

SUMMARY
Genetically encoded fluorescent biosensors are powerful tools for monitoring biochemical activities in live cells, but their multiplexing capacity is limited by the available spectral space. We overcome this problem by developing a set of barcoding proteins that can generate over 100 barcodes and are spectrally separable from commonly used biosensors. Mixtures of barcoded cells expressing different biosensors are simultaneously imaged and analyzed by deep learning models to achieve massively multiplexed tracking of signaling events. Importantly, different biosensors in cell mixtures show highly coordinated activities, thus facilitating the delineation of their temporal relationship. Simultaneous tracking of multiple biosensors in the receptor tyrosine kinase signaling network reveals distinct mechanisms of effector adaptation, cell autonomous and non-autonomous effects of KRAS mutations, as well as complex interactions in the network. Biosensor barcoding presents a scalable method to expand multiplexing capabilities for deciphering the complexity of signaling networks and their interactions between cells.
Graphical abstract

In brief
Genetically encoded barcodes that uniquely identify cells expressing a particular fluorescent biosensor enable the simultaneous use of a broader set of biosensors in mixed cell populations and dynamic imaging of many cellular responses in a single experiment to reduce inter-experimental variability when interrogating complex biological interactions, such as the downstream consequences of receptor tyrosine kinase activation, with the help of image-based deep learning models for automated barcode unmixing.
INTRODUCTION
To understand the complex regulatory relationship between signaling, metabolic, and other biochemical activities in cells, it is often necessary to study their dynamics under a multitude of perturbation conditions. Genetically encoded fluorescent biosensors offer a versatile tool to continuously monitor a wide range of biochemical activities in live cells while revealing cell-to-cell variability that is masked in ensemble measurements (Newman et al., 2011; Zhang et al., 2002). A major drawback of fluorescent biosensors is their limited multiplexing capability due to the broad emission spectra of fluorescent proteins (FPs) and limited availability of spectral space (Welch et al., 2011). Efforts have been directed toward expanding the spectral range by developing far-red/infrared fluorophores, replacing two fluorophore biosensors (e.g., those based on fluorescence resonance energy transfer [FRET]) with single-fluorophore designs, and computational multiplexing to reveal the spatiotemporal relationship between biosensors (Chernov et al., 2017; Grant et al., 2008; Machacek et al., 2009; Marston et al., 2020; Mehta et al., 2018; Regot et al., 2014). Despite these improvements, no more than a handful of biosensors can be imaged concurrently. To overcome this limitation, we developed a “biosensor barcoding” method for highly multiplexed tracking of fluorescent biosensors. The key idea is to label cells with barcoding proteins made of different fluorophores targeted to distinct subcellular locations. Combinations of barcoding proteins are then used to generate barcodes that uniquely identify the cells expressing specific biosensors (Figures 1A–1C). To track multiple activities in parallel, mixed populations of barcoded cells are imaged in a time-lapse microscopy experiment. Activities from cells with the same barcode are pooled together to obtain the temporal profile of the corresponding biosensor (Figures 1D–1F; Video S1). Our method is compatible with any biosensor with emission wavelengths between 450 nm and 550 nm, including the majority of existing FRET or single fluorophore biosensors.
Figure 1. Schematic of biosensor barcoding.
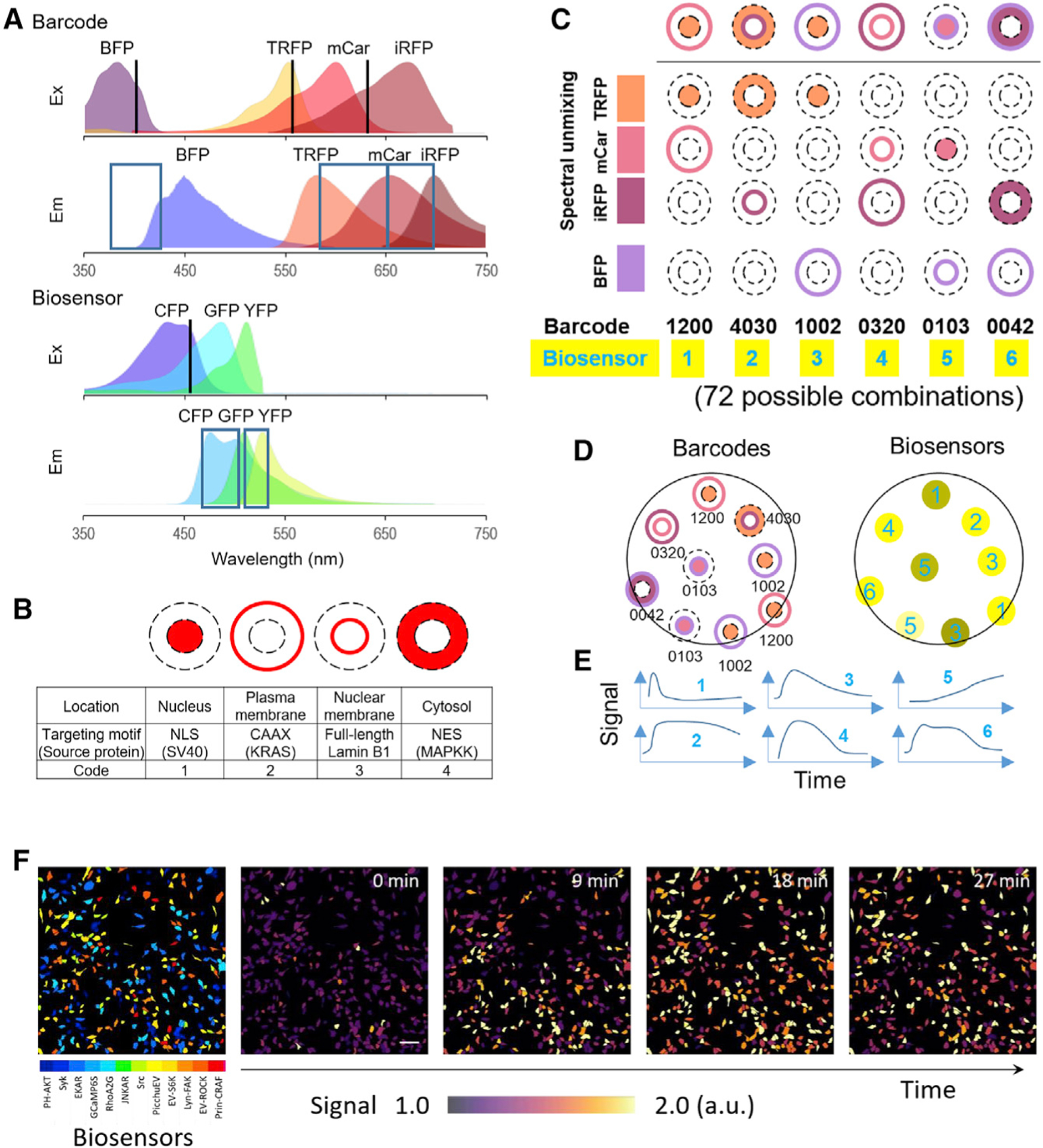
(A) Excitation (Ex) and emission (Em) spectra of barcoding proteins (EBFP2, TagRFP, mCardinal, and iRFP702) and biosensors. Excitation laser lines (405/561/633 nm for barcodes; 458 nm for biosensors) and the corresponding ranges of emission detection are indicated by black lines and blue boxes, respectively. The profiles of different FPs were generated using the FPbase Spectra Viewer (Lambert, 2019).
(B) Targeting sites for barcoding proteins.
(C) A pair of barcoding proteins (different colors, different targeting sites) are co-expressed with a fluorescent biosensor in the cell. Barcode digits from left to right represent: TagRFP, mCardinal, iRFP702, and BFP targeted to nucleus (1), plasma membrane (2), nuclear membrane (3), cytosol (4), or non-expressed (0). For example, 1200 denotes TagRFP at location 1 (nucleus), mCardinal at location 2 (plasma membrane), and no iRFP702 or BFP.
(D) Cells are mixed and imaged for barcodes using spectral detectors and for biosensors using CFP/YFP channels. The identity of the biosensor can be inferred by linear unmixing of the barcode spectral images.
(E) Activities corresponding to the same barcodes are averaged to obtain the temporal profile of each biosensor.
(F) Representative images of HeLa cells expressing different biosensors identified by barcodes and changes of biosensor signals over time upon treatment with EGF. Scale bar, 100 μm.
RESULTS
Barcoding cells
In theory, N different barcoding proteins can be combined to create 2N different barcodes, assuming binary expression (i.e., not expressed or expressed). In reality, the expression of barcoding proteins is a continuum, and low-expressors may be hard to distinguish from nonexpressors, thus precluding the use of all combinations. To ensure robust barcode identification, we only included combinations of two barcoding proteins that were (1) of different colors; and (2) targeted to different subcellular locations. Barcoding proteins also need to be spectrally separable from biosensors. A large number of fluorescent biosensors are based on detecting changes in (1) the intracellular localization or intensity of a single fluorophore, in many cases green FP (GFP), or (2) the FRET efficiency between a donor and an acceptor, most commonly cyan and yellow FPs (CFP and YFP) (Greenwald et al., 2018; Terai et al., 2019). Therefore, the cyan-green-yellow range of the emission spectrum (~450–550 nm) covers a vast number of existing fluorescent biosensors (Greenwald et al., 2018).
We first tested whether red or far-red FPs (550–700 nm; red FPs for brevity) targeted to different subcellular locations can be used as barcoding proteins. To this end, we constructed plasmids encoding sequences of TagRFP (Shaner et al., 2008), mCardinal (Chu et al., 2014), or iRFP702 (Shcherbakova and Verkhusha, 2013) fused to targeting motifs for localizing to (1) the nucleus; (2) the plasma membrane; (3) the nuclear membrane; or (4) the cytoplasm (Figures 1A and 1B). We took spectral images of HeLa cells expressing pairs of barcoding proteins under a confocal microscope equipped with a spectral detector. Knowing the reference emission profile of individual fluorophores, the contribution of each can be determined by linear unmixing (Figure S1A). To further increase the number of barcodes, we included BFP (EBFP2) (Ai et al., 2007) as the fourth fluorophore because (1) it is not excited by the 458 nm laser used for biosensors; and (2) a properly chosen spectral range (400–430 nm) detects the emission from BFP but not CFP used for biosensors (Figures 1A and S1B). Using four FPs targeted to four subcellular locations, a total of (4 × 3/2) × 4 × 3 = 72 barcodes can be generated.
Tracking multiple biosensor activities in barcoded cells
We selected 24 FRET or single-fluorophore biosensors that report the activities of 14 distinct targets in various subcellular compartments, including kinases (AMPK, ERK, p38, JNK, PKC, FAK, Src, and PI3K), G-proteins (Gαi1, Gαi2, and Gαi3), calcineurin, RhoA GTPase, and calcium (Table S1). We coexpressed each biosensor with a unique pair of barcoding proteins in HeLa cells by transient transfection. Cells with different biosensor/barcode combinations were then mixed and imaged simultaneously. We obtained the activities for individual biosensors by averaging across cells with corresponding barcodes, identified by linear unmixing of spectral images as described above. We stimulated cells with six pharmacological agents including 2-deoxyglucose (2DG), anisomycin, epidermal growth factor (EGF), ionomycin, phorbol-12,13-dibutyrate (PDBu), and UK14304, which have been used to test these biosensors in previous studies (Table S1). We analyzed the responses of all 24 biosensors to the agents and vehicle control (Figure 2A; Table S2). The averaged biosensor activities responded to previously reported stimuli (Figure 2A, black boxes), but wide variations were noted between different cells expressing the same biosensor (Figure 2B). Moreover, different biosensors that detect the same molecular activity may display distinct kinetics. For example, translocation-based ERKKTR (Regot et al., 2014) and FRET-based EKAR (Harvey et al., 2008) both report ERK activation, but the kinetics of ERKKTR was delayed compared to that of EKAR, consistent with previous observations (Figure 2C) (Pargett et al., 2017; Yang et al., 2018).
Figure 2. Multiplexed real-time tracking of biosensor activities in barcoded cells.

(A) Mixed population of barcoded HeLa cells expressing 24 FRET (blue) and non-FRET (green) biosensors (Table S1) were stimulated with the indicated activators (2DG, 10 mM 2-deoxyglucose; ANI, 1 μg/mL anisomycin; EGF, 100 ng/mL EGF; ION, 1 μM ionomycin; PDB, 200 nM phorbol-12,13-dibutyrate; and UK, 10 μM UK14304). For each barcode, the activities were normalized to pre-stimulus levels across cells of the corresponding barcode and adjusted for its dynamic range (Table S2). Black boxes indicate expected responses to known activators. Red boxes indicate additional responses.
(B) Individual cell traces of EKAR and ERKKTR activities normalized to pre-stimulus levels. Thick blue lines represent the average activities.
(C) Comparison of activation kinetics of EKAR and ERKKTR obtained from mixed barcoded cells. Activities are scaled to the peak responses and represent the mean of 46 (EKAR) and 22 (EKRKTR) cells.
See also Table S3.
To validate the results derived from mixed barcoded cells, we compared the responses to those from a homogeneous population of cells expressing single biosensors. The kinetics of the responses obtained from single-cell populations were in general agreement with those from mixed-cell populations (Table S2). Because all responses initiated between 3–9 min after stimulation, we compared their magnitude at 9 min and found that for the majority (75%) of biosensors, no significant difference was noted between the two groups (Table S2). Analysis of responses 15 min after stimulation produced a similar result (Table S2). For biosensors that showed statistically significant differences, the responses were smaller in the mixed population group in about half of the cases and larger in the remainder. Such variability is expected among experimental replicates with identical protocols (see the section “Synchrony among biosensors in barcoded cell mixtures” below). To rule out cross-transfection between different barcoded cells (e.g., through carryover of transfection material orexosomes), we mixed cells transfected with single barcoding proteins of different colors and targeting sites. In over 600 cells, no instance of double expression was found, suggesting that cross-transfection is an extremely unlikely event (Figures S1C and S1D).
In addition to the responses to known activators, our analyses revealed unanticipated responses, such as ionomycin-induced activation of AMPK, JNK, p38, RhoA, and PKC, as well as PDBu-induced activation of ERK and p38 (Figure 2A, red boxes). These observations are supported by earlier reports based on a variety of assays in different cell types (Table S3). Taken together, these results demonstrate that (1) the barcodes allow for correct identification of cells expressing different biosensors and are compatible with biosensor imaging without affecting biosensor responses; (2) the technique reveals variations between cells and kinetics of biosensors; and (3) multiplexed biosensor imaging can facilitate comprehensive identification and kinetic characterization of multiple cellular activities.
Deep learning models for barcode reading
To facilitate the identification of barcoded cells, we developed deep learning models for automated barcode reading. Using deep convolutional neural network models trained on curated images, we classified a testing set of linearly unmixed images of cells expressing pairs of barcoding proteins. The average accuracy was 97%, but varied from 50% to 100% for individual barcodes mostly due to suboptimal unmixing of two red FPs. To overcome this problem, we adopted a new barcoding strategy by pairing BFP with one of the three red FPs targeted to any of the four subcellular locations, generating a total of 4 × 4 × 3 = 48 different barcodes. We replaced TagRFP with mCherry, which was brighter but harder to unmix with mCardinal in the original barcoding scheme (Figure 3A).
Figure 3. Barcode reading using deep learning models.
