

Abstract
Background
The unfolded protein response (UPR) served as a vital role in the progression of tumors, but the molecule mechanisms of UPR in bladder cancer (BLCA) have been not fully investigated.
Methods
We identified differentially expressed unfolded protein response-related genes (UPRRGs) between BLCA samples and normal bladder samples in the Cancer Genome Atlas (TCGA) database. Univariate Cox analysis and the least absolute shrinkage and selection operator penalized Cox regression analysis were used to construct a prognostic signature in the TCGA set. We implemented the validation of the prognostic signature in GSE13507 from the Gene Expression Omnibus database. The ESTIMATE, CIBERSORT, and ssGSEA algorithms were used to explore the correlation between the prognostic signature and immune cells infiltration as well as key immune checkpoints (PD-1, PD-L1, CTLA-4, and HAVCR2). GDSC database analyses were conducted to investigate the chemotherapy sensitivity among different groups. GSEA analysis was used to explore the potential mechanisms of UPR-based signature.
Results
A prognostic signature comprising of seven genes (CALR, CRYAB, DNAJB4, KDELR3, CREB3L3, HSPB6, and FBXO6) was constructed to predict the outcome of BLCA. Based on the UPRRGs signature, the patients with BLCA could be classified into low-risk groups and high-risk groups. Patients with BLCA in the low-risk groups showed the more favorable outcomes than those in the high-risk groups, which was verified in GSE13507 set. This signature could serve as an autocephalous prognostic factor in BLCA. A nomogram based on risk score and clinical characteristics was established to predict the over survival of BLCA patients. Furthermore, the signature was closely related to immune checkpoints (PD-L1, CTLA-4, and HAVCR2) and immune cells infiltration including CD8+ T cells, follicular helper T cells, activated dendritic cells, and M2 macrophages. GSEA analysis indicated that immune and carcinogenic pathways were enriched in high-risk group.
Conclusions
We identified a novel unfolded protein response-related gene signature which could predict the over survival, immune microenvironment, and chemotherapy response of patients with bladder cancer.
Keywords: Unfolded protein response, Bladder cancer, Prognostic, Biomarker
We first comprehensive investigated the expression, protein–protein interaction, and prognostic value of unfolded protein response-related gene in bladder cancer.
We constructed and validated a novel prognostic signature based on seven unfolded protein response-related genes, which was an independent prognostic factors in bladder cancer.
We revealed the correlation between the prognostic signature and tumor immunosuppressive microenvironment.
We investigated the relationship between the prognostic signature and sensitivity of common chemotherapy drugs.
Introduction
Bladder cancer (BLCA) is one of the most common genitourinary cancers and leads to unfavorable outcomes and unsatisfactory quality of life worldwide. Smoking and occupational exposures are closely associated with the tumorigenesis and progression of BLCA [1]. At present, surgical treatment remains the main therapeutic strategy for patients with BLCA. Despite several achievements in surgical equipment and systemic therapy, incidence and mortality of BLCA remain unacceptable [2]. Therefore, investigating the molecular mechanisms and identifying novel biomarkers are of enormous clinical significance to BLCA research and management.
The unfolded protein response (UPR), a prevalent biological phenomenon in eukaryotic cells, is an intracellular signaling pathway of adaptive cell self-protection [3]. Endoplasmic reticulum (ER) homeostasis is constantly threatened by physiological demands and pathological insults such as ER calcium depletion, hypoxia, altered glycosylation, nutrient deprivation, oxidative stress, and DNA damage, which can disrupt the protein folding process and subsequently result in accumulation of unfolded or misfolded proteins in the ER [3–6]. To dispose the accumulation of the unfolded or misfolded proteins, a series of signal transduction pathways, also collectively known as UPR, were activated in the ER, to maintain ER homeostasis through altering transcriptional and translational programs [7–10]. Several studies have confirmed that UPR has been implicated in the tumorigenesis owing to the rapid proliferation of tumor cells [5, 11, 12]. The research progress of targeted UPR in cancer therapy has also attracted increasing attention [7, 13–16].
In the present study, we investigated the mRNA expression profiles of UPR-related genes from several public databases and established a novel UPR-related gene signature for predicting the outcomes and chemotherapy sensitivity of patients with BLCA. Furthermore, we also found that the UPR-based signature was closely correlated with immune cell infiltration and key immune checkpoints.
Materials and methods
Data sources
Unfolded protein response-related genes (UPRRGs) were obtained from UALCAN (ualcan.path.uab.edu/home) database. The mRNA sequencing and clinical data for patients with BLCA were downloaded from The Cancer Genome Atlas (TCGA, https://portal.gdc.cancer.gov/) database and included data for 19 normal bladder samples and 414 BLCA samples. In addition, GSE13507 [17], containing 165 primary BLCA samples, was used for verification studies.
Identification of differentially expressed UPRRGs
We used limma package in R software to identify differentially expressed UPRRGs (DEUPRRGs) in the TCGA-BLCA dataset. UPRGs with |log2fold change (FC)|> 1 and false discovery rate (FDR) < 0.05 were considered significantly differentially expressed.
Gene ontology and KEGG analyses
To further explore the potential molecular mechanisms in which the DEUPRRGs were involved, Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses of DELRGs were performed with packages (ggplot2, org.Hs.eg.db, enrichplot, and clusterProfiler) in R software. P < 0.05 was considered statistically significant.
Protein–protein interaction (PPI) network
The STRING database (http://www.string-db.org/) was applied to acquire protein–protein interaction (PPI) information related to DEUPRRGs. Cytoscape software (version 3.7.2) was utilized to establish and visualize the PPI network.
Construction and validation of the prognostic signature based on DEUPRRGs
To identify the DEUPRRGs associated with prognosis in patients with BLCA in the TCGA dataset, univariate Cox regression analysis was performed. Only DEUPRRGs with p < 0.05 were selected for subsequent construction of a risk signature with the least absolute shrinkage and selection operator (LASSO) Cox regression model through packages (glmnet and survival) in R software. The risk score was calculated with the following formula: risk scores= (where X is the coefficient of the prognosis-related DEUPRRGs and Y is the expression of the relevant gene). BLCA patients were categorized into two groups (high-risk and low-risk) based on the median risk score. Kaplan–Meier analysis was used to compare the survival of BLCA patients between the two groups. Time-dependent receiver operating characteristic (ROC) curves were applied to evaluate the reliability of the constructed signature for predicting prognosis. In addition, GSE13507 was used to verify the performance of this prognostic signature via the same method.
Construction of a nomogram
We investigated the correlation between clinicopathological characteristics and the prognostic signature. Univariate and multivariate Cox regression analyses were performed to ascertain whether this prognostic signature was an independent prognostic indicator. To predict the overall survival of BLAC patients at 3 and 5 years, we constructed a nomogram composed of risk scores and clinical variables with R package (rms). The consistency index was used to assess the accuracy of the nomogram. The calibration curve was utilized to visualize the performance of the nomogram.
Gene set enrichment analysis
Gene set enrichment analysis (GSEA) was conducted to further explore the potential molecular mechanisms among different groups. FDR < 5% and p < 0.05 was considered statistically significant.
Immune infiltration analyses
Given the importance of tumor immune microenvironment, ESTIMATE algorithm was conducted to evaluate the stromal score, ESTIMATE score, and immune score and investigate the relationship between risk score and tumor microenvironment. The Cell type Identification By Estimating Relative Subsets Of RNA Transcripts (CIBERSORT) algorithm was used to identify the immune cell fractions of 22 distinct leukocyte subsets between different groups. Furthermore, we also performed single-sample gene set enrichment analysis (ssGSEA) to evaluate the differences in immune-related pathways between the two groups via package (gsva) in R software. P values < 0.05 were considered statistically significant.
Chemotherapy sensitivity prediction
To explore the difference of chemotherapy sensitivity between different groups, we used GDSC database to estimate the half maximal inhibitory concentration (IC50) of chemotherapy drugs for predicting the sensitivity of chemotherapy drugs by using the package (pRRophetic). P < 0.05 were considered statistically significant.
Validation of seven prognostic genes
The mRNA expression and promoter methylation level of the seven UPR genes in the prognostic signature in normal tissues and cancer tissues were examined in the UALCAN database (http://ualcan.path.uab.edu/index.html).
Immune infiltrates analysis of seven prognostic genes
The Tumor IMmune Estimation Resource (TIMER, https://cistrome.shinyapps.io/timer/) database was used to explore the relationship between the immune cell abundances and the seven prognostic genes in BLCA. To further validate the correlation of seven genes with immune cells markers, the Gene Expression Profiling Interactive Analysis (GEPIA, https://gepia.cancer-pku.cn) database was used. Statistical analysis was conducted by Spearman’s correlation.
Statistical analyses
All these statistical analyses were conducted in R, version 3.6.2. Gene expression, clinical characteristics, immune cell infiltration, and risk score were compared between different groups through the Wilcoxon test. The difference in survival among different groups was evaluated by Kaplan–Meier curve. Independent prognostic analysis was performed by univariate and multivariate Cox regression. P < 0.05 was considered statistically significant.
Results
Differentially expressed UPRRGs and functions
We acquired 251 UPRRGs from the UALCAN database and analyzed the expression of these genes in BLCA samples compared with normal bladder tissues in the TCGA dataset. Forty-four UPRRGs were considered as differentially expressed genes in BLCA with the criteria of |log2 FC|> 1 and FDR < 0.05, including 21 downregulated genes and 23 upregulated genes (Fig. 1A, B). The results of GO analyses showed that the most enriched GO terms in the biological processes were response to topologically incorrect protein, response to unfolded protein, response to endoplasmic reticulum stress, cellular response to unfolded protein, and endoplasmic reticulum unfolded protein response (Fig. 2A). In the cellular component category, the DEUPRRGs were mainly enriched in endoplasmic reticulum lumen, collagen-containing extracellular matrix, inclusion body, and chaperone complex. In the molecular function category, chaperone binding, unfolded protein binding, cAMP response element binding, structural constituent of eye lens, and glycolipid binding were enriched in these DEUPRRGs. In the KEGG pathway analyses, the results showed that the DEUPRRGs were mainly involved in protein processing in the endoplasmic reticulum, human T-cell leukemia virus 1 infection, the p53 signaling pathway, and longevity regulating pathway (Fig. 2B).
Fig. 1.

