

Summary
Robust protocols are required to investigate in vitro the molecular mechanisms that control astrocyte metabolism and pro-inflammatory activities. In the present protocol, we describe step by step the isolation and culture of primary murine astrocytes from neonatal brains, followed by their genetic manipulation with siRNA. We further describe cytokine activation of the cultured astrocytes for the analysis of their pro-inflammatory responses, and the oxygen consumption analysis to assess their metabolic function.
For complete details on the use and execution of this protocol, please refer to Chao et al. (2019), Clark et al. (2021), and Rothhammer et al. (2018).
Subject areas: Cell culture, Cell isolation, Immunology, Metabolism, Neuroscience
Graphical abstract
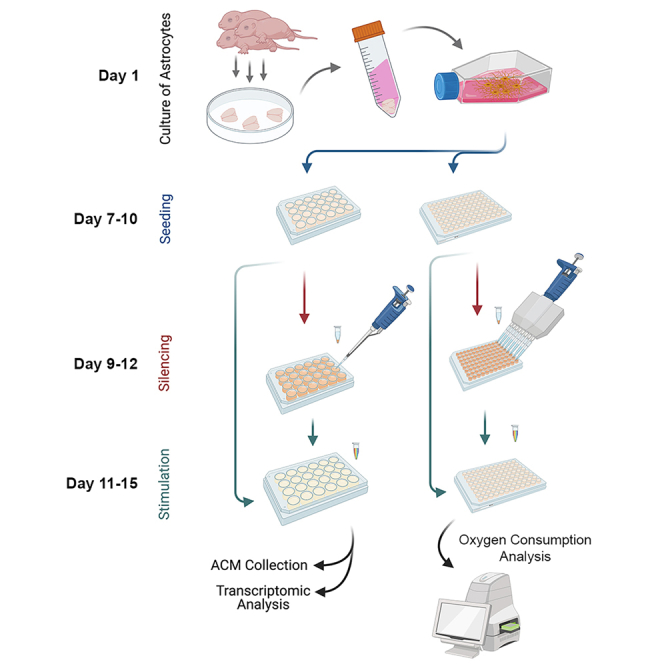
Highlights
-
•
Primary murine astrocyte culture establishment
-
•
Effective knockdown studies on primary astrocytes in vitro
-
•
Stimulation with cytokines for downstream assays
-
•
Oxygen consumption analysis to assess metabolic function
Robust protocols are required to investigate in vitro the molecular mechanisms that control astrocyte metabolism and pro-inflammatory activities. In the present protocol, we describe step by step the isolation and culture of primary murine astrocytes from neonatal brains, followed by their genetic manipulation with siRNA. We further describe cytokine activation of the cultured astrocytes for the analysis of their pro-inflammatory responses, and the oxygen consumption analysis to assess their metabolic function.
Before you begin
Institutional permission and must be granted in advance. Work with your institution’s Institutional Animal Care and Use Committee (IACUC) to get the necessary approvals and always follow their guidelines for pups euthanasia (see note 3).
Coating of flasks and plates with poly-L-lysin
Timing: 30 min
Note: Work under sterile conditions.
-
1.
Coat 75 cm2 flasks (T75) or plates with Poly-L-lysin. Add 5–8 mL of Poly-L lysine to each flask. For plates use the volumes stated in Table 1.
-
2.
Incubate for at least 10 min at Room Temperature (20°C–24°C).
Alternatives: incubate at 37°C to ensure maximal coating or 4°C overnight (ON, 16 h).
-
3.
Remove Poly-L-Lysine. Wash once with PBS or HBSS and let the flask dry while proceeding with next steps.
Table 1.
