

Abstract
In the kidney, a set of proteins expressed in the epithelial cells of the thick ascending loop of Henle and the distal convoluted tubule directly or indirectly play important roles in the regulation of serum magnesium levels. Magnesium reabsorption in the thick ascending loop of Henle occurs through a passive paracellular pathway, while in the distal convoluted tubule, the final magnesium concentration is established through an active transcellular pathway. The players involved in magnesium reabsorption include proteins with diverse functions including tight junction proteins, cation and anion channels, sodium chloride cotransporter, calcium-sensing receptor, epidermal growth factor, cyclin M2, sodium potassium adenosine triphosphatase subunits, transcription factors, a serine protease, and proteins involved in mitochondrial function. Mutations in the genes that encode these proteins impair their function and cause different rare diseases associated with hypomagnesemia, which may lead to muscle cramps, fatigue, epileptic seizures, intellectual disability, cardiac arrhythmias, and chronic kidney disease. The purpose of this review is to describe the clinical and genetic characteristics of these hereditary kidney diseases and the current research findings on the pathophysiological basis of these diseases.
Keywords: Hypomagnesemia, Magnesium handling, Mutation, Rare disease, Renal tubulopathies
Introduction
Magnesium (Mg2+), the second-most abundant intracellular cation, is an indispensable ion for many cellular functions including energy metabolism and nucleic acid and protein synthesis [1]. Mg2+ is also a regulator of sodium, potassium, and calcium channels. Therefore, serum Mg2+ levels, which are usually 0.70 to 1.1 mmol/L, need to be precisely controlled. Hypomagnesemia is defined as serum Mg2+ level below 0.7 mmol/L. The predominant laboratory tests used for the diagnosis of hypomagnesemia are the serum Mg2+ concentration and the 24-hour urinary Mg2+ tests. In cases in which the serum Mg2+ level is low, a 24-hour urine Mg2+ higher than 24 mg/day suggests renal Mg2+ wasting as the cause of the hypomagnesemia, while values lower than 24 mg/day indicate deficient Mg2+ intake and/or gastrointestinal losses. Magnesium homeostasis is determined by intestinal absorption, renal reabsorption, and storage in bone. In the kidney and intestine, these processes involve a combination of paracellular and transcellular epithelial transport routes. Hypomagnesemia may cause muscle cramps, fatigue, appetite loss, and disruptions in calcium and potassium homeostasis [1]. Acute hypomagnesemia may lead to more serious consequences like epileptic seizures, intellectual disability, and cardiac arrhythmias. Causes of hypomagnesemia include type 2 diabetes, gastrointestinal diseases, alcoholism, use of diuretics or other medications, dietary deficiency, and genetic defects. Over the last two decades, clinical and genetic studies of patients with rare hereditary disorders of Mg2+ handling have enabled the identification of important components of epithelial Mg2+ transport in the kidney [2].
After passing the glomerular filter, 90% to 95% of the filtered Mg2+ is subsequently reabsorbed along the nephron [1]. Approximately 10% to 25% of the Mg2+ is reabsorbed by the proximal tubule (PT), 50% to 70% is reabsorbed by the thick ascending limb of the loop of Henle (TAL), and 5% to 10% is reabsorbed by the distal convoluted tubule (DCT) (Fig. 1). In the PT, Mg2+ reabsorption occurs in a passive paracellular mode. The mechanisms that control this process are unknown. In the TAL, Mg2+ reabsorption also takes place through a passive paracellular pathway, which is facilitated by tight junction proteins claudin-16 and claudin-19 (Fig. 2) [3,4]. The basolateral calcium-sensing receptor (CaSR) controls the paracellular transport of calcium (Ca2+) and Mg2+ by regulating the claudin-16/claudin-19 channel function through a signaling pathway [5]. In contrast, the main K+ secretory channel in the kidney, the renal outer medullary K+ channel (ROMK), which is located in the apical membrane of the TAL, plays a key role in the generation of the lumen-positive potential in TAL [1]. The renal chloride (Cl–) channel ClC-Kb, which is located in the basolateral membrane of the TAL and also the DCT, facilitates the efflux of Cl– to the interstitium [6]. The DCT establishes the final Mg2+ concentration through active transcellular reabsorption, which is highly regulated, via transient receptor potential melastatin type 6 (TRPM6) Mg2+ channels located in the apical membrane [7] (Fig. 3). TRPM6 needs to form heterotetramers with its close homolog TRPM7 to function [8]. The protein(s) that facilitates Mg2+ efflux from the basolateral side to the blood compartment has not been identified [9]. Cyclin M2 (CNNM2) and solute carrier family member A1 (SLC41A1) have been proposed as likely candidates for this function, but this subject is still under debate [10–12]. Other proteins involved, albeit indirectly, in Mg2+ transport in the DCT include epidermal growth factor (EGF), a hormone that regulates the expression of TRPM6 on the apical membrane, basolateral Na+, K+-adenosine triphosphatase (ATPase), and K+ channel Kir4.1, and the apical K+ channel Kv1.1 and Na+ Cl– cotransporter (NCC) [13–16]. The last four proteins participate in the generation of the driving force needed for Mg2+ transport [9]. Mutations in any of the above proteins, except SLC41A1, which are involved directly or indirectly in Mg2+ transport, have been shown to cause hypomagnesemia (Table 1).
Figure 1. Schematic drawing of Mg2+ reabsorption by different segments of the nephron.
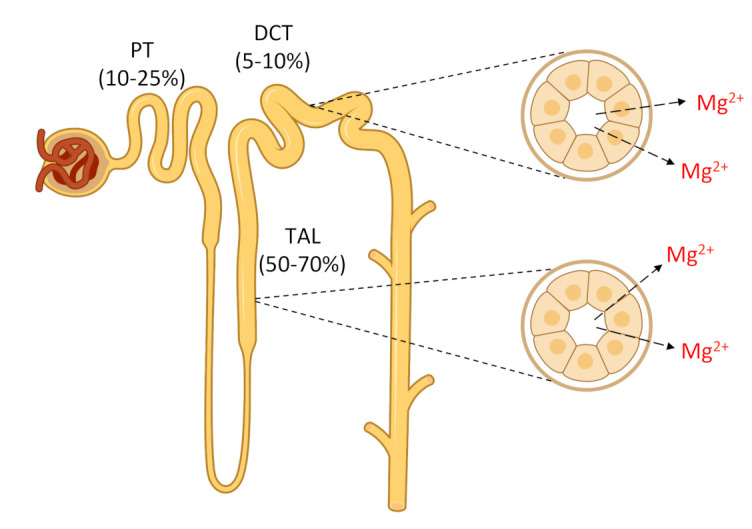
Mg2+ is filtered at the glomerulus. In the proximal tubule (PT), about 10% to 25% of the initially filtered load of Mg2+ is reabsorbed through a passive paracellular route that remains poorly understood. The thick ascending limb of the loop of Henle (TAL) reabsorbs approximately 50% to 70% of the filtered load of Mg2+ by a passive paracellular transport. The remaining 5% to 10% of the filtered Mg2+ is reabsorbed in the distal convoluted tubule (DCT) through an active transcellular route. Approximately 95% of filtered Mg2+ is reabsorbed along the nephron.
Figure 2. Reabsorption of Mg2+ in the TAL.
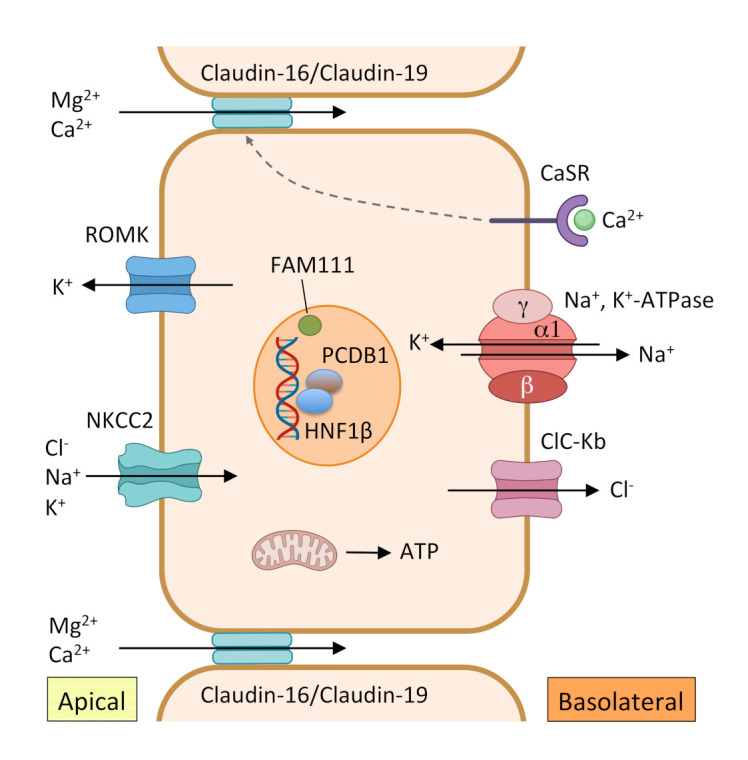
In this segment of the nephron, Mg2+ reabsorption takes place through a passive paracellular pathway via claudin-16/claudin-19 channels located in the tight junctions. This transport depends on the driving force generated by the joint actions of Na+, K+-adenosine triphosphatase (ATPase), and Cl– channel ClC-Kb, both located in the basolateral membrane, and Na+-K+-2Cl-cotransporter (NKCC2) and renal outer medullary K+ channel (ROMK), the main K+ secretory channel in the kidney, located in the apical membrane. Basolateral calcium-sensing receptor (CaSR) indirectly regulates the paracellular divalent cation transport and the transcellular NaCl transport in the TAL.