Differentially expressed UPRRGs in TCGA-BLCA set. A Heatmap showed differentially expressed UPRRGs; B volcano presented of differentially expressed UPRRGs, red showed the over-expression of UPR gene, and green showed the down-expression of UPR gene, FC: fold change, fdr: false discovery rate
Fig. 2.

Enrichment analysis of differentially expressed UPRRGs. A GO analysis including biological process, cellular component, and molecular function; B KEGG analysis
PPI network
To better understand the roles of these DEUPRRGs in regulating BLCA, a PPI network was constructed through the utilization of the STRING database. Then we used Cytoscape software to analyze and visualize the PPI network, which contained 33 nodes and 57 edges (Fig. 3).
Fig. 3.

Protein–protein interaction network
Construction and validation of a SLERGs-based signature
We used univariate Cox regression analysis to identify the DEUPRRGs closely correlated with the overall survival of BLCA patients. Eleven DEUPRRGs were selected for subsequent analyses (p < 0.05) (Fig. 4).We successfully constructed a prognostic model consisting of 7 DEUPRGs (CALR, CRYAB, DNAJB4, KDELR3, CREB3L3, HSPB6, and FBXO6) by performing LASSO Cox regression analysis based on the 11 candidate genes. The risk score was calculated as follows: Risk Score = CALR expression × 0.3265 + DNAJB4 expression × 0.2036 + CRYAB expression × 0.0609 + KDELR3 expression × 0.0715 − CREB3L3 expression × 0.5633 − FBXO6 expression × 0.3616 + HSPB6 expression × 0.0096. BLCA patients were classified into high- and low-risk groups based on the median risk score. The Kaplan–Meier curves showed that patients in the low-risk group had better outcomes than those in the high-risk group (Fig. 5A). The area under the ROC (AUC) curve value of the constructed model was 0.710 at 5 years (Fig. 5D). The survival status of each patient was represented on dot plots, which showed that patients in the low-risk group had more favorable outcomes than those in the high-risk group (Fig. 5B). Figure 5C indicates that the high-risk group highly expressed CRYAB, DNAJB4, HSPB6, KDELR3, and CALR, while the low-risk group highly expressed FBXO6 and CREB3L3. We also verified the prognostic capacity of this prognostic signature in the Gene Expression Omnibus (GEO) dataset, showing consistent results between GEO dataset and TCGA dataset (Fig. 5E, F, H). These results indicated acceptable performance of the prognostic signature.
Fig. 4.
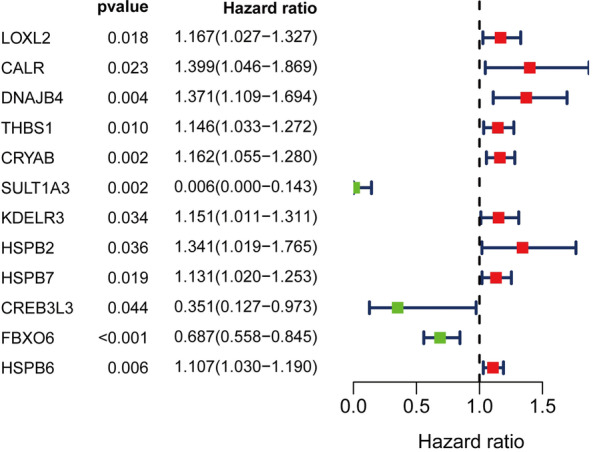
Univariate Cox regression analysis of differentially expressed UPRRGs
Fig. 5.