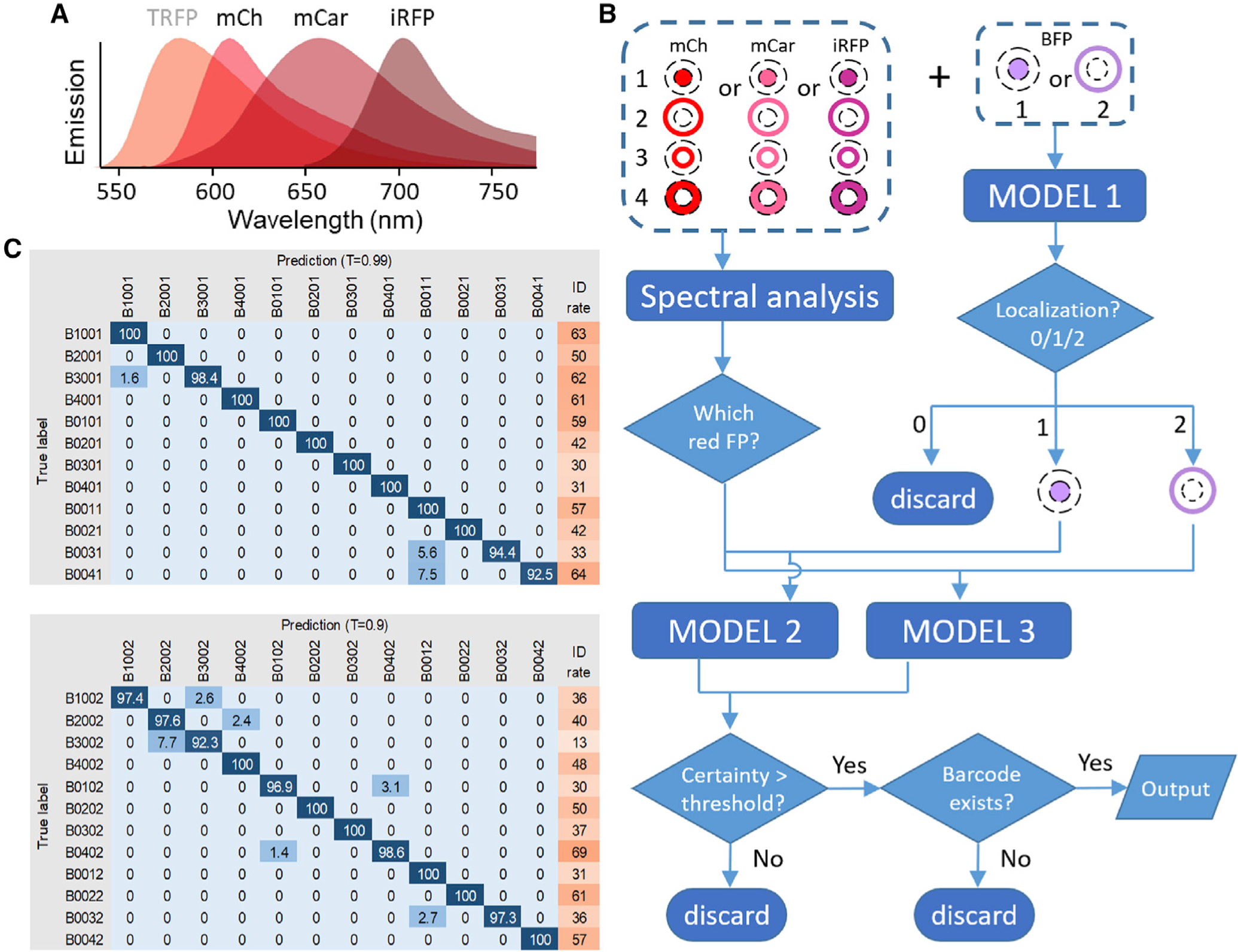
(A) Emission spectra of mCherry (mCh), mCardinal (mCar), and iRFP702 (iRFP) used for barcode reading by deep learning models. TagRFP (TRFP), used in the original barcoding scheme (Figure 1), was replaced by mCherry.
(B) Cells were barcoded by one of the red FPs targeted to four subcellular locations plus BFP targeted to the nucleus or the plasma membrane. The BFP image was classified by model 1. For the red barcoding proteins, spectral profiles were used to determine which of the three unmixed channels corresponded to the correct FP. The unmixed red FP image was then combined with BFP image and classified by model 2 or model 3 depending on whether BFP signal was in the nucleus or membrane. The output was filtered by a certainty threshold and checked for the existence of the barcode.
(C) The confusion matrix and identification rate (%) for all barcodes identified by model 2 (top) and model 3 (bottom) under indicated certainty thresholds. In general, higher certainty thresholds lead to increased accuracy but decreased identification rate. The four numbers in the barcode denote mCherry, mCardinal, iRFP702, and BFP localized to the nucleus (1), plasma membrane (2), nuclear membrane (3), cytosol (4), or non-expressed (0), respectively.
To test the new barcoding scheme for classification by deep learning models, we coexpressed BFP targeted to either the nucleus or plasma membrane along with a red FP targeted to four different subcellular locations in HeLa cells. Spectral images were acquired and unmixed using the reference spectra of mCherry, mCardinal, and iRFP702 to determine the identity of the red FP. The BFP image, once classified, could serve as a reference for red-FP localization. Therefore, we trained three deep learning models to classify (1) BFP images; (2) red FP images combined with nuclear BFP; and (3) red FP images combined with plasma membrane BFP (Figure 3B). We tested the performance of these models on images of barcoded cells and achieved an average accuracy of 99% (range, 92.5%–100%) (Figure 3C). The cause of misidentification included saturating or dim fluorescence signals, the presence of a second cell in the region selected for analysis, and unhealthy or apoptotic cells. The time to analyze one imaging experiment (~300 cells) was less than 10 s using deep learning models (Intel Core i5-7500 CPU @ 3.40 GHz, 3.41 GHz, 32 GB RAM). For comparison, human reading of the same number of barcodes took ~2 h. Together, these results demonstrated that deep learning models can achieve high efficiency and accuracy in barcode reading.
Expanding the barcoding toolkit
We next sought to extend the success with barcoding HeLa cells to other cell lines, including 293T, MCF7, U87MG, SiHa, U2OS, and A6L, which varied in morphology and transfection efficiency. High transfection efficiency was achieved in 293T, MCF7, and U2OS cells. Most barcodes in these cells were readily identifiable (Figures 4A, 4B, and S2A). Interestingly, in some U2OS cells, plasma membrane-targeted FPs appeared as a broad “haze” that resembled cytosolic signals (e.g., the D2E3 cell in Figure S2A). 3D reconstruction of z-stacks from these cells revealed a “cowboy hat” morphology with extended protrusion of the membrane around the cell periphery (Figure S2B). Although the broad membrane signals might appear cytosolic when examined in isolation, they were still distinct from the cytosolic signal in the same cell (e.g., the D2E4 cell in Figure S2A).
Figure 4. Expanding the barcoding toolkit.
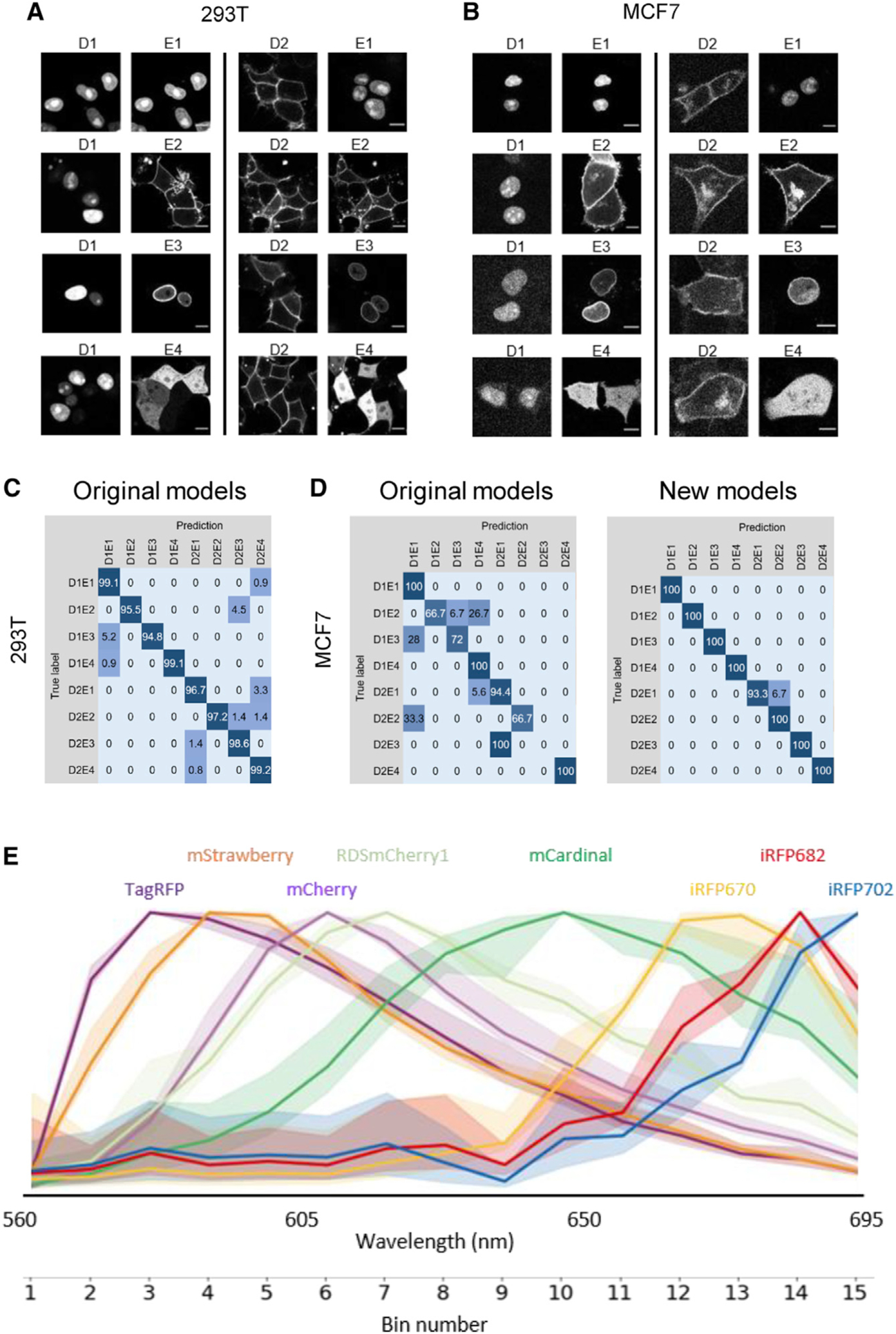
(A and B) 293T and MCF7 cells transfected with BFP targeted to the nucleus (D1) or plasma membrane (D2) along with mCherry targeted to one of the four subcellular locations (E1, nucleus; E2, plasma membrane; E3, nuclear membrane; and E4, cytosol). Scale bars, 10 μm.
(C) Confusion matrix (%) of 293T cells barcoded with nuclear or cytoplasmic BFP plus mCherry targeted to one of the four subcellular locations using the deep learning models trained on HeLa cells.
(D) Confusion matrix (%) of barcoded MCF7 cells using the deep learning models trained on HeLa cells (original models, left) and new models trained on MCF7 cell images (right). See also Figure S2 for U2OS cells.
(E) Emission profiles of red FPs acquired on a Zeiss LSM 780 microscope equipped with a GaAsP detector. For each fluorophore, the curve represents the mean of 100 HeLa cells expressing the nucleus-targeted version of the FP, with the full range of variation shown as shaded area. See also Table S4.
We applied deep learning models to classify images of barcoded 293T, MCF7, and U2OS cells. The models trained on HeLa images correctly identified 97.8% (range, 94.8%–100%) of barcoded 293T cells (Figure 4C). However, the accuracy was lower for MCF7 (90.1%) and U2OS (81.4%) (Figures 4D and S2C), in part due to the aforementioned broad membrane signals. We therefore trained new models using MCF7 and U2OS images, which achieved accuracy of 99.2% (range, 93.3%–100%) and 98.1% (range, 92.7%–100%) for MCF7 and U2OS cells, respectively (Figures 4D and S2C). These results demonstrate that barcode reading by deep learning models can be generalized to different cell lines.
To overcome the low transfection efficiency in U87MG, SiHa, and A6L cells, we generated lentiviral vectors encoding barcoding proteins using the Gateway recombination system (Figure S3A). Co-transduction of these cells with pairs of lentiviruses led to robust expression of barcoding proteins that were localized to the expected subcellular sites (Figures S3B–S3D). In addition to barcoding a broader range of cell lines, the more stable expression of lentiviral vectors compared to transient transfections will allow for long-term imaging experiments.
We reasoned that the number of barcodes in the new scheme can be easily scaled up with additional red FPs that are spectrally separable. In this scheme, red FPs were identified by their peak emissions based on the spectral profile acquired using a Zeiss GaAsP detector in 15 bins (of 9 nm each) between 560 and 695 nm. For example, the peaks of mCherry, mCardinal, and iRFP702 fell within bins 6, 10, and 15, respectively (Figure 4E). Using the spectral information from FPbase (Lambert, 2019) as a guide, we selected additional red FPs including LSS-mKate2, mStrawberry, RDSmCherry1, mPlum, mNeptune, mNeptune2.5, iRFP670, and iRFP682 for testing and acquired their emission profiles by imaging HeLa cells expressing nucleus-targeted versions of these FPs (Figure 4E). Eight of the FPs (TagRFP, mStrawberry, mCherry, RDSmCherry1, mCardinal, iRFP670, iRFP682, and iRFP702) showed distinct emission peaks in all cells examined (Figure 4E; Table S4). With four different subcellular localizations, the combination of BFP and one of the eight red FPs can generate a total of 4 × 4 × 8 = 128 different barcodes. Besides tracking an even greater number of biosensors, the additional fluorophores can compensate for the possible loss of usable subcellular locations in cells with more challenging morphologies.
Synchrony among biosensors in barcoded cell mixtures
A common problem associated with comparing or combining biosensor measurements from multiple experiments is the variability caused by factors such as subtle changes in cell density or temperature. We reasoned that such variability can be minimized by subjecting mixed populations of barcoded cells to a common physicochemical environment, allowing accurate comparison of different biosensors. To test this idea, we examined the results of ten experimental replicates in which barcoded cells expressing PH-AKT, EKAR, nuc-EKAR, Src, and Lyn-FAK biosensors were stimulated with EGF. Despite identical settings, variations in the timing and amplitude of the responses were noted (Figure 5A). We fitted individual traces to a biphasic exponential model to obtain the response initiation time (RIT) of the responses (Figures 5B and S4). Comparison of the RITs of individual experiments and those averaged across ten experiments indicated that different biosensors were synchronized in the same experiment despite significant variabilities between experiments (Figure 5C).
Figure 5. Synchrony between biosensors in mixed barcoded cells.