ATP, adenosine triphosphate; PCBD1, pterin-4 α-carbinolamine dehydratase; HNF1β, hepatocyte nuclear factor-1 beta; TAL, thick ascending limb of the loop of Henle.
Figure 3. Reabsorption of Mg2+ in the DCT.
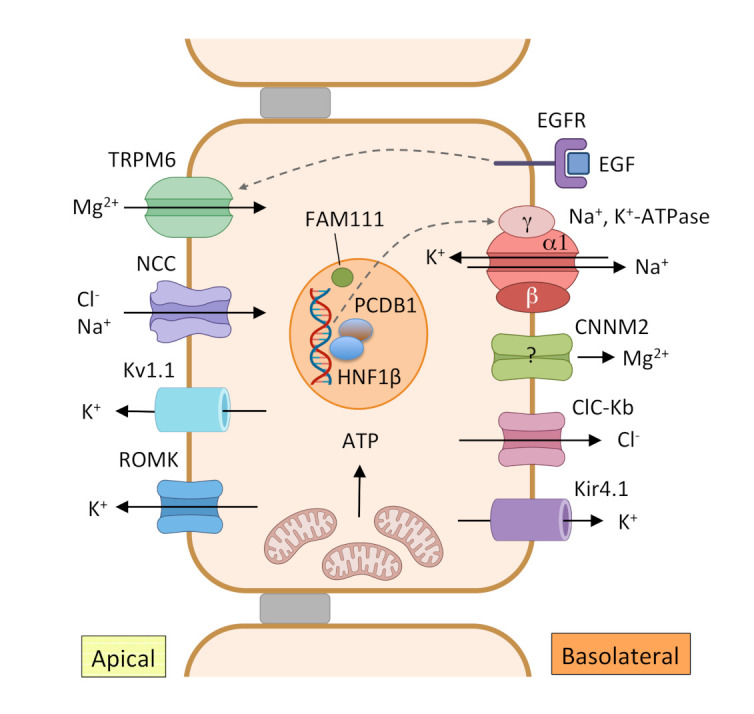
In this tubular segment, Mg2+ is reabsorbed through a transcellular pathway. The apical Mg2+ channel transient receptor potential melastatin type 6 (TRPM6) facilitates transport of Mg2+ from the lumen to the epithelial cell. The driving force needed for Mg2+ transport is generated by the joint actions of Na+, K+-adenosine triphosphatase (ATPase), the Cl– channel ClC-Kb and the K+ channel Kir4.1 in the basolateral membrane and Na+-Cl– cotransporter (NCC) and K+ secretory channels Kv1.1 and renal outer medullary K+ channel (ROMK) in the apical membrane. Epidermal growth factor (EGF) is implicated in the regulation of Mg2+ reabsorption through TRPM6. HNF1β with coactivator pterin-4 α-carbinolamine dehydratase (PCBD1) increase transcription of the γ subunit of the Na+, K+-ATPase. Cyclin M2 (CNNM2) is a candidate for basolateral extrusion of Mg2+ or a regulator of Mg2+ transport, but this has not been experimentally confirmed.
ATP, adenosine triphosphate; DCT, distal convoluted tubule; EGFR, epidermal growth factor receptor; HNF1β, hepatocyte nuclear factor-1 beta.
Table 1.
Inherited diseases associated with hypomagnesemia
| Disorder | Associated gene | Proteina | Inheritance | OMIM # |
|---|---|---|---|---|
| Group 1 | ||||
| FHHNC type 1 | CLDN16 | Claudin-16 | AR | 248250 |
| FHHNC type 2 | CLDN19 | Claudin-19 | AR | 248190 |
| Hypomagnesemia with secondary hypocalcemia | TRPM6 | Mg2+ channel TRPM6 | AR | 602014 |
| Isolated recessive hypomagnesemia | EGF | Epidermal growth factor | AR | 611718 |
| NISBD2 | EGFR | Epidermal growth factor receptor | AR | 616069 |
| HSMR type 1 | CNNM2 | Cyclin M2 | AD/AR | 616418/613882 |
| Group 2 | ||||
| Bartter syndrome type 3 | CLCNKB | Cl– channel ClC-Kb | AR | 607364 |
| Gitelman syndrome | SLC12A3 | Na+ Cl– cotransporter | AR | 263800 |
| Autosomal dominant hypocalcemia with hypercalciuria | CASR | Calcium-sensing receptor CaSR | AD | 601198 |
| Episodic ataxia type 1 | KCNA1 | Voltage-gated K+ channel Kv1.1 | AD | 160120 |
| Isolated dominant hypomagnesemia | FXYD2 | γ subunit of Na+, K+-ATPase | AD | 154020 |
| HSMR type 2 | ATP1A1 | α1 subunit of Na+, K+-ATPase | AD | 618314 |
| EAST/SeSAME syndrome | KCNJ10 | K+ channel Kir4.1 | AR | 612780 |
| ADTKD-HNF1B | HNF1B | Transcription factor HNF1β | AD | 137920 |
| TNHP | PCBD1 | PCBD1, dimerization cofactor of HNF1β | AR | 264070 |
| Kenny-Caffey syndrome type 2 | FAM111A | Nuclear serine protease FAM111A | AD | 127000 |
| Group 3 | ||||
| HHH syndrome | MT-TI | Isoleucine tRNA | Mitochondrial | 500005 |
| Kearns-Sayre syndrome | Large deletions | - | Mitochondrial | 530000 |
| HUPRA syndrome | SARS2 | seryl-tRNA synthetase | AR | 613845 |
AD, autosomal dominant; ADTKD-HNF1B, autosomal dominant tubulointerstitial kidney disease subtype HNF1B; AR, autosomal recessive; EAST/SeSAME, epilepsy, ataxia, sensorineural deafness and tubulopathy syndrome/seizures, sensorineural deafness, ataxia, mental retardation and electrolyte imbalance; FHHNC, familial hypomagnesemia with hypercalciuria and nephrocalcinosis; HHH, hypertension, hypercholesterolemia and hypomagnesemia; HNF1B, hepatocyte nuclear factor-1 beta; HSMR, hypomagnesemia, seizures, and mental retardation disorder; HUPRA, hyperuricemia, pulmonary hypertension, renal failure and alkalosis; NISBD2, neonatal inflammatory skin and bowel disease type 2; TNHP, transient neonatal hyperphenylalaninemia and primapterinuria; tRNA, transfer RNA.
All the genes associated with hereditary hypomagnesemias encode proteins, except MT-TI, which encodes a tRNA.
In this review, we present the current knowledge of hereditary kidney diseases associated with hypomagnesemia. We discuss the clinical characteristics and genetic information for each disease and describe the pathophysiological basis that has been proposed for some of the diseases, although in general these remain incompletely understood. We classified hypomagnesemias in three groups according to the implicated genes (Table 1). Group 1 includes hypomagnesemias associated with genes that encode proteins directly involved in Mg2+ transport and the regulatory proteins. Group 2 contains hypomagnesemias associated with genes encoding proteins involved in transport of other ions or their regulators, which indirectly affect Mg2+ handling. Group 3 includes hypomagnesemias associated with genes required for mitochondrial function, which also indirectly result in Mg2+ loss.
Group 1 hypomagnesemias
Familial hypomagnesemia with hypercalciuria and nephrocalcinosis types 1 and 2
Familial hypomagnesemia with hypercalciuria and nephrocalcinosis (FHHNC) is a rare autosomal recessive tubular disorder characterized by excessive urinary loss of Mg2+ and Ca2+, bilateral nephrocalcinosis, and progressive chronic kidney disease (CKD) [17]. FHHNC patients typically present during early childhood or before adolescence with recurrent urinary tract infections, polyuria, polydipsia, nephrolithiasis, and failure to thrive [18–21]. FHHNC patients may show a pronounced decline in glomerular filtration rate at the time of diagnosis, and approximately one-third of cases progress to chronic renal failure during childhood or adolescence [21,22]. In contrast to patients with other hypomagnesemias, FHHNC patients have high serum levels of parathyroid hormone (PTH) before the onset of chronic renal failure [1,19,23]. In some cases, patients present amelogenesis imperfecta [24,25]. Clinical signs of severe hypomagnesemia such as seizures and muscular tetany are rare.
FHHNC is caused by recessive mutations in CLDN16 (FHHNC type 1, OMIM #248250) or CLDN19 (FHHNC type 2, OMIM #248190) [3,4]. Patients with mutations in CLDN19 also present ocular abnormalities such as severe myopia, nystagmus, and macular colobamata [4,21,26]. CLDN16 and CLDN19 encode the tight junction proteins claudin-16 and claudin-19, respectively, which are strongly expressed in the kidney [3,4]. Claudin-19 is also expressed in peripheral neurons and retina [4,27]. Claudin-16 and claudin-19 form heteromeric paracellular cation channels in the TAL that regulate Ca2+ and Mg2+ transport [28,29]. The reabsorption of Mg2+ is greatly dependent on the transepithelial potential as a driving force, which is created by a transepithelial NaCl concentration gradient. CLDN16 and CLDN19 mutations have been shown to impair the permeating function of these channels, resulting in a reduced lumen-positive potential and the simultaneous loss of the driving force for Mg2+ reabsorption [22,28].
