

Abstract
Highly effective CFTR modulator drug therapy is increasingly available to those with cystic fibrosis. Multiple observational research studies are now being conducted to better understand the impacts of this important therapeutic milestone on long-term outcomes, patient care needs, and future research priorities. PROMISE is a large, multi-disciplinary academic study focused on the broad impacts of starting elexacaftor/tezacaftor/ivacaftor in the US population age 6 years and older. The many areas of investigation and rationale for each are discussed by organ systems, along with recognition of remaining important questions that will not be addressed by this study alone. Knowledge gained through this and multiple complementary studies around the world will help to understand important health outcomes, clinical care priorities, and research needs for a large majority of people treated with these or similarly effective medications targeting the primary cellular impairment in cystic fibrosis.
Background
Clinical care and research in cystic fibrosis (CF) have been heavily influenced by the emergence of highly effective modulator drug therapies (HEMT) that target the CF transmembrane conductance regulator protein (CFTR). These CFTR modulators increase CFTR function by improving the processing, trafficking, and gating of aberrant proteins in the cell. Results show that CFTR modulators significantly, and at times dramatically, improve health outcomes and quality of life1, 2. Herein we highlight a breadth of observational research studies now being conducted to understand the impact of HEMT regimens across a spectrum of disease manifestations. This work is organized within the PROMISE study (NCT04038047) in the United States and sponsored by the Cystic Fibrosis Foundation, independent of the manufacturer. Nearly 700 individuals will ultimately participate to contribute prospective research visits through 2 years of elexacaftor/tezacaftor/ivacaftor (ETI) treatment. This is similar to other recent post-approval trials (i.e. GOAL; NCT01521338, PROSPECT; NCT02477319), but PROMISE will expand upon key questions that have arisen in the modulator era. In the spirit of multi-disciplinary coordination, 8 sub-studies within PROMISE will be conducted by specialty experts. We describe below the rationale, objectives, and deliverables, while highlighting key questions that will remain to be addressed by further research and other investigative efforts (e.g. RECOVER; NCT04602468, BEGIN; NCT04509050) equally important in understanding the future for PwCF treated with modulators. This is a unique and time sensitive opportunity to understand how effective CFTR restoration alters the disease state as a large majority of PwCF begin HEMT. PROMISE, as one of several studies being conducted within the US CF Therapeutics Development Network (TDN), will broadly clarify remaining needs and priorities for clinical care and research in individuals with CF treated with HEMT. As with similar other studies supported by the CF Foundation and conducted in the TDN, upon study completion and publication of primary study results, de-identified datasets will be available through the CFF TDN Data Archive. Researchers may apply to the CF TDN for use of de-identified data from the archive for research purposes, and must receive appropriate IRB approval before data is sent from the Archive. In PROMISE, biospecimens are also being aliquoted and carefully preserved for future investigator-initiated requests through the centralized CF Foundation Biorepository. When approved, these samples can be linked with de-identified data from the Archive.
Current status of highly effective CFTR modulator therapy
CFTR modulators were discovered on the basis of high throughput screening campaigns using cellular models of chloride transport, followed by medicinal chemistry to optimize drug-like properties3. Clinical proof of concept was first established using ivacaftor to potentiate CFTR activity of CFTR gating mutations, such as G551D, and subsequently with other CFTR mutations with retained surface expression but inadequate function4. As gating mutations were relatively responsive to small molecule therapy because of retained cell surface expression, activity in vivo was pronounced, helping to establish the potential for CFTR modulators to substantially alter clinical disease5. The more common F508del CFTR mutation proved to be a more challenging target, requiring both a corrector to increase CFTR cell surface expression and a CFTR potentiator to augment activity of the rescued channel6. While both lumacaftor/ivacaftor and tezacaftor/ivacaftor proved efficacious, bioactivity and clinical effects fell short of the standard established by ivacaftor in G551D, necessitating further research in modulators targeting F508del7. Most recently, a combination of tezacaftor and elexacaftor as correctors to more fully restore F508del CFTR expression, and ivacaftor to augment function, produced results in vitro and in clinical trials that met or exceeded the HEMT benchmark8. Consequently, phase 3 testing showed improved sweat chloride indicative of CFTR activity that approaches the diagnostic threshold of CF, substantial improvements in lung function -- including those transitioning from antecedent tezacaftor-ivacaftor, improved nutrition, lower rates of pulmonary exacerbation, and better self-reported quality of life1. Importantly, efficacy was also established in individuals with a single responsive F508del mutation, a population for whom previous CFTR modulator therapy was unsuccessful2. The development of ETI has helped establish the era of HEMT for a large majority of PwCF, and an understanding of the long-term effects of ETI and its influence on a broad array of clinical outcome measures is just beginning to emerge9.
The PROMISE Study and Core Assessments
PROMISE enrolls those prescribed ETI according to the FDA-approved label, which included PwCF age 12 years and older at the start of the study in late 2019 and at least 1 F508del mutation. The study is conducted at 56 CF research sites across the US and will follow participants for the first 2 years of ETI use. The study has fully enrolled 487 participants, 98% of whom successfully started ETI. Approximately 50% are homozygous for F508del, the majority of whom transitioned to ETI from a 2-drug modulator combination (i.e. tezacaftor/ivacaftor or lumacaftor/ivacaftor). The remaining study participants have 1 copy of F508del and largely have no prior modulator use, except for 35 who also have a G551D allele and previously used ivacaftor monotherapy (Table 1).
Table 1:
Genotype and prior modulator drug use in the PROMISE Study participants
| Genotype | modulator drug use when starting ETI | ||||
|---|---|---|---|---|---|
| None n=238 | IVA n=34 | LUM/IVA n=64 | TEZ/IVA n=151 | Total n=487 | |
| F508del homozygous | 27 (11%) | 0 (0%) | 63 (98%) | 146 (97%) | 236 (48%) |
| F508del plus minimal function mutation* | 192 (81%) | 0 (0%) | 1 (2%) | 2 (1%) | 195 (40%) |
| F508del plus G551D | 2 (1%) | 33 (97%) | 0 (0%) | 0 (0%) | 35 (7%) |
| F508del plus other mutation | 17 (7%) | 1 (3%) | 0 (0%) | 3 (2%) | 21 (4%) |
IVA: ivacaftor; LUM: lumacaftor; TEZ: tezacaftor
as defined in clinical trials of ETI
Research sites have been organized to focus on a shared group of assessments (i.e. ‘Core’ outcome measures) plus additional targeted outcomes distributed to a subgroup of sites and aligned with organ-system sub-studies (Figure 1). Core measurements include lung function by spirometry (e.g. percent predicted forced expiratory volume in 1 second (ppFEV1)), body mass index (BMI), respiratory symptom-based quality of life, and change in sweat chloride concentration (SwCl). In anticipation of FDA approval for ETI soon to include children 6 to 11 years old, a large pediatric sub-study was added. A separate but related study (BEGIN) will enroll those in infancy to age 5 years old and will evaluate the natural history of CF in young children prior to modulator use, followed by research measuring the impact of ETI in these children who generally have less established end organ damage.
Figure 1.
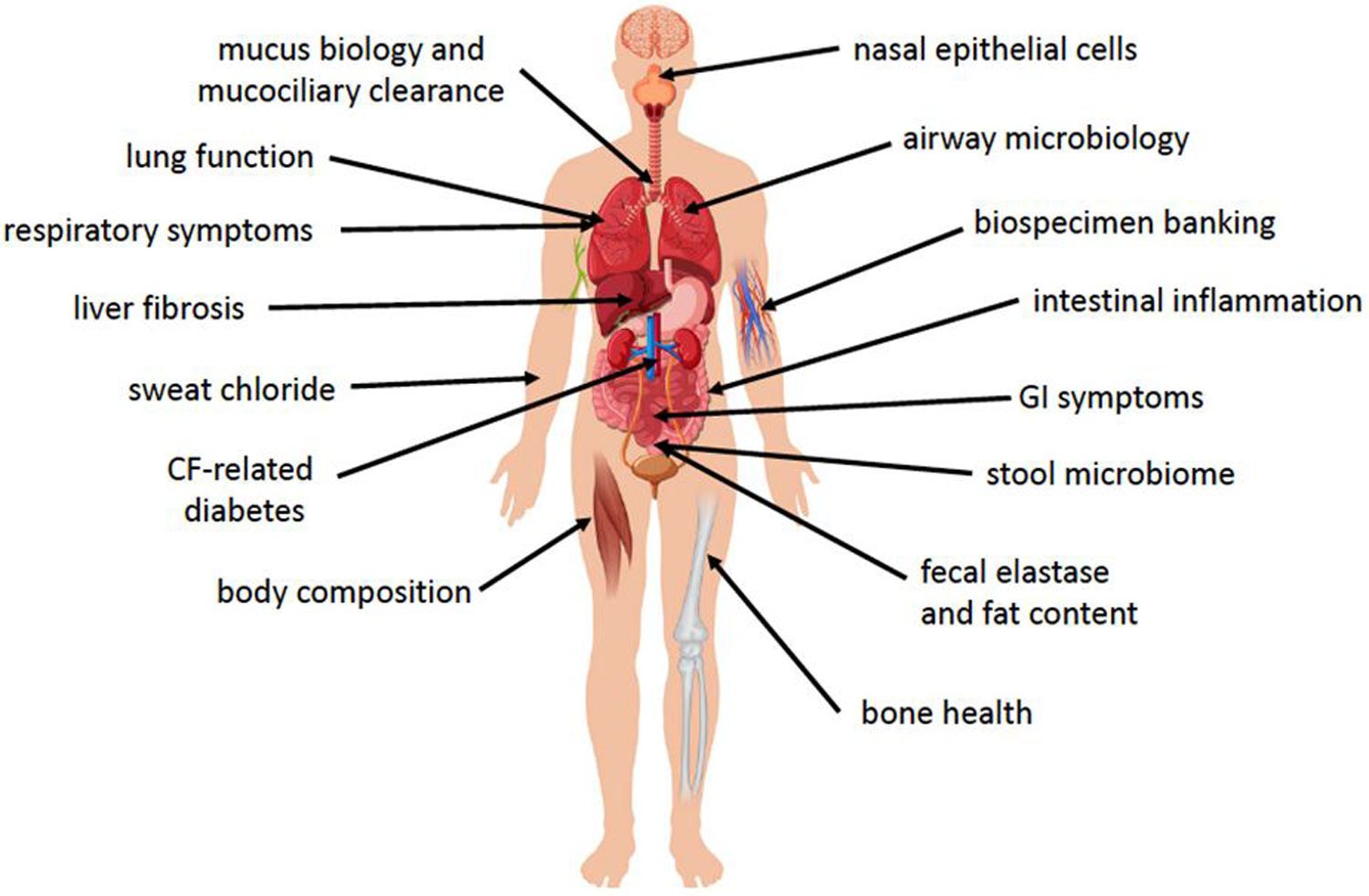
Organ systems being evaluated in the PROMISE study.
A wide range of organ systems and disease manifestations are being studied during the multi-year observational period. This will allow us to better understand the impacts of ETI therapy in clinical use and where it may be most beneficial to focus or prioritize ongoing therapeutic and research goals in those treated with highly effective modulator drugs.
The Core analyses of PROMISE are typical outcomes for CFTR modulator studies and will be important to test the clinical effectiveness of ETI in a real-world population. Apropos, only 48% of those enrolled in PROMISE met key eligibility criteria for the pivotal trials, mostly due to an upper limit inclusion criterion for ppFEV1. Linking these core data to the outcomes measured in the sub-studies allows investigators to test for associations with improvements in SwCl, respiratory symptoms, or lung function, and to explore potential biomarkers of disease status, response to therapy, or progression over time. A central feature of PROMISE is biospecimen collection for immediate use and banking (Table 2) to address unanticipated questions conceived by the broader CF community. SwCl, as a biomarker of CFTR function, will be serially measured during the 2 years in PROMISE. It is particularly interesting when considering those transitioning from other CFTR modulator regimens or those already using HEMT in the form of ivacaftor. A separate study (CHEC-SC NCT03350828) is focusing on SwCl on a population basis in a much larger-scale cross-sectional study of individuals beginning or transitioning between CFTR modulator therapies.
Table 2:
Primary and secondary investigational areas of focus sub-studies within PROMISE
| Focus Area | Key Outcomes of Interest | Additional Biomarkers or Assays |
|---|---|---|
| Clinical Changes | lung function (FEV1), respiratory symptoms, sweat chloride, BMI | |
| Mucus Biology | mucociliary clearance, percent solids in sputum | Sputum viscosity, exhaled breath condensate pH and sialic acid:urea ratio |
| Microbiology | P. aeruginosa and S. aureus sputum density, stool and respiratory microbiota, stool metagenome | S. maltophilia, A. xylosoxidans, and Burkholderia species, PCR vs. culture-based results, bacterial lineage stability |
| Inflammation and Host Response | Sputum free neutrophil elastase, serum hsCRP | Sputum IL-1B, IL-6, IL-8, IL-10, TNF-a, IFN-g, calprotectin, serum HMGB-1, calprotectin |
| Gastroenterology | GI symptoms (PAGI-SYM, PAC-SYM, PAC-QOL), intestinal pH and transit time | Bristol stool score, fecal calprotectin, elastase, steatocrit. Serum IRT, intestinal fatty acid binding protein, 7-hydroxy-4-cholesten-3-one, FGF-19. Urine prostaglandin E2 metabolites |
| Endocrinology | Effect on glucose metabolism/control (glucose excursion, peak, glucose tolerance category), insulin use and dosage. Lumbar and hip bone mineral density. Whole body lean body mass | Insulin, glucagon, incretin activities, free fatty acid flux and glucose disposal. Additional measures of bone mass, mineral content, and density. Additional measures of body composition, IGF-1, and IGF-BP3. |
| Liver disease | Transient elastography, serum C4, and FGF-19 | Liver disease classification, GGTP, FIB4, and APRI |
| Nasal airway epithelial cell function | Forskolin-stimulated Isc | Airway surface liquid depth, in vitro ciliary functioning and mucus viscosity |
| Pediatric (age 6–11y) participants | Lung clearance index, sweat chloride, FEV1, airway microbiology from sputum and oropharyngeal swab collection, respiratory and GI symptoms, BMI | |
| Biospecimen collection for future research | Historical and prospective data, serum, plasma, blood leukocytes, urine, bacterial colonies and bacterial DNA, stool, processed sputum, expanded nasal airway epithelial cells | |
Several important research questions will be left unanswered by PROMISE and are broadly relevant to HEMT, including the impact of HEMT on structural lung disease (e.g. CT or other imaging modalities), adherence to prescribed medications over time and its relationship to effectiveness, and the impact of ETI on CF-related sinus disease. Impacts on mental health and female fertility are not systematically collected. Additionally, while this study will record hospitalizations and outpatient antibiotic use, it is not designed to systematically capture all acute pulmonary exacerbation events. PROMISE does not include an untreated comparison group, which complicates estimation of impact on long-term outcomes such as lung function decline. Instead, analyses will be performed comparing participants to their historical data in the CF Foundation National Patient Registry10.
Mucus Biology and Mucociliary Clearance
CFTR dysfunction leads to progressive changes in airway mucus that result in its retention. Although restoration of CFTR activity with ivacaftor significantly improved mucociliary clearance (MCC)11, the combination of lumacaftor and ivacaftor in F508del homozygous individuals did not have meaningful effects on MCC. While this likely reflects different levels of CFTR restoration, little work has been done to investigate the biochemical and biophysical links between CFTR activation and MCC in humans. Understanding how restoration of CFTR improves MCC, whether due to improvements in mucus hydration, pH, viscosity/rheology, or a combination of these factors, could drive future drug development decisions.
In the PROMISE Mucus/MCC Sub-study, MCC will be measured in people with CF in those with mild to moderate pulmonary disease (FEV1 ≧ 40%) using a standardized gamma scintigraphy method12. We expect to observe substantial improvements in MCC measurements, which will provide a key functional endpoint against which novel and mechanistic outcome measures will be compared. Using induced sputum sampling, we will evaluate the effects of ETI on mucus hydration (percent solids) and rheologic properties. Exhaled breath condensate (EBC) collections will also be performed to measure pH, given the role of CFTR role in bicarbonate secretion, and mucus sialic acid:urea ratio as an index of mucin concentration13. Coupling these outcomes could yield insights into the mechanism by which MCC improves and its relationship to severity of underlying airway obstruction or degree of lung function improvement. Further, thresholds of CFTR activity sufficient to alter mucus viscosity and transport may be established.
Because MCC measurements require specialized research capabilities, we also hope to determine whether any of the described ex vivo assays might suitably substitute for MCC. This study may also help us understand whether ETI has fully corrected the underlying mechanisms of mucociliary dysfunction, and may elucidate treatment targets that guide alternative therapies that complement CFTR modulation, such as dornase alfa and hypertonic saline.
Airway Microbiology
Questions remain about the effects of modulators on chronic lung infections, a cardinal manifestation of CF. This has not been a focus of development efforts, and most knowledge comes from independent studies that have produced inconsistent results. For example, studies based on registry data showed ivacaftor decreased the prevalence of culture positivity for P. aeruginosa14–16. However, prospective studies using PCR and 16s rRNA gene sequencing have generally shown minimal changes in sputum microbiota. In contrast, a smaller study using quantitative culture found that ivacaftor rapidly reduced P. aeruginosa density and lung inflammation17. Interestingly, P. aeruginosa density rebounded in the second year of treatment, and many of the P. aeruginosa strains present before ivacaftor persisted for years after treatment despite sustained reductions in sweat chloride, raising questions about the durabilty of HEMT-related effects that need to be addresssed.
The PROMISE microbiology substudy examines nearly 250 participants with collection at 1, 3, 6, 12, and 24 month time points. Larger numbers will increase the probability of generating robust findings on CF pathogens and help identify target populations most likely to experience HEMT-related effects in microbiology. The serial timepoints will define changes in pathogen density over time. PROMISE collects induced sputum, addressing decrements in sputum production after modulator treatment. The use of a CLIA-certified laboratory for centralized analysis that occurs without freezing is advantagous for reliability and consistency. Work comparing culture and PCR measurements of P. aeruginosa showed that culture detected larger changes after ivacaftor or antibiotic treatment, perhaps because PCR measures both live and dead organisms17, 18. We will measure quantitative qPCR of targeted species, as well as 16S rRNA gene sequencing to measure a broader composition of microbial DNA reflective of both lower and upper airway components in sputum. Genome sequencing studies will determine if pathogen strains present pre-treatment persist. Sequencing may also identify bacterial adaptations to modulator-induced changes in the lung environment, and determine how modulators affect the composition of genetically-diversified pathogen populations.
One future challenge will be defining causal relationships between changes in respiratory microbiology and other important disease manifestations. For example, decreased pathogen density could reduce lung inflammation, increase mucociliary clearance, or both. Likewise, primary improvements in inflammation or mucociliary clearance could significantly alter airway microbiology. PROMISE will not examine residual host defense defects that may be present and impact bacterial survival after ETI. Similarly, testing the effects of combining antibiotics with modulator initiation to improve bacterial eradication will require a separate effort.
Effects of HEMT on Pulmonary and Systemic Inflammation
Inflammation is a hallmark feature of CF and plays a key role in the progression of lung injury. Previous studies that have investigated the effects of ivacaftor on airway inflammation have yielded mixed results. In the GOAL study, there were no significant changes in sputum markers of inflammation (free neutrophil elastase activity, α1-antitrypsin, secretory leukoprotease inhibitor, IL-1β, IL-6, IL-8) following 6 months of ivacaftor therapy in two distinct cohorts: one small group with milder lung disease assessed by sputum induction16, and a second with more impaired lung function who spontaneously expectorated sputum before and after initiation of therapy19. In contrast, a separate study found that ivacaftor markedly reduced sputum markers of inflammation (neutrophil elastase activity, IL-1β, IL-8) during the first year of treatment in a cohort of 12 adults with the G551D mutation, most of whom were chronically infected with P. aeruginosa17. These data suggest that at least in people with established lung disease, HEMT is unlikely to resolve inflammation. The PROMISE study provides an opportunity to determine the effects of restoration of CFTR function on airway and systemic inflammation, critical downstream consequences of impaired CFTR function. Individuals achieving normal or near-normal sweat chloride concentrations, may be a particularly interesting group in which to evaluate the extent to which inflammation is improved.
In PROMISE, induced sputum will be collected from up to 200 participants to detect changes from baseline in two important markers of neutrophilic inflammation (free neutrophil elastase, calprotectin), plus a predetermined cytokine panel will be serially examined through two years of treatment. Since sputum from these participants will also be evaluated for microbiologic outcomes on the same schedule, correlations between airway inflammation and infection can be assessed. This large, paired study population provides a real opportunity to understand key features of CF lung disease after starting HEMT.
Changes in systemic measures of inflammation were also measured in a subset of the GOAL cohort treated with ivacaftor20. Circulating HMGB-1 and calprotectin decreased significantly at 1 and 6 months after treatment initiation compared to baseline. In PROMISE, changes in serum high sensitivity C reactive protein, calprotectin, and HMGB-1 will be investigated. Importantly, if inflammation persists in the face of partial restoration of CFTR activity, this finding will support the need for adjunctive anti-inflammatory therapy in pwCF. Future studies will need to investigate how HEMT modulates the function of innate (neutrophils and macrophages) and adaptive (B and T lymphocytes) immune cells and whether HEMT alters the aberrant and dysregulated inflammatory responses by the airway epithelium during viral infections or other stimuli.
Gastrointestinal Disease and Symptoms
Gastrointestinal (GI) disease is another central feature of cystic fibrosis21. Malabsorption associated with pancreatic insufficiency, abnormal intestinal secretions, dysmotility, and chronic inflammation are all present in CF, and many individuals experience malnutrition and disordered growth22. Abdominal pain, bloating, abnormal stools, constipation, and gastro-esophogeal reflux are found in as many as 80% of pwCF23. These problems impact quality of life and survival24–26. Determining what impact HEMT has on these gastrointestinal problems is critical, especially as supportive care is inadequate for many pwCF.
Previous data demonstrated improvement in small bowel pH with ivacaftor in individuals with the G551D mutation27, but no change in transit time. Data collected on gastrointestinal motility profiles and intestinal pH in participants treated with ETI will be important to understanding its impact on GI ailments, particularly when correlations between symptoms, motility, and luminal pH are discerned.
Five areas of cystic fibrosis-related gastrointestinal disease will be evaluated in the PROMISE study: inflammation, malabsorption, dysmotility, luminal pH and symptomatology. These impact weight maintenance and growth. The target population includes all participants for assessment of symptoms and quality of life, and subgroups for examining inflammation, non-pancreatic malabsorption, exocrine pancreatic function, intestinal pH and dysmotility. Symptomatology and quality of life related to gastrointestinal disease are measured throughout the 2-year study using the Patient Reported Outcomes used in the GALAXY study (NCT03801993), while inflammation and exocrine pancreatic function are measured using fecal testing.
The fecal samples to be collected during PROMISE will also be analyzed for changes in the microbiome. Compared with the lung, the microbiology of the CF gastrointestinal (GI) tract remains relatively undefined; however, a growing number of studies have identified a CF dysbiosis that correlates with measures of GI inflammation, impaired nutrition and growth, and even the risk of respiratory exacerbations28–31. A study of 16 PwCF with pancreatic exocrine insufficiency31 identified concurrent improvements in fecal measures of dysbiosis and inflammation with ivacaftor treatment; another recent study of 12 pancreatic-sufficient PwCF demonstrated no such changes with ivacaftor32. The large size, frequent sample collection, and diversity of functional and symptomatic outcomes planned in the PROMISE study will define the role of the GI microbiome with much greater resolution. Similar to other areas of focus in PROMISE, additional follow up beyond two years may be necessary to understand the full GI effects of HEMT.
CF-Related Liver Disease
Liver involvement beyond laboratory-based safety monitoring has not been systematically studied in clinical trials or prior post-approval studies of CFTR modulator drugs. The primary focus has been on safety as determined by serum transaminases and bilirubin. Recent progress in the understanding of CF liver disease and its relationship to serum and imaging markers of liver fibrosis enabled the inclusion of markers of liver disease in PROMISE. Notably, liver disease is a leading source of morbidity and mortality in PwCF and may become increasingly important as pulmonary complications improve33, 34. Potential targets for therapeutic impact and related biomarkers chosen in PROMISE are shown in Table 2.
Several studies have demonstrated that AST to platelet ratio (APRI) is a good marker of hepatic fibrosis35. Recently transient elastography, which measures liver stiffness by ultrasound, was also shown to correlate well with hepatic fibrosis in several liver diseases36 and CF37. Thus, we will measure if hepatic fibrosis is a potential, yet unrecognized, therapeutic target when studying the clinical impact of ETI. Improvement in hepatic fibrosis as determined by APRI and transient elastography has been shown with successful treatment of viral hepatitis C38 and B39. One challenge in CF is the anticipated slow improvement in hepatic fibrosis. We chose a 2-year time frame for the evaluation of changes in markers of fibrosis, liver stiffness and liver classification as studies in viral hepatitis suggest that highly effective antiviral therapy can meaningfully impact these measures in 2–4 years. There is a gradual increase in liver stiffness in CF with age. We chose to focus on the rate of increase in liver stiffness, hypothesizing that we will see no increase in liver stiffness over 2 years of treatment. There is a significant need for an earlier (1–3 month) biomarker for changes in hepatic physiology.
Standard phenotypic characterization of CF liver involvement using phenotypic classification40 was selected as a target as it is clinically relevant to care in CF. Exploratory biomarkers FGF19 and 7α hydroxycholest-4-en-3-one (C4) have associated with improved bile flow and reduced malabsorption in other liver diseases41–43. CFTR is integral to bile acid flow, and improvement of CFTR function in the biliary epithelium would be expected to enhance bile flow and decrease bile acid malabsorption.
A key question regarding ETI in CF liver disease is whether it can reduce or prevent the development of advanced liver disease with portal hypertension that affects about 7% of PwCF. Testing this question may require drug initiation at an early age44 and better predictive biomarkers or long-term follow up. Information collected in this and other studies will also help guide future studies testing alternative therapeutic strategies.