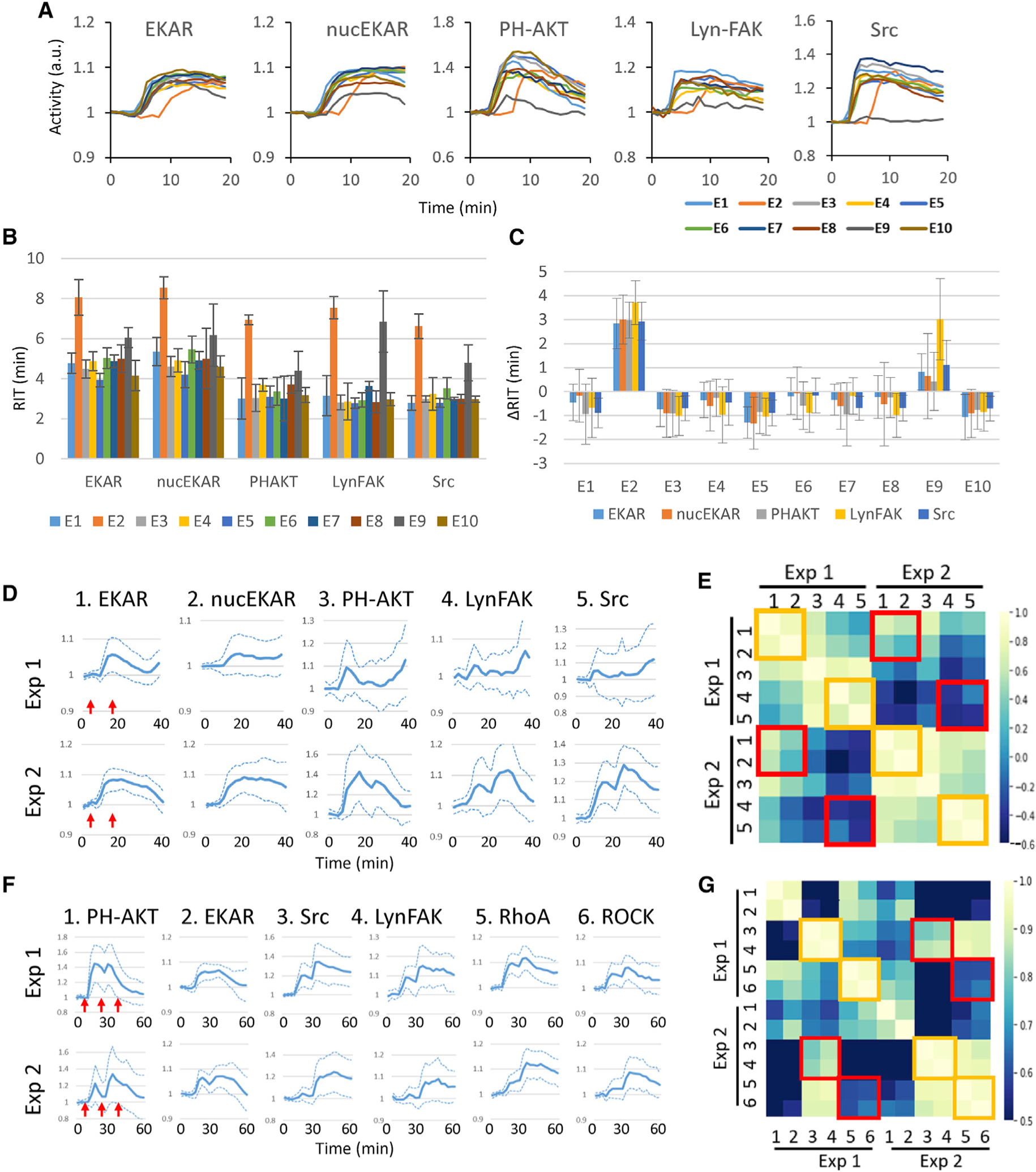
(A) Responses of biosensors obtained from mixed barcoded cells stimulated with 10 ng/mL EGF, added 2 min after imaging started. Traces of mean responses are shown in different colors for ten experimental repeats (E1–E10).
(B) Response initiation time (RIT) for each biosensor based on the traces in (A) (see STAR Methods and Figure S4). Error bars, 95% confidence interval.
(C) ΔRIT, defined as individual RIT minus the average RIT across the ten experiments for each biosensor. Error bars, 95% confidence interval.
(D) Biosensor responses from mixed barcoded cells stimulated with 0.5 and 10 ng/mL EGF at 4 and 18 min (red arrows), respectively. Traces of mean ± SD from two experiments are shown.
(E) Cross-correlation between the responses of different biosensors between the same and different experiments from (D).
(F) Biosensor responses from mixed barcoded cells stimulated with 1, 10, and 100 ng/mL EGF at 6, 21, and 36 min (red arrows), respectively. Traces of mean ± SD from two experiments are shown.
(G) Cross-correlation between the responses of different biosensors between the same and different experiments from (F). In (E) and (G), boxes indicate pairs of biosensors detecting the same proteins (ERK) or one protein and its immediate effector (Src/FAK or RhoA/ROCK) from the same (yellow boxes) or different (red boxes) experiments.
See also Data S1.
We next examined the responses to more complex stimulation schemes by treating barcoded cells expressing EKAR, nucEKAR, PH-AKT, Lyn-FAK, and the Src biosensor sequentially with 0.5 and 10 ng/mL EGF. In two experimental repeats, the response to the second stimulus was delayed in one but immediate in the other (Figure 5D). Cross-correlation analyses showed higher similarities within the same experiment than between experiments (Figure 5E). Notably, in the same experiment, highest correlation occurred between the two ERK biosensors, EKAR and nucEKAR, and between biosensors for FAK and Src, which are known to activate each other through direct interactions (Frame et al., 2010) (Figure 5E, yellow boxes). The correlation between these two pairs of biosensors was much lower across different experiments (Figure 5E, red boxes). In a second set of experiments in which cells were stimulated with 1, 10, and 100 ng/mL EGF (Figure 5F), a high correlation was noted between biosensors for Src and FAK and between those for RhoA (Fritz et al., 2013) and ROCK (Li et al., 2017) within the same experiment (Figure 5G, yellow boxes) but not between two experimental repeats (Figure 5G, red boxes). Note that ROCK is a downstream effector of RhoA through direct binding (Amano et al., 2010). Therefore, the strong correlation between biosensor pairs is consistent with known interactions between signaling proteins. Together, the above results suggest a high degree of synchronization among the activities of different biosensors in the same experiment and significant variations of the same biosensor across different experiments.
Interrogating the kinetics of signaling networks
We next investigated the responses of the receptor tyrosine kinase (RTK) signaling network to EGF and inhibitors. After testing 28 biosensors for their responsiveness to EGF (Table S1), we selected 10 responders that detected the activity of the RTK/EGF receptor (EGFR) as well as its multiple downstream effectors, including PI3K, Syk, ERK, calcium, RhoA, Src, S6K, FAK, and ROCK (Figure 6A). We used biosensor barcoding to simultaneously record their responses to 100 ng/mL EGF, and the results from two representative experiments are shown in Figure 6B. PicchuEV, an RTK biosensor (Komatsu et al., 2011), showed a gradual increase in activity before reaching a plateau at ~12 min after stimulation. Interestingly, the downstream biosensors displayed varying degrees of adaptation after reaching the peak level. Importantly, the adaptation of all downstream effectors except S6K started while PicchuEV activity was still increasing. To rule out the possibility that RTK inactivation was not detected by PicchuEV in real time, we treated EGF-stimulated cells with the EGFR inhibitor gefitinib and found immediate decrease in PicchuEV activity (Figure S5A). Therefore, the plateauing of the PicchuEV signal indicated continued RTK activation, and downstream adaptation was not caused by RTK inactivation.
Figure 6. Responses to RTK stimulation.
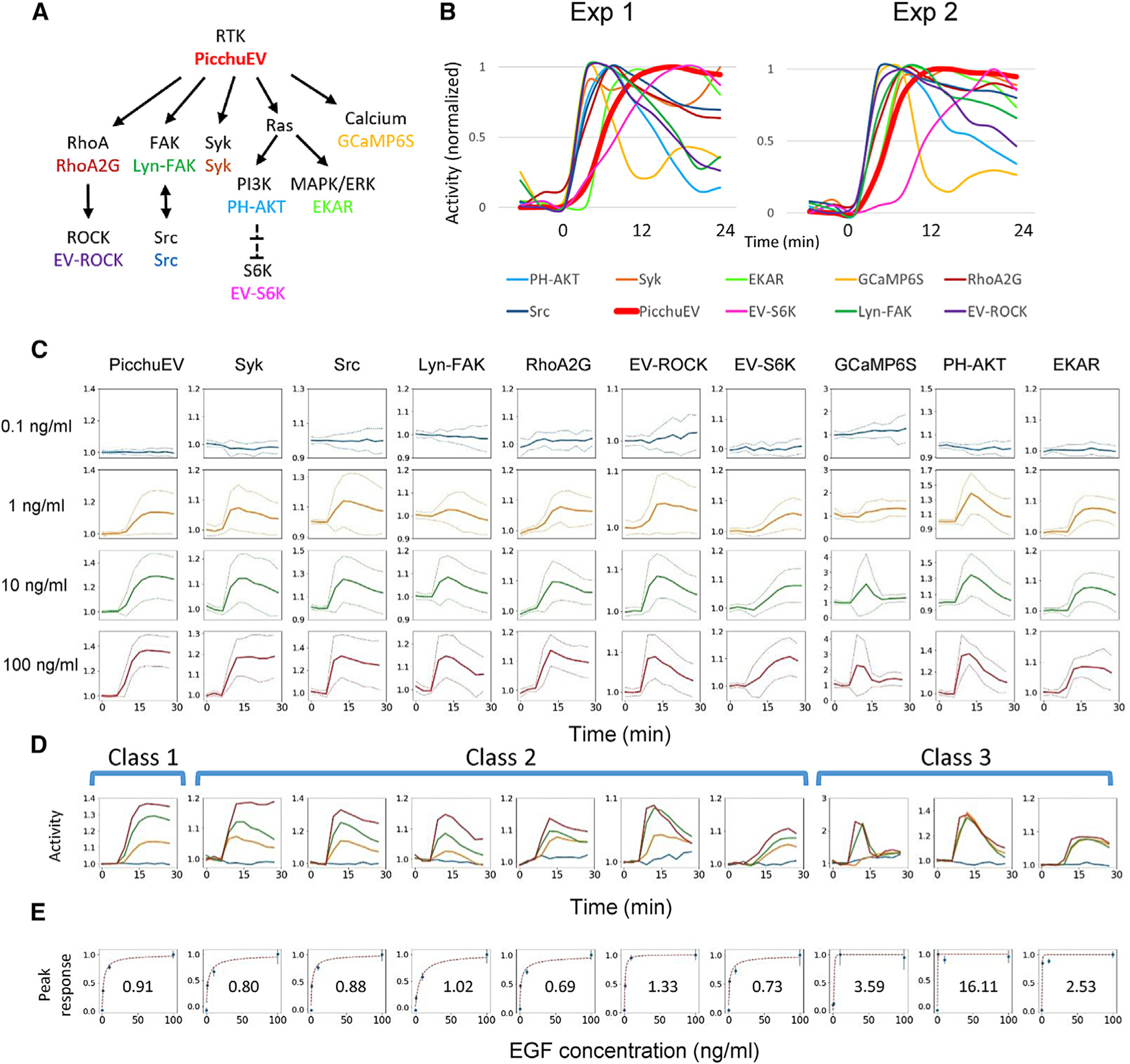
(A) 10 biosensors (in color) and their targets mapped to the RTK signaling network.
(B) Responses scaled to the peak levels of the 10 biosensors to 100 ng/mL EGF stimulation at 0 min in mixed barcoded cells from two independent experiments.
(C) Responses of biosensors in barcoded cells to four concentrations of EGF, applied at 6 min. The vertical axes represent biosensor activity (mean ± SD of cells from 5 independent experiments) normalized to the prestimulus levels
(D) Activities in (C) plotted on the same graph. The biosensors were grouped into three classes based on their kinetic and dose-response features (see text).
(E) Peak responses (mean ± SEM) at different EGF doses, normalized to the peak levels induced by 100 ng/mL EGF, fit to a Hill equation (red dashed line, with the Hill coefficient shown).
See also Figures S5 and S6.
We investigated the responses to different concentrations of EGF. As expected, higher EGF generally induced greater responses (Figure 6C). However, the temporal profile of the responses displayed unique features that fell into three classes: (1) stimulus-dependent plateau; (2) stimulus-dependent adaptation; and (3) all-or-none (Figure 6D). Class I included only one biosensor, PicchuEV, which reached a plateau level determined by the stimulus level. Class 2 included Syk, Src, FAK, RhoA2G, EV-ROCK, and S6K, which reached a stimulus-dependent peak level followed by adaptation at various rates. (Note that the seemingly non-adapting response of Syk at the highest EGF dose was caused by a delayed second peak induced by continued receptor activation in some experiments, e.g., Exp 1 in Figure 6B.) Class 3 included GCaMP6S, PH-AKT, and EKAR, which responded with a stimulus-independent peak to EGF above a threshold level. To determine the level of cooperativity for different responses, we fit the peak response versus EGF concentration to a Hill equation (Figure 6E). Whereas class 1 and 2 responses had Hill coefficients close to 1 and thus were noncooperative, class 3 responses showed strong cooperativity with high Hill coefficients, consistent with previous studies on PI3K and ERK signaling (Huang and Ferrell, 1996; Shindo et al., 2016; Zhan et al., 2020).
To check whether partial responses may result from averaging across all-or-none responders, we examined the responses of individual cells to escalating doses of EGF. For class 1 and 2 biosensors, the responses triggered by 1 ng/mL EGF were smaller than those triggered by 10 ng/mL EGF (Figure S5B; note that 100 ng/mL EGF failed to trigger a third response in most cells likely due to previous responses). In contrast, the responses of class 3 biosensors were independent of EGF doses. Therefore, the dose response features of different classes arose at the level of individual cells.
We noticed that EGF triggered transient spreading of cells driven by actin polymerization, as detected by recruitment of LifeAct (Riedl et al., 2008) to the protrusions (not shown). To test the possible involvement of actin in adaptation, we pretreated cells with jasplakinolide or latrunculin B, which stabilizes or inhibits actin polymerization, respectively. Neither jasplakinolide nor latrunculin B blocked the adaptation of class 2 and class 3 responses (Figures S5C and S5D), suggesting that actin dynamics are not required for adaptation. Interestingly, the responses of RhoA2G, EV-ROCK, and GCaMP6S were greatly diminished, suggesting a possible role of actin in RhoA, ROCK, and calcium activation.
We reasoned that the kinetic and dose response features of different biosensors reflect the topology of regulatory interactions. Specifically, in class 1 responses, the dose-dependent plateaus can be described by a simple binding model (Figure S6A). In class 2 responses, adaptation to continued stimulation can result from either negative feedback (NFB) or incoherent feedforward loop (IFFL) mechanisms (Hoeller et al., 2014; Ma et al., 2009). In both NFB and IFFL models, larger stimuli trigger responses with greater amplitude, but in IFFL, the rate of decay is faster in multiple simulation settings, leading to shorter overall responses (Iglesias and Shi, 2014; Tu and Rappel, 2018). Because class 2 responses did not show a stimulus-dependent change in the decay rate of adaptation (Figure 6D), we chose the NFB model in our simulation (Figure S6B), although an IFFL mechanism cannot be completely ruled out. Finally, for class 3 responses, the all-or-none features are characteristic of excitable networks, which generally involve a positive feedback, or autocatalytic loop, that drives a full response once a threshold is crossed, as well as a delayed negative feedback loop that shuts off the response (Xiong et al., 2010) (Figure S6C). We carried out computational simulations of mathematical models representing different network topologies. With appropriate selection of model parameters, we were able to recapitulate the experimentally observed dose response kinetics.