Hypomagnesemia with secondary hypocalcemia
Hypomagnesemia with secondary hypocalcemia (HSH, OMIM #602014) is a rare autosomal recessive disorder characterized by severe hypomagnesemia associated with hypocalcemia. The levels of serum Mg2+ in patients with HSH are usually much lower than in patients with other types of hereditary hypomagnesemias [30]. The disease usually presents in early infancy, with neurological symptoms including tetany and severe seizures that are refractory to anticonvulsant therapy [30,31]. HSH patients have very low serum levels of PTH. Persistent low levels of serum Mg2+ likely cause secondary hypocalcemia by inhibiting PTH secretion and inducing resistance at the receptor sites [30]. The main defect in HSH is a reduction in intestinal Mg2+ absorption, which is in contrast to all other identified hereditary hypomagnesemias, along with reduced Mg2+ reabsorption in the DCT with renal Mg2+ wasting [31,32].
HSH is caused by recessive loss-of-function mutations in the TRPM6 gene, which encodes the TRPM6 cation channel [31,32]. TRPM6 is predominantly expressed in the apical membrane of the intestinal and renal DCT epithelial cells, where it is involved in Mg2+ reabsorption [7]. Its channel activity and expression are regulated by several factors including EGF and adenosine triphosphate (ATP) [14]. TRPM6 mutations identified in HSH patients disrupt Mg2+ conductance through the channel, both in the colon and DCT, causing hypomagnesemia [7]. However, the mechanisms leading to this disease are not entirely known. TRPM6 interacts with its close homolog TRPM7 to form functional heteromeric TRPM6/TRPM7 Mg2+ channels [8]. TRPM6 and TRPM7 contain a transmembrane ion channel segment covalently joined to a cytosolic serine/threonine protein kinase domain at the carboxy terminus. The kinase domain is cleaved from the channel segment and, after its translocation to the nucleus, it regulates the transcription of many genes involved in development [33]. Therefore, it has been suggested that TRPM6 may play a much wider role in the cell than only in Mg2+ homeostasis [33].
Isolated recessive hypomagnesemia
Isolated recessive renal hypomagnesemia is a rare disorder characterized by hypomagnesemia and normocalciuria [34]. Hypomagnesemia is due to renal Mg2+ wasting. Patients show seizures and neurodevelopmental delay during childhood. Only two affected girls from a consanguineous family have been reported, and no other biochemical abnormalities were identified in these patients. A homozygous missense mutation in the EGF gene coding for pro-EGF was identified as the underlying genetic defect [34]. Pro-EGF is a type I membrane-bound precursor protein that is proteolytically cleaved to generate the soluble EGF peptide hormone. EGF binds with high affinity to the renal epidermal growth factor receptor (EGFR) at the basolateral membrane of the DCT. EGFR activation triggers a signaling cascade leading to an increase of TRPM6 channels on the apical membrane and increased Mg2+ reabsorption [14]. The disease-causing EGF mutation results in diminished sorting of pro-EGF, preventing adequate secretion of the EGF hormone [34]. This leads to inadequate stimulation of the EGFR, and therefore insufficient activation of the TRPM6 channel, which results in reduced reabsorption of Mg2+.
Neonatal inflammatory skin and bowel disease type 2
Using whole-exome sequencing, a rare homozygous missense mutation (p.Gly428Asp) in EGFR was identified in a child with an inflammatory syndrome affecting the skin, bowel, and lungs (OMIM #616069) [35]. The pregnancy was complicated by polyhydramnios and the child was born prematurely. Laboratory tests revealed low serum levels of magnesium. The child showed failure to thrive and died at 2.5 years of age from widespread cutaneous and pulmonary infections in addition to electrolyte imbalance. Results of a skin biopsy and immunofluorescence microscopy studies revealed that the mutation p.Gly428Asp reduces EGFR plasma membrane localization [35].
Hypomagnesemia, seizures, and mental retardation disorder type 1
Hypomagnesemia, seizures, and mental retardation disorder (HSMR) type 1 is a complex rare condition characterized by renal Mg2+ loss that results in hypomagnesemia, infantile or juvenile epileptic seizures, and intellectual disability [36,37]. Patients also show autistic features, aggressive behavior, variable degrees of delayed psychomotor development, speech limitations, impaired motor skills, and in some cases obesity [37,38]. HSMR type 1 is caused by loss-of-function mutations in the CNNM2 gene, which encodes the transmembrane protein CNNM2 [36–38]. Most HSMR type 1 patients carry heterozygous mutations that are generated de novo or inherited in an autosomal dominant pattern (OMIM #616418) [36–38]. However, a recessive mode of inheritance has been reported for several families [37,39]. Patients with recessive CNNM2 mutations show a severe phenotype, including brain malformations, refractory epilepsy, and acute intellectual disability (OMIM #613882).
CNNM2 is expressed in many organs and tissues including brain and kidney. In the kidney, CNNM2 is predominantly expressed at the basolateral membrane of the DCT cells, where it is associated with Mg2+ reabsorption [36,40]. Whether CNNM2 is itself an Mg2+ transporter or a regulator of Mg2+ transport is unclear [11]. However, CNNM2 pathogenic mutations reduce its expression in the plasma membrane, resulting in defective Mg2+ reabsorption and Mg2+ wasting [11,38]. Furthermore, recent studies revealed that Mg2+-ATP binding to the intracellular C-terminus of CNNM2 is required for protein dimerization and Mg2+ efflux [41]. Disease-causing mutations that are located in the Mg2+-ATP-binding site abolish ATP binding and Mg2+ efflux activity. The basis of the neurological defects remains unknown.
Group 2 hypomagnesemias
Bartter syndrome type 3
Bartter syndrome (BS) includes a group of several tubulopathies characterized by renal salt wasting, hypokalemia, hypochloremic metabolic alkalosis, hyperreninemia, hyperaldosteronism, and low to normal blood pressure [42,43]. Patients usually present during the first years of life with failure to thrive, polyuria, and polydipsia. The main pathogenic mechanism in these tubulopathies is defective salt reabsorption predominantly in the TAL. Five different types of BS have been identified based on the gene involved [43]. Patients with BS type 3 or classic BS (OMIM #607364) develop hypomagnesemia during childhood or later in life [44]. This disorder is characterized by a great clinical variability, and there is a correlation between the severity of mutations and younger age at diagnosis [44,45].
BS type 3 is caused by recessive loss-of-function mutations of the CLCNKB gene, which encodes the kidney-specific Cl– channel ClC-Kb that is involved in NaCl reabsorption in the renal tubule [6]. The ClC-Kb protein is expressed in the basolateral membrane of epithelial cells in the TAL and the DCT. In these tubular segments, Cl– exits the cell through ClC-Ka and ClC-Kb channels. Mutations in CLCNKB alter the intracellular Cl– regulation, which subsequently interferes with the generation of the lumen-positive potential and results in salt wasting and possibly hypomagnesemia.
Gitelman syndrome
Gitelman syndrome (GS, OMIM #263800) is an autosomal recessive salt-losing tubulopathy characterized by hypokalemic metabolic alkalosis, low or normal blood pressure, hypocalciuria, and hypomagnesemia with renal Mg2+ wasting [46]. GS is the most common cause of hereditary hypomagnesemia and is usually detected during adolescence or adulthood. GS may be asymptomatic or associated with mild symptoms including chronic fatigue, muscle weakness, thirst, salt craving, nocturia, and cramps, which can considerably reduce the quality of life [47]. Severe complications such as cardiac arrhythmias have been reported in some cases [46].
GS is caused by recessive inactivating mutations of the SLC12A3 gene encoding the thiazide-sensitive NCC, which is localized in the apical membrane of DCT cells, where it plays a fundamental role in the reabsorption of Na+ and Cl– [48,49]. NCC mutations lead to reduced reabsorption, which results in Na+ and Cl– wasting, hypovolemia, and subsequent hyperaldosteronism with metabolic alkalosis [50]. The pathogenesis of hypocalciuria and hypomagnesemia may be explained by the compensatory paracellular reabsorption of Na+ and Ca2+ in the PT due to volume reduction and by decreased apical expression of TRPM6 [51]. There is phenotypic variability in GS, including in patients with the same SLC12A3 mutation.
Autosomal dominant hypocalcemia with hypercalciuria
Autosomal dominant hypocalcemia with hypercalciuria (ADHH, OMIM #601198) is a rare disorder of calcium homeostasis characterized by variable levels of hypocalcemia and low or normal serum levels of PTH [52]. Patients also present with hypomagnesemia, hypermagnesuria, hyperphosphatemia, and hypercalciuria [52,53]. Hypocalcemia is a derived effect of hypomagnesemia as a result of parathyroid failure or PTH resistance [30]. ADHH patients may develop hypocalcemic symptoms (paresthesias, carpopedal spasm, and seizures), and some have renal and basal ganglia calcifications, but others are asymptomatic [52].
ADHH is caused by heterozygous gain-of-function mutations in the CASR gene [52,54]. This gene encodes the extracellular CaSR, a G protein-coupled receptor that is highly expressed in parathyroid glands and kidneys [53]. In the kidney, CaSR is highly expressed in the basolateral membrane of the TAL, where it regulates Ca2+ reabsorption independently of PTH [53,55]. A signaling pathway including two microRNAs and the tight junction proteins claudin-14 and claudin-16 mediates the effect of CaSR on renal Ca2+ and Mg2+ excretion [5]. Activating mutations of CaSR increase the expression of claudin-14, which binds to claudin-16 and blocks the cation permeability of the claudin-16/claudin-19 channel.