The correlation between the prognostic signature and outcome of BLCA patients. A–E Kaplan–Meier survival analysis of BLCA patients between high-risk groups and low-risk groups in TCGA set (A) and GSE13507 set (E); B–F distribution of survival status based on the median risk score in TCGA set (B) and GSE13507 set (F); C–G heatmap displayed the seven prognostic genes in TCGA set (C) and GSE13507 set (G); D–H time-independent receiver operating characteristic (ROC) analysis of risk scores predicting the overall survival in TCGA set (D) and GSE13507 set (H)
Construction of a nomogram
Univariate and multivariate Cox proportional hazard models were performed to identify the prognostic variables. Based on the univariate analysis, age (HR 1.037; 95% CI 1.020–1.055; p < 0.001), TNM stage (HR 1.783; 95% CI 1.440–2.207; p < 0 0.001), T stage (HR 1.569; 95% CI 1.233–1.998; p < 0 0.001), N stage (HR 1.548; 95% CI 1.317–1.819; p < 0.001), and risk score (HR 3.556; 95% CI 2.315–5.462; p < 0.001) were believed to be related to unfavorable outcomes (Fig. 6A). In the multivariate analysis, age (HR 1.034; 95% CI 1.016–1.052; p < 0.001) and risk score (HR 2.724; 95% CI 1.751–4.238; p < 0.001) could serve as independent prognostic factors (Fig. 6B). To further predict the outcomes of BLCA patients, we constructed a nomogram consisting of the risk score and age. The nomogram predicted the 3- and 5-year survival rates of patients with BLCA (Fig. 7A). The calibration curve showed that the actual patient outcomes were in keeping with the predicted outcomes (Fig. 7B, C). The C index of the nomogram was 0.645, which indicated the definite predictive ability of the nomogram.
Fig. 6.

The prognostic signature was an independent prognostic factor for BLCA in TCGA set. A The correlations between the risk score for OS and clinicopathological factors by univariate Cox regression analysis; B the correlations between the risk score for OS and clinicopathological factors by multivariate Cox regression analysis; C ROC curves of the clinical characteristics and risk score
Fig. 7.

Construction of a nomogram. A Nomogram for predicting 3‐ or 5‐year OS; B the calibration plots for predicting 3‐year OS; C the calibration plots for predicting 5‐year OS
The correlation between the prognostic signature and clinical characteristics
We also explored the relationship between the prognostic signature and clinical characteristics. The results showed that the elevated risk score was closely correlated with several inferior clinical characteristics (TNM stage, T stage, N stage, and grade) (Fig. 8A, B). With an increase in risk score, the expression levels of CALR, CRYAB, DNAJB4, KDELR3, and HSPB6 were increased, while the expression levels of FBXO6 and CREB3L3 were decreased. In addition, the elevated expressions of CALR, CRYAB, DNAJB4, KDELR3, and HSPB6 were associated with inferior clinical characteristics, while the under-expressions of FBXO6 and CREB3L3 were associated with superior clinical characteristics. Furthermore, the signature was related to unfavorable prognosis in certain subgroups stratified by sex (male or female), age (> 65 years) or (≤ 65 years), N stage (N0), TNM stage (I-II) or (III-IV), grade (high), and T stage (T3-T4) (Fig. 9). However, the signature showed no differences in other subgroups of N stage (N1–N2–N3), grade (low), and T stage (T1–T2).
Fig. 8.

The correlation between the signature and clinical characteristics. A Heatmap showed the correlation between the risk scores and clinicopathological factors. The elevated risk score was closely correlated with several inferior clinical characteristics (TNM stage, T stage, N stage, and grade). P: 0.05 > * > 0.01 > ** > 0.001 > ***. B Boxplot showed the correlation between the risk scores and clinicopathological factors
Fig. 9.

Subgroup analysis
GSEA
We performed GSEA to explore the signaling pathways that might be closely enriched among two groups. The results of GSEA indicated that focal adhesion, dilated cardiomyopathy, ECM receptor interaction, TGF-β signaling pathway, GAP junction, pathway in cancer, calcium signaling pathway, MAPK signaling pathway, chemokine signaling pathway, GnRH signaling pathway, and hedgehog signaling pathway were mainly enriched in high-risk group (Fig. 10A). In contrast, oxidative phosphorylation, retinol metabolism, fatty acid metabolism, linoleic acid metabolism, tyrosine metabolism, PPAR signaling pathway, and Parkinson's disease were mainly enriched in low-risk group (Fig. 10B).
Fig. 10.

