

Abstract
Fanconi anemia (FA) is a chromosome instability syndrome with congenital abnormalities, cancer predisposition and bone marrow failure (BMF). While hematopoietic stem and progenitor cell (HSPC) transplantation is the recommended therapy, new therapies are needed for FA patients without suitable donors. BMF in FA is caused, at least in part, by a hyperactive growth suppressive TGFβ pathway, regulated by the TGFβ1, TGFβ2, and TGFβ2, and TGFβ3 ligands. Accordingly, the TGFβ pathway is an attractive therapeutic target for FA. While inhibition of TGFβ1 and TGFβ3 promotes blood cell expansion, inhibition of TGFβ2 is known to suppress hematopoiesis. Here, we report the effects of AVID200, a potent TGFβ1 and TGFβ3 specific inhibitor, on FA hematopoiesis. AVID200 promoted the survival of murine FA HSPCs in vitro. AVID200 also promoted in vitro the survival of human HSPCs from patients with FA, with the strongest effect in patients progressing to severe aplastic anemia or myelodysplastic syndrome (MDS). Previous studies have indicated that the toxic upregulation of the Non-Homologous End Joining (NHEJ) pathway accounts, at least in part, for the poor growth of FA HSPCs. AVID200 downregulated the expression of NHEJ related genes and reduced DNA damage in primary FA HSPCs in vitro and in in vivo models. Collectively, AVID200 shows activity in FA mouse and human preclinical models. AVID200 may therefore provide a therapeutic approach to improving BMF in FA.
Keywords: TGFβ, Fanconi Anemia, Bone Marrow Failure
Graphical Abstract
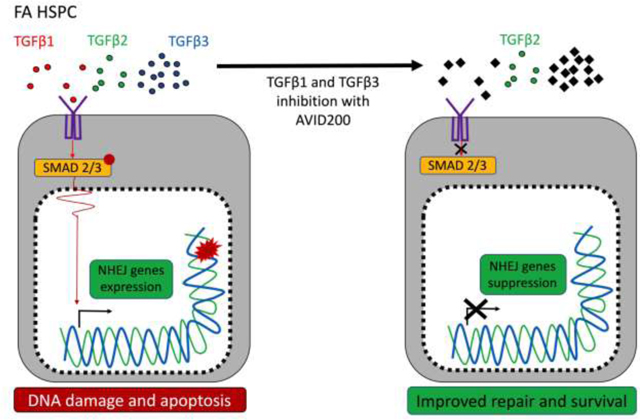
INTRODUCTION
Fanconi anemia (FA) is a chromosome instability syndrome characterized by congenital malformations, cancer predisposition, and childhood onset of bone marrow failure (BMF) [1–3]. FA is the most common inherited BMF syndrome, and BMF occurs due to a defect in the maintenance of the hematopoietic stem and progenitor cell (HSPC) pool [4, 5]. HSPC pool attrition in FA patients underlies the development of aplastic anemia (AA) as well as the strong predisposition to myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) [1, 6, 7].
FA is caused by inherited bi-allelic mutations in any of the twenty-three FA genes (FANCA to FANCW) [7–9] whose protein products coordinately repair DNA interstrand crosslinks (ICLs). ICLs interfere with the progression of replication forks during S phase of the cell cycle [2]. ICLs are detected by the FA core complex, and they are unhooked by endonucleases associated with the FANCD2/I complex. The resulting double strand break (DSB) is ultimately repaired by the downstream homologous recombination repair (HRR) machinery [10, 11]. The final step in DSB repair is coordinated by downstream FA gene products, including FANCS/BRCA1 and FANCD1/BRCA2 [12].
A defective FA pathway has been linked to the accumulation of DNA damage and to the hyperactivation of growth suppressive pathways in the HSPC pool. On the one hand, DNA damage activates the p53 pathway which hyperactivates p21 and arrests FA HSPCs in the G1 phase of the cell cycle [4]. On the other hand, a hyperactive TGFβ pathway contributes to the BMF in FA by promoting the toxic upregulation of non-homologous end-joining (NHEJ), leading to the accumulation of misrepaired DNA damage, unresolved chromosome radials, and HSPC growth suppression [13]. TGFβ pathway inhibitors downregulate the NHEJ pathway genes and upregulate the high-fidelity HRR genes, thus promoting the survival and proliferation of mouse and human FA HSPCs both in in vitro and in vivo models [13].
Three different TGFβ ligands - namely, TGFβ1, TGFβ2, and TGFβ3, regulate the TGFβ pathway [14]. The three TGFβ isoforms remain in the extracellular space as inactive TGFβ dimers until their cleavage and activation by the matrix metalloproteinases, MMP2 and MMP9, or by integrin-mediated mechanisms [15].
The TGFβ pathway is dysregulated in several human diseases, including idiopathic pulmonary fibrosis, [16, 17], myelofibrosis [18, 19], MDS [20], AML [21] and other types of cancers [22]. Drugs have been developed for targeting components of this pathway, including small-molecule inhibitors, monoclonal antibodies, antisense oligonucleotides, and vaccines, all of which have been analyzed in preclinical models. TGFβ pathway inhibitors are effective in eliminating cancer stem cells in mouse models of glioblastoma [23] and chronic myeloid leukemia [24] and can block metastasis in mouse breast cancer models [25].
TGFβ inhibitors have moved into clinical trials, particularly for the treatment of fibrosis and cancer. These agents include the monoclonal antibody Fresolimumab, the antisense oligonucleotide Trabedersen [26], the small molecule inhibitor Galunisertib developed by Eli Lilly against TGFβRI [27, 28], TGFβ ligand trap [29] and Losartan, a pre-existing antihypertensive drug that inhibits the angiotensin type II receptor but also blocks TGFβ signaling [30] More recently, TGFβ pathway inhibitors have been shown to boost immune-response in immunotherapy trials [31, 32]. Such is the case for M7824, a bifunctional fusion protein composed of a monoclonal antibody against programmed death ligand 1 (PD-L1) fused to a TGF-β trap [33].
Although inhibitors targeting the three types of TGFβ ligands or the TGFβ receptors have been shown to promote hematopoiesis (e.g. SD208, 1D11, Galunisertib) [13, 34], inhibition of the TGFβ2 ligand is less desirable since its inhibition induces cardiotoxicity and metastasis [35]. Also, TGFβ2 positively regulates hematopoiesis [36], and its inhibition could in principle accelerate BMF in FA.
In the current study, we employed preclinical models to evaluate the activity of AVID200, a TGFβ ligand trap, on murine and human FA HSPCs. AVID200, constructed by fusing the TGFβR ectodomain to a IgG Fc region, is a potent TGFβ trap, with 1000 times more potency against TGFβ1 and TGFβ3 and minimal activity against TGFβ2 [37]. AVID200 has been shown to be an efficient TGFβ pathway inhibitor in preclinical models of Shwachman-Diamond syndrome that is also characterized by BMF [38]. Bone marrow plasma from FA patients contained reduced levels of active TGFβ1 ligand, significantly reduced levels of active TGFβ2 ligand and higher levels of active TGFβ3 ligand, compared to healthy controls. In vitro testing of the response to AVID200 in HSPC cells, derived from FA patients at different stages of the disease, showed that AVID200 promotes hematopoietic clonogenic growth in primary FA samples. Additional experiments using HSPCs from FA mouse models and FA patient-derived lymphoblastoid cell lines demonstrated that mechanistically AVID200 suppresses the toxic overexpression of the NHEJ transcriptional program and promotes efficient DNA repair.
METHODS
Isolation of mouse HSPCs and LT-HSC
Bone marrow cells were harvested from tibia and femurs of mice by gentle flushing with HBSS++ buffer [Hanks balanced salt solution (10–547F, Lonza) + HEPES (BP299–100, Fisher Scientific) + Fetal bovine serum (F2442, Sigma) + penicillin-streptomycin (15140–122, GIBCO)]. Samples were filtered through a 70 μM filter to obtain a single cell suspension and Lin− enrichment was performed with lineage negative selection using the lineage cell depletion kit (130–090-858, Miltenyi). For isolation of LSK cells and LT-HSCs, Lin− cells were incubated with biotin-labeled lineage antibody cocktail from BD containing a mixture of antibodies against CD3, CD11b, CD19, B220, Gr-1 and Ter119 (51–09082J, BD Pharmingen), followed by staining with streptavidin-PE secondary antibody (554061, BD). LSK cells were identified as Lin-Sca-1+c-kit+ using PE-Cy7-Sca (Clone D7, 558162, BD Biosciences) and APC-c-kit (Clone ACK2, 135108, BD Biosciences) antibodies; whereas LT-HSCs were recognized by being LSK and CD150+CD48- using PE-Cy7-Sca (Clone D7, 558162, BD Biosciences), APC-c-kit (Clone ACK2, 135108, BD Biosciences), Pacific Blue-CD150 (Clone TC15–12F12.2, 115924, Biolegend) and APC-Cy7-CD48 (Clone HM48–1, 47–0481-82, e-Bioscience). Cells were sorted using a BD FACSAria cell sorter.
Western blotting for detection of phospho-SMAD2/3
Isolated Lin− cells were expanded during 24 h in serum-free medium StemSpan SFEM (09600, StemCell Technologies) containing 1% penicillin/streptomycin (15140–122, GIBCO), 2% L-glutamine (25030–081GIBCO), 100 ng/ml TPO (315–14-10UG, Peprotech) and 100 ng/ml SCF (250–03-10UG, Peprotech). Cells were exposed during 2 h to TGFβ1 (5 ng/ml), TGFβ2 (5 ng/ml) or TGFβ3 (5 ng/ml) along with AVID200 (25 ng/ml). Whole cell lysates were prepared using RIPA cell lysis buffer (9803, Cell Signaling) and 1 mM PMSF (8553s, Cell Signaling) and western blots were performed using the SMAD2/3 (86855, Cell Signalling), phospho-SMAD2 (ab3849, Millipore), phospho-SMAD2/3 (8828s, Cell Signaling) and β-actin (3700s, Cell Signalling) antibodies. A representative blot is shown.