Volumes of Poly-L lysine used to coat different sizes of plates
| Culture plate | Volume per well of PLL for precoating |
|---|---|
| 12 well | 0.5 mL |
| 24 well | 0.25 mL |
| 48 well | 0.2 mL |
| 96 well Seahorse | 0.1–0.05 mL |
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| Poly-L-lysine | Sigma-Aldrich | # P4707 |
| DPBS no calcium no magnesium | Gibco | # 14190144 |
| HBSS no calcium no magnesium | Gibco | # 14170112 |
| 75 cm2/250mL Flasks | Falcon | # 353136 |
| 0.25% Trypsin-EDTA | Gibco | # 25200072 |
| 70 micron Nylon Cell Strainers | Fisherbrand | # 22-363-548 |
| DMEM F12 with Glutamax | Gibco | #10565042 |
| DMEM no glucose no glutamine | Gibco | #A1443001 |
| OptiMEM | Gibco | #31985070 |
| Fetal Bovine Serum - heat inactivated (FBS-HI) | Gibco | #10-438-026 |
| Penicillin-Streptomycin (10,000 U/mL) (P/S) | Gibco | #15140122 |
| HEPES 1M | Gibco | #15630080 |
| N1 Medium Supplement | Sigma-Aldrich | #N6530 |
| Glucose Solution 200 g/L | Gibco | #A2494001 |
| MEM Non-Essential Amino Acids Solution (NEAA) | Gibco | #11140050 |
| Bovine Albumin Fraction V (7.5% solution) | Gibco | #15260037 |
| INTERFERin | Polyplus Transfection | #409 |
| Buffer RLT | Qiagen | #79216 |
| recombinant mouse IFNg | R&D Systems | #485-MI-100 |
| recombinant mouse TNFa | R&D Systems | #410-MT-010 |
| recombinant mouse IL1b | R&D Systems | #401-ML-005 |
| recombinant mouse IFNb | R&D Systems | #8234-MB-010/CF |
| recombinant human TGFb | Miltenyi Biotec | #130-095-067 |
| recombinant human TGFa | R&D Systems | #239-A-100 |
| recombinant mouse VEGF-B | R&D Systems | #751-VE-025 |
| recombinant mouse IL-17 | R&D Systems | #421-ML-025/CF |
| Complement Component C1q Native Protein | MyBioSource | #MBS143105 |
| recombinant mouse IL-10 | R&D Systems | #417-ML-005 |
| recombinant GM-CSF | PeproTech | #315-03 |
| XF DMEM medium, pH 7.4 | Agilent Technologies | 103575-100 |
| XF 1.0 M Glucose solution | Agilent Technologies | 103577-100 |
| XF 100 mM Pyruvate solution | Agilent Technologies | 103578-100 |
| XF 200 mM Glutamine solution | Agilent Technologies | #103579-100 |
| Oligonucleotides | ||
| SMARTpool siRNAs | Dharmacon | N/A |
| ON-TARGETplus Non-targeting Pool | Dharmacon | D-001810-10 |
| Critical commercial assays | ||
| Seahorse XF Cell Mito Stress Test Kit | Agilent Technologies | #103015-100 |
| CyQUANT Cell Proliferation Assay Kit, for cells in culture | Thermo Fisher Scientific | #C7026 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J 1–3 days old mouse pups | The Jackson Laboratory | #000664 |
| Other | ||
| Conical 50 and 15 mL tubes | Falcon | #352196 #352070 |
| 70 μm cell strainers | Corning | #431751 |
| Serological pipets | Greiner Bio-One | #606180 #607180 |
| Tissue Culture Plates | Corning | #3596 #3526 |
| Centrifuge | N/A | N/A |
| Incubator | N/A | N/A |
| Shaker with controlled temperature | VWR | #76407-108 |
| XFe96 Seahorse analyzer | Agilent Technologies | N/A |
Materials and equipment
Alternatives: The list of reagents described in the key resources table are the ones tested and available in our laboratory. Equivalent chemicals, plasticware, media, from different vendors may be suitable alternatives and can be tested by interested users.
Note: Always prewarm the media to 37°C before adding it to the cell cultures. All media can be prepared and stored at 4°C for up to 6 months. Avoid long periods of incubation of media bottles at 37°C to prevent the loss of properties. It is recommended aliquoting the media to avoid repeated heating of large volumes.
