

Summary
Here, we present the miRMut protocol to annotate mutations found in miRNA genes based on whole-exome sequencing (WES) or whole-genome sequencing (WGS) results. The pipeline assigns mutation characteristics, including miRNA gene IDs (miRBase and MirGeneDB), mutation localization within the miRNA precursor structure, potential RNA-binding motif disruption, the ascription of mutation according to Human Genome Variation Society (HGVS) nomenclature, and miRNA gene characteristics, such as miRNA gene confidence and miRNA arm balance. The pipeline includes creating tabular and graphical summaries.
For complete details on the use and execution of this protocol, please refer to Urbanek-Trzeciak et al. (2020).
Subject areas: Bioinformatics, Sequence analysis, Cancer, Genetics, High Throughput Screening, Molecular Biology, Systems biology
Graphical abstract
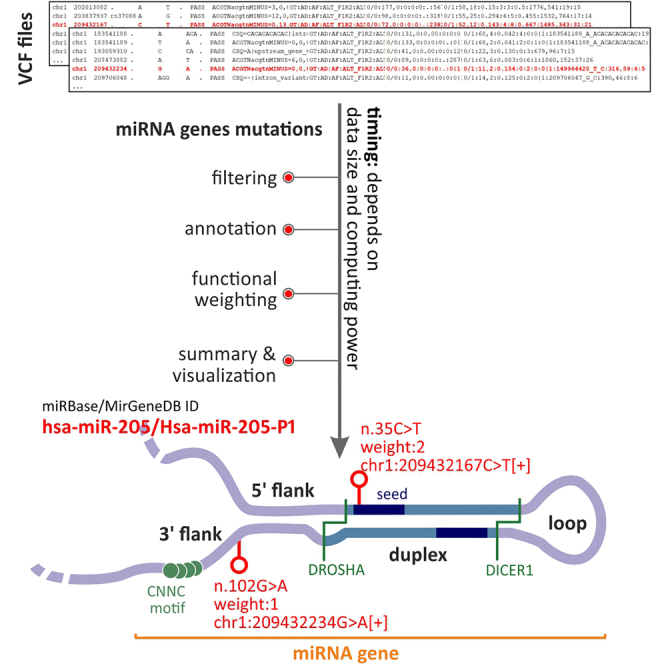
Highlights
-
•
miRMut is a software to annotate mutations in miRNA genes
-
•
miRMut utilizes VCF or CSV files generated based on WGS or WES results
-
•
miRMut assigns different mutation characteristics and potential functional impact
-
•
The output is presented in tabular and graphical summaries
Here, we present the miRMut protocol to annotate mutations found in miRNA genes based on whole-exome sequencing (WES) or whole-genome sequencing (WGS) results. The pipeline assigns mutation characteristics, including miRNA gene IDs (miRBase and MirGeneDB), mutation localization within the miRNA precursor structure, potential RNA-binding motif disruption, the ascription of mutation according to Human Genome Variation Society (HGVS) nomenclature, and miRNA gene characteristics, such as miRNA gene confidence and miRNA arm balance. The pipeline includes creating tabular and graphical summaries.
Before you begin
Most genetic studies and, therefore, developed software have focused on the protein-coding part of the genome. In recent years, noncoding variants found in promoters, untranslated regions (UTRs), introns, or microRNAs have also started to be recognized as potential disease drivers (Elliott and Larsson, 2021; Rheinbay et al., 2020; Tan, 2020). The importance of mutations found in the noncoding portion of the genome suggests that new tools for annotation need to be developed. miRNA gene mutation annotation may be used for (i) prioritization of variants detected in miRNA genes and (ii) analysis of functional variant enrichment, e.g., for identification of positive selection signals or identification of cancer-driving genes as it is commonly used in protein-coding sequences (based on the ratio of missense to synonymous mutations ascertained to be more functional and neutral, respectively). There are numerous tools for annotation of coding variants (e.g., ANNOVAR (Annotate Variation), VEP (Ensembl Variant Effect Predictor), SIFT (Sorting Intolerant From Tolerant), and PMUT (Predicting pathological MUtations)) (Ejigu and Jung, 2020; Pabinger et al., 2014; Shameer et al., 2016), but miRMut is, to our knowledge, the first annotation tool for mutations in miRNA genes.
Originally, the pipeline was used utilizing compressed variant call format (VCF.GZ) files from The Cancer Genome Atlas (TCGA) project containing somatic mutation data from >10,000 cancer samples from 33 cancer types (Urbanek-Trzeciak et al., 2020) (project ID: 16565; phs000178.v11.p8). As miRNA genes, we defined pre-miRNA-coding sequences extended upstream and downstream by 25 nucleotides. The pre-miRNA-coding sequences were reconstructed as described previously (Galka-Marciniak et al., 2019).
Note: The user should already have performed WES/WGS and created a variant call format (VCF) or VCF.GZ files with a list of the identified variants/mutations, which serve as the main input files of the protocol. The input files may also contain mutations outside of miRNA genes, which are filtered out in subsequent steps of the procedure. The user may also use files generated previously in any external project. The generation of VCF/VCF.GZ files based on unmapped sequence reads can be performed with the use of the genome analysis toolkit (GATK) developed by the Broad Institute (Cibulskis et al., 2013; Garcia et al., 2020; Van der Auwera, 2020). Alternatively, the user may use a comma-separated values (CSV) file containing mutations as described in more detail in the section Adding miRNA-specific information to specific mutations.
Note: The scripts assume that all needed quality filtering was done before, while creating VCF/VCF.GZ files. If multiple sequencings were performed or multiple files were generated for a single sample (as occurs in some TCGA somatic mutation files), they should be combined, or the issue should be resolved otherwise to avoid distortion. An example solution for TCGA file merging is described in the troubleshooting section under problem 1.
Reference files
Reference files for human miRNA genes (hg38 and hg19) are available at the GitHub repository and within supplementary files, and we recommend using these files. Reference files include information on miRNA gene coordinates (coordinates_{genome version}.bed), miRNA characteristics (arm balance and high confidence miRNAs), miRNA gene subregions [flanking sequence 5′ (flanking-5), duplex before seed (pre-seed), seed (seed), duplex after seed (post-seed), loop, and flanking sequence 3’ (flanking-3)], and gene orientation (localizations_{genome version}.csv) as well as miRBase-based precursor sequences (coordinates_with_seq_{genome version}.bed).