Phosphoflow for analysis of phospho-SMAD2/3
Mouse Lin− cells were exposed for 30 min to 5 ng/ml of mouse recombinant TGFβ1 (7666-MB-005/CF, R&D Biosystems) and 0.2 ng/ml of AVID200 (Forbius) or 1D11 (10 μg/mL) for 1 h. Mouse LSK cells were cultured for 24 h in presence of 0.2 ng/ml of AVID200 (Forbius). After culture Lin− and LSK cells were gently fixed with pre-warmed BD Phosflow Lyse/Fix Buffer 1x (558049, BD Bioscience) and incubated for 10 min at 37°C, washed with BD PermWash Buffer (51–2091KZ, BD Bioscience), permeabilized while vortexing by slowly adding cold BD PermBuffer III (558050, BD Bioscience) and incubated 30 min on ice. Cells were washed with BD PermWash buffer and incubated with PE mouse anti-Smad2/3 antibody (562586, BD Bioscience) during 30 min at RT. Cells were washed with BD PermWash buffer and run in a CytoFLEX flow cytometer (Beckman Coulter). Data were analyzed using FLOWJO (FLOWJO, LLC) version 10.4.2.
Survival assays with mouse bone marrow cells
For clonogenic assays,20,000 Lin− cells per condition (in triplicate) and 2000 sorted LSK cells per condition (in triplicate) were plated in Methocult GF M3434 methylcellulose (03444,Stem Cell Technologies) and cultured for 7 days. The TGFβ inhibitors such as AVID200 (Forbius), SD208 (S7071, Sigma-Aldrich) and 1D11 (Genzyme) were added in graded concentrations in the cultures. For acetaldehyde treatments, bone marrow cells were exposed to increasing doses of acetaldehyde (00070–100ML, Sigma-Aldrich) for 4 h, washed and then cultured in Methocult GF M3434 methylcellulose. The hematopoietic colonies were scored after 7 days of culture at 37 °C and 5% CO2.
For proliferation assays, sorted LSK cells were cultured in serum-free medium StemSpan SFEM (09600, StemCell Technologies) containing 1% penicillin/streptomycin (15140–122, GIBCO), 2% L-glutamine (25030–081GIBCO), 100 ng/ml TPO (315–14-10UG, Peprotech) and 100 ng/ml SCF (250–03-10UG, Peprotech). After 48 h in culture, DNA damage was assessed by immunofluorescence staining the cells to detect γH2AX foci formation (2577s, Cell Signaling) as described [13].
Viral transduction of CD34+ cells
Isolated human CD34+ cells were cultured in StemSpan SFEMII (09655, Stem Cell Technologies,) with 100ng/ml of all recombinant human cytokines SCF (300–07, Peprotech), TPO (300–18, Peprotech), Flt3 (300–19, Peprotech) and IL-6 (200–06, Peprotech) at a density of 1–2 million cells/ml in non-tissue culture (non-TC) treated plates for 36 hours. After initial culture, cells were plated in non-TC treated 96 well plates with a density of 1–2×10^5 cells in 100–150 μl of new media with 8 μg/ml polybrene (TR-1003-G, Sigma) and shRNA producing lentivirus targeting human FANCD2. A MOI of 50 was used for the scrambled shRNA and a MOI of 100 was used for the targeted shRNAs. Plates were spun down at 2300 rpm for 30 minutes at RT after addition of the viral prep. New media was added after 12–16 hours of incubation. Selection media with 1ug/ml puromycin (MIR 5940, MirusBio) was added to cultures 12–24 hours after viral infection. Puromycin selection was applied during 72 hours. Puromycin resistant cells were used for assays with FA-like cells.
Human HSPC isolation and culture
Whole bone marrow was obtained from FA patients after informed consent of sample usage for research. Fresh healthy bone marrow samples were purchased from Lonza (1M-105, Lonza). Red blood cell lysis was performed by incubating the samples with Ammonium Chloride (07800, StemCell Technologies) for 10 min on ice followed by washing with PBS. After red blood cell lysis, Lin- cells were enriched from mononuclear cells (MNCs) by negative selection using the EasySep kit (19056, StemCell Technologies), according to manufacturer’s instructions.
Clonogenic potential of human HSPC was assessed in CFU assays by plating 3000 HSPCs per triplicate in human methylcellulose MethoCult H4434 Classic (04434, StemCell Technologies). Colonies were quantified and classified 14 days after culture. Pictures were taken with the STEMvision System (StemCell Technologies). Proliferation potential of human HSCPs was assessed by culturing human Lin− cells in 96 well plates in Serum-Free Expansion Medium StemSpan SFEM (09600, StemCell Technologies) supplemented with recombinant human hematopoietic cytokines: TPO (100 ng/ml) (AF-300–18, Peprotech), Flt-3 (100 ng/ml) (AF-300–19, Peprotech), SCF (100 ng/ml) (AF-300–07, Peprotech) and IL-6 (20 ng/ml) (AF-200–06, Peprotech) for 7 days.
Cytokines quantification
Plasma was collected from human whole bone marrow blood and subjected to a bead-based Multiplex Immunoassay for detection of human cytokines TGFβ1, TGFβ2, TGFβ3 using the discovery assay of Eve Technologies (Eve Technologies Corporation, Canada). This multiplexing technology is based on color-coded antibody-coupled polystyrene beads. The bead analyzer Bio-Plex 200 (BIORAD) was used for detection and results were quantified according to a standard curve.
Immunofluorescence
Sorted LT-HSCs were cytospun into SuperFrost slides (12–550-15, Fisher Scientific) and fixed for 15 min in 4% paraformaldehyde (30525–89-4, Electron Microscopy Sciences) in PBS. Cells were then permeabilized in 0.2% Triton-X-100/PBS and incubated for 1 h in blocking buffer (1% FCS/0.1% Triton-X-100/PBS). For γH2AX staining, cells were incubated with rabbit anti-mouse phospho-histone H2A.X (Ser 139) antibody (2577s, Cell signaling) and incubated with a secondary antibody anti rabbit Alexa Fluor-488 (20E3, Cell Signaling) diluted 1:200 in 1% FCS/PBS and incubated in a humidified chamber overnight. The following day slides were washed three times for 5 min in PBS, counterstained and mounted with ProLong Gold antifade reagent with DAPI (P36931, Life Technologies). Images were taken with a Zeiss Imager fluorescent microscope. Slides were assessed for γH2AX foci using Cell Profiler (Open cell image analysis software).
Quantitative real-time PCR array and targeted RNAseq
RNA was extracted from LSK cells using the micro RNA extraction kit (74034, QIAGEN) and from lymphoblast cell lines using the Mini RNA extraction kit (74134, QIAGEN) following the manufacturer’s instructions. RNA quality was assessed using an Agilent Bioanalyzer and RNA Nano Chip in the Biopolymers Facility of Harvard Medical School.
For gene expression analysis of LSK cells, cDNA was synthesized using the RT2 PreAMP cDNA synthesis kit (33045, QIAGEN), target sequences were amplified using the RT2 PreAMP Pathway primer Mix (PBM-029Z, 30241, QIAGEN) and qPCR Master Mix was RT2 SYBR® Green (330533, QIAGEN). Multiplex real-time PCR was performed with the Mouse DNA damage signaling pathway PCR array (PAMM-029ZA, QIAGEN) following manufacturer’s instructions. PCR was performed in a QuantStudio 7 Flex real Time machine (Life Technologies). For the gene expression analysis in EUFA316 cells, a QIAseq Targeted RNA panel targeted was designed (CRHS-10510Z-219–12, 333022, QIAGEN) following manufacturer’s instructions. Index assignment was done using the QIAseq Targeted RNA 96-index I kit (333117, QIAGEN). Libraries were sequenced in a Illumina NextSeq 500 Mid Output flow cell and run in an Illumina NextSeq 500 sequencer in the Biopolymers Facility of Harvard Medical School.
Survival assays
For survival assays, EUFA316+EV and EUFA316+G cells were exposed to increasing doses of acetaldehyde during 4 h, washed and seeded at a density of 1×103 cells per well in 96-well plates. Cell viability was assessed after 5 days of culture using the Cell Titer-Glo® Luminescent Cell Viability Assay (G7573, Promega).
Chromosome aberration analysis
EUFA316+EV and EUFA316+G cell lines were pre-treated with 0.2 ng/ml of AVID200 (Forbius) during 24 h and then exposed to MMC (20 ng/ml) during 48 h and harvested. Alternatively, the cell lines were pre-treated with 0.2 ng/ml of AVID200 (Forbius) during 24 h, exposed for 4 h to acetaldehyde (2 mM), washed and allowed to recover during 48 h. Cells were harvested using KaryoMAX® Colcemid® Solution (15210–040, GIBCO) for 1 h, treated with hypotonic solution for 20 min and fixed with methanol: glacial acetic acid solution (3:1) (both from Fisher) solution. Samples were washed 3 times with methanol (A412–500, Fisher):acetic acid (A38S-500, Fisher) solution, cells were dropped into slides and stained with Giemsa staining solution (Fisher). Chromosome breaks and radial figures were scored in 50 metaphases per condition using conventional cytogenetic criteria.
Karyotyping and FISH analysis of bone marrow and blood
BM blood samples were cultivated following standard cytogenetic procedures. Fluorescent in situ hybridization (FISH) was performed on fresh bone marrow samples using Vysis probes LSI 7q31 (D7S486) spectrum orange/ CEP7 spectrum green, LSI 1p36 spectrum orange/LSI 1q25 spectrum green, LSI BCL6 (ABR) Dual color (SG/SO) Break apart (all from Abbott Molecular). For G banding 25 metaphase spreads were analyzed per patient. A cytogenetic clone with chromosome gain was reported when the same chromosome gains were discovered in at least 2 cells, a cytogenetic clone with chromosome lost was reported when the same chromosome abnormality was found in at least three cells. For FISH 1000 cells were scored and the finding of >5% cells with the same abnormality was considered a cytogenetic clone. Cytogenetic clone definition and karyotype formulas were according to the International System for Chromosome Nomenclature (ISCN) 2016 guidelines.