Complete DMEM F12
| Reagent | Final concentration | Amount |
|---|---|---|
| DMEM F12 | 1× | 435 mL |
| FBS-H1 | 10% | 50 mL |
| P/S 10,000 U/mL | 100 U/mL | 5 mL |
| HEPES 1M | 100 mM | 5 mL |
| NEAA 100× | 1× | 5 mL |
| Total | n/a | 500 mL |
N1- DMEM F12
| Reagent | Final concentration | Amount |
|---|---|---|
| DMEM F12 | 1× | 475 mL |
| N1 Medium Supplement 100× | 1× | 5 mL |
| BSA Fraction V 7.5% | 0.075% | 5 mL |
| P/S 10,000 U/mL | 100 U/mL | 5 mL |
| HEPES 1M | 100 mM | 5 mL |
| NEAA 100× | 1× | 5 mL |
| Total | n/a | 500 mL |
Low Glucose- DMEM
| Reagent | Final concentration | Amount |
|---|---|---|
| DMEM no glucose no glutamine | 1× | 98 mL |
| Glucose 200 g/L | 1 mM | 0.09 mL |
| P/S 10,000 U/mL | 100 U/mL | 1 mL |
| HEPES 1M | 100 mM | 1 mL |
| Total | n/a | 100 mL |
Step-by-step method details
Culture of astrocytes
This protocol describes how to obtain a pure culture of neonatal astrocytes from brains from mouse pups 1–3 days old. Older mice could be use but the yield of the process is reduced significantly. We recommend using 6–8 brain cortices per T75 flask to be cultured. Numbers and surface area can be adjusted if the researcher choses to use whole brains or older mice.
Note: Culture of astrocytes from adult mouse brain has been reviewed elsewhere (Sun et al., 2017). Please refer to this same publication for micrographs of postnatal astrocyte cultures similar to the ones described here.
Alternatives: Whole brains instead of cortices could be used. However, Astrocytes from different brain regions are heterogeneous, which may lead to increased variability in the results across experiments.
Note: Always work under sterile conditions.
-
1.
Prepare sterile dissection tools and place them into 70% Ethanol.
-
2.
Prepare one 50 mL tube with 10–15 mL of 4°C HBSS per Flask to be cultured, place them on ice.
-
3.
Prepare 10 cm petri dishes with 5–8 mL of 4°C HBSS and place them on ice.
-
4.
Anesthetize pups.
-
5.
Euthanize each pup by decapitation. Gently extract the brain by performing two lateral incisions at the back of the head. Avoid touching the brains and gently drop them facing up into the petri dishes.
Note: Always follow your institution’s IACUC guidelines for anesthesia and euthanasia procedures
-
6.
Using magnification system if needed, gently separate the cortices from the rest of the brain. Add cortices to the 50 mL tubes with HBSS previously prepared
-
7.
Repeat steps 5 and 6 until done with all the pups
-
8.
Once Cortices have been pooled in the 50 mL tubes (5–8/tube) spin them down 400 or 300 × g (RCF) for 5 min at 4°C
-
9.
Remove the supernatant carefully to avoid moving the cortices.
-
10.
Add 5 mL of 0.25% Trypsin-EDTA, pipette up and down at least 6 times with a 5 or 10 mL pipet to gently disaggregate the tissue.
-
11.
Incubate for 15 min at 37°C in a cell culture incubator.
-
12.
Add 10 mL of Complete DMEM/F12 to stop the reaction. Invert the tube
-
13.
Place a 70 micron cell strainer on top of a new 50 mL tube and filter the suspension by gravity flow. Help the flow through with a syringe plunger if necessary.
-
14.
Centrifuge the flow-through at 400 × g for 5 min
-
15.
Remove supernatant
-
16.
Resuspend pellet in 12–15 mL of Complete DMEM/F12.
-
17.
Add to the precoated T75 flask
-
18.
Incubate at 37°C 5% CO2 and 85%–95% humidity.
-
19.
Change media the next day or the latest 48 h later
-
20.
Replace media every 2–3 days
-
21.
Astrocytes will be confluent and rid of debris by day 7–10.
Note: Media can be replaced every 2–3 days for around a month and the cells will still be fine. Old cultures will just have more microglia growing on them (see next steps).
Alternatives: Instead of using Complete DMEM, one can use N1-DMEM to avoid presence of FBS which could impact astrocyte biology. Astrocytes typically grow twice as slow in N1-DMEM and we recommend starting with more cell density (Like 10–12 cortices per T75 flask). Expect to have a confluent culture after about 2 or 3 weeks of culture. Change media as described above.