Delineating cell autonomous and non-autonomous interactions of signaling networks
The ability to barcode different cell populations in a mixture allows for the discrimination between cell autonomous and non-autonomous interactions. As a demonstration, we investigated the effects of mutant KRAS, one of the most frequent drivers of cancer. Taking advantage of the membrane localization of KRAS by farnesylation, we used BFP-fused KRAS as a barcoding protein. Expression of BFP-fused wild-type KRAS had no effect on the activity of the RAS effector ERK, whereas expression of BFP-fused oncogenic (G12V) and dominant-negative (S17N) mutants of KRAS led to persistently elevated and suppressed ERK activities, respectively, suggesting that BFP fusion did not affect the function of KRAS and its mutants (Figure S7A).
We used biosensor barcoding to examine the responses of HeLa cells expressing KRAS(G12V). Compared to the stereotypical EGF responses of control cells, KRAS(G12V)-expressing cells had smaller responses that were often followed by a second peak (Figure 7A) and showed significant variability between experiments (Figure S7B). Next, we mixed barcoded control and KRAS(G12V) cells before stimulation with EGF. Interestingly, the control cells in the mixture displayed smaller, biphasic responses (Figure 7B, top) with increased experimental variability (Figure S7C), suggesting cell non-autonomous effects mediated by KRAS(G12V) cells. The responses of KRAS(G12V) cells were similar regardless of the presence or absence of control cells (Figure 7B, bottom). We surmised that KRAS(G12V) may exert their effect through metalloproteinases of the MMP or ADAM families, which have been shown to release EGFR ligands that mediate the propagation of ERK activation across cell populations (Aoki et al., 2013). To test this hypothesis, we pretreated the control-KRAS(G12V) mixture with the MMP/ADAM inhibitor (MMPi) TAPI-1 before stimulation. Indeed, the effects of KRAS(G12V) cells on the control cells were largely eliminated (Figure 7C). Because cells in the same mix are synchronized (Figure 5), we reasoned that the “raw activity” of mixed control and KRAS(G12V) cells can be directly compared using non-normalized FP fluorescence (Figure 7D). As expected, EKAR showed a high basal activity that was not further activated by EGF in KRAS(G12V) cells, apparently due to direct activation by the constitutively active KRAS mutant. Interestingly, the activities were altered in several other biosensors in KRAS(G12V) cells. We verified the effects of KRAS(G12V) on FAK and ERK by immunoblotting (Figure S7D). Together, these results reveal the distinct effects of KRAS(G12V) on different nodes in the RTK signaling network through both cell-intrinsic and paracrine mechanisms.
Figure 7. Interactions between pathways and cells.
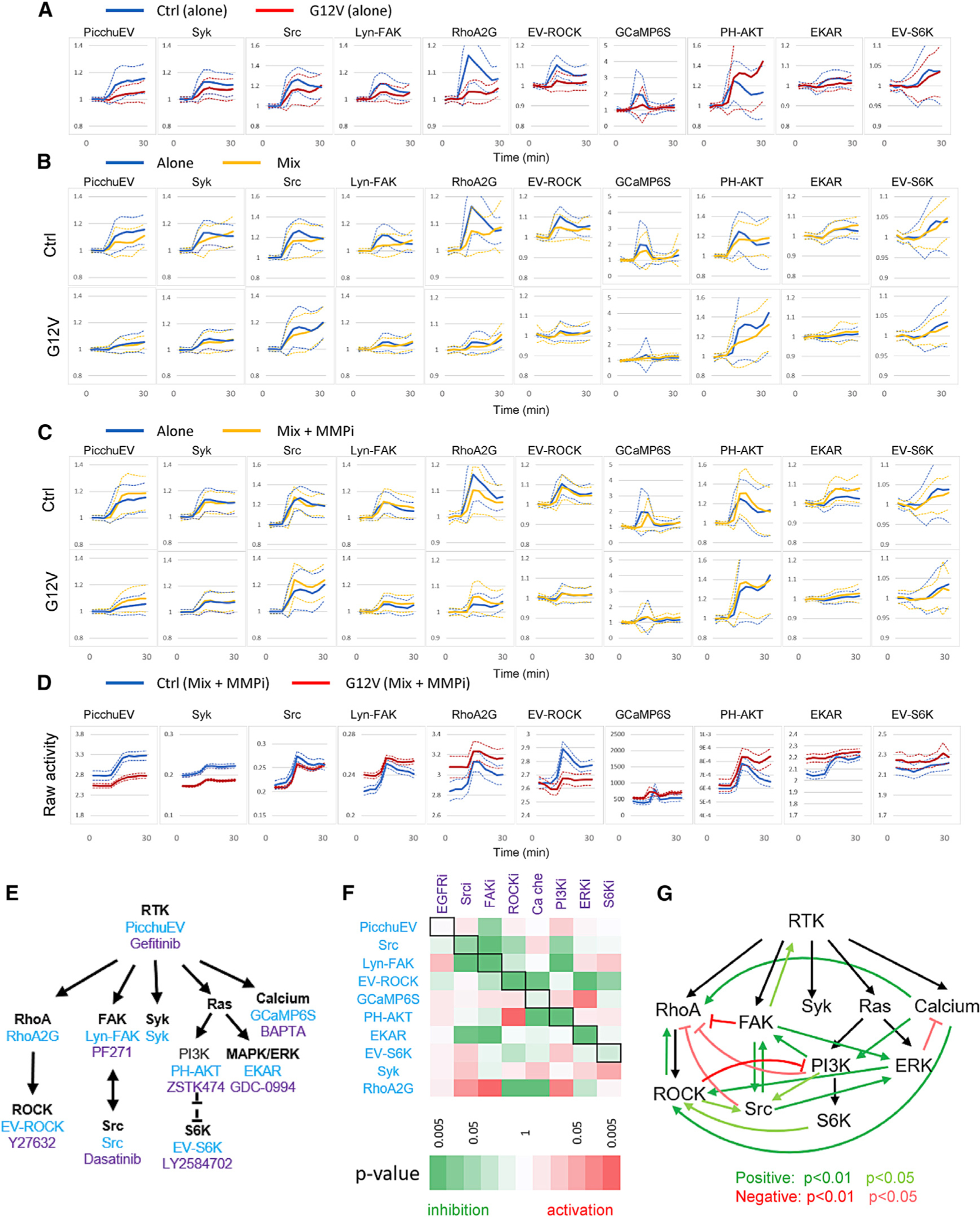
(A) Normalized responses (mean ± SD) of control cells alone and KRAS(G12V) cells alone to 100 ng/mL EGF stimulation added at 6 min.
(B) Normalized responses (mean ± SD) of control and KRAS(G12V) cells in a 1:1 mix to 100 ng/mL EGF (yellow). The responses of control and KRAS(G12V) cells when they were separately stimulated with 100 ng/mL EGF were plotted for comparison (blue). See also Figure S7.
(C) Normalized responses (mean ± SD) of control and KRAS(G12V) cells in a 1:1 mix to 100 ng/mL EGF with pretreatment of 10 μM TAPI-1 (MMPi, matrix metalloproteinase inhibitor) for 1 h prior to the experiments (yellow). The responses of control and KRAS(G12V) cells when they were separately stimulated with 100 ng/mL EGF were plotted for comparison (blue).
(D) Comparison of raw activities (mean ± SEM) of control and KRAS(G12V) cells in a 1:1 mix pretreated with 10 μM TAPI-1 (MMPi) and stimulated with 100 ng/mL EGF. The raw activities represent mean ± SEM of YFP/CFP (for PicchuEV, RhoA-2G, EV-ROCK, EKAR, and EV-S6K), CFP/YFP (Syk, Src, and Lyn-FAK), cytosolic fluorescence (GCaMP6S), or 1/cytosolic fluorescence (PH-AKT).
(E) Biosensors (blue) and inhibitors (purple) targeting the RTK signaling network.
(F) Inhibition matrix showing the effect of inhibitors on biosensor activities. The effect of an inhibitor on a biosensor was calculated by integrating the activity for all time points after the inhibitor was added minus that of DMSO control. Black boxes denote biosensors corresponding to the targets of inhibitors. Positive and negative effects are shown in red and green, respectively, and the statistical significance is represented by the color scale. See also Table S5.
(G) Feedback loops inferred from the inhibition matrix. Positive (green) or negative (red) interactions were assigned when an inhibitor caused an inhibition or activation, respectively, of another node in the network.
We interrogated the interactions within the RTK signaling network by tracking the biosensor responses to a panel of inhibitors targeting different nodes (Figure 7E; Table S5). To quantify the effects of each inhibitor, we integrated the changes in biosensor activity over time, and compared the effects to those of the vehicle control. In addition to the expected targets, several inhibitors altered the activities of other nodes in the network (Figure 7F). Inhibitory and activating effects of the inhibitors on each node suggest positive and negative interactions, respectively (Figure 7G). It should be noted that some of these interactions may be indirect or may result from off-target effects of the inhibitors, and are likely cell-type specific. Nevertheless, the results highlight the complex regulatory relationships between nodes in the RTK signaling network and represent a powerful strategy to discover unknown interactions within signaling networks in cells of interest. Together, our results demonstrated the power of biosensor barcoding in delineating both cell autonomous and non-autonomous interactions within signaling networks.
DISCUSSION
There has been an increasing need to track multiple biosensors simultaneously for various applications. In one study, three different cell populations expressing biosensors targeted to distinct subcellular locations were co-cultured for simultaneous imaging (Fujita et al., 2014). In this strategy, the multiplicity is limited to the number of sites that can be robustly distinguished in microscopic images. To our knowledge, the only generalizable approach for multiplexing biosensor imaging is the recently reported “signaling reporter islands” method, in which different biosensors are clustered at different spots in cells to allow for spatial separation of their signals (Linghu et al., 2020). However, the method is technically demanding, requiring laborious post-imaging sample processing and analysis to identify the biosensor in each cluster, and is incompatible with translocation based biosensors. The need to engineer and test individual scaffolds used for clustering also makes this method not easily scalable. More generally, one concern about expressing multiple biosensors in the same cells is the possible interaction or interference between the biosensors. Our biosensor barcoding method overcomes these problems by providing a simple way to simultaneously track large numbers of existing fluorescent biosensors, whether they are based on FRET, intensity, or translocation, as long as their emission falls within the CFP to YFP (450 nm to 550 nm) spectral range.
High-throughput imaging can also be used to track large numbers of biosensors in parallel (Chapnick et al., 2019; Kuchenov et al., 2016). However, we demonstrated substantial variation in biosensor activities obtained from physically segregated cell populations. Importantly, we found that mixing cells expressing different biosensors synchronized their responses to perturbations, as demonstrated by the coordinated activities within the same imaging experiment but not between experimental replicates. The cause of variation between experiments is unclear in most cases but likely involves a combination of subtle differences in physical, chemical, and biological factors such as temperature, cell density, nutrient levels, or metabolic states of cells. In addition to the synchronization of biosensor activities, the ability of our method to identify distinct cell populations in a mixture is hard to achieve using other approaches, and will find broad applications in studying cell-cell interactions.
Using biosensor barcoding, we found distinct kinetic properties of multiple downstream pathways in the RTK signaling network. In particular, the all-or-none responses of PH-AKT and EKAR to increasing EGF stimuli add to a growing list of evidence for excitability of the Ras-PI3K-ERK signaling network, including wave-like propagation of activities, refractoriness to repeated stimuli, and characteristic spatiotemporal responses to disruptions in wave patterns (De Simone et al., 2021; Fukushima et al., 2019; van Haastert et al., 2017; Hiratsuka et al., 2015; Yang et al., 2016, 2018; Zhan et al., 2020). Calcium waves have also been reported to display features of excitable systems in various tissues (Gelens et al., 2014). Interestingly, we found that calcium responses were also all-or-none but with a higher threshold compared to PI3K and ERK responses, suggesting that calcium signaling belongs to a different excitable network. Moreover, although extensive crosstalk exists between Ras-PI3K-ERK signaling and other downstream effectors of RTKs, the responses of FAK, Src, Syk, and Rho GTPase were not excitable but showed dose-dependent adaptation that likely involves negative feedback mechanisms. The molecular basis of the features displayed by different classes of effectors will be key to understanding the structure and function of the RTK signaling network, which is the target of many clinically approved therapeutics.
Given the ever-growing list of fluorescent biosensors, with a recent review listing over a thousand designs for ~170 cellular targets including enzymes, voltage, ions, and metabolites (Greenwald et al., 2018), we envision our method to find a wide range of applications, such as reconstructing complex molecular networks, delineating the signaling interaction between different cell types, and identifying molecular pathways targeted by pharmacological agents, some examples of which are shown in Figure 7. In addition, biosensor barcoding can facilitate the development of new biosensors through side-by-side comparison of the sensitivity and dynamic range of multiple biosensor designs. Newly developed biosensors will then further expand the biosensor barcoding technique. Although we only demonstrated the use of barcoding for CFP-YFP FRET- and GFP-based biosensors, our technique can be readily adapted to other types of fluorescent or bioluminescent biosensors with similar emission spectra.
Limitations of study
The rich kinetic data from simultaneous recording of signaling activities will inform the formulation of more comprehensive network models, which hold promise for predicting the responses to genetic or pharmacological perturbations with greater accuracy. In this study, we used simultaneous tracking of biosensors to construct mathematical models that captured salient features of the RTK signaling responses to different concentrations of EGF. For each downstream effector, a model of the receptor-effector pair was created and fitted using synchronized data from both, but different effectors were modeled separately. Similarly, an interaction map was built from the integrated response of biosensors to inhibitors, but a quantitative model describing the temporal changes of all measured responses has yet to be achieved. To take full advantage of the synchronized recording, it is theoretically possible to encompass all effectors in a comprehensive model that is constrained by the kinetic data. One hurdle is that as the number of interactions increases, the complexity of the model rises greatly and finding unique parameter values and model structures becomes difficult. Moreover, the models assume a linear correspondence between the biosensor signal and its target activity, which may not have been fully validated across the measured range of some biosensors. Nevertheless, we envision that the vast amount of data obtained from parallel recording of large numbers of biosensors upon various perturbations will motivate further development of computational tools for quantitative modeling of complex biological networks.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Chuan-Hsiang Huang (chaung29@jhmi.edu).
Materials availability
Plasmids generated in this study will be deposited to Addgene (https://www.addgene.org).
Data and code availability
All data supporting the findings of the current study are available within the article and its Supplemental Information files or from the corresponding authors upon reasonable request. The codes used in the study are provided as a ZIP file (Data S1).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
HeLa, HEK293T, MCF7, and U2OS cells, purchased from ATCC, were grown at 37°C and 5% CO2 in DMEM high glucose medium (GIBCO, #11965092) supplemented with 10% FBS (Corning Cellgro, 35–010-CV), 1 mM sodium pyruvate (GIBCO, #11360070), and 1X nonessential amino acids (GIBCO, #11140076). Transient transfections were performed using GenJet In Vitro DNA Transfection Reagent ver. II (SignaGen, #SL100499), GenJet In Vitro DNA Transfection Reagent for MCF7 cells (SignaGen, #SL100489-MCF7), or GenJet In Vitro DNA Transfection Reagent for U2OS cells (SignaGen, #SL100489-OS) following the manufacturer’s instructions. For biosensor barcoding, 2×105 cells were seeded in 12-well plates and allowed to attach overnight. Cells in each well were transfected with a pair of barcoding proteins and one biosensor, using a total of 0.75 μg plasmid per well. The next day, cells were harvested from each well with Accutase (Corning, 25–058-CI) and mixed together. The resulting cell mixture was then seeded into 35 mm glass-bottom dishes (Mattek, P35GC-0–14-C) at 7×105 cells per dish and incubated at 37°C and 5% CO2 overnight. The cells were starved in serum-free, phenol red-free DMEM (GIBCO, #21063029) for 1 h before imaging experiments.