GSEA analysis between high-risk group (A) and low-risk group (B)
Immune cell infiltration analyses
To further investigate the relationship between risk score and immune cell infiltration, ESTIMATE algorithm was first performed. We found that stromal score, immune score, and ESTIMATE score were remarkably higher in high-risk group than those in low-risk group (Fig. 11A). In addition, the results of the CIBERSORT algorithm showed the significant differences in the proportions of distinct leukocyte subsets between different groups. The proportions of CD8+ T cells, follicular helper T cells, and activated dendritic cells were higher in the low-risk group, whereas those of M0 macrophages, M2 macrophages, and neutrophils were higher in the high-risk group (Fig. 11B). Furthermore, the ssGSEA results indicated that a majority of immune-related pathways had higher enrichment scores in the high-risk group than in the low-risk group. Cytolytic activity, inflammation promotion, MHC class I, type I IFN response, and type II IFN response were not significantly different between the two groups (Fig. 11C). CIBERSORT results showed that immune suppressive cells were mainly enriched in high-risk group (Fig. 12A). To verify the immunosuppressive status of high-risk group, we further investigated the expression of immune suppressive molecules among different groups. The results indicated that immune suppressive molecules were elevated in high-risk group, suggesting that patients of high-risk group have inferior activities of anticancer immune response (Fig. 12B). Furthermore, we also analyzed the expression of key immune checkpoints (PDCD1, PD-L1(CD274), CTLA-4, and HAVCR2) and found that PD-L1, CTLA-4, and HAVCR2 were upregulated in high-risk group (Fig. 12C). In addition, chemokines implicated in immunosuppressive process (IL-10, TGF-β1, TGF-β2, TGF-β3) were also elevated upregulated in high-risk group (Fig. 12D). All these results indicated that the unfavorable outcomes of high-risk patients might be owing to the immunosuppressive microenvironment.
Fig. 11.

Immune cells infiltration and correlated pathways between high-risk groups and low-risk groups. A Immune microenvironment analysis between high-risk groups and low-risk groups by ESTIMATE; B immune cells infiltration between high-risk groups and low-risk groups by CIBERSORT; C immune-related pathways between high-risk groups and low-risk groups by ssGSEA. P: 0.05 > * > 0.01 > ** > 0.001 > ***
Fig. 12.

The correlation between immune microenvironment and the prognostic signature. A Immune suppressive cells between high-risk groups and low-risk groups; B immune suppressive molecules between high-risk groups and low-risk groups; C key immune checkpoints between high-risk groups and low-risk groups; D immunosuppressive chemokines between high-risk groups and low-risk groups. P: 0.05 > * > 0.01 > ** > 0.001 > ***
TIMER analysis
The TIMER database was used to investigate the correlation between the mRNA expressions of CALR, CREB3L3, HSPB6, FBXO6, CRYAB, DNAJB4, KDELR3, and the abundances of immune cells in BLCA (Fig. 13). The expression level of CALR showed significant correlations with the abundances of CD8+ T cells (cor = 0.235, p = 5.33e−6), CD4+ T cell (cor = 0.113, p = 3.05e−2), macrophages (cor = 0.257, p = 6.22e−7), neutrophils (cor = 0.264, p = 3.51e−7), and dendritic cells (cor = 0.352, p = 5.33e−12). DNAJB4 was closely related to the CD8+ T cells (cor = 0.312, p = 9.98e−10), CD4+ T cell (cor = 0.244, p = 0.244e−6), macrophages (cor = 0.252, p = 1.02e−7), neutrophils (cor = 0.411, p = 2.92e−16), and dendritic cells (cor = 0.315, p = 7.19e−10). CRYAB showed strong correlations with B cells (cor = − 0.11, p = 3.66e−2), CD4+ T cells (cor = 0.16, p = 2.17e−3), macrophages (cor = 0.327, p = 1.45e−10), and dendritic cells (cor = 0.106, p = 4.35e−2). The expression of KDELR3 displayed significant correlation with the abundance of CD8+ T cells (cor = 0.116, p = 2.64e−2), macrophages (cor = 0.399, p = 2.17e−15), and dendritic cells (cor = 0.164, p = 1.62e−3). CREB3L3 was only related to CD4+ T cells (cor = 0.125, p = 1.17e−2). FBXO6 showed significant correlation with the abundances of CD8+ T cells (cor = 0.133, p = 1.11e−2), neutrophils (cor = 0.357, p = 2.36e−12), and dendritic cells (cor = 0.29, p = 1.58e−8). HSPB6 showed strong correlations with the abundances of CD4+ T cells (cor = 0.132, p = 1.13e−2), macrophages (cor = 0.4, p = 1.78e−15), and dendritic cells (cor = -0.137, p = 8.63e−3). Meanwhile, we found that seven UPRGs CNVs were correlated with several immune cells (Fig. 14). We also revealed the correlation between seven UPRGs and immune marker genes of immune cells by using GEPIA database. The results suggested that seven UPRGs were also related to immune marker genes using the GEPIA database (Table 1).
Fig. 13.

Immune infiltrates analysis of seven prognostic genes by TIMER database
Fig. 14.