Episodic ataxia type 1
Loss-of-function mutations in the KCNA1 gene are typically associated with an autosomal dominant neurological disorder called episodic ataxia type 1 (EA1, OMIM #160120), which is characterized by recurring episodes of ataxia and myokymia from early childhood [13]. The clinical phenotype in EA1 patients can include seizures, epilepsy, and, in some cases, paroxysmal kinesigenic dyskinesia, cataplexy, myokymia, and hypomagnesemia [56]. These symptoms can appear alone or in combination with EA1. A genotype-phenotype correlation analysis in a large cohort of EA1 patients revealed high inter- and intrafamilial variability of symptoms, but the penetrance of hypomagnesemia has not been evaluated [57].
The KCNA1 gene encodes the α subunit of the voltage-gated potassium channel Kv1.1, which is abundantly expressed in specific neurons and plays an important role in regulating neuronal excitability in the central and peripheral nervous system [57]. In the kidney, KCNA1 is exclusively expressed at the apical membrane of the DCT cells alongside the Mg2+ transporter TRPM6 [58]. Previous studies suggested that potassium (K+) secretion via Kv1.1 provides the electrochemical gradient needed for Mg2+ reabsorption by the TRPM6 channel [58,59]. Interestingly, two specific KCNA1 heterozygous mutations, p.Asn255Asp and p.Leu328Val, have been associated with hypomagnesemia, leading to muscle cramps and tetanic episodes [58,59]. Electrophysiological analyses showed that both amino acid substitutions result in nonfunctional Kv1.1 channels [58–60]. The reduction in the K+ conductance depolarizes the apical membrane of DCT cells, reducing the electrical driving force and leading to renal Mg2+ loss. The frequency of hypomagnesemia in patients with KCNA1 mutations may be greater than reported, since serum Mg2+ levels have not been examined in all EA1 patients. Additional research is needed to understand the association of KCNA1 mutations with hypomagnesemia.
Isolated dominant hypomagnesemia
Isolated dominant hypomagnesemia is a rare autosomal dominant disorder characterized by hypomagnesemia, hypocalciuria, and occasionally chondrocalcinosis (OMIM #154020) [61]. Some patients suffer from muscle cramps, episodes of convulsions, or chondrocalcinosis [61,62]. This disease has been identified in only three families who carry the same missense mutation, p.Gly41Arg, in the FXYD2 gene and appear to be descendants of a common ancestor [62,63]. FXYD2 encodes the kidney-specific regulatory γ-subunit of basolateral Na+, K+-ATPase, which is composed of a catalytic α subunit and an auxiliary β subunit. The Na+, K+-ATPase complex maintains the electrochemical gradients of Na+ and K+ across the basolateral plasma membrane that provide the driving force for transepithelial Mg2+ transport particularly in the DCT [15]. There are two splice variants of the γ-subunit: FXYD2a, which is expressed mainly in the TAL and PT, and FXYD2b, which is expressed exclusively in the basolateral membrane of the DCT and collecting duct [64]. Expression studies showed that the p.Gly41Arg mutation causes incorrect trafficking of the mutant γ FXYD2b subunit, preventing its interaction with the α and β Na+, K+-ATPase subunits [62]. This leads to destabilization and reduction of Na+, K+-ATPase activity in the DCT, which eventually results in decreased Mg2+ uptake and renal Mg2+ loss.
Hypomagnesemia, seizures, and mental retardation disorder type 2
Recently, three children from non-consanguineous families were reported who presented with generalized seizures in infancy associated with severe intellectual disability, massive renal Mg2+ wasting, and hypomagnesemia (HSMR type 2; OMIM #618314) [65]. Other findings included significant developmental delay and limited motor skills. Mutational analysis identified heterozygous de novo mutations in the ATP1A1 gene encoding the catalytic α1 subunit of the Na+, K+-ATPase [65]. This ubiquitously expressed subunit is the major α isoform in the kidney and is present in practically all cell types of the central nervous system [15]. Dominant and de novo mutations in ATP1A1 are also the cause of autosomal dominant Charcot-Marie-Tooth disease type 2 (CMT2) [66]. To the best of our knowledge, serum Mg2+ levels have not been studied in CMT2 patients. Functional studies of the mutant Na+, K+-ATPase α1 subunits revealed loss of pump activity and anomalous cation permeability, leading to membrane depolarization [65]. These discoveries emphasize the essential task of the catalytic α1 subunit of the Na+, K+-ATPase in renal tubular Mg2+ transport and neuronal activity. Since ATP1A1 is expressed in the TAL and DCT, it could affect transport in both tubular segments.
Epilepsy, ataxia, sensorineural deafness, and tubulopathy/seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance syndrome
Epilepsy, ataxia, sensorineural deafness, and tubulopathy (EAST) syndrome or seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME) syndrome (OMIM #612780) is an autosomal recessive disease characterized by early-onset epilepsy, delayed psychomotor development, ataxia, sensorineural deafness, and a salt-wasting tubulopathy with or without mental retardation [67,68]. The renal phenotype develops during the course of the disease and comprises polyuria, hypokalemia, metabolic alkalosis, hypocalciuria, and hypomagnesemia [69,70]. Plasma renin and aldosterone levels are increased and blood pressure is at the low end of the normal range. Urinary findings include K+, Mg2+, and Na+ wasting.
This complex disorder is caused by loss-of-function mutations in the KCNJ10 gene encoding Kir4.1, one of the components of the inwardly rectifying K+ channel Kir4.1/Kir5.1 [16,67,68]. KCNJ10 is mainly expressed in glial cells of the brain, the stria vascularis of the inner ear, and the kidney [69]. The renal phenotype of patients with EAST/SeSAME syndrome closely resembles that of patients with GS, suggesting that KCNJ10 mutations mainly impair transport in the DCT. In the kidney, Kir4.1 is expressed in the basolateral membrane of the DCT and is involved in K+ recycling, which is needed for the activity of Na+, K+-ATPase, and generation of a negative transmembrane potential [16]. Mutations that inactivate Kir4.1 function lead to a depolarization of the basolateral membrane and to a reduction of the driving force for anion channels and Na+-coupled exchangers. This alteration in membrane voltage could also affect other transport processes, such as those for Cl– and Mg2+, which could explain the Mg2+ loss detected in patients with EAST/SeSAME syndrome.
Autosomal dominant tubulointerstitial kidney disease caused by HNF1B mutations
Autosomal dominant tubulointerstitial kidney disease (ADTKD) comprises a group of rare kidney disorders characterized by tubular damage and interstitial fibrosis without glomerular lesions [71]. Affected individuals usually develop CKD and end-stage renal disease in adulthood. Heterozygous mutations in several genes cause ADTKD, and this disease is subdivided into several subtypes based on the mutated gene [71].
ADTKD subtype HNF1B (ADTKD-HNF1B, OMIM #137920) is associated with hypomagnesemia [72,73]. The phenotypes detected in ADTKD-HNF1B patients are very heterogeneous and may appear during pregnancy, in childhood, or in adulthood [73]. Symptoms include renal cysts, kidney malformations, abnormalities of the genital tract and liver, and maturity-onset diabetes of the young (MODY) type 5 [72,74]. Hypomagnesemia and hypermagnesuria are observed in approximately 50% of patients [72,73].
ADTKD-HNF1B is caused by heterozygous mutations of the HNF1B gene, which encodes the developmentally regulated transcription factor hepatocyte nuclear factor-1β (HNF1β) [72,74,75]. These mutations are inherited in a dominant inheritance pattern or appear de novo. HNF1β regulates tissue-specific gene expression in epithelial cells of several organs, including the kidneys, pancreas, liver, and urogenital tract [76]. In the adult kidney, HNF1β is expressed in epithelial cells of all tubular segments. However, its role in renal Mg2+ reabsorption seems to take place in the DCT, since hypomagnesemia in ADTKD-HNF1B patients is frequently accompanied by hypocalciuria [72,73]. Several transcriptional targets of HNF1β have been identified, including the FXYD2 gene [72], which is involved in reabsorption of Mg2+ in the DCT, as described above. Therefore, inactivating mutations of HNF1β would lead to reduced expression of FXYD2 and, consequently, to renal Mg2+ wasting and hypomagnesemia [72]. HNF1β also regulates the transcription of KCNJ10, a gene that can affect Mg2+ transport in the DCT, as described above [77].
Transient neonatal hyperphenylalaninemia and primapterinuria
Transient neonatal hyperphenylalaninemia and primapterinuria (TNHP, OMIM #264070) is an autosomal recessive disorder characterized by mild transient hyperphenylalaninemia and elevated urinary levels of 7-biopterin [78]. Affected individuals are asymptomatic and exhibit normal psychomotor development. A follow-up study of TNHP patients showed that patients may also develop hypomagnesemia with renal Mg2+ wasting and MODY type diabetes [79]. TNHP is caused by mutations in the PCBD1 gene, which encodes the bifunctional protein pterin-4 α-carbinolamine dehydratase (PCBD1). PCBD1 functions as a cytosolic enzyme that is implicated in the regeneration of the essential cofactor tetrahydrobiopterin as well as a coactivator of HNF1β-mediated transcription within the nucleus [80]. Gene expression studies combined with immunohistochemical analysis showed that in the kidney, PCBD1 is expressed predominantly in the DCT [79]. PCBD1 interacts with the dimerization domain of HNF1β and regulates the formation of a transcriptionally active tetrameric complex [81]. Binding of PCBD1 to HNF1β stimulates the FXYD2 promoter in the DCT, and PCBD1 mutations identified in TNHP patients cause defective dimerization and degradation of the PCBD1 protein, leading to decreased FXYD2 promoter activity [79]. The reduced expression of FXYD2 would cause hypomagnesemia in these patients.
