

Abstract
Chronic kidney disease (CKD) is associated with increased risk of cardiovascular (CV) events, and the disease burden is rising rapidly. An important contributor to CV events and CKD progression is high blood pressure (BP). The main mechanisms of hypertension in early and advanced CKD are renin-angiotensin system activation and volume overload, respectively. Sodium retention is well known as a factor for high BP in CKD. However, a BP increase in response to total body sodium or volume overload can be limited by neurohormonal modulation. Recent clinical trial data favoring intensive BP lowering in CKD imply that the balance between volume and neurohormonal control could be revisited with respect to the safety and efficacy of strict volume control when using antihypertensive medications. In hemodialysis patients, the role of more liberal use of antihypertensive medications with the concept of functional dry weight for intensive BP control must be studied.
Keywords: Antihypertensive medication, Chronic kidney disease, Diuretics, Hemodialysis, Hypertension, Renin-angiotensin system, Sodium
Introduction
In chronic kidney disease (CKD), cardiovascular (CV) risk is increased linearly by 7% per 10 mL/min/1.73 m2 decrease in glomerular filtration rate (GFR) [1]. Death from CV disease (CVD) is much more common than that from progression to end-stage renal disease (ESRD) [2]. In addition, CKD is a poor prognostic factor in CVD patients [3].
CKD is defined as persistently elevated urine albumin excretion (≥30 mg/g [3 mg/mmoL] creatinine [Cr]), persistently reduced estimated GFR (eGFR < 60 mL/min per 1.73 m2), or both for greater than 3 months, in accordance with current Kidney Disease: Improving Global Outcomes (KDIGO) guidelines [4]. Kidney damage in many diseases usually can be ascertained by the presence of albuminuria, defined as albumin-to-Cr ratio of >30 mg/g in two of three spot urine specimens based on KDIGO guidelines [5]. In addition to chronicity, demonstration of damaged renal parenchyma is mandatory for diagnosis of CKD stages 1 and 2. For CKD stage 3 or higher, chronic decreased kidney function as determined by GFR of <60 mL/min/1.73 m2 is sufficient for diagnosis regardless of the presence of renal damage.
Approximately one-third of CKD stage 3 and less than one-half of CKD stage 4 patients have albuminuria [6–8]. Increased blood pressure (BP) aggravates renal damage and proteinuria, which results in more rapid loss of kidney function. This decreased renal function contributes to higher BP, completing a vicious cycle [9]. Moreover, increased BP not only exacerbates renal function deterioration but also damages CV systems.
The differences in the rationale or approach between prevention of renal progression and CVD are frequently discussed among cardiologists, hypertension specialists, and nephrologists. This article will review recent updates in BP control in CKD with a major focus on CVD prevention in addition to renal outcomes with viewpoints of salt retention or volume status control and the use of diuretics and non-diuretics as antihypertensive medications (AHMs) for intensive BP control. This review will not cover specific target BPs according to specific disease populations of CKD.
Updates on tissue or skin sodium
In addition to classic pressure natriuresis relationships, recent views highlighted that sodium balance is largely dependent on neurohormonal modulation [10]. Accumulation of sodium in soft tissue measured by skin and/or muscle sodium has been demonstrated to be a buffer between sodium intake and changes in volume status, which was regarded as an explanation of the time lag or loose connection between sodium intake and BP response as shown in Fig. 1 [11]. Titze et al. [12] reported that glycosaminoglycan (GAG) in tissue in the epidermal skin layer plays a role as a sodium buffer, assuming that GAG binding to sodium ions makes it osmotically inactive. The skin buffer concept by hypertonicity is useful to explain the time lag or discrepancy between salt overload and BP increase.
Figure 1. Hypothetical distribution of skin sodium according to tonicity of skin sodium.
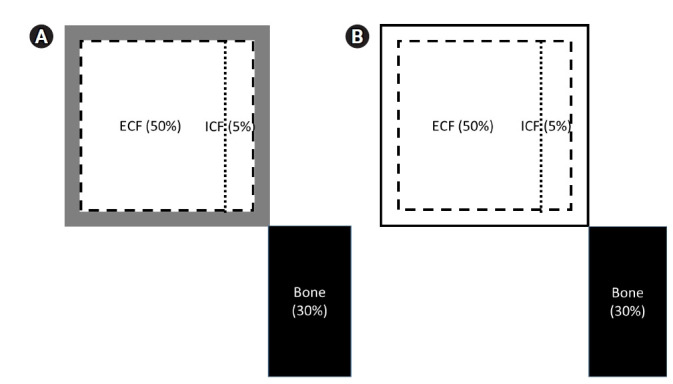
(A) A three-compartment model with hypertonic sodium in the skin, muscle, or artery that could buffer the exchangeable sodium overload. Exact mechanisms on how sodium could be concentrated in the skin remain unknown. (B) A two-compartment model with isotonic skin sodium exhibiting distribution in the other interstitial tissues or edema. The dotted line indicates a sodium gradient across the cell membrane. The broken line means sodium distribution by Starling force. The separated bone compartment suggests different exchangeability kinetics from the other.
ECF, extracellular fluid; ICF, intracellular fluid.
However, a recent study by Rossitto G et al. [13] showed that sodium concentration in the skin is importantly isotonic but could be mistakenly identified as hypertonic due to technical reasons during skin magnetic resonance imaging. In this respect, the amount of skin sodium could be regarded as an extension of the expansion of extracellular fluid (ECF) volume or interstitial edema. Inflammation was explained not only through hypertonic stress, but also by the biomechanical stress of edema. A high salt-related increase in peripheral resistance was found to be due to compression from perivascular swelling or edema both in terms of vasoconstriction and vasodilation [13].
Hypertonic or nonosmolar sodium accumulation in tissue compartments can be denied when sodium in tissues is isotonic and a reflection of ECF volume [14]. However, isotonic edema is also associated with increased GAG and biomechanical stress as well as crosstalk between GAG and macrophages. This results in prohypertensive effects in the kidney, vasculature, and brain [15]. Angiotensin II (Ag II) also has proinflammatory effects through macrophage infiltration in the renal interstitium, leading to sodium retention and BP elevation, as well as renal damage mediated by T lymphocytes [16]. The direct connection between high sodium intake and Ag II induction, both of which are crucial components of the salt-induced hypertension model, seems to be weak [16].
In CKD, a diverse level of skin or tissue sodium was reported, which now could be interpreted as silent edema or volume expansion by high salt intake. In a prospective observational study performed in a CKD population, high salt intake measured by 24-hour urine sodium excretion was significantly correlated with CKD progression and CVD events [17]. The harmful effect of sodium intake seems to be more obvious in CKD compared to the general population [18]. Systematic reviews have exhibited that dietary sodium reduction demonstrate short-term reductions in BP in CKD populations [19]. However, it is necessary to have more clinical data evaluating the effects of dietary sodium reduction on CV events in CKD populations [4]. It is essential to be reminded of inconsistencies among studies examining the relationship of dietary sodium intake with health outcomes in persons with diabetes, suggesting that the effects of dietary sodium intake changes on health benefits and harms depend on different causes and severities of CKD [4,19–21]. The clinical evidence on whether so-called salt toxicity is independent of BP changes is debated [22].
There is also ample, recent evidence to indicate that physical factors are clearly the subordinates of the neurohumoral mechanisms. Essentially, the question is then whether the normal physiological situation is best described as (i) neurohumoral modulation of the pressure natriuresis mechanism, or (ii) neurohumoral control occasionally modulated by pressure natriuresis. The latter possibility appears attractive [10].