METHOD DETAILS
Plasmids
Biosensors
Plasmids for biosensors were purchased from Addgene (see Table S1). ERKKTR, p38KTR and JNKKTR genes were further cloned into pEGFP-N1 (CloneTech) via XhoI/SaII and BamHI restriction sites using the following primers:
5′-CAAgtcgacATGAAGGGCCGAAAGCCTC-3′ and 5′-CAAggatccccGGATGGGAATTGAAAGCTGGACT-3′ for ERKKTR, 5′-CAA;ctcgagATGCGTAAGCCAGATCTCCG-3′ and 5′-CAAggatccccGCTGGACTGGAGGGTCAG-3′ for p38KTR; 5′-CAActcgagATGAGTAACCCTAAGATCCTAAAACAGAG-3′ and 5′-CAAggatccccGCTGGACTGGAGGGTCAG-3′ for JNKKTR.
Barcoding proteins
The original set of barcoding proteins were derived from five fluorescent proteins (designated A: TagRFP, B: mCardinal, C: iRFP702, D: BFP, and E: mCherry) linked to targeting sequences of four subcellular locations (designated 1: nucleus, 2: plasma membrane, 3: nuclear membrane, and 4: cytoplasm). Thus, a total of 16 barcoding proteins were generated, each represented by a letter-number combination (e.g., B1 indicates mCardinal targeted to the nucleus). To construct barcoding proteins, the following fragments were joined together by overlapping PCR: 1) fluorescent protein sequence; 2) spacer (TCTGGCAGCGGAGGCTCTGGAGGC); and 3) targeting sequence. The following plasmids were used as templates for the fluorescent proteins: H2B-TagRFP (Addgene #99271, a gift from Philipp Keller), mCardinal-H2B-C-10 (Addgene #56162) (Chu et al., 2014), piRFP702-N1 (Addgene #45456) (Shcherbakova and Verkhusha, 2013), EBFP2-Nucleus-7 (Addgene #55249, a gift from Michael Davidson), and pmCherry-NLS (Addgene #39319) (Micutkova et al., 2012). The targeting sequences for the nucleus, plasma membrane, nuclear membrane, and cytoplasm were derived from the NLS of SV40, CAAX of K-Ras, lamin B1, and the NES of MAPKK, respectively. The sequence ATGGGATGTATAAAATC AAAAGGGAAAGACAGC, derived from the Lyn kinase, was used as an alternative plasma membrane targeting motif in some cells. Additional fluorophores used for further expanding the barcoding proteins are shown in Table S4.
Chemical reagents
Stocks of 200 μM phorbol-12,13-dibutyrate (PDBu, EMD Millipore, #524390), 1 mg/mL anisomycin (Sigma-Aldrich, A9789), 10 mM UK14304 (Sigma-Aldrich, U104), 10 mM yohimbine (Sigma-Aldrich, Y3125), 1 mM gefitinib (Cayman, #13166), 1 mM ionomycin (Peprotech, #5608212), 2 mM jasplakinolide (Cayman, #11705), 25 mM latrunculin B (Enzo Life Sciences, BML-T110–0001), 10 mM PF562271 (AdipoGen, SYN-1064), 10 mM ZSTK474 (Cell Signaling, #13213), 10 mM dasatinib (Cayman, #11498), 10 mM GDC-0994 (APExBIO Technology, B5817), 1 mM LY2584702 (Selleck, S7698), 10 mM BAPTA-AM (Selleck, S7534) were prepared by dissolving the chemicals in DMSO; 10 mM Y27632 (Enzo Life Sciences, #ALX-270–333) in water. Stocks were diluted to the indicated final concentrations in the culture medium. The EGF stock solution was prepared by dissolving EGF (Sigma-Aldrich, E9644) in 10 mM acetic acid to a final concentration of 1 mg/ml. All drug stocks were stored at −20°C. 2-Deoxyglucose (2-DG, MilliporeSigma, #D8375) was dissolved in culture medium to 100 mM and used immediately.
Lentivirus production and transduction
Lentiviral plasmids were constructed using the Gateway Recombination Cloning Technology (Figure S3). Entry vectors were generated by integrating targeting sequence into pDONR221 (Invitrogen, #12536017) with Gateway BP Clonase ll Enzyme Mix (Invitrogen, #11789100). To generate fluorescent protein sequence-inserted destination vectors, the upstream sequence of the attR1 sequence of pLex307 (Addgene, #41392) was first modified from 5′-GCTAGCATCGATTGATCA-3′ to 5′-GCTAGCTTAATTAAGGGCATATGATCGATGGATCA-3′ to insert PacI and NheI restriction enzyme sites and to remove an in-frame stop codon in the BclI restriction enzyme site. The fluorescent protein sequence was subsequently cloned into the vector via PacI and NheI sites. The final expression vectors were generated by transferring the targeting sequence from the entry vector to the destination vector with Gateway LR Clonase ll Enzyme Mix (Invitrogen, #11791100).
For lentivirus production, 4×106 HEK293T were seeded in a 10-cm dish with DMEM supplemented with 10% FBS and allowed to attach overnight. Cells were transfected with 1.25 μg of pMD2.G (Addgene, #12259), 3.75 μg of psPAX2 (Addgene, #12260), and 5 μg of lentiviral plasmids using GenJet In Vitro DNA Transfection Reagent ver. II, and the cell culture medium was replaced with fresh medium 5 h after transfection. Lentiviral supernatants were harvested 24 h after transfection and filtered through a 0.45 μm PVDF filter (Millipore, #SLHVM33RS).
For lentiviral transduction of U87MG, SiHa, and A6L, cells were seeded into an 8-well chamber (2.5×104 cells) with DMEM supplemented with 10% FBS and allowed to attach overnight. Cells were transduced with lentivirus in medium containing 8 μg/ml polybrene (Millipore, #TR-1003-G). Cells were incubated at 37°C and 5% CO2 for 48 h before imaging.
Microscopy
Imaging experiments were carried out on a Zeiss LSM 780 or 880 single-point laser-scanning microscope (Zeiss AxioObserver with 780 or 880-Quasar confocal module; 34-channel spectral, high-sensitivity gallium arsenide phosphide (GaAsP) detectors) with a motorized stage for capturing multiple viewfields controlled by Zen software as previously described (Huang et al., 2013). Live-cell imaging was carried out in a temperature/humidity/CO2-regulated chamber. To image barcodes, spectral images for red-far red barcoding proteins were acquired between 560 and 695 nm at 8.9 nm windows using Lambda Mode under 561 nm and 633 nm illumination. Reference spectra for TagRFP, mCherry, mCardinal, and iRFP702 were acquired by imaging HeLa cells expressing H2B-TagRFP under 561 nm excitation, H2B-mCherry and H2B-mCardinal under both 561 and 633 nm excitation, and H2B-iRFP702 under 633 nm excitation. Since the BFP emission spectrum is well separated from those of the red-far red fluorophores (Figure 1A), no unmixing is required for BFP images, which were therefore acquired in the channel mode. To avoid bleedthrough from CFP and YFP used in FRET-based biosensors, we collected BFP emission in the 370–430 nm range under 405 nm excitation. To image biosensors, CFP (458–499 nm) and YFP (508–543 nm) emissions under 458 nm illumination were obtained. This setting, while optimized for detecting CFP-YFP FRET biosensors, also captures GFP-based biosensors due to the overlapping spectra of GFP and YFP. Using a single imaging setting for both types of biosensors is convenient, and it reduces cell exposure to illumination, therefore minimizing phototoxicity. Time-lapse images of the biosensors were taken at a rate of one frame every three minutes unless specified. Cells in 2 mL DMEM (GIBCO #21063029) were stimulated by adding signaling activators or inhibitors (200 μL for 2DG and 20 μL for all other reagents) at the indicated time points.
Image analysis
Analysis of barcodes by linear unmixing of spectral images
Using the linear unmixing function in ZEN Software, spectral images of cells expressing pairs of barcoding proteins acquired under 561 nm illumination were unmixed using TagRFP and mCardinal reference spectra, whereas spectral images acquired under 633 μm illumination were unmixed with mCardinal and iRFP702 spectra. The unmixed images for TagRFP, mCardinal, and iRFP702 as well as images of BFP were then combined using NIH ImageJ and Fiji (Schindelin et al., 2012; Schneider et al., 2012) and visually inspected for the expression and localization of each fluorophore. In the majority of cases (> 90%) the two expressed barcoding proteins could be unambiguously identified (Figure S1). Cells with ambiguous barcodes were excluded from the analysis of biosensors. For simplicity, barcodes were represented by four numbers that denoted the expression and location of TagRFP, mCardinal, iRFP702, and BFP (0: no expression; 1: nucleus; 2: plasma membrane, 3; nuclear membrane, and 4: cytoplasm; see Figure 1C). For the second barcoding scheme in which one of the three red FPs (mCherry, mCardinal, or iRFP702) was combined with BFP (Figure 3), spectral images acquired under 633 nm excitation were unmixed using the reference spectra of mCherry, mCardinal, and iRFP702 followed by processing in NIH ImageJ and Fiji as described above.
Analysis of biosensors in mixed populations of barcoded cells
Images of biosensors were processed and analyzed with NIH ImageJ and Fiji (Schindelin et al., 2012; Schneider et al., 2012). The barcode of each cell, determined as described above, allows for identification of the biosensor expressed by the cell. To measure the activities of FRET-based biosensors, the mean intensity of YFP over the entire cell was divided by that of CFP for each frame. For ERKKTR, p38KTR and JNKKTR, the mean intensity of YFP in the nucleus was divided by that of a cytoplasmic region. For PH-AKT, the mean intensity of YFP in an intracellular region was measured. For GCaMP6S and GCaMP6S-PM, the mean intensity of YFP over the entire cell was measured. The activities for every frame were then normalized to the average of those from the prestimulus frames. Normalized activities from cells with the same barcode were then pooled together to calculate the mean and standard deviation (SD) for the corresponding biosensor.
Barcode image classification by deep learning models
Image datasets
To create training and testing sets for deep learning models, we manually segmented processed barcode images (see Image Analysis above) comprising four channels (BFP, mCherry, mCardinal, and iRFP702), two of which contained “real” signals of an FP targeted to one of four distinct subcellular locations (the nucleus, plasma membrane, nuclear membrane, or cytosol), in ImageJ (Schindelin et al., 2012). Low quality or ambiguous images were discarded. Images from channels that did not correspond to a barcoding protein were included in the “none” class, thus generating a total of five image classes. Since mCherry, mCardinal, and iRFP702 images were obtained by linear unmixing of spectral images (see above), they sometimes contained noise and bleedthrough signals from other channels. We therefore performed a “mutual exclusion” correction, whereby we compared the mCherry, mCardinal, and iRFP702 images pixelwise and set the pixels from the two lower intensity channels to 0. This is because after unmixing, the channel with the real signal generally had the highest intensity. We used a script created in Python (Python Software Foundation. Python Language Reference, version 3.7, https://www.python.org) to carry out “mutual exclusion” correction followed by rescaling the pixel values to be between 0 and 1 and resizing to 150 pixels by 150 pixels. The final dataset contained 8,946 images (2,000 nucleus, 1,304 plasma membrane, 819 nuclear membrane, 1,724 cytosol, and 3,099 none), which were split into training (6,516 images), validation (1,630 images), and test sets (800 images).
Deep Convolutional Neural Network
We trained three deep convolutional neural network models using Keras (Chollet, 2018) with the TensorFlow (Abadi et al., 2015) backend: one model to classify BFP images and two models to classify red FP images with a known BFP reference marker. The model for BFP image classification had six layers, including four convolutional layers and two fully connected layers. We used 32, 64, 128, and 128 for the number of units in the convolutional layers and 512 and 3 in the fully connected layers. The two networks for red FP image classification had seven layers, including four convolutional layers and three fully connected layers. We used 32, 64, 128, and 128 for the number of units in the convolutional layers and 512, 128 and 5 in the fully connected layers. We used 3 × 3 convolutional filters for every convolutional layer and 2 × 2 max pooling with step size 2 after every convolutional layer. For the activation function, we used rectified linear units for every layer except for the last layer, in which we used Softmax for the 3-class and 5-class classifiers. We included a Dropout (with a rate of 0.5) between the last convolutional layer and fully connected layer to prevent overfitting (Srivastava et al., 2014). We used the RMSprop (Hinton et al., 2012) or Adam (Kingma and Ba, 2014) (initial learning_rate = 0.001, beta_1 = 0.9, beta_2 = 0.999) optimizer with a batch of 32 training images to minimize the cross-entropy loss. The weights were initialized with the default Glorot initializer provided by Keras (Glorot and Bengio, 2010). The maximum output of the softmax function was used for thresholding during the barcode identification analysis.
We used the preprocessed images as described above to train the BFP models. For the two red FP classification models, images from the red FP channels were combined with either nuclear or plasma membrane BFP images to form a two-channel image resized to 150 pixels × 150 pixels. Before inputting the images into the neural network, we augmented the training images (using ImageGenerator) by randomly shifting, shearing, horizontal/vertical flipping, or rotating the images. The models were trained for ~100 epochs, selected according to validation loss, and subsequently tested on the test data.
Fitting responses to kinetic models
Suppose that y(i,j)(tk) denotes the normalized biosensor level for biosensor i, cell j at time tk. These time-dependent traces were fit to a model:
in two different ways. In the first, one set of parameters a1, …, a4 and t0 was selected to minimize the sum of all the (squared) differences between the and all the traces; i.e.
This was done for ten individual experiments. Furthermore, for each specific cell-biosensor pair, a set of coefficients was obtained that minimized
Minimization was carried out using nonlinear least-squares optimization in MATLAB (MathWorks, Natick, MA) using the “fit” command of the Curve Fitting Toolbox (version 2020b). Sample values are given in Figure S4.
Simulation of RTK network responses to EGF stimulation
The experimental mean data from Figure 6D were fitted to one of three models, according to the respective classes. The first, a receptor/ligand binding model described by R + L ⇋ C is given by the differential equation
| (1) |
The four parameters, k1, k2, k3 and t0, were obtained by fitting the experimental data to the simulation using a custom-based script in MATLAB that relies on fminsearch for the nonlinear optimization and ode45 to solve for the differential equation. Here, L is the stimulus strength (0.1, 1, 10 and 100 ng/ml) and is the normalized activity of the biosensor (minus one, so that the prestimulus level is zero). This model was used to fit the experimental data for PicchuEV.
The second model represents the combined activity of a receptor model (as in Equation 1) and a negative feedback loop as follows:
In this model, the two species, X and Y, exist in two states, and the transitions between the two states is mediated by Michaelis-Menten kinetics. The various parameters, k1, …, k13 and t0, were obtained as in the first model, fitting the level of X to the biosensor data. This model was used to describe Syk, Lyn-FAK, RhoA2G, SRC, EVROCK and EV-S6K.