Kenny-Caffey syndrome type 2
Kenny-Caffey syndrome type 2 (KCS type 2, #OMIM 127000) is characterized by severe short stature, impaired skeletal development, eye abnormalities, hypomagnesemia, and hypoparathyroidism [82,83]. KCS type 2 patients may also have frequent episodes of low Ca2+ levels in serum triggered by hypoparathyroidism. This multisystem disease is caused by heterozygous missense mutations in the FAM111A gene, which encodes the nuclear trypsin-like serine protease FAM111A [82,83]. FAM111A is involved in the regulation of PTH production, calcium homeostasis, bone development and growth, but the specific mechanisms have not been determined [82,83]. FAM111A mutations identified in patients usually appear de novo, but some cases with autosomal dominant inheritance have been described [84]. These mutations affect the peptidase domain of FAM111A and may impair its catalytic activity [83,85].
FAM111A, first identified as an antiviral restriction factor, is ubiquitously expressed, and its nuclear localization suggests that it might be involved in transcriptional regulation [82,83,86]. FAM111A mutations result in hyperactivation of the FAM111A intrinsic protease activity, which could cause abnormal degradation of DNA-binding proteins or FAM111A depletion through hyper-autoproteolytic cleavage [87–89]. Hyperactive FAM111A is cytotoxic, disrupting nuclear structure and pore distribution in a protease-dependent manner [89]. Targets of FAM111A protease activity have been recently identified and include nucleoporins and the associated germinal center-associated nuclear protein transcription/replication factor [89]. Hypomagnesemia in KCS type 2 patients could be attributed to degradation of transcription factors involved in magnesium homeostasis. Tan et al. [90] suggested a potential role of FAM111A in regulation of CaSR, since KCS type 2 shares phenotypic characteristics with ADHH-like hypocalcemia and hypoparathyroidism.
Group 3 hypomagnesemias
Mitochondria are abundant in kidneys, as the kidney requires high amounts of energy to enable the reabsorption of ions in different tubular segments [91]. Mitochondrial dysfunction leads to reduced ATP synthesis and loss of renal function. Because the function and biogenesis of mitochondria are under the genetic control of both mitochondrial DNA (mtDNA) and nuclear DNA, mutations in either genome can be the cause of disease [92]. mtDNA mutations are usually inherited from the patient’s mother but they can also appear de novo. The clinical features of these patients can be very variable since they depend not only on the type of mutation but also on the number of mitochondria affected [92,93]. Mitochondrial diseases manifest in infancy and are multisystemic, and there are no effective treatment options for the patients. Some mitochondrial diseases are associated with hypomagnesemia but the mechanism underlying how they cause hypomagnesemia remains unsolved. These diseases may affect the TAL, the DCT, or both.
Hypertension, hypercholesterolemia, and hypomagnesemia
This disorder was identified in a large pedigree and includes hypertension, hypercholesterolemia, and hypomagnesemia (OMIM #500005) [94]. Affected individuals also have hypocalciuria and hypokalemia. Each of these traits was transmitted via the maternal lineage with a pattern indicating mitochondrial inheritance. Other clinical characteristics include migraine headache, sensorineural hearing loss, and hypertrophic cardiomyopathy, all usually associated with mitochondrial dysfunction. Sequence analysis of the mitochondrial genome in the maternal lineage of the pedigree identified a C to T transition in the MT-TI gene, which encodes isoleucine transfer RNA (tRNAIle) [94]. Further analysis of affected members revealed that all copies of the mtDNA contain this mutation. The affected C is located immediately 5’ to the tRNAIle anticodon, and biochemical studies showed that the substitution of T significantly weakens ribosome binding [95]. These studies suggest that this mutation results in the loss of mitochondrial function. Hypomagnesemia associated with hypocalciuria points to a primary defect in the DCT [96].
Kearns-Sayre syndrome
Kearns-Sayre syndrome (KSS, OMIM #530000) is a rare progressive multisystem disease characterized by ophthalmoparesis/ptosis and pigmentary retinopathy. The onset is typically before 20 years of age, and patients typically present one of the following: cerebellar ataxia, cardiac conduction defects, deafness, short stature, cognitive involvement, tremor, and cardiomyopathy [97]. Some cases develop hypoparathyroidism and renal tubular dysfunction resulting in severe hypomagnesemia, hypocalcemia with hypermagnesuria, and hypokalemia [98,99]. KSS is caused by large deletions in mtDNA, which can reduce considerably ATP production [93]. In most cases, the deletions arise de novo but some are transmitted through maternal inheritance. Patients’ cells usually contain a mixture of wild-type and mutant mtDNA molecules in variable quantities, which is critical in determining the level of cellular dysfunction [92]. A defect in Mg2+ reabsorption in the TAL or DCT, due to ATP depletion, could be the cause of hypomagnesemia in these patients.
Hyperuricemia, pulmonary hypertension, renal failure, and alkalosis syndrome
Hyperuricemia, pulmonary hypertension, renal failure and alkalosis (HUPRA) syndrome (OMIM #613845) is an autosomal recessive disease characterized by early-onset progressive renal failure, hyperuricemia, metabolic alkalosis, pulmonary hypertension, developmental delay, and, in some cases, hypomagnesemia [100]. This rare disorder has been diagnosed in only six children from three families and it is caused by homozygous missense mutation in SARS2 on chromosome 19, which encodes the mitochondrial seryl-tRNA synthetase (SARS2) [100,101]. SARS2 catalyzes the serine aminoacylation of two mitochondrial tRNAs. Only two mutations have been identified in patients with HUPRA syndrome, p.Asp390Gly and p.Arg402His. The p.Asp390Gly mutation significantly reduces the aminoacylation of one mitochondrial tRNA leading to its degradation, which would cause alterations in the synthesis of mitochondrial proteins and consequently in energy supply [100]. Decreased energy production may account for diminished Na+, K+-ATPase activity in the TAL and DCT, which could explain the salt wasting and hypomagnesemia observed in HUPRA patients.
Concluding remarks
Mg2+ is an essential ion that plays a key role in the regulation of many cellular functions. In the kidney, proteins expressed in the apical and basolateral membranes of the TAL and DCT maintain serum Mg2+ levels within a narrow physiological range. Mutations in the genes that encode these proteins cause different types of hypomagnesemia. Consequently, serum Mg2+ levels should be determined in patients presenting with seizures, muscle cramps, and arrhythmias.
In this review, we have described the current research findings on hereditary hypomagnesemias. Some of these diseases share common characteristics and thus precise diagnosis requires the identification of the causative mutation. Therefore, we suggest a diagnostic approach for clinicians based on previously reported data [2,102] (Fig. 4). Many questions are still unanswered. For instance, the underlying mechanisms for hypomagnesemia in most hereditary diseases are still unclear. In addition, the physiological functions of CNNM2 and SLC41A1 remain to be clarified. The identification of the protein(s) that facilitate the basolateral extrusion of Mg2+ will greatly improve our understanding of Mg2+ transport in the DCT. A better knowledge of the pathophysiology of these diseases is needed to develop specific therapeutic strategies. So far, the only therapeutic option to treat hypomagnesemia in patients with GS, BS type 3, EAST/SeSAME, HSH, or isolated dominant hypomagnesemia is essentially supportive and involves oral or intravenous magnesium supplementation. However, this approach has been found to be unsuccessful in patients with other hypomagnesemias like FHHNC, HSMR, and ADTKD-HNF1B.
Figure 4. Flowchart for the diagnosis of inherited hypomagnesemias.

Group 3 hypomagnesemias were not included because the clinical features of these patients are extremely variable.
ADHH, autosomal dominant hypocalcemia with hypercalciuria; ADTKD-HNF1B, autosomal dominant tubulointerstitial kidney disease subtype HNF1B; BS3, Bartter syndrome type 3; EA1, episodic ataxia type 1; EAST, epilepsy, ataxia, sensorineural deafness, and tubulopathy; FHHNC1, familial hypomagnesemia with hypercalciuria and nephrocalcinosis type 1; FHHNC2, familial hypomagnesemia with hypercalciuria and nephrocalcinosis type 2; GS, Gitelman syndrome; HNF1B, hepatocyte nuclear factor-1 beta; HSH, hypomagnesemia with secondary hypocalcemia; HSMR1, hypomagnesemia, seizure, and mental retardation disorder type 1; HSMR2, hypomagnesemia, seizures, and mental retardation disorder type 2; IDH, isolated dominant hypomagnesemia; IRH, isolated recessive hypomagnesemia; KCS2, Kenny-Caffey syndrome type 2; MODY, maturity-onset diabetes of the young; NISBD2, neonatal inflammatory skin and bowel disease type 2; SeSAME, seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance; TNHP, transient neonatal hyperphenylalaninemia and primapterinuria.
aThese clinical signs do not appear in all patients.