Seeding of astrocytes
Once cells are confluent and uniform, healthy and without major debris, they can be seeded in multi well plates for downstream analyses. Please refer to Table 2 for cell numbers to be used in each type of multi well plate.
-
22.
Shake T75 flask for 30 min 180–200 rpm at 37°C
Note: If the shaker doesn’t have CO2 influx, it is convenient to seal the lid of flasks so they can keep the CO2 levels inside.
-
23.
Remove media and add 10 mL of fresh medium
-
24.
Shake for 1.5–2 h more at 200 rpm at 37°C. Check under the microscope for the detachment of microglia.
-
25.
Remove media
-
26.
Rinse astrocytes once with HBSS
-
27.
Add 4 mL of 0.25% Trypsin-EDTA and shake them at 37°C checking them every 2 min until monolayer has detached.
-
28.
Add 10 mL of complete DMEM/F12 to stop the reaction
-
29.
Pipet thoughtfully and transfer to a 50 mL tube.
-
30.
Centrifuge at 400 × g for 5 min
-
31.
Resuspend the pellet in 10 mL of Complete DMEM/F12 and count the cells
-
32.
Seed the cells according to numbers of Table 2
-
33.
Incubate at 37°C, 5% CO2 and 85%–95% humidity for 24 or 48 h until the cells look flat again.
Note: For problems with cell attachment refer to troubleshooting 1.
Note: For problems with culture contamination refer to troubleshooting 2.
Note: Media from steps 23 and 25 will contain microglia; if you want to use them for downstream analyses, we recommend you purify them using positive selection for CD11b+ cells or any other marker of your choice. You could do that by FACS or magnetic beads-based separation.
Table 2.
Astrocyte optimal seeding densities
| Culture plate | Volume of media per well | Number of cells |
|---|---|---|
| 12 well | 1.5 mL | 4–5 × 105 |
| 24 well | 1 mL | 2–2.5 × 105 |
| 48 well | 0.5 mL | 105 |
| 96 well Seahorse | 0.1–0.2 mL | 30,000–40,000 |
Continue to step 34 for astrocyte silencing or directly to step 40 for astrocyte stimulation.
Silencing of astrocytes
In this section we describe how to silence primary murine astrocytes with siRNAs.
Note: Always use the microscope to confirm that cells are healthy and attached before starting the protocol. User should determine the best siRNA sequence for their target molecule of interest. As a general guidance, we used DHARMACON smart pools.
-
34.
Mix INTERFERin reagent with the desired siRNAs in Opti-MEM following Table 3.
-
35.
Incubate the mix 10 min at room temperature.
-
36.
Change the media of the astrocytes to fresh new media (Complete DMEM or N1-DMEM) following volumes on Table 3.
-
37.
Add the mix of INTERFERin, siRNAs and Opti-MEM dropwise to the astrocyte’s wells and incubate at 37°C.
Table 3.
Knockdown of astrocytes
| Culture plate | Opti-MEM volume | INTERFERin volume | Amount of siRNA | Volume of DMEM per well |
|---|---|---|---|---|
| 24 well | 100 μL | 2 μL | 0.6 pmoles | 500 μL |
| 48 well | 75 μL | 1 μL | 0.3 pmoles | 300 μL |
| 96 well Seahorse | 50 μL | 0.5 μL | 0.17 pmoles | 100 μL |
If viability of astrocytes is critically reduced, go to troubleshooting 3.
-
38.
After 48 h incubation, downstream analysis can be performed (see following sections)
-
39.
Knockdown efficiency can be confirmed by qPCR or western blot, it should be detected from 24 to 96 h after transfection.
Note: If target molecule expression is not reduced, go to troubleshooting 4.
Note: In our experience, astrocytes are quite hard to transfect or transduce. Researchers could explore the possibility of transducing with lentiviruses or electroporating or nucleofecting the astrocytes if the above transfection with siRNAs does not end in the expected results.
Proceed to the following section or oxygen consumption analysis section as desired.
Cytokine stimulation and Astrocyte Conditioned Media collection
In this section we describe how to stimulate primary murine astrocytes with cytokines in vitro and how to proceed with gene expression analysis and/or Astrocyte Conditioned Media (ACM) collection.
Researchers can choose to do the following steps after seeding the astrocytes (step 33) or after silencing (step 39).
-
40.
When cells are ready, change the media to fresh media (Complete DMEM or N1-DMEM) with the desired final concentration of the required cytokines. See Table 4 as a guidance for concentrations (Chao et al., 2019; Rothhammer et al., 2018; Sanmarco et al., 2021)
Note: This is general guidance, but researchers must set up the optimal condition for specific cytokines and combinations. Proceed to step 41 or 43
Note: On use of small molecules or chemical inhibitors: If the researcher needs to treat astrocytes with some sort of drug, we recommend doing that 30 min prior the cytokine stimulation (Chao et al., 2019; Clark et al., 2021) and keep the drug in the media after cytokine addition if the drug effects are reversible or if the incubation time is going to be long.
Table 4.
Cytokines for astrocytes stimulation
| Cytokine | Final concentration |
|---|---|
| mouse IFNγ | 100 ng/mL |
| mouse TNF | 5–50 ng/mL |
| mouse IFNβ | 500 IU/mL |
| mouse IL1β | 10–100 ng/mL |
| human TGFβ | 5 ng/mL |
| human TGFα | 0.1 ng/mL |
| mouse VEGF-B | 10 ng/mL |
| mouse IL-17 | 10 ng/mL |
| mouse C1q | 100 ng/mL |
| mouse IL-10 | 100 ng/mL |
-
41.
Incubate at 37°C for 6–48 h for gene expression analysis.
-
42.
Wash the cells gently once with HBSS, aspirate HBSS and lysate the cells with Buffer RLT (Qiagen), store at −20 or −80°C for later RNA extraction (Qiagen RNeasy kit) or proceed directly with RNA extraction and desired gene expression analysis (RT-qPCR…).
-
43.
For ACM collection, incubate the cells at 37°C with media with cytokines for 8 h to ON.
-
44.
Wash the cells gently at least twice with HBSS to get rid of any trace of cytokines.
-
45.
Replace with fresh media without cytokines and incubate at 37°C for 2 days.
-
46.
Collect the ACM to be used from downstream studies like Neurotoxicity assay or measurement of cytokine production by ELISA.
Note: ACM media can be aliquoted and frozen at −80°C, however for certain downstream procedures it is better to use it fresh.
Oxygen consumption analysis
In this part of the protocol, we describe oxygen consumption analysis performed on primary murine astrocytes with a XFe96 Seahorse analyzer (Agilent Technologies).
CRITICAL: Change astrocyte media to Low Glucose-DMEM
-
47.
Incubate at 37°C for 24 or 48 h (this a starvation step)
-
48.
Stimulate the astrocytes, if needed, following steps 40 and 41.
-
49.
The day before performing the assay make sure you hydrate the sensor cartridge in Seahorse XF Calibrant.
-
50.
Start Mito Stress assay with Seahorse XF Cell Mito Stress Test Kit (#103015-100, Agilent Technologies). Strictly follow the manufacturers’ manual (Mito_Stress_Test_Kit_Guide). With this system, oxygen consumption rate (OCR) is quantified real-time after sequential addition of 2 mM Oligomycin, 1 mM FCCP and 5 mM Rotenone/Antimycin A.
Note: Preparation and performance of Seahorse analysis is very well explained in the Seahorse XF Cell Mito Stress Test Kit User Guide. Please refer to it and strictly follow the protocol.
-
51.
Once Seahorse run is done carefully remove the plate from the machine
-
52.
Aspirate media, wash cells once with HBSS and make sure under the microscope that the cells are still attached to the wells and looking healthy.
Note: If cells are no longer attached or look severely distressed, refer to troubleshooting 5.
-
53.
Aspirate all the liquid and freeze the plate immediately at −80°C for at least over night before proceeding. Plates can be store for up to 4 weeks.
-
54.
Thaw the cell plates at room temperature.
-
55.
Quantify the cells using CyQUANT cell proliferation assay kit (#C7026, Invitrogen) following the protocol for Cell Number Determination (CyQUANT_manual). We recommend using a cell number standard curve with previously frozen aliquots of known astrocyte numbers. This is an easy and reliable method.
Alternatives: Likewise, we describe here for the XF Cell Mito Stress Test, other Agilent seahorse tests can be run following the same pre- and post-Seahorse analysis steps described here.
Alternatives: Normalization can be performed with other techniques like direct number of cells quantification by DAPI staining and imaging. However, normalization to protein amount (with BCA or Bradford assays for example) is extremely discouraged as the poly-L-lysine in the plate would interfere with measurements.
Expected outcomes
As mentioned above, we recommend plating 6–8 brain cortices per 75 cm2 flask. That will give rise to roughly 5 to 6 million mature astrocytes per flask.
When using this protocol for siRNA-based knockdown, in most cases we achieved > 50% gene silencing as determined by mRNA levels (Figure 1); these levels were sufficient to affect astrocyte responses (Chao et al., 2019). However, for some genes this protocol did not result in significant knockdown.
Figure 1.

siRNA silencing efficiency example
(A and B) Bsg gene expression (A) and protein levels (B) after siRNA knockdown in astrocytes in vitro (unpaired t test, error bars represent SEM).
Reprinted with permission from Chao et al. (2019).
We and others have shown that astrocytes can acquire a pro-inflammatory phenotype following activation with multiple cytokines including TNF, IFNγ, IL1β, C1q (Figure 2) (Chao et al., 2019; Clark et al., 2021; Liddelow et al., 2017; Rothhammer et al., 2016, 2018; Wheeler et al., 2019, 2020). Interestingly, some cytokines can also induce an anti-inflammatory astrocyte phenotype (Sanmarco et al., 2021). Thus, we encourage researchers to carefully choose cytokine of interest for their investigation.
Figure 2.

Astrocytes stimulation example
mRNA expression determined by qPCR of proinflammatory genes after different stimuli including TNF and IFNg cytokines. (unpaired t test, compared to corresponding condition between WT and MAVS−/− astrocytes, error bars represent SEM).
Reprinted with permission from Chao et al. (2019).
Limitations
Researchers should consider that this protocol may result in some small microglia contamination in astrocytes cultures. Hence, we recommend the addition of a flow cytometry-based cell sorting step before astrocyte seeding to completely remove microglia. The caveat of this stage is that sometimes astrocytes will take longer to look good after seeding.
The protocol described here only applies to neonatal mice astrocytes, which are easier to culture and more broadly used in the field. Adult astrocytes can be also cultured from adult brain after myelin removal, but cultures typically take around 3 weeks to be established.
Troubleshooting
Problem 1
Cells did not attach or only a few did, and they do not look flat (step 33).
Potential solution
Probably Poly-L lysine coating did not work or was insufficient. There is not a real solution at this step of the protocol and unfortunately your cells are lost at this point. Next time incubate the Poly-L lysine for longer period of time.
Another possibility is that the cells have died due to contaminations (see problem 2).
As a general practice, if you are planning to treat your cells with some sort of drug, wait till they are seeded and attached before initiating treatment.
Problem 2
Cells are contaminated right after the seeding or during some time of the culture (step 33).
Potential solution
Discard the culture immediately and decontaminate the incubator. We also recommend discarding the media used to culture those specific samples to avoid possible cross contaminations.
If cells were contaminated right after their isolation, consider next time improve your sterile manipulation immersing the dissection tools in 70% ethanol. The use of a tissue culture hood is recommended at every step.
Problem 3
Viability is drastically reduced after siRNA treatment (step 37).
Potential solution
Replace the media after 4 h of incubation with siRNAs/INTERFERin mix. This hopefully will maximize cell viability. We never experienced reduced viability, but knockdown of certain molecules can be harmful to astrocytes.
Problem 4
Gene or Protein of interest expression is not reduced after siRNA treatment (step 39).
Potential solution
Change to an alternative siRNA sequence for the same gene or try to adjust the silencing window.
Problem 5
Cells did suffer or did not survive after Seahorse assay (step 53).
Potential solution
Try to optimize cell seeding density as well as how long you wait to perform the assay after seeding the cells. Make sure next time you coat the seahorse plate with fresh Poly-L lysine. If possible, coat the plates ON at 4°C.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Francisco J. Quintana, fquintana@rics.bwh.harvard.edu.
Materials availability
This study did not generate new unique reagents.