Alkaline comet assay
Sorted LT-HSCs were mixed with low-melting-temperature agarose, plated on slides and incubated at 4°C overnight in lysis solution using the CometAssay kit (4250–050-K, Trevigen) and following manufacturers’ instructions. Next day cells were subject to a current voltage of 12 V washed and stained with SybrGreen dye (S7567, Invitrogen). As a DNA damage positive control, some cells were exposed to 10 Gy of irradiation using a 137Cs radiation source (model RS2000, Rad source) with a dose rate of 1 Gy min. Pictures were taken with a Zeiss Imager Z1 fluorescence microscope and olive-tail moment analysis was performed using the OpenComet plugin in Image J software.
Patients and Clinical correlations of CFU data
Data from FA patients were obtained from the clinical records of the Boston Children’ Hospital and Instituto Nacional de Pediatría, Mexico. Absolute neutrophil count (ANC), Platelet count (PC), and Hemoglobin (Hb) levels were used for classifying the patients as having mild aplastic anemia if ANC <1,500/mm3, PC 150,000–50,000/mm3 and Hb≥8 g/dL; moderate aplastic anemia if ANC <1,000/mm3, PC <50,000/mm3 and Hb<8 g/dL; and severe aplastic anemia if ANC <500/mm3, PC <30,000/mm3 and Hb<8 g/dL.
Statistics
Normality was assessed using the D’Agostino-Pearson, Shapiro-Wilk and Kolmogorov Smirnoff normality tests. 2-way A OVA and Turkey’s multiple comparisons test were used for detection of differences between experimental groups. TGFβ levels in bone marrow plasma were compared using unpaired t test with Welch correction. γH2AX and Olive tail moment were analyzed using the Kruskal-Wallis test. Colony counts in CFU assays were transformed to fold-change by dividing the average colonies in the AVID200 treated sample by the average in the untreated sample and correlated to clinical data presented in Table 1. CFU fold change in primary samples exposed to AVID200 were compared using Student’s t-Test and Wilcoxon matched-pairs signed rank test. Graphpad Prism 8 was used for all statistical analysis.
Table 1.
FA patients included in this study
| Study ID | Sex | Age | FA subtype | Ethnicity/Origin | Aplastic anemia | Cytogenetic clone | Karyotype in BM | Peripheral blood T-cell mosaicism |
|---|---|---|---|---|---|---|---|---|
| 1 | F | 8.2 | FANCA | Mexican mestizo | No aplastic anemia | No | 44,XX,t(4;13)(q34;q1 2.1),−20,22[1]/45,XX,- 5[1]/44,XX,−5,- 15[1]/43,XX,−6,−14,- 15[1]/45,XX,- 9[1]/46,XX,inv(9)(p11 q12)[1]/45,XX,10[1]/44,XX,−11,- 22[1]/44,XX,−12,20[1]/46,XX,inv(18)(p 11.2q12.2[1]/46,XX [10] | No |
| 2 | M | 17.8 | Unknown | Mexican mestizo | No aplastic anemia | Yes | 45,XY,- 16[3]/46,XY[12] | No |
| 3 | M | 10.3 | FANCA | No aplastic anemia | No | 46,XY[20] | Yes | |
| 4 | M | 2.7 | FANCC | Arabic | No aplastic anemia | No | 46,XY[20] | Yes |
| 5 | F | 3.1 | FANCC | White | Mild aplastic anemia | No | 46,XX[20] | No |
| 6 | F | 12.0 | FANCC | Arabic | Mild aplastic anemia | Yes | 46,XX,del(7)(q31q 36)[5]/46,XX [15]. FISH: del7q (19.5% of cells) | Yes |
| 7 | M | 7.8 | FANCC | Arabic | Mild aplastic anemia | No | 46,XY[20] | Yes |
| 8 | M | 26.5 | FANCA | White | Mild aplastic anemia | Yes | 46,XY,dup(12)(q1 5q13)[6]/ 46,XY[14] | No |
| 9 | M | 9.2 | FANCC | Mild aplastic anemia | Yes | 46,XY. FISH: del13q (35.5% of cells) | No | |
| 10 | F | 6.9 | FANCA | Mexican mestizo | Mild aplastic anemia | No | 46,XX [20] | No |
| 11 | M | 8.6 | Unknown | White | Moderate aplastic anemia | No | 46,XY[20] | No |
| 12 | F | 10.9 | FANCA | Mexican mestizo | Moderate aplastic anemia | No | 46,XX [20] | No |
| 13 | F | 15.4 | Unknown | Mexican mestizo | Severe aplastic anemia | Yes | 45,XX- 7[10]/46,XX[10] | No |
| 14 | F | 8.3 | FANCG | Mexican mestizo | Severe aplastic anemia | No | 46,XX [20] | No |
| 15 | F | 12.0 | Unknown | Mexican mestizo | Severe aplastic anemia | Unknown | No growth | No |
| 16 | M | 10.2 | Unknown | Mexican mestizo | Severe aplastic anemia | No | 45,XY,- 2[1]/45,XY,−18[1]/ 46,XY[18] | No |
| 17 | F | 8.8 | Unknown | Mexican mestizo | Severe aplastic anemia | No | 46,XX [20] | No |
| 18 | M | 8.2 | Unknown | Mexican mestizo | Severe aplastic anemia | No | 46,XY[20] | No |
| 19 | M | 8.6 | FANCA | Mexican mestizo | Severe aplastic anemia | No | No clone detected by FISH | No |
| 20 | M | 10.1 | FANCA | Mexican mestizo | Severe aplastic anemia | No | No clone detected by FISH | No |
| 21 | M | 5.6 | Unknown | Mexican mestizo | Severe aplastic anemia | No | No clone detected by FISH | No |
Study approval
Experimental procedures were approved by the Animal Care and Use Committee of the Dana Farber Cancer Institute. Written informed consent was received from participants prior to inclusion in the study.
RESULTS
TGFβ Inhibition by AVID200 results in enhanced murine HSPC colony growth
We have previously shown that inhibition of TGFβ by a neutralizing antibody rescues the growth defects in murine FA bone marrow (10). In the current work we tested the specificity of AVID200 for sparing the activity of TGFβ2. We confirmed that AVID200 reduces the activity of the TGFβ1 and TGFβ3 ligands but not TGFβ2 on murine progenitors and HSPCs, as determined by reduced SMAD2/3 phosphorylation in cells exposed in vitro to the three members of the TGFβ ligand family (Figure 1A and Supplementary Figure 1A–B). We next compared the performance of AVID200 in promoting hematopoietic colony formation with respect to other known pharmacological inhibitors of TGFβ pathway in murine FA bone marrow. Lineage negative (Lin−) bone marrow cells from WT and Fancd2−/− mice were cultured in a methylcellulose medium in presence of either a small molecule inhibitor of TGFβ Receptor I (SD208), a neutralizing antibody against TGFβ ligands (1D11), or AVID200 (a TGFβ1 and TGFβ3 molecular trap), and clonogenic growth was determined (Figure 1B and Supplementary Figure 1C). All TGFβ pathway inhibitors including AVID200 promoted the clonogenic growth of bone marrow progenitors from Fancd2−/− mice (Figure 1B). Colony size was increased after AVID200 treatment. Increased proliferation and increased colony size after AVID200 treatment was confirmed by solubilization of the hematopoietic colonies and quantification of cell numbers after the culture (Supplementary Figure 1D).
Figure 1. Inhibition of the TGFβ pathway promotes clonogenic growth of HSPCs from FA mice.

A) Western blots of the lysates from murine bone marrow cells. Lin− cells from bone marrow of wild-type mice were cultured for 2 h in presence of TGFβ1 (5 ng/ml), TGFβ2 (5 ng/ml) or TGFβ3 (5 ng/ml) with or without AVID200. Note that two types of antibodies were used to detect the phospho-SMAD2 levels. One of the antibodies was used only against phospho-Smad2 (p-Smad2) whereas the other one was against both phosphor-Smad2 and Smad3 (p-Smad2/3). A representative blot is shown.
B) TGFβ pathway inhibitors promote colony formation of murine Fancd2−/− HSPCs. Lin− cells from wild-type (WT) or Fancd2−/− mice were cultured for 7 days in methylcellulose medium with inhibitors of the TGFβ pathway, namely, SD208 (10 μM), 1D11 (10 μg/mL) and AVID200 (0.2 ng/mL). Hematopoietic colonies were quantified (n=3).
p-values of 0.01 to 0.05 were considered significant (*), p-values of 0.001 to 0.01 were considered very significant (**) and p-values of <0.001 were considered extremely significant (****). Data in (B) are represented as mean ± SEM. See also Supplementary Figure 1.
TGFβ inhibition by AVID200 rescues the clonogenic defects of primary FA-patient derived bone marrow
We next tested the effects of AVID200 on primary human cells. Using a lentivirus expressing a short-hairpin RNA (shRNA) against FANCD2, we generated FA-like HSPCs using cord blood CD34+ cells. Two different shRNAs against FANCD2, expressed in primary cord blood CD34+ cells, reduced the relative abundance of FANCD2 transcript (Supplementary Figure 2A). As expected, primary human FA-like HSPCs exhibited reduced clonogenic potential, and AVID200 rescued these clonogenic growth defects in a dose dependent manner (Figure 2A and Supplementary Figure 2B). Similar to mouse CFU, the colony size was increased after AVID200 treatment. The increase in colony size and increased proliferation of FA-like cells after AVI200 treatment was confirmed by solubilization of the hematopoietic colonies and quantification of cell numbers after the culture (Supplementary Figure 2C).
Figure 2. Inhibition of the TGFβ pathway with AVID200 rescues clonogenic defects of primary bone marrow from FA patients.

A) TGFβ pathway inhibitors promote cell growth (colony formation) of primary human FA-like cells. Healthy human cord blood CD34+ cells were infected with two different lentivirus expressing a shRNA against the human FANCD2 gene, thus generating FA-like cells. Human FA-like cells were then exposed to increasing concentrations of AVID200 (0.06, 0.2, 0.6, and 1.8 ng/mL) or SD208 (1 μM), as a positive control of TGFβ pathway inhibition, colony formation was assessed after 14 days of culture in methylcellulose medium (n=3).