Footnotes
Conflict of interest
All authors have no conflicts of interest to declare.
Funding
This work was financially supported by the “Instituto de Salud Carlos III-Subdirección General de Evaluación y Fomento de la Investigación” and the European Regional Development Fund “Another way to build Europe” (grant number PI20/00652, Project RenalTube).
Disclosure
Figures were created using the web application BioRender.com.
Authors’ contributions
Conceptualization: All authors
Writing original draft: FCM, APR
Review and editing: All authors
All authors read and approved the final manuscript.
References
- 1.de Baaij JH, Hoenderop JG, Bindels RJ. Magnesium in man: implications for health and disease. Physiol Rev. 2015;95:1–46. doi: 10.1152/physrev.00012.2014. [DOI] [PubMed] [Google Scholar]
- 2.Viering DH, de Baaij JH, Walsh SB, Kleta R, Bockenhauer D. Genetic causes of hypomagnesemia, a clinical overview. Pediatr Nephrol. 2017;32:1123–1135. doi: 10.1007/s00467-016-3416-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Simon DB, Lu Y, Choate KA, et al. Paracellin-1, a renal tight junction protein required for paracellular Mg2+resorption. Science. 1999;285:103–106. doi: 10.1126/science.285.5424.103. [DOI] [PubMed] [Google Scholar]
- 4.Konrad M, Schaller A, Seelow D, et al. Mutations in the tight-junction gene claudin 19 (CLDN19) are associated with renal magnesium wasting, renal failure, and severe ocular involvement. Am J Hum Genet. 2006;79:949–957. doi: 10.1086/508617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gong Y, Renigunta V, Himmerkus N, et al. Claudin-14 regulates renal Ca⁺⁺ transport in response to CaSR signalling via a novel microRNA pathway. EMBO J. 2012;31:1999–2012. doi: 10.1038/emboj.2012.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Simon DB, Bindra RS, Mansfield TA, et al. Mutations in the chloride channel gene, CLCNKB, cause Bartter’s syndrome type III. Nat Genet. 1997;17:171–178. doi: 10.1038/ng1097-171. [DOI] [PubMed] [Google Scholar]
- 7.Voets T, Nilius B, Hoefs S, et al. TRPM6 forms the Mg2+influx channel involved in intestinal and renal Mg2+ absorption. J Biol Chem. 2004;279:19–25. doi: 10.1074/jbc.M311201200. [DOI] [PubMed] [Google Scholar]
- 8.Chubanov V, Waldegger S, Mederos y Schnitzler M, et al. Disruption of TRPM6/TRPM7 complex formation by a mutation in the TRPM6 gene causes hypomagnesemia with secondary hypocalcemia. Proc Natl Acad Sci U S A. 2004;101:2894–2899. doi: 10.1073/pnas.0305252101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Franken GA, Adella A, Bindels RJ, de Baaij JH. Mechanisms coupling sodium and magnesium reabsorption in the distal convoluted tubule of the kidney. Acta Physiol (Oxf) 2021;231:e13528. doi: 10.1111/apha.13528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kolisek M, Nestler A, Vormann J, Schweigel-Röntgen M. Human gene SLC41A1 encodes for the Na+/Mg2+ exchanger. Am J Physiol Cell Physiol. 2012;302:C318–C326. doi: 10.1152/ajpcell.00289.2011. [DOI] [PubMed] [Google Scholar]
- 11.Arjona FJ, de Baaij JH. CrossTalk opposing view: CNNM proteins are not Na+ /Mg2+ exchangers but Mg2+ transport regulators playing a central role in transepithelial Mg2+ (re)absorption. J Physiol. 2018;596:747–750. doi: 10.1113/JP275249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arjona FJ, Latta F, Mohammed SG, et al. SLC41A1 is essential for magnesium homeostasis in vivo. Pflugers Arch. 2019;471:845–860. doi: 10.1007/s00424-018-2234-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Browne DL, Gancher ST, Nutt JG, et al. Episodic ataxia/myokymia syndrome is associated with point mutations in the human potassium channel gene, KCNA1. Nat Genet. 1994;8:136–140. doi: 10.1038/ng1094-136. [DOI] [PubMed] [Google Scholar]
- 14.Thebault S, Alexander RT, Tiel Groenestege WM, Hoenderop JG, Bindels RJ. EGF increases TRPM6 activity and surface expression. J Am Soc Nephrol. 2009;20:78–85. doi: 10.1681/ASN.2008030327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mayan H, Farfel Z, Karlish SJ. Renal Mg handling, FXYD2 and the central role of the Na,K-ATPase. Physiol Rep. 2018;6:e13843. doi: 10.14814/phy2.13843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Su XT, Ellison DH, Wang WH. Kir4.1/Kir5.1 in the DCT plays a role in the regulation of renal K + excretion. Am J Physiol Renal Physiol. 2019;316:F582–F586. doi: 10.1152/ajprenal.00412.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Claverie-Martin F. Familial hypomagnesaemia with hypercalciuria and nephrocalcinosis: clinical and molecular characteristics. Clin Kidney J. 2015;8:656–664. doi: 10.1093/ckj/sfv081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Praga M, Vara J, González-Parra E, et al. Familial hypomagnesemia with hypercalciuria and nephrocalcinosis. Kidney Int. 1995;47:1419–1425. doi: 10.1038/ki.1995.199. [DOI] [PubMed] [Google Scholar]
- 19.Weber S, Schneider L, Peters M, et al. Novel paracellin-1 mutations in 25 families with familial hypomagnesemia with hypercalciuria and nephrocalcinosis. J Am Soc Nephrol. 2001;12:1872–1881. doi: 10.1681/ASN.V1291872. [DOI] [PubMed] [Google Scholar]
- 20.Blanchard A, Jeunemaitre X, Coudol P, et al. Paracellin-1 is critical for magnesium and calcium reabsorption in the human thick ascending limb of Henle. Kidney Int. 2001;59:2206–2215. doi: 10.1046/j.1523-1755.2001.00736.x. [DOI] [PubMed] [Google Scholar]
- 21.Claverie-Martín F, García-Nieto V, Loris C, et al. Claudin-19 mutations and clinical phenotype in Spanish patients with familial hypomagnesemia with hypercalciuria and nephrocalcinosis. PLoS One. 2013;8:e53151. doi: 10.1371/journal.pone.0053151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Konrad M, Hou J, Weber S, et al. CLDN16 genotype predicts renal decline in familial hypomagnesemia with hypercalciuria and nephrocalcinosis. J Am Soc Nephrol. 2008;19:171–181. doi: 10.1681/ASN.2007060709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vall-Palomar M, Burballa C, Claverie-Martín F, Meseguer A, Ariceta G. Heterogeneity is a common ground in familial hypomagnesemia with hypercalciuria and nephrocalcinosis caused by CLDN19 gene mutations. J Nephrol . 2021;34:2053–2062. doi: 10.1007/s40620-021-01054-6. [DOI] [PubMed] [Google Scholar]
- 24.Yamaguti PM, Neves FA, Hotton D, et al. Amelogenesis imperfecta in familial hypomagnesaemia and hypercalciuria with nephrocalcinosis caused by CLDN19 gene mutations. J Med Genet. 2017;54:26–37. doi: 10.1136/jmedgenet-2016-103956. [DOI] [PubMed] [Google Scholar]
- 25.Bardet C, Courson F, Wu Y, et al. Claudin-16 deficiency impairs tight junction function in ameloblasts, leading to abnormal enamel formation. J Bone Miner Res. 2016;31:498–513. doi: 10.1002/jbmr.2726. [DOI] [PubMed] [Google Scholar]
- 26.Godron A, Harambat J, Boccio V, et al. Familial hypomagnesemia with hypercalciuria and nephrocalcinosis: phenotype-genotype correlation and outcome in 32 patients with CLDN16 or CLDN19 mutations. Clin J Am Soc Nephrol. 2012;7:801–809. doi: 10.2215/CJN.12841211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miyamoto T, Morita K, Takemoto D, et al. Tight junctions in Schwann cells of peripheral myelinated axons: a lesson from claudin-19-deficient mice. J Cell Biol. 2005;169:527–538. doi: 10.1083/jcb.200501154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hou J, Renigunta A, Konrad M, et al. Claudin-16 and claudin-19 interact and form a cation-selective tight junction complex. J Clin Invest. 2008;118:619–628. doi: 10.1172/JCI33970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hou J, Renigunta A, Gomes AS, et al. Claudin-16 and claudin-19 interaction is required for their assembly into tight junctions and for renal reabsorption of magnesium. Proc Natl Acad Sci U S A. 2009;106:15350–15355. doi: 10.1073/pnas.0907724106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schlingmann KP, Sassen MC, Weber S, et al. Novel TRPM6 mutations in 21 families with primary hypomagnesemia and secondary hypocalcemia. J Am Soc Nephrol. 2005;16:3061–3069. doi: 10.1681/ASN.2004110989. [DOI] [PubMed] [Google Scholar]
- 31.Schlingmann KP, Weber S, Peters M, et al. Hypomagnesemia with secondary hypocalcemia is caused by mutations in TRPM6, a new member of the TRPM gene family. Nat Genet. 2002;31:166–170. doi: 10.1038/ng889. [DOI] [PubMed] [Google Scholar]
- 32.Walder RY, Landau D, Meyer P, et al. Mutation of TRPM6 causes familial hypomagnesemia with secondary hypocalcemia. Nat Genet. 2002;31:171–174. doi: 10.1038/ng901. [DOI] [PubMed] [Google Scholar]