The third model represents the combined activity of a receptor model (as in Equation 1) and an activator-inhibitor excitable network (Xiong et al., 2010):
The various parameters, k1, …, k10 and t0, were obtained as in the previous model, fitting the level of U to the biosensor data. This model was used to fit the data for GCaMP6S, EKAR and PH-AKT.
The various parameter values are given in the following table. Note that, except for t0, which is given in minutes, all other parameters are non-dimensional.
| Biosensor | t 0 | k 1 | k 2 | k 3 | k 4 | k 5 | k 6 | k 7 | k 8 | k 9 | k 10 | k 11 | k 12 | k 13 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PicchuEV | 8.338 | 0.368 | 0.134 | 2.251 | ||||||||||
| GCaMP6S | 4.916 | 0.484 | 0.114 | 6.043 | 0.461 | 2.306 | 2.167 | 0.8560 | 1.3493 | 0.0960 | 0.1251 | |||
| EKAR | 7.582 | 0.741 | 0.178 | 0.361 | 1.503 | 1.035 | 0.130 | 0.0341 | 0.0799 | 2.24 × 10−5 | 0.2741 | |||
| PH-AKT | 3.497 | 0.973 | 0.108 | 0.335 | 0.691 | 0.456 | 0.40 | 0.1314 | 0.2136 | 1.25 × 10−12 | 0.6303 | |||
| EV-ROCK | 5.609 | 2.007 | 0.190 | 3.550 | 0.110 | 1.566 | 0.628 | 0.070 | 0.040 | 1.42 × 10−4 | 1.440 | 1.83 × 10−9 | 0.071 | 0.079 |
| EV-S6K | 7.701 | 0.165 | 0.137 | 1.939 | 0.675 | 0.210 | 0.046 | 0.592 | 0.877 | 0.367 | 0.387 | 0.204 | 0.109 | 1.370 |
| Lyn-FAK | 5.967 | 4.817 | 0.647 | 18.29 | 0.241 | 0.054 | 6.171 | 2.300 | 1.891 | 1.06 × 10−6 | 4.235 | 7.158 | 7.946 × 10−3 | 0.159 |
| RhoA2G | 5.362 | 0.476 | 0.196 | 3.961 | 0.221 | 0.488 | 1.203 | 1.657 | 0.487 | 0.547 | 0.978 | 8.44 × 10−4 | 0.067 | 0.044 |
| SRC | 5.403 | 0.652 | 0.175 | 3.279 | 0.582 | 0.450 | 1.208 | 0.697 | 0.405 | 3.38 × 10−7 | 0.260 | 7.01 × 10−4 | 0.064 | 0.135 |
| Syk | 6.565 | 0.826 | 0.112 | 1.964 | 0.854 | 0.070 | 1.083 | 0.500 | 1.170 | 2.49 × 10−5 | 0.271 | 5.20 × 10−4 | 0.045 | 0.074 |
Hill equation fitting
To perform fitting of the Hill equation, we took the peak values of the responses of each biosensor at four different concentrations (0.1, 1, 10, 100 ng/ml) of EGF stimulation, scaled it to the maximum response, and fitted it to
where K is the concentration of half responses, x is the measurement, and n is the Hill coefficient. The fitting is performed with the curve_fit tool in the Python library “scipy.optimize” with nonlinear least-squares regression.
QUANTIFICATION AND STATISTICAL ANALYSIS
Python and Microsoft Excel were used for statistical analysis and generating graphs. Specifically, Python Seaborn package was used for plotting Figures 5E and 5G and Python matplotlib.pyplot package for Figures 6C–6E. For imaging experiments involving mixed barcoded cells, at least three independent experiments were carried out on different days, and statistics were derived by aggregating the number of samples noted in each figure legend across independent experiments. Mean ± SD was reported as indicated in the figure legends. Statistical significance and p-values for comparing the responses of single and mixed cell populations (Table S2) were determined using two-tailed unpaired Welch’s t test for comparison between the two groups.
Supplementary Material
Figure S1. Images of barcoded cells, related to Figure 1
(A-B) Examples of HeLa cells expressing pairs of red/far-red barcoding proteins (A) and HeLa cells expressing BFP and one of the three red/far-red FPs (B). Spectral images of cells expressing pairs of barcoding proteins targeted to different subcellular locations were unmixed to obtain three images corresponding to TagRFP, mCardinal, and iRFP702, respectively. BFP was imaged separately. The numbers on top of every set of images denote barcodes as defined in Figure 1C legend. 20 out of 72 possible barcodes are shown. Scale bars: 10 μm.
(C-D) Absence of cross-transfection in mixed barcoded cells. (C) Representative images of HeLa cells transfected with BFP targeted to the nucleus (D1) or plasma membrane (D2) mixed with cells transfected with one of the red FPs (A: TagRFP; B: mCardinal; C: iRFP702; E: mCherry) localized to different locations (1: nucleus; 2: cell membrane; 3: nuclear membrane; 4: cytosol). (D) Collage of 12 viewing fields from a single microscopy experiment showing HeLa cells transfected with plasma membrane-targeted BFP (cyan) mixed with cells transfected with nuclear TagRFP (red). Scale bars: 50 μm.
Figure S2. Barcoding U2OS cells, related to Figure 4
(A) Representative images of U2OS cells transfected with BFP targeted to the nucleus (D1) or plasma membrane (D2) along with mCherry targeted to one of the four subcellular locations (E1: nucleus; E2: plasma membrane; E3: nuclear membrane; E4: cytosol). Lyn sequence was used for plasma membrane targeting in U2OS cells. Scale bars: 10 μm.
(B) 3D reconstruction from z-stacks of a U2OS cell with broad membrane signal of Lyn-CFP showing the “cowboy hat” morphology (left). A cell with sharply localized membrane signal (right) is shown for comparison. Scale bars: 10 μm.
(C) Confusion matrix (%) of U2OS cells barcoded with nuclear or cytoplasmic BFP plus mCherry targeted to one of the four subcellular locations using the deep learning models trained on HeLa cells (original models, left) and new models trained on U2OS cell images (right).
Figure S3. Lentiviral vectors for barcoding, related to STAR Methods
(A) Schematic of lentiviral vector construction.
(B-D) Representative images of U87MG (B), SiHa (C), and A6L (D) cells co-transduced with lentiviral vectors expressing BFP targeted to the nucleus (D1) or plasma membrane (D2) along with mCardinal targeted to one of the four subcellular locations (B1: nucleus; B2: plasma membrane; B3: nuclear membrane; B4: cytosol). Scale bars: 10 μm.
Figure S4. Fitting responses to kinetic models, related to Figure 5 and STAR Methods
(A) General form of the kinetic model used to fit the biosensor responses to EGF stimulation.
(B, C) Best-fit curves (red) and observed responses (open circles) of individual cells expressing EKAR (B) and nucEKAR (C) from E1 of Figure 4A.
(D, E) Statistics of the parameters and graphs showing all individual traces (thin black), average data (bold black) ± SD (blue), and data fit (red) for EKAR (D) and nucEKAR (E). See STAR Methods.
Figure S5. Response to different patterns of stimulation and cytoskeletal inhibitors, related to Figure 6
(A) Responses to transient and continued EGF stimulation. (Top) Responses of biosensors obtained from mixed barcoded cells treated with 100 ng/ml EGF at 6 min and 1 μM gefitinib (EGFRi) at 12 min. (Bottom) Responses to EGF without EGFRi (adapted from Figure 6C) are shown for comparison. The activities are normalized to the pre-stimulation level. Values represent mean (solid lines) ± SD (dashed lines).
(B) Responses to low and high EGF doses in individual cells. Barcoded HeLa cells expressing different biosensors were stimulated with 1, 10 and 100 ng/ml EGF at 6, 21, and 34 min, respectively. Traces of normalized activity from five representative cells expressing indicated biosensors are shown for each biosensor. Red arrows indicate the timing of the three stimuli.
(C) Mixed barcoded cells were pretreated with 5 μM jasplakinolide for 30 min, and then stimulated with 10 ng/ml EGF at the indicated time point.
(D) Mixed barcoded cells were pretreated with 5 μM latrunculin B for 30 min, and then stimulated with 100 ng/ml EGF at the indicated time point. In (C) and (D), the signals are normalized to the pre-stimulation level, and values represent mean (solid lines) ± SD (dashed lines).
Figure S6. Network topology of different classes of activities in RTK signaling network, related to Figure 6
(A-C) Computer simulation (solid lines) of the responses to different doses of stimuli based on a simple binding model for class 1 (A), negative feedback loop for class 2 (B), and excitable network for class 3 (C) biosensors. Schematics of the models are shown, with the variable plotted in the graphs in red. The models were fitted to the mean responses of HeLa cells to different concentrations of EGF (data points represented by dots). See STAR Methods for details of model fitting.
Figure S7. Effects of KRAS mutants, related to Figure 7
(A) EKAR activities (mean ± SD of YFP/CFP ratio) of mixture of four cell populations: 1) HeLa (Ctrl); 2) HeLa:KRAS(WT), 3) HeLa:KRAS(G12V), and 4) HeLa:KRAS(S17N) stimulated with 100 ng/ml EGF at 6 min.
(B,C) Responses of HeLa:KRAS(G12V) cells (B), and mixture of HeLa (Ctrl) and HeLa:KRAS(G12V) cells (C) to 100 ng/ml EGF added at 6 min. In each set of experiments, traces of the same color are derived from the same experiment (e.g., all the green traces from Ctrl and G12V in (C) are derived from the same cell mixture). The values represent the mean normalized to pre-stimulation levels.
(D) Immunoblots of phospho-FAK, phospho-ERK, and GAPDH using samples from control and KRAS(G12V) cells before and 15 min after stimulation with 100 ng/ml EGF. Error bars: SD. **p < 0.05 by Student’s t test (n = 3).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| p44/42 MAP kinase (phosphorylated Erk1/2) antibody | Cell Signaling | Cat# 9101, RRID:AB_331646 |
| Phospho-FAK (Tyr397) (D20B1) Rabbit mAb antibody | Cell Signaling | Cat# 8556, RRID:AB_10891442 |
| Rabbit Anti-GAPDH Monoclonal Antibody, Unconjugated, Clone 14C10 | Cell Signaling | Cat# 2118, RRID:AB_561053 |
| Chemicals, peptides, and recombinant proteins | ||
| Phorbol-12,13-dibutyrate (PDBu) | EMD Millipore | #524390 |
| Anisomycin | Sigma-Aldrich | A9789 |
| UK14304 | Sigma-Aldrich | U104 |
| Yohimbine | Sigma-Aldrich | Y3125 |
| Gefitinib | Cayman | #13166 |
| Ionomycin | Peprotech | #5608212 |
| Jasplakinolide | Cayman | #11705 |
| Latrunculin B | Enzo Life Sciences | BML-T110–0001 |
| PF562271 | AdipoGen | SYN-1064 |
| ZSTK474 | Cell Signaling | #13213 |
| Dasatinib | Cayman | #11498 |
| GDC-0994 | APExBIO Technology | B5817 |
| LY2584702 | Selleck | S7698 |
| BAPTA-AM | Selleck | S7534 |
| Y27632 | Enzo Life Sciences | ALX-270–333 |
| EGF | Sigma-Aldrich | E9644 |
| 2-Deoxyglucose (2-DG) | Sigma-Aldrich | D8375 |
| Experimental models: Cell lines | ||
| Human: A6L | L. Wood Lab (JHU) | N/A |
| Human: HEK293T | C. Hung Lab (JHU) | N/A |
| Human: HeLa | M. Iijima Lab (JHU) | N/A |
| Human: MCF7 | C. Hung Lab (JHU) | N/A |
| Human: SiHa | C. Hung Lab (JHU) | N/A |
| Human: U2OS | ATCC | HTB-96 |
| Human: U87MG | M. Iijima Lab (JHU) | N/A |
| Oligonucleotides | ||
| Primer for ERKKTR (forward): CAAgtcgacATGAAGGGCCGAAAGCCTC | This paper | N/A |
| Primer for ERKKTR (reverse): CAAggatccccGGATGGGAATTGAAAGCTGGACT | This paper | N/A |
| Primer for p38KTR (forward): CAActcgagATGCGTAAGCCAGATCTCCG | This paper | N/A |
| Primer for p38KTR (reverse): CAAggatccccGCTGGACTGGAGGGTCAG | This paper | N/A |
| Primer for JNKKTR (forward): CAActcgagATGAGTAACCCTAAGATCCTAAAACAGAG | This paper | N/A |
| Primer for JNKKTR (reverse): CAAggatccccGCTGGACTGGAGGGTCAG | This paper | N/A |
| Recombinant DNA | ||
| AMPKAR | Tsou et al., 2011 | Addgene plasmid #35097 |
| AMPKAR-EV | Konagaya et al., 2017 | Addgene plasmid #105241 |
| cyto-ABKAR | Miyamoto et al., 2015 | Addgene plasmid #61510 |
| LAMP1-ABKAR | Miyamoto et al., 2015 | Addgene plasmid #65068 |
| CaNAR2 | Mehta et al., 2014 | Addgene plasmid #64728 |
| ER-CaNAR2 | Mehta et al., 2014 | Addgene plasmid #64732 |
| PM-CaNAR2 | Mehta et al., 2014 | Addgene plasmid #64730 |
| GCaMP6S | Chen et al., 2013 | Addgene plasmid #40753 |
| GCaMP6S-PM | Tsai et al., 2014 | Addgene plasmid #52228 |
| EKAR | Harvey et al., 2008 | Addgene plasmid #18679 |
| Nuc-EKAR | Harvey et al., 2008 | Addgene plasmid #18681 |
| Lyn-EKAR | This paper | N/A |
| ERKKTR | Regot et al., 2014 | Addgene plasmid #59150 |
| Cyto-FAK | Seong et al., 2011 | Addgene plasmid #78300 |
| Lyn-FAK | Seong et al., 2011 | Addgene plasmid #78299 |
| Src | Ouyang et al., 2008 | Addgene plasmid #78302 |
| Gαi1 | van Unen et al., 2016 | Addgene plasmid #69623 |
| Gαi2 | van Unen et al., 2016 | Addgene plasmid #69624 |
| Gαi3 | van Unen et al., 2016 | Addgene plasmid #69625 |
| JNKAR | Fosbrink et al., 2010 | Addgene plasmid #61625 |
| JNKKTR | Regot et al., 2014 | Addgene plasmid #59151 |
| p38KTR | Regot et al., 2014 | Addgene plasmid #59152 |
| PH-AKT | Watton and Downward, 1999 | N/A |
| AKTAR2 | Zhou et al., 2015 | Addgene plasmid #64932 |
| CKAR | Violin et al., 2003 | Addgene plasmid #14860 |
| PM-CKAR | Violin et al., 2003 | Addgene plasmid #14862 |
| RhoA-2G | Fritz et al., 2013 | Addgene plasmid #40176 |
| Rac1-2G | Fritz et al., 2015 | Addgene plasmid #66110 |
| Cdc42–2G | Martin et al., 2016 | Addgene plasmid #68814 |
| LifeAct | Riedl et al., 2008 | Addgene plasmid #54610 |
| Raichu-HRas | Komatsu et al., 2011 | N/A |
| Raichu-KRas | Komatsu et al., 2011 | N/A |
| Raichu-Rras | Takaya et al., 2007 | N/A |
| BKAR | Kunkel et al., 2005 | Addgene plasmid #14875 |
| TORCAR | Zhou et al., 2015 | Addgene plasmid #64927 |
| Prin-BRaf | Terai and Matsuda, 2006 | N/A |
| Prin-CRAF | Terai and Matsuda, 2005 | N/A |
| EV-RSK | Komatsu et al., 2011 | N/A |
| EV-ROCK | Li et al., 2017 | N/A |
| EV-S6K | Komatsu et al., 2011 | N/A |
| EV-EGFR (PicchuEV(CrkII)) | Komatsu et al., 2011 | N/A |
| Syk biosensor | Xiang et al., 2011 | Addgene plasmid #125729 |
| pEGFP-N1 | CloneTech | N/A |
| EBFP2-Nucleus-7 | gift from Michael Davidson | Addgene plasmid #55249 |
| H2B-TagRFP | gift from Philipp Keller | Addgene plasmid #99271 |
| pmCherry-NLS | Micutkova et al., 2012 | Addgene #39319 |
| mCardinal-H2B-C-10 | Chu et al., 2014 | Addgene plasmid #56162 |
| mCardinal-N1 | Chu et al., 2014 | Addgene #54590 |
| piRFP702-N1 | Shcherbakova and Verkhusha, 2013 | Addgene plasmid #45456 |
| piRFP682-N1 | Shcherbakova and Verkhusha, 2013 | Addgene plasmid #45459 |
| pNLS-iRFP670 | Shcherbakova and Verkhusha, 2013 | Addgene plasmid #45466 |
| mNeptune2-C1 | gift from Michael Davidson | Addgene plasmid #54836 |
| mNeptune2.5 | Chu et al., 2014 | Addgene plasmid #51310 |
| mPlum-C1 | Kremers et al., 2009 | Addgene plasmid #54839 |
| RDSmCherry1 | Shen et al., 2017 | Addgene plasmid #89987 |
| LSS-mKate2-C1 | Piatkevich et al., 2010 | Addgene plasmid #31869 |
| mStrawberry-N1 | Shaner et al., 2004 | Addgene #54644 |
| pDONR221 | Invitrogen | #12536017 |
| pLex307 | gift from David Root | Addgene plasmid #41392 |
| pMD2.G | gift from Didier Trono | Addgene plasmid #12259 |
| psPAX2 | gift from Didier Trono | Addgene plasmid #12260 |
| Software and algorithms | ||
| Zen Microscopy Software | Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software/zen.html |
| ImageJ/Fiji | Schindelin et al., 2012; Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| Python | Python Software Foundation | https://www.python.org |
| Keras | Chollet, 2018 | https://keras.io/ |
| TensorFlow | Abadi et al., 2015 | https://www.tensorflow.org/ |
| Dropout | Srivastava et al., 2014 | N/A |
| RMSprop | Hinton et al., 2012 | N/A |
| Adam | Kingma and Ba, 2014 | N/A |
| Glorot Initializer | Glorot and Bengio, 2010 | N/A |
| MATLAB | MathWorks | https://www.mathworks.com/products/matlab.html |
Highlights.