Coordinates_{genome version}.bed file includes coordinates of miRNA genes in the following structure:
| chr1 | 17343 | 17456 | hsa-mir-6859–1 |
| chr1 | 30384 | 30483 | hsa-mir-1302–2 |
| chr1 | 187865 | 187978 | hsa-mir-6859–2 |
Each coordinate is in a new line and has a tab separator between the following columns: chromosome, start, stop, and miRNA_gene_ID. No header should be present in the file. Start and stop coordinates are both inclusive.
Coordinates_with_seq_{genome version}.bed file has a structure as coordinates_{genome version}.bed file with an additional column (last) with the sequences of the genomic regions. The file is necessary for the prediction of disruptions of sequence motifs caused by the mutations.
Localizations_{genome version}.csv file is a CSV file with columns as follows (for a more precise column description, please see Tables 2 and S1): chrom (chromosome, e.g., chr1), name (name of miRNA region, e.g., hsa-mir-6859–1_flanking-3), ID (miRBase miRNA ID, e.g., MI0022705), start (first position (inclusive) of region, e.g., 17344), stop (last position (inclusive) of region, e.g., 17368), orientation (miRNA gene orientation, e.g., -), based_on_coordinates (information, if the region coordinates were calculated based on miRBase coordinates or only one mature miRNA was defined in miRBase and coordinates must have been predicted by the script, e.g., yes), arm (side of miRNA precursor (or loop) in which mutation will be located, i.e., 5p, 3p, or loop), type (region type, i.e., flanking sequence 5′ (flanking-5), duplex before seed (pre-seed), seed (seed), duplex after seed (post-seed), loop, and flanking sequence 3’ (flanking-3)), pre_name (miRBase pre-miRNA name, i.e., hsa-mir-6859-1), start_pre_build (miRBase start coordinate), stop_pre_build (miRBase stop coordinate), confidence (as high-confidence miRNA genes, we considered genes of miRNA precursors annotated as “high confidence” in miRBase and/or deposited in MirGeneDB v2.0), balance (according to the number of reads reported for the particular pre-miRNA arm (miRBase), the analyzed precursors were classified into one of 3 categories as follows: (i) generating mature miRNA predominantly from the 5p arm (≥90% of reads from the 5p arm) - 5p; (ii) generating mature miRNA predominantly from the 3p arm (≥90% of reads from the 3p arm) - 3p; and (iii) balanced (>10% of reads from each arm) - both; if no reads were available, the balance was assigned as unknown), mirgenedb_ID (ID from MirGeneDB if available, i.e., Hsa-Mir-8-P2a_pre)
Note: In the reference files, we excluded miRNAs (n = 9) that were problematic in the context of the second strand and, therefore, reliable structure prediction (hsa-mir-4489, hsa-mir-4539, hsa-mir-1469, hsa-mir-657, hsa-mir-4325, hsa-mir-548bb, hsa-mir-4285, hsa-mir-548o, and hsa-mir-601). All of the excluded miRNAs had low confidence according to miRBase and were not annotated in the MirGeneDB database.
Note: The reference files were primarily prepared based on miRBase v.22.1 (Kozomara and Griffiths-Jones, 2014; Kozomara et al., 2019) and MirGeneDB v2.0 (Fromm et al., 2020) for the human genome (hg38). Hg19 files (coordinates_hg19.bed, coordinates_with_seq_hg19.bed, and localizations_hg19.csv) were prepared as liftover from hg38 files, and some of the miRNAs (n=8) were removed during liftover (hsa-mir-6859–2, hsa-mir-10401 (3 genomic coordinates), hsa-mir-486–1, hsa-mir-486–2, hsa-mir-1234, hsa-mir-4477b, hsa-mir-4477a, and hsa-mir-532). For other releases and/or other organisms, please refer to the troubleshooting section under problem 2.
Table 2.
Mutation list column description in alphabetical order.
| Column name | Column description | Possible values |
|---|---|---|
| alg | algorithm used for detection of mutations | e.g.,: mutect2, somatic sniper |
| alt | alternative allele | e.g.,: G, A, GAA |
| arm | in which part of miRNA precursor mutation is located | 5p, loop, 3p |
| balance | arm from which the main miRNA is generated; defined based on deep sequencing reads (miRBase), [for details see Urbanek-Trzeciak et al. (2020)] | 5p, 3p, both, undefined |
| based_on_coordinates | indicates whether used coordinates were defined based on miRBase annotations or were predicted based on miRNA structure | yes, no |
| chrom | chromosome | e.g.,: chr1, chrX |
| confidence | as high-confidence miRNA genes, we considered genes coding for miRNA precursors annotated as “high confidence” in miRBase and/or deposited in MirGeneDB v2.0 | High, Low |
| cut_region | whether the mutation is within Drosha or Dicer cut region | 0 (no), 1 (yes) |
| duplex | whether the mutation is within duplex | 0 (no), 1 (yes) |
| from_start | nucleotide position from start of miRNA region in which mutation is located (e.g., within the seed, 5′ flanking sequence) | e.g.,: 1, 3 |
| from_end | nucleotide position from end of miRNA region in which mutation is located (e.g., within the seed, 5′ flanking sequence) | e.g.,: -1, -25 |
| hgvs | noncoding HGVS nomenclature of the mutation in the reference to the miRBase coordinates ∗Note that annotation of some indels may not match the latest HGVS recommendations; for multimutations (multiple changes in a single position in one sample) HGVS nomenclature is not available |
e.g.,: n.90G>A, n.67+21G>A, n.1–15delA |
| hgvs_g | genomic HGVS nomenclature of the mutation; for multimutations HGVS is not available | e.g.,: NC_000010.11:g.17845137C>A, NC_000001.11:g.1167872_1167873insT |
| ID | miRBase ID | e.g.,: MI0000737 |
| indiv_name | sample identifier | e.g.,: sample01 |
| mirgenedb_ID | mirGeneDB ID | e.g.,: Hsa-Mir-8-P1a_pre |
| motifs | whether any motif was disturbed as a result of the mutation | 0 (no), 1 (yes) |
| motifs_{} | information for each checked motif if there was the loss of a motif as a result of the mutation | -, loss |
| mutation_type | mutation type: substitution, insertion, deletion, insertion-deletion, multimutation | subst, ins, del, indel, multimutation |
| name | miRNA region name built by pre-miRNA name and precursor region connected by an underscore | e.g.,: hsa-mir-200a_flanking-5, hsa-mir-511_post-seed |
| no_of_loc | internal check column | 1 |
| orientation | miRNA gene orientation | +, - |
| pos | genomic position of the mutation | e.g.,: 1167859, 20633667 |
| pre_name | precursor name | e.g.,: hsa-mir-200a, hsa-mir-6084 |
| ref | reference allele | e.g.,: C, GCA |
| seed | whether mutation is located within seed | 0 (no), 1 (yes) |
| start | miRNA region start coordinate | e.g.,: 1167853, 20633667 |
| start_pre_build | miRBase start coordinate | e.g.,: 1167863, 20633679 |
| stop | miRNA region stop coordinate | e.g.,: 1167877, 20633691 |
| stop_pre_build | miRBase stop coordinate | e.g.,: 1167952, 20633788 |
| type | region of miRNA precursor | flanking-5, pre-seed, seed, post-seed, loop, flanking-3 |
| type_of_subst | substitution type, n.a. for non-substitution mutations | transition, transversion, n.a. |
| weight | weight score | 1, 1.5, 2 |
A description of all the columns present in transitional or summary files is included in Table S1.