- 33.Krapivinsky G, Krapivinsky L, Renthal NE, Santa-Cruz A, Manasian Y, Clapham DE. Histone phosphorylation by TRPM6’s cleaved kinase attenuates adjacent arginine methylation to regulate gene expression. Proc Natl Acad Sci U S A. 2017;114:E7092–E7100. doi: 10.1073/pnas.1708427114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Groenestege WM, Thébault S, van der Wijst J, et al. Impaired basolateral sorting of pro-EGF causes isolated recessive renal hypomagnesemia. J Clin Invest. 2007;117:2260–2267. doi: 10.1172/JCI31680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Campbell P, Morton PE, Takeichi T, et al. Epithelial inflammation resulting from an inherited loss-of-function mutation in EGFR. J Invest Dermatol. 2014;134:2570–2578. doi: 10.1038/jid.2014.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stuiver M, Lainez S, Will C, et al. CNNM2, encoding a basolateral protein required for renal Mg2+ handling, is mutated in dominant hypomagnesemia. Am J Hum Genet. 2011;88:333–343. doi: 10.1016/j.ajhg.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arjona FJ, de Baaij JH, Schlingmann KP, et al. CNNM2 mutations cause impaired brain development and seizures in patients with hypomagnesemia. PLoS Genet. 2014;10:e1004267. doi: 10.1371/journal.pgen.1004267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Franken GA, Müller D, Mignot C, et al. The phenotypic and genetic spectrum of patients with heterozygous mutations in cyclin M2 (CNNM2) Hum Mutat. 2021;42:473–486. doi: 10.1002/humu.24182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Accogli A, Scala M, Calcagno A, et al. CNNM2 homozygous mutations cause severe refractory hypomagnesemia, epileptic encephalopathy and brain malformations. Eur J Med Genet. 2019;62:198–203. doi: 10.1016/j.ejmg.2018.07.014. [DOI] [PubMed] [Google Scholar]
- 40.de Baaij JH, Stuiver M, Meij IC, et al. Membrane topology and intracellular processing of cyclin M2 (CNNM2) J Biol Chem. 2012;287:13644–13655. doi: 10.1074/jbc.M112.342204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen YS, Kozlov G, Fakih R, et al. Mg 2+-ATP sensing in CNNM, a putative magnesium transporter. Structure. 2020;28:324–335. doi: 10.1016/j.str.2019.11.016. [DOI] [PubMed] [Google Scholar]
- 42.Rodríguez-Soriano J. Bartter and related syndromes: the puzzle is almost solved. Pediatr Nephrol. 1998;12:315–327. doi: 10.1007/s004670050461. [DOI] [PubMed] [Google Scholar]
- 43.Konrad M, Nijenhuis T, Ariceta G, et al. Diagnosis and management of Bartter syndrome: executive summary of the consensus and recommendations from the European Rare Kidney Disease Reference Network Working Group for Tubular Disorders. Kidney Int. 2021;99:324–335. doi: 10.1016/j.kint.2020.10.035. [DOI] [PubMed] [Google Scholar]
- 44.Seys E, Andrini O, Keck M, et al. Clinical and genetic spectrum of Bartter syndrome type 3. J Am Soc Nephrol. 2017;28:2540–2552. doi: 10.1681/ASN.2016101057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Konrad M, Vollmer M, Lemmink HH, et al. Mutations in the chloride channel gene CLCNKB as a cause of classic Bartter syndrome. J Am Soc Nephrol. 2000;11:1449–1459. doi: 10.1681/ASN.V1181449. [DOI] [PubMed] [Google Scholar]
- 46.Blanchard A, Bockenhauer D, Bolignano D, et al. Gitelman syndrome: consensus and guidance from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2017;91:24–33. doi: 10.1016/j.kint.2016.09.046. [DOI] [PubMed] [Google Scholar]
- 47.Cruz DN, Shaer AJ, Bia MJ, Lifton RP, Simon DB; Yale Gitelman’s and Bartter’s Syndrome Collaborative Study Group. Gitelman’s syndrome revisited: an evaluation of symptoms and health-related quality of life. Kidney Int. 2001;59:710–717. doi: 10.1046/j.1523-1755.2001.059002710.x. [DOI] [PubMed] [Google Scholar]
- 48.Simon DB, Nelson-Williams C, Bia MJ, et al. Gitelman’s variant of Bartter’s syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat Genet. 1996;12:24–30. doi: 10.1038/ng0196-24. [DOI] [PubMed] [Google Scholar]
- 49.Vargas-Poussou R, Dahan K, Kahila D, et al. Spectrum of mutations in Gitelman syndrome. J Am Soc Nephrol. 2011;22:693–703. doi: 10.1681/ASN.2010090907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McCormick JA, Ellison DH. Distal convoluted tubule. Compr Physiol. 2015;5:45–98. doi: 10.1002/cphy.c140002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nijenhuis T, Vallon V, van der Kemp AW, Loffing J, Hoenderop JG, Bindels RJ. Enhanced passive Ca2+ reabsorption and reduced Mg2+ channel abundance explains thiazide-induced hypocalciuria and hypomagnesemia. J Clin Invest. 2005;115:1651–1658. doi: 10.1172/JCI24134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pearce SH, Williamson C, Kifor O, et al. A familial syndrome of hypocalcemia with hypercalciuria due to mutations in the calcium-sensing receptor. N Engl J Med. 1996;335:1115–1122. doi: 10.1056/NEJM199610103351505. [DOI] [PubMed] [Google Scholar]
- 53.Hannan FM, Kallay E, Chang W, Brandi ML, Thakker RV. The calcium-sensing receptor in physiology and in calcitropic and noncalcitropic diseases. Nat Rev Endocrinol. 2018;15:33–51. doi: 10.1038/s41574-018-0115-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pollak MR, Brown EM, Estep HL, et al. Autosomal dominant hypocalcaemia caused by a Ca(2+)-sensing receptor gene mutation. Nat Genet. 1994;8:303–307. doi: 10.1038/ng1194-303. [DOI] [PubMed] [Google Scholar]
- 55.Loupy A, Ramakrishnan SK, Wootla B, et al. PTH-independent regulation of blood calcium concentration by the calcium-sensing receptor. J Clin Invest. 2012;122:3355–3367. doi: 10.1172/JCI57407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Paulhus K, Ammerman L, Glasscock E. Clinical spectrum of KCNA1 mutations: new insights into episodic ataxia and epilepsy comorbidity. Int J Mol Sci. 2020;21:2802. doi: 10.3390/ijms21082802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Graves TD, Cha YH, Hahn AF, et al. Episodic ataxia type 1: clinical characterization, quality of life and genotype-phenotype correlation. Brain. 2014;137(Pt 4):1009–1018. doi: 10.1093/brain/awu012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Glaudemans B, van der Wijst J, Scola RH, et al. A missense mutation in the Kv1.1 voltage-gated potassium channel-encoding gene KCNA1 is linked to human autosomal dominant hypomagnesemia. J Clin Invest. 2009;119:936–942. doi: 10.1172/JCI36948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.van der Wijst J, Konrad M, Verkaart SA, et al. A de novo KCNA1 mutation in a patient with tetany and hypomagnesemia. Nephron. 2018;139:359–366. doi: 10.1159/000488954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.van der Wijst J, Glaudemans B, Venselaar H, et al. Functional analysis of the Kv1.1 N255D mutation associated with autosomal dominant hypomagnesemia. J Biol Chem. 2010;285:171–178. doi: 10.1074/jbc.M109.041517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Geven WB, Monnens LA, Willems JL, Buijs W, Hamel CJ. Isolated autosomal recessive renal magnesium loss in two sisters. Clin Genet. 1987;32:398–402. doi: 10.1111/j.1399-0004.1987.tb03157.x. [DOI] [PubMed] [Google Scholar]
- 62.Meij IC, Koenderink JB, van Bokhoven H, et al. Dominant isolated renal magnesium loss is caused by misrouting of the Na(+),K(+)-ATPase gamma-subunit. Nat Genet. 2000;26:265–266. doi: 10.1038/81543. [DOI] [PubMed] [Google Scholar]
- 63.de Baaij JH, Dorresteijn EM, Hennekam EA, et al. Recurrent FXYD2 p.Gly41Arg mutation in patients with isolated dominant hypomagnesaemia. Nephrol Dial Transplant. 2015;30:952–957. doi: 10.1093/ndt/gfv014. [DOI] [PubMed] [Google Scholar]
- 64.Arystarkhova E, Sweadner KJ. Splice variants of the gamma subunit (FXYD2) and their significance in regulation of the Na, K-ATPase in kidney. J Bioenerg Biomembr. 2005;37:381–386. doi: 10.1007/s10863-005-9475-y. [DOI] [PubMed] [Google Scholar]