Large numbers of fluorescent biosensors can be concurrently tracked in barcoded cells
Biosensor activities are synchronized in mixed populations of barcoded cells
Deep learning models facilitate image analysis for biosensor barcoding
Simultaneous biosensor tracking reveals signaling network structures and interactions
ACKNOWLEDGMENTS
We would like to thank Hoku West-Foyle and Takanari Inoue for helpful discussions, as well as Michiyuki Matsuda for plasmids of PicchuEV, Prin-BRAF, Prin-CRAF, EV-RSK, EV-ROCK, and EV-S6K. Zeiss LSM 780 and 880 confocal microscopes were purchased with NIH grants S10OD016374 and S10OD023548, respectively. This work was supported by NIH (K22CA212060 and R01GM136711 to C.-H.H.), Cervical Cancer SPORE P50CA098252 (Career Development Award to J.-M.Y., Pilot Project Award to C.-H.H.), the W.W. Smith Charitable Trust (C1901 to C.-H.H.), and the Sol Goldman Pancreatic Cancer Research Center (to C.-H.H.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.cell.2021.11.005.
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPORTING CITATIONS
The following references appear in the Supplemental Information: Bondeva et al. (2002); Chatila et al. (1989); Chen and Tan (1998); Elzi et al. (2001); Guzeloglu et al. (2004); Jimenez-Vargas et al. (2018); Meus et al. (2017); Rao et al. (2001); Sample et al. (2015).
REFERENCES
- Abadi M, Agarwal A, Barham P, Brevdo E, Chen Z, Citro C, Corrado GS, Davis A, Dean J, Devin M, et al. (2015). TensorFlow: Large-scale machine learning on heterogeneous systems. arXiv, arXiv:1603.04467. [Google Scholar]
- Ai H-W, Shaner NC, Cheng Z, Tsien RY, and Campbell RE (2007). Exploration of new chromophore structures leads to the identification of improved blue fluorescent proteins. Biochemistry 46, 5904–5910. [DOI] [PubMed] [Google Scholar]
- Amano M, Nakayama M, and Kaibuchi K (2010). Rho-kinase/ROCK: A key regulator of the cytoskeleton and cell polarity. Cytoskeleton (Hoboken) 67, 545–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki K, Kumagai Y, Sakurai A, Komatsu N, Fujita Y, Shionyu C, and Matsuda M (2013). Stochastic ERK activation induced by noise and cell-to-cell propagation regulates cell density-dependent proliferation. Mol. Cell 52, 529–540. [DOI] [PubMed] [Google Scholar]
- Bondeva T, Balla A, Várnai P, and Balla T (2002). Structural determinants of Ras-Raf interaction analyzed in live cells. Mol. Biol. Cell 13, 2323–2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapnick DA, Bunker E, Liu X, and Old WM (2019). Temporal metabolite, ion, and enzyme activity profiling using fluorescence microscopy and genetically encoded biosensors. In High-Throughput Metabolomics: Methods and Protocols, D’Alessandro A, ed. (Springer; New York: ), pp. 343–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatila T, Silverman L, Miller R, and Geha R (1989). Mechanisms of T cell activation by the calcium ionophore ionomycin. J. Immunol 143, 1283–1289. [PubMed] [Google Scholar]
- Chen YR, and Tan TH (1998). Inhibition of the c-Jun N-terminal kinase (JNK) signaling pathway by curcumin. Oncogene 17, 173–178. [DOI] [PubMed] [Google Scholar]
- Chen T-W, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, Schreiter ER, Kerr RA, Orger MB, Jayaraman V, et al. (2013). Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499, 295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernov KG, Redchuk TA, Omelina ES, and Verkhusha VV (2017). Near-Infrared Fluorescent Proteins, Biosensors, and Optogenetic Tools Engineered from Phytochromes. Chem. Rev 117, 6423–6446. [DOI] [PubMed] [Google Scholar]
- Chollet F (2018). Keras: The Python Deep Learning Library (Astrophysics Source Code Library)
- Chu J, Haynes RD, Corbel SY, Li P, González-González E, Burg JS, Ataie NJ, Lam AJ, Cranfill PJ, Baird MA, et al. (2014). Non-invasive intravital imaging of cellular differentiation with a bright red-excitable fluorescent protein. Nat. Methods 11, 572–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Simone A, Evanitsky MN, Hayden L, Cox BD, Wang J, Tornini VA, Ou J, Chao A, Poss KD, and Di Talia S (2021). Control of osteoblast regeneration by a train of Erk activity waves. Nature 590, 129–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elzi DJ, Bjornsen AJ, MacKenzie T, Wyman TH, and Silliman CC (2001). Ionomycin causes activation of p38 and p42/44 mitogen-activated protein kinases in human neutrophils. Am. J. Physiol. Cell Physiol 281, C350–C360. [DOI] [PubMed] [Google Scholar]
- Fosbrink M, Aye-Han N-N, Cheong R, Levchenko A, and Zhang J (2010). Visualization of JNK activity dynamics with a genetically encoded fluorescent biosensor. Proc. Natl. Acad. Sci. USA 107, 5459–5464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frame MC, Patel H, Serrels B, Lietha D, and Eck MJ (2010). The FERM domain: organizing the structure and function of FAK. Nat. Rev. Mol. Cell Biol 11, 802–814. [DOI] [PubMed] [Google Scholar]
- Fritz RD, Letzelter M, Reimann A, Martin K, Fusco L, Ritsma L, Ponsioen B, Fluri E, Schulte-Merker S, van Rheenen J, and Pertz O (2013). A versatile toolkit to produce sensitive FRET biosensors to visualize signaling in time and space. Sci. Signal 6, rs12. [DOI] [PubMed] [Google Scholar]
- Fritz RD, Menshykau D, Martin K, Reimann A, Pontelli V, and Pertz O (2015). SrGAP2-Dependent Integration of Membrane Geometry and Slit-Robo-Repulsive Cues Regulates Fibroblast Contact Inhibition of Locomotion. Dev. Cell 35, 78–92. [DOI] [PubMed] [Google Scholar]
- Fujita Y, Komatsu N, Matsuda M, and Aoki K (2014). Fluorescence resonance energy transfer based quantitative analysis of feedforward and feedback loops in epidermal growth factor receptor signaling and the sensitivity to molecular targeting drugs. FEBS J 281, 3177–3192. [DOI] [PubMed] [Google Scholar]
- Fukushima S, Matsuoka S, and Ueda M (2019). Excitable dynamics of Ras triggers spontaneous symmetry breaking of PIP3 signaling in motile cells. J. Cell Sci 132, jcs224121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelens L, Anderson GA, and Ferrell JE Jr. (2014). Spatial trigger waves: positive feedback gets you a long way. Mol. Biol. Cell 25, 3486–3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glorot X, and Bengio Y (2010). Understanding the difficulty of training deep feedforward neural networks. J. Mach. Learn. Res 9, 249–256. [Google Scholar]
- Grant DM, Zhang W, McGhee EJ, Bunney TD, Talbot CB, Kumar S, Munro I, Dunsby C, Neil MAA, Katan M, and French PM (2008). Multiplexed FRET to image multiple signaling events in live cells. Biophys. J 95, L69–L71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwald EC, Mehta S, and Zhang J (2018). Genetically Encoded Fluorescent Biosensors Illuminate the Spatiotemporal Regulation of Signaling Networks. Chem. Rev 118, 11707–11794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzeloglu A, Subramaniam P, Michel F, and Thatcher WW (2004). Interferon-tau induces degradation of prostaglandin H synthase-2 messenger RNA in bovine endometrial cells through a transcription-dependent mechanism. Biol. Reprod 71, 170–176. [DOI] [PubMed] [Google Scholar]
- Harvey CD, Ehrhardt AG, Cellurale C, Zhong H, Yasuda R, Davis RJ, and Svoboda K (2008). A genetically encoded fluorescent sensor of ERK activity. Proc. Natl. Acad. Sci. USA 105, 19264–19269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinton G, Srivastava N, and Swersky K (2012). Neural networks for machine learning lecture 6a overview of mini-batch gradient descent https://www.cs.toronto.edu/~tijmen/csc321/slides/lecture_slides_lec6.pdf.