Acknowledgments
We thank all members of the Quintana laboratory for helpful advice and discussions as well as Eduardo Balsa and Mari-Carmen Fernandez-Aguera for their advice on oxygen consumption assays. This work was supported by grants NS102807, ES02530, ES029136, AI126880 and AI149699 from the NIH; RG4111A1 from the National Multiple Sclerosis Society (to F.J.Q.), and PA-1604-08459 from the International Progressive MS Alliance (to F.J.Q.). C.G.-V. was supported by an Alfonso Martín Escudero Foundation postdoctoral fellowship and by a postdoctoral fellowship (ALTF 610-2017) from the European Molecular Biology Organization.
Author contributions
C.G.V. and F.J.Q. wrote the manuscript. F.J.Q. designed and supervised the study and edited the manuscript.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Cristina Gutiérrez-Vázquez, Email: cgutierrezvazquez@gmail.com.
Francisco J. Quintana, Email: fquintana@rics.bwh.harvard.edu.
Data and code availability
This study did not generate any unique datasets or code.
References
- Chao C.C., Gutierrez-Vazquez C., Rothhammer V., Mayo L., Wheeler M.A., Tjon E.C., Zandee S.E.J., Blain M., de Lima K.A., Takenaka M.C., et al. Metabolic control of astrocyte pathogenic activity via cPLA2-MAVS. Cell. 2019;179:1483–1498.e22. doi: 10.1016/j.cell.2019.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark I.C., Gutierrez-Vazquez C., Wheeler M.A., Li Z., Rothhammer V., Linnerbauer M., Sanmarco L.M., Guo L., Blain M., Zandee S.E.J., et al. Barcoded viral tracing of single-cell interactions in central nervous system inflammation. Science. 2021;372:eabf1230. doi: 10.1126/science.abf1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow S.A., Guttenplan K.A., Clarke L.E., Bennett F.C., Bohlen C.J., Schirmer L., Bennett M.L., Münch A.E., Chung W.S., Peterson T.C., et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541:481–487. doi: 10.1038/nature21029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothhammer V., Borucki D.M., Tjon E.C., Takenaka M.C., Chao C.-C., Ardura-Fabregat A., de Lima K.A., Gutiérrez-Vázquez C., Hewson P., Staszewski O., et al. Microglial control of astrocytes in response to microbial metabolites. Nature. 2018;557:724–728. doi: 10.1038/s41586-018-0119-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothhammer V., Mascanfroni I.D., Bunse L., Takenaka M.C., Kenison J.E., Mayo L., Chao C.C., Patel B., Yan R., Blain M., et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat. Med. 2016;22:586–597. doi: 10.1038/nm.4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanmarco L.M., Wheeler M.A., Gutierrez-Vazquez C., Polonio C.M., Linnerbauer M., Pinho-Ribeiro F.A., Li Z., Giovannoni F., Batterman K.V., Scalisi G., et al. Gut-licensed IFNgamma(+) NK cells drive LAMP1(+)TRAIL(+) anti-inflammatory astrocytes. Nature. 2021;590:473–479. doi: 10.1038/s41586-020-03116-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X., Hu X., Wang D., Yuan Y., Qin S., Tan Z., Gu Y., Huang X., He C., Su Z. Establishment and characterization of primary astrocyte culture from adult mouse brain. Brain Res. Bull. 2017;132:10–19. doi: 10.1016/j.brainresbull.2017.05.002. [DOI] [PubMed] [Google Scholar]
- Wheeler M.A., Clark I.C., Tjon E.C., Li Z., Zandee S.E.J., Couturier C.P., Watson B.R., Scalisi G., Alkwai S., Rothhammer V., et al. MAFG-driven astrocytes promote CNS inflammation. Nature. 2020;578:593–599. doi: 10.1038/s41586-020-1999-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler M.A., Jaronen M., Covacu R., Zandee S.E.J., Scalisi G., Rothhammer V., Tjon E.C., Chao C.C., Kenison J.E., Blain M., et al. Environmental control of astrocyte pathogenic activities in CNS inflammation. Cell. 2019;176:581–596.e18. doi: 10.1016/j.cell.2018.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate any unique datasets or code.