Endocrine-related Disorders
With improved life-expectancy in individuals with CF, endocrine complications, including diabetes, bone disease, and reduced linear growth, are increasingly important medical considerations. Prevalence of CF-related diabetes (CFRD) increases with age and is present in nearly 50% of adults45. CF-related bone disease (CFBD) is also common, manifesting as decreased bone density and increased fracture rates46, and can be present even in childhood47. Additionally, shorter stature occurs with severe CFTR class mutations10.
Understanding how HEMT impacts insulin requirements in people with CFRD and progression to CFRD is critically important. Reductions in pancreatic islet mass, local and systemic inflammation, impaired intestinal secretion of incretins, and disrupted insulin secretion may all contribute to developing CFRD48–50. Improved insulin secretion, lower rates of progression to CFRD, and CFRD resolution were observed with ivacaftor5, 51, 52 although the impact of lumacaftor/ivacaftor on glucose tolerance and insulin secretion remains unresolved. PROMISE is prospectively collecting data on insulin requirements, hypoglycemia, and extended, multi-sample oral glucose tolerance tests performed at baseline, 12-, and 24-months to examine glucose tolerance/excursion, insulin secretory rates, hepatic insulin clearance, and incretin and glucagon secretion.
CFBD appears multifactorial and varies upon individuals: nutritional compromise, reduced physical activity, reduced lean body mass, systemic inflammation, hypogonadism, and systemic glucocorticoid therapy all likely have roles46. Data implicate both direct and indirect roles of CFTR in bone health and mineralization46. Complicating studies of bone health in youth, shorter stature and pubertal lag may inflate deficits in bone mineral density (BMD). Dual-energy X-ray absorptiometry (DXA) will be obtained for BMD and body composition annually. In youth, annualized bone accretion will be adjusted for growth, pubertal status, linear growth, and lean body mass (LBM) accrual, and in both youth and adults, changes in BMD-Z will be examined. Poor linear growth is independently associated with higher mortality53. Linear growth improvements occur with ivacaftor therapy and are anticipated with therapeutic CFTR correction during PROMISE. Age, gender, and pubertal status specific growth velocity Z-scores will be determined and compared with baseline growth velocity and to normative data. Greater LBM is hypothesized to contribute to the positive associations between nutritional status and pulmonary function54. CFTR modulator therapy is associated with weight gain and BMI increases5. Whole body DXA will be leveraged to assess LBM and fat mass changes in the context of BMI. Additionally, we will evaluate endocrine growth factors throughout the range of the study.
While PROMISE is positioned to provide important data on the impact of HEMT across a range of important endocrine-related CF disease manifestations, the durability of these effects will need to be considered in future studies. Similarly, the extent to which early HEMT initiation can preserve beta-cell mass and function or prevent rather than reverse abnormalities will require long-term studies with HEMT from a young age.
Advancing Personalized Outcome Measures
While it is established that the majority of those treated will garner benefit from ETI therapy, variability is expected in both effects on CFTR function and clinical status. Previously, work in the GOAL/GOALe2 (NCT01521338) and additional observational studies55–57 has demonstrated that respiratory epithelial tissues cultured at air-liquid interface (ALI) may act as an effective surrogate for an individual patient’s response to highly effective modulators. The PROMISE study will test the hypothesis that grouped and individual responses to CFTR restoration in vitro (e.g. short circuit current activity, increased airway surface layer depth, or other functional parameters determined by micro-optical tomography)58, 59 in human nasal epithelial (HNE) cells collected and cultured in the laboratory may serve as a biomarker for clinical outcomes. We will also test correlations of groups defined by genetic mutations.
Through this work, we hope to affirm that in vitro measures conducted using cultured primary nasal airway epithelial cells collected from participants correlate with clinical and other outcomes, thereby supporting HNE as a useful biomarker for clinical response to ETI. Second, we intend to establish a robust biorepository of these tissues to perform future investigations, including use of nasal spheroids as an alternative measure to quantify ion transport and fluid secretion60, genome-wide association studies for variability in response to ETI therapy, and other investigator-initiated projects. Findings will allow for more robust mechanistic understanding of the interplay of CFTR rescue and mucociliary clearance and serve as a validation for future use of HNE-based analyses as a predictive tool for CFTR modulators
Considerations for development in pediatric populations
Compared to adolescent and adults, pwCF 6 to 11 years of age generally have limited structural lung damage, relatively preserved lung function, and lower prevalence of chronic Pseudomonas infection. Thus, ETI may have greater potential to modify the natural history of CF in younger age groups. The PROMISE Pediatric sub-study (PROMISE PEDS; NCT04613128) will evaluate longitudinal outcomes associated with ETI initiation in children ages 6–11, providing an important step in our understanding of whether HEMT will be sufficient to control CF disease progression.
Similar to the parent PROMISE study, PROMISE PEDS will evaluate the short- and long-term impact of ETI on lung function, microbiology, nutritional status, sweat chloride, and patient-reported outcomes over 2 years. The primary outcome measure will be the lung clearance index (LCI) as measured by nitrogen multiple breath washout (MBW); spirometry will also be performed. LCI has been shown to be more sensitive than FEV1 to early CF lung disease, with a robust response to modulator therapies in several studies61–63.
CFTR modulator therapy may alter the lower airway milieu for microbial pathogens in several ways, including improvement in mucus clearance, stabilization or reversal of structural disease, and/or alteration of neutrophilic airways inflammation. As spontaneous sputum production is often not feasible in this age range, both induced sputum and oropharyngeal samples will be obtained for microbiologic analysis. Oropharyngeal cultures will ensure longitudinal microbiologic data in children unable to consistently produce sputum even with sputum induction. Key microbiologic outcomes will include reduction in sputum pathogen density and eradication of specific pathogens, particularly Pseudomonas aeruginosa and Staphylococcus aureus. Changes in the respiratory microbiota will be measured longitudinally. Associations between changes in lung function, airway microbiology and markers of airway inflammation will be explored. Respiratory samples will also be banked for future studies. Effects of modulator therapy on nutritional status and gastrointestinal health will be captured through longitudinal changes in BMI and GI-related patient-reported outcome measures, and ultimately compared with older cohorts.
Conclusions
The approval of ETI has established a new paradigm in care and research for the majority of PwCF. The PROMISE Study will capture important longitudinal data and biospecimens from these individuals, including those with the G551D CFTR mutation transitioning from ivacaftor monotherapy to ETI. This rapidly evolving landscape supports a rationale to study the clinical and translational impact of ETI across several organ systems, including its effects on novel biological assays and emerging biomarkers, fundamentally improving our understanding of future CF care needs and emerging therapeutic priorities. Large in scope, this US-based collaborative research will also complement other critical research programs around the world as ETI approval expands to other countries and younger individuals. Important questions of adherence to modulator drugs or pre-existing therapies64 and the impacts of starting HEMT early in life require attention and are being addressed by other post-approval studies like BEGIN, RECOVER, SIMPLIFY (NCT04378153), QUEST (NCT04320381), CF-STORM, HERO, and others. Collectively, we are positioned to comprehensively understand the real-world use and impact of ETI, which is likely to represent a key milestone in the treatment of this disease.
Acknowledgement:
This work is supported by the CF Foundation, Bethesda, MD USA.
Contributor Information
Dave P. Nichols, Seattle Children’s Research Institute, Department of Pediatrics, University of Washington
Scott H. Donaldson, Department of Medicine, Univ. North Carolina at Chapel Hill
Carla A. Frederick, Jacobs School of Medicine and Biomedical Sciences of the University of Buffalo
Steven D. Freedman, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA
Daniel Gelfond, Western New York Pediatric Gastroenterology.
Lucas R Hoffman, Seattle Children’s Research Institute, Department of Pediatrics and Microbiology, University of Washington.
Andrea Kelly, Department of Endocrinology, University of Pennsylvania.
Michael R. Narkewicz, Digestive Health Institute, Children’s Hospital Colorado and University of Colorado School of Medicine, Aurora, CO
Jessica E. Pittman, Department of Pediatrics, Washington University School of Medicine
Felix Ratjen, Department of Pediatrics, University of Toronto.
Margaret Rosenfeld, Seattle Children’s Research Institute, Department of Pediatrics and Epidemiology, University of Washington.
Scott D. Sagel, Department of Pediatrics, Children’s Hospital Colorado and University of Colorado School of Medicine, Aurora, CO.
Sarah Jane Schwarzenberg, University of Minnesota Masonic Children’s Hospital, Minneapolis, MN.
Pradeep K. Singh, Department of Microbiology, University of Washington
George M Solomon, Gregory Fleming James Cystic Fibrosis Research Center, University of Alabama at Birmingham, Birmingham, AL.
Michael S Stalvey, Gregory Fleming James Cystic Fibrosis Research Center, University of Alabama at Birmingham, Birmingham, AL.
Shannon Kirby, Therapeutics Development Network Coordinating Center, Seattle Children’s Hospital.
Jill M. VanDalfsen, Therapeutics Development Network Coordinating Center, Seattle Children’s Hospital
John P Clancy, Cystic Fibrosis Foundation.
Steven M Rowe, Gregory Fleming James Cystic Fibrosis Research Center, University of Alabama at Birmingham, Birmingham, AL.
References
- 1.Heijerman HGM, McKone EF, Downey DG, et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, phase 3 trial. Lancet. November 23 2019;394(10212):1940–1948. doi: 10.1016/S0140-6736(19)32597-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Middleton PG, Mall MA, Drevinek P, et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N Engl J Med. November 7 2019;381(19):1809–1819. doi: 10.1056/NEJMoa1908639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Van Goor F, Hadida S, Grootenhuis PD, et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci U S A. November 3 2009;106(44):18825–30. doi:0904709106[pii] 10.1073/pnas.0904709106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Accurso FJ, Rowe SM, Clancy JP, et al. Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N Engl J Med. November 18 2010;363(21):1991–2003. doi: 10.1056/NEJMoa0909825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ramsey BW, Davies J, McElvaney NG, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. November 03 2011;365(18):1663–72. doi: 10.1056/NEJMoa1105185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van Goor F, Hadida S, Grootenhuis PD, et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proceedings of the National Academy of Sciences of the United States of America. November 15 2011;108(46):18843–8. doi: 10.1073/pnas.1105787108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taylor-Cousar JL, Mall MA, Ramsey BW, et al. Clinical development of triple-combination CFTR modulators for cystic fibrosis patients with one or two F508del alleles. ERJ Open Res. April 2019;5(2)doi: 10.1183/23120541.00082-2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Keating D, Marigowda G, Burr L, et al. VX-445-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N Engl J Med. October 25 2018;379(17):1612–1620. doi: 10.1056/NEJMoa1807120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mall MA, Mayer-Hamblett N, Rowe SM. Cystic Fibrosis: Emergence of Highly Effective Targeted Therapeutics and Potential Clinical Implications. Am J Respir Crit Care Med. May 15 2020;201(10):1193–1208. doi: 10.1164/rccm.201910-1943SO [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.CF Foundation. Cystic Fibrosis Foundation Patient Registry 2019 Annual Data Report 2020.
- 11.Donaldson SH, Laube BL, Corcoran TE, et al. Effect of ivacaftor on mucociliary clearance and clinical outcomes in cystic fibrosis patients with G551D-CFTR. JCI Insight. December 20 2018;3(24)doi: 10.1172/jci.insight.122695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Donaldson SH, Corcoran TE, Laube BL, Bennett WD. Mucociliary clearance as an outcome measure for cystic fibrosis clinical research. Proc Am Thorac Soc. August 1 2007;4(4):399–405. [DOI] [PubMed] [Google Scholar]
- 13.Esther CR Jr., Hill DB, Button B, et al. Sialic acid-to-urea ratio as a measure of airway surface hydration. Am J Physiol Lung Cell Mol Physiol. March 1 2017;312(3):L398–L404. doi: 10.1152/ajplung.00398.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guimbellot JS, Baines A, Paynter A, et al. Long term clinical effectiveness of ivacaftor in people with the G551D CFTR mutation. J Cyst Fibros. November 25 2020;doi: 10.1016/j.jcf.2020.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heltshe SL, Mayer-Hamblett N, Burns JL, et al. Pseudomonas aeruginosa in cystic fibrosis patients with G551D-CFTR treated with ivacaftor. Clin Infect Dis. March 1 2015;60(5):703–12. doi: 10.1093/cid/ciu944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rowe SM, Heltshe SL, Gonska T, et al. Clinical mechanism of the cystic fibrosis transmembrane conductance regulator potentiator ivacaftor in G551D-mediated cystic fibrosis. Am J Respir Crit Care Med. July 15 2014;190(2):175–84. doi: 10.1164/rccm.201404-0703OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hisert KB, Heltshe SL, Pope C, et al. Restoring Cystic Fibrosis Transmembrane Conductance Regulator Function Reduces Airway Bacteria and Inflammation in People with Cystic Fibrosis and Chronic Lung Infections. Am J Respir Crit Care Med. June 15 2017;195(12):1617–1628. doi: 10.1164/rccm.201609-1954OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nelson MT, Wolter DJ, Eng A, et al. Maintenance tobramycin primarily affects untargeted bacteria in the CF sputum microbiome. Thorax. September 2020;75(9):780–790. doi: 10.1136/thoraxjnl-2019-214187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harris JK, Wagner BD, Zemanick ET, et al. Changes in Airway Microbiome and Inflammation with Ivacaftor Treatment in Patients with Cystic Fibrosis and the G551D Mutation. Annals of the American Thoracic Society. February 2020;17(2):212–220. doi: 10.1513/AnnalsATS.201907-493OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoppe JEW BW; Rowe SM; Heltshe S; Sagel SD Changes in circulating proteins with ivacaftor treatment in G551D subjects. In: 31st Annual North American Cystic Fibrosis Conference. Wiley; 2017:318. [Google Scholar]
- 21.Borowitz D, Gelfond D. Intestinal complications of cystic fibrosis. Curr Opin Pulm Med. November 2013;19(6):676–80. doi: 10.1097/MCP.0b013e3283659ef2 [DOI] [PubMed] [Google Scholar]
- 22.Borowitz D Update on the evaluation of pancreatic exocrine status in cystic fibrosis. Curr Opin Pulm Med. November 2005;11(6):524–7. doi: 10.1097/01.mcp.0000181474.08058.b3 [DOI] [PubMed] [Google Scholar]
- 23.Tabori H, Arnold C, Jaudszus A, et al. Abdominal symptoms in cystic fibrosis and their relation to genotype, history, clinical and laboratory findings. PLoS One. 2017;12(5):e0174463. doi: 10.1371/journal.pone.0174463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Habib AR, Manji J, Wilcox PG, Javer AR, Buxton JA, Quon BS. A systematic review of factors associated with health-related quality of life in adolescents and adults with cystic fibrosis. Annals of the American Thoracic Society. March 2015;12(3):420–8. doi: 10.1513/AnnalsATS.201408-393OC [DOI] [PubMed] [Google Scholar]
- 25.Rowbotham NJ, Smith S, Leighton PA, et al. The top 10 research priorities in cystic fibrosis developed by a partnership between people with CF and healthcare providers. Thorax. April 2018;73(4):388–390. doi: 10.1136/thoraxjnl-2017-210473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yen EH, Quinton H, Borowitz D. Better nutritional status in early childhood is associated with improved clinical outcomes and survival in patients with cystic fibrosis. J Pediatr. March 2013;162(3):530–535 e1. doi: 10.1016/j.jpeds.2012.08.040 [DOI] [PubMed] [Google Scholar]
- 27.Gelfond D, Heltshe S, Ma C, et al. Impact of CFTR Modulation on Intestinal pH, Motility, and Clinical Outcomes in Patients With Cystic Fibrosis and the G551D Mutation. Clin Transl Gastroenterol. March 16 2017;8(3):e81. doi: 10.1038/ctg.2017.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoen AG, Li J, Moulton LA, et al. Associations between Gut Microbial Colonization in Early Life and Respiratory Outcomes in Cystic Fibrosis. J Pediatr. July 2015;167(1):138–47 e1–3. doi: 10.1016/j.jpeds.2015.02.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hayden HS, Eng A, Pope CE, et al. Fecal dysbiosis in infants with cystic fibrosis is associated with early linear growth failure. Nat Med. February 2020;26(2):215–221. doi: 10.1038/s41591-019-0714-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Loman BR, Shrestha CL, Thompson R, et al. Age and environmental exposures influence the fecal bacteriome of young children with cystic fibrosis. Pediatr Pulmonol. July 2020;55(7):1661–1670. doi: 10.1002/ppul.24766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ooi CY, Syed SA, Rossi L, et al. Impact of CFTR modulation with Ivacaftor on Gut Microbiota and Intestinal Inflammation. Sci Rep. December 13 2018;8(1):17834. doi: 10.1038/s41598-018-36364-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pope CE, Vo AT, Hayden HS, et al. Changes in fecal microbiota with CFTR modulator therapy: A pilot study. J Cyst Fibros. December 31 2020;doi: 10.1016/j.jcf.2020.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pals FH, Verkade HJ, Gulmans VAM, et al. Cirrhosis associated with decreased survival and a 10-year lower median age at death of cystic fibrosis patients in the Netherlands. J Cyst Fibros. May 2019;18(3):385–389. doi: 10.1016/j.jcf.2018.11.009 [DOI] [PubMed] [Google Scholar]
- 34.Boelle PY, Debray D, Guillot L, Clement A, Corvol H, French CFMGSI. Cystic Fibrosis Liver Disease: Outcomes and Risk Factors in a Large Cohort of French Patients. Hepatology. April 2019;69(4):1648–1656. doi: 10.1002/hep.30148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leung DH, Khan M, Minard CG, et al. Aspartate aminotransferase to platelet ratio and fibrosis-4 as biomarkers in biopsy-validated pediatric cystic fibrosis liver disease. Hepatology. November 2015;62(5):1576–83. doi: 10.1002/hep.28016 [DOI] [PubMed] [Google Scholar]
- 36.Baranova A, Lal P, Birerdinc A, Younossi ZM. Non-invasive markers for hepatic fibrosis. BMC Gastroenterol. August 17 2011;11:91. doi: 10.1186/1471-230X-11-91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lewindon PJ, Puertolas-Lopez MV, Ramm LE, et al. Accuracy of Transient Elastography Data Combined With APRI in Detection and Staging of Liver Disease in Pediatric Patients With Cystic Fibrosis. Clin Gastroenterol Hepatol. March 15 2019;doi: 10.1016/j.cgh.2019.03.015 [DOI] [PubMed] [Google Scholar]
- 38.Elsharkawy A, Alem SA, Fouad R, et al. Changes in liver stiffness measurements and fibrosis scores following sofosbuvir based treatment regimens without interferon. J Gastroenterol Hepatol. September 2017;32(9):1624–1630. doi: 10.1111/jgh.13758 [DOI] [PubMed] [Google Scholar]
- 39.Liu R, Guo J, Lu Y, et al. Changes in APRI and FIB-4 in HBeAg-negative treatment-naive chronic hepatitis B patients with significant liver histological lesions receiving 5-year entecavir therapy. Clin Exp Med. August 2019;19(3):309–320. doi: 10.1007/s10238-019-00560-z [DOI] [PubMed] [Google Scholar]
- 40.Flass T, Narkewicz MR. Cirrhosis and other liver disease in cystic fibrosis. J Cyst Fibros. December 19 2013;12:116–24. doi: 10.1016/j.jcf.2012.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li Z, Lin B, Lin G, et al. Circulating FGF19 closely correlates with bile acid synthesis and cholestasis in patients with primary biliary cirrhosis. PLoS One. 2017;12(6):e0178580. doi: 10.1371/journal.pone.0178580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu WY, Xie DM, Zhu GQ, et al. Targeting fibroblast growth factor 19 in liver disease: a potential biomarker and therapeutic target. Expert Opin Ther Targets. May 2015;19(5):675–85. doi: 10.1517/14728222.2014.997711 [DOI] [PubMed] [Google Scholar]
- 43.Lenicek M, Juklova M, Zelenka J, et al. Improved HPLC analysis of serum 7alpha-hydroxycholest-4-en-3-one, a marker of bile acid malabsorption. Clin Chem. June 2008;54(6):1087–8. doi: 10.1373/clinchem.2007.100107 [DOI] [PubMed] [Google Scholar]
- 44.Leung DH, Ye W, Molleston JP, et al. Baseline Ultrasound and Clinical Correlates in Children with Cystic Fibrosis. J Pediatr. October 2015;167(4):862–868 e2. doi: 10.1016/j.jpeds.2015.06.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moran A, Becker D, Casella SJ, et al. Epidemiology, pathophysiology, and prognostic implications of cystic fibrosis-related diabetes: a technical review. Diabetes Care. December 2010;33(12):2677–83. doi: 10.2337/dc10-1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stalvey MS, Clines GA. Cystic fibrosis-related bone disease: insights into a growing problem. Curr Opin Endocrinol Diabetes Obes. December 2013;20(6):547–52. doi: 10.1097/01.med.0000436191.87727.ec [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kelly A, Schall J, Stallings VA, Zemel BS. Trabecular and cortical bone deficits are present in children and adolescents with cystic fibrosis. Bone. September 2016;90:7–14. doi: 10.1016/j.bone.2016.04.030 [DOI] [PubMed] [Google Scholar]
- 48.Guo JH, Chen H, Ruan YC, et al. Glucose-induced electrical activities and insulin secretion in pancreatic islet β-cells are modulated by CFTR. Nat Commun. July 2014;5:4420. doi: 10.1038/ncomms5420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hillman M, Eriksson L, Mared L, Helgesson K, Landin-Olsson M. Reduced levels of active GLP-1 in patients with cystic fibrosis with and without diabetes mellitus. J Cyst Fibros. March 2012;11(2):144–9. doi: 10.1016/j.jcf.2011.11.001 [DOI] [PubMed] [Google Scholar]
- 50.Hart NJ, Aramandla R, Poffenberger G, et al. Cystic fibrosis-related diabetes is caused by islet loss and inflammation. JCI Insight. April 2018;3(8)doi: 10.1172/jci.insight.98240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bellin MD, Laguna T, Leschyshyn J, et al. Insulin secretion improves in cystic fibrosis following ivacaftor correction of CFTR: a small pilot study. Pediatr Diabetes. September 2013;14(6):417–21. doi: 10.1111/pedi.12026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Volkova N, Moy K, Evans J, et al. Disease progression in patients with cystic fibrosis treated with ivacaftor: Data from national US and UK registries. J Cyst Fibros. January 2020;19(1):68–79. doi: 10.1016/j.jcf.2019.05.015 [DOI] [PubMed] [Google Scholar]
- 53.Vieni G, Faraci S, Collura M, et al. Stunting is an independent predictor of mortality in patients with cystic fibrosis. Clinical nutrition. June 2013;32(3):382–5. doi: 10.1016/j.clnu.2012.08.017 [DOI] [PubMed] [Google Scholar]
- 54.Pedreira CC, Robert RG, Dalton V, et al. Association of body composition and lung function in children with cystic fibrosis. Pediatr Pulmonol. March 2005;39(3):276–80. doi: 10.1002/ppul.20162 [DOI] [PubMed] [Google Scholar]
- 55.Debley JS, Barrow KA, Rich LM, Singh P, McKone EF, Nichols DP. Correlation between Ivacaftor-induced CFTR Activation in Airway Epithelial Cells and Improved Lung Function: A Proof-of-Concept Study. Ann Am Thorac Soc. August 2020;17(8):1024–1027. doi: 10.1513/AnnalsATS.202001-082RL [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pranke IM, Hatton A, Simonin J, et al. Correction of CFTR function in nasal epithelial cells from cystic fibrosis patients predicts improvement of respiratory function by CFTR modulators. Sci Rep. August 7 2017;7(1):7375. doi: 10.1038/s41598-017-07504-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Solomon G, Liu Z, Baines A, et al. IN VITRO RESPONSES OF G551D AND R117H HUMAN NASAL EPITHELIAL CELLS CORRELATES TO CLINICAL IMPROVEMENT TO IVACAFTOR In: North American CF Conference Denver, CO. 2018; [Google Scholar]
- 58.Birket SE, Chu KK, Houser GH, et al. Combination therapy with cystic fibrosis transmembrane conductance regulator modulators augment the airway functional microanatomy. Am J Physiol Lung Cell Mol Physiol. May 15 2016;310(10):L928–39. doi: 10.1152/ajplung.00395.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Peabody JE, Shei RJ, Bermingham BM, et al. Seeing cilia: imaging modalities for ciliary motion and clinical connections. Am J Physiol Lung Cell Mol Physiol. June 1 2018;314(6):L909–L921. doi: 10.1152/ajplung.00556.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu Z, Anderson JD, Deng L, et al. Human Nasal Epithelial Organoids for Therapeutic Development in Cystic Fibrosis. Genes (Basel). May 29 2020;11(6)doi: 10.3390/genes11060603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Davies J, Sheridan H, Bell N, et al. Assessment of clinical response to ivacaftor with lung clearance index in cystic fibrosis patients with a G551D-CFTR mutation and preserved spirometry: a randomised controlled trial. Lancet Respir Med. October 2013;1(8):630–638. doi: 10.1016/S2213-2600(13)70182-6 [DOI] [PubMed] [Google Scholar]
- 62.Ratjen F, Hug C, Marigowda G, et al. Efficacy and safety of lumacaftor and ivacaftor in patients aged 6–11 years with cystic fibrosis homozygous for F508del-CFTR: a randomised, placebo-controlled phase 3 trial. Lancet Respir Med. July 2017;5(7):557–567. doi: 10.1016/S2213-2600(17)30215-1 [DOI] [PubMed] [Google Scholar]
- 63.Shaw M, Khan U, Clancy JP, et al. Changes in LCI in F508del/F508del patients treated with lumacaftor/ivacaftor: Results from the prospect study. J Cyst Fibros. June 6 2020;doi: 10.1016/j.jcf.2020.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mayer-Hamblett N, Nichols DP, Odem-Davis K, et al. Evaluating the Impact of Stopping Chronic Therapies after Modulator Drug Therapy in Cystic Fibrosis: The SIMPLIFY Study Design. Ann Am Thorac Soc. January 19 2021;doi: 10.1513/AnnalsATS.202010-1336SD [DOI] [PMC free article] [PubMed] [Google Scholar]