

Abstract
Purpose
To develop drug-combination nanoparticles (DcNPs) composed of hydrophilic gemcitabine (G) and hydrophobic paclitaxel (T) and deliver both drugs to metastatic cancer cells.
Methods
GT DcNPs were evaluated based on particle size and drug association efficiency (AE%). The effect of DcNP on GT plasma time-course and tissue distribution was characterized in mice and a pharmacokinetic model was developed. A GT distribution study into cancer nodules (derived from 4 T1 cells) was performed.
Results
An optimized GT DcNP composition (d = 59.2 nm ±9.2 nm) was found to be suitable for IV formulation. Plasma exposure of G and T were enhanced 61-fold and 3.8-fold when given in DcNP form compared to the conventional formulation, respectively. Mechanism based pharmacokinetic modeling and simulation show that both G and T remain highly associated to DcNPs in vivo (G: 98%, T:75%). GT DcNPs have minimal distribution to healthy organs with selective distribution and retention in tumor burdened tissue. Tumor bearing lungs had a 5-fold higher tissue-to-plasma ratio of gemcitabine in GT DcNPs compared to healthy lungs.
Conclusions
DcNPs can deliver hydrophilic G and hydrophobic T together to cancer nodules and produce long acting exposure, likely due to stable GT association to DcNPs in vivo.
Keywords: breast cancer, mechanism based pharmacokinetic modeling, nanoparticles, pharmacokinetics, tumor distribution
INTRODUCTION
With improved diagnosis, novel targeted therapies and hormone-based therapies, the odds of 5-year survival in early-stage breast cancer patients is high at approximately 95% (1). However, almost 30% of these patients progress to metastatic disease and subsequently, 5-year survival rates are low at 27% (2). Once they reach the metastatic stage and drug resistance emerges, early-stage hormone receptor targeted treatments are no longer effective. Thus, metastatic patients are often treated with highly potent but toxic (and dose-limiting) chemotherapy. Under those circumstances, drug combination regimens are prescribed as a combination of two or more chemotherapeutics to maximize cancer cell death and overcome drug resistance. These regimens are typically based on anthracyclines (i.e. doxorubicin, daunorubicin, epirubicin) or taxanes (i.e. paclitaxel, docetaxel) in combination with other agents. Neither taxanes nor anthracyclines are superior to one another, but metastatic patients will likely have a limited duration of treatment with anthracyclines due to the cumulative lifetime risk of cardiac toxicity (3). This cumulative cardiotoxicity risk is inherent to anthracycline therapy. Once patients have reached their lifetime anthracycline dose, they can no longer be treated with doxorubicin, daunorubicin or epirubicin without the risk of heart failure. To overcome anthracycline dependent dose-limiting cardiotoxicity, taxane combinations with gemcitabine are used. Gemcitabine and paclitaxel (GT, Gemcitabine = G, Paclitaxel = T) is a combination regimen with a 41.4% response rate in breast cancer, similar to that of anthracycline combinations (4). However, GT is given as sequential infusions (T over 3 h followed by G over 30 to 60 min) to minimize adverse events, which also reduces the time where both G and T circulate in plasma at pharmacologically relevant concentrations.
When G is given as a single agent, it requires intracellular phosphorylation to a tri-phosphate form to mediate cytotoxicity. In patients with leukemia, the concentration of G triphosphate in cancer cells is proportional to the plasma concentration of G up to 3 μg/mL (5). At higher plasma concentrations of G, the tri-phosphate levels no longer increase above 3 μg/mL; thus this target concentration is currently used for G in plasma (6). The target therapeutic plasma concentrations of T were determined by establishing the threshold concentrations for neutropenia (0.09 μg/mL) with the intent to maximize T dosing before adverse events occur (7). Despite having target therapeutic plasma concentrations, there is only a 2-h window in which G and T circulate above those concentrations under the current recommended dose and time sequences. This is because of the varying physicochemical and pharmacokinetic profiles of GT. Longer or simultaneous infusions have been attempted in clinic, but poor patient tolerability and the physical incompatibility of GT limit these approaches (8). Given the physicochemical and clinical characteristics of GT, the pharmacologic effect of this regimen can be improved by synchronizing distribution and retention of both drugs in target tissues to maximize therapeutic benefit. This way, a lower overall dose may be needed to eliminate cancer cells and healthy tissues will be less exposed to drug, potentially reducing off-target toxicity. One approach to producing intracellular GT synchronization in target tissues is using a drug carrier that enables accumulation in the target tissues and cells, and in turn reduce the presence of free drugs in plasma. By reducing the fraction of free drug in plasma, systemic toxicity associated with high concentrations of cytotoxic drugs in off-target tissues is minimized (9).
To achieve the synchronized delivery of GT to target cells, drug delivery systems can be used carry multiple chemotherapeutic agents as a single particle. However, water soluble G (logP = −1.4) and water insoluble T (logP = 3) are difficult to co-formulate with existing formulation strategies. Drug delivery systems such as liposomes (100 nm to several microns in diameter) or small polymeric nanoparticles (<10 nm in diameter) may, in some cases, mitigate systemic toxicity by reducing the high concentration of free drugs that cause toxicity. However, targeting these particles to cancer cells is a challenge. Biological barriers such as the reticuloendothelial system can sequester liposomes (>200 nm) into the liver and spleen for elimination. Thus, premature clearance prevents liposomal drugs from reaching target cells. Small polymeric nanoparticles or micelles (<10 nm) can undergo renal filtration and elimination by the kidney, leading to short plasma half-life and limited effect.
Recently, our team has developed a method to co-formulate hydrophobic and hydrophilic drugs together in a stable manner as a drug combination particle suitable for injectable dosage form. This novel drug-combination delivery platform uses a simple approach to combine chemically distinct (e.g., water soluble and insoluble) drugs and stabilize them with lipid excipients to generate a drug combination nanoparticle (DcNP) in suspension. DcNPs have been used to deliver a combination of 3 or more water soluble and insoluble anti-HIV drugs in non-human primates (NHP). After subcutaneous administration in NHPs, anti-HIV DcNPs were shown to produce long-acting drug concentrations in lymphocytes and lymph nodes as well as in plasma (10–13). This DcNP platform has been successfully applied to a range of HIV antiviral drugs, including tenofovir (logP = −3.4), lopinavir (logP = 4.69), atazanavir (logP = 4.5) and ritonavir (logP = 3.9) (12,14). The long acting plasma circulation of anti-HIV DcNPs for up to 4 weeks suggests that these particles have specific characteristics that allow them to overcome biological barriers such as sequestration in healthy (e.g. liver and kidney) organs or premature elimination.
Thus, the goal of this study is to determine whether water soluble G and water insoluble T can be stably assembled together in DcNP form and allow synchronization of both drugs in metastatic breast cancer burdened tissue. We have stabilized GT in DcNP form and transformed GT from a current short-acting combination therapy into a long-acting combination therapy in target tissues and cells. In addition, we have used pharmacokinetic modeling and simulations as a novel approach to distinguish DcNP associated and dissociated fractions of GT in plasma. By doing so, we can begin to understand how the fraction of drug association to DcNP in vivo impacts the overall pharmacokinetics and exposure of GT when formulated together in DcNP dosage form.
MATERIALS AND METHODS
Materials
G (>99%) and T (>99.5%) were purchased from LC Laboratories (Woburn, Massachusetts). 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N- [amino (polyethylene glycol)-2000] (ammonium salt) (DSPE-PEG2000) (GMP grade) were purchased from Cordon Pharma (Liestal, Switzerland). Anhydrous ethanol was purchased from Decon Pharmaceuticals (King of Prussia, PA). All other reagents used were of analytical grade or higher.
Preparation and Characterization of Gemcitabine and Paclitaxel (GT) Drug Combination Particles
T (0.7 mg/mL) and G (7 mg/mL) were solubilized together in hot ethanol (60°C) with DSPC (25 mg/mL) and mPEG2000-DSPE (10 mg/mL) in a round bottom flask. The total concentration of solutes (drugs + excipients) in ethanol was 5% w/v. Solvent was removed by rotary evaporation followed by vacuum desiccation. The dry film was removed from the round bottom flask and triturated to achieve a uniform dry powder. Dry powder was rehydrated in 0.45% NaCl with 20 mM NaHCO3 buffer at 70°C and a pH of 7.4 to achieve a nominal concentration of 100 mM total lipids. Particle size reduction was achieved through bath sonication (Avanti Polar Lipids, Inc. Alabaster, AL) (5 min on, 5 min off, 3 cycles). Particle size and zeta potential were determined by photon correlation spectroscopy using a NICOMP 380 ZLS (Particle Sizing Systems, Santa Barbara, CA). The pH of the DcNP suspension was measured using MQuant pH-indicator test strips (Supelco|Sigma Aldrich, St. Louis, MO). The osmolarity of GT DcNPs was measured using a Vapro osmometer (Wescor Inc., Logan, UT).
The morphology of GT DcNPs was investigated compared to a liposome control using transmission electron microscopy (TEM) with negative staining. Liposome controls were formed by dissolving egg L-α-phosphatidylcholine (EPC) (Avanti polar lipids, Inc. Alabaster, AL) in chloroform. Chloroform was removed via rotary evaporation and the dry lipid film was rehydrated in normal saline. The rehydrated lipid film was then extruded through 100 nm pores to yield a liposome suspension. Sample suspensions containing either GT DcNPs or liposomes were transferred onto a TEM grid (copper grid, 300-mesh, coated with carbon and Formvar film). Particles from the sample suspensions were allowed to settle onto the grid and excess suspension was removed by filter paper after 5 min. The grid was then stained with 4 μL 5% uranyl acetate. After one minute, excess staining solution was removed by filter paper and the grid was air-dried. All images were acquired on a Tecnai G2 F20 electron microscope (FEI, Hillsboro, OR) operating at 200 kV.
Drug association efficiency (AE%) was determined by dialyzing 100 μl of the DcNP suspension (6–8 k MWCO) against 1000 × volume (100 mL, pH = 7.4) of bicarbonate buffered saline for 4 h at room temperature. Drugs were extracted by acetonitrile and drug concentrations in pre and post- dialysis DcNP suspensions were measured with a Shimadzu HPLC-UV system (Kyoto, Japan). Chromatographic separation was achieved using a KinetexC18 column (100 Å, 5 μm, 4.6 mm × 100 mm) (Phenomenex, Torrance, CA). The flow rate was set to 1.0 mL/min with a 10 μl sample injection volume. The mobile phase for separation consisted of pump A (Acetonitrile) and B (10 mM Ammonium Acetate in water). The gradient program used was as follows: pump B was set to 40%, and increased to 100% over 5 min. The wavelength for detection of gemcitabine and paclitaxel was 254 nm. AE% was calculated as the ratio of pre- over post-dialysis concentrations of G or T.
Preparation of Gemcitabine and Paclitaxel (GT) Combination in Cremophor EL Suspension (CrEL)
To prepare an equivalent GT drug combination for use as a control formulation, T was first dissolved in ethanol (20 mg/mL). To make a stable suspension, the 20 mg/mL T was diluted with Cremophor EL [1:1, (v/v)] (Sigma-Aldrich, St. Louis, MO). The resultant T in suspension was further diluted 10-fold with PBS containing pre-dissolved G (hydrochloride salt, 12.65 mg/mL). The final concentrations of drug combination in suspension were 10 mg/ml G and 1 mg/ml T. The control drug combination in CrEL suspension was used in animal studies within the same day of preparation due to instability.
Pharmacokinetic Study of Gemcitabine and Paclitaxel (GT) in DcNPs Compared to CrEL
Animal studies were conducted in accordance with the University of Washington Institute of Animal Care and Use Committee (IACUC) approved protocol number 2372–06 and federal guidelines. Five- or six-week old female BALB/c mice were purchased from The Jackson Laboratory (Bar Harbor, Maine) and housed in an animal research facility for at least one week before use. Mice were approximately 8 to 9 weeks old for the pharmacokinetic studies. Mice were administered GT either as DcNP or CrEL suspensions intravenously by tail vein injection at 50/5 mg/kg (G/T) in a 100 μl bolus volume (G ~ 10 mg/mL, T ~ 1 mg/mL). Blood was collected through retro-orbital bleeding at 5, 60, 180, 360, 1440 (24 h), and 2880 min (48 h) for DcNP and at 5, 60, 120, 180 and 360 min for the GT CrEL formulation. Each mouse represented a single biological replicate and 3 mice were used to estimate the mean plasma concentration time course of G and T at each time point. Necropsies were performed on each animal to harvest respective tissues for tissue distribution studies.
Drug Extraction from Plasma and Tissues
Protein precipitation was used to extract G, 2′,2′-difluorodeoxyuridine (dFdU), and T from plasma or tissue homogenates. Briefly, 50 μL of sample was transferred into 1.5 mL tubes with or without dilution by blank matrix to an appropriate concentration range. Samples were spiked with internal standards followed by the addition of 9 volumes of acetonitrile (450 μL). Samples were then vortexed for 6 min and centrifuged at 4°C for 15 min at 14000 rpm. The supernatant was removed and dried under nitrogen at 40°C. The dried samples were reconstituted to 50 μL containing 20% methanol and 80% water.
Quantification of Gemcitabine, Paclitaxel and dFdU by LC-MS/MS
Drugs extracted from biological matrices such as plasma or tissue homogenates were quantified by a Shimadzu HPLC system coupled to a 3200 QTRAP mass spectrometer (Applied Biosystems, Grand Island, NY). The HPLC system consisted of two Shimadzu LC-20A pumps, a DGU-20A5 degasser, and a Shimadzu SIL-20 AC HT autosampler. The mass spectrometer was equipped with an electrospray ionization (ESI) TurboIonSpray source. The system was operated with Analyst software, version 1.5.2 (ABSciex, Framingham, MA). Chromatographic separation of G and T was achieved using a Synergi column (100 × 2.0 mm; 4-μm particle size) (Phenomenex, Torrance, CA) with an inline C8 guard column (4.0 × 2.0 mm) also from Phenomenex. The flow rate was set to 0.5 mL/min with a 5 μl sample injection volume. The mobile phase for separation consisted of pump A (20 mM Ammonium Acetate in water) and B (Reagent Alcohol). The gradient program used was as follows: pump B was maintained at 20% for 1.0 min, then increased to 97% at 2.0 min, held at 97% until 3.0 min, ramped to 3% by 4.0 min and held until 5.5 min. The needle was washed with isopropanol after each injection. Analytes were monitored using multiple-reaction monitoring (MRM) for positive ions. The following ion transitions were monitored: gemcitabine, m/z 264.066 → 112.000; dFdU, m/z 265.084 → 113.200; paclitaxel, m/z 854.266 → 286.200; a stable labeled isotope of gemcitabine (C8C13 H12ClF2N15N2O4) (m/z 267.067 → 115.100) was used as an internal standard for gemcitabine and dFdU; docetaxel (m/z 830.312 → 549.3) was used as an internal standard for paclitaxel.
Estimating the Maximum Dissociated Fraction of Gemcitabine and Paclitaxel In Vivo when Administered as a DcNP
To estimate a maximum dissociated fraction (fdiss. max) of GT from DcNPs in vivo, we first utilized a non-compartmental approach based on the plasma AUCs of equi-molar injections of DcNP and CrEL formulations (15):
In Eq. 1, we assume that systemic clearance of G or T administered as DcNPs only occurs after drug dissociates from the particles, and dissociated drug has the same clearance pathways as the free drug control (CrEL). This non compartmental approach also assumes that drug clearance occurs solely in the central compartment. For our two drugs of interest, T is mainly metabolized in the liver by CYP3A4 and CYP2C8 but G is metabolized by ubiquitous cytidine deaminase (CDA) (16). To understand the impact of the ubiquitous metabolism of G on its pharmacokinetic profile, we used the plasma concentration of the primary metabolite of G, dFdU, as a biomarker for dissociated drug. For Eq. 2, we also assume that G conversion to dFdU exhibits linear kinetics. The equation below then provides an estimate of the fraction of the G dose metabolized to dFdU. If the calculated fraction metabolized (fdiss. G→dFdU) from Eq. 2 equals the fdiss. max from Eq. 1, we can surmise that once G is released from DcNPs at all locations in the body it exchanges readily with the circulating G and subsequently undergoes metabolism (i.e., metabolism of all dissociated drug occurs in the central compartment). However, if some or all of the drug released from DcNPs in peripheral tissue is immediately metabolized to dFdU without exchanging with G in circulation, we would then expect the fdiss. G→dFdU estimates from Eq. 2 to exceed the fdiss. max from Eq. 1 (i.e, dissociated drug is metabolized in both the central and peripheral compartments).
Which can then be simplified to:
G AUC0→∞, CrEL is the plasma AUC of G given in free drug form. G AUC0→∞, DcNP is the total AUCs of associated and dissociated G given in DcNP form. dFdU AUC0→∞, CrEL is the AUC of dFdU formed after administration of free G. dFdU AUC0→∞, DcNP is the AUC of dFdU after dosing G in DcNP, which reflects the amount of dissociated G released from DcNPs and available for metabolic conversion. fdiss. G→dFdU represents the fraction of DcNP-dose that is released as free drug and available for conversion to dFdU by CDA.
The assumptions underlying Eq. 2 are as follows: [1] Dissociated (free) G, but not associated G, is readily available for metabolism by CDA. [2] Dissociated G is rapidly and extensively metabolized to dFdU by CDA (17,18). [3] The free fraction of dissociated G in plasma and tissue is independent of drug concentration (i.e., protein binding does not change with G concentration). A detailed pharmacokinetic model for the associated and dissociated species will be presented next to simulate the in vivo association of both drugs.
Mechanism-Based Pharmacokinetic Modeling (MBPK) to Estimate DcNP Associated and Dissociated Fractions of GT
To further evaluate the strong in vivo association of both G and T to DcNPs, we have utilized a mechanism-based pharmacokinetic model (MBPK) to simulate the plasma time course of the associated and dissociated drugs. A previously developed MBPK model for long-acting anti-HIV DcNP (11,15) was adapted for the present analysis of G and T pharmacokinetics following DcNP administration. This extended MBPK or MBPK2 model was initially described and validated in non-human primates given subcutaneous and intravenous injections of DcNPs containing 3 anti-HIV drugs. The systemic portion of the MBPK2 model was slightly modified to fit the time course of total drug plasma concentrations (i.e. DcNP-associated plus dissociated drug species) after DcNP injection and that of plasma dissociated drug concentrations after CrEL suspension (15).
Briefly, the model adopted here for GT is composed of two sub-models representing DcNP-associated drug and dissociated drug species. Each sub-model has a central compartment and a peripheral compartment. A visual representation of the model is presented in Fig. 1. The observed plasma concentration is the sum of DcNP-associated and dissociated species since they cannot be measured separately. The dissociated drug pharmacokinetic parameters, k1,2, k2,1 and k0,1, were determined by fitting the dissociated drug sub-model to the observed plasma concentration-time data for G and T after injection of CrEL (free-drug) suspension. These parameters were then fixed and the linked sub-models were fit to the observed total concentrations for G and T obtained after injection of the DcNP-dose. The model reasonably assumed that the pharmacokinetics of drugs released from particles will be the same as that of the free-drug (CrEL) control. We assumed that G and T are released from the particle at independent rates (k1,3) in the central compartment and only dissociated drugs are subject to clearance from the system (k0,1). The model input was the dose, which was assumed to be 100% associated for T based on the high in vitro association (95%) and corroborated by Eq. 1 (>75%, See results). Although G in vitro association was 9%, we assumed that G was also 100% associated per the in vivo results from Eq. 1 (>98%, See results). In fact, when 9% G association (based on in vitro dialysis under sink conditions) was assigned to the DcNP-dose, the model fit did not converge due to gross underprediction of the observed plasma G concentration-time data. The peripheral compartment features purely drug exchange between plasma and a group of slowly equilibrating tissues (k4,3, k3,4). In other words, the DcNP-associated drug is cleared only via its release from the particles in the central compartment. Thus, we expect Eq.1 will provide a reasonable boundary estimate for the dissociated fraction after DcNP-associated drug injection and should be close to the estimate from the modeling. The model predictions fit the observed total plasma drug concentration-time data well (R2 > 0.9 for both G and T). MBPK modeling and parameter estimations were performed using SAAM II v2.3 (The Epsilon Group, Charlottesville, VA).
Fig. 1.
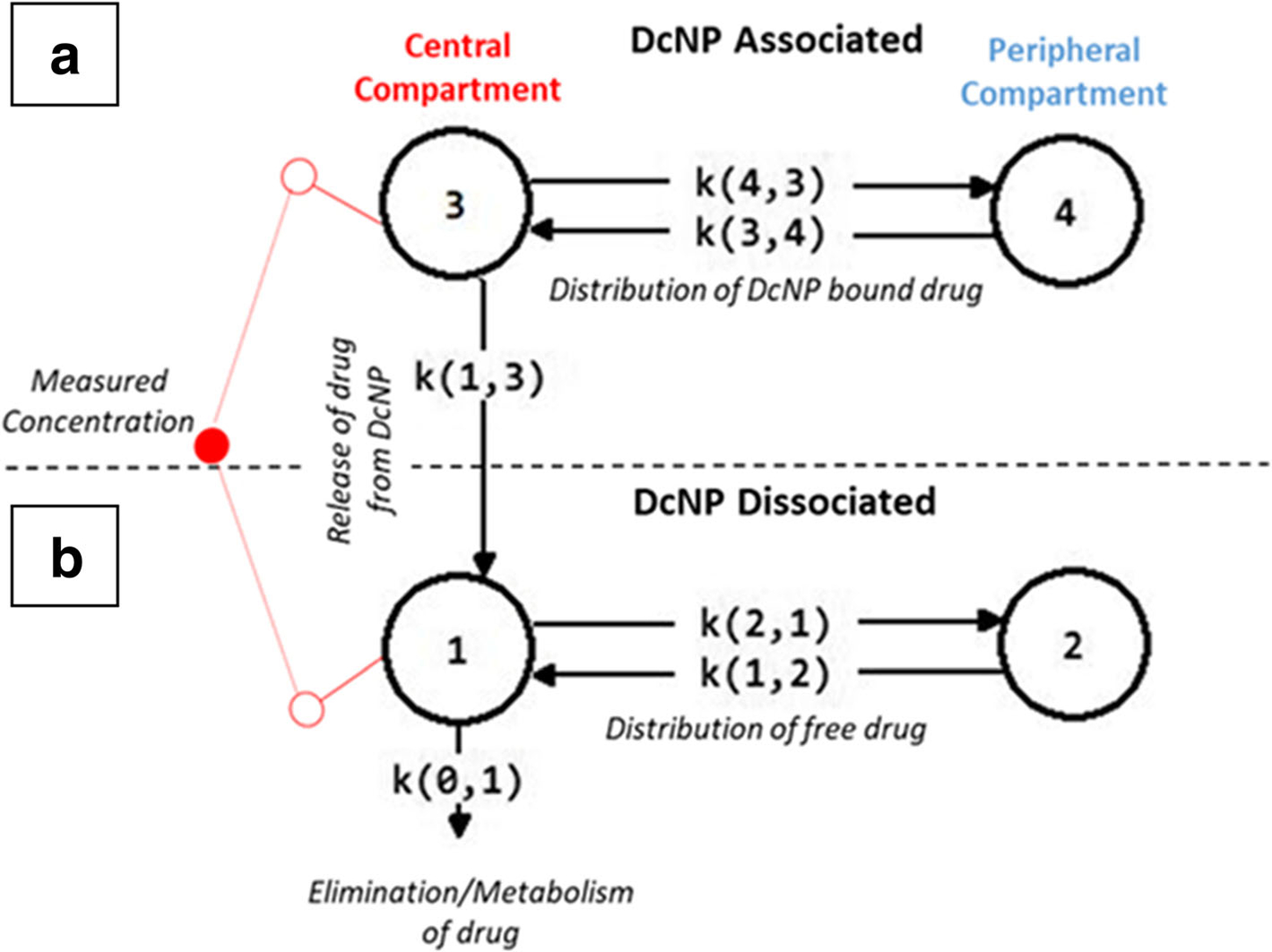
Schematic representation of a mechanism-based pharmacokinetic model for DcNP associated and dissociated gemcitabine and paclitaxel in plasma after IV dosing. A Mechanism-based pharmacokinetic (MBPK) model was developed to describe the association and dissociation of drug from DcNPs in plasma. The model features two parts: (a) the behavior of the fraction of gemcitabine or paclitaxel associated to DcNPs and their distribution to peripheral compartments. (b) The behavior of the fraction of DcNP dissociated gemcitabine or paclitaxel in plasma including distribution into peripheral compartments and clearance as dissociated drug. The DcNP associated and dissociated models are linked by the release parameter k1,3 in the central compartment. After dissociation through the release parameter, gemcitabine and paclitaxel are assumed to behave as they would in their free drug form as presented in the conventional CrEL dosage form control.
Establishment of Metastatic Nodules in Lungs
Female BALB/c mice were injected with 2 × 105 4T1 metastatic breast cancer cells that express luciferase as a marker (referred to as 4T1-luc cells and kindly provided by Dr. Stanley Riddell from the Fred Hutchinson Cancer Research Center) intravenously by tail vein on day 0. Stable expression of luciferase in these cells allows for bioluminescent monitoring of tumor growth and metastasis (19). Over a 14-day period, mouse behavior and health conditions were monitored daily and body weight measurements were taken every 2 days. On day 14, mice were administered 150 mg/kg D-luciferin through intraperitoneal injections 10 to 15 min before in vivo imaging to confirm establishment of tumor nodules in lungs. The bioluminescence imaging was acquired through a XENOGEN IVIS 200 imaging system (PerkinElmer, Waltham, MA). The bioluminescence imaging parameters for live mice were set as follows: field of view, 24; excitation filter, closed; emission filter, open; exposure time, 180 s; binning factor, 4; f/stop, 2. Bioluminescence intensity from mice were integrated using Live Image software (PerkinElmer, Waltham, MA). Mice were then intravenously administered GT in DcNPs at a dose of 50 mg/kg G and 5 mg/kg T or the same dose of drug in control suspension (CrEL) and sacrificed at fixed time points.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism 7.04 (GraphPad Software Inc., San Diego, CA). Statistical comparisons were performed using 2-sided t-tests with Welch’s correction for unequal variances. Significance probability α was set at 0.05. Pharmacokinetic parameters from non-compartmental analysis were calculated using the trapezoidal rule and relevant pharmacokinetic equations shown in Table II.
Table II.
Effect of DcNP Formulation on Pharmacokinetic Parameters of Gemcitabine and Paclitaxel Administered Together in GT DcNP or Control Suspension
| Parameter | Units | Gemcitabine |
Ratio | Paclitaxel |
Ratio | ||
|---|---|---|---|---|---|---|---|
| Control | DcNP | Control | DcNP | ||||
| AUC0 to ∞ a | μg min/ml | 920.8 | 56,218.6 | 61.0 | 156.5 | 588.8 | 3.8 |
| T1/2,app b | hr | 1.6 | 13.7 | 8.6 | 1.8 | 2.0 | 1.1 |
| C0 c | μg/mL | 165.1 | 181.4 | 1.1 | 17.7 | 13.9 | 0.8 |
| Dose/AUCd | mL/h | 65.2 | 1.1 | 0.02 | 38.3 | 10.2 | 0.3 |
| Vsse | mL | 16.6 | 12.1 | 0.7 | 35.6 | 10.0 | 0.3 |
| MRTf | hr | 0.25 | 11.3 | 45.2 | 1.0 | 1.0 | 1.0 |
Pharmacokinetic parameter estimates were derived from the data shown in Fig. 1
Non compartmental analysis was used to estimate area under the curve (AUC) from 0 to ∞ of gemcitabine and paclitaxel in DcNP and CrEL control
Apparent half-life was estimated from the slope of the last 3 time points collected in each condition
Concentration at time 0 was back extrapolated from the first time point in each condition
Dose/AUC is shown for the relative apparent clearances of gemcitabine and paclitaxel
Vss was calculated using Dose*AUMC/AUC2 extrapolated to infinity
Mean residence time (MRT) was calculated by dividing the area under the moment curve (AUMC) by the AUC from 0 to infinity
RESULTS
Preparation and Characterization of Injectable GT Combination in Drug Combination Nanoparticle Form
To determine whether two chemically distinct cancer drugs of interest (G, logP = −1.4 and T, logP = 3) can be formulated together in a single particle suitable for IV injection, we have evaluated several lipid excipient combinations that exhibit amphipathic properties. Hydrophobic T may interact with the acyl chains of phospholipids while the hydrophilic G may interact with the pegylated periphery of lipids when drugs and lipid excipients are assembled together. We found that two lipid excipients in the composition were able to stabilize GT in a DcNP suspension. In the treatment of metastatic breast cancer, G and T are infused in a weight by weight ratio of 14.3 to 1 (G-to-T) per cycle of chemotherapy. This clinical drug ratio was used as a starting point for DcNP optimization. A DcNP composition with 10:1 (w/w) G-to-T ratio containing two lipid excipients (DSPC, DSPE-PEG2000, 9:1 m/m) was able to produce a stable DcNP suspension with a total drug to total lipid ratio of 1:12 (w/w) for use as an IV injectable dosage form. With the intent to streamline scale-up processes, the preparation procedure was designed to minimize processes such as removal of unbound drug. This process appears to be robust and reproducible with consistent AE% of drugs (see below). We have further evaluated product characteristics including degree of drug association (AE%) to DcNP in suspension under sink conditions and batch to batch variability in size, drug concentration and association efficiency. These data are presented in Table I. The pH, zeta potential, osmolarity and morphology of GT DcNPs were also characterized.
Table I.
Characterization and Batch-To-Batch Variability of GT DcNPs
| Batch | Size (nm)a | AE%b |
[Drug Conc] (mg/mL) |
||
|---|---|---|---|---|---|
| PTX | GEM | PTX | GEM | ||
| 1 | 49.2 ± 35 | 95 | 7 | 1 | 13.9 |
| 2 | 59.9 ± 44 | 97 | 9 | 1.2 | 13.9 |
| 3 | 69.7 ± 47 | 97 | 10 | 1.3 | 15.3 |
| 4 | 68.7 ± 46 | 97 | 10 | 1.1 | 14.5 |
| 5 | 48.5 ± 30 | 92 | 8 | 0.7 | 14 |
| Average | 59.2 | 96 | 9 | 1.1 | 14.3 |
| S.D | 9.2 | 2 | 1 | 0.2 | 0.5 |
Mean particle diameter was determined by photon correlation spectroscopy and presented as the mean ± standard deviation
Association efficiency of gemcitabine (Gem) and paclitaxel (PTX) was determined by dialysis under sink conditions as described in Materials and Methods
A total of 5 batches were tested for formulation process and quality with respect to reproducibility. The mean particle size for all 5 batches was 59.2 ± 9.2 nm and suitable for IV dosing. The degree of drug association to DcNP was measured and expressed as AE% measured under sink conditions in buffered normal saline. For G, the average AE% for 5 batches was 9 ± 1; for T, the average AE% for 5 batches was 96 ± 2. The pH and osmolarity of GT DcNPs in suspension were consistent across batches and measured to be pH = 8.0 and 355 milliosmoles, respectively. Both of these parameters fall within acceptable boundaries for IV injectable products. The average zeta potential of GT DcNPs was measured to be −16.4 mV. The morphology of GT DcNPs was investigated using transmission electron microscopy (TEM). GT DcNPs have a distinct, discoid-like shape (Fig. 2, a) with no apparent bilayer structure. In contrast, the conventional liposome controls (Fig. 2, b) are observed to have lipid vesicles with enclosed bilayer membranes.
Fig. 2.

Structural morphology of GT DcNPs by electron microscopy. The morphology of GT DcNPs was evaluated using negatively stained transmission election microscopy and compared against conventional liposomes. Electron microscopy of GT DcNPs (Panel a) exhibits a discoid morphology with no evidence of bilayer structure (dashed arrows). In comparison, conventional liposome controls (Panel b) exhibit typical spherical structures with visible bilayer membranes (solid arrows).
Collectively, these data indicate that GT DcNPs are suitable for injectable dosage forms and can be prepared in a reproducible manner. The final composition for GT DcNPs had nominal concentrations of 16 mg/ml G and 1.6 mg/ml T. The GT DcNP injectable dosage form was subsequently diluted to 10 mg/mL and 1 mg/mL for use in pharmacokinetic studies in mice.
Effect of DcNP Formulation on Gemcitabine and Paclitaxel Plasma Time Course and Pharmacokinetics
We next investigated the effects of DcNP formulation on G and T in vivo. Due to the limited solubility of T, we used a Cremophor EL (referred to as CrEL) suspension to stabilize the GT control dosage form for IV administration. Although this CrEL-based micellar formulation may not fully represent a free and soluble T control, it does represent the clinical dosage form of T (Taxol). Thus, we compared intravenously administered G and T (50/5 mg/kg G/T) to mice in either DcNP or control CrEL-solubilized form. The total drug concentrations of G and T were determined in plasma at indicated time points. These data are presented as a plasma concentration time course comparison in Fig. 3. Pharmacokinetic parameter estimates are presented in Table II.
Fig. 3.

Association of GT to DcNPs increases the concentration of GT in plasma over time compared to CrEl control suspension. Gemcitabine (50 mg/kg, Panel a) administered as a DcNP (Dashed, ○) substantially increases the plasma circulation levels in healthy BALB/c mice (n = 3 per time point) measured at identical time points to the CrEL control (Cremophor El/saline suspension, solid line, ●). The plasma concentration of paclitaxel (5 mg/kg, Panel b) was also increased in plasma relative to the control suspension but a smaller effect is observed. Paclitaxel levels fall below the LLOQ of our LC-MS/MS assay after 6 h. The LLOQ of gemcitabine or paclitaxel is represented as dotted lines for both panels.
As shown in Fig. 3, the plasma drug concentration time course of G and T were substantially different when administered as DcNPs in comparison to CrEL control dosage form. Based on their respective in vitro AE% (9% for G and 95% for T under sink conditions), we would expect a greater difference in exposure for T than G. To the contrary, we found much greater enhancement in plasma concentrations of G than T when comparing DcNP to that of the CrEL control (Fig. 3a and b). For example, 3 h after DcNP dosing, mean plasma G concentration was 470 times greater than at the same time point for mice treated with the control CrEL (41,065 ng/mL vs 87.28 ng/mL at 3 h, p < 0.05). For T, there was also a higher plasma drug concentration in those treated with DcNP compared to the CrEL control; however, the formulation effect was more modest. At 3 h, the plasma T concentration for DcNP was 3.3x greater than CrEL (642.9 ng/mL vs 193.6 ng/mL at 3-h, p < 0.05). By 24 h, the plasma G and T concentrations in mice treated with the CrEL control formulation fell below the detection limit (LLOD). In contrast, for the test group treated with DcNP dosage form, persistent G concentrations in plasma were detected for the entire 48 h study in mice administered DcNPs (Fig. 3a). No T concentrations were detected in plasma after 6 h in mice likely due to its much lower dose (50 mg vs 5 mg/kg G to T ratio in DcNP formulation) (Fig. 3b).
The plasma drug concentration time course was further analyzed using non-compartmental analysis. The pharmacokinetic parameters are presented in Table II. The total exposure or area under the curve (AUC) of G is increased by 61-fold when administered as a GT DcNP compared to the control CrEL suspension (56,218.6 vs 920.8 μg*min/mL). Since both DcNP and control groups received the same dose of G (50 mg/kg), the apparent clearance (represented by dose/AUC) for the DcNP cohort decreased reflective of the increase in exposure (1.1 vs. 65.2 mL/h). No major change is observed in the concentration of G in plasma at time 0 (C0) after administration in DcNP or control suspension (181.4 versus 165.1 μg/mL). The apparent half-life (t1/2 app) increased 8.6-fold when administered in DcNP form compared to the control suspension (13.7 vs 1.6 h), reflecting a change in the long-acting plasma time-course that extended the apparent terminal slope of G. There was a limited reduction in volume of distribution at steady state for G in DcNP and free forms (12.1 mL vs 16.6 mL). Mean residence time for G, or the average time G molecules stay in the body, increases when given in DcNP form (11.3 vs. 0.25 h). If we consider the relative change in exposure from G administered in DcNP form versus free form, it is unlikely that G is only 9% associated as indicated by in vitro dialysis under sink conditions. Alternatively, if we assume that systemic clearance of G administered in DcNP form can only occur after dissociation and the clearance mechanisms do not differ between free and DcNP form, then Eq. 1 can provide an in vivo estimate of the maximum dissociated fraction (fdiss. max) of G. This estimate is calculated to be 1.6% and suggests that G is mostly associated in vivo.
For T, the total exposure or AUC is increased by 3.8-fold when administered as a DcNP versus a CrEL control suspension. Both groups were administered the same dose of T (5 mg/kg) and apparent clearance decreased reflective of the increase in exposure (10.2 vs 38.3 mL/h). Interestingly, the volume of distribution at steady state changed in concert with clearance when given in DcNP or control form (10 mL vs 35.6 mL) and to a greater degree than G. No major change is observed in the initial concentration of T in plasma when administered as either the CrEL suspension or DcNP. In contrast to G, no change is observed in the apparent half-life or mean residence time of T when given in DcNP or control suspension (2.0 vs 1.8 h; 1.0 vs 1.0 h, respectively) (Table II). Using the same assumption stated above, the in vivo association of T can be estimated by Eq. 1. The fdiss. max of T is calculated to be 26.6%.
The changes to the in vivo behavior of GT administered as a DcNP compared to their conventionally solubilized control suggests that DcNPs are reasonably stable in vivo. If DcNPs degraded rapidly in plasma after IV administration, we would expect drug to release from the particles and behave like the CrEL control suspension. Instead, we observe a remarkable enhancement in G circulation in plasma for up to 48 h and a lesser but notable enhancement with T. Collectively, these data suggest that large fractions of both G and T remain associated to DcNPs after IV administration. This effect is surprisingly more remarkable for water soluble G, which was initially predicted to readily dissociate in blood according to in vitro predictions in buffer.
Effect of DcNP on Gemcitabine Metabolism to dFdU and Estimation of Dissociated Drug
To further investigate the discrepancy between low in vitro G association and the much higher than anticipated overall drug exposure in mice attributed to DcNP (Fig. 3 and Table II), we next evaluated metabolic conversion of G to 2′,2′-difluorodeoxyuridine (dFdU) in cells and tissues. In this instance, we assume that when G is bound to DcNPs, it is not accessible to CDA, the primary metabolizing enzyme of G that is present in plasma and peripheral tissues. Due to the ubiquitous expression of the CDA enzyme and its ability to convert G to dFdU in peripheral tissues, dissociated drug that undergoes immediate metabolism in the periphery is not directly accounted for using Eq. 1 (which is based on plasma G levels). With these assumptions, we utilize this primary metabolite of G (dFdU) as a marker for the fraction of G that dissociates from DcNPs, which subsequently undergoes metabolism in all tissues (following free G to dFdU conversion kinetics) as per Eq. 2. In our studies with IV injections, we assume that G in the control CrEL formulation is 100% freely soluble and available to CDA for conversion to dFdU in the body. The elimination of dFdU from the body occurs primarily through the kidneys and we assume that all dFdU formed in the body (peripheral tissues) will readily return to the central compartment for renal elimination. Under these conditions, when G is stably associated with DcNPs in the body, G is not available for conversion to dFdU. When G dissociates from the particle in either the central or peripheral compartment, it is then free to interact with CDA and undergo metabolism to dFdU.
The results in Fig. 4a show the time course of G and dFdU after IV administration of GT in the CrEL control dosage form. The plasma concentrations of the primary metabolite (dFdU) rose rapidly and reached concentrations equivalent to G within 15 min (11,836.7 ng/mL and 12,023.5 ng/mL, respectively) and in 3 h the concentration of dFdU in plasma was 50x times higher than G (4343.2 ng/mL vs 87.3 ng/mL). Over time, this gap became larger with the G to dFdU ratio falling to 0.01 in 6 h. In contrast, when G was given as a GT DcNP dosage form the plasma concentration of dFdU did not reach the G levels throughout 48 h of study (Fig. 4b). At 3 h, plasma dFdU is 3.4x lower than G. The variation in G/dFdU ratios over time can be seen in Fig. 4c with open circles representing the CrEL control and closed circles representing DcNPs. Based on these data (Fig. 4) and Eq. 2, the estimated fraction of dissociated G accessible for metabolism (fdiss. G→dFdU) is calculated to be 8%. In other words, 92% of G in DcNP dosage form is not accessible for dFdU conversion after IV GT-DcNP administration. The higher dissociated fraction of G derived from dFdU analysis is higher than the 1.6% estimate provided by Eq. 1 and suggests that peripheral metabolism competes with the redistribution of dissociated drug back into systemic circulation. Nevertheless, both estimates collectively point to the majority of G (>92%) remaining associated to DcNPs in vivo and being likely inaccessible by CDA for metabolic conversion.
Fig. 4.
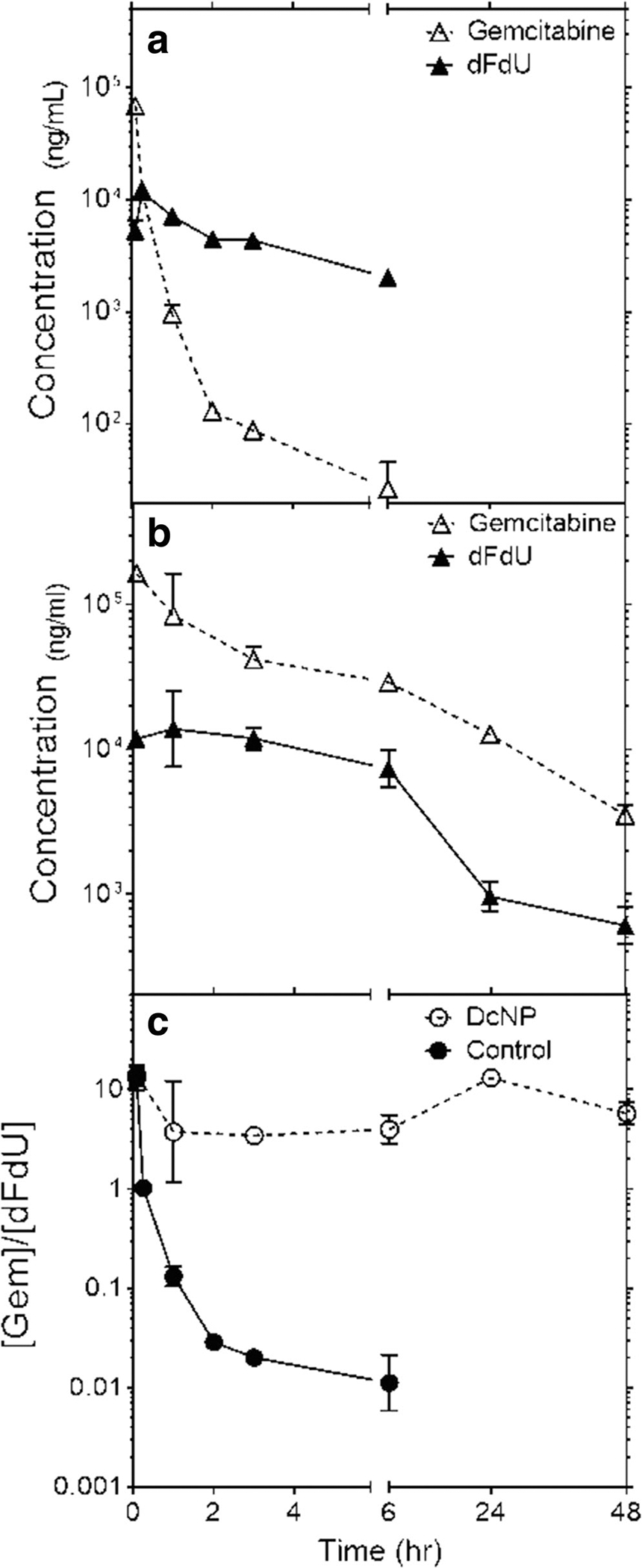
Effect of DcNP formulation on dFdU formation over time compared to CrEL control. Panel (a) shows the plasma time course of gemcitabine (Δ) and its metabolite dFdU (▲) in control soluble gemcitabine (50 mg/kg; in CrEL) dosage form. Panel (b) represents the plasma time course of mice treated with gemcitabine in GT DcNP at equivalent doses to the soluble control; the symbols are the same as those represented in Panel (a). The ratios of gemcitabine to dFdU over time for mice treated with gemcitabine are presented in Panel C comparing gemcitabine in a DcNP (○) or CrEL (●) control dosage form.
These data are consistent with our initial premise that GT DcNP particle are sufficiently stable in vivo, and that G association to DcNPs is high throughout the course of the pharmacokinetic study. Additionally, the plasma time course of G and dFdU show that dFdU kinetics are elimination rate limited when given in free form and changes to formation-rate limited (or release-rate limited) kinetics when administered in DcNP form. DcNPs enabling a shift to release-rate limited kinetics may essentially act as an extended infusion of G and may be beneficial for therapeutic effect (20).
Mechanism-Based Pharmacokinetic Simulations of DcNP-Associated and Dissociated Drug Time-Courses
To further understand the effect of DcNP on the pharmacokinetic behavior of GT, we next employed a mechanism-based pharmacokinetic model (MBPK) to simulate the time course of DcNP-associated and dissociated drugs in plasma. The MBPK model adapted for GT-DcNPs is based on a validated MBPK model developed for long-acting HIV drug combination nanoparticles tested in non-human primates (15). Experimentally, we can measure total drug concentrations in plasma (i.e. DcNP-associated plus dissociated), but we cannot distinguish between DcNP-associated and dissociated drugs. Due to these limitations, direct comparisons of the pharmacokinetics of GT when administered in DcNP or CrEL control form are difficult to interpret. The MBPK model presented in Fig. 1 utilizes data from both DcNP and CrEL control formulation treated animals to estimate plasma concentrations of DcNP-associated and dissociated drug species over time and their respective time-averaged fractions in vivo. After intravenous administration as a DcNP, GT can theoretically exist in at least two species: DcNP-associated drug and dissociated drug. The latter can be reasonably assumed to distribute and be cleared as free G and T after administration of CrEL control formulation. By using the experimental data from the CrEL control group as an anchor for dissociated drug, the model simulates the contribution of DcNP association to the observed increase in plasma concentrations and is reported in Fig. 5.
Fig. 5.
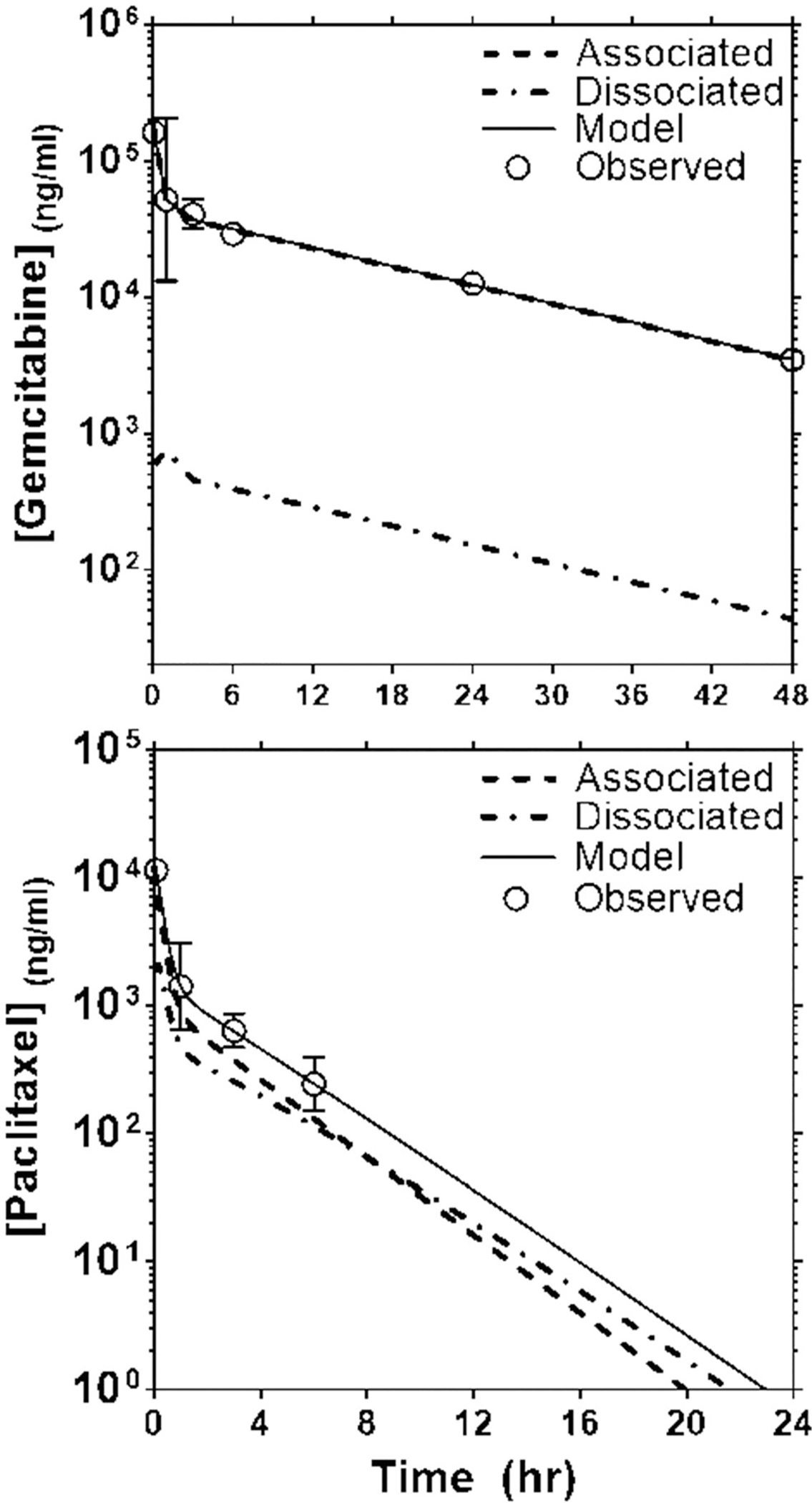
Validation of an MBPK model predicted concentration time curve for gemcitabine and paclitaxel with experimental data in mouse plasma after intravenous administration of GT DcNPs. Panel (a) represents the gemcitabine plasma time course of associated and dissociated fractions of drug. The experimental data are presented in open circles (○) with an SD error bar. The MBPK model simulated values are plotted as a continuous solid line. The dotted lines represent the DcNP dissociated gemcitabine concentration over time, simulated by the model. Panel (b) presents the experimental data and simulated DcNP associated and dissociated fractions over time for paclitaxel. The symbol and line representations for Panel (b) are the same as for Panel (a). It is interesting to note that the total simulated plasma concentrations and the DcNP associated species of gemcitabine overlap with most of the gemcitabine remaining DcNP associated throughout the study period.
Analysis of MBPK Structure and Assumptions
Individual distribution, metabolism and elimination of G and T are well characterized. G undergoes rapid and complete deamination to inactive metabolite (dFdU) by ubiquitous CDA. T is metabolized in the liver primarily by CYP3A4 and CYP2C8 with subsequent excretion of metabolites into the bile. In this model, the k0,1 term represents the aggregate elimination processes of G and T through their independent described pathways. A peripheral compartment for both G and T was added and distribution was parameterized with the k1,2 and k2,1 terms (21,22). With these considerations, we then added a parallel compartment to represent DcNP associated drug and assumed the following: [1] at the moment of injection, G and T are both completely associated to DcNPs, but are released at different rates; [2] apart from release, there is no other mechanism of clearance for drug bound to the DcNP; [3] when either G or T has been released from DcNPs their pharmacokinