uniquely positioned to mediate synaptic plasticity, forming a molecular and cellular basis for learning and memory [38]. Abbarrent NMDAR activities can otherwise cause synapse loss and synaptic disruption [63]. The toxicity is principally mediated by excessive Ca2+ entry via over-activated NMDARs [41, 64]. Meanwhile, how the NMDAR activity is persistantly altered in the aging brain and its regulatory mechanism in AD progression are poorly understood. Current research in this area has exclusively examined GluN1 and GluN2; little attention has been paid to a possible role of GluN3 in AD pathophysiology.
In rodents, the GluN3A expression is high in the developing brain but lower in adults [65, 66]. Our previous data indicate that despite this drop off, GluN3A remains prevalent in the adult mouse brain and exerts significant neuroprotective effects against excitotoxicity after ischemic stroke among other important functional roles [45, 67, 68]. In humans the distribution of GluN3A is significant and widespread in both fetal and adult brains [69, 70]. It was identified that there are “native NR1-NR2A/B-NR3A assemblies in adult human CNS”, and GluN1 (NR1) and GluN3 (NR3) “behave similarly with regard to receptor assembly and trafficking” [70]. Thus, GluN3A can play vital regulatory roles in both young and adult human physiology and pathophysiology.
Human studies have identified that GluN3 encoding genes GRIN3A and GRIN3B are highly heterogeneous; around 2–10% people worldwide carry mutated GRIN3 variants including null alleles with complete deficiency (GRIN3A mutates in ~2% Han-Chinese and Japanese, no information available for GRIN3A in other countries; GRIN3B variants in ≥10% of populations in European-Americans and some other countries) [71–73]. A clinical study of AD patients investigated genetic risk factors based on GRIN3A and GRIN3B alleles [74]. Two single nucleotide polymorphisms, 3104 G/A (rs10989563) and 3723 G/A (rs3739722) in GRIN3A and 2 GRIN3B polymorphisms were probed and the genetic variation of the GRIN3A was suggested to be a risk factor for AD [74]. It is worth mentioning that the percentage of people with GRIN3 mutations is in close proximity to the prevalence of AD (1.6 – 6.4%) among general populations in different countries [75]. Nevertheless, there is no further information about a possible correlation between mutation/dysfunction of GRIN3A/GRIN3B and AD.
The FDA approved use of MEM as a treatment for moderate-to-severe AD provides a support for the clinical significance of NMDAR regulation in AD [76–78]. The essential role played by NMDARs in early Ca2+ dysregulation suggest that MEM could delay or halt the progression of early AD. Indeed, several clinical trials in mild AD/dementia patients support this prediction although this approach is still under debate [79–85]. In this investigation, we examined a primary pathogenic role of persistent NMDAR hyperactivity mediated by the deficiency of its inhibitory subunit GluN3A in AD development, leading to cognition decline well before the formation of Aβ plagues and tau pathology. Data from the GluN3A KO mouse shed lights on the mystery of what is the initiating mechanism or the mysterious switch for a chronic pro-AD Ca2+ dysregulation and whether it is possible to delay, ameliorate or prevent AD progression at an early phase of the disease.
PART II: CONSOLIDATED REULTS AND STUDY DESIGN
2.1. Consolidated results and study design
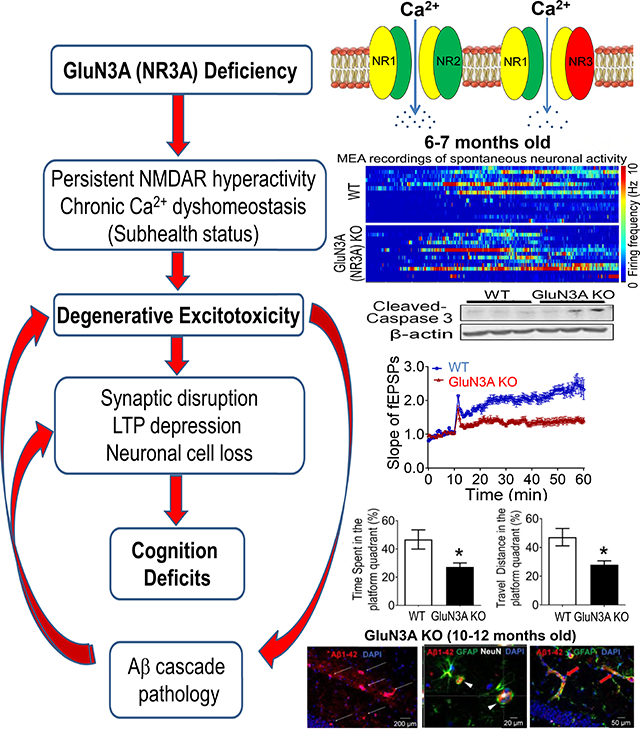
In brain slices from GluN3A KO mice, we performed MultiElectrode Array (MEA) recordings and tow-photon Ca2+ imaging. Comparing to young adult GluN3A KO brains, the aging KO brain exhibited hyperactivities of spontaneous firing accompanied with moderate but sustained elevations of resting [Ca2+]i. Meanwhile, the aging KO brain showed reduced presynaptic protein synapsin and impaired synaptic plasticity in the hippocampus compared to wild type (WT) age-matched controls. Significant increases of Ca2+-dependent signaling proteins such as CamKII and PKCα were identified in the hippocampus and/or cortex. Inflammatory activities such as increased TNF-α, reactive astrocytes and microglia cells persisted in the aging brain of GluN3A KO mice but not in normal aging controls. In age-dependent manners, GluN3A deficiency culminated in neuronal loss due to programmed cell death and recapitulated the disease symptoms including early onset of olfactory dysfunction followed months later by neuropsychological and cognitive deficits, which were not seen in WT controls. AD pathological hallmarks, i.e. Aβ production/deposition and tau hyperphosphorylation developed in the cortex and hippocampus in a much-delayed fashion several months after the onset of cognition decline. Consistent with the hypothesis that NMDAR hyperactivity is a pathogenic factor, MEM treatment at a clinically relevant dose (10 mg/kg/day in drinking water for 3 months) starting at the preclinical stage of 3-months old prevented or attenuated psychological and cognitive symptoms in GluN3A KO mice compared to vehicle controls. Of clinical significance, Western blot analysis of AD patient’s brain samples revealed lower levels of GluN3A in the cortex compared to normal aging subjects. We propose that a chronic state of hyperexcitability-induced “degenerative excitotoxicity” due to the GluN3A deficiency is sufficient to initiate AD evolution in an age-dependent manner. The long-term GluN3 modulation of NMDARs begins early in life, which implies early biomarkers, a wide therapeutic window for intervention, and potential targets for sporadic AD. The GluN3A KO mouse provides a novel model for studying the Ca2+ hypothesis in SAD development. The information of degenerative excitotoxicity obtained from this SAD mouse model may help to better understand other neurodegenerative diseases involving disrupted Ca2+ homeostasis.
PART III: DETAILED METHODS AND RESULTS
3.1. Methods and animals
GluN3A KO mice and wild type (WT) counterparts were tested. There was no change in the expression of other NMDAR subunits [74]. Detailed information of these mice was described previously [57, 62]. Genotyping of animals was performed as needed [67, 86]. Detailed experimental methods are provided in Supplemental Materials.
3.2. Neuronal hyperactivity and impairments of synaptic plasticity in the GluN3A KO brain
In brain slices from adult GluN3A KO mice of 6–12-months old (roughly equal to 35–60 years old humans), firing of hippocampal neurons were recorded using the MultiElectrode Array (MEA) system. Spontaneous neuronal activities were monitored for 10 min in 58 hippocampal locations. GluN3A KO hippocampal neurons indeed displayed constant higher frequencies of firings than that in age-matched WT brains (Fig. 1A–1B).
Figure 1. Neuronal hyperactivity and impaired synaptic plasticity in the GluN3A KO brain.
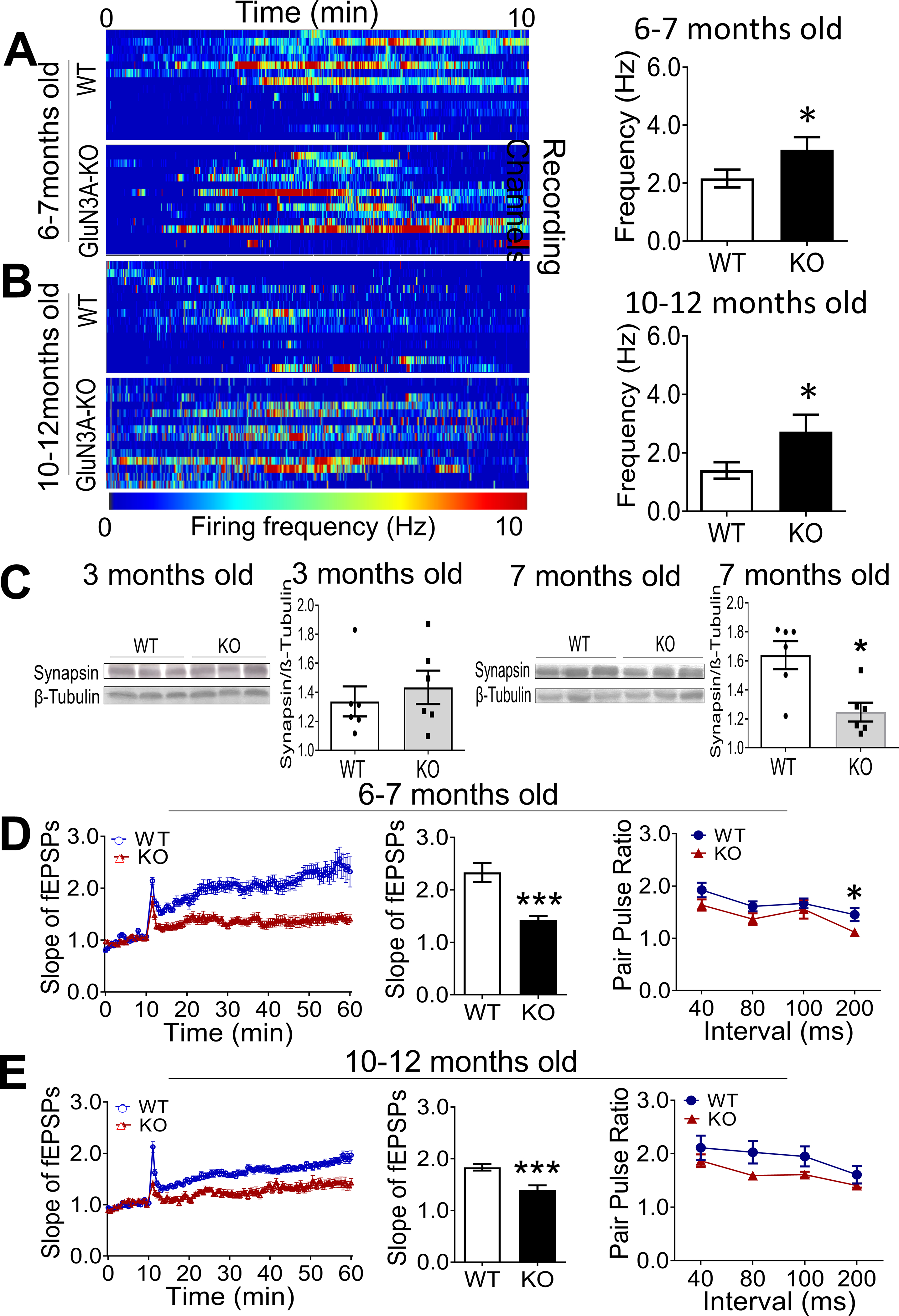
A MultiElectrode Array (MEA) system with 60-electrodes was used to record spontaneous activities and fEPSP in the brain slices containing hippocampus of WT and GluN3A KO mice of different ages. A and B. Heatmaps generated by the frequency of spontaneous spikes over time in the regular aCSF, are representative recording channels in brain slices from adult (6–7-month old) and aging (10–12-month old) mice. After stabilization of the recording for 10 min, spontaneous spikes was monitored for 10 min. Bar graphs show that the average frequency was significantly higher in GluN3A KO slices of the two age groups compared to age-matched WT slices (n=4 animals in WT group and 5 in KO group; *P<0.05 vs. WT; Student’s t-test). C. Western blot analysis of the presynaptic protein synapsin in the hippocampus. The expression of synapsin in 3 months old mice was similar between the two groups. Significant reduction of synapsin was seen in GluN3A KO mice at 7-month age compared to WT controls. D and E. In brain slices from 6–7 (C) and 10–12-month (D) old mice, LTP of fEPSPs in the hippocampus was induced by the typical high frequency stimulation (HFS; 100 Hz, 1 sec) in the CA1 region. The magnitude of LTP in GluN3A KO slices was significantly and constant smaller than that in WT slices. Bar graphs show quantified results at the 50 min time point. On the right, the two-line graphs show paired-pulse facilitation (PPF) induced by stimuli of various inter-stimulus intervals, which is a reflection of presynaptic plasticity. In Two-way ANAVA analysis, the main factor of genetic background (WT vs. KO) showed significant difference (C; *P<0.05), suggested significantly reduced synaptic facilitation/plasticity in GluN3A KO slices at 6–7 months compared to age-matched WT controls. No statistical significance was found between GluN3A KO and WT at 10–12 months old (n=5 animals per group; ***P<0.001, *P<0.05 vs. WT; Two-way ANOVA).
Synaptic disruption is a key pathological step in the early development of AD and proposed to be a Ca2+-dependent cellular degeneration in AD [87]. Western blotting showed that in brains from 3-months old mice, there was no difference in the expression level of the presynaptic protein synapsin between GluN3A KO group and age-matched WT group (Fig. 1C). At 7 months of age, however, a significantly lower level of synapsin was detected in the hippocampus of GluN3A KO mice comparing to WT controls (Fig. 1C). There was no significant difference in the expression of synaptophysin and PSD-95 between WT and KO brains (Suppl. Fig. 1A and 1B).
We previously reported that young adult GluN3A KO mice (2–4-month old) exhibited enhanced LTP compared to age-matched WT mice [68]. To examine whether the chronic hyperexcitability state and the synapsin decrease with aging might disrupt synaptic functions, LTP in the hippocampal slice from aging GluN3A KO mice (6–7-month old) was recorded. In striking contrary to young animals, LTP in the hippocampus of older ages was evidently suppressed (Fig. 1D). The impaired LTP induction persisted in brain slices of aging GluN3A KO mice of 9–12-month old (Fig. 1E). Paired-pulse facilitation (PPF) was recorded to detect whether the deteriorated synaptic plasticity was attributable to a pre-synaptic abnormality. The PPF, induced by various inter-stimulus intervals, confirmed a defect in synaptic facilitation of transmitter release in the hippocampus of aging GluN3A KO mice (6–12-month old) compared to age-matched WT brains (Fig. 1D and 1E).
3.3. Disrupted Ca2+ homeostasis in the GluN3A KO brain
To verify whether the chronic hyperactivity of NMDARs might disrupt tonic Ca2+ homeostasis, the basal level of [Ca2+]i in brain slices was measured using Fura-2-AM two-photon imaging (Fig. 2A–2B). Average [Ca2+]i values of hippocampal CA1 and dented gyrus (DG) cells of GluN3A KO mice were generally higher, but statistically not significant compared to WT cells due to large variations (Table 1). In the Fura-2 ratio measurement, the value in DG neurons of GluN3A KO mice (10–12-month old) was significantly greater than that of WT neurons (Fig. 2B). We compared the percentage of cells with [Ca2+]i higher than 200 nM. The percentage of high [Ca2+]i cells showed a strong indication of increase in CA1 of 6–7-month old GluN3A KO mice (P=0.058 vs. WT). High [Ca2+]i cells in DG of 10–12-month old GluN3A KO mice was significantly increased compared to age-matched WT mice (Fig. 2C).
Figure 2. Elevated intracellular Ca2+ and Ca2+-dependent signaling in the GluN3A KO brain.
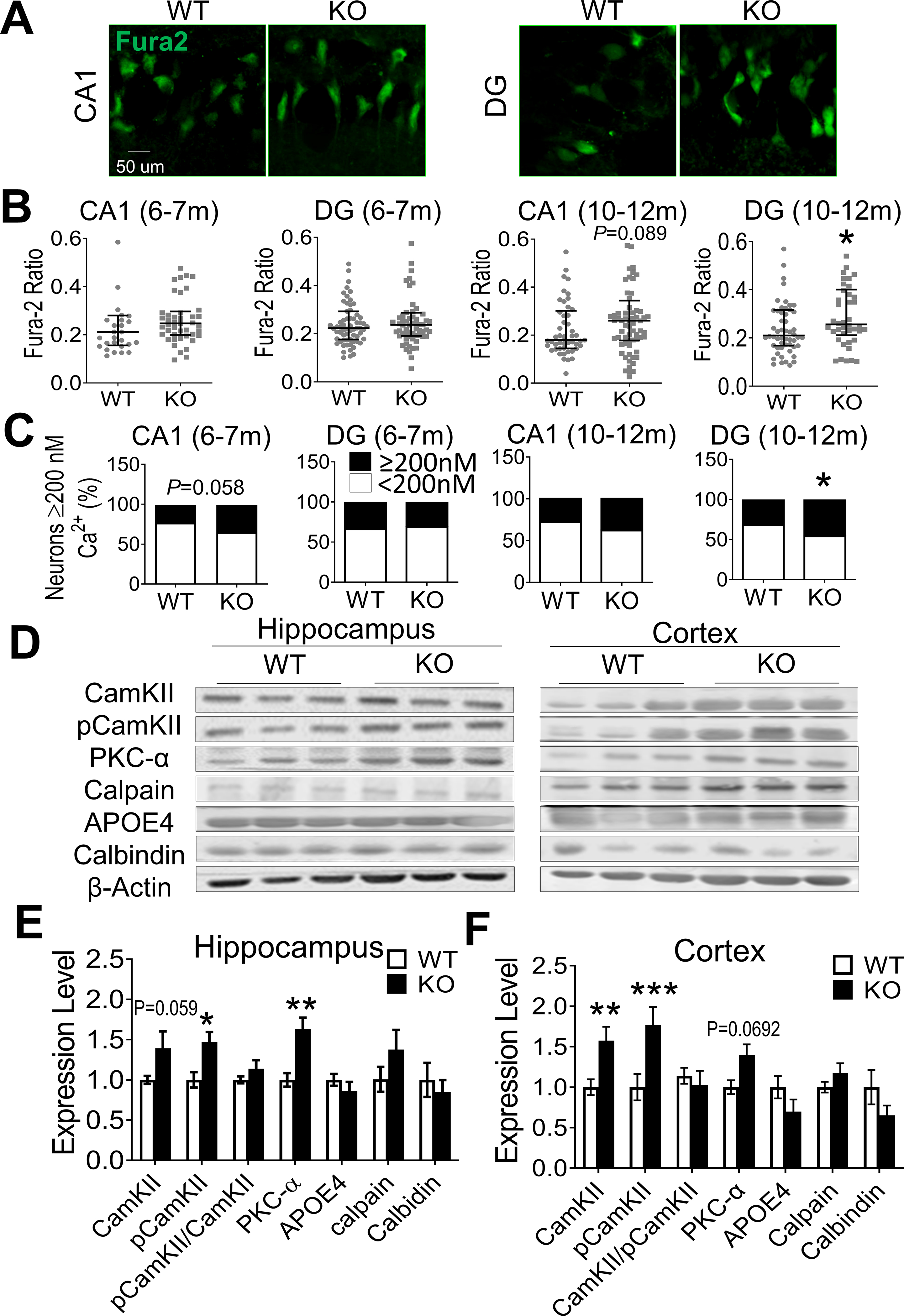
Intracellular free Ca2+ concentration [Ca2+]i and Ca2+-dependent signaling proteins were measured in the hippocampus and cortex of WT and GluN3A KO mice of 6–12-month old. A. In situ Ca2+ imaging using Fura-2AM was performed under a two-photon microscope. Images show Fura-2AM labeled CA1 and DG neurons in brain slices from WT and GluN3A KO mice. B. The Fura-2AM ratio measurements of [Ca2+]i in WT and GluN3A KO neurons at 6–7 month and 10–12-month old n=4 and 5 for WT and KO mice of 6–7-months group, respectively; n=4 animals for both WT and KO mice of 10–12-months group; *P<0.05 vs. WT; Mann-Whitney U test). C. The percentage of CA1 and DG cells with [Ca2+]i above and below 200 nM in total cells measured. CA1 neurons of 6–7-months old GluN3A KO mice showed a strong trend of greater percentage of high Ca2+ neurons compared to WT cells (P=0.05). At the age of 10–12-month old, the percentage of high Ca2+ cells was significantly larger than that in WT mice (animal numbers were the same as in B; *P<0.05 vs. WT; Chi-Squared test). D. Western blotting of hippocampus and cortex tissues from aging WT and GluN3A KO mice (10–12-month old). E and F. Summary bar graphs of Western blot data. In the hippocampus, the expression of pCaMKII and PKC-α increased significantly in GluN3A KO mice while CaMKII and calpain showed trends of increase (E). In the cortex of GluN3A KO mice, the expression of CaMKII and pCaMKII was significantly higher while PKC-α remained tentatively higher compared to levels in WT (n=6 animals per group; *P<0.05 vs. WT, **P<0.01 vs. WT; Two-way ANOVA).
Table 1.
Basal intracellular Ca2+ concentration in WT and GluN3A KO neurons
| Brain region | Age | WT [Ca2+]i (nM) | N | GluN3A KO [Ca2+]i (nM) | N | P value |
|---|---|---|---|---|---|---|
| CA1 | 6–7 month | 71.6±39.3 | 51 cells/4 mice | 133.3±31.5 | 51 cells/5 mice | 0.09 |
| 10–12 month | 61.7±26.1 | 53 cells/4 mice | 107.6±24.5 | 57 cells/4 mice | 0.19 | |
| DG | 6–7 month | 72.2±31.2 | 51 cells/4 mice | 153.0±43.1 | 51 cells/5 mice | 0.12 |
| 10–12 month | 62.4±24.9 | 53 cells/4 mice | 109.7±24.3 | 57 cells/4 mice | 0.17 |
The unpaired Student t test was performed in statistical analysis.
3.4. Ca2+-dependent molecular signaling and inflammatory factors in GluN3A KO mice
In the hippocampus of GluN3A KO mice of 6–7-month old, the ratio of phosphorylated Ca2+-calmodulin-dependent kinase II (pCaMKII) against total CaMKII was significantly elevated, while the expression of several Ca2+-dependent signals such as PKCα, calpain, and calbindin all showed trends of increases (Suppl. Fig. 1C and 1D). At older ages of 10–12-month old, the expression of pCaMKII and PKCα remained significantly higher or showed strong trends in the hippocampus and cortex of GluN3A KO mice comparing to WT controls (Fig. 2D–2F). Thus, the GluN3A KO brain presented a unique environment of persistent hyperexcitability, subtle but significant elevation of basal [Ca2+]i, impaired synaptic plasticity of pre-synaptic defect, and specific Ca2+-dependent signaling activities.
Disruption of Ca2+ homeostasis is associated with chronic inflammation, inducing productions of inflammatory cytokines and nitric oxide (NO)[88]. The pro-inflammatory factor TNFα significantly increased in the hippocampus of GluN3A KO mice of 6- and 12-month old, although interleukin-1β (IL-1β) and CCL2 (MCP) were unaltered (Fig. 3A–3C). The level of IL-6, an anti-inflammatory factor, was not changed at younger ages, but markedly decreased at 12-month old (Fig. 3D). GluN3A KO mice of 12-month-old displayed enhanced expression of inducible NO synthase (iNOS) (Fig. 3E). The NF-κB signaling, an upstream pathway in neuroinflammation[89], showed an early upregulation with increased NF-κB (p65) in the GluN3 KO hippocampus (Fig. 3F). Meanwhile, more GFAP-positive astrocytes and IBA-1-positive microglia were observed in the hippocampus of GluN3A KO mice of 6–12-month old (Fig. 3G–3H).
Figure 3. Inflammation and neuronal loss in the GluN3A KO brain.
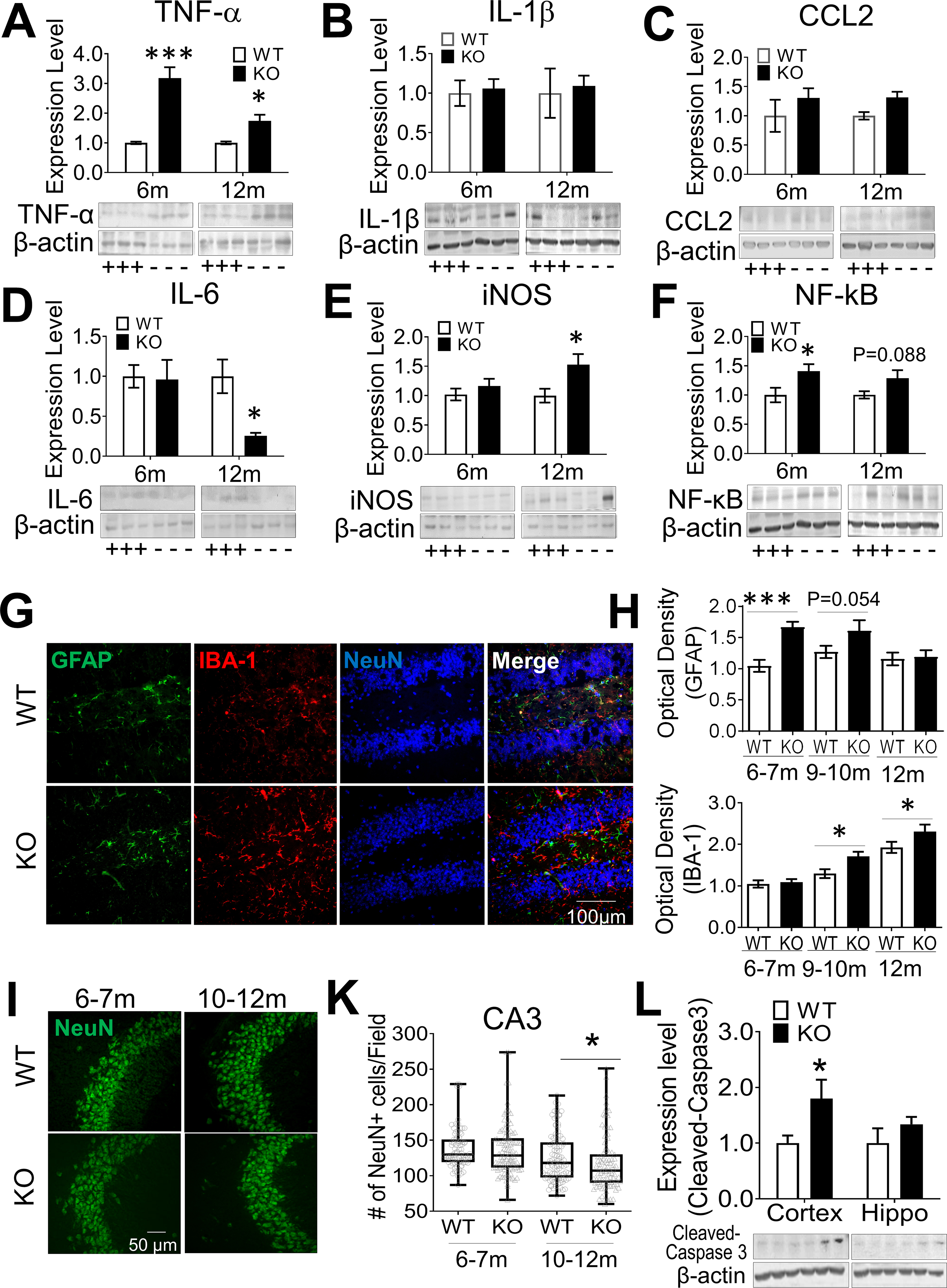
Chronic inflammation was examined in the hippocampus of WT and GluN3A KO mice of 6–12-month old. Western blot analysis was performed to measure the expression level of inflammatory factors. A-E. Quantified results showed significantly increased expression of TNFα, and iNOS in GluN3A KO mice (marked with “-“), compared to age-matched WT controls (marked with “+”). Meanwhile, the expression level of CCL2 and IL-1β remained unchanged and anti-inflammatory IL-6 was largely reduced in the GluN3A KO mice. F. The enhanced expression of NF-κB suggested the involvement of the NF-κB pathway in the chronic inflammation (n=4 animals in all groups; *P<0.05, **P<0.01, ***P<0.001 vs. WT controls; Two-way ANOVA). G. Representative GFAP and IBA-1 staining in the hippocampus of GluN3A KO and WT brains aged at 10–12-month. H. Increased numbers of GFAP-positive astrocytes and IBA-1-positive-microglia cells were detected in the hippocampus of GluN3A KO mice, compared to the age-matched controls (n=3 animals in all groups; *P<0.05, ***P<0.001 vs. WT controls; Two-way ANOVA ). I and K. NeuN staining and cell counting were performed to assess neuronal cell loss. At younger ages of 6–7-month old, no significant reduction of neuronal cells was detected in the CA1 and CA3 regions (also see Suppl. Fig. 2B–2C). At the elder age of 10–12-month old, the number of NeuN-positive cells in the CA3 of GluN3A KO mice was significantly less compared to WT mice (n=5 animals in WT group, 7 in KO group; Median ± Interquartile range; *P<0.05 vs. WT, Mann-Whitney U test). L. Western blotting for the expression of cleaved-caspase 3 in the hippocampus and cortex of WT and GluN3A KO mice at 10–12-month old. Cleaved caspase-3 was significantly increased in the cortex of GluN3A KO mice (n=8 per group; *P<0.05 vs. WT; Two-way ANOVA). See Suppl. Fig. 5B–5C for the pro-caspase 3 result.
3.5. Neuronal loss in the aging GluN3A KO brain
We previously reported neurodegeneration of olfactory sensory neurons in GluN3A KO mice of 2–3-month old [67]. In the present investigation, significant TUNEL-positive cells were detected in the olfactory bulb of 10–12-month old GluN3A KO mice but not in age-matched WT controls (Suppl. Fig. 2A). In adult (6–7-month old) mice of both groups, there was no significant cell death in the hippocampus (Suppl. Fig. 2B–2C, Fig. 3I–3K) and cortex (data not shown). In aging (10–12-month old) GluN3A KO mice, however, NeuN-positive cells significantly decreased in the CA3 region (Fig. 3I–3K). Western blotting identified a moderate but significant increase of apoptotic marker cleaved caspase-3 in the GluN3A KO cortex of 10–12-month old compared to age-matched WT mice (Fig. 3L). The pro-caspase-3 level was unchanged (Suppl. Fig. 2D–2E).
3.6. Olfactory dysfunction and psychological alterations in young adult GluN3A KO mice
Olfactory deficit is an early symptom of neurodegenerative diseases including AD [67]. GluN3A KO mice of different ages were subjected to the olfactory habituation/dishabituation test. When exposed to sequential odors, WT mice increased sniff behavior when different odors were presented, while this increase was absent in GluN3A KO mice of 2–5-month old (Suppl. Fig. 3A–3B). In the buried food test, GluN3A KO mice of 5- and 7-month old took significantly longer latency to find buried food compared to WT mice (Suppl. Fig.3C).
AD patients often develop psychological problems such as anxiety and social barriers [80]. The open field test was used to assess anxious-like behavior [68]. Although WT and GluN3A KO mice of 3-month old behaved normally, aging GluN3A KO mice (7–12-month old) traveled much less in the center area (Suppl. Fig. 3D). In the sociability assay of the three-chamber test, WT mice displayed a preference to visit the chamber hosting a naïve mouse (Suppl. Fig. 3E) and sniffed significantly longer around the stranger (Suppl. Fig. 3F). GluN3A KO mice of 5–12-month old lost these preferences (Suppl. Fig. 3E–3F). In the social novelty test, WT animals spent significantly more time with the novel stranger than with the familiar mouse, whereas GluN3A KO mice showed no preference (Suppl. Fig. 3G–3H).
3.7. Cognition decline in an age-dependent manner in GluN3A KO mice
Progressive memory loss and learning difficulty are hallmarks of AD and dementia. Spatial learning and memory functions were tested using the Morris water maze test [68, 86]. At adult ages of 5–6-month old, GluN3A KO mice started to show slower learning during the 5-day training, although they did not yet have significant difficulties in finding the platform compared to WT mice in the probe trial (Fig. 4A). Significant spatial working memory impairment was seen in aging GluN3A KO mice around and after 7-month old, showing significant less time and travel distance in the platform quarter compared to WT mice (Fig. 4B–4C).
Figure 4. Development of cognition decline in aging GluN3A KO mice.
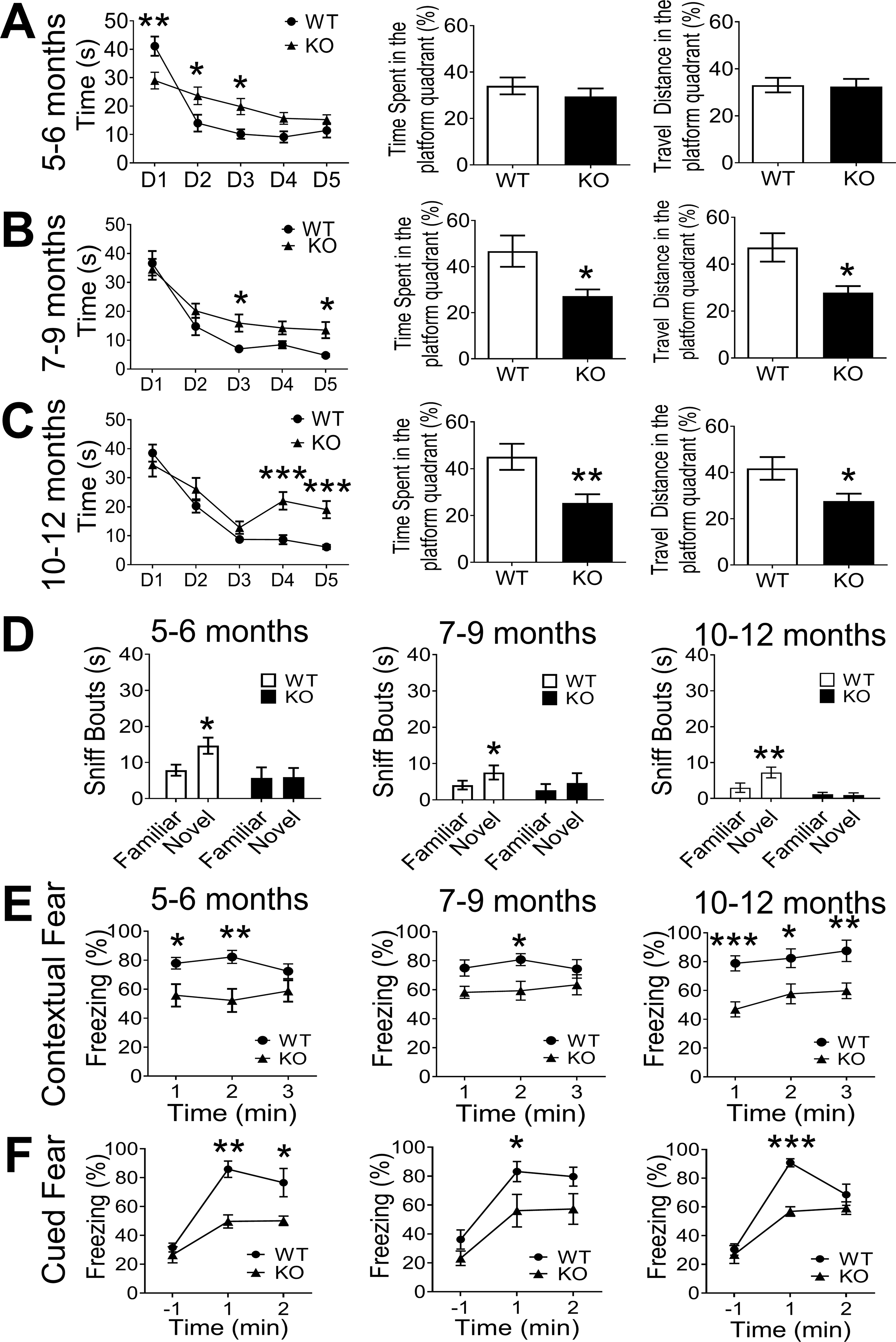
Cognitive function was tested in WT and Glu N3A KO mice at ages of 5 to 12-month old. A. Spatial learning and memory ability was tested using the Morris water maze test. At adult ages of 5–6-month old, GluN3A KO mice started to show slower learning ability during the 5 day trial protocol, yet the bar graphs indicates that, one day after the training, GluN3A KO mice did not have difficulties in memorizing the location of the platform. B. GluN3A KO mice of 7–9-month old developed slower learning activity during the 5-day training. The time spent and travel distances in the platform quarter of these animals were significantly less compared to WT mice. C. The water maze test was performed in mice at ages of 10–12-month old. GluN3A KO mice suffered significant difficulties in the spatial memory shown as a flat learning curve during the last 3 days of the training period. The bar graphs show marked deteriorations in identifying the platform quarter one day after training compared to WT mice (5–6m: n=12 in WT and n=12 in KO; 7–9m: n=12 in WT and n=9 in KO; 10–12m: n=14 in WT and n=7 in KO; *P<0.05, **P<0.01, ***P<0.001 vs. WT controls; Two-way ANOVA for training curve and Student’s t-test for memory recall test). D. In the novel object test, the mouse was allowed to explore 2 identical objects. On the test day, one of the training objects was replaced with a novel object. WT mice sniffed significantly more on the novel object at 1 hr after training, suggesting memory of previous object. GluN3A KO mice showed no preference on these objects (5–6m: n=8 in WT and n=8 in KO; 7–9m: n=8 in WT and n=8 in KO; 10–12m: n=9 in WT and n=8 in KO; *P<0.05, **P<0.01 vs. familiar object; Two-way ANOVA). E. In the context test, the test mouse was reintroduced to the conditioning chamber and recorded for 3 min without stimulation to evaluate the contextual fear. Comparing to age-matched WT mice, GluN3A KO mice aged at 5–9-month old showed significantly less freeze behavior during the first 2 min. Elderly GluN3A KO mice, moreover, had much less freeze behavior during the whole 3-min period. (5–6m: n=11 in WT and n=9 in KO; 7–9m: n=11 in WT and n=13 in KO; 10–12m: n=10 in WT and n=12 in KO; *P<0.05, **P<0.01, ***P<0.001 vs. WT controls; Two-way ANOVA. The main factor of genetic background (WT vs. KO) is significantly different across the three age groups). F. In the cued text, the test mice were introduced to a new chamber with different context background for 3 min, followed by the 30s tone cue (US). The cued fear was accessed by evaluating the freeze behavior after US. GluN3A KO mice aged at 5–12m showed significant less freezing for 2 minutes after US compared to age-matched WT ones. Thus, GluN3A KO mice developed poorer contextual and cued conditioning fear memory (5–6m: n=7 in WT and n=5 in KO; 7–9m: n=8 in WT and n=6 in KO; 10–12m: n=4 in WT and n=9 in KO; *P<0.05, **P<0.01, ***P<0.001 vs. WT controls; Two-way ANOVA and main factor of genetic background (WT vs. KO) is significantly different across the three age groups).
In the novel object recognition test, which requires acquisition, storage and recall of recognition memory of familiar objects [68], WT mice always exhibited preference of novel object over familiar ones, while GluN3A KO mice failed to show such preference in the retention task 1 hr after training (Fig. 4D). The impaired recognition memory was detected as early as 5-month old and continued with aging. The contextual and cued memory was evaluated by scoring freezing behavior 24 hrs after mice were trained with the conditioned stimulus-unconditioned stimulus (CS–US) pairing. Twenty-four hrs after conditioning, GluN3A KO mice of ≥5-month old presented significantly less freezing behavior than age-matched WT mice after reintroducing to the conditioning context and tone cue, suggesting poor fear memory (Fig. 4E–4F). GluN3A KO mice did not show deficit in short-term spatial memory in the Y maze spontaneous alternation test (Suppl. Fig. 4).
3.8. Spontaneously developed amyloid and tau pathology in the elderly GluN3A KO brain
There was little Aβ deposition in the 3-month old young adult brains of WT and GluN3A KO mice (Suppl. Fig. 5A). Immunohistochemical assays using the Aβ antibody against 1–42 peptides detected a few Aβ spots in the CA1 region of 6–7-month old GluN3A KO mice, which is not significant compared to age-matched WT controls (Fig. 5A–5B). Significant depositions of endogenous Aβ overlaid with neurons, reactive astrocytes and vasculatures were readily visible in the hippocampus of GluN3A KO mice of 10–15-month old of both sexes but not in age-matched WT mice (Fig. 5A–5C).
Figure 5. Amyloid and tau pathology in the aging GluN3A KO brain.
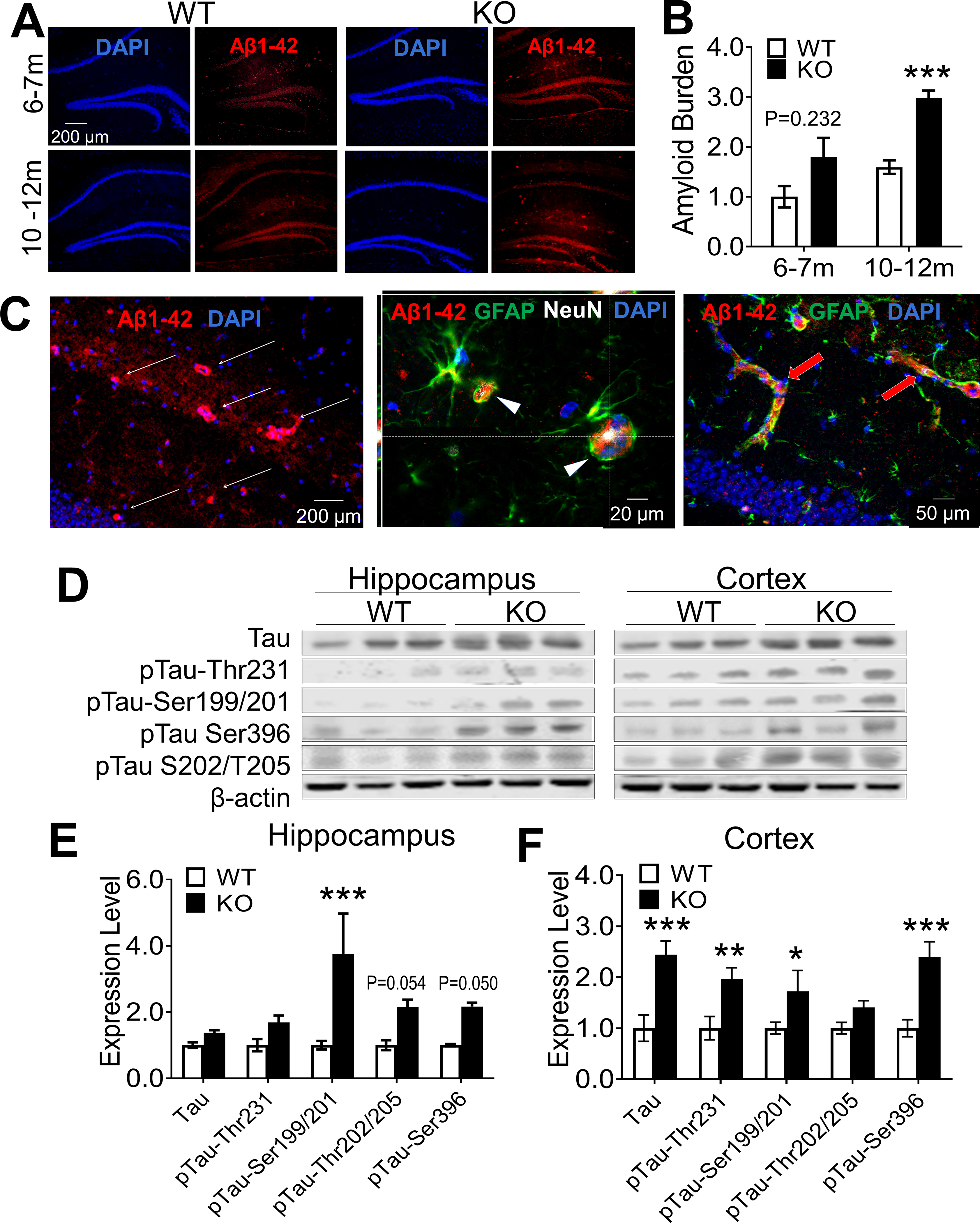
Aβ deposition and tau protein phosphorylation were examined in WT and GluN3A KO brains of 3 to 12 months old. A. Immunohistochemical staining using the amyloid antibody against the 1–42 peptide revealed Aβ depositions in the CA1 and DG of GluN3A KO mice of 6–12-month old (for negative result in the 3-month old brain, see Suppl. Fig. 5A). B. Significant Aβ accumulations were detected in the GluN3A KO brain of 10–12 months old (n=5 in WT and 6 in KO group; ***P<0.001 vs. WT; Two-way ANOVA). C. Images from brain section staining of female GluN3A KO mice of 15-months old. Deposition of Aβ 1–42 was seen in the hippocampus of GluN3A KO mice. The middle 3-D image shows co-labeling of Aβ (red) and NeuN (white) in the same cells, suggestive of intracellular deposition of Aβ in neuronal cells (arrowhead). Hypertrophic reactive astrocytes positive to GFAP staining (green) are frequently seen adjacent to Aβ-positive cells as seen in AD brains. The yellow color of GFAP-positive cells (green) also indicates Aβ deposition in astrocytes. The image on right shows Aβ distribution along vessel-like structures (red arrows). D. Western blot was applied to measure tau phosphorylation, including changes of some common phosphorylation sites of the tau protein. E and F. Summarized Western blot measurements of tau protein and phosphorylated forms in 10–12-month old mice. The phosphorylation at several common sites was significantly increased or tentatively increased in the hippocampus of GluN3A KO mice compared to levels in WT. Phosphorylation levels of most tau proteins tested was significantly increased in the cortex of GluN3A KO mice (n=6 per group; *P<0.05, **P<0.01, ***P<0.001 vs. WT; Two-way ANOVA and the main factor of genetic background (WT vs. KO) is significantly different in both hippocampus and cortex (E-F, P<0.001). For analysis of the ratio of p-tau against total proteins see Suppl. Fig. 6E–6F.
In Western blot analysis, the expression of amyloid precursor protein (APP) was similar between WT and GluN3A KO mice at 6–7-month old, while significantly increased APP was detected in the hippocampus of GluN3A KO mice of 10–12-month old (Suppl. Fig. 5B–5C). Meanwhile, no increases of tau phosphorylation were found in the hippocampus and cortex of younger GluN3A KO mice of 6–7-month old, with one exception that the total tau expression noticeably increased in the cortex (Suppl. Fig. 6A–6D). In 10–12-month old GluN3A KO brains, the total tau protein remained to be elevated in the cortex, and hyperphosphorylation was detected in several common phosphorylation sites of tau protein in both the hippocampus and cortex (Fig. 5D–5F). A further analysis on the ratio of p-tau against the total tau verified hyperphosphorylation of pTau-Ser199/201 in the hippocampus of GluN3A KO mice (Suppl. Fig. 6E–6F). Thus, Aβ and tau pathology emerged several months after cognition decline (~5 months old) in the GluN3A null animal.
3.9. Age-dependent AD-like symptoms in female GluN3A KO mice
Female GluN3A KO mice aged at 2–5-month old showed poor performance than WT females in the buried food and olfactory habituation/dishabituation test (Suppl. Fig. 7A–7B). In the water maze test, female GluN3A KO mice aged at 7–10-month displayed slower learning in the training phase (Suppl. Fig. 7C). Consistently, female GluN3A KO mice developed impaired recognition/visual memory and contextual fear memory detectable in the novel object and fear conditioning tests (Suppl. Fig. 7D–7E).
3.10. Early memantine treatment prevented olfactory and cognitive deficits of GluN3A KO mice
In adult GluN3A KO mice of 3-month-old when olfactory dysfunction was detected but no cognition deficits, MEM daily treatment was started at a clinically relevant dosage (10 mg/kg/day in drinking water) [90]. Age-matched GluN3A KO mice and WT mice received drug-free water as vehicle and normal controls, respectively. After 3-month treatments, the olfactory odor discrimination function of GluN3A KO mice in the MEM group recovered to the normal level of WT mice (Fig. 6A). In the Morris water maze test, MEM-treated mice performed better than vehicle controls in the 5-day training (Fig. 6B). The time spent and travel distance of MEM-treated mice in the platform quadrant tended to be marginal longer compared to vehicle controls (Fig. 6C–6D). In the fear conditioning test, MEM-treated KO mice displayed much improved memory of contextual fear compared to vehicle control mice (Fig. 6E). Cued fear memory was not improved by MEM (Fig. 6F). MEM did not show benefit in the novel objective recognition test that involves visual function and recognition memory [91](Fig. 6G), and no improvements in the three-chamber social test (Fig. 6H–6I) or anxious behavior in the open field test (data not shown).
Figure 6. Early and daily memantine treatment alleviated the development of AD-associated behaviors in GluN3A KO mice.
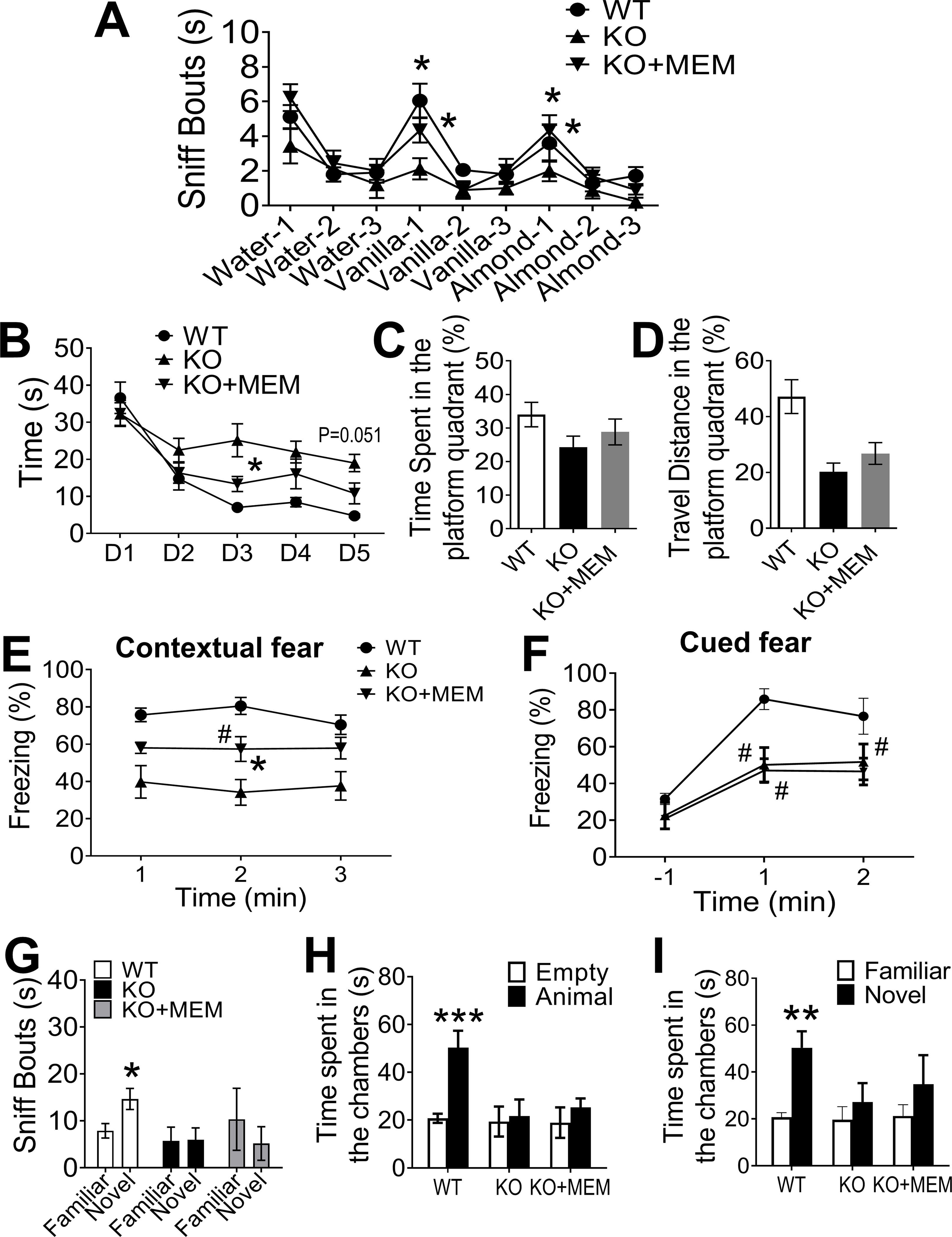
To test effects of daily chronic MEM treatments in GluN3A KO mice, MEM (10 mg/kg/day in drinking water) or vehicle control (regular water) was started from the preclinical stage of 3-month old when olfactory dysfunction had emerged but no detectable cognitive deficits. Functional and behavioral tests were performed after the daily treatment for 3 months. A. In the olfactory habituation/dishabituation test, the vehicle group of GluN3A KO mice continued to be insensitivity to the initial exposure to vanilla and almond. The MEM group, however, showed significant improvements in this test; the treatment basically reversed the functional deficit to the WT control level (WT: n=19, KO: n=9, KO+MEM: n=9; *P<0.05 KO+MEM vs. KO; One-way ANOVA). B. During the 5-day training of the water maze test, the chronic 3-month MEM treatment significantly improved the learning curve closer to WT control (WT: n=7, KO: n=8, KO+MEM: n=9; *P<0.05, KO+MEM vs. KO. Two-way ANOVA; the main factor between KO control and treatment groups showed significant difference (P<0.001). C and D. One day after the 5-day training period, GluN3A KO mice received the MEM treatment appeared to perform better (more time and travel distance in the platform quadrant), but the change was not statistically significant (same experimental groups as in B; One-way ANOVA). E and F. In the fear conditioning memory test, WT mice exhibited high rate (~80%) of freeze reaction due to normal contextual memory while GluN3A KO mice exhibited a low (~40%) freeze response as a result from losing the memory. The 3-month MEM treatment significantly improved the contextual memory of GluN3A KO mice, demonstrated by a significantly higher rate (~60%) of freeze behavior. Meanwhile, the cued memory was not improved in the GluN3A mice with MEM treatment (WT: n=10, KO: n=8, KO+MEM: n=8; *P<0.05 KO+MEM vs. KO; #P<0.05 KO+MEM vs. WT; Two-way ANOVA). G. MEM did not improve visual/recognition memory of GluN3A KO mice in the novel object test (WT: n=8, KO: n=8, KO+MEM: n=8). H and I. In the three-chamber test, the MEM treatment had no effect on the sociability of GluN3A KO mice (G); there appeared a trend of improvement in social novelty behavior with no statistical significance (H) (WT: n=9, KO: n=8, KO+MEM: n=9; ***P<0.001, animal vs. empty chamber, **P<0.01 Novel vs. Familiar object; Two-way ANOVA).
3.11. Reduced GluN3A expression in the brain of Alzheimer’s disease patients
We inspected NMDAR subunit levels in human brain tissues of AD patients and normal aging subjects. In Western blot assay of the frontal lobe, the expression of GluN1, GluN2A, GluN2B and GluN3A subunits showed no significant difference between AD and normal brains (Suppl. Fig. 8A–8B). In the occipital cortex, the GluN2A, 2B and GluN3A expressions were significantly lower in the AD brain while the GluN1 level was similar between AD and normal brains (Suppl. Fig. 8A and 8C).
3.12. Discussion, hypotheses and future direction
Previous investigations on the role of NMDARs in AD pathophysiology have exclusively focused on GluN1 and GluN2 subunits for how they might be affected by the amyloid cascade [92, 93]. We show for the first time that deficiency of GluN3A alone progressively evolves a series of AD-associated pathophysiology and functional deficits before detectable Aβ/tau pathologies. Unlike many transgenic AD mouse models, the GluN3A KO mouse develops virtually all major pathophysiology and age-dependent psychological, cognitive symptoms seen in AD patients. AD-related alterations in the human brain begin years before obvious signs of the disease (i.e. preclinical stage)[94]. The spontaneous progression was also evolved in the GluN3A KO mouse, with enhanced neuronal activities, marginally elevated resting [Ca2+]i, selective increases of Ca2+-regulated signaling molecules such as CamKII, PKCα, and calpain. The lasting disruption of Ca2+ homeostasis and activation of specific cellular signaling pathways generate a chronic pro-AD environment in the brain. Although the GluN3A deficiency induced [Ca2+]i increases did not cause instantaneous acute excitotoxicity, the persistent hyperexcitability of glutamatergic neurons ought to imposes a tonic Ca2+ burden conferring a subhealth status that slowly leads to degenerative excitotoxicity. Both male and female GluN3A KO mice similarly develop early onset olfactory dysfunction followed by dementia symptoms and thereafter Aβ/tau pathology. Supporting that continual hyperactivity of NMDARs is a risk/causal factor of AD, daily MEM treatment in GluN3A KO mice starting from the preclinical stage for 3 months ameliorated cognitive decline and even restored previously lost olfactory function. The clinical significance of the GluN3A-associated mechanism is supported by the human data showing reduced GluN3A protein level in the postmortem AD brain.
GluN3A KO mice are viable and fertile. In general, they breed normally and do not show significant phenotypic difference from WT littermates up to young adult age. After reaching adulthood, functional tests in these animals start to detect impairments in locomotor, olfactory and social activities [67, 68, 95]. Interestingly, on the other hand, enhanced learning/memory and greater LTP induction in hippocampal neurons were detected as a unique feature in early adulthood of GluN3A KO mice [68], which is consistent with hyperactivation of NMDARs before advancing to degenerative excitotoxicity. Beyond this age point (around and after 5-month old in this mouse model), a series of psychological and cognitive deficits as well as depressed LTP gradually cultivate with aging in the absence of GluN3A. It will be interesting for an epidemiological study to examine whether similar unique biphasic cognitive function and behavioral features exist during the lifespan of people who bear mutated GRIN3 genes or those having been diagnosed with AD or dementia.
GluN3A was observed in the pre- and post-synaptic components of glutamatergic neurons and may regulate transmitter release [96]. In GluN3A KO brain slices, we detected a pre-synaptic deficit in synaptic plasticity in PPF recordings. This is highly consistent with the significant decrease of presynaptic proteins synapsin in the aging GluN3A KO brain. Other studies perceived the location of GluN3A to both extrasynaptic and peri-synaptic membrane domains [61]. This complexity implies that both synaptic and extrasynaptic NMDAR activity must be kept in check for normal brain function. It will be intriguing to delineate the relative role of GluN3A across different synaptic locations in AD development.
The molecular regulation and long-term modulation of NMDAR activity in AD are poorly understood. Based on the popular belief that changes in Ca2+ signaling are secondary to deleterious actions of Aβ/tau/PS pathogenesis [97], investigations testing the role of Ca2+ in AD have been predominantly focused on the relationship between Aβ/tau/PS and Ca2+ regulation. Specifically, investigations have closely inspected interactions between the amyloidogenic pathway with membrane channels, receptors and intracellular Ca2+ regulatory proteins in the ER and mitochondria [25, 98–100]. Information from these investigations is valuable for understanding FAD and the later stage of SAD, but may not be relevant to the pathogenesis of SAD. Most previous studies on Ca2+ dysregulation were performed in transgenic mouse models resembling FAD [25, 98–100]. These observations do not resolve the clinical disconnect between Aβ pathology and cognitive status of aging/aged people. Whether disruption of Ca2+ homeostasis is a cause of AD development in an Aβ-independent manner has rarely been questioned or explored. This is likely due to the domination of the amyloid hypothesis and the lack of evidence of an Aβ-independent casual mechanism responsible for chronic Ca2+ dyshomeostasis and AD development.
Our results support the Ca2+ hypothesis for AD in terms of that NMDAR-mediated Ca2+ dyshomeostasis appears to generate a chronic state of degenerative excitotoxicity leading to AD development and progression. In addition, we provide the first clear demonstration that a sustained Ca2+ dyshomeostasis and the resulted final common degenerative pathway is due to the deficiency of the NMDAR inhibitory subunit GluN3A. Departing from current view of the Ca2+ hypothesis, we show that the disruption of Ca2+ regulation is an upstream initiating pathophysiology before and throughout AD progression, but not triggered by the production of any known FAD genes. The amyloid cascade hypothesis has implied that amyloid pathology must precede the clinical symptoms. This is neither the case for sporadic AD development in many AD patients nor in the GluN3A KO mouse. During the early progression in the SAD brain, there was no evidence of amyloid/tau pathology or alterations of expression levels of APP and ApoE. There were also no evidence supporting crosstalk between the amyloid cascade and Ca2+ regulation during this period. Nevertheless, Aβ deposition and tau hyperphosphorylation did occur later in the GluN3A null brain, in harmony with the definition of AD. Presenilin was not measured in these experiments, it may be necessary to delineate whether the production of PS1/PS2 and/or their mutation may contribute to Ca2+-mediated degenerative excitotoxicity.
There are other mechanisms that may affect sustained Ca2+ homeostasis. Theoretically, any chronic aberrant cellular and subcellular activities or dysfunction may cause lasting hyperactivity of glutamatergic neurons resulting in sustained Ca2+ dysregulation. A few possible examples may include increased expression or function of NMDAR GluN1 and GluN2 subunits, impaired Ca2+ buffering systems in cellular organelles including the ER and mitochondria [25, 98–100], soluble Aβ oligomers [101], aberrant neuronal and non-neuronal activities [100], etc. Our observation does not exclude interplays or interactions among signaling pathways such as mitochondrial dysfunction during the AD process. Optimal and constant energy supply is crucial for Ca2+ homeostasis and optimal performance of neuronal cells. Chronic burden on energy metabolism may generate a subhealth condition leading to hypoglycemia-like stress followed by neural dysfunction and degeneration. Future investigations should test whether and how these alterations induce the subhealth condition in early and middle AD stages independent of the amyloid cascade and develop possible interventions during different stages of the lifelong process.
There are clear distinctions between degenerative excitotoxicity and acute excitotoxicity in ischemic stroke. These may include 1) temporal differences that an ischemic attack causes sudden eruption of massive Ca2+ influx that is far beyond the capacity of intracellular Ca2+ buffering pools within a time scale of minutes to hours, leading directly to necrotic death (the dominate form of cell death in stroke) and activation of delayed programmed death cascades in following days if the cells survive from the initial attack [102–104]. In the case of AD, there is no rapid collapse of Ca2+ homeostasis while the cellular Ca2+ regulatory systems are continuously activated to maintain the resting [Ca2+]i close to the normal level while gradually losing the capability under the chronic stress; 2) the extent of Ca2+ elevation is dramatic after an ischemic insult (from low nM concentrations up to at least μM levels) while degenerative excitotoxicity is associated with much more moderate Ca2+ elevation (≥200 nM range) that is not immediately lethal but chronically stressful and detrimental via downstream signaling; 3) the spatial control of cellular Ca2+ in acute excitotoxicity is generally wiped out by Ca2+ overflow while during degenerative excitotoxicity intracellular Ca2+ pools and buffering systems may retain spatial specificity in subcellular locations, at full strength and then partially, for an extended time period (months in rodents and years in humans). Localized Ca2+ signals in dendritic spines and processes are central to proper neuronal activity and synaptic functioning [105, 106]; disruption of localized Ca2+ regulation can be a characteristic of degenerative excitotoxicity and requires further investigations with imaging tools; 4) related to the above features, the type of neuronal injury and cell death is significantly different. Necrosis is the dominant form of acute neuronal damage. On the other hand, degenerative excitotoxicity involves delayed and continuous programmed deteriorations caused by inflammatory factors, metabolic stress, apoptosis, autophagy, and possibly Ca2+-dependent necrotic component. Most likely, the prolonged process allows activation of multiple signaling pathways and injury cascades in the same cells, ultimately evolving to a form of hybrid (mixed) cell death as seen chronically after stroke and energy deficiency [48, 103, 107]. Both neuronal and non-neuronal cells may be affected directly or indirectly by degenerative excitotoxity throughout AD progression, which needs to be examined in future investigations; 5) in ischemic stroke, Ca2+ dysregulation is largely restricted to the ischemic region, while in the AD brain, sustained dysregulation of Ca2+ homeostasis may affect multiple brain regions and leads to a global interruption in cellular/structural functions [108]; 6) degenerative excitotoxicity may selectively affect vulnerable brain regions and specific neuronal subtypes due to different GluN3A expression and unique patterns of Ca2+ signaling alterations that closely tie to disease symptoms. Thus, regional and cell type specificity may explain pathogenesis for other neurodegenerative diseases such as Parkinson’s disease [22, 108]. The possibility that NMDAR-mediated, Ca2+ dyshomeostasis-induced degenerative excitotoxicity may have broader implications in neuronal system failure beyond AD and dementia may be considered and explored.
We examined NMDAR subunit expression in human brain samples from AD patients. This initial effort was limited to two cortical regions, yet we still detected a significantly reduced GluN3A protein level in the occipital lobe (OL), where visual information is processed [109]. Visual impairment is a common symptom of AD and prevalently reported as an early sign of the disease [110]. In our study, object recognition deficits were detected in GluN3A KO mice, suggesting impaired vision memory and a potential damage of occipital-temporal connections. The significance of observed reductions of GluN3A and GluN2 levels in the occipital cortex merits further investigations, while possible changes of GluN3A expression in other AD brain regions such as the hippocampus should be examined.
Considering that 2–10% of humans bear mutated/deficient GRIN3 genes, and similar rates of AD prevalence worldwide [71–73, 75], it appears necessary to study GRIN3A variants in different human populations. The relationship between GRIN3A mutations/low GluN3A level to AD should be examined and genetic approaches for the GluN3A deficiency may be developed. As a first step, a genetic survey of GRIN3A variants and GluN3A expression should be performed in AD patients. Alternative/additional strategies for NMDAR modulations to overcome degenerative excitotoxicity should also be considered. Meanwhile, channelopathy of other Ca2+ permeable channels can play roles in the process and remains to be further investigated.
It is recognized that treatments that show even moderate effect in a key domain of AD (cognition, function/behavior) can have significant clinical impacts. Current FDA guideline and clinical trials of MEM therapy are focused on symptomatic treatments for moderate-to-severe AD/dementia, with the Mini–Mental-State-Examination (MMSE) score <20. According to the new evidence in the Ca2+ hypothesis, this late stage therapy misses the early pathogenic phase of the disease. More clinical trials with MEM monotherapy in preclinical/mild AD/dementia patients and pre-symptomatic people susceptible to AD should be performed to validate the therapeutic benefits observed in the SAD model. This can be done with early biomarkers/neuroimaging combined with NMDAR assays and/or olfactory deficits in high risk populations or older people with MMSE score of 21–26. This approach may provide an evidence-based anti-dementia/anti-AD treatment to delay and ameliorate the disease progression. Future basic and clinical research should evaluate how MEM prevents AD/dementia pathology at the cellular and molecular levels. The therapeutic window as well as long-term effects of this treatment should be verified to improve the therapeutic efficacy.
CONCLUSIONS
This investigation provides the first evidence to suggest that a sporadic AD cascade can be initiated by the deficiency of the single NMDAR regulatory subunit GluN3A. The result strongly supports the Ca2+ hypothesis that a lifelong moderate but sustained Ca2+ overload is a causal pathogenic mechanism of sporadic AD. The GluN3A KO mouse provides a unique animal model closer to sporadic features in most AD patients; and will be useful for mechanistic and therapeutic investigations. We believe the concepts of subhealth status in seemingly healthy individuals and degenerative excitotoxicity in the AD development will help to better explain complicated and sustained conditions in SAD and other neurodegenerative diseases. The therapeutic benefits from preventive MEM treatment are consistent evidence for the pathogenic role of NMDAR hyperactivity and implicate an exciting possibility to delay, ameliorate, or even prevent AD symptoms from early phases of the disease. In addition to pharmacological approaches, genetic strategies may be explored to target the modulation/manipulation of the GluN3 gene GRIN3A. Aberrant glutamate release and impaired clearance are potential mechanisms for hyperactivation of NMDARs. More research should focus on glutamate and NMDA induced chronic hyperexcitability and the regulation of sustained Ca2+ homeostasis as a unique pathogenic mechanism independent from the production of Aβ or the interaction with amyloid cascades.
Supplementary Material
Acknowledgments:
This study was supported by NIH grants NS057255 (SPY),NS099596 (LW and SPY), NS091585 (LW),VA Merit grant RX001473 (SPY), AHA Pre-doctoral Fellowship Award AHA 0840110N (MQJ),the O. Wayne Rollins Endowment fund (SPY) and John E. Steinhaus Endowment fund (LW). It was also supported by an NIH equipment grant S10 OD021773 (KB). We acknowledge the Brain Tissue Bank in the Goizueta Alzheimer’s Disease Research Center at Emory University for providing human brain samples in this investigation. We also acknowledge the Viral Vector Core of Emory Neuroscience NINDS Core Facilities supported by an NIH grant P30NS055077. We appreciate the editing and proofread of the manuscript by Takira Estaba during the final revision.
Abbreviations:
- AD
Alzheimer’s disease
- APP
Amyloid precursor protein
- ApoE
Apolipoprotein E
- aCSF
Artificial cerebrospinal fluid
- Aβ
β-amyloid
- CaMKII
Ca2+-calmodulin-dependent kinase II
- CNS
Central nervous system
- fEPSP
Field excitatory postsynaptic potentials
- HFS
High frequency stimulation
- IACUC
Institutional Animal Care and Use Committee
- IL-1β
Interleukin 1β
- IL-6
Interleukin 6
- IL-10
Interleukin 10
- [Ca2+]I
Intracellular free calcium concentration
- KO
Knockout
- LTP
Long-term potentiation
- MEM
Memantine
- MMSE
Mini–Mental State Examination
- MCI
Mild/moderate cognitive impairment
- MEA
MultiElectrode Array
- NIH
National Institutes of Health
- NF-κB
Nuclear factor kappa B
- NO
Nitric oxide
- NMDA receptor
N-methyl-D-aspartate receptor
- OL
Occipital lobe
- PPF
Paired-pulse facilitation
- PACCH
Pro-AD condition of chronic hyperexcitability
- TNFα
Tumor necrotic factor α
- WT
Wild type
Footnotes
Competing interests:
The authors report no competing interests.
References
- [1].Huang Y, Mucke L. Alzheimer mechanisms and therapeutic strategies. Cell. 2012;148:1204–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Selkoe DJ. Amyloid beta protein precursor and the pathogenesis of Alzheimer’s disease. Cell. 1989;58:611–2. [DOI] [PubMed] [Google Scholar]
- [3].Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256:184–5. [DOI] [PubMed] [Google Scholar]
- [4].Caselli RJ, Knopman DS, Bu G. An agnostic reevaluation of the amyloid cascade hypothesis of Alzheimer’s disease pathogenesis: The role of APP homeostasis. Alzheimers Dement. 2020;16:1582–90. [DOI] [PubMed] [Google Scholar]
- [5].Tanzi RE, Gusella JF, Watkins PC, Bruns GA, St George-Hyslop P, Van Keuren ML, et al. Amyloid beta protein gene: cDNA, mRNA distribution, and genetic linkage near the Alzheimer locus. Science. 1987;235:880–4. [DOI] [PubMed] [Google Scholar]
- [6].Thinakaran G The role of presenilins in Alzheimer’s disease. J Clin Invest. 1999;104:1321–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Karch CM, Cruchaga C, Goate AM. Alzheimer’s disease genetics: from the bench to the clinic. Neuron. 2014;83:11–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Alzheimer’s Association Calcium Hypothesis W. 2020 Alzheimer’s disease facts and figures. Alzheimers Dement 2020;Epub ahead of print. :doi: 10.1002/alz.12068. [DOI] [Google Scholar]
- [9].Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–6. [DOI] [PubMed] [Google Scholar]
- [10].Kazee AM, Johnson EM. Alzheimer’s Disease Pathology in Non-Demented Elderly. J Alzheimers Dis. 1998;1:81–9. [DOI] [PubMed] [Google Scholar]
- [11].Schmitt FA, Davis DG, Wekstein DR, Smith CD, Ashford JW, Markesbery WR. “Preclinical” AD revisited: neuropathology of cognitively normal older adults. Neurology. 2000;55:370–6. [DOI] [PubMed] [Google Scholar]
- [12].Goldman WP, Price JL, Storandt M, Grant EA, McKeel DW Jr., Rubin EH, et al. Absence of cognitive impairment or decline in preclinical Alzheimer’s disease. Neurology. 2001;56:361–7. [DOI] [PubMed] [Google Scholar]
- [13].Herrup K The case for rejecting the amyloid cascade hypothesis. Nat Neurosci. 2015;18:794–9. [DOI] [PubMed] [Google Scholar]
- [14].Ricciarelli R, Fedele E. The Amyloid Cascade Hypothesis in Alzheimer’s Disease: It’s Time to Change Our Mind. Curr Neuropharmacol. 2017;15:926–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kametani F, Hasegawa M. Reconsideration of Amyloid Hypothesis and Tau Hypothesis in Alzheimer’s Disease. Front Neurosci. 2018;12:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Morris GP, Clark IA, Vissel B. Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer’s disease. Acta Neuropathol Commun. 2014;2:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Liu PP, Xie Y, Meng XY, Kang JS. History and progress of hypotheses and clinical trials for Alzheimer’s disease. Signal Transduct Target Ther. 2019;4:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bezprozvanny I, Mattson MP. Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci. 2008;31:454–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Marx J Alzheimer’s disease. Fresh evidence points to an old suspect: calcium. Science. 2007;318:384–5. [DOI] [PubMed] [Google Scholar]
- [20].Canzoniero LM, Snider BJ. Calcium in Alzheimer’s disease pathogenesis: too much, too little or in the wrong place? J Alzheimers Dis. 2005;8:147–54; discussion 209–15. [DOI] [PubMed] [Google Scholar]
- [21].Alzheimer’s Association Calcium Hypothesis Work Group. Calcium Hypothesis of Alzheimer’s disease and brain aging: A framework for integrating new evidence into a comprehensive theory of pathogenesis. Alzheimers Dement. 2017;13:178–82 e17. [DOI] [PubMed] [Google Scholar]
- [22].Khachaturian ZS. Hypothesis on the regulation of cytosol calcium concentration and the aging brain. Neurobiol Aging. 1987;8:345–6. [DOI] [PubMed] [Google Scholar]
- [23].Khachaturian ZS. Calcium, membranes, aging, and Alzheimer’s disease. Introduction and overview. Ann N Y Acad Sci. 1989;568:1–4. [DOI] [PubMed] [Google Scholar]
- [24].Khachaturian ZS. Calcium hypothesis of Alzheimer’s disease and brain aging. Ann N Y Acad Sci. 1994;747:1–11. [DOI] [PubMed] [Google Scholar]
- [25].LaFerla FM. Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat Rev Neurosci. 2002;3:862–72. [DOI] [PubMed] [Google Scholar]
- [26].Demuro A, Parker I, Stutzmann GE. Calcium signaling and amyloid toxicity in Alzheimer disease. J Biol Chem. 2010;285:12463–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Dreses-Werringloer U, Lambert JC, Vingtdeux V, Zhao H, Vais H, Siebert A, et al. A polymorphism in CALHM1 influences Ca2+ homeostasis, Abeta levels, and Alzheimer’s disease risk. Cell. 2008;133:1149–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Yin J, VanDongen AM. Enhanced Neuronal Activity and Asynchronous Calcium Transients Revealed in a 3D Organoid Model of Alzheimer’s Disease. ACS Biomater Sci Eng. 2021;7:254–64. [DOI] [PubMed] [Google Scholar]
- [29].Heck A, Fastenrath M, Coynel D, Auschra B, Bickel H, Freytag V, et al. Genetic Analysis of Association Between Calcium Signaling and Hippocampal Activation, Memory Performance in the Young and Old, and Risk for Sporadic Alzheimer Disease. JAMA Psychiatry. 2015;72:1029–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Rubio-Moscardo F, Seto-Salvia N, Pera M, Bosch-Morato M, Plata C, Belbin O, et al. Rare variants in calcium homeostasis modulator 1 (CALHM1) found in early onset Alzheimer’s disease patients alter calcium homeostasis. PLoS One. 2013;8:e74203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Bootman MD. Calcium signaling. Cold Spring Harb Perspect Biol. 2012;4:a011171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kawamoto EM, Vivar C, Camandola S. Physiology and pathology of calcium signaling in the brain. Front Pharmacol. 2012;3:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Koizumi S, Bootman MD, Bobanovic LK, Schell MJ, Berridge MJ, Lipp P. Characterization of elementary Ca2+ release signals in NGF-differentiated PC12 cells and hippocampal neurons. Neuron. 1999;22:125–37. [DOI] [PubMed] [Google Scholar]
- [34].Choi DW. Excitotoxic cell death. J Neurobiol. 1992;23:1261–76. [DOI] [PubMed] [Google Scholar]
- [35].Stutzmann GE. RyR2 calcium channels in the spotlight-I’m ready for my close up, Dr. Alzheimer! Cell Calcium. 2021;94:102342. [DOI] [PubMed] [Google Scholar]
- [36].Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol. 2000;1:11–21. [DOI] [PubMed] [Google Scholar]
- [37].Greger IH, Mayer ML. Structural biology of glutamate receptor ion channels: towards an understanding of mechanism. Curr Opin Struct Biol. 2019;57:185–95. [DOI] [PubMed] [Google Scholar]
- [38].Morris RG. NMDA receptors and memory encoding. Neuropharmacology. 2013;74:32–40. [DOI] [PubMed] [Google Scholar]
- [39].Vyklicky V, Korinek M, Smejkalova T, Balik A, Krausova B, Kaniakova M, et al. Structure, function, and pharmacology of NMDA receptor channels. Physiol Res. 2014;63:S191–203. [DOI] [PubMed] [Google Scholar]
- [40].Choi DW, Koh JY, Peters S. Pharmacology of glutamate neurotoxicity in cortical cell culture: attenuation by NMDA antagonists. J Neurosci. 1988;8:185–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Michaels RL, Rothman SM. Glutamate neurotoxicity in vitro: antagonist pharmacology and intracellular calcium concentrations. J Neurosci. 1990;10:283–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Choi DW. Excitotoxicity: Still Hammering the Ischemic Brain in 2020. Front Neurosci. 2020;14:579953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Rajdev S, Reynolds IJ. Calcium green-5N, a novel fluorescent probe for monitoring high intracellular free Ca2+ concentrations associated with glutamate excitotoxicity in cultured rat brain neurons. Neurosci Lett. 1993;162:149–52. [DOI] [PubMed] [Google Scholar]
- [44].Kiedrowski L N-methyl-D-aspartate excitotoxicity: relationships among plasma membrane potential, Na(+)/Ca(2+) exchange, mitochondrial Ca(2+) overload, and cytoplasmic concentrations of Ca(2+), H(+), and K(+). Mol Pharmacol. 1999;56:619–32. [DOI] [PubMed] [Google Scholar]
- [45].Lee JH, Wei ZZ, Chen D, Gu X, Wei L, Yu SP. A neuroprotective role of the NMDA receptor subunit GluN3A (NR3A) in ischemic stroke of the adult mouse. Am J Physiol Cell Physiol. 2015;308:C570–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015;14:388–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Marttinen M, Paananen J, Neme A, Mitra V, Takalo M, Natunen T, et al. A multiomic approach to characterize the temporal sequence in Alzheimer’s disease-related pathology. Neurobiol Dis. 2019;124:454–68. [DOI] [PubMed] [Google Scholar]
- [48].Xiao AY, Wang XQ, Yang A, Yu SP. Slight impairment of Na+,K+-ATPase synergistically aggravates ceramide- and beta-amyloid-induced apoptosis in cortical neurons. Brain Res. 2002;955:253–9. [DOI] [PubMed] [Google Scholar]
- [49].Peric A, Annaert W. Early etiology of Alzheimer’s disease: tipping the balance toward autophagy or endosomal dysfunction? Acta Neuropathol. 2015;129:363–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Li G, Xie F, Yan S, Hu X, Jin B, Wang J, et al. Subhealth: definition, criteria for diagnosis and potential prevalence in the central region of China. BMC Public Health. 2013;13:446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Texido L, Martin-Satue M, Alberdi E, Solsona C, Matute C. Amyloid beta peptide oligomers directly activate NMDA receptors. Cell Calcium. 2011;49:184–90. [DOI] [PubMed] [Google Scholar]
- [52].Arispe N, Rojas E, Pollard HB. Alzheimer disease amyloid beta protein forms calcium channels in bilayer membranes: blockade by tromethamine and aluminum. Proc Natl Acad Sci U S A. 1993;90:567–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].McDaid J, Mustaly-Kalimi S, Stutzmann GE. Ca(2+) Dyshomeostasis Disrupts Neuronal and Synaptic Function in Alzheimer’s Disease. Cells. 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Sobolevsky AI. Structure and gating of tetrameric glutamate receptors. J Physiol. 2015;593:29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Paoletti P Molecular basis of NMDA receptor functional diversity. Eur J Neurosci. 2011;33:1351–65. [DOI] [PubMed] [Google Scholar]
- [56].Kohr G NMDA receptor function: subunit composition versus spatial distribution. Cell Tissue Res. 2006;326:439–46. [DOI] [PubMed] [Google Scholar]
- [57].Das S, Sasaki YF, Rothe T, Premkumar LS, Takasu M, Crandall JE, et al. Increased NMDA current and spine density in mice lacking the NMDA receptor subunit NR3A. Nature. 1998;393:377–81. [DOI] [PubMed] [Google Scholar]
- [58].Sasaki YF, Rothe T, Premkumar LS, Das S, Cui J, Talantova MV, et al. Characterization and comparison of the NR3A subunit of the NMDA receptor in recombinant systems and primary cortical neurons. J Neurophysiol. 2002;87:2052–63. [DOI] [PubMed] [Google Scholar]
- [59].Matsuda K, Fletcher M, Kamiya Y, Yuzaki M. Specific assembly with the NMDA receptor 3B subunit controls surface expression and calcium permeability of NMDA receptors. J Neurosci. 2003;23:10064–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Tong G, Takahashi H, Tu S, Shin Y, Talantova M, Zago W, et al. Modulation of NMDA receptor properties and synaptic transmission by the NR3A subunit in mouse hippocampal and cerebrocortical neurons. J Neurophysiol. 2008;99:122–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Perez-Otano I, Lujan R, Tavalin SJ, Plomann M, Modregger J, Liu XB, et al. Endocytosis and synaptic removal of NR3A-containing NMDA receptors by PACSIN1/syndapin1. Nat Neurosci. 2006;9:611–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Henson MA, Roberts AC, Perez-Otano I, Philpot BD. Influence of the NR3A subunit on NMDA receptor functions. Prog Neurobiol. 2010;91:23–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Talantova M, Sanz-Blasco S, Zhang X, Xia P, Akhtar MW, Okamoto S, et al. Abeta induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc Natl Acad Sci U S A. 2013;110:E2518–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Choi DW. Ionic dependence of glutamate neurotoxicity. J Neurosci. 1987;7:369–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Sucher NJ, Akbarian S, Chi CL, Leclerc CL, Awobuluyi M, Deitcher DL, et al. Developmental and regional expression pattern of a novel NMDA receptor-like subunit (NMDAR-L) in the rodent brain. J Neurosci. 1995;15:6509–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Wong HK, Liu XB, Matos MF, Chan SF, Perez-Otano I, Boysen M, et al. Temporal and regional expression of NMDA receptor subunit NR3A in the mammalian brain. J Comp Neurol. 2002;450:303–17. [DOI] [PubMed] [Google Scholar]
- [67].Lee JH, Wei L, Deveau TC, Gu X, Yu SP. Expression of the NMDA receptor subunit GluN3A (NR3A) in the olfactory system and its regulatory role on olfaction in the adult mouse. Brain Struct Funct. 2016;221:3259–73. [DOI] [PubMed] [Google Scholar]
- [68].Mohamad O, Song M, Wei L, Yu SP. Regulatory roles of the NMDA receptor GluN3A subunit in locomotion, pain perception and cognitive functions in adult mice. J Physiol. 2013;591:149–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Eriksson M, Nilsson A, Froelich-Fabre S, Akesson E, Dunker J, Seiger A, et al. Cloning and expression of the human N-methyl-D-aspartate receptor subunit NR3A. Neurosci Lett. 2002;321:177–81. [DOI] [PubMed] [Google Scholar]
- [70].Nilsson A, Eriksson M, Muly EC, Akesson E, Samuelsson EB, Bogdanovic N, et al. Analysis of NR3A receptor subunits in human native NMDA receptors. Brain Res. 2007;1186:102–12. [DOI] [PubMed] [Google Scholar]
- [71].Tarabeux J, Kebir O, Gauthier J, Hamdan FF, Xiong L, Piton A, et al. Rare mutations in N-methyl-D-aspartate glutamate receptors in autism spectrum disorders and schizophrenia. Transl Psychiatry. 2011;1:e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Niemann S, Kanki H, Fukui Y, Takao K, Fukaya M, Hynynen MN, et al. Genetic ablation of NMDA receptor subunit NR3B in mouse reveals motoneuronal and nonmotoneuronal phenotypes. Eur J Neurosci. 2007;26:1407–20. [DOI] [PubMed] [Google Scholar]
- [73].Takata A, Iwayama Y, Fukuo Y, Ikeda M, Okochi T, Maekawa M, et al. A population-specific uncommon variant in GRIN3A associated with schizophrenia. Biol Psychiatry. 2013;73:532–9. [DOI] [PubMed] [Google Scholar]
- [74].Liu HP, Lin WY, Liu SH, Wang WF, Tsai CH, Wu BT, et al. Genetic variation in N-methyl-D-aspartate receptor subunit NR3A but not NR3B influences susceptibility to Alzheimer’s disease. Dement Geriatr Cogn Disord. 2009;28:521–7. [DOI] [PubMed] [Google Scholar]
- [75].Qiu C, Kivipelto M, von Strauss E. Epidemiology of Alzheimer’s disease: occurrence, determinants, and strategies toward intervention. Dialogues Clin Neurosci. 2009;11:111–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Lipton SA. Paradigm shift in NMDA receptor antagonist drug development: molecular mechanism of uncompetitive inhibition by memantine in the treatment of Alzheimer’s disease and other neurologic disorders. J Alzheimers Dis. 2004;6:S61–74. [DOI] [PubMed] [Google Scholar]
- [77].Winblad B, Jones RW, Wirth Y, Stoffler A, Mobius HJ. Memantine in moderate to severe Alzheimer’s disease: a meta-analysis of randomised clinical trials. Dement Geriatr Cogn Disord. 2007;24:20–7. [DOI] [PubMed] [Google Scholar]
- [78].Gauthier S, Loft H, Cummings J. Improvement in behavioural symptoms in patients with moderate to severe Alzheimer’s disease by memantine: a pooled data analysis. Int J Geriatr Psychiatry. 2008;23:537–45. [DOI] [PubMed] [Google Scholar]
- [79].Modrego PJ, Fayed N, Errea JM, Rios C, Pina MA, Sarasa M. Memantine versus donepezil in mild to moderate Alzheimer’s disease: a randomized trial with magnetic resonance spectroscopy. Eur J Neurol. 2010;17:405–12. [DOI] [PubMed] [Google Scholar]
- [80].Zhang N, Wei C, Du H, Shi FD, Cheng Y. The Effect of Memantine on Cognitive Function and Behavioral and Psychological Symptoms in Mild-to-Moderate Alzheimer’s Disease Patients. Dement Geriatr Cogn Disord. 2015;40:85–93. [DOI] [PubMed] [Google Scholar]
- [81].Peskind ER, Potkin SG, Pomara N, Ott BR, Graham SM, Olin JT, et al. Memantine treatment in mild to moderate Alzheimer disease: a 24-week randomized, controlled trial. Am J Geriatr Psychiatry. 2006;14:704–15. [DOI] [PubMed] [Google Scholar]
- [82].Pomara N, Ott BR, Peskind E, Resnick EM. Memantine treatment of cognitive symptoms in mild to moderate Alzheimer disease: secondary analyses from a placebo-controlled randomized trial. Alzheimer Dis Assoc Disord. 2007;21:60–4. [DOI] [PubMed] [Google Scholar]
- [83].Orgogozo JM, Rigaud AS, Stoffler A, Mobius HJ, Forette F. Efficacy and safety of memantine in patients with mild to moderate vascular dementia: a randomized, placebo-controlled trial (MMM 300). Stroke. 2002;33:1834–9. [DOI] [PubMed] [Google Scholar]
- [84].Ilhan Algin D, Dagli Atalay S, Ozkan S, Ozbabalik Adapinar D, Ak Sivrioz I. Memantine improves semantic memory in patients with amnestic mild cognitive impairment: A single-photon emission computed tomography study. J Int Med Res. 2017;45:2053–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Bakchine S, Loft H. Memantine treatment in patients with mild to moderate Alzheimer’s disease: results of a randomised, double-blind, placebo-controlled 6-month study. J Alzheimers Dis. 2007;11:471–9. [DOI] [PubMed] [Google Scholar]
- [86].Mohamad O, Song M, Wei L, Yu SP. Regulatory roles of the NMDA receptor GluN3A subunit in locomotion, pain perception and cognitive functions in adult mice. J Physiol. 2013;591:149–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Chakroborty S, Hill ES, Christian DT, Helfrich R, Riley S, Schneider C, et al. Reduced presynaptic vesicle stores mediate cellular and network plasticity defects in an early-stage mouse model of Alzheimer’s disease. Mol Neurodegener. 2019;14:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Sama DM, Norris CM. Calcium dysregulation and neuroinflammation: discrete and integrated mechanisms for age-related synaptic dysfunction. Ageing Res Rev. 2013;12:982–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Ju Hwang C, Choi DY, Park MH, Hong JT. NF-kappaB as a Key Mediator of Brain Inflammation in Alzheimer’s Disease. CNS Neurol Disord Drug Targets. 2019;18:3–10. [DOI] [PubMed] [Google Scholar]
- [90].Devi L, Ohno M. Cognitive benefits of memantine in Alzheimer’s 5XFAD model mice decline during advanced disease stages. Pharmacol Biochem Behav. 2016;144:60–6. [DOI] [PubMed] [Google Scholar]
- [91].Lueptow LM. Novel Object Recognition Test for the Investigation of Learning and Memory in Mice. J Vis Exp. 2017;126:55718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Kodis EJ, Choi S, Swanson E, Ferreira G, Bloom GS. N-methyl-D-aspartate receptor-mediated calcium influx connects amyloid-beta oligomers to ectopic neuronal cell cycle reentry in Alzheimer’s disease. Alzheimers Dement. 2018;14:1302–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Evans CE, Thomas RS, Freeman TJ, Hvoslef-Eide M, Good MA, Kidd EJ. Selective reduction of APP-BACE1 activity improves memory via NMDA-NR2B receptor-mediated mechanisms in aged PDAPP mice. Neurobiol Aging. 2019;75:136–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Epelbaum S, Genthon R, Cavedo E, Habert MO, Lamari F, Gagliardi G, et al. Preclinical Alzheimer’s disease: A systematic review of the cohorts underlying the concept. Alzheimers Dement. 2017;13:454–67. [DOI] [PubMed] [Google Scholar]
- [95].Lee JH, Zhang JY, Wei ZZ, Yu SP. Impaired social behaviors and minimized oxytocin signaling of the adult mice deficient in the N-methyl-d-aspartate receptor GluN3A subunit. Exp Neurol. 2018;305:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Olivero G, Vergassola M, Cisani F, Usai C, Pittaluga A. Immuno-Pharmacological Characterization of Presynaptic GluN3A-Containing NMDA Autoreceptors: Relevance to Anti-NMDA Receptor Autoimmune Diseases. Mol Neurobiol. 2019;56:6142–55. [DOI] [PubMed] [Google Scholar]
- [97].Wallace J Calcium dysregulation, and lithium treatment to forestall Alzheimer’s disease - a merging of hypotheses. Cell Calcium. 2014;55:175–81. [DOI] [PubMed] [Google Scholar]
- [98].Angulo SL, Henzi T, Neymotin SA, Suarez MD, Lytton WW, Schwaller B, et al. Amyloid pathology-produced unexpected modifications of calcium homeostasis in hippocampal subicular dendrites. Alzheimers Dement. 2020;16:251–61. [DOI] [PubMed] [Google Scholar]
- [99].Tu H, Nelson O, Bezprozvanny A, Wang Z, Lee SF, Hao YH, et al. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer’s disease-linked mutations. Cell. 2006;126:981–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Stutzmann GE. The pathogenesis of Alzheimers disease is it a lifelong “calciumopathy”? Neuroscientist. 2007;13:546–59. [DOI] [PubMed] [Google Scholar]
- [101].Hector A, Brouillette J. Hyperactivity Induced by Soluble Amyloid-beta Oligomers in the Early Stages of Alzheimer’s Disease. Front Mol Neurosci. 2020;13:600084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Wei L, Han BH, Li Y, Keogh CL, Holtzman DM, Yu SP. Cell death mechanism and protective effect of erythropoietin after focal ischemia in the whisker-barrel cortex of neonatal rats. J Pharmacol Exp Ther. 2006;317:109–16. [DOI] [PubMed] [Google Scholar]
- [103].Wei L, Ying DJ, Cui L, Langsdorf J, Yu S. Necrosis, apoptosis and hybrid death in the cortex and thalamus after barrel cortex ischemia in rats. Brain Res. 2004;1022:54–61. [DOI] [PubMed] [Google Scholar]
- [104].Song M, Yu SP. Ionic regulation of cell volume changes and cell death after ischemic stroke. Transl Stroke Res. 2014;5:17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Zucker RS. Calcium- and activity-dependent synaptic plasticity. Curr Opin Neurobiol. 1999;9:305–13. [DOI] [PubMed] [Google Scholar]
- [106].Higley MJ, Sabatini BL. Calcium signaling in dendrites and spines: practical and functional considerations. Neuron. 2008;59:902–13. [DOI] [PubMed] [Google Scholar]
- [107].Xiao AY, Wei L, Xia S, Rothman S, Yu SP. Ionic mechanism of ouabain-induced concurrent apoptosis and necrosis in individual cultured cortical neurons. J Neurosci. 2002;22:1350–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Schrank S, Barrington N, Stutzmann GE. Calcium-Handling Defects and Neurodegenerative Disease. Cold Spring Harb Perspect Biol. 2020;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Kusne Y, Wolf AB, Townley K, Conway M, Peyman GA. Visual system manifestations of Alzheimer’s disease. Acta Ophthalmol. 2017;95:e668–e76. [DOI] [PubMed] [Google Scholar]
- [110].Lenoir H, Sieroff E. Visual perceptual disorders in Alzheimer’s disease. Geriatr Psychol Neuropsychiatr Vieil. 2019;17:307–16. [DOI] [PubMed] [Google Scholar]
- [111].Zhong W, Yuan Y, Gu X, Kim SI, Chin R, Loye M, et al. Neuropsychological Deficits Chronically Developed after Focal Ischemic Stroke and Beneficial Effects of Pharmacological Hypothermia in the Mouse. Aging Dis. 2020;11:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Yu SP, Tung JK, Wei ZZ, Chen D, Berglund K, Zhong W, et al. Optochemogenetic Stimulation of Transplanted iPS-NPCs Enhances Neuronal Repair and Functional Recovery after Ischemic Stroke. J Neurosci. 2019;39:6571–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Yang M, Crawley JN. Simple behavioral assessment of mouse olfaction. Curr Protoc Neurosci. 2009;Chapter 8:Unit 8 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.