The relationship between immune cells infiltration and CNVs of seven prognostic genes by TIMER database. P: 0.05 > * > 0.01 > ** > 0.001 > ***
Table 1.
The correlation between seven UPR genes and immune cell markers
| Description | Gene markers | CRYAB | CALR | DNAJB4 | HSPB6 | KDELR3 | FBXO6 | CREB3L3 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GEPIA | |||||||||||||||
| R | P | R | P | R | P | R | P | R | P | R | P | R | P | ||
| B cell | CD19 | 0.41 | *** | 0.12 | * | 0.13 | ** | 0.46 | *** | 0.27 | *** | 0.24 | *** | 0.15 | ** |
| CD79A | 0.42 | *** | 0.056 | ns | 0.14 | ** | 0.46 | *** | 0.25 | *** | 0.18 | *** | 0.11 | * | |
| MS4A1 | 0.36 | *** | − 0.018 | ns | 0.17 | *** | 0.46 | *** | 0.19 | *** | 0.13 | * | 0.16 | ** | |
| CD8+ T cell | CD8A | 0.28 | *** | 0.24 | *** | 0.3 | *** | 0.18 | *** | 0.18 | *** | 0.46 | *** | 0.1 | * |
| CD8B | 0.23 | *** | 0.27 | *** | 0.25 | *** | 0.19 | *** | 0.16 | ** | 0.39 | *** | 0.08 | ns | |
| IL2RA | 0.45 | *** | 0.31 | *** | 0.41 | *** | 0.32 | *** | 0.3 | *** | 0.38 | *** | 0.12 | * | |
| M1 macrophages | PTGS2 | 0.16 | ** | 0.11 | * | 0.34 | *** | 0.12 | * | 0.22 | *** | 0.13 | * | 0.072 | ns |
| NOS2 | 0.12 | * | − 0.081 | ns | 0.1 | * | 0.26 | *** | 0.11 | * | 0.14 | ** | 0.13 | ** | |
| M2 macrophages | CD163 | 0.54 | *** | 0.36 | *** | 0.35 | *** | 0.44 | *** | 0.42 | *** | 0.34 | *** | 0.094 | ns |
| CD209 | 0.52 | *** | 0.29 | *** | 0.43 | *** | 0.39 | *** | 0.33 | *** | 0.31 | *** | 0.085 | ns | |
| MRC1 | 0.51 | *** | 0.28 | *** | 0.39 | *** | 0.4 | *** | 0.39 | *** | 0.29 | *** | 0.11 | * | |
| M0 macrophages | CD14 | 0.56 | *** | 0.35 | *** | 0.38 | *** | 0.39 | *** | 0.31 | *** | 0.33 | *** | 0.057 | ns |
| CD33 | 0.53 | *** | 0.31 | *** | 0.34 | *** | 0.45 | *** | 0.33 | *** | 0.29 | *** | 0.11 | * | |
| ITGAX | 0.53 | *** | 0.26 | *** | 0.41 | *** | 0.41 | *** | 0.36 | *** | 0.34 | *** | 0.12 | * | |
| TAM | CCL2 | 0.54 | *** | 0.23 | *** | 0.34 | *** | 0.52 | *** | 0.41 | *** | 0.27 | *** | 0.14 | ** |
| CD68 | 0.31 | *** | 0.29 | *** | 0.44 | *** | 0.14 | ** | 0.11 | * | 0.37 | *** | 0.0091 | ns | |
| CD86 | 0.5 | *** | 0.31 | *** | 0.45 | *** | 0.32 | *** | 0.26 | *** | 0.38 | *** | 0.083 | ns | |
| Neutrophil | CD55 | 0.13 | ** | 0.014 | ns | 0.21 | *** | 0.14 | ** | 0.34 | *** | 0.016 | ns | 0.07 | ns |
| FCGR3A | 0.49 | *** | 0.37 | *** | 0.4 | *** | 0.34 | *** | 0.39 | *** | 0.39 | *** | 0.094 | ns | |
| ITGAM | 0.5 | *** | 0.33 | *** | 0.43 | *** | 0.39 | *** | 0.37 | *** | 0.35 | *** | 0.15 | ** | |
| Natural killer cell | CD7 | 0.35 | *** | 0.27 | *** | 0.29 | *** | 0.24 | *** | 0.19 | *** | 0.47 | *** | 0.082 | ns |
| KIR3DL1 | 0.19 | *** | 0.16 | ** | 0.19 | *** | 0.14 | ** | 0.14 | ** | 0.33 | *** | 0.11 | * | |
| Dendritic cell | CD1C | 0.37 | *** | 0.0072 | ns | 0.26 | *** | 0.36 | *** | 0.0078 | ns | 0.018 | ns | 0.048 | ns |
| THBD | 0.25 | *** | 0.057 | ns | 0.32 | *** | 0.048 | ns | − 0.038 | ns | 0.16 | ** | − 0.17 | *** | |
| T cell exhaustion | CTLA4 | 0.36 | *** | 0.26 | *** | 0.31 | *** | 0.2 | *** | 0.19 | *** | 0.43 | *** | 0.12 | * |
| LAG3 | 0.31 | *** | 0.36 | *** | 0.28 | *** | 0.15 | ** | 0.21 | *** | 0.47 | *** | 0.12 | * | |
| PD-1 | 0.32 | *** | 0.27 | *** | 0.29 | *** | 0.23 | *** | 0.21 | *** | 0.48 | *** | 15 | ** | |
| Treg | FOXP3 | 0.39 | *** | 0.24 | *** | 0.38 | *** | 0.29 | *** | 0.23 | *** | 0.39 | *** | 0.14 | ** |
| IL7R | 0.51 | *** | 0.27 | *** | 0.42 | *** | 0.35 | *** | 0.35 | *** | 0.3 | *** | 0.075 | ns | |
| NT5E | 0.4 | *** | 0.25 | *** | 0.39 | *** | 0.18 | *** | 0.27 | *** | 0.16 | ** | − 0.024 | ns | |
| Th1 cell | CCR1 | 0.4 | *** | 0.36 | *** | 0.36 | *** | 0.32 | *** | 0.41 | *** | 0.34 | *** | 0.12 | * |
| CCR5 | 0.37 | *** | 0.3 | *** | 0.35 | *** | 0.28 | *** | 0.29 | *** | 0.43 | *** | 0.15 | ** | |
| IL12RB1 | 0.37 | *** | 0.28 | *** | 0.34 | *** | 0.25 | *** | 0.22 | *** | 0.46 | *** | 0.11 | * | |
| Th2 cell | CCR4 | 0.35 | *** | 0.081 | ns | 0.38 | *** | 0.39 | *** | 0.25 | *** | 0.26 | *** | 0.17 | *** |
| CCR8 | 0.34 | *** | 0.14 | ** | 0.46 | *** | 0.25 | *** | 0.28 | *** | 0.31 | *** | 0.18 | *** | |
***P < 0.001, **P < 0.01, *P < 0.05, ns
Validation of the mRNA expression of 7 prognostic genes
UALCAN database was used to examine the mRNA expression and promoter methylation level of seven UPR genes (Fig. 15). The results of UALCAN database analysis showed that the mRNA expression levels of CALR, CREB3L3, FBOX6, and KDELR3 were increased in cancer tissues compared with normal tissues, while the mRNA expression levels of CRYAB, DNAJB4, and HSPB6 were decreased in BLCA. Furthermore, the elevated expressions of CALR, CREB3L3, FBOX6, and KDELR3 and the down-expressions of CRYAB, and HSPB6 might be correlated with promoter methylation. However, we did not find the correlation between the mRNA expression and promoter methylation level of DNAJB4. All these results were consistent with previous results.
Fig. 15.

Validation of the mRNA expression and promoter methylation of seven prognostic genes
Correlation between risk score and chemotherapy sensitivity
In the current research, we used GDSC database to predict the response to common chemotherapy drugs by estimating the differences of IC50 among different groups. We found that patients with high risk score were more sensitive to several chemotherapy drugs, including cisplatin, rapamycin, cyclopamine, and bleomycin, while patients with low risk score were more sensitive to metformin and methotrexate (Fig. 16). In addition, there was no obvious difference in sensitivity of gemcitabine, mitomycin C, and doxorubicin among two groups.
Fig. 16.

Chemotherapy sensitivity prediction between different groups
Discussion
Bladder cancer (BLCA) is a highly heterogeneous malignancy that develops via a complex multistep biological process. Accumulating evidence has demonstrated the crucial role of the UPR in the progression and outcome of various types of cancer, including BLCA [12, 18, 19]. The UPR was mainly mediated by three ER transmembrane protein sensors: inositol requiring kinase 1 (IRE1), protein kinase RNA-like endoplasmic reticulum kinase (PERK), and activating transcription factor 6 (ATF6) [20]. Silencing OTUB1 can markedly suppress the proliferation and migration of BLCA cells through activating ATF6 signaling [18]. Most of studies focused on the effect of UPR in cancer progression and metastasis, and few studies have investigated the prognostic value of UPR-related genes in cancers, especially in BLCA. Recently, mounting evidence has confirmed that multiple genes model is being used to predict outcomes and therapeutic effect seems to have high credibility [21–24].
Hence, in the current study, we conducted comprehensive bioinformatics approach to investigate the expression patterns of 251 UPR-related genes in BLCA and their correlation with OS. We identified a prognostic signature consisting of 7 UPRGs correlated with progression and outcomes of patients with BLCA and verified the novel prognostic signature in an external dataset GSE13507. Univariate and multivariate Cox regression analysis suggested that the signature based on 7 UPRGs was independent prognostic factor for survival in BLCA patients. Multivariate ROC analysis showed that risk scores were superior to traditional prognostic factors in predicting outcomes of patients with BLCA. In addition, UPR-based signature was closely associated with inferior clinical characteristics. Subgroups analyses stratified by clinical characteristics also indicated that patients with low risk scores had longer survival than those with high risk scores. A nomogram incorporating clinical characteristics and risk score was established to predict the outcomes of BLCA patients.
Among the seven UPRGs in the prognostic signature that we established, calreticulin (CALR), with a high ability for Ca2+ binding, has been implicated in the homeostasis of Ca2+ and protein folding as well as cell adhesion [25]. Several studies have also shown that CALR was associated with immune responses and apoptosis [26, 27]. Mounting evidence suggested that CALR expression was correlated with carcinogenesis, progression, outcome, and drug resistance as well as epithelial–mesenchymal transition [28]. Alpha-crystallin B (CRYAB), a member of the small heat-shock protein family with the functions of suppressing protein aggregation and promoting protein refolding, has been reported to inhibit the tumorigenic phenotypes of BLCA cells through suppression of the PI3K/AKT and ERK signaling pathway [29]. DnaJ homolog subfamily B member 4 (DNAJB4), also called HLJ1, has been proven to serve as a tumor suppressor role in many types of cancer including lung cancer, melanoma, and breast cancer [30–32]. Genetic deletion of KDELR3 was found to inhibit the lung metastasis of melanoma cells [33]. CREB3L3, an ER stress-associated transcription factor, was shown to be related to the proliferation of HBV-associated HCC cells by regulating the PI3K/Akt and AMPK signaling pathways [34]. Studies have confirmed the critical function of HSPB6 in the endothelial proliferation and migration of various cancers [35, 36]. Overexpression of HSPB6 suppressed BLCA T24 cell migration to a certain level but had no effect on T24 cell proliferation [37]. F-box protein 6 (FBXO6), a vital component of the ubiquitin protein ligase complex, has been reported to be involved in the invasion and sensitivity of cisplatin [38]. Wang et al. reported that FBXO6 might be a CD8+ T cell infiltration-promoting factor and improve anti-PD1 drug resistance in BLCA patients [39].
Previous studies have confirmed that UPR was highly correlated with immune cell infiltration. In addition, accumulating evidence has also confirmed that immune cell infiltration was closely associated with the development, progression, and prognosis as well as the treatment of BLCA. Therefore, the stromal score, ESTIMATE score, and immune score of BLCA samples were estimated by making use of the ESTIMATE algorithm. We found that the risk score was positively correlated with immune score, stromal score, and ESTIMATE score. Subsequently, we further used the CIBERSORT algorithm and ssGSEA to explore the relationship between the prognostic signature and immune cell infiltration. The results indicated that the signature based on 7 UPRGs was closely associated with immune cell infiltration. The low-risk group had a higher proportion of CD8+ T cells than high-risk groups, which was in line with previous research showing that the infiltration level of CD8+ T cells was positively related to favorable prognosis [39, 40]. In contrast, M2 macrophages and neutrophils had higher proportions in the high-risk group than low-risk group, which was also in accordance with previous studies that M2 macrophages and neutrophils were related to unfavorable outcomes [41–43]. In addition, cytokines (IL-10, TGF-β1, TGF-β2, TGF-β3) involved in immunosuppressive process and immune checkpoints (PD-L1, CTLA-4, and HAVCR2) were elevated in high-risk group, which indicated the immunosuppression microenvironment in high-risk group. These results exhibited the potential of UPR signature in predicting immune microenvironment of BLCA, which might improve the immunotherapeutic effect of tumor.
We further investigated the potential mechanisms among different groups and found that immune and carcinogenic pathways were enriched in high-risk group, which might explain the unfavorable prognosis and immune-suppressive status of patients in high-risk group, while the metabolism-associated pathways were highly enriched in low-risk groups. In addition, patients with high risk score might benefit from several chemotherapy drugs, including cisplatin, rapamycin, cyclopamine, and bleomycin, while patients with low risk score might benefit from the treatments of metformin and methotrexate.
In brief, UPR plays a critical role in the occurrence and progression of tumors. Our study demonstrated the value of a set of UPRGs as a prognostic biomarker in BLCA. Nevertheless, as a retrospective study performed through bioinformatics analyses, this study inevitably has several disadvantages. In addition, the biological mechanisms of these prognostic UPRGs remain to be further confirmed by relevant experiments and clinical evidence.
Conclusion
In summary, we established and validated a risk model based on seven UPRGs for predicting the prognosis, immune microenvironment, and chemotherapy response of patients with BLCA, which might have reliable potential for clinical application in BLCA.
Acknowledgements
Not applicable.
Authors' contributions
KZ, LX, and WD designed the research study. LX and WD took responsibility for statistical analyses. KZ wrote the manuscript. WG and BF evaluated and revised the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by grants from the National Natural Science Foundation of China (No: 81960512).
Availability of data and materials
The raw data of this study are derived from the TCGA database (https://portal.gdc.cancer.gov/) and GEO data portal (https://www.ncbi.nlm.nih.gov/geo/), which are publicly available databases.
Declarations
Ethics approval and consent to participate.
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Ke Zhu, Liu Xiaoqiang, and Wen Deng contributed equally to this work
Contributor Information
Gongxian Wang, Email: wanggx-mr@126.com.
Bin Fu, Email: urofubin@sina.com.
References
- 1.Kamat AM, Hahn NM, Efstathiou JA, Lerner SP, Malmström P-U, Choi W, et al. Bladder cancer. The Lancet. 2016;388(10061):2796–2810. doi: 10.1016/S0140-6736(16)30512-8. [DOI] [PubMed] [Google Scholar]
- 2.Patel VG, Oh WK, Galsky MD. Treatment of muscle-invasive and advanced bladder cancer in 2020. CA Cancer J Clin. 2020;70(5):404–423. doi: 10.3322/caac.21631. [DOI] [PubMed] [Google Scholar]
- 3.Sidrauski C, Chapman R, Walter P. The unfolded protein response: an intracellular signalling pathway with many surprising features. Trends Cell Biol. 1998;8(6):245–249. doi: 10.1016/s0962-8924(98)01267-7. [DOI] [PubMed] [Google Scholar]
- 4.Kaufman RJ. Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev. 1999;13(10):1211–1233. doi: 10.1101/gad.13.10.1211. [DOI] [PubMed] [Google Scholar]
- 5.Li X, Zhang K, Li Z. Unfolded protein response in cancer: the Physician’s perspective. J Hematol Oncol. 2011;4(1):8. doi: 10.1186/1756-8722-4-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cao SS, Kaufman RJ. Unfolded protein response. Curr Biol. 2012;22(16):R622–R626. doi: 10.1016/j.cub.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 7.Wang G, Yang Z-Q, Zhang K. Endoplasmic reticulum stress response in cancer: molecular mechanism and therapeutic potential. Am J Transl Res. 2010;2(1):65–74. [PMC free article] [PubMed] [Google Scholar]
- 8.Ron D. Translational control in the endoplasmic reticulum stress response. J Clin Invest. 2002;110(10):1383–1388. doi: 10.1172/JCI16784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, et al. Translational Control Is Required for the Unfolded Protein Response and In Vivo Glucose Homeostasis. Mol Cell juin. 2001;7(6):1165–1176. doi: 10.1016/s1097-2765(01)00265-9. [DOI] [PubMed] [Google Scholar]
- 10.Mori K. Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell mai. 2000;101(5):451–454. doi: 10.1016/s0092-8674(00)80855-7. [DOI] [PubMed] [Google Scholar]
- 11.Bobrovnikova-Marjon E, Grigoriadou C, Pytel D, Zhang F, Ye J, Koumenis C, et al. PERK promotes cancer cell proliferation and tumor growth by limiting oxidative DNA damage. Oncogene. 2010;29(27):3881–3895. doi: 10.1038/onc.2010.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li C, Huang Y, Fan Q, Quan H, Dong Y, Nie M, et al. p97/VCP is highly expressed in the stem-like cells of breast cancer and controls cancer stemness partly through the unfolded protein response. Cell Death Dis avr. 2021;12(4):286. doi: 10.1038/s41419-021-03555-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Siegelin MD, Dohi T, Raskett CM, Orlowski GM, Powers CM, Gilbert CA, et al. Exploiting the mitochondrial unfolded protein response for cancer therapy in mice and human cells. J Clin Invest. 2011;121(4):1349–1360. doi: 10.1172/JCI44855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tameire F, Verginadis II, Koumenis C. Cell intrinsic and extrinsic activators of the unfolded protein response in cancer: mechanisms and targets for therapy. Semin Cancer Biol. 2015;33:3–15. doi: 10.1016/j.semcancer.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rufo N, Garg AD, Agostinis P. The unfolded protein response in immunogenic cell death and cancer immunotherapy. Trends Cancer sept. 2017;3(9):643–658. doi: 10.1016/j.trecan.2017.07.002. [DOI] [PubMed] [Google Scholar]
- 16.Martelli AM, Paganelli F, Chiarini F, Evangelisti C, McCubrey JA. The unfolded protein response: a novel therapeutic target in acute leukemias. Cancers. 2020;12(2):333. doi: 10.3390/cancers12020333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim W-J, Kim E-J, Kim S-K, Kim Y-J, Ha Y-S, Jeong P, et al. Predictive value of progression-related gene classifier in primary non-muscle invasive bladder cancer. Mol Cancer. 2010;9:3. doi: 10.1186/1476-4598-9-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang H-H, Li C, Ren J-W, Liu L, Du X-H, Gao J, et al. OTUB1 facilitates bladder cancer progression by stabilizing ATF6 in response to endoplasmic reticulum stress. Cancer Sci. 2021;112(6):2199–2209. doi: 10.1111/cas.14876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hua Y-Q, Zhang K, Sheng J, Ning Z-Y, Li Y, Shi W, et al. NUCB1 suppresses growth and shows additive effects with gemcitabine in pancreatic ductal adenocarcinoma via the unfolded protein response. Front Cell Dev Biol. 2021;9:641836. doi: 10.3389/fcell.2021.641836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Madden E, Logue SE, Healy SJ, Manie S, Samali A. The role of the unfolded protein response in cancer progression: from oncogenesis to chemoresistance. Biol Cell. 2019;111(1):1–17. doi: 10.1111/boc.201800050. [DOI] [PubMed] [Google Scholar]