B) Levels of free TGFβ1, TGFβ2 and TGFβ3 ligands in primary bone marrow plasma from FA patients (n=12) and healthy donors (n=5). Levels of free TGFβ2 are significantly reduced in BM plasma from FA patients, whereas levels of free TGFβ3 are significantly increased in FA patients.
C) Clonogenic growth of bone marrow progenitors isolated from FA patients (n=19) and healthy donors (n=3). Primaryprogenitor cells were isolated from bone marrow of 21 FA patients or 3 healthy donors and cultured in triplicates in complete methylcellulose medium with or without AVID200 (0.2 ng/mL) for 14 days and clonogenic growth was assessed for total colonies, myeloid colonies and erythroid colonies, patient 1 and 15 behaved as outliers and were removed from statistical analysis. Left panel shows raw average CFU-C numbers per sample from normal donor or FA patient. Right panel shows the fold change in colony numbers in the AVID200 treated samples after normalization with respect to the vehicle-treated control. Comparisons in both raw CFU-C numbers and fold change converted numbers were made by matching each vehicle-treated sample with its respective AVID200 treated sample per patient (matching pair).
D) Representative photomicrograph showing improved clonogenic growth of a FA bone marrow sample after AVID200 exposure in vitro in comparison to a bone marrow sample from a healthy donor control. Erythroid colonies are indicated in red, Myeloid colonies are indicated in yellow, multipotent colonies are indicated in blue.
E) Correlation between the degree of anemia in FA patients and response to AVID200 showing that the response to AVID200 is significantly better in the bone marrow from FA patients with severe bone marrow failure than the bone marrow from patients with mild anemia. Left panel. Raw average CFU-C numbers per patient’s sample. Right panel. Absolute mean colony numbers were normalized to fold-change and FA patients were classified according to the severity of their aplastic anemia.
F) Correlation between the presence of a cytogenetic clone in FA patients and the response to AVID200 showing that patients with chromosomal clonal abnormalities, detected by bone marrow karyotype or FISH, responded better to AVID200 treatment in vitro. Left panel. Raw average CFU-C number per patient’s sample. Right panel. For comparisons the absolute mean colony numbers were normalized to fold-change and FA patients were classified according to bone marrow karyotypes performed the day of progenitor cells isolation and culture.
p-values of 0.01 to 0.05 were considered significant (*), p-values of 0.001 to 0.01 were considered very significant (**) and p-values of <0.001 were considered extremely significant (***). Data in (A) are represented as mean ± SEM, data in (B), (C), (E) and (F) are presented as boxplots. See also Supplementary Figure 2.
The TGFβ ligand family is composed of three members, TGFβ1, β2, and β3, which are secreted into the extracellular milieu as inactive homodimers that become active monomers once they are cleaved by specific extracellular metalloproteases or integrins [15]. Active TGFβ monomers have the capacity to bind to the TGFβ receptor I and to activate signal transduction pathways, culminating in changes in gene expression [22]. Overexpression of the TGFβ pathway genes is evident in mouse and human FA bone marrow [13], however, elevated levels of TGFβ ligands have not been reported in primary FA bone marrow-derived serum or plasma. Using an immunoassay, we observed an imbalance in the levels of TGFβ ligands in the plasma collected from the bone marrow of 12 FA patients. There was a reduction in the level of active (free) TGFβ1, a significant reduction in the level of active TGFβ2 and an increase in level of active TGFβ3 in bone marrow plasma of FA patients (Figure 2B). As TGFβ1 and TGFβ3 are growth suppressive for HSPCs, the significantly increased levels of active TGFβ3 in the FA BM plasma may activate the growth suppressive pathways accounting for BMF in FA patients. Reduced levels of active TGFβ2 may account for the ineffective stimulation of hematopoiesis in FA. The levels of total TGFβ ligands have a distribution similar to the levels of active TGFβ ligands, meaning increased TGFβ3, and reduced TGFβ1 and TGFβ2 in FA samples (Supplementary Figure 2D).
To determine whether specific inhibition of elevated TGFβ3 in FA bone marrow can rescue the bone marrow defects, we next assessed the effects of AVID200 in primary HSPCs from FA patients. Bone marrow Lin− cells were cultured for seven days in the presence of growth factors, and cell proliferation was determined. AVID200 enhanced proliferation of HSPCs from 7 out of 9 FA samples. FA patients at different clinical stages of their disease were included in this study. As expected, only low numbers of HSPCs were obtained from FA patients with severe aplastic anemia (samples 10, 13, 17 and 21). Accordingly, only a single measurement (sm) of survival was possible for these patients (Supplementary Figure 2E). We next determined the clonogenic potential of primary Lin− cells from 20 FA patients by culturing them in methylcellulose medium in the presence or absence of AVID200. An overall significant increase in total colony numbers was observed with AVID200 in most of the FA samples (Figure 2C). AVID200 increased also the myeloid and erythroid clonogenic potential of the bone marrow from FA patients (Figure 2C). A representative photomicrograph shows the improved CFU-C capacity of FA samples cultured with AVID200 (Figure 2D). Response to AVID200 in the bone marrow of individual FA patients is presented in the supplementary Figures (Supplementary Figure 2F, 2G and 2H). Although the triplicate cultures were not possible from all the samples, a tendency of increased colony numbers was observed with AVID200 in the bone marrow from most of the FA patients. Importantly, the samples which did not exhibit an overall colony improvement, showed an improvement in either the myeloid or erythroid colony formation (Supplementary Figure 2G and 2H).
Clinical information from FA patients (Table 1) was used to derive correlations with AVID200 response. The samples from patients with advanced aplastic anemia displayed the highest fold change in response to AVID200, whereas patients with mild anemia showed only a modest response to AVID200 (Figure 2E). Interestingly, samples derived from patients with cytogenetic evidence of clonal cytogenetic abnormalities were among the best responders to AVID200 (Figure 2F). Collectively, inhibition of TGFβ1 and TGFβ3 by AVID200 promoted the clonogenic potential of primary bone marrow progenitors from FA patients.
TGFβ inhibition by AVID200 rescues DNA repair defects of FA HSPCs
We have previously shown that TGFβ pathway inhibitors downregulate error-prone NHEJ pathway gene expression and upregulate high-fidelity HRR gene expression [13]. We therefore next asked whether specific inhibition of TGFβ1 and TGFβ3 will have similar consequences.
Since efficient DNA repair may improve the cellular survival, we challenged murine Fancd2−/− Lin− cells (Figure 3A) and Fancg−/− Lin− cells (Supplementary Figure 3A) by exposure to acetaldehyde, a DNA damaging agent to which FA cells are hypersensitive [39, 40]. As expected, the in vitro clonogenic potential of Fancd2−/− and Fancg−/− cells was reduced upon exposure to acetaldehyde, in contrast to WT cells (Figure 3A and Supplementary Figure 3A). Acetaldehyde hypersensitivity of Fancd2−/− and Fancg−/− cells was significantly rescued with AVID200 pretreatment. In addition, AVID200 rescued the acetaldehyde sensitivity of murine Fancd2−/− HSPCs (Lin−Sca-1+c-Kit+, LSK cells) and improved their proliferation (Supplementary Figure 3B). AVID200 also reduced acetaldehyde-induced DNA damage in LSK cells from Fancd2−/− mice as assessed by two assays, reduction in the amount of γH2AX foci (Figure 3B) and reduction in the amount of BrdU foci (Supplementary Figure 3C, 3D), in this assay the thymidine analogue BrdU is incorporated into the DNA of cycling cells, and after induction of DNA damage the amount of single stranded DNA formation at not-rejoined DSBs can be quantified by using an antibody that only recognizes BrdU in single stranded DNA but not in double stranded DNA.
Figure 3. Inhibition of the TGFβ pathway with AVID200 rescues genotoxicity in FA mouse models.

A) AVID200 improves the survival of mouse FA bone marrow after exposure to acetaldehyde. Lin− cells from bone marrow of wild-type (WT) or Fancd2−/− mice were pretreated with AVID200 (0.2 ng/mL) and exposed to acetaldehyde (2 mM) for 4 hrs. After washing, the cells were cultured in complete methylcellulose medium with and without AVID200 for 7 days and survival of the hematopoietic progenitors was determined by quantification of hematopoietic colonies (n=3).
B) AVID200 reduces acetaldehyde-induced DNA damage in HSPCs from bone marrow of FA mice. LSK cells from wild-type (WT) or Fancd2−/− mice were pretreated with AVID200 (0.2 ng/mL) for 24 h and exposed to acetaldehyde (2 mM) for 4 h. After washing, the cells were allowed to recover for 48 h in StemSpan Medium supplemented with cytokines TPO, SCF, Flt3 ligand and L-Glutamine. The cells were then analyzed for γH2AX foci by immunofluorescence. 100 cells were counted per condition (n=3).
C) AVID200 modifies the gene expression profile of DNA repair related genes in mouse LSK cells. LSK cells from wild-type (WT) or Fancd2−/− mice were exposed to AVID200 (0.2 ng/mL) for 48 hrs and gene expression profile was analyzed. Note the downregulation of Trp53bp1, Prkdc and Trp53, in cells from Fancd2−/− mice (n=3).
D) AVID200 prevents polyinosinic:polycytidylic acid (pI:pC)-induced DNA damage in LT-HSCs from bone marrow of FA mice in vivo. Wild-type (WT) and Fancd2−/− mice (n=3 mice per group) were co-injected with water, pI:pC (5 mg/kg), AVID200 (5 mg/kg) or a combination of both. LT-HSCs were isolated and DNA damage was assessed by immunofluorescence staining of γH2AX foci, left panel shows the amount of γH2AX foci per cell 48 h after injection, whereas right panel shows the amount of γH2AX foci per cell after 30 days of 2 injections per week (n=5 mice per group).
(E) Representative pictures of γH2AX foci in LT-HSC are shown. At least 50 cells from each group were scored for γH2AX foci.
p-values of 0.01 to 0.05 were considered significant (*), p-values of 0.001 to 0.01 were considered very significant (**) and p-values of <0.001 were considered extremely significant (***, ****). Data in (A) and (B) are represented as mean ± SEM, data in (C) are presented as scatter plot, data in (D) are presented as boxplot. See also Supplementary Figure 3.