Installing prerequisites
Please refer to the wiki pages of GitHub repository for more details on prerequisites and installation. In particular, additional options of setup using Docker (see also problem 3) or using conda environments (see also problem 5) are presented there.
-
1.Downloading or cloning GitHub repository
-
a.Clone GitHub: https://github.com/martynaut/mirnome-mutations repository, e.g., through ssh protocol using the command in a terminal:
-
a.
The repository can be also downloaded as a zip file and unpacked in a chosen directory.
A copy of the repository (release version v1.0.1, Zenodo: https://doi.org/10.5281/zenodo.5574501) is available in Data S1 (however, the users are strongly encouraged to use GitHub version of the software, where the newest version will be available).
-
2.Installing Python prerequisites
-
a.The user is encouraged to use a virtual environment for the project to avoid Python library conflicts among projects, but it is not a requirement. Example steps for conda virtual environment are shown in the troubleshooting section under problem 5.
-
b.Change working directory to the repository folder:>cd mirnome-mutations
-
c.Install Python prerequisites from requirements.txt file. The scripts were primarily tested and are recommended to run using Python 3.8. Run command:>pip install -r requirements.txtNote: The scripts were tested on Ubuntu (20.04.3 LTS) and MacOS (Big Sur) with both Python 3.7 and Python 3.8 clean environments; therefore, in the commands presented in the manuscript, we use Unix notation of paths. Due to the use of specific Python libraries (e.g., hgvs), it is not currently straightforward to use the miRMut pipeline on Windows.
-
a.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| TCGA data | Campbell et al. (2020) | https://gdc.cancer.gov/ |
| miRBase | Kozomara and Griffiths-Jones (2014) and Kozomara et al. (2019) | http://www.mirbase.org/index.shtml |
| MirGeneDB | Fromm et al. (2015, 2020) | https://mirgenedb.org/ |
| Software and algorithms | ||
| ViennaRNA | Lorenz et al. (2011) | https://www.tbi.univie.ac.at/RNA/#download |
| miRMut code | GitHub | GitHub: https://github.com/martynaut/mirnome-mutations |
| miRMut Docker image | Docker Hub | https://hub.docker.com/repository/docker/martynaut/mirmut |
Step-by-step method details
We prepared a GitHub repository containing all the necessary reference files, scripts, and example inputs and outputs for the method presented below. The repository wiki pages contain information on installation and running miRMut. The repository is found at
GitHub: https://github.com/martynaut/mirnome-mutations.
To follow the step-by-step instructions, users are encouraged to download the whole repository, including the example input files. For instructions on how to clone the repository and install the required Python libraries, please refer to the “installing prerequisites” section in “before you begin”.
It is expected that users will run the entire process all at once, but it is also possible to run the steps independently (1 - filtering miRNA gene mutations, 2 – processing mutation list, 3 - adding miRNA-specific information to specific mutations, 4 - calculating mutation weights, 5 - generating mutation CSV summaries, and 6 - generating mutation visualizations). Here, we describe how to run the script all at once, and later how to run each step separately. For default to run the whole procedure, the user should run the following command within the repository:
>python3 run_mirnaome_analysis.py \
/path/to/input/folder/with/vcfs \
/path/to/output/folder/ \
/path/to/reference/files/coordinates.bed \
/path/to/reference/files/localizations.csv \
/path/to/reference/files/coordinates_with_seq.bed
To run the script on example files provided in the repository, run the following command:
>python3 run_mirnaome_analysis.py \ input_files/input_vcf/example_vcf_set \
output_folder/user_example_output_from_vcf \
reference_files/coordinates_hg38.bed \
reference_files/localizations_hg38.csv \
reference_files/coordinates_with_seq_hg38.bed
The already calculated output for this command can be found within the repository in the output_folder/example_output_from_vcf folder.
Non-default values for all optional arguments used and described in each of the six steps can also be defined for a full run as follows (for parameters, please refer to Table 1):
>python3 run_mirnaome_analysis.py \
/path/to/input/folder/with/vcfs \
/path/to/output/folder/ \
/path/to/reference/files/coordinates.bed \
/path/to/reference/files/localizations.csv \
/path/to/reference/files/coordinates_with_seq.bed \
-m 1 -f 1
Table 1.
All command line parameters for the miRMut scripts
| Parameter | Function | Scripts that accepts the parameter |
|---|---|---|
| --from_step, -s | Accepts a number value from 1 to 6. Starts analysis from chosen step (inclusive), where: 1 – Filtering miRNA gene mutations 2 – Processing mutation list 3 – Adding miRNA-specific information to specific mutations 4 – Calculating mutation weights and HGVS nomenclature 5 – Generating summaries 6 – Generating visualization of mutations Note that previous steps need to be run before as each step depends on output files from earlier steps. Note that --from_step parameter is overwritten when --csv_file parameter is used if defined --from_step value is lower than 3. |
run_mirnaome_analysis.py |
| --end_step, -es | Accepts a number value from 1 to 6. Stops the analysis on the chosen step (inclusive). For numbers description see --from_step parameter. |
run_mirnaome_analysis.py |
| --include_merger, -m | Accepts values 0 (disabled) or 1 (enabled). It must be enabled if analysed VCF/VCF.GZ files were created with multiple algorithms (e.g., MuSE, MuTect2, etc.). If the merger of algorithms is enabled, mutations are grouped by position, individual ID of patient, and mutation type (reference and alternative allele), and if the same mutation is found in files generated by different algorithms, it is treated as one mutation. This transformation prevents counting the same mutation multiple times. If the merger of algorithms is enabled, the “indiv_name” has to be defined in the VCF/VCF.GZ files. |
run_mirnaome_analysis.py, merge_algorithms.py |
| --include_filtering, -f | Accepts values 0 (disabled) or 1 (enabled). If filtering is enabled, for each “results_{file type}.csv” file, “results_{file type}_eval.csv” is created with an additional column “eval”, which contains information if the mutation passes filtering as defined below. If filtering is enabled, the following criteria are taken into account (only if available in VCF/VCF.GZ files):
|
run_mirnaome_analysis.py, merge_algorithms.py |
| --csv_file, -c | Accepts path to a CSV file. The parameter is used when the miRMut is to be run on a CSV file instead of VCF/VCF.GZ files. The CSV file needs to be formatted as defined in adding miRNA-specific information to specific mutations section. When used with run_mirnaome_analysis.py script, the input folder argument will not be used (but needs to be defined), and the script will automatically start from step 3 (unless a later step is defined using --from_step parameter), skipping the first two steps. |
run_mirnaome_analysis.py, add_mirna_info.py |
| --pass_arg, -p | Accepts values 0 (disabled) or 1 (enabled). When PASS filtering is enabled using --pass_arg parameter only mutations with PASS value in the FILTER column from VCF/VCF.GZ files are included in the analysis. Although there is this option, it is recommended to do VCF/VCF.GZ filtering prior to the miRMut usage. |
run_mirnaome_analysis.py, extract_results_for_mirnaome.py |
| --weight_filter, -w | Accepts values 1, 1.5 and 2. The default value is set to 1. It filters out only mutations with selected weight or higher. It is done on the 4th step of the script and affects files with hgvs nomenclature, summaries and figures. |
run_mirnaome_analysis.py, add_weghts.py |
If the script is run but not completed, the user may define from which step analysis should start using the “from_step” (“-s”) optional argument, e.g.,
>python3 run_mirnaome_analysis.py \
/path/to/input/folder/with/vcfs \
/path/to/output/folder/ \
/path/to/reference/files/coordinates.bed \
/path/to/reference/files/localizations.csv \
/path/to/reference/files/coordinates_with_seq.bed \
-s 2
The command will start the analysis from step 2 of the procedure.
The end step of the analysis can also be defined (e.g., if the full analysis is not yet wanted) using the “end_step” (“-es”) optional argument as follows:
>python3 run_mirnaome_analysis.py \
/path/to/input/folder/with/vcfs \
/path/to/output/folder/ \
/path/to/reference/files/coordinates.bed \
/path/to/reference/files/localizations.csv \
/path/to/reference/files/coordinates_with_seq.bed \
-s 2 \
-es 2
To run analyses of the example files, the reusable bash file with Python commands (run_test.sh, available in the repository) may be used. It can be executed by running the following command in a terminal:
>./run_test.sh
Note: The TCGA analysis in Urbanek-Trzeciak et al. (2020) was run with -m, -f and -p flags. For details, please refer to the original manuscript. The descriptions of the parameters can be found in Table 1.
Note: For 1500 VCF.GZ files (1.61GB) on a PC with Ubuntu 20.04.3 LTS (8GB RAM; 3.20GHz CPU) it took about 2h40 to process complete miRMut analysis (step 1 took approx. 1h40, step 2 – 20s, step 3 – 5s, step 4 – 10min, step 5 – 1s, step 6 – 50min).
Filtering miRNA gene mutations
Timing: hours to days; factors that affect timing include the number and size of samples as well as available computing resources.
Within the first step from the provided VCF and/or compressed VCF.GZ files, mutations within miRNA gene coordinates are filtered and gathered in a CSV file. This is usually the longest step of the procedure. To run this step, the coordinates_{genome version}.bed reference file, which is described in the “before you begin” section, is required.
Additionally, the user needs to provide a path to the folder with VCF/VCF.GZ files (all files may be in a single folder but subfolders are allowed) and a path to the folder where output files should be saved (if the folder does not exist, it will be created; and if it exists, the current content of the folder will be removed).
-
1.
To run only the first step of the script, use one of the following commands:
>python3 run_mirnaome_analysis.py \
/path/to/input/folder/with/vcfs \
/path/to/output/folder/ \
/path/to/reference/files/coordinates.bed \
/path/to/reference/files/localizations.csv \
/path/to/reference/files/coordinates_with_seq.bed \
-s 1\
-es 1
or
>python3 extract_results_for_mirnaome.py \
/path/to/input/folder/with/vcfs \
/path/to/output/folder/ \
/path/to/reference/files/coordinates.bed
There are several output files saved in the defined output folder from this part of the protocol: files_summary.csv, which summarizes files included in the analysis, depending on information included in the VCF/VCF.GZ files, it includes individual/sample names and IDs, tumor/normal sample names and IDs, and types of algorithms used for mutation detection; results_{file type}.csv, file is created for each file type, where “file type” is an algorithm name used for VCF file creation (if the information is available in the VCF file), e.g., MuTect or MuSE.
Note: If all VCF files are created with a single algorithm, only one results file will be created. If multiple algorithms are used (e.g., 4 algorithms in TCGA data), multiple files will be created. In the second step of the procedure, the user may choose to merge mutations identified by different algorithms.
Note: In the pipeline, it is assumed that all needed filtering has been completed during VCF/VCF.GZ files preparation. However, during this step, it is possible to filter only mutations that passed all filters (has FILTER assigned to PASS value), which can be achieved by using the optional argument “-p” as follows:
>python3 extract_results_for_mirnaome.py \
/path/to/input/folder/with/vcfs \
/path/to/output/folder/ \
/path/to/calculated/input/files/coordinates.bed \
-p 1
As we encourage users to filter VCF files prior to use of miRMut software, by default, the filtering of only PASS variants is disabled.
Note: If no VCF/VCF.GZ files are found in the input directory, the pipeline will be terminated.
IMPORTANT: Please note that if the defined output folder does exist, the current content of the folder will be removed by the script.
Processing mutation list
The second step of the pipeline involves processing the mutation list, including the following two optional transformations: a merger of mutations identified by different algorithms (if more than one algorithm is used for mutation detection); and additional internal filtering of mutations as defined below. The optional transformations are disabled on default, but they may be enabled using two flags (“-m” and “-f”) as shown below. The flags might be used in a similar way when running the whole pipeline at once.
If the merger of algorithms is enabled, mutations are grouped by position, individual ID of patient, and mutation type (reference and alternative allele), and if the same mutation is found by different algorithms, it is treated as one mutation. This transformation prevents counting the same mutation multiple times.
If additional internal filtering is enabled, the following criteria are taken into account (only if available in VCF/VCF.GZ files): SSC (Somatic Score) > 30 - parameter determined by VarScan2 and SomaticSniper; BQ (Base Quality) of an alternative allele in tumor sample > 20 - parameter determined by MuSE; QSS (Sum of base quality scores for each allele) of an alternative allele in tumor samples divided by alternative allele count in tumor samples > 20 - parameter determined by MuTect2, at least two mutation-supporting reads in a tumor sample (if no mutation-supporting read was detected in the corresponding normal sample) OR at least 5 × higher frequency of mutation-supporting reads in the tumor sample than in the corresponding normal sample.
Please note that some of the filtering criteria are strictly algorithm-related and are not relevant in other projects.
-
2.To run this step of the script, use one of the following proposed commands enabling needed options:
-
a.Second step without merging and filtering>python3 run_mirnaome_analysis.py \/path/to/input/folder/with/vcfs \/path/to/output/folder/ \/path/to/reference/files/coordinates.bed\/path/to/reference/files/localizations.csv \/path/to/reference/files/coordinates_with_seq.bed \-s 2 \-es 2or>python3 merge_algorithms.py /path/to/output/folder/
-
b.Second step of the script enabling both merger of various algorithms and filtering of mutations>python3 run_mirnaome_analysis.py \/path/to/input/folder/with/vcfs \/path/to/output/folder/ \/path/to/reference/files/coordinates.bed \/path/to/reference/files/localizations.csv \/path/to/reference/files/coordinates_with_seq.bed \-s 2 \-es 2 \-m 1 \-f 1or>python3 merge_algorithms.py /path/to/output/folder/ \-m 1 \-f 1This step generates multiple files. If filtering is enabled, for each results_{file type}.csv file, results_{file type}_eval.csv is created with an additional column “eval”, which contains information if the mutation passes filtering as defined above. Additionally, all_mutations.csv file is created, including list of mutations described by chromosome (chrom), nucleotide position (pos), reference allele (ref), alternative allele (alt), and if available, individual name (indiv_name), reference allele count in normal sample (norm_ref_count), alternative allele count in normal sample (norm_alt_count), reference allele count in tumor sample (tumor_ref_count) alternative allele count in tumor sample (tumor_alt_count), and algorithm used (alg).Note: If VCF/VCF.GZ files were created with multiple algorithms but the merger of algorithms is not enabled, the analysis will be terminated on this step with a relevant message. Similarly, if no mutations in miRNA genes were found in defined input files, the script will be terminated.IMPORTANT: If the merger of algorithms is enabled, the “indiv_name” has to be defined in the VCF/VCF.GZ files.
-
a.
Adding miRNA-specific information to specific mutations
During this step, miRNA mutations listed in the “all_mutations.csv” file generated in the previous steps are annotated. The annotation is based mainly on the information gathered in the “localizations_{genome version}.csv'' files. The following pieces of information are annotated based on the mutation position: miRNA ID (from miRBase and MirGeneDB), miRNA name, miRNA region (flanking sequence, loop, and mature miRNA), miRNA confidence (information if miRNA found in miRBase is believed to be true miRNA), and balance (5′miRNA or 3′miRNA depending on the main miRNA generated from a particular miRNA gene based on miRBase data). Additionally, mutation type is defined (substitution/insertion/deletion) if relevant, and substitution type is defined if relevant.
Alternatively, the user may run this part of the script (and following steps) on an external CSV file. The CSV file needs to contain the following columns: chrom (with chromosome defined as, e.g., chr1), pos (with nucleotide position of a mutation in the chromosome), ref (reference allele), alt (alternative allele), and indiv_name (with sample ID; the script will accept empty values here but to enable all summaries sample ID is recommended). To execute the script on a CSV file, please see step 3 (last option). The example_csv.csv example CSV file is provided in the repository in the input_files/input_csv folder.
-
3.
To run this step of the script, use one of the following commands:
>python3 run_mirnaome_analysis.py \
/path/to/input/folder/with/vcfs \
/path/to/output/folder/ \
/path/to/reference/files/coordinates.bed \
/path/to/reference/files/localizations.csv \
/path/to/reference/files/coordinates_with_seq.bed \
-s 3 \
-es 3
or
>python3 add_mirna_info.py /path/to/output/folder/ \
/path/to/reference/files/localizations.csv
Alternatively, this step may be run on any CSV file fulfilling the requirements defined earlier, using option “-c”:
>python3 add_mirna_info.py /path/to/output/folder/ \
/path/to/reference/files/localizations.csv \
-c /path/to/csv/file/with/mutations.csv
This step generates the all_mutations_with_localization.csv file, which includes a list of mutations based on the all_mutations.csv file (or user-defined file with mutations) with annotated information.
Note: It is possible to run the whole script at once on a CSV file using the “-c” optional argument as follows:
>python3 run_mirnaome_analysis.py \
/path/to/input/folder \
/path/to/output/folder/ \
/path/to/reference/files/coordinates.bed \
/path/to/reference/files/localizations.csv \
/path/to/reference/files/coordinates_with_seq.bed \
-c /path/to/csv/file/with/mutations.csv
In this case, the input folder argument will not be used (but needs to be defined), and the script will automatically start from step 3, skipping the first two steps.
Calculating mutation weights and HGVS nomenclature
During the fourth step, the potential functional impact of each mutation is evaluated based on the mutation characteristics and location. A mutation receives a higher score (weight) if it is located within the DROSHA or DICER1 (key endonucleases involved in miRNA biogenesis) cleavage site, is located within a miRNA duplex, especially in the seed region, and is located in binding sites of proteins known to play role in miRNA biogenesis.
The scoring is as follows: seed region mutation - weight 2; cleavage sites, duplex (excluding seed region) or disturbance of protein binding motif - weight 1.5; all other mutations - weight 1.
Additionally, during this step, mutation designation according to the HGVS nomenclature is defined. Both genomic (using chromosomal position) and noncoding DNA coordinate (using miRBase pre-miRNA coordinate) designations are calculated.
-
4.
To run this step of the script, use one of the following commands:
>python3 run_mirnaome_analysis.py \ /path/to/input/folder/with/vcfs \
/path/to/output/folder/ \
/path/to/reference/files/coordinates.bed \
/path/to/reference/files/localizations.csv \
/path/to/reference/files/coordinates_with_seq.bed \
-s 4 \
-es 4
or
>python3 add_weights.py \
/path/to/output/folder/ \
/path/to/reference/files/coordinates_with_seq.bed
This step generates the three files: all_mutations_with_weights.csv, which includes a list of mutations with additional information on motifs and weights; all_mutations_with_hgvs.csv, which includes all information as the previous file and genomic HGVS designation; all_mutations_with_n_hgvs.csv, which includes all information as the previous file and miRBase HGVS designation.
Note: If a mutation is predicted to disturb more than one functional site, the highest weight is counted.
Note: Sequence motifs analyzed within the script can be found in the add_weights_helpers.py file in the repository. The sequence motif calculations were based on previously published scripts (Urbanek-Trzeciak et al., 2018).
Generating summaries
In this step, files summarizing mutations in miRNA genes are generated, including summaries from the miRNA gene perspective, specific mutation perspective, and complex mutation (explained below) perspective.
-
5.
To run this step of the script, use one of the following commands:
>python3 run_mirnaome_analysis.py \
/path/to/input/folder/with/vcfs \
/path/to/output/folder/ \
/path/to/reference/files/coordinates.bed \
/path/to/reference/files/localizations.csv \
/path/to/reference/files/coordinates_with_seq.bed \
-s 5 \
-es 5
or
>python3 distinct_occur.py \
/path/to/output/folder/
This step generates the three files: occur.csv file, which reports the number of mutations found in each miRNA gene in the analyzed cohort, for each miRNA gene (characterized by chromosome, pre-miRNA name, and miRNA miRBase ID), the count of unique samples with at least one mutation (indiv_name_nunique), count of all mutations (indiv_name_count), and count of unique mutation positions (pos_nunique) are provided, additionally, complex mutations, if identified in a particular miRNA, are reported; distinct_mutations.csv file, which summarizes distinct mutations (genome positions with specific alteration), including the number of patients/samples with the mutation, if available, the sum of reads supporting reference and alternative alleles are also provided; complex_mutations.csv file, which includes information on complex mutations defined as multiple nucleotide alterations found in a single miRNA gene in a single sample/patient (please note: if the “indiv_name” (sample identifier) is not defined, this file will be inutile as all mutations will be treated as found within a single sample). The file shows a number of mutations (pos_count) and unique mutation positions (pos_nunique) found in each miRNA gene (characterized by chromosome, pre-miRNA name, and miRNA miRBase ID) in each sample (indiv_name) and if any of the mutations are complex.
IMPORTANT: Do not use complex_mutations.csv if the individual name (indiv_name) is not defined (empty or nonunique) in the VCF/VCF.GZ or CSV files. In such a case, the “if_complex” column in the occur.csv file is also inutile.
Generating visualization of mutations
During this step, figures graphically presenting mutations on a schematic structure of miRNA genes are generated. For visualization of mutations and to enable examination of sequence variants in the context of miRNA precursor structures, we superimposed the identified variants on the consensus miRNA precursor structure and categorized them according to localization in the miRNA gene subregions. Generated figures depict (i) all found variants (Figure 1A), (ii) variants found in miRNA genes with the dominant 3′ miRNA, (iii) variants found in miRNA genes with the dominant 5′ miRNA, and (iv) balanced miRNAs. Similar figures are generated separately for each miRNA gene with at least one mutation in the provided dataset (example shown in Figure 1B). All figures are generated as Scalable Vector Graphics (SVG) files to enable further editing in graphic software.
-
6.
To run the sixth step of the script, use one of the following commands:
>python3 run_mirnaome_analysis.py \
/path/to/input/folder/with/vcfs \
/path/to/output/folder/ \
/path/to/reference/files/coordinates.bed \
/path/to/reference/files/localizations.csv \
/path/to/reference/files/coordinates_with_seq.bed \
-s 6 \
-es 6
Figure 1.

Localization of mutations detected by the script
(A) All mutations found in the pan-cancer cohort in TCGA (Urbanek-Trzeciak et al., 2020).
(B) Mutations found within miR-205 in the pan-cancer cohort in TCGA (Urbanek-Trzeciak et al., 2020). miRNA duplex positions are indicated in blue, and seed regions are indicated in dark blue. The flanking regions and terminal positions of the apical loop are indicated in gray. The numbers in the lower right corner represent the number of plotted mutations (upper) and the number of mutated miRNA genes (lower; not provided for mutations shown within a single miRNA gene). If present, sequence variants localized beyond position 22 in longer mature miRNAs are cumulatively shown at position 22. The plot shows mutations within six positions of the loop (first 3 and last 3 nucleotides). The number of the remaining loop mutations is indicated within the loop.
or
>python3 mutation_loc_figures.py \
/path/to/output/folder/
Expected outcomes
Steps 1–5 from the step-by-step method generate CSV files. The output files are briefly described in each step, and example outputs are provided in the GitHub repository in the “output_folder” folder. A description of each column found in all CSV files is included in Tables 2 and S1.
Step 6 generates figures. Figure 1 shows example output figures, which partially resembles Figures 2 and S3 in our companion paper by Urbanek-Trzeciak et al. (2020). Example figures may also be found in the “output_folder” in the GitHub repository.
Limitations
The protocol was tested on several VCFs and VCF.GZ files generated with different pipelines; however, taking into account the diversity of this file type, some files may not be feasible. In such a case, the solution may be the conversion of the data to the CSV format (as described above in step 3 - adding miRNA-specific information to specific mutations section.
With increasing knowledge of miRNA biogenesis and function, including verified miRNA-target interactions, the assessment of mutation functional impact may be further developed.
miRMut was tested and is supported on Ubuntu (tested on Ubuntu 20.04.3 LTS) and MacOS (tested on Big Sur) systems.
Troubleshooting
Problem 1
In some projects, multiple sequencing is performed on a single sample, e.g., TCGA project (see before you begin). Treating such samples separately is not recommended as some mutations specific for the sample (patient or tumor) may be redundant and, in some cases, generate artifacts in downstream analyses. However, in unique situations, e.g., if sections of the same cancer sample are tested in the context of cancer heterogeneity, the particular sections may need to be considered separate samples.
Potential solution
Here, we provide a script that merges VCF.GZ files. The script was prepared to be compatible with VCF.GZ files provided by TCGA consortium and may not be suitable for other files.
The script checks all the VCF.GZ files gathered in the input directory, defines which samples have multiple files available, merges the files, saves the files in the output folder, and moves all unique VCF.GZ files to the output folder. Alternatively, the user may force the script to create a copy of the input files (instead of moving them), keeping the original files intact in the input directory.
This step assumes that the user cloned the repository of the project as described in the previous sections.
To run the script, use one of the following commands:
>python3 merge_vcf_files.py \
/path/to/input/folder/with/vcfs \
/path/to/new/output/folder/
If the user wants to copy unchanged files instead of moving them, the command is as follows with parameter -c defined:
>python3 merge_vcf_files.py \
/path/to/input/folder/with/vcfs \
/path/to/new/output/folder/ \
-c 1
There are several output files in addition to merged and copied/moved VCF.GZ files saved in the defined output folder: files_summary_before_merging.csv - summary of files in the defined input directory before merger, including the path to the files, sample identifier (indiv_name, indiv_ID, sample_ID_tumor_name, sample_ID_tumor_aliQ, sample_ID_normal_name, sample_ID_normal_aliQ), and algorithm used to create VCF.GZ file (type_of_file); files_summary_count_per_patient_before_merging.csv - count of files per sample using the same algorithm; not_unique_patients.csv – a fragment of the files_summary_count_per_patient_before_merging.csv file with nonunique samples; do_not_use.txt - list of paths to files of nonunique samples that are used to create merged files.
IMPORTANT: Please note that if the defined output folder does exist, the current content of the folder will be removed by the script.
Problem 2
The VCF/VCF.GZ files were prepared on a different genome version for the human genome or for another organism (see reference files section).
Potential solution
Reference files may be prepared based on the reference files available within the repository and described in the “reference files” section for any organism and genome version. Alternatively, we provide a script that creates reference files based on files from miRBase and MirGeneDB. The GitHub repository needs to be cloned as defined in the “installing prerequisites” section.
To install ViennaRNA, there are two options. Option 1 is to install from source code, in which case you should download ViennaRNA distribution from https://www.tbi.univie.ac.at/RNA/#download to a selected folder and install ViennaRNA using the following instructions:
>tar -zxvf ViennaRNA-2.4.18.tar.gz
>cd ViennaRNA-2.4.18
>./configure
>make
>sudo make install
Option 2 is to install by pip. Within your Python environment, run:
>pip install viennarna
After installing ViennaRNA, you need to prepare input files from miRBase and MirGeneDB. From miRBase, download the following files (from ftp site for chosen miRBase release https://www.mirbase.org/ftp.shtml): hairpin.fa, hsa.gff3.txt, confidence.txt, confidence_score.txt, aliases.txt, and mirna_chromosome_build.txt, From MirGeneDB, download the following file (https://mirgenedb.org/download): hsa.gff (Genomic coordinates gff file).
Run prepare_localization_file.py script in a similar way as shown below on the downloaded files:
>python3 prepare_localization_file.py \
/path/to/ViennaRNA-2.4.11 \
/path/to/hairpin.fa \
/path/to/hsa.gff3.txt \
/path/to/confidence.txt \
/path/to/confidence_score.txt \
/path/to/aliases.txt \
/path/to/mirna_chromosome_build.txt \
/path/to/hsa.gff \
/path/to/output/folder
Note: One may also use the Docker image where the Vienna package is already pre-installed (for details see problem 3).
Problem 3
Running into dependency error while installing Python packages from requirements.txt (see installing prerequisites section).
Potential solution
When using conda (Python environment management system) for virtual environment handling, the user may encounter dependency error if the base Python has libraries depending on CPython installed before. It is caused by how conda is handling the base space and the fact that the HGVS library depends on CPython. For more details please refer to the conda GitHub issues (GitHub: https://github.com/conda/conda/issues/7173). The possible solution would be to use virtualenv instead of conda for virtual environment handling as virtualenv does not have this issue. However, acknowledging that conda has several advantages over virtualenv it is also possible to use Python 3.7 instead of 3.8 (if Python 3.8 is the user’s base Python version) for miRMut.
Alternatively, the user can use the provided Docker image. The latest Docker image can be found in the Dockerhub: https://hub.docker.com/repository/docker/martynaut/mirmut. The Docker image contains Python 3.8 with all required Python libraries installed, including ViennaRNA. We recommend using docker-compose.yml file to run the Docker image to automatically copy the output files to the host location. Example docker-compose.yml file is available within the miRMut GitHub repository.
Using the docker-compose.yml file, the Docker image can be run using a command:
>docker-compose run app
Problem 4
Low quality mutation calls (see before you begin section).
Potential solution
Our protocol starts with the list of already called variants (VCF files) and it is assumed that all needed filtering has been completed during VCF/VCF.GZ files preparation. But for the reliability of the analysis, it is worth paying attention to the quality of the sequencing, and reliability of mutation calls affected among others by the depth of coverage. This should be noted that the coverage below 10× or 30× in the case of sequencing of normal and tumor samples, respectively, may not be sufficient and many variants may not be properly identified or missed (Xiao et al., 2021). To filter out mutations that are likely false positives users may use --pass_arg and --include_filtering parameters in the miRMut pipeline. When PASS filtering is enabled only mutations with PASS value in the FILTER column from VCF/VCF.GZ files are included in the analysis. Similarly, when the additional internal filtering is enabled (which includes the parameters described in step 2 “processing mutation list”), only mutations that fulfill the criteria are included in the analysis. For details please refer to the flag descriptions in Table 1.
Problem 5
The user has a different Python version installed than recommended in the manuscript (see installing prerequisites section).
Potential solution
The scripts were tested on Python 3.7 and 3.8, it is not recommended to use earlier versions and Python 3.9 as not all libraries may work in the environment. If the user is using a different version of Python by default, we recommend using conda for clean environments with specific Python versions.
To create a clean environment with required libraries follow the instructions:
1. Download and install conda (for details please see https://conda.io/projects/conda/en/latest/user-guide/install/index.html)
2. Create a new conda environment with Python 3.8:
>conda create --name mirmut_env python=3.8
3. Activate virtual environment
>conda activate mirmut_env
4. Install all needed libraries from requirements file (working directory needs to be the repository folder)
>pip install -r requirements.txt
5. Perform all needed analyses using miRMut
6. After working with miRMut deactivate virtual environment
>conda deactivate
Next time using miRMut, the user only needs to activate the environment (step 3), run analyses (step 5), and deactivate the environment (step 6).
For more detail on using conda virtual environments see: https://conda.io/projects/conda/en/latest/user-guide/getting-started.html#managing-environments.
Alternatively, use the prepared Docker image with miRMut software with all installed dependencies. For the details, please see the problem 3 section.
If the user encounters any problem not addressed in the troubleshooting section, please create an issue in the miRMut GitHub repository.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Paulina Galka-Marciniak (pgalka@ibch.poznan.pl).
Materials availability
This study did not generate new unique reagents.
Acknowledgments
The results published here are based upon data generated by TCGA project (project ID: 16565) at the following research network: https://www.cancer.gov/tcga. This work was supported by research grants from the Polish National Science Centre [2016/22/A/NZ2/00184 and 2020/39/B/NZ5/01970 (to P.K.); 2015/17/N/NZ3/03629 (to M.O.U-T.); and 2020/39/D/NZ2/03106 (to P.G-M.)]
Author contributions
Protocol Design and Conceptualization – P.K., P.G-M., and M.O.U-T.; Pipeline Execution and Documentation - M.O.U-T; Pipeline testing P.G-M. and M.O.U-T.; Manuscript Writing - Review and Editing - P.G-M., M.O.U-T., and P.K.; Funding Acquisition and Supervision - P.K., P.G-M., and M.O.U-T.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2021.101023.
Contributor Information
Martyna O. Urbanek-Trzeciak, Email: murbanek@ibch.poznan.pl.
Paulina Galka-Marciniak, Email: pgalka@ibch.poznan.pl.
Supplemental information
Data and code availability
The code is available at GitHub repository: GitHub: https://github.com/martynaut/mirnome-mutations. Example input and output files are provided. To rerun full TCGA-based pan-cancer analysis as shown in Urbanek-Trzeciak et al. (2020), a request to TCGA consortium needs to be made.
References
- Campbell P.J., Getz G., Korbel J.O., Stuart J.M., Jennings J.L., Stein L.D., Perry M.D., Nahal-Bose H.K., Ouellette B.F.F., Li C.H., et al. Pan-cancer analysis of whole genomes. Nature. 2020;578:82–93. [Google Scholar]
- Cibulskis K., Lawrence M.S., Carter S.L., Sivachenko A., Jaffe D., Sougnez C., Gabriel S., Meyerson M., Lander E.S., Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ejigu G.F., Jung J. Review on the computational genome annotation of sequences obtained by next-generation sequencing. Biology. 2020;9:295. doi: 10.3390/biology9090295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott K., Larsson E. Non-coding driver mutations in human cancer. Nat. Rev. Cancer. 2021;21:500–509. doi: 10.1038/s41568-021-00371-z. [DOI] [PubMed] [Google Scholar]
- Fromm B., Billipp T., Peck L.E., Johansen M., Tarver J.E., King B.L., Newcomb J.M., Sempere L.F., Flatmark K., Hovig E., et al. A uniform system for the annotation of vertebrate microrna genes and the evolution of the human microRNAome. Annu. Rev. Genet. 2015;49:213–242. doi: 10.1146/annurev-genet-120213-092023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromm B., Domanska D., Høye E., Ovchinnikov V., Kang W., Aparicio-Puerta E., Johansen M., Flatmark K., Mathelier A., Hovig E., et al. MirGeneDB 2.0: the metazoan microRNA complement. Nucleic Acids Res. 2020;48:D132–D141. doi: 10.1093/nar/gkz885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galka-Marciniak P., Urbanek-Trzeciak M.O., Nawrocka P.M., Dutkiewicz A., Giefing M., Lewandowska M.A., Kozlowski P. Somatic mutations in miRNA genes in lung cancer—potential functional consequences of non-coding sequence variants. Cancers. 2019;11:793. doi: 10.3390/cancers11060793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia M., Juhos S., Larsson M., Olason P.I., Martin M., Eisfeldt J., DiLorenzo S., Sandgren J., Ståhl T.D.D., Ewels P., et al. Sarek: a portable workflow for whole-genome sequencing analysis of germline and somatic variants. F1000Res. 2020;9:63. doi: 10.12688/f1000research.16665.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozomara A., Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014;42:D68–D73. doi: 10.1093/nar/gkt1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozomara A., Birgaoanu M., Griffiths-Jones S. miRBase: from microRNA sequences to function. Nucleic Acids Res. 2019;47:D155–D162. doi: 10.1093/nar/gky1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz R., Bernhart S.H., Höner zu Siederdissen C., Tafer H., Flamm C., Stadler P.F., Hofacker I.L. ViennaRNA package 2.0. Algorithms Mol. Biol. 2011;6:26. doi: 10.1186/1748-7188-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pabinger S., Dander A., Fischer M., Snajder R., Sperk M., Efremova M., Krabichler B., Speicher M.R., Zschocke J., Trajanoski Z. A survey of tools for variant analysis of next-generation genome sequencing data. Brief. Bioinform. 2014;15:256–278. doi: 10.1093/bib/bbs086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rheinbay E., Nielsen M.M., Abascal F., Wala J.A., Shapira O., Tiao G., Hornshøj H., Hess J.M., Juul R.I., Lin Z., et al. Analyses of non-coding somatic drivers in 2,658 cancer whole genomes. Nature. 2020;578:102–111. doi: 10.1038/s41586-020-1965-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shameer K., Tripathi L.P., Kalari K.R., Dudley J.T., Sowdhamini R. Interpreting functional effects of coding variants: challenges in proteome-scale prediction, annotation and assessment. Brief Bioinform. 2016;17:841–862. doi: 10.1093/bib/bbv084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan H. Somatic mutation in noncoding regions: the sound of silence. EBioMedicine. 2020;61:103084. doi: 10.1016/j.ebiom.2020.103084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbanek-Trzeciak M.O., Jaworska E., Krzyzosiak W.J. miRNAmotif-A tool for the prediction of pre-miRNA−Protein interactions. Int. J. Mol. Sci. 2018;19:4075. doi: 10.3390/ijms19124075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbanek-Trzeciak M.O., Galka-Marciniak P., Nawrocka P.M., Kowal E., Szwec S., Giefing M., Kozlowski P. Pan-cancer analysis of somatic mutations in miRNA genes. EBioMedicine. 2020;61:103051. doi: 10.1016/j.ebiom.2020.103051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Auwera G.A. O’Reilly Media; 2020. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra. [Google Scholar]
- Xiao W., Ren L., Chen Z., Fang L.T., Zhao Y., Lack J., Guan M., Zhu B., Jaeger E., Kerrigan L., et al. Toward best practice in cancer mutation detection with whole-genome and whole-exome sequencing. Nat. Biotechnol. 2021;39:1141–1150. doi: 10.1038/s41587-021-00994-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The code is available at GitHub repository: GitHub: https://github.com/martynaut/mirnome-mutations. Example input and output files are provided. To rerun full TCGA-based pan-cancer analysis as shown in Urbanek-Trzeciak et al. (2020), a request to TCGA consortium needs to be made.