- 21.Qi W, Zhang Q. Development and clinical validation of a 3-miRNA signature to predict prognosis of gastric cancer. PeerJ. 2021;9:e10462. doi: 10.7717/peerj.10462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu Y, Liu Y, He A, Guan B, He S, Zhang C, et al. Identification of the Six-RNA-binding protein signature for prognosis prediction in bladder cancer. Front Genet. 2020;11:992. doi: 10.3389/fgene.2020.00992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang L, Chen S, Wang B, Su Y, Li S, Liu G, et al. An eight-long noncoding RNA expression signature for colorectal cancer patients’ prognosis. J Cell Biochem. 2019;120(4):5636–5643. doi: 10.1002/jcb.27847. [DOI] [PubMed] [Google Scholar]
- 24.Gu Y, Chen G, Lin P, Cheng J, Huang Z, Luo J, et al. Development and validation of an immune prognostic classifier for clear cell renal cell carcinoma. Cancer Biomark. 2020;27(2):265–275. doi: 10.3233/CBM-191017. [DOI] [PubMed] [Google Scholar]
- 25.Lu Y-C, Weng W-C, Lee H. Functional roles of calreticulin in cancer biology. BioMed Res Int. 2015;2015:526524. doi: 10.1155/2015/526524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fujiwara Y, Tsunedomi R, Yoshimura K, Matsukuma S, Fujiwara N, Nishiyama M, et al. Pancreatic cancer stem-like cells with high calreticulin expression associated with immune surveillance. Pancreas. 2021;50(3):405–413. doi: 10.1097/MPA.0000000000001772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fucikova J, Spisek R, Kroemer G, Galluzzi L. Calreticulin and cancer. Cell Res. 2021;31(1):5–16. doi: 10.1038/s41422-020-0383-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kasikova L, Hensler M, Truxova I, Skapa P, Laco J, Belicova L, et al. Calreticulin exposure correlates with robust adaptive antitumor immunity and favorable prognosis in ovarian carcinoma patients. J Immunother Cancer. 2019;7:312. doi: 10.1186/s40425-019-0781-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ruan H, Li Y, Wang X, Sun B, Fang W, Jiang S, et al. CRYAB inhibits migration and invasion of bladder cancer cells through the PI3K/AKT and ERK pathways. Jpn J Clin Oncol. 2020;50(3):254–260. doi: 10.1093/jjco/hyz172. [DOI] [PubMed] [Google Scholar]
- 30.Miao W, Li L, Wang Y. A targeted proteomic approach for heat shock proteins reveals DNAJB4 as a suppressor for melanoma metastasis. Anal Chem. 2018;90(11):6835–6842. doi: 10.1021/acs.analchem.8b00986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mo L, Liu J, Yang Z, Gong X, Meng F, Zou R, et al. DNAJB4 identified as a potential breast cancer marker: evidence from bioinformatics analysis and basic experiments. Gland Surg. 2020;9(6):1955–1972. doi: 10.21037/gs-20-431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen C-H, Chang W-H, Su K-Y, Ku W-H, Chang G-C, Hong Q-S, et al. HLJ1 is an endogenous Src inhibitor suppressing cancer progression through dual mechanisms. Oncogene. 2016;35(43):5674–5685. doi: 10.1038/onc.2016.106. [DOI] [PubMed] [Google Scholar]
- 33.Marie KL, Sassano A, Yang HH, Michalowski AM, Michael HT, Guo T, et al. Melanoblast transcriptome analysis reveals pathways promoting melanoma metastasis. Nat Commun. 2020;11(1):333. doi: 10.1038/s41467-019-14085-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vecchi C, Montosi G, Garuti C, Corradini E, Sabelli M, Canali S, et al. Gluconeogenic signals regulate iron homeostasis via hepcidin in mice. Gastroenterology. 2014;146(4):1060–1069.e3. doi: 10.1053/j.gastro.2013.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang X, Wang X, Zhu H, Kranias EG, Tang Y, Peng T, et al. Hsp20 Functions as a novel cardiokine in promoting angiogenesis via activation of VEGFR2. Qin G, rédacteur. PLoS ONE. 2012;7(3):e32765. doi: 10.1371/journal.pone.0032765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen S, Huang H, Yao J, Pan L, Ma H. Heat shock protein B6 potently increases non-small cell lung cancer growth. Mol Med Rep. 2014;10(2):677–682. doi: 10.3892/mmr.2014.2240. [DOI] [PubMed] [Google Scholar]
- 37.Chen Y, Xu T, Xie F, Wang L, Liang Z, Li D, et al. Evaluating the biological functions of the prognostic genes identified by the Pathology Atlas in bladder cancer. Oncol Rep. 2021;45(1):191–201. doi: 10.3892/or.2020.7853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hong X, Huang H, Qiu X, Ding Z, Feng X, Zhu Y, et al. Targeting posttranslational modifications of RIOK1 inhibits the progression of colorectal and gastric cancers. Elife. 2018;7:e29511. doi: 10.7554/eLife.29511. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 39.Wang Y, Yan K, Lin J, Liu Y, Wang J, Li X, et al. CD8+ T cell co-expressed genes correlate with clinical phenotype and microenvironments of urothelial cancer. Front Oncol. 2020;10:553399. doi: 10.3389/fonc.2020.553399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lin C-T, Tung C-L, Tsai Y-S, Shen C-H, Jou Y-C, Yu M-T, et al. Prognostic relevance of preoperative circulating CD8-positive lymphocytes in the urinary bladder recurrence of urothelial carcinoma. Urol Oncol Semin Orig Investig. 2012;30(5):680–687. doi: 10.1016/j.urolonc.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 41.Sharifi L, Nowroozi MR, Amini E, Arami MK, Ayati M, Mohsenzadegan M. A review on the role of M2 macrophages in bladder cancer; pathophysiology and targeting. Int Immunopharmacol. 2019;76:105880. doi: 10.1016/j.intimp.2019.105880. [DOI] [PubMed] [Google Scholar]
- 42.Lv Y, Jin P, Chen Z, Zhang P. Characterization of hazard infiltrating immune cells and relative risk genes in bladder urothelial carcinoma. Am J Transl Res. 2020;12(11):7510–7527. [PMC free article] [PubMed] [Google Scholar]
- 43.Xue Y, Tong L, Liu F, Liu A, Zeng S, Xiong Q, et al. Tumor-infiltrating M2 macrophages driven by specific genomic alterations are associated with prognosis in bladder cancer. Oncol Rep. 2019;42(2):581–594. doi: 10.3892/or.2019.7196. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The raw data of this study are derived from the TCGA database (https://portal.gdc.cancer.gov/) and GEO data portal (https://www.ncbi.nlm.nih.gov/geo/), which are publicly available databases.