- 65.Schlingmann KP, Bandulik S, Mammen C, et al. Germline de novo mutations in ATP1A1 cause renal hypomagnesemia, refractory seizures, and intellectual disability. Am J Hum Genet. 2018;103:808–816. doi: 10.1016/j.ajhg.2018.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lassuthova P, Rebelo AP, Ravenscroft G, et al. Mutations in ATP1A1 cause dominant charcot-marie-tooth type 2. Am J Hum Genet. 2018;102:505–514. doi: 10.1016/j.ajhg.2018.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bockenhauer D, Feather S, Stanescu HC, et al. Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N Engl J Med. 2009;360:1960–1970. doi: 10.1056/NEJMoa0810276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Scholl UI, Choi M, Liu T, et al. Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc Natl Acad Sci U S A. 2009;106:5842–5847. doi: 10.1073/pnas.0901749106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bandulik S, Schmidt K, Bockenhauer D, et al. The salt-wasting phenotype of EAST syndrome, a disease with multifaceted symptoms linked to the KCNJ10 K+ channel. Pflugers Arch. 2011;461:423–435. doi: 10.1007/s00424-010-0915-0. [DOI] [PubMed] [Google Scholar]
- 70.Scholl UI, Dave HB, Lu M, et al. SeSAME/EAST syndrome--phenotypic variability and delayed activity of the distal convoluted tubule. Pediatr Nephrol. 2012;27:2081–2090. doi: 10.1007/s00467-012-2219-4. [DOI] [PubMed] [Google Scholar]
- 71.Devuyst O, Olinger E, Weber S, et al. Autosomal dominant tubulointerstitial kidney disease. Nat Rev Dis Primers. 2019;5:60. doi: 10.1038/s41572-019-0109-9. [DOI] [PubMed] [Google Scholar]
- 72.Adalat S, Woolf AS, Johnstone KA, et al. HNF1B mutations associate with hypomagnesemia and renal magnesium wasting. J Am Soc Nephrol. 2009;20:1123–1131. doi: 10.1681/ASN.2008060633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Faguer S, Decramer S, Chassaing N, et al. Diagnosis, management, and prognosis of HNF1B nephropathy in adulthood. Kidney Int. 2011 Oct;80:768–776. doi: 10.1038/ki.2011.225. [DOI] [PubMed] [Google Scholar]
- 74.Horikawa Y, Iwasaki N, Hara M, et al. Mutation in hepatocyte nuclear factor-1 beta gene (TCF2) associated with MODY. Nat Genet. 1997;17:384–385. doi: 10.1038/ng1297-384. [DOI] [PubMed] [Google Scholar]
- 75.Bingham C, Bulman MP, Ellard S, et al. Mutations in the hepatocyte nuclear factor-1beta gene are associated with familial hypoplastic glomerulocystic kidney disease. Am J Hum Genet. 2001;68:219–224. doi: 10.1086/316945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ferrè S, Igarashi P. New insights into the role of HNF-1β in kidney (patho)physiology. Pediatr Nephrol. 2019;34:1325–1335. doi: 10.1007/s00467-018-3990-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kompatscher A, de Baaij JHF, Aboudehen K, et al. Loss of transcriptional activation of the potassium channel Kir5.1 by HNF1β drives autosomal dominant tubulointerstitial kidney disease. Kidney Int. 2017;92:1145–1156. doi: 10.1016/j.kint.2017.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Thöny B, Neuheiser F, Kierat L, et al. Hyperphenylalaninemia with high levels of 7-biopterin is associated with mutations in the PCBD gene encoding the bifunctional protein pterin-4a-carbinolamine dehydratase and transcriptional coactivator (DCoH) Am J Hum Genet. 1998;62:1302–1311. doi: 10.1086/301887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ferrè S, de Baaij JH, Ferreira P, et al. Mutations in PCBD1 cause hypomagnesemia and renal magnesium wasting. J Am Soc Nephrol. 2014;25:574–586. doi: 10.1681/ASN.2013040337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Johnen G, Kaufman S. Studies on the enzymatic and transcriptional activity of the dimerization cofactor for hepatocyte nuclear factor 1. Proc Natl Acad Sci U S A. 1997;94:13469–13474. doi: 10.1073/pnas.94.25.13469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mendel DB, Khavari PA, Conley PB, et al. Characterization of a cofactor that regulates dimerization of a mammalian homeodomain protein. Science. 1991;254:1762–1767. doi: 10.1126/science.1763325. [DOI] [PubMed] [Google Scholar]
- 82.Unger S, Górna MW, Le Béchec A, et al. FAM111A mutations result in hypoparathyroidism and impaired skeletal development. Am J Hum Genet. 2013;92:990–995. doi: 10.1016/j.ajhg.2013.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Isojima T, Doi K, Mitsui J, et al. A recurrent de novo FAM111A mutation causes Kenny-Caffey syndrome type 2. J Bone Miner Res. 2014;29:992–998. doi: 10.1002/jbmr.2091. [DOI] [PubMed] [Google Scholar]
- 84.Nikkel SM, Ahmed A, Smith A, Marcadier J, Bulman DE, Boycott KM. Mother-to-daughter transmission of Kenny-Caffey syndrome associated with the recurrent, dominant FAM111A mutation p.Arg569His. Clin Genet. 2014;86:394–395. doi: 10.1111/cge.12290. [DOI] [PubMed] [Google Scholar]
- 85.Cheng SS, Chan PK, Luk HM, Mok MT, Lo IF. Adult Chinese twins with Kenny-Caffey syndrome type 2: a potential age-dependent phenotype and review of literature. Am J Med Genet A. 2021;185:636–646. doi: 10.1002/ajmg.a.61991. [DOI] [PubMed] [Google Scholar]
- 86.Fine DA, Rozenblatt-Rosen O, Padi M, et al. Identification of FAM111A as an SV40 host range restriction and adenovirus helper factor. PLoS Pathog. 2012;8:e1002949. doi: 10.1371/journal.ppat.1002949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kojima Y, Machida Y, Palani S, et al. FAM111A protects replication forks from protein obstacles via its trypsin-like domain. Nat Commun. 2020;11:1318. doi: 10.1038/s41467-020-15170-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hoffmann S, Pentakota S, Mund A, et al. FAM111 protease activity undermines cellular fitness and is amplified by gain-of-function mutations in human disease. EMBO Rep. 2020;21:e50662. doi: 10.15252/embr.202050662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nie M, Oravcová M, Jami-Alahmadi Y, et al. FAM111A induces nuclear dysfunction in disease and viral restriction. EMBO Rep. 2021;22:e50803. doi: 10.15252/embr.202050803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tan RS, Lee CH, Dimke H, Todd Alexander R. The role of calcium-sensing receptor signaling in regulating transepithelial calcium transport. Exp Biol Med (Maywood) 2021;24:2407–2419. doi: 10.1177/15353702211010415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bhargava P, Schnellmann RG. Mitochondrial energetics in the kidney. Nat Rev Nephrol. 2017;13:629–646. doi: 10.1038/nrneph.2017.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gorman GS, Chinnery PF, DiMauro S, et al. Mitochondrial diseases. Nat Rev Dis Primers. 2016;2:16080. doi: 10.1038/nrdp.2016.80. [DOI] [PubMed] [Google Scholar]
- 93.Govers LP, Toka HR, Hariri A, Walsh SB, Bockenhauer D. Mitochondrial DNA mutations in renal disease: an overview. Pediatr Nephrol. 2021;36:9–17. doi: 10.1007/s00467-019-04404-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wilson FH, Hariri A, Farhi A, et al. A cluster of metabolic defects caused by mutation in a mitochondrial tRNA. Science. 2004;306:1190–1194. doi: 10.1126/science.1102521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ashraf SS, Ansari G, Guenther R, Sochacka E, Malkiewicz A, Agris PF. The uridine in “U-turn”: contributions to tRNA-ribosomal binding. RNA. 1999;5:503–511. doi: 10.1017/s1355838299981931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Konrad M, Schlingmann KP, Gudermann T. Insights into the molecular nature of magnesium homeostasis. Am J Physiol Renal Physiol. 2004;286:F599–F605. doi: 10.1152/ajprenal.00312.2003. [DOI] [PubMed] [Google Scholar]
- 97.Mancuso M, Orsucci D, Angelini C, et al. Redefining phenotypes associated with mitochondrial DNA single deletion. J Neurol. 2015;262:1301–1309. doi: 10.1007/s00415-015-7710-y. [DOI] [PubMed] [Google Scholar]
- 98.Katsanos KH, Elisaf M, Bairaktari E, Tsianos EV. Severe hypomagnesemia and hypoparathyroidism in Kearns-Sayre syndrome. Am J Nephrol. 2001;21:150–153. doi: 10.1159/000046239. [DOI] [PubMed] [Google Scholar]
- 99.Emma F, Pizzini C, Tessa A, et al. “Bartter-like” phenotype in Kearns-Sayre syndrome. Pediatr Nephrol. 2006;21:355–360. doi: 10.1007/s00467-005-2092-5. [DOI] [PubMed] [Google Scholar]
- 100.Belostotsky R, Ben-Shalom E, Rinat C, et al. Mutations in the mitochondrial seryl-tRNA synthetase cause hyperuricemia, pulmonary hypertension, renal failure in infancy and alkalosis, HUPRA syndrome. Am J Hum Genet. 2011;88:193–200. doi: 10.1016/j.ajhg.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rivera H, Martín-Hernández E, Delmiro A, et al. A new mutation in the gene encoding mitochondrial seryl-tRNA synthetase as a cause of HUPRA syndrome. BMC Nephrol. 2013;14:195. doi: 10.1186/1471-2369-14-195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Horinouchi T, Nozu K, Kamiyoshi N, et al. Diagnostic strategy for inherited hypomagnesemia. Clin Exp Nephrol. 2017;21:1003–1010. doi: 10.1007/s10157-017-1396-7. [DOI] [PubMed] [Google Scholar]