- Hiratsuka T, Fujita Y, Naoki H, Aoki K, Kamioka Y, and Matsuda M (2015). Intercellular propagation of extracellular signal-regulated kinase activation revealed by in vivo imaging of mouse skin. eLife 4, e05178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeller O, Gong D, and Weiner OD (2014). How to understand and outwit adaptation. Dev. Cell 28, 607–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CY, and Ferrell JE Jr. (1996). Ultrasensitivity in the mitogen-activated protein kinase cascade. Proc. Natl. Acad. Sci. USA 93, 10078–10083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C-H, Tang M, Shi C, Iglesias PA, and Devreotes PN (2013). An excitable signal integrator couples to an idling cytoskeletal oscillator to drive cell migration. Nat. Cell Biol 15, 1307–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iglesias PA, and Shi C (2014). Comparison of adaptation motifs: temporal, stochastic and spatial responses. IET Syst. Biol 8, 268–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Vargas NN, Pattison LA, Zhao P, Lieu T, Latorre R, Jensen DD, Castro J, Aurelio L, Le GT, Flynn B, et al. (2018). Protease-activated receptor-2 in endosomes signals persistent pain of irritable bowel syndrome. Proc. Natl. Acad. Sci. USA 115, E7438–E7447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingma DP, and Ba J (2014). Adam: A Method for Stochastic Optimization. arXiv, arXiv:1412.6980. [Google Scholar]
- Komatsu N, Aoki K, Yamada M, Yukinaga H, Fujita Y, Kamioka Y, and Matsuda M (2011). Development of an optimized backbone of FRET biosensors for kinases and GTPases. Mol. Biol. Cell 22, 4647–4656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konagaya Y, Terai K, Hirao Y, Takakura K, Imajo M, Kamioka Y, Sasaoka N, Kakizuka A, Sumiyama K, Asano T, and Matsuda M (2017). A Highly Sensitive FRET Biosensor for AMPK Exhibits Heterogeneous AMPK Responses among Cells and Organs. Cell Rep 21, 2628–2638. [DOI] [PubMed] [Google Scholar]
- Kremers G-J, Hazelwood KL, Murphy CS, Davidson MW, and Piston DW (2009). Photoconversion in orange and red fluorescent proteins. Nat. Methods 6, 355–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchenov D, Laketa V, Stein F, Salopiata F, Klingmüller U, and Schultz C (2016). High-Content Imaging Platform for Profiling Intracellular Signaling Network Activity in Living Cells. Cell Chem. Biol 23, 1550–1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel MT, Ni Q, Tsien RY, Zhang J, and Newton AC (2005). Spatiotemporal dynamics of protein kinase B/Akt signaling revealed by a genetically encoded fluorescent reporter. J. Biol. Chem 280, 5581–5587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert TJ (2019). FPbase: a community-editable fluorescent protein database. Nat. Methods 16, 277–278. [DOI] [PubMed] [Google Scholar]
- Li C, Imanishi A, Komatsu N, Terai K, Amano M, Kaibuchi K, and Matsuda M (2017). A FRET Biosensor for ROCK Based on a Consensus Substrate Sequence Identified by KISS Technology. Cell Struct. Funct 42, 1–13. [DOI] [PubMed] [Google Scholar]
- Linghu C, Johnson SL, Valdes PA, Shemesh OA, Park WM, Park D, Piatkevich KD, Wassie AT, Liu Y, An B, et al. (2020). Spatial Multiplexing of Fluorescent Reporters for Imaging Signaling Network Dynamics. Cell 183, 1682–1698.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W, Trusina A, El-Samad H, Lim WA, and Tang C (2009). Defining network topologies that can achieve biochemical adaptation. Cell 138, 760–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machacek M, Hodgson L, Welch C, Elliott H, Pertz O, Nalbant P, Abell A, Johnson GL, Hahn KM, and Danuser G (2009). Coordination of Rho GTPase activities during cell protrusion. Nature 461, 99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marston DJ, Vilela M, Huh J, Ren J, Azoitei ML, Glekas G, Danuser G, Sondek J, and Hahn KM (2020). Multiplexed GTPase and GEF biosensor imaging enables network connectivity analysis. Nat. Chem. Biol 16, 826–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin K, Reimann A, Fritz RD, Ryu H, Jeon NL, and Pertz O (2016). Spatio-temporal co-ordination of RhoA, Rac1 and Cdc42 activation during prototypical edge protrusion and retraction dynamics. Sci. Rep 6, 21901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta S, Aye-Han N-N, Ganesan A, Oldach L, Gorshkov K, and Zhang J (2014). Calmodulin-controlled spatial decoding of oscillatory Ca2+ signals by calcineurin. eLife 3, e03765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta S, Zhang Y, Roth RH, Zhang J-F, Mo A, Tenner B, Huganir RL, and Zhang J (2018). Single-fluorophore biosensors for sensitive and multiplexed detection of signalling activities. Nat. Cell Biol 20, 1215–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meus M-A, Hertig V, Villeneuve L, Jasmin J-F, and Calderone A (2017). Nestin Expressed by Pre-Existing Cardiomyocytes Recapitulated in Part an Embryonic Phenotype; Suppressive Role of p38 MAPK. J. Cell. Physiol 232, 1717–1727. [DOI] [PubMed] [Google Scholar]
- Micutkova L, Hermann M, Offterdinger M, Hess MW, Matscheski A, Pircher H, Mück C, Ebner H-L, Laich A, Ferrando-May E, et al. (2012). Analysis of the cellular uptake and nuclear delivery of insulin-like growth factor binding protein-3 in human osteosarcoma cells. Int. J. Cancer 130, 1544–1557. [DOI] [PubMed] [Google Scholar]
- Miyamoto T, Rho E, Sample V, Akano H, Magari M, Ueno T, Gorshkov K, Chen M, Tokumitsu H, Zhang J, and Inoue T (2015). Compartmentalized AMPK signaling illuminated by genetically encoded molecular sensors and actuators. Cell Rep 11, 657–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman RH, Fosbrink MD, and Zhang J (2011). Genetically encodable fluorescent biosensors for tracking signaling dynamics in living cells. Chem. Rev 111, 3614–3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang M, Sun J, Chien S, and Wang Y (2008). Determination of hierarchical relationship of Src and Rac at subcellular locations with FRET biosensors. Proc. Natl. Acad. Sci. USA 105, 14353–14358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pargett M, Gillies TE, Teragawa CK, Sparta B, and Albeck JG (2017). Single-Cell Imaging of ERK Signaling Using Fluorescent Biosensors. Methods Mol. Biol 1636, 35–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piatkevich KD, Hulit J, Subach OM, Wu B, Abdulla A, Segall JE, and Verkhusha VV (2010). Monomeric red fluorescent proteins with a large Stokes shift. Proc. Natl. Acad. Sci. USA 107, 5369–5374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao JN, Li L, Golovina VA, Platoshyn O, Strauch ED, Yuan JX, and Wang JY (2001). Ca2+-RhoA signaling pathway required for polyamine-dependent intestinal epithelial cell migration. Am. J. Physiol. Cell Physiol 280, C993–C1007. [DOI] [PubMed] [Google Scholar]
- Regot S, Hughey JJ, Bajar BT, Carrasco S, and Covert MW (2014). High-sensitivity measurements of multiple kinase activities in live single cells. Cell 157, 1724–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedl J, Crevenna AH, Kessenbrock K, Yu JH, Neukirchen D, Bista M, Bradke F, Jenne D, Holak TA, Werb Z, et al. (2008). Lifeact: a versatile marker to visualize F-actin. Nat. Methods 5, 605–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sample V, Ramamurthy S, Gorshkov K, Ronnett GV, and Zhang J (2015). Polarized activities of AMPK and BRSK in primary hippocampal neurons. Mol. Biol. Cell 26, 1935–1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seong J, Ouyang M, Kim T, Sun J, Wen P-C, Lu S, Zhuo Y, Llewellyn NM, Schlaepfer DD, Guan J-L, et al. (2011). Detection of focal adhesion kinase activation at membrane microdomains by fluorescence resonance energy transfer. Nat. Commun 2, 406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner NC, Campbell RE, Steinbach PA, Giepmans BNG, Palmer AE, and Tsien RY (2004). Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol 22, 1567–1572. [DOI] [PubMed] [Google Scholar]
- Shaner NC, Lin MZ, McKeown MR, Steinbach PA, Hazelwood KL, Davidson MW, and Tsien RY (2008). Improving the photostability of bright monomeric orange and red fluorescent proteins. Nat. Methods 5, 545–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcherbakova DM, and Verkhusha VV (2013). Near-infrared fluorescent proteins for multicolor in vivo imaging. Nat. Methods 10, 751–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Chen Y, Wu J, Shaner NC, and Campbell RE (2017). Engineering of mCherry variants with long Stokes shift, red-shifted fluorescence, and low cytotoxicity. PLoS ONE 12, e0171257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shindo Y, Iwamoto K, Mouri K, Hibino K, Tomita M, Kosako H, Sako Y, and Takahashi K (2016). Conversion of graded phosphorylation into switch-like nuclear translocation via autoregulatory mechanisms in ERK signalling. Nat. Commun 7, 10485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava N, Hinton G, Krizhevsky A, Sutskever I, and Salakhutdinov R (2014). Dropout: A Simple Way to Prevent Neural Networks from Overfitting. J. Mach. Learn. Res 15, 1929–1958. [Google Scholar]
- Takaya A, Kamio T, Masuda M, Mochizuki N, Sawa H, Sato M, Nagashima K, Mizutani A, Matsuno A, Kiyokawa E, and Matsuda M (2007). R-Ras regulates exocytosis by Rgl2/Rlf-mediated activation of RalA on endosomes. Mol. Biol. Cell 18, 1850–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terai K, and Matsuda M (2005). Ras binding opens c-Raf to expose the docking site for mitogen-activated protein kinase kinase. EMBO Rep 6, 251–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terai K, and Matsuda M (2006). The amino-terminal B-Raf-specific region mediates calcium-dependent homo- and hetero-dimerization of Raf. EMBO J 25, 3556–3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terai K, Imanishi A, Li C, and Matsuda M (2019). Two decades of genetically encoded biosensors based on Förster resonance energy transfer. Cell Struct. Funct 44, 153–169. [DOI] [PubMed] [Google Scholar]
- Tsai F-C, Seki A, Yang HW, Hayer A, Carrasco S, Malmersjö S, and Meyer T (2014). A polarized Ca2+, diacylglycerol and STIM1 signalling system regulates directed cell migration. Nat. Cell Biol 16, 133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou P, Zheng B, Hsu C-H, Sasaki AT, and Cantley LC (2011). A fluorescent reporter of AMPK activity and cellular energy stress. Cell Metab 13, 476–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu Y, and Rappel WJ (2018). Adaptation in Living Systems. Annu. Rev. Condens. Matter Phys 9, 183–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Haastert PJM, Keizer-Gunnink I, and Kortholt A (2017). Coupled excitable Ras and F-actin activation mediates spontaneous pseudopod formation and directed cell movement. Mol. Biol. Cell 28, 922–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Unen J, Stumpf AD, Schmid B, Reinhard NR, Hordijk PL, Hoffmann C, Gadella TWJ Jr., and Goedhart J (2016). A New Generation of FRET Sensors for Robust Measurement of Gαi1, Gαi2 and Gαi3 Activation Kinetics in Single Cells. PLoS ONE 11, e0146789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Violin JD, Zhang J, Tsien RY, and Newton AC (2003). A genetically encoded fluorescent reporter reveals oscillatory phosphorylation by protein kinase C. J. Cell Biol 161, 899–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watton SJ, and Downward J (1999). Akt/PKB localisation and 3′ phosphoinositide generation at sites of epithelial cell-matrix and cell-cell interaction. Curr. Biol 9, 433–436. [DOI] [PubMed] [Google Scholar]
- Welch CM, Elliott H, Danuser G, and Hahn KM (2011). Imaging the coordination of multiple signalling activities in living cells. Nat. Rev. Mol. Cell Biol 12, 749–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang X, Sun J, Wu J, He H-T, Wang Y, and Zhu C (2011). A FRET-Based Biosensor for Imaging SYK Activities in Living Cells. Cell. Mol. Bioeng 4, 670–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y, Huang C-H, Iglesias PA, and Devreotes PN (2010). Cells navigate with a local-excitation, global-inhibition-biased excitable network. Proc. Natl. Acad. Sci. USA 107, 17079–17086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HW, Collins SR, and Meyer T (2016). Locally excitable Cdc42 signals steer cells during chemotaxis. Nat. Cell Biol 18, 191–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J-M, Bhattacharya S, West-Foyle H, Hung C-F, Wu T-C, Iglesias PA, and Huang C-H (2018). Integrating chemical and mechanical signals through dynamic coupling between cellular protrusions and pulsed ERK activation. Nat. Commun 9, 4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan H, Bhattacharya S, Cai H, Iglesias PA, Huang C-H, and Devreotes PN (2020). An Excitable Ras/PI3K/ERK Signaling Network Controls Migration and Oncogenic Transformation in Epithelial Cells. Dev. Cell 54, 608–623.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Campbell RE, Ting AY, and Tsien RY (2002). Creating new fluorescent probes for cell biology. Nat. Rev. Mol. Cell Biol 3, 906–918. [DOI] [PubMed] [Google Scholar]
- Zhou X, Clister TL, Lowry PR, Seldin MM, Wong GW, and Zhang J (2015). Dynamic Visualization of mTORC1 Activity in Living Cells. Cell Rep 10, 1767–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Images of barcoded cells, related to Figure 1
(A-B) Examples of HeLa cells expressing pairs of red/far-red barcoding proteins (A) and HeLa cells expressing BFP and one of the three red/far-red FPs (B). Spectral images of cells expressing pairs of barcoding proteins targeted to different subcellular locations were unmixed to obtain three images corresponding to TagRFP, mCardinal, and iRFP702, respectively. BFP was imaged separately. The numbers on top of every set of images denote barcodes as defined in Figure 1C legend. 20 out of 72 possible barcodes are shown. Scale bars: 10 μm.
(C-D) Absence of cross-transfection in mixed barcoded cells. (C) Representative images of HeLa cells transfected with BFP targeted to the nucleus (D1) or plasma membrane (D2) mixed with cells transfected with one of the red FPs (A: TagRFP; B: mCardinal; C: iRFP702; E: mCherry) localized to different locations (1: nucleus; 2: cell membrane; 3: nuclear membrane; 4: cytosol). (D) Collage of 12 viewing fields from a single microscopy experiment showing HeLa cells transfected with plasma membrane-targeted BFP (cyan) mixed with cells transfected with nuclear TagRFP (red). Scale bars: 50 μm.
Figure S2. Barcoding U2OS cells, related to Figure 4
(A) Representative images of U2OS cells transfected with BFP targeted to the nucleus (D1) or plasma membrane (D2) along with mCherry targeted to one of the four subcellular locations (E1: nucleus; E2: plasma membrane; E3: nuclear membrane; E4: cytosol). Lyn sequence was used for plasma membrane targeting in U2OS cells. Scale bars: 10 μm.
(B) 3D reconstruction from z-stacks of a U2OS cell with broad membrane signal of Lyn-CFP showing the “cowboy hat” morphology (left). A cell with sharply localized membrane signal (right) is shown for comparison. Scale bars: 10 μm.
(C) Confusion matrix (%) of U2OS cells barcoded with nuclear or cytoplasmic BFP plus mCherry targeted to one of the four subcellular locations using the deep learning models trained on HeLa cells (original models, left) and new models trained on U2OS cell images (right).
Figure S3. Lentiviral vectors for barcoding, related to STAR Methods
(A) Schematic of lentiviral vector construction.
(B-D) Representative images of U87MG (B), SiHa (C), and A6L (D) cells co-transduced with lentiviral vectors expressing BFP targeted to the nucleus (D1) or plasma membrane (D2) along with mCardinal targeted to one of the four subcellular locations (B1: nucleus; B2: plasma membrane; B3: nuclear membrane; B4: cytosol). Scale bars: 10 μm.
Figure S4. Fitting responses to kinetic models, related to Figure 5 and STAR Methods
(A) General form of the kinetic model used to fit the biosensor responses to EGF stimulation.
(B, C) Best-fit curves (red) and observed responses (open circles) of individual cells expressing EKAR (B) and nucEKAR (C) from E1 of Figure 4A.
(D, E) Statistics of the parameters and graphs showing all individual traces (thin black), average data (bold black) ± SD (blue), and data fit (red) for EKAR (D) and nucEKAR (E). See STAR Methods.
Figure S5. Response to different patterns of stimulation and cytoskeletal inhibitors, related to Figure 6
(A) Responses to transient and continued EGF stimulation. (Top) Responses of biosensors obtained from mixed barcoded cells treated with 100 ng/ml EGF at 6 min and 1 μM gefitinib (EGFRi) at 12 min. (Bottom) Responses to EGF without EGFRi (adapted from Figure 6C) are shown for comparison. The activities are normalized to the pre-stimulation level. Values represent mean (solid lines) ± SD (dashed lines).
(B) Responses to low and high EGF doses in individual cells. Barcoded HeLa cells expressing different biosensors were stimulated with 1, 10 and 100 ng/ml EGF at 6, 21, and 34 min, respectively. Traces of normalized activity from five representative cells expressing indicated biosensors are shown for each biosensor. Red arrows indicate the timing of the three stimuli.
(C) Mixed barcoded cells were pretreated with 5 μM jasplakinolide for 30 min, and then stimulated with 10 ng/ml EGF at the indicated time point.
(D) Mixed barcoded cells were pretreated with 5 μM latrunculin B for 30 min, and then stimulated with 100 ng/ml EGF at the indicated time point. In (C) and (D), the signals are normalized to the pre-stimulation level, and values represent mean (solid lines) ± SD (dashed lines).
Figure S6. Network topology of different classes of activities in RTK signaling network, related to Figure 6
(A-C) Computer simulation (solid lines) of the responses to different doses of stimuli based on a simple binding model for class 1 (A), negative feedback loop for class 2 (B), and excitable network for class 3 (C) biosensors. Schematics of the models are shown, with the variable plotted in the graphs in red. The models were fitted to the mean responses of HeLa cells to different concentrations of EGF (data points represented by dots). See STAR Methods for details of model fitting.
Figure S7. Effects of KRAS mutants, related to Figure 7
(A) EKAR activities (mean ± SD of YFP/CFP ratio) of mixture of four cell populations: 1) HeLa (Ctrl); 2) HeLa:KRAS(WT), 3) HeLa:KRAS(G12V), and 4) HeLa:KRAS(S17N) stimulated with 100 ng/ml EGF at 6 min.
(B,C) Responses of HeLa:KRAS(G12V) cells (B), and mixture of HeLa (Ctrl) and HeLa:KRAS(G12V) cells (C) to 100 ng/ml EGF added at 6 min. In each set of experiments, traces of the same color are derived from the same experiment (e.g., all the green traces from Ctrl and G12V in (C) are derived from the same cell mixture). The values represent the mean normalized to pre-stimulation levels.
(D) Immunoblots of phospho-FAK, phospho-ERK, and GAPDH using samples from control and KRAS(G12V) cells before and 15 min after stimulation with 100 ng/ml EGF. Error bars: SD. **p < 0.05 by Student’s t test (n = 3).
Data Availability Statement
All data supporting the findings of the current study are available within the article and its Supplemental Information files or from the corresponding authors upon reasonable request. The codes used in the study are provided as a ZIP file (Data S1).