One of the best characterized mechanisms of action of the TGFβ pathway is the regulation of gene expression through its downstream effectors [22], We therefore hypothesized that AVID200 rescues DNA damage and improves survival of FA cells by modulating the gene expression profile of DNA repair genes. Murine Fancd2−/− LSK cells were therefore exposed in vitro to AVID200, and changes in expression of a panel of genes involved in DNA repair and genome maintenance were evaluated by a multiplex real time-PCR assay. Remarkably, AVID200 induced a switch in the gene expression profile of Fancd2−/− LSK cells (Figure 3C), resulting in downregulation of genes involved in NHEJ, namely, Prkdc (encoding for DNA-PK) and Trp53bp1 (encoding for 53bp1) [41, 42]. These results suggest that AVID200 inhibits the expression of NHEJ genes, thus allowing the repair of DSBs by HR mechanisms without interference by NHEJ. Moreover, improved DNA repair capacity was predicted to reduce p53 pathway activation. Accordingly, the Trp53 gene (encoding for p53) was also downregulated after AVID200 treatment, thereby reducing cell cycle checkpoint activation and allowing for enhanced cell proliferation.
Although, FA mice do not exhibit bone marrow failure spontaneously, BMF can be induced by exposure of mice to a physiological stress such as polyinosinic-polycytidylic acid (pI:pC). pI:pC is a potent inductor of the type 1 interferon response [43]. pI:pC activates the HSCs in FA mice and induces DNA damage resulting into BMF [43]. We therefore next asked whether physiological stress-induced DNA damage in HSCs can be reduced by AVID200. WT and Fancd2−/− mice were exposed to pI:pC and AVID200, and DNA damage was assessed in LT-HSCs after 48 hrs (Figure 3D left panel) and after 30 days (Figure 3D right panel). As expected, pI:pC treatment caused DNA damage in LT-HSCs and increased levels of γH2AX foci. AVID200 reduced the pI:pC-induced γH2AX foci in LT-HSCs from both WT and Fancd2−/− mice (Figure 3D and 3E), even after 30 days of continuous in vivo exposure. AVID200 also reduced the DNA damage in LT-HSCs, as assessed by the comet assay (Supplementary Figure 3E and 3F). Taken together, AVID200 reduces physiological stress-induced and acetaldehyde-induced DNA damage in HSCs from Fancd2−/− mice.
TGFβ inhibition by AVID200 reduces chromosomal aberrations in FA lymphoblast cells
Although we observed an improvement in colony formation and survival in primary bone marrow from FA patients, the low number of primary HSPCs from FA patients limited extensive studies with AVID200. Therefore, we next assessed AVID200 in a human-lymphoblast FA cell line EUFA316+EV derived from a FANCG patient. As expected, the EUFA316+EV cells were more sensitive than its corrected counterpart (EUFA316+G cells) to increasing concentrations of acetaldehyde, and AVID200 rescued this sensitivity (Figure 4A). Acetaldehyde also induced chromosomal breaks and radials in EUFA316+EV cells. Remarkably, AVID200 significantly reduced the frequency of these chromosomal aberrations (Supplementary Figure 4A), AVID200 also significantly reduced the frequency of Mitomycin C (MMC)- induced chromosomal aberrations (Figure 4B).
Figure 4. Inhibition of the TGFβ pathway with AVID200 rescues genotoxicity in FA-derived human cell lines.

A) AVID200 improves the survival of FANCG-deficient human FA lymphoblast cells in presence of acetaldehyde. FANCG-deficient parental cells (EUFA316+EV) and FANCG-corrected cells (EUFA316+FANCG) were exposed to graded concentration of acetaldehyde and AVID200 (0.2 ng/mL) for 6 days and survival was determined by CellTiter Glo reagent (n=12 per condition).
B) AVID200 pretreatment reduces the frequency of Mitomycin C (MMC)-induced chromosomal aberrations in FANCG-deficient cells. EUFA316+EV cells and EUFA316+FANCG cells were pre-treated with AVID200 before exposure to MMC and metaphase spreads of the chromosomes were scored for chromosomal aberrations. Quantification of the MMC-induced chromosomal aberrations is shown in the left panel (n=3). A representative metaphase from the EUFA316+EV cell line treated with MMC is shown in the right panel, red arrow indicates a radial figure.
C) Volcano plot showing DNA repair-related differentially expressed genes of the FANCG-deficient cell line (EUFA316+EV) relative to the FANCG-corrected cell line (EUFA316+FANCG). The log fold change in the EUFA316+EV cell line versus the EUFA316+G cell line is represented on the x-axis. The y-axis shows the Log10 of the p value. Downregulated genes with a p value of 0.05 are indicated in blue, whereas upregulated genes with a p value of 0.05 are indicated in red (n=3).
D) Volcano plot showing DNA repair-related differentially expressed genes of the FANCG-deficient cell line (EUFA316+EV) after 24 h exposure to AVID200 (0.2 ng/mL). The log fold change in the EUFA316+EV + AVID200 cell line versus the untreated cell line is represented on the x-axis. The y-axis shows the Log10 of the p value. Downregulated genes with a p value of 0.05 are indicated in blue, whereas upregulated genes with a p value of 0.05 are indicated in red (n=3).
p-values of 0.01 to 0.05 were considered significant (*), p-values of 0.001 to 0.01 were considered very significant (**) and p-values of <0.001 were considered extremely significant (***, ****). Data in (A) are represented as mean ± SEM, data in (B) are presented as boxplot, data in (C) and (D) are presented as volcano plots. See also Supplementary Figure 4.
TGFβ inhibition by AVID200 reduces NHEJ gene expression in FA lymphoblast cells
Using bone marrow HSPCs from Fancd2−/− mouse model, we observed that AVID200 reduces the DNA damage and promotes cell survival, consistent with a reduction in the expression of NHEJ genes. We followed a similar approach and determined whether AVID200 modifies the gene expression profile of the EUFA316 +EV cells using a custom RNAseq panel containing DNA repair gene targets.
Interestingly, the alternative error-prone DNA repair genes, POLQ [44] and RAD52 [45], and NHEJ genes such as TP53BP1 [41], were differentially expressed in the EUFA316+EV cell line, in comparison to the corrected EUFA316+G cells (Figure 4C). Changes in gene expression profile of the EUFA316+EV cells after 24 h of exposure to AVID200 were therefore assessed (Figure 4D). Of note, several genes switch their transcriptional expression in response to AVID200, depending on the presence or absence of a functional FA pathway. Specifically, in EUFA316+EV cells, TERF1, RAD51, TOPBP1, ATRIP, LTBP2, MAD2L2 and DDB2 were downregulated whereas SIRT1, BLM, H2AFX, and DDB1 genes were upregulated after AVID200 exposure. The downregulation of expression of the MAD2L2 gene, which encodes the REV7 protein, is noteworthy. REV7 protein, a subunit of the Shieldin complex, is critical for suppressing double strand break resection and promoting NHEJ activity [46]. Downregulation of MAD2L2 promotes efficient HR repair, since the Shieldin complex becomes dismantled and NHEJ is mitigated [46–48]. As AVID200 rescues the chromosomal abnormalities including radial chromosomes, the reduced expression of genes encoding components of the NHEJ pathway, including REV7, may provide a mechanism by which AVID200 promotes efficient DNA repair and survival in FA cells.
DISCUSSION
FA pathway deficiency is known to result in a hyperactivation of the growth suppressive TGFβ pathway [13]. TGFβ is a master regulator of cellular proliferation during embryogenesis and in adult tissue homeostasis [49] and it contributes, at least in part, to the BMF of FA [13] and Shwachman-Diamond syndrome [38]. TGFβ inhibits the growth of many cellular types, including epithelial, endothelial, hematopoietic, and immune cells. In the current study, we have shown that in contrast to healthy controls, the BM plasma from FA patients has increased concentrations of total and active TGFβ3 ligand, a potent inhibitor of HSPCs proliferation, and decreased concentrations of total and active TGFβ1 and TGFβ2 ligands, a positive regulator of HSPCs maintenance [36]. This imbalance of TGFβ ligand isoforms contributes to the BMF in FA and offers a therapeutic strategy.
Using a murine FA-deficient HSPCs, we screened several inhibitors of the TGFβ pathway and assessed their capacity to promote HSPCs clonogenic growth in vitro. These molecules included inhibitors of the TGFβ RI receptor and inhibitors of the TGFβ ligands themselves. Each drug promoted the growth and clonogenic potential of the FA HSPCs, thus confirming that a hyperactive TGFβ pathway has a growth suppressive effect on FA hematopoiesis. Importantly, TGFβ2, but not TGFβ1 or TGFβ3, is involved in multiple developmental processes of heart and great vessels [50], and is also a positive regulator of adult HSCs [36]. Complete inhibition of the TGFβ pathway might therefore have negative consequences, including unintentional stimulation of dormant tumor cells [35] or adverse side effects, such as inflammation, autoimmunity, or cardiotoxicity [50–52]. These adverse effects could have major consequences for pediatric FA patients who require novel therapies for their progressing BMF.
Using AVID200, a potent TGFβ trap against TGFβ1 and β3, with minimal activity against TGFβ2, we have performed the first preclinical studies avoiding the potential negative side effects that TGFβ2 inhibition might have. We proved that AVID200 promotes cell proliferation and/or clonogenic survival in a large number of FA primary bone marrow samples, with the strongest effect on HSPCs from patients with severe aplastic anemia and in patients with cytogenetic evidence of clonal chromosome abnormalities.
In our studies, mouse Fancd2−/− and Fancg−/− HSPCs, and the human FANCG deficient lymphoblast cells, were rescued from acetaldehyde toxicity when they were concomitantly treated or pre-treated in vitro with AVID200. AVID200 also reduced the DNA damage foci and chromosomal aberrations. A mouse in vivo assay confirmed that the DNA damage burden caused by pI:pC-induced physiological stress in the LT-HSC compartment [43] can be dramatically reduced when AVID200 is concomitantly administered with pI:pC treatment in mice. Importantly, here we demonstrate that genotoxicity induced by endogenous aldehyde and physiological stress can be lowered by AVID200, which might result from changes in expression of genes mediating DNA repair.
TGFβ pathway is known to play a role in maintaining genomic stability by regulating the transcription of genes involved in DNA damage response [53]. Importantly, TGFβ pathway hyperactivity promotes the expression of alternative error-prone DNA repair genes [54]. Accordingly, inhibitions of TGFβ1 and TGFβ3 with AVID200 might modulate this transcriptional landscape. The improved survival of FA cells by AVID200 may therefore be a consequence of improved DNA repair capacity. Gene expression analysis in mouse Fancd2−/− HSPCs and in a human FANCG deficient lymphoblast cells, showed that indeed AVID200 effectively modifies the expression profile of DNA repair genes, frequently downregulating the expression of error-prone NHEJ genes.
TGFβ pathway activation can promote genomic instability by downregulating RAD51 protein expression [55] or by reducing the efficiency of DNA DSB repair by downregulating the expression of various homologous recombination repair genes, such as BLM, BRCA2, FANCF, NBN, RAD50, RDM1, WRN, ATM and ATR [54]. The response of cells to TGFβ pathway signaling, and thus the response to its inhibition, is highly context dependent [22]. Indeed, we observed differences in the subsets of DNA repair genes, either downregulated or upregulated, depending on the cell type in which the TGFβ pathway is inhibited. We consistently found NHEJ genes to be downregulated in FA models upon AVID200 treatment; however, the specific genes were not the same in every system studied. For example, Tp53bp1 (coding for 53bp1) and Prkdc (coding for DNA-PK) were downregulated in murine HSPCs, whereas MAD2L2/REV7 was downregulated in the human FANCG deficient lymphoblast cell line. TERF1 and Terf1, telomere maintenance associated genes [56] in human and mouse respectively, were genes consistently downregulated by AVID200, suggesting that AVID200 may reduce on DNA damage in FA cells by downregulation of this gene, a hypothesis that warrants further study. Although we did not explore epigenetic modifications in the regulatory regions controlling the expression of DNA repair genes, we speculate that AVID200 might modify the chromatin state of NHEJ genes or interfere with the recruitment of critical transcription factors to the promoters of these genes, opening new avenues of exploration.
Although TGFβ1 levels appear not to be high in FA, so as to have a crucial role in downstream activation of the TGFβ pathway, AVID200 has the advantage in the FA patient setting of inhibiting both TGFβ1 and TGFβ3 function while, at the same time, preserving the positive functions of TGFβ2. AVID200 is currently in phase 1 clinical trials in patients with advanced or metastatic solid tumor and its safety has been evaluated in several preclinical models. Our results open the possibility of using AVID200 as a more precise therapy for FA patients, avoiding the use of broad inhibitors of the TGFβ family, which are multifunctional and highly context-dependent signaling molecules.
Supplementary Material
Supplementary Figure 1. Related to Figure 1. AVID200 inhibits TGFβ pathway signaling in primary mouse bone marrow cells. A) Reduced activation of the TGFβ pathway by AVID200 in mouse bone marrow progenitors. Murine bone marrow Lin− progenitors were exposed to TGFβ1 (5 ng/mL) along with AVID200 (0.2 ng/mL) or 1D11 (10 μg/mL) as a positive control and the cells were stained with phospho-SMAD2/3 antibody. Phospho-SMAD2/3 staining was then detected by flow cytometry. Representative FACS plots for staining in wild-type (WT) cells are shown. B) Quantitation of the staining intensity as mean fluorescence value (MFI) in the Lin− cells from WT and Fancd2−/− mice is shown in panel B. C) Raw total colony numbers from colony formation assay shown in Figure 1B. Lin− cells from wild-type (WT) or Fancd2−/− mice were cultured for 7 days in methylcellulose medium with inhibitors of the TGFβ pathway, namely, SD208 (10 μM), 1D11 (10 μg/mL) and AVID200 (0.2 ng/mL). Hematopoietic colonies were quantified (n=3). D) Cell numbers after solubilization of hematopoietic colonies in methylcellulose medium. Lin− cells from WT and Fancd2−/− mice were cultured in methylcellulose medium for 7 days in the presence or absence of AVID200 (0.2 ng/mL), colonies quantified, and methylcellulose was solubilized with PBS. The number of cells per well was quantified and plotted (n=3). p-values of 0.01 to 0.05 were considered significant (*) and p-values of 0.001 to 0.01 were considered very significant (**). Data in (C) are represented as mean ± SEM, data in (D) are presented as boxplots.
Supplementary Figure 2. Related to Figure 2. AVID200 enhances clonogenic growth of myeloid and erythroid progenitors from bone marrow of FA patients. A) FANCD2 gene expression analysis by Real time PCR showing the knockdown efficiency of sh1 FANCD2 and sh3 FANCD2 in human cord blood CD34+ cells. Error bars represent S.E.M. n=3. B) Raw total colony numbers from colony formation assay for primary human FA-like CD34+ cells shown in Figure 2A. Human FA-like CD34+ cells were then exposed to increasing concentrations of AVID200 (0.06, 0.2, 0.6, and 1.8 ng/mL) or SD208 (1 μM), as a positive control of TGFβ pathway inhibition, colony formation was assessed after 14 days of culture in methylcellulose medium (n=3). C) Cell numbers after solubilization of hematopoietic colonies in methylcellulose medium. FA-like CD34+ cells were cultured in methylcellulose medium for 14 days in the presence or absence of AVID220 (0.2 ng/mL), colonies quantified, and methylcellulose was solubilized with PBS. The number of cells per well was quantified and plotted (n=3). D) Total TGFβ1, TGFβ2 and TGFβ3 ligand levels in the bone marrow plasma of FA patients (n=12) and normal donors (n=5). Levels of total TGFβ1 and TGFβ2 are significantly reduced in BM plasma from FA patients, whereas levels of total TGFβ3 are significantly increased in FA patients. E) AVID200 promotes the proliferation of bone marrow progenitors from FA patients. Progenitor cells isolated from bone marrow of 9 FA patients and 3 healthy donors were cultured in StemSpan medium with TPO (100 ng/ml) (AF-300–18, Peprotech), Flt-3 (100 ng/ml) (AF-300–19, Peprotech), SCF (100 ng/ml) (AF-300–07, Peprotech) and IL-6 (20 ng/ml) (AF-200–06, Peprotech) for 7 days and live cells were counted. Note that healthy bone marrow cells did not respond or had a poor response to AVID200 treatment. sm= single measurement in samples with very few progenitors. ns=non-significant. F) Primary progenitor cells isolated from bone marrow of 20 FA patients or 3 healthy donors were cultured in triplicates in complete methylcellulose medium with or without AVID200 (0.2 ng/mL) for 14 days. Total number of hematopoietic colonies (total CFU) including myeloid colonies as well as erythroid colonies are shown per sample. G) Myeloid CFU-C production of bone marrow from individual FA patients or normal donor in response to AVID200 after 14 days of culture in complete methylcellulose medium with or without AVID200 (0.2 ng/mL). H) Erythroid CFU-C production of bone marrow from individual FA patient or normal donor in response to AVID200 after 14 days of culture in complete methylcellulose medium with or without AVID200 (0.2 ng/mL). p-values of 0.01 to 0.05 were considered significant (*), p-values of 0.001 to 0.01 were considered very significant (**) and p-values of <0.001 were considered extremely significant (***). Data in (A), (E), (F), (G) and (H) are presented as bar graphs, data in (B are presented as mean ± SEM, data in (D) are presented as boxplots.
Supplementary Figure 3. Related to Figure 3. AVID200 rescues genotoxicity in bone marrow progenitors from FA mice. A) Lin− cells from the bone marrow of wild-type or Fancg−/− mice were pretreated with AVID200 (0.2 ng/mL) and exposed to acetaldehyde (2 mM) for 4 h. The survival of cells was determined by clonogenic assay after culturing them in methylcellulose medium for 7 days (n=3). B) LSK cells from the bone marrow of wild-type or Fancd2−/− mice were pretreated with AVID200 (0.2 ng/mL), exposed to acetaldehyde (2 mM) for 4 h and then allowed to recover for 7 days in StemSpan medium enriched with cytokines SCF, Flt3 ligand, TPO and L-Glutamine. Survival of the cells was determined by counting the number of live cells after 7 days of culture (n=3). C) AVID200 reduces the amount of acetaldehyde-induced unrepaired single stranded DNA in bone marrow cells. LSK cells from bone marrow of wild-type (WT) or Fancd2−/− mice were exposed to acetaldehyde and BrdU along with AVID200 as indicated in the schematic in panel C and the amount of unrepaired single stranded DNA was measured by immunofluorescence as BrdU foci. Note that exposure to acetaldehyde increases the number of unrepaired single stranded DNA, measured as BrdU foci, in Fancd2−/− LSK cells. AVID200 reduces the amount of unrepaired single stranded DNA, as demonstrated by the reduction of BrdU foci. C) Representative images of BrdU foci in LSK cells are shown in panel D. At least 30 cells were counted for BrdU foci. E) AVID200 reduces pI:pC-induced DNA damage in LT-HSCs of Fancd2−/− mice. WT or Fancd2−/− mice were co-injected with pI:pC (5 mg/kg) and AVI200 (5 mg/kg), LT-HSCs were isolated after 48 h of the treatment and comet assay was performed to analyze DNA damage. Quantitation of the Olive tail moment in the LT-HSC is shown in panel E. F) Representative images of the pI:pC-induced alkaline comets in LT-HSCs are shown in panel F. p-values of 0.01 to 0.05 were considered significant (*), p-values of 0.001 to 0.01 were considered very significant (**) and p-values of <0.001 were considered extremely significant (***, ****). Data in (A) and (B) are presented as mean ± SEM, data in (C) and (E) are presented as boxplots.
Supplementary Figure 4. Related to Figure 4. AVID200 reduces the acetaldehyde-induced chromosomal aberrations in FANCG deficient lymphoblast cells. A) EUFA316+EV cells and EUFA316+FANCG cells were pre-treated with AVID200 before exposure to acetaldehyde. The cells were then allowed to recover, and metaphase spreads of chromosomes were scored for chromosomal abnormalities (n=3). Data are presented as bar graphs.
Highlights.
AVID200 traps TGFβ1 and TGFβ3 ligands and improves hematopoiesis in FA models
The TGFβ3 ligand is overexpressed in the bone marrow plasma of FA patients
AVID200 improves the survival of HSPCs from FA patients with severe aplastic anemia
AVID200 improves the survival of HSPCs from FA patients with clonal hematopoiesis
AVID200 rescues genotoxicity in a FA mouse model and human cell lines
ACKNOWLEDGMENTS
This research was supported by grants from the U.S. National Institutes of Health (R37HL052725, P01HL048546), the Leukemia and Lymphoma Society (6237–13), and the Fanconi Anemia Research Fund (to ADD); SEP-CONACYT (243102) and PAPIIT (IA202615) to Sara Frías. We thank Forbius (Formation Biologics), Inc for providing the AVID200 used for the experiments in this study.
Footnotes
CONFLICT OF INTEREST AND DISCLOSURE
A.D. D’Andrea is a consultant/advisory board member for Lilly Oncology, Merck-EMD Serono, Intellia Therapeutics, Sierra Oncology, Cyteir Therapeutics, Third Rock Ventures, AstraZeneca, Ideaya Inc., Cedilla Therapeutics Inc., a stockholder in Ideaya Inc., Cedilla Therapeutics Inc., and Cyteir, and reports receiving commercial research grants from Lilly Oncology and Merck-EMD Serono. Other authors declare no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Shimamura A. and Alter BP, Pathophysiology and management of inherited bone marrow failure syndromes. Blood Rev, 2010. 24(3): p. 101–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ceccaldi R, Sarangi P, and D’Andrea AD, The Fanconi anaemia pathway: new players and new functions. Nat Rev Mol Cell Biol, 2016. 17(6): p. 337–49. [DOI] [PubMed] [Google Scholar]
- 3.Tischkowitz M. and Dokal I, Fanconi anaemia and leukaemia–clinical and molecular aspects. Br. J. Haematol, 2004. 126(2): p. 176–191. [DOI] [PubMed] [Google Scholar]
- 4.Ceccaldi R, et al. , Bone marrow failure in Fanconi anemia is triggered by an exacerbated p53/p21 DNA damage response that impairs hematopoietic stem and progenitor cells. Cell Stem Cell, 2012. 11(1): p. 36–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garaycoechea JI and Patel KJ, Why does the bone marrow fail in Fanconi anemia? Blood, 2014. 123(1): p. 26–34. [DOI] [PubMed] [Google Scholar]
- 6.Savage SA and Walsh MF, Myelodysplastic Syndrome, Acute Myeloid Leukemia, and Cancer Surveillance in Fanconi Anemia. Hematol Oncol Clin North Am, 2018. 32(4): p. 657–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wegman-Ostrosky T. and Savage SA, The genomics of inherited bone marrow failure: from mechanism to the clinic. Br J Haematol, 2017. 177(4): p. 526–542. [DOI] [PubMed] [Google Scholar]
- 8.Knies K, et al. , Biallelic mutations in the ubiquitin ligase RFWD3 cause Fanconi anemia. J Clin Invest, 2017. 127(8): p. 3013–3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rodriguez A. and D’Andrea A, Fanconi anemia pathway. Curr Biol, 2017. 27(18): p. R986–R988. [DOI] [PubMed] [Google Scholar]
- 10.Clauson C, Scharer OD, and Niedernhofer L, Advances in understanding the complex mechanisms of DNA interstrand cross-link repair. Cold Spring Harb Perspect Biol, 2013. 5(10): p. a012732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kottemann MC and Smogorzewska A, Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature, 2013. 493(7432): p. 356–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.D’Andrea AD, Susceptibility pathways in Fanconi’s anemia and breast cancer. N Engl J Med, 2010. 362(20): p. 1909–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang H, et al. , TGF-beta Inhibition Rescues Hematopoietic Stem Cell Defects and Bone Marrow Failure in Fanconi Anemia. Cell Stem Cell, 2016. 18(5): p. 668–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Millan FA, et al. , Embryonic gene expression patterns of TGF beta 1, beta 2 and beta 3 suggest different developmental functions in vivo. Development, 1991. 111(1): p. 131–43. [DOI] [PubMed] [Google Scholar]
- 15.Worthington JJ, Klementowicz JE, and Travis MA, TGFbeta: a sleeping giant awoken by integrins. Trends Biochem Sci, 2011. 36(1): p. 47–54. [DOI] [PubMed] [Google Scholar]
- 16.Khalil N, et al. , TGF-beta 1, but not TGF-beta 2 or TGF-beta 3, is differentially present in epithelial cells of advanced pulmonary fibrosis: an immunohistochemical study. Am J Respir Cell Mol Biol, 1996. 14(2): p. 131–8. [DOI] [PubMed] [Google Scholar]
- 17.Willis BC, et al. , Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: potential role in idiopathic pulmonary fibrosis. Am J Pathol, 2005. 166(5): p. 1321–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Agarwal A, et al. , Bone marrow fibrosis in primary myelofibrosis: pathogenic mechanisms and the role of TGF-beta. Stem Cell Investig, 2016. 3: p. 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vannucchi AM, et al. , A pathobiologic pathway linking thrombopoietin, GATA-1, and TGF-beta1 in the development of myelofibrosis. Blood, 2005. 105(9): p. 3493–501. [DOI] [PubMed] [Google Scholar]
- 20.Bachegowda L, et al. , Signal transduction inhibitors in treatment of myelodysplastic syndromes. J Hematol Oncol, 2013. 6: p. 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gong Y, et al. , Megakaryocyte-derived excessive transforming growth factor beta1 inhibits proliferation of normal hematopoietic stem cells in acute myeloid leukemia. Exp Hematol, 2018. 60: p. 40–46 e2. [DOI] [PubMed] [Google Scholar]
- 22.David CJ and Massague J, Contextual determinants of TGFbeta action in development, immunity and cancer. Nat Rev Mol Cell Biol, 2018. 19(7): p. 419–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anido J, et al. , TGF-beta Receptor Inhibitors Target the CD44(high)/Id1(high) Glioma-Initiating Cell Population in Human Glioblastoma. Cancer Cell, 2010. 18(6): p. 655–68. [DOI] [PubMed] [Google Scholar]
- 24.Naka K, et al. , TGF-beta-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature, 2010. 463(7281): p. 676–80. [DOI] [PubMed] [Google Scholar]
- 25.Xie F, et al. , FAF1 phosphorylation by AKT accumulates TGF-beta type II receptor and drives breast cancer metastasis. Nat Commun, 2017. 8: p. 15021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bogdahn U, et al. , Targeted therapy for high-grade glioma with the TGF-beta2 inhibitor trabedersen: results of a randomized and controlled phase IIb study. Neuro Oncol, 2011. 13(1): p. 132–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ikeda M, et al. , Phase 1b study of galunisertib in combination with gemcitabine in Japanese patients with metastatic or locally advanced pancreatic cancer. Cancer Chemother Pharmacol, 2017. 79(6): p. 1169–1177. [DOI] [PubMed] [Google Scholar]
- 28.Herbertz S, et al. , Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des Devel Ther, 2015. 9: p. 4479–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Komrokji R, et al. , Sotatercept with long-term extension for the treatment of anaemia in patients with lower-risk myelodysplastic syndromes: a phase 2, dose-ranging trial. Lancet Haematol, 2018. 5(2): p. e63–e72. [DOI] [PubMed] [Google Scholar]
- 30.Habashi JP, et al. , Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science, 2006. 312(5770): p. 117–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bialkowski L, et al. , Immune checkpoint blockade combined with IL-6 and TGF-beta inhibition improves the therapeutic outcome of mRNA-based immunotherapy. Int J Cancer, 2018. 143(3): p. 686–698. [DOI] [PubMed] [Google Scholar]
- 32.Mariathasan S, et al. , TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature, 2018. 554(7693): p. 544–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Strauss J, et al. , Phase I Trial of M7824 (MSB0011359C), a Bifunctional Fusion Protein Targeting PD-L1 and TGFbeta, in Advanced Solid Tumors. Clin Cancer Res, 2018. 24(6): p. 1287–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao X, et al. , TGF-beta inhibitors stimulate red blood cell production by enhancing self-renewal of BFU-E erythroid progenitors. Blood, 2016. 128(23): p. 2637–2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bragado P, et al. , TGF-beta2 dictates disseminated tumour cell fate in target organs through TGF-beta-RIII and p38alpha/beta signalling. Nat Cell Biol, 2013. 15(11): p. 1351–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Langer JC, et al. , Quantitative trait analysis reveals transforming growth factor-beta2 as a positive regulator of early hematopoietic progenitor and stem cell function. J Exp Med, 2004. 199(1): p. 5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thwaites M KJ, Tremblay G, O’Connor-McCourt M, AVID200: A Novel TGF-β Inhibitor for the Treatment of Anemia Associated with Myelodysplastic Syndromes. Blood 2017. 130(Suppl 1): p. 2532. [Google Scholar]
- 38.Joyce CE, et al. , TGFbeta signaling underlies hematopoietic dysfunction and bone marrow failure in Shwachman-Diamond Syndrome. J Clin Invest, 2019. 129(9): p. 3821–3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garaycoechea JI, et al. , Genotoxic consequences of endogenous aldehydes on mouse haematopoietic stem cell function. Nature, 2012. 489(7417): p. 571–5. [DOI] [PubMed] [Google Scholar]
- 40.Garaycoechea JI, et al. , Alcohol and endogenous aldehydes damage chromosomes and mutate stem cells. Nature, 2018. 553(7687): p. 171–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Adamo A, et al. , Preventing nonhomologous end joining suppresses DNA repair defects of Fanconi anemia. Mol Cell, 2010. 39(1): p. 25–35. [DOI] [PubMed] [Google Scholar]
- 42.Pace P, et al. , Ku70 corrupts DNA repair in the absence of the Fanconi anemia pathway. Science, 2010. 329(5988): p. 219–23. [DOI] [PubMed] [Google Scholar]
- 43.Walter D, et al. , Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature, 2015. 520(7548): p. 549–52. [DOI] [PubMed] [Google Scholar]
- 44.Ceccaldi R, et al. , Homologous-recombination-deficient tumours are dependent on Poltheta-mediated repair. Nature, 2015. 518(7538): p. 258–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sallmyr A. and Tomkinson AE, Repair of DNA double-strand breaks by mammalian alternative end-joining pathways. J Biol Chem, 2018. 293(27): p. 10536–10546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ghezraoui H, et al. , 53BP1 cooperation with the REV7-shieldin complex underpins DNA structure-specific NHEJ. Nature, 2018. 560(7716): p. 122–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Noordermeer SM, et al. , The shieldin complex mediates 53BP1-dependent DNA repair. Nature, 2018. 560(7716): p. 117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dev H, et al. , Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat Cell Biol, 2018. 20(8): p. 954–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Akhurst RJ and Hata A, Targeting the TGFbeta signalling pathway in disease. Nat Rev Drug Discov, 2012. 11(10): p. 790–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bartram U, et al. , Double-outlet right ventricle and overriding tricuspid valve reflect disturbances of looping, myocardialization, endocardial cushion differentiation, and apoptosis in TGF-beta(2)-knockout mice. Circulation, 2001. 103(22): p. 2745–52. [DOI] [PubMed] [Google Scholar]
- 51.Shull MM, et al. , Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature, 1992. 359(6397): p. 693–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Anderton MJ, et al. , Induction of heart valve lesions by small-molecule ALK5 inhibitors. Toxicol Pathol, 2011. 39(6): p. 916–24. [DOI] [PubMed] [Google Scholar]
- 53.Barcellos-Hoff MH and Cucinotta FA, New tricks for an old fox: impact of TGFbeta on the DNA damage response and genomic stability. Sci Signal, 2014. 7(341): p. re5. [DOI] [PubMed] [Google Scholar]
- 54.Pal D, et al. , TGF-beta reduces DNA ds-break repair mechanisms to heighten genetic diversity and adaptability of CD44+/CD24- cancer cells. Elife, 2017. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kanamoto T, et al. , Functional proteomics of transforming growth factor-beta1-stimulated Mv1Lu epithelial cells: Rad51 as a target of TGFbeta1-dependent regulation of DNA repair. EMBO J, 2002. 21(5): p. 1219–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hockemeyer D. and Collins K, Control of telomerase action at human telomeres. Nat Struct Mol Biol, 2015. 22(11): p. 848–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Related to Figure 1. AVID200 inhibits TGFβ pathway signaling in primary mouse bone marrow cells. A) Reduced activation of the TGFβ pathway by AVID200 in mouse bone marrow progenitors. Murine bone marrow Lin− progenitors were exposed to TGFβ1 (5 ng/mL) along with AVID200 (0.2 ng/mL) or 1D11 (10 μg/mL) as a positive control and the cells were stained with phospho-SMAD2/3 antibody. Phospho-SMAD2/3 staining was then detected by flow cytometry. Representative FACS plots for staining in wild-type (WT) cells are shown. B) Quantitation of the staining intensity as mean fluorescence value (MFI) in the Lin− cells from WT and Fancd2−/− mice is shown in panel B. C) Raw total colony numbers from colony formation assay shown in Figure 1B. Lin− cells from wild-type (WT) or Fancd2−/− mice were cultured for 7 days in methylcellulose medium with inhibitors of the TGFβ pathway, namely, SD208 (10 μM), 1D11 (10 μg/mL) and AVID200 (0.2 ng/mL). Hematopoietic colonies were quantified (n=3). D) Cell numbers after solubilization of hematopoietic colonies in methylcellulose medium. Lin− cells from WT and Fancd2−/− mice were cultured in methylcellulose medium for 7 days in the presence or absence of AVID200 (0.2 ng/mL), colonies quantified, and methylcellulose was solubilized with PBS. The number of cells per well was quantified and plotted (n=3). p-values of 0.01 to 0.05 were considered significant (*) and p-values of 0.001 to 0.01 were considered very significant (**). Data in (C) are represented as mean ± SEM, data in (D) are presented as boxplots.
Supplementary Figure 2. Related to Figure 2. AVID200 enhances clonogenic growth of myeloid and erythroid progenitors from bone marrow of FA patients. A) FANCD2 gene expression analysis by Real time PCR showing the knockdown efficiency of sh1 FANCD2 and sh3 FANCD2 in human cord blood CD34+ cells. Error bars represent S.E.M. n=3. B) Raw total colony numbers from colony formation assay for primary human FA-like CD34+ cells shown in Figure 2A. Human FA-like CD34+ cells were then exposed to increasing concentrations of AVID200 (0.06, 0.2, 0.6, and 1.8 ng/mL) or SD208 (1 μM), as a positive control of TGFβ pathway inhibition, colony formation was assessed after 14 days of culture in methylcellulose medium (n=3). C) Cell numbers after solubilization of hematopoietic colonies in methylcellulose medium. FA-like CD34+ cells were cultured in methylcellulose medium for 14 days in the presence or absence of AVID220 (0.2 ng/mL), colonies quantified, and methylcellulose was solubilized with PBS. The number of cells per well was quantified and plotted (n=3). D) Total TGFβ1, TGFβ2 and TGFβ3 ligand levels in the bone marrow plasma of FA patients (n=12) and normal donors (n=5). Levels of total TGFβ1 and TGFβ2 are significantly reduced in BM plasma from FA patients, whereas levels of total TGFβ3 are significantly increased in FA patients. E) AVID200 promotes the proliferation of bone marrow progenitors from FA patients. Progenitor cells isolated from bone marrow of 9 FA patients and 3 healthy donors were cultured in StemSpan medium with TPO (100 ng/ml) (AF-300–18, Peprotech), Flt-3 (100 ng/ml) (AF-300–19, Peprotech), SCF (100 ng/ml) (AF-300–07, Peprotech) and IL-6 (20 ng/ml) (AF-200–06, Peprotech) for 7 days and live cells were counted. Note that healthy bone marrow cells did not respond or had a poor response to AVID200 treatment. sm= single measurement in samples with very few progenitors. ns=non-significant. F) Primary progenitor cells isolated from bone marrow of 20 FA patients or 3 healthy donors were cultured in triplicates in complete methylcellulose medium with or without AVID200 (0.2 ng/mL) for 14 days. Total number of hematopoietic colonies (total CFU) including myeloid colonies as well as erythroid colonies are shown per sample. G) Myeloid CFU-C production of bone marrow from individual FA patients or normal donor in response to AVID200 after 14 days of culture in complete methylcellulose medium with or without AVID200 (0.2 ng/mL). H) Erythroid CFU-C production of bone marrow from individual FA patient or normal donor in response to AVID200 after 14 days of culture in complete methylcellulose medium with or without AVID200 (0.2 ng/mL). p-values of 0.01 to 0.05 were considered significant (*), p-values of 0.001 to 0.01 were considered very significant (**) and p-values of <0.001 were considered extremely significant (***). Data in (A), (E), (F), (G) and (H) are presented as bar graphs, data in (B are presented as mean ± SEM, data in (D) are presented as boxplots.
Supplementary Figure 3. Related to Figure 3. AVID200 rescues genotoxicity in bone marrow progenitors from FA mice. A) Lin− cells from the bone marrow of wild-type or Fancg−/− mice were pretreated with AVID200 (0.2 ng/mL) and exposed to acetaldehyde (2 mM) for 4 h. The survival of cells was determined by clonogenic assay after culturing them in methylcellulose medium for 7 days (n=3). B) LSK cells from the bone marrow of wild-type or Fancd2−/− mice were pretreated with AVID200 (0.2 ng/mL), exposed to acetaldehyde (2 mM) for 4 h and then allowed to recover for 7 days in StemSpan medium enriched with cytokines SCF, Flt3 ligand, TPO and L-Glutamine. Survival of the cells was determined by counting the number of live cells after 7 days of culture (n=3). C) AVID200 reduces the amount of acetaldehyde-induced unrepaired single stranded DNA in bone marrow cells. LSK cells from bone marrow of wild-type (WT) or Fancd2−/− mice were exposed to acetaldehyde and BrdU along with AVID200 as indicated in the schematic in panel C and the amount of unrepaired single stranded DNA was measured by immunofluorescence as BrdU foci. Note that exposure to acetaldehyde increases the number of unrepaired single stranded DNA, measured as BrdU foci, in Fancd2−/− LSK cells. AVID200 reduces the amount of unrepaired single stranded DNA, as demonstrated by the reduction of BrdU foci. C) Representative images of BrdU foci in LSK cells are shown in panel D. At least 30 cells were counted for BrdU foci. E) AVID200 reduces pI:pC-induced DNA damage in LT-HSCs of Fancd2−/− mice. WT or Fancd2−/− mice were co-injected with pI:pC (5 mg/kg) and AVI200 (5 mg/kg), LT-HSCs were isolated after 48 h of the treatment and comet assay was performed to analyze DNA damage. Quantitation of the Olive tail moment in the LT-HSC is shown in panel E. F) Representative images of the pI:pC-induced alkaline comets in LT-HSCs are shown in panel F. p-values of 0.01 to 0.05 were considered significant (*), p-values of 0.001 to 0.01 were considered very significant (**) and p-values of <0.001 were considered extremely significant (***, ****). Data in (A) and (B) are presented as mean ± SEM, data in (C) and (E) are presented as boxplots.
Supplementary Figure 4. Related to Figure 4. AVID200 reduces the acetaldehyde-induced chromosomal aberrations in FANCG deficient lymphoblast cells. A) EUFA316+EV cells and EUFA316+FANCG cells were pre-treated with AVID200 before exposure to acetaldehyde. The cells were then allowed to recover, and metaphase spreads of chromosomes were scored for chromosomal abnormalities (n=3). Data are presented as bar graphs.