Renal salt excretion in sodium balance under normal conditions
Slight changes in osmolarity by salt intake, inducing immediate movement of water from the intracellular to the extracellular compartments, thirst, and secretion of antidiuretic hormones resulted in increasing and maintaining ECF volumes almost without apparent changes in sodium concentration [23]. Even small volume increases resulted in relatively large pressure elevation through the secondary increase in total peripheral resistance [24].
When a bout of dietary salt is loaded in a normal subject, approximately half is excreted on day 1. With thirst and renal water resorption, body weight and ECF volume increases are associated with a variable degree of BP changes according to individual salt sensitivity [25]. After 3 to 4 days when the sodium balance becomes zero, the original steady state is recovered as long as no more bout of salt was maintained [25,26]. Although the exact details are being debated, renal excretion of sodium has been found to be related to the function of ECF volume and BP. According to Walser’s analysis, the time constant of the relationship between ECF volume and renal salt excretion determines the speed at which an individual can adapt to a change in dietary intake [27]. Daily urinary sodium excretion is proportional to the time constant and the amount of sodium excess. While the time constant is approximately 0.79 per day–1 in normal subjects, it appears to be reduced by aging and CKD, indicating that it takes longer than 3 to 4 days for the kidney to recover its sodium balance to zero [27]. Sodium retention might be similar to the setting when a drug in maintenance dose is repeated within the elimination half-life so that increased sodium amount or volume will be maintained.
Sodium retention, volume overload, and hypertension in chronic kidney disease
The sodium excretion rate appears to be reduced by aging and CKD, and the mechanisms of decreased sodium excretion are a mixture of reduced glomerular filtration of sodium and increased tubular reabsorption of sodium independently, or both situations in combination. CKD is an important contributor to salt sensitivity. In a small study for male CKD patients on sequential low salt and high salt diets, the salt sensitivity index was calculated by the increase in mean arterial pressure in mmHg divided by the increase in 24-hour sodium excretion in mEq/day, which is the inversed slope of the classic pressure natriuresis relationship [28]. In this study, the log of the salt sensitivity index was linearly associated with Cr clearance (r =–0.89, intercept –0.74). For example, the salt sensitivity index can be calculated as 1.4 mmHg / (100 mEq of sodium per day) at a Cr clearance of 75 mL/min and was increased to 2.0 mmHg and 3.6 mmHg at a Cr clearance of 50 mL/min and 25 mL/min, respectively. In an animal study, high dietary sodium intake in CKD contributes to sodium retention through aldosterone-independent activation of the mineralocorticoid receptor-mediated through small GTPase Rac1 [29,30]. In addition to Rac1, high dietary salt in salt-sensitive rodents appears to increase serum and glucocorticoid-induced kinase 1 independent of aldosterone, which could be another pathway through which dietary salt directly increases distal nephron sodium reabsorption [30,31]. ECF volume expansion through a repeated sufficient amount of salt intake within the half time that the sodium balance reaches zero can lead to a compensatory decrease in tubular reabsorption of sodium, reestablishment of the steady state of sodium balance, a variable degree of hypertension, and accompanied with or without other manifestations of ECF volume expansion. Once the new steady state is achieved, tubular reabsorption of sodium reaches almost the original sodium resorption status before increased salt intake, producing infinitely small error and infinite gain of control (correction/error) where the correction is the change of tubular resorption of sodium during the process [32].
In an insightful and comprehensive study by Essig et al. [33], CKD stage 3 exhibited higher BP with ECF volume expansion, even with greater use of AHM, compared to CKD stage 1. Compared to CKD stage 2, CKD stage 3 exhibited comparable BP and ECF volume expansion. This was the case even with more AHM including diuretics and less sodium excretion compared to CKD stage 2. The steady status of CKD stages 2 and 3 in Fig. 2 did not directly exhibit the dynamics such as salt sensitivity index for each stage due to the limitation of the cross-sectional study, but it suggested that the required amount of salt restriction and required AHM should be increased to allow for comparable levels of BP and ECF volume excesses (Fig. 2).
Figure 2. A clinical example of the steady-state relationships among SBP, eGFR, urinary sodium excretion, and eECF according to the stage of CKD.
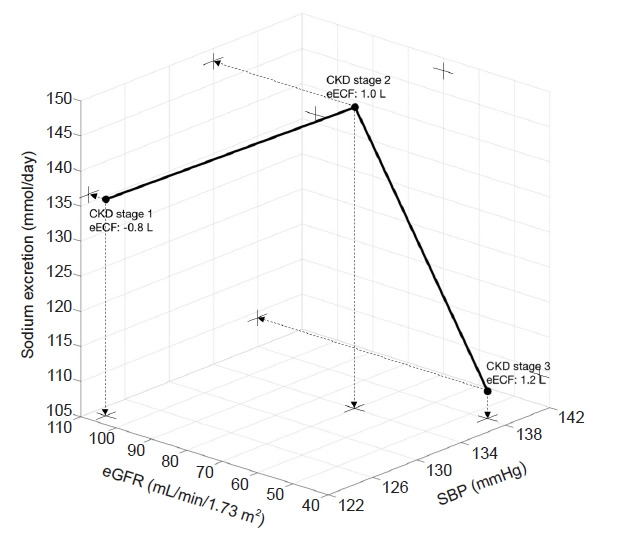
All of the relationships between sodium excretion and SBP, the relationship between eGFR and sodium excretion, and the relationship between sodium excretion and eECF are not linear. Linear relationships were observed only among eGFR, eECF, and SBP. Crosses indicate projections on the two-dimensional planes from the dot points in the three-dimensional space (data from the tables in Essig et al. [33]).
CKD, chronic kidney disease; eECF, extracellular fluid excess; eGFR, estimated glomerular filtration; SBP, systolic blood pressure.
In general, the prevalence of masked hypertension (MH) is higher in CKD compared to the general population and is related to low eGFR and proteinuria [34]. The prevalence of normal, MH, and sustained hypertension in CKD stages 1 and 2 was reported as 58%, 27%, and 15%, respectively [35]. MH with elevated nighttime BP is reported to be associated with target organ damage [35]. Even in CKD stages 1 and 2, it is reasonable to have a high level of suspicion of MH in patients with prehypertension. To detect MH through increased nighttime BP in CKD stages 1 and 2, ambulatory BP monitoring (ABPM) seems to be the best method. Moreover, the proportion of CKD patients with masked uncontrolled hypertension and sustained uncontrolled hypertension is expected to increase when using a lower systolic BP (SBP) threshold as per the recommendation of recent guidelines [4].
The association of MH and lower eGFR was observed only in patients with increased nighttime BP even though the mechanisms of increased MH in CKD are multifactorial [35]. Meanwhile, the mechanisms of increased nighttime BP in CKD are salt sensitivity, increased sympathetic activity, and proteinuria [36].
Management of hypertension in chronic kidney disease
Role of diuretics and volume control
ECF volume overload was reported as a group in early CKD, but routine assessments on an individual patient level are not practical [33]. The existence of an ECF volume increase can be detected by noticing impressive BP reduction when diuretics are added and/or salt was restricted specifically in a situation of uncontrolled high BP through the use of AHMs other than diuretics.
There is insufficient data on the role of diuretics as the first-line therapy for the management of hypertension in CKD populations, and several guidelines on hypertension have shown different opinions and views on the use of diuretics [4]. Previously, National Institute for Health and Care Excellence (NICE) guidelines recommended that diuretics be used in non-proteinuric CKD [37] as a first-line therapy. Since albuminuria is a common indicator to diagnose CKD stages 1 and 2, and some patients of CKD stages 3 and 4 do not possess albuminuria [7], the first-line use of diuretics seems to be more useful in CKD stages 3 and 4 with salt retention and reduced GFR. The role of diuretics in CKD stages 3 and 4 is increasing, and more potent loop diuretics and some thiazide-like diuretics such as chlorthalidone, metolazone, and indapamide appear to be effective for optimal BP control [4]. However, because the glomerular stretch in the nephron level triggers a vicious cycle of CKD progression even in CKD stages 3 and 4, renin-angiotensin system blockade (RASB) could theoretically have a major role as a first-line therapy, even in non-proteinuric CKD stage 3 and 4 patients [37,38].
Despite an increase in ECF volume in CKD, most guidelines prefer an RASB as the first-line therapy as long as BP is well controlled. The ACC/AHA (American College of the Cardiology/American Heart Association) 2017 guidelines recommend an angiotensin-converting enzyme (ACE) inhibitor for CKD and emphasize initial combination therapy and a target BP of <130/80 mmHg [39,40] so that second-line drugs can be used as initial combination therapy. As the second-line drug, the National Kidney Foundation’s Kidney Disease Outcomes Quality Initiative (KDOQI) guidelines recommend diuretics [32], but ESH-ESC (European Society of Hypertension/European Society of Cardiology) 2018 guidelines advocated for calcium channel blockers (CCB) [41]. As for CCB, it was reported to be associated with mortality in CKD patients with glomerulonephritis [42]. But in a meta-analysis, CCB has similar effects to RASB in terms of long-term BP, mortality, heart failure, stroke or cerebrovascular events, and renal function [43]. For comparable BP levels and salt intake, there can be a different volume status according to the proportion of diuretics and other AHMs (Fig. 3A vs. 3B). These discrepancies call to attention the need for more individualized approaches balanced by volume status, side effects, CVD risk, and comorbid CVD profiles to select second-line drugs as well as further studies demonstrating clinical evidence. For example, increased BP and/or ECF volume is the main factor for left ventricular hypertrophy (LVH) in CKD [44]. In CKD stages 3 and 4, additional furosemide on top of an RASB exhibited a greater regression of LVH [45]. Similarly, among patients treated with an ACE inhibitor or angiotensin II receptor blocker, the combination of salt restriction and a diuretic can provide a greater antiproteinuric effect as well as improved BP reduction than either intervention alone [46]. Therefore, RASB will be the basis for antihypertensive drug therapy in CKD, and the choice between diuretics and CCB should be individualized.
Figure 3. Spectrum of therapeutic options for AHM vs. diuretics or UF in CKD (A, B, and F) and the status at the end of the hemodialysis session (C, D, and E).
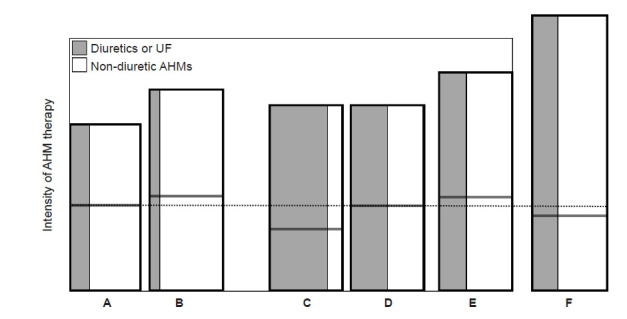
(A) CKD, with normovolemia. (B) CKD, with permissible hypervolemia, less active use of diuretics. (C) Hemodialysis, absolute dry weight, hypovolemia with minimum use of AHM. (D) Hemodialysis, normovolemia. (E) Hemodialysis, functional dry weight or permissible hypervolemia. (F) CKD, intensive blood pressure (BP) control with permissible hypovolemia, more active use of diuretics. The achieved BP levels among A–E in the box are conventional and comparable. Relative intensity of antihypertensive therapy was depicted by the area of the rectangles. Compared to C, D–E conditions require more non-diuretics AHM to achieve the comparable level. F is the condition in need of intensive BP control to achieve the lower target BPs on the basis of permissible hypovolemia through the greater active use of diuretics or UF. The dotted line indicates the ideal status of extracellular volume or normovolemia. Each horizontal solid line represents the volume status achieved by diuretics or UF.
AHM, antihypertensive medications other than diuretics; CKD, chronic kidney disease; UF, ultrafiltration.
For additional use of diuretics in CKD, there was a recent observational study showing the benefit of spironolactone in CVD and renal outcomes in CKD stages 3 and 4 [47]. However, the risk of hyperkalemia should be more preemptively managed through the use of dietary and/or pharmacologic interventions using potassium-wasting diuretics and oral potassium binders [48]. More options for diuretics such as a higher dose of loop diuretics in twice a day use or in combination with chlorthalidone and spironolactone could be considered for optimal BP control [48,49].
Newer drugs associated with volume control
As the elderly population rapidly increases, there are more and more cardiorenal syndrome patients with CKD. From the randomized controlled trial (RCT) for heart failure in which volume control by diuretics is an essential component in standard drug therapy, two interesting drugs potentially related to volume control or sodium excretion are more and more frequently indicated in heart failure with CKD. First, the sodium glucose co-transporter 2 inhibitor (SGLT2I) has several mechanisms to protect the heart and kidneys. Among them, sodium excretion in the proximal tubule followed by increased sodium delivery to the macula densa and tubuloglomerular feedback results in a short-term increase in the excretion of renal sodium [50]. BP reduction as the one of the important mechanisms of cardiorenal protection has been proven in CKD patients [51,52]. Although precise interactions or differential effects between loop diuretics and SGLT2I require further studies or analyses, it was reported in terms of heart failure that SGLT2I prevents the increase of the dose of loop diuretics, and that it can overcome the resistance of loop diuretics [53]. Second, theoretically, the natriuretic components of angiotensin receptor neprilysin inhibitor (ARNI) could facilitate renal excretion of sodium. In CKD patients, neprilysin inhibitor components have stable pharmacokinetic properties, and the reduction in loop diuretic dose was reported in RCT for heart failure [54]. Since diabetes and cardiorenal syndrome are frequent comorbidities in CKD, the roles of SGLT2I and ARNI in volume control require further analysis.
Intensive blood pressure control in chronic kidney disease
KDOQI guidelines underscore the purpose of antihypertensive therapy for prevention of both CKD progression and CVD [32]. In a meta-analysis, lower achieved BP (SBP < 120 mmHg) in those with CKD reported a smaller risk reduction for CV events compared to patients without CKD [55]. The impact of SPRINT is important in terms of clinical evidence in CKD, and the results from SPRINT were supported by subsequent meta-analysis examining death exclusively in the CKD subgroups of RCTs, which found a benefit of lower achieved or target BP [4,56]. In the KDIGO 2021 guidelines, it was suggested that adults with high BP and CKD be treated with a target SBP of <120 mmHg. Despite this intensive target BP, some risk-benefit ratios need to be individualized, and intensive BP control could be harmful, for examples, when BP was measured in a non-standardized manner, when it is uncertain if the patient has a silent coronary obstruction, when the patient cannot tolerate intensive target BP or is extremely old (>90 years old), and when patient is bed-ridden or when life expectancy is limited.
Practically, in general, CKD stage 3 and 4 patients are very concerned about GFR decline in terms of fear of dialysis. First of all, it is very difficult for a patient to understand that higher GFR associated with higher BP could indicate a poor prognosis and that hyperfiltration or glomerular stretch at the level of the nephron is harmful. The acute decline of GFR especially by RASB, a functional side effect, could be uncomfortable for patients. Clearly informing patients of this potential issue and warning that 10% to 20% of the initial Cr increase is normal and reversible seems to be essential for patient adherence [57]. In some CKD patients who have comorbidities such as heart failure or coronary artery disease that requires the use of RASB to reduce CV mortality, an initial Cr increase more than >30% could be acceptable [58].
The initial decline in GFR has become a greater challenge for physicians because intensive BP lowering is increasingly considered for better CVD outcomes. After all, recommendations not to retry RASB in cases of an initial Cr increase of >30% and in cases of failure to return to baseline after dosage reduction or cessation do not seem to have a solid scientific basis. Avoidance of an initial Cr increase more than >30% could be regarded as a consistent application of the “primum non nocere” principle and a compromise to alleviate patient apprehension [32,59]. For this issue, volume status and a preexisting or combination regimen of RASB and diuretics vs. CCB seems to be differently related to the degree of initial decline [60]. In SPRINT, the difference in the rate of such (eGFR) decline was very small with comparable biomarker changes, even though initial decline during the first 6 months was significantly faster in the intensive group compared with the standard group [5,61,62]. Moreover, RASB in patients with advanced CKD was reported to be rather safe [63], even though there are studies from selected small populations reporting that habitual use does not appear to be beneficial and might even be harmful [63,64]. Despite the risk reduction of dialysis therapy (27.9% vs. 36.1%), stopping RASB was associated with a higher absolute 5-year risk of all-cause mortality (54.5% vs. 40.9%) and major adverse CV events (59.5% vs. 47.6%) [65].
Strict volume control for intensive BP control could have increased risk of hypovolemia. Hypovolemia and/or hypotension are the most common factors for acute kidney injury (AKI). The nonrecovery of kidney function following an episode of AKI is a major contributing factor for the prevalence of CKD and the progression of CKD to an advanced stage [66,67]. Hypovolemic patients in CKD stages 3 and 4 reported to have lower BP and lower AHM use including diuretics. This means that, in some CKD patients, patient-related factors such as fewer comorbidities, decreased lean body mass, anemia, and other unknown causes might be more important in determining volume status than is use of diuretics [68]. Diuretic use was associated with poor renal outcomes independently of BP, volume status, and other covariates [68]. Since patient factor-driven hypovolemia is associated with lower BP, whether inducing minimal hypovolemia by diuretics therapy to comparable level to the patient factor-driven hypovolemia could be beneficial or not for intensive BP control needs future study (Fig. 3F). The role of sodium restriction or weight reduction in the context of intensive BP control also requires further studies.
Blood pressure variability in hypertension management in chronic kidney disease
There are several types of BP variabilities (BPV) such as visit-to-visit BPV in clinics, day-to-day BPV with home BP monitoring (HBPM), and short-term BPV with ABPM. The mechanisms for increased BPV in CKD are largely unknown, but impaired baroreceptor sensitivity, altered sympathetic nervous system activity, oxidative stress, inflammation, and increased arterial stiffness were suggested [69]. Few studies have been performed to correlate high sodium intake and BPV [70].
The Spanish ABPM registry showed that BPV increases as CKD progresses from stage 1 to 5 [71], and short‐term systolic BPV by ABPM was associated with renal outcome independently of 24-hour BP in a CKD population with a mean eGFR of 50 mL/min/1.73 m2 [72].
For clinic BP, among 114,900 patients with CKD, BPV was associated with all-cause mortality, hemorrhagic stroke, ESKD, and heart failure [73]. There were also studies showing that systolic BPV predicts the risk of death, but not CKD progression to dialysis in CKD [74].
However, few studies have reported on the therapeutic implications of BPV in CKD. CCB was reported to be associated with lower BPV in CKD compared to beta-blockers or RASB [75,76]. In SPRINT, similar patterns were observed, with lower BPV in participants receiving chlorthalidone and CCB and higher BPV variability among those on RASB [73].
There could also be a marked reduction in GFR when starting RASB in CKD despite the slight BP reduction. Diuretics or RASB and diuretics in combination are more commonly associated with side effects and a worse BPV profile than CCB in CKD [77,78].
BPV can affect variability in GFR because volume changes and impaired GFR autoregulation can be common in CKD, and variability in GFR can predict CV outcomes [79] as well as renal outcomes [80]. However, since studies of GFR variability have not reported the relationship between BPV and GFR variability, BPV could be a significant confounding factor.
Blood pressure control in hemodialysis patients
This review confined to BP control under a standard reimbursement protocol with a thrice a week hemodialysis (HD) protocol spanning about 4 hours seems to be limited for adopting various dialysis protocols for better BP control.
For the volume status description, dry weight is defined operationally as the lowest postdialysis weight with minimal signs or symptoms of hypovolemia even though the exact definition of dry weight remains uncertain or multiple definitions have been suggested [81]. Theoretically, reducing volume overload by ultrafiltration (UF) is such efficient measure to manage BP as to stop the use of AHMs in up to 90% of patients undergoing HD even though the long-term consequences have not been studied, as shown in Fig. 3C [82,83]. The finding that 36% of normovolemia and 54% of hypervolemia patients in whom AHM were prescribed in 45% and 54%, respectively, as schematized in Figs. 3D and 3E, were hypertensive suggests a huge gap in real practice, with more frequent AHM use and low probability of hypovolemia [84]. This finding suggests that the dry weight concept when used in an effort to simply avoid or stop AHM for BP control must be reconsidered [85]. There might be a concept of “functional dry weight” permitting greater use of AHM instead of “absolute dry weight” as long as BP is controlled [86]. In fact, AHM can be considered the first-line to lowering BP in patients receiving HD [87]. It is reasonable to choose AHMs based on patient characteristics, CV indications, and availability as well as intradialytic BP patterns with regard to drug dialyzability and elimination routes [88,89]. In theory, drugs with hepatic clearance could provide stable antihypertensive effects, and dialyzable drugs would be better in lowering BPs increased by volume uptick that will be removed during dialysis simultaneously with volume. However, an optimal combination strategy of drugs with hepatic and renal clearances in which dose reduction is required remains unknown (Fig. 4). It is very important to limit interdialysis weight gain (IDWG), which results in higher BP levels and BPV requiring additional AHMs.
Figure 4. Conceptual framework for antihypertensive drug prescription according to elimination route, dialyzability, and administration timing.
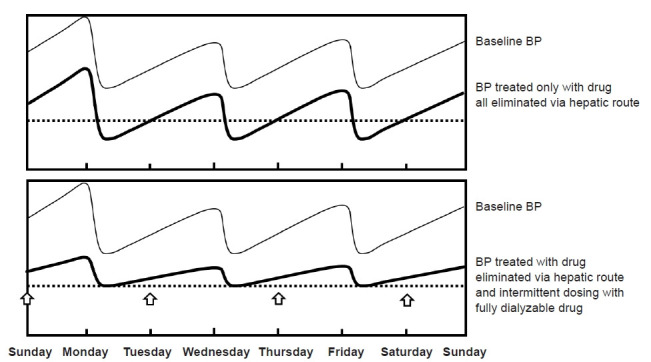
Each arrow represents the start of the fully dialyzable drug. (A) The blood pressure (BP) treated solely by antihypertensive drugs eliminated through the hepatic route. Since dialysis cannot reduce the drug concentration, active treatment targeting interdialytic BP could increase the risk of intradialytic hypotension. (B) It shows that BP treated by both antihypertensive drugs eliminated by the hepatic route targeting the BP in dry weight and fully dialyzable drugs targeting the BP increased during the interdialytic period and could be fully removed by dialysis without affecting the BP in dry weight. The dotted line represents the threshold for intradialytic hypotension.
Optimal predialysis BP for mortality risk was 130 to 160 mmHg when adjusted for confounding factors including AHM [90]. BP measurement methods seem to be limited to demonstrate the relationship between volume status and BP because HBPM or ABPM could predict CVD outcomes better than casual BP in HD patients and because BPV during inter and intradialytic periods was much higher in HD patients compared to non-dialysis patients [91]. Therefore, for more reliable BP control, reference methods to monitor BP should be HBPM or ABPM, and further studies for optimal BP thresholds to prevent hard outcomes are necessary. Therapeutic implications of BPV in HD patients are not established but, in a large-scale observational study, lower visit-to-visit BPV was associated with greater UF volume, dry weight attainment, and AHM other than β-blockers or RASB, suggesting the roles of volume control and CCB for more stable BP control [92].
Volume overload and high BP are the most important contributors to LVH. In HD patients, there are other remarkable factors related to LVH such as anemia, arterial-venous dialysis accesses with high cardiac output, arterial stiffness, and bone mineral hormones [93]. LVH and accompanied cardiac fibrosis are strongly associated with cardiac diastolic dysfunction in which high left ventricular (LV) filling pressure is required to maintain LV end-diastolic volume that will be transferred to stroke volume by LV contraction. LVH are strongly associated with CV prognosis in HD patients, and a 10% decrease in LV mass predicts a 28% decrease in CV outcomes [94,95]. It should be essential to achieve individualized target BPs for prevention of severe LVH or diastolic dysfunction to prevent intradialytic hypotension (IDH) and CVD until the proven BP target beneficial for LVH is available.
Establishing a balance between AHMs and volume control is most challenging for treatment of severe IDH. If an effective AHM prescription during the phase of advanced CKD stage is available in a patient, the same AHM regimen with a UF amount equivalent to the role of diuretics during the CKD phase seems reasonable to be maintained during HD (Fig. 3D) [88]. The so-called “wet strategy” with some degree of permissive hypervolemia with profuse use of AHM as long as the target BP is achieved caring of residual renal function might be an option for IDH [96]. It is mandatory to decrease IDWG for which sodium restriction to the level to avoid IDH concomitantly with ultrafiltration rate adjustment is needed [97]. Routine increase in IDWG to avoid IDH is not recommended.
Conclusion
In conclusion, sodium retention can be demonstrated in both early and late CKD. The BP response to sodium retention could be modulated by neurohormones to maintain stable BP to a certain limit. RAS seems to be involved in a common pathophysiology for nephron injury in CKD with reduced renal mass. Recent clinical trial data favoring intensive BP lowering in CKD imply that the balance between volume and neurohormonal control could be shifted by using more AHMs other than diuretics than previously believed. More liberal use of AHMs could be allowed for effective BP control and CV protection even after HD is initiated with the concept of functional dry weight.
Footnotes
Conflicts of interest
All authors have no conflicts of interest to declare.
Authors’ contributions
Conceptualization: JS, CHL
Visualization: JS, CHL
Writing–original draft: JS
Writing–review & editing: CHL, JS
All authors read and approved the final manuscript.
References
- 1.Manjunath G, Tighiouart H, Ibrahim H, et al. Level of kidney function as a risk factor for atherosclerotic cardiovascular outcomes in the community. J Am Coll Cardiol. 2003;41:47–55. doi: 10.1016/s0735-1097(02)02663-3. [DOI] [PubMed] [Google Scholar]
- 2.Gargiulo R, Suhail F, Lerma EV. Cardiovascular disease and chronic kidney disease. Dis Mon. 2015;61:403–413. doi: 10.1016/j.disamonth.2015.07.005. [DOI] [PubMed] [Google Scholar]
- 3.Kim SM, Tripathy DR, Park SW, et al. Impact of chronic kidney disease on clinical outcomes in diabetic patients undergoing percutaneous coronary intervention in the era of newer-generation drug-eluting stents. Korean Circ J. 2017;47:222–230. doi: 10.4070/kcj.2016.0312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kidney Disease: Improving Global Outcomes (KDIGO) Blood Pressure Work Group KDIGO 2021 clinical practice guideline for the management of blood pressure in chronic kidney disease. Kidney Int. 2021;99(3S):S1–S87. doi: 10.1016/j.kint.2020.11.003. [DOI] [PubMed] [Google Scholar]
- 5.Cheung AK, Chang TI, Cushman WC, et al. Blood pressure in chronic kidney disease: conclusions from a Kidney Disease: improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2019;95:1027–1036. doi: 10.1016/j.kint.2018.12.025. [DOI] [PubMed] [Google Scholar]
- 6.Levey AS, de Jong PE, Coresh J, et al. The definition, classification, and prognosis of chronic kidney disease: a KDIGO Controversies Conference report. Kidney Int. 2011;80:17–28. doi: 10.1038/ki.2010.483. [DOI] [PubMed] [Google Scholar]
- 7.Coresh J, Selvin E, Stevens LA, et al. Prevalence of chronic kidney disease in the United States. JAMA. 2007;298:2038–2047. doi: 10.1001/jama.298.17.2038. [DOI] [PubMed] [Google Scholar]
- 8.Coresh J, Astor BC, Greene T, Eknoyan G, Levey AS. Prevalence of chronic kidney disease and decreased kidney function in the adult US population: Third National Health and Nutrition Examination Survey. Am J Kidney Dis. 2003;41:1–12. doi: 10.1053/ajkd.2003.50007. [DOI] [PubMed] [Google Scholar]
- 9.Ku E, Lee BJ, Wei J, Weir MR. Hypertension in CKD: core curriculum 2019. Am J Kidney Dis. 2019;74:120–131. doi: 10.1053/j.ajkd.2018.12.044. [DOI] [PubMed] [Google Scholar]
- 10.Bie P. Mechanisms of sodium balance: total body sodium, surrogate variables, and renal sodium excretion. Am J Physiol Regul Integr Comp Physiol. 2018;315:R945–R962. doi: 10.1152/ajpregu.00363.2017. [DOI] [PubMed] [Google Scholar]
- 11.Rakova N, Jüttner K, Dahlmann A, et al. Long-term space flight simulation reveals infradian rhythmicity in human Na(+) balance. Cell Metab. 2013;17:125–131. doi: 10.1016/j.cmet.2012.11.013. [DOI] [PubMed] [Google Scholar]
- 12.Titze J, Shakibaei M, Schafflhuber M, et al. Glycosaminoglycan polymerization may enable osmotically inactive Na+ storage in the skin. Am J Physiol Heart Circ Physiol. 2004;287:H203–H208. doi: 10.1152/ajpheart.01237.2003. [DOI] [PubMed] [Google Scholar]
- 13.Rossitto G, Mary S, Chen JY, et al. Tissue sodium excess is not hypertonic and reflects extracellular volume expansion. Nat Commun. 2020;11:4222. doi: 10.1038/s41467-020-17820-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oppelaar JJ, Vogt L. Body fluid-independent effects of dietary salt consumption in chronic kidney disease. Nutrients. 2019;11:2779. doi: 10.3390/nu11112779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodríguez-Iturbe B, Franco M, Tapia E, Quiroz Y, Johnson RJ. Renal inflammation, autoimmunity and salt-sensitive hypertension. Clin Exp Pharmacol Physiol. 2012;39:96–103. doi: 10.1111/j.1440-1681.2011.05482.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ozawa Y, Kobori H, Suzaki Y, Navar LG. Sustained renal interstitial macrophage infiltration following chronic angiotensin II infusions. Am J Physiol Renal Physiol. 2007;292:F330–F339. doi: 10.1152/ajprenal.00059.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.He J, Mills KT, Appel LJ, et al. Urinary sodium and potassium excretion and CKD progression. J Am Soc Nephrol. 2016;27:1202–1212. doi: 10.1681/ASN.2015010022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mente A, O’Donnell M, Rangarajan S, et al. Urinary sodium excretion, blood pressure, cardiovascular disease, and mortality: a community-level prospective epidemiological cohort study. Lancet. 2018;392:496–506. doi: 10.1016/S0140-6736(18)31376-X. [DOI] [PubMed] [Google Scholar]
- 19.McMahon EJ, Campbell KL, Bauer JD, Mudge DW. Altered dietary salt intake for people with chronic kidney disease. Cochrane Database Syst Rev. 2015;(2):CD010070. doi: 10.1002/14651858.CD010070.pub2. [DOI] [PubMed] [Google Scholar]
- 20.Ekinci EI, Clarke S, Thomas MC, et al. Dietary salt intake and mortality in patients with type 2 diabetes. Diabetes Care. 2011;34:703–709. doi: 10.2337/dc10-1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mills KT, Chen J, Yang W, et al. Sodium excretion and the risk of cardiovascular disease in patients with chronic kidney disease. JAMA. 2016;315:2200–2210. doi: 10.1001/jama.2016.4447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hung SC, Lai YS, Kuo KL, Tarng DC. Volume overload and adverse outcomes in chronic kidney disease: clinical observational and animal studies. J Am Heart Assoc. 2015;4:e001918. doi: 10.1161/JAHA.115.001918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Borrelli S, Provenzano M, Gagliardi I, et al. Sodium intake and chronic kidney disease. Int J Mol Sci. 2020;21:4744. doi: 10.3390/ijms21134744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guyton AC. Kidneys and fluids in pressure regulation: small volume but large pressure changes. Hypertension. 1992;19(1 Suppl):I2–I8. doi: 10.1161/01.hyp.19.1_suppl.i2. [DOI] [PubMed] [Google Scholar]
- 25.Titze J, Dahlmann A, Lerchl K, et al. Spooky sodium balance. Kidney Int. 2014;85:759–767. doi: 10.1038/ki.2013.367. [DOI] [PubMed] [Google Scholar]
- 26.Strauss MB, Lamdin E, Smith WP, Bleifer DJ. Surfeit and deficit of sodium; a kinetic concept of sodium excretion. AMA Arch Intern Med. 1958;102:527–536. doi: 10.1001/archinte.1958.00260210013003. [DOI] [PubMed] [Google Scholar]
- 27.Walser M. Phenomenological analysis of renal regulation of sodium and potassium balance. Kidney Int. 1985;27:837–841. doi: 10.1038/ki.1985.89. [DOI] [PubMed] [Google Scholar]
- 28.Koomans HA, Roos JC, Boer P, Geyskes GG, Mees EJ. Salt sensitivity of blood pressure in chronic renal failure. Evidence for renal control of body fluid distribution in man. Hypertension. 1982;4:190–197. doi: 10.1161/01.hyp.4.2.190. [DOI] [PubMed] [Google Scholar]
- 29.Shibata S, Nagase M, Yoshida S, et al. Modification of mineralocorticoid receptor function by Rac1 GTPase: implication in proteinuric kidney disease. Nat Med. 2008;14:1370–1376. doi: 10.1038/nm.1879. [DOI] [PubMed] [Google Scholar]
- 30.Bovée DM, Cuevas CA, Zietse R, Danser AH, Mirabito Colafella KM, Hoorn EJ. Salt-sensitive hypertension in chronic kidney disease: distal tubular mechanisms. Am J Physiol Renal Physiol. 2020;319:F729–F745. doi: 10.1152/ajprenal.00407.2020. [DOI] [PubMed] [Google Scholar]
- 31.Farjah M, Roxas BP, Geenen DL, Danziger RS. Dietary salt regulates renal SGK1 abundance: relevance to salt sensitivity in the Dahl rat. Hypertension. 2003;41:874–878. doi: 10.1161/01.HYP.0000063885.48344.EA. [DOI] [PubMed] [Google Scholar]
- 32.Kidney Disease Outcomes Quality Initiative (K/DOQI) K/DOQI clinical practice guidelines on hypertension and antihypertensive agents in chronic kidney disease. Am J Kidney Dis. 2004;43(5 Suppl 1):S1–S290. [PubMed] [Google Scholar]
- 33.Essig M, Escoubet B, de Zuttere D, et al. Cardiovascular remodelling and extracellular fluid excess in early stages of chronic kidney disease. Nephrol Dial Transplant. 2008;23:239–248. doi: 10.1093/ndt/gfm542. [DOI] [PubMed] [Google Scholar]
- 34.Agarwal R, Pappas MK, Sinha AD. Masked uncontrolled hypertension in CKD. J Am Soc Nephrol. 2016;27:924–932. doi: 10.1681/ASN.2015030243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Drawz PE, Alper AB, Anderson AH, et al. Masked hypertension and elevated nighttime blood pressure in CKD: prevalence and association with target organ damage. Clin J Am Soc Nephrol. 2016;11:642–652. doi: 10.2215/CJN.08530815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sinha AD, Agarwal R. The complex relationship between CKD and ambulatory blood pressure patterns. Adv Chronic Kidney Dis. 2015;22:102–107. doi: 10.1053/j.ackd.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.National Institute for Health and Care Excellence (NICE) Chronic kidney disease in adults: assessment and management [CG182] London: NICE; 2014. [PubMed] [Google Scholar]
- 38.Kario K. Key points of the 2019 Japanese Society of Hypertension Guidelines for the Management of Hypertension. Korean Circ J. 2019;49:1123–1135. doi: 10.4070/kcj.2019.0246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Whelton PK, Carey RM, Aronow WS, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension. 2018;71:e13–e115. doi: 10.1161/HYP.0000000000000065. [DOI] [PubMed] [Google Scholar]
- 40.You SC, Jung S, Swerdel JN, et al. Comparison of first-line dual combination treatments in hypertension: real-world evidence from multinational heterogeneous cohorts. Korean Circ J. 2020;50:52–68. doi: 10.4070/kcj.2019.0173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Williams B, Mancia G, Spiering W, et al. 2018 ESC/ESH Guidelines for the management of arterial hypertension. Eur Heart J. 2018;39:3021–3104. doi: 10.1093/eurheartj/ehy339. [DOI] [PubMed] [Google Scholar]
- 42.Haider DG, Sauter T, Lindner G, et al. Use of calcium channel blockers is associated with mortality in patients with chronic kidney disease. Kidney Blood Press Res. 2015;40:630–637. doi: 10.1159/000368539. [DOI] [PubMed] [Google Scholar]
- 43.Lin YC, Lin JW, Wu MS, Chen KC, Peng CC, Kang YN. Effects of calcium channel blockers comparing to angiotensin-converting enzyme inhibitors and angiotensin receptor blockers in patients with hypertension and chronic kidney disease stage 3 to 5 and dialysis: a systematic review and meta-analysis. PLoS One. 2017;12:e0188975. doi: 10.1371/journal.pone.0188975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Franco Palacios CR, Goyal P, Thompson AM, Deschaine B. Systolic blood pressure values might further risk-stratify the adverse outcomes of LVH in older patients with chronic kidney disease. Clin Hypertens. 2016;22:21. doi: 10.1186/s40885-016-0056-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zamboli P, De Nicola L, Minutolo R, et al. Effect of furosemide on left ventricular mass in non-dialysis chronic kidney disease patients: a randomized controlled trial. Nephrol Dial Transplant. 2011;26:1575–1583. doi: 10.1093/ndt/gfq565. [DOI] [PubMed] [Google Scholar]
- 46.Vogt L, Waanders F, Boomsma F, de Zeeuw D, Navis G. Effects of dietary sodium and hydrochlorothiazide on the antiproteinuric efficacy of losartan. J Am Soc Nephrol. 2008;19:999–1007. doi: 10.1681/ASN.2007060693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang CT, Kor CT, Hsieh YP. Long-term effects of spironolactone on kidney function and hyperkalemia-associated hospitalization in patients with chronic kidney disease. J Clin Med. 2018;7:459. doi: 10.3390/jcm7110459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Agarwal R, Rossignol P, Romero A, et al. Patiromer versus placebo to enable spironolactone use in patients with resistant hypertension and chronic kidney disease (AMBER): a phase 2, randomised, double-blind, placebo-controlled trial. Lancet. 2019;394:1540–1550. doi: 10.1016/S0140-6736(19)32135-X. [DOI] [PubMed] [Google Scholar]
- 49.Ellison DH. Clinical pharmacology in diuretic use. Clin J Am Soc Nephrol. 2019;14:1248–1257. doi: 10.2215/CJN.09630818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.He Y, Pachori A, Chen P, et al. Glucosuric, renal and haemodynamic effects of licogliflozin, a dual inhibitor of sodium-glucose co-transporter-1 and sodium-glucose co-transporter-2, in patients with chronic kidney disease: a randomized trial. Diabetes Obes Metab. 2021;23:1182–1190. doi: 10.1111/dom.14327. [DOI] [PubMed] [Google Scholar]
- 51.Kobayashi K, Toyoda M, Hatori N, et al. Sodium-glucose cotransporter 2 inhibitor-induced reduction in the mean arterial pressure improved renal composite outcomes in type 2 diabetes mellitus patients with chronic kidney disease: a propensity score-matched model analysis in Japan. J Diabetes Investig. 2021;12:1408–1416. doi: 10.1111/jdi.13491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rhee JJ, Jardine MJ, Chertow GM, Mahaffey KW. Dedicated kidney disease-focused outcome trials with sodium-glucose cotransporter-2 inhibitors: lessons from CREDENCE and expectations from DAPA-HF, DAPA-CKD, and EMPA-KIDNEY. Diabetes Obes Metab. 2020;22(Suppl 1):46–54. doi: 10.1111/dom.13987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grodin JL, Tang WH. Sodium-glucose cotransporter-2 inhibitors and loop diuretics for heart failure: priming the natriuretic and metabolic reserve of the kidney. Circulation. 2020;142:1055–1058. doi: 10.1161/CIRCULATIONAHA.120.048057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vardeny O, Claggett B, Kachadourian J, et al. Reduced loop diuretic use in patients taking sacubitril/valsartan compared with enalapril: the PARADIGM-HF trial. Eur J Heart Fail. 2019;21:337–341. doi: 10.1002/ejhf.1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ettehad D, Emdin CA, Kiran A, et al. Blood pressure lowering for prevention of cardiovascular disease and death: a systematic review and meta-analysis. Lancet. 2016;387:957–967. doi: 10.1016/S0140-6736(15)01225-8. [DOI] [PubMed] [Google Scholar]
- 56.Malhotra R, Nguyen HA, Benavente O, et al. Association between more intensive vs less intensive blood pressure lowering and risk of mortality in chronic kidney disease stages 3 to 5: a systematic review and meta-analysis. JAMA Intern Med. 2017;177:1498–1505. doi: 10.1001/jamainternmed.2017.4377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schmidt M, Mansfield KE, Bhaskaran K, et al. Serum creatinine elevation after renin-angiotensin system blockade and long term cardiorenal risks: cohort study. BMJ. 2017;356:j791. doi: 10.1136/bmj.j791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hirsch S, Hirsch J, Bhatt U, Rovin BH. Tolerating increases in the serum creatinine following aggressive treatment of chronic kidney disease, hypertension and proteinuria: pre-renal success. Am J Nephrol. 2012;36:430–437. doi: 10.1159/000343453. [DOI] [PubMed] [Google Scholar]
- 59.Ruggenenti P, Remuzzi G. Dealing with renin-angiotensin inhibitors, don’t mind serum creatinine. Am J Nephrol. 2012;36:427–429. doi: 10.1159/000343455. [DOI] [PubMed] [Google Scholar]
- 60.Hoshida S, Nakagawa T, Shinoda Y, Inui H, Watanabe T. Analysis of the difference in the pressure-natriuresis relationship based on the class of medication using spot urine tests in hypertensive patients. J Hypertens. 2014;3:1000154. [Google Scholar]
- 61.Malhotra R, Katz R, Jotwani V, et al. Estimated GFR variability and risk of cardiovascular events and mortality in SPRINT (Systolic Blood Pressure Intervention Trial) Am J Kidney Dis. 2021;78:48–56. doi: 10.1053/j.ajkd.2020.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Beddhu S, Shen J, Cheung AK, et al. Implications of early decline in eGFR due to intensive BP control for cardiovascular outcomes in SPRINT. J Am Soc Nephrol. 2019;30:1523–1533. doi: 10.1681/ASN.2018121261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Arora N, Katz R, Bansal N. ACE inhibitor/angiotensin receptor blocker use patterns in advanced CKD and risk of kidney failure and death. Kidney Med. 2020;2:248–257. doi: 10.1016/j.xkme.2019.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Oh YJ, Kim SM, Shin BC, et al. The impact of renin-angiotensin system blockade on renal outcomes and mortality in pre-dialysis patients with advanced chronic kidney disease. PLoS One. 2017;12:e0170874. doi: 10.1371/journal.pone.0170874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fu EL, Evans M, Clase CM, et al. Stopping renin-angiotensin system inhibitors in patients with advanced CKD and risk of adverse outcomes: a nationwide study. J Am Soc Nephrol. 2021;32:424–435. doi: 10.1681/ASN.2020050682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.D’hoore E, Neirynck N, Schepers E, et al. Chronic kidney disease progression is mainly associated with non-recovery of acute kidney injury. J Nephrol. 2015;28:709–716. doi: 10.1007/s40620-015-0181-5. [DOI] [PubMed] [Google Scholar]
- 67.Chua HR, Wong WK, Ong VH, et al. Extended mortality and chronic kidney disease after septic acute kidney injury. J Intensive Care Med. 2020;35:527–535. doi: 10.1177/0885066618764617. [DOI] [PubMed] [Google Scholar]
- 68.Khan YH, Sarriff A, Adnan AS, Khan AH, Mallhi TH. Chronic kidney disease, fluid overload and diuretics: a complicated triangle. PLoS One. 2016;11:e0159335. doi: 10.1371/journal.pone.0159335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Velasquez MT, Beddhu S, Nobakht E, Rahman M, Raj DS. Ambulatory blood pressure in chronic kidney disease: ready for prime time? Kidney Int Rep. 2016;1:94–104. doi: 10.1016/j.ekir.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cai W, Lang M, Jiang X, et al. Correlation among high salt intake, blood pressure variability, and target organ damage in patients with essential hypertension: study protocol clinical trial (SPIRIT compliant) Medicine (Baltimore) 2020;99:e19548. doi: 10.1097/MD.0000000000019548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sarafidis PA, Ruilope LM, Loutradis C, et al. Blood pressure variability increases with advancing chronic kidney disease stage: a cross-sectional analysis of 16 546 hypertensive patients. J Hypertens. 2018;36:1076–1085. doi: 10.1097/HJH.0000000000001670. [DOI] [PubMed] [Google Scholar]
- 72.Wang Q, Wang Y, Wang J, Zhang L, Zhao MH, C‐STRIDE (Chinese Cohort Study of Chronic Kidney Disease) Short-term systolic blood pressure variability and kidney disease progression in patients with chronic kidney disease: results from C-STRIDE. J Am Heart Assoc. 2020;9:e015359. doi: 10.1161/JAHA.120.015359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chang TI, Tabada GH, Yang J, Tan TC, Go AS. Visit-to-visit variability of blood pressure and death, end-stage renal disease, and cardiovascular events in patients with chronic kidney disease. J Hypertens. 2016;34:244–252. doi: 10.1097/HJH.0000000000000779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mallamaci F, Tripepi G, D’Arrigo G, et al. Blood pressure variability, mortality, and cardiovascular outcomes in CKD patients. Clin J Am Soc Nephrol. 2019;14:233–240. doi: 10.2215/CJN.04030318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Murphy D, Drawz PE. Blood pressure variability in CKD: treatable or hypertension's homocysteine? Clin J Am Soc Nephrol. 2019;14:175–177. doi: 10.2215/CJN.14991218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Di Iorio B, Pota A, Sirico ML, et al. Blood pressure variability and outcomes in chronic kidney disease. Nephrol Dial Transplant. 2012;27:4404–4410. doi: 10.1093/ndt/gfs328. [DOI] [PubMed] [Google Scholar]
- 77.Gregg LP, Hedayati SS, Yang H, et al. Association of blood pressure variability and diuretics with cardiovascular events in patients with chronic kidney disease stages 1-5. Hypertension. 2021;77:948–959. doi: 10.1161/HYPERTENSIONAHA.120.16117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jamerson K, Weber MA, Bakris GL, et al. Benazepril plus amlodipine or hydrochlorothiazide for hypertension in high-risk patients. N Engl J Med. 2008;359:2417–2428. doi: 10.1056/NEJMoa0806182. [DOI] [PubMed] [Google Scholar]
- 79.Perkins RM, Tang X, Bengier AC, Kirchner HL, Bucaloiu ID. Variability in estimated glomerular filtration rate is an independent risk factor for death among patients with stage 3 chronic kidney disease. Kidney Int. 2012;82:1332–1338. doi: 10.1038/ki.2012.281. [DOI] [PubMed] [Google Scholar]
- 80.Chen SC, Lin MY, Huang TH, et al. Variability in estimated glomerular filtration rate by area under the curve predicts renal outcomes in chronic kidney disease. ScientificWorldJournal. 2014;2014:802037. doi: 10.1155/2014/802037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sinha AD, Agarwal R. Can chronic volume overload be recognized and prevented in hemodialysis patients?: the pitfalls of the clinical examination in assessing volume status. Semin Dial. 2009;22:480–482. doi: 10.1111/j.1525-139X.2009.00641.x. [DOI] [PubMed] [Google Scholar]
- 82.Ekinci C, Karabork M, Siriopol D, Dincer N, Covic A, Kanbay M. Effects of volume overload and current techniques for the assessment of fluid status in patients with renal disease. Blood Purif. 2018;46:34–47. doi: 10.1159/000487702. [DOI] [PubMed] [Google Scholar]
- 83.Ok E, Asci G, Chazot C, Ozkahya M, Mees EJ. Controversies and problems of volume control and hypertension in haemodialysis. Lancet. 2016;388:285–293. doi: 10.1016/S0140-6736(16)30389-0. [DOI] [PubMed] [Google Scholar]
- 84.Kalainy S, Reid R, Jindal K, Pannu N, Braam B. Fluid volume expansion and depletion in hemodialysis patients lack association with clinical parameters. Can J Kidney Health Dis. 2015;2:54. doi: 10.1186/s40697-015-0090-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Agarwal R, Weir MR. Dry-weight: a concept revisited in an effort to avoid medication-directed approaches for blood pressure control in hemodialysis patients. Clin J Am Soc Nephrol. 2010;5:1255–1260. doi: 10.2215/CJN.01760210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.McIntyre CW, Salerno FR. Diagnosis and treatment of intradialytic hypotension in maintenance hemodialysis patients. Clin J Am Soc Nephrol. 2018;13:486–489. doi: 10.2215/CJN.11131017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Flythe JE, Chang TI, Gallagher MP, et al. Blood pressure and volume management in dialysis: conclusions from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2020;97:861–876. doi: 10.1016/j.kint.2020.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sinha AD, Agarwal R. Clinical pharmacology of antihypertensive therapy for the treatment of hypertension in CKD. Clin J Am Soc Nephrol. 2019;14:757–764. doi: 10.2215/CJN.04330418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tieu A, Velenosi TJ, Kucey AS, Weir MA, Urquhart BL. β-Blocker dialyzability in maintenance hemodialysis patients: a randomized clinical trial. Clin J Am Soc Nephrol. 2018;13:604–611. doi: 10.2215/CJN.07470717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zoccali C, Moissl U, Chazot C, et al. Chronic fluid overload and mortality in ESRD. J Am Soc Nephrol. 2017;28:2491–2497. doi: 10.1681/ASN.2016121341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bansal N, McCulloch CE, Lin F, et al. Blood pressure and risk of cardiovascular events in patients on chronic hemodialysis: the CRIC Study (Chronic Renal Insufficiency Cohort) Hypertension. 2017;70:435–443. doi: 10.1161/HYPERTENSIONAHA.117.09091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Shafi T, Sozio SM, Bandeen-Roche KJ, et al. Predialysis systolic BP variability and outcomes in hemodialysis patients. J Am Soc Nephrol. 2014;25:799–809. doi: 10.1681/ASN.2013060667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Charytan D. Is left ventricular hypertrophy a modifiable risk factor in end-stage renal disease. Curr Opin Nephrol Hypertens. 2014;23:578–585. doi: 10.1097/MNH.0000000000000067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zoccali C, Benedetto FA, Mallamaci F, et al. Left ventricular mass monitoring in the follow-up of dialysis patients: prognostic value of left ventricular hypertrophy progression. Kidney Int. 2004;65:1492–1498. doi: 10.1111/j.1523-1755.2004.00530.x. [DOI] [PubMed] [Google Scholar]
- 95.London GM, Pannier B, Guerin AP, et al. Alterations of left ventricular hypertrophy in and survival of patients receiving hemodialysis: follow-up of an interventional study. J Am Soc Nephrol. 2001;12:2759–2767. doi: 10.1681/ASN.V12122759. [DOI] [PubMed] [Google Scholar]
- 96.Donciu MD, Lumita V, Covic A. In: Cardio-renal clinical challenges. Goldsmith D, Covic A, Spaak J, editors. Switzerland: Springer International Publishing, Cham; 2015. Chapter 12. Volume overload in ckd: pathophysiology, assessment techniques, consequences and treatment; pp. 119–114. [Google Scholar]
- 97.Kayikcioglu M, Tumuklu M, Ozkahya M, et al. The benefit of salt restriction in the treatment of end-stage renal disease by haemodialysis. Nephrol Dial Transplant. 2009;24:956–962. doi: 10.1093/ndt/gfn599. [DOI] [PubMed] [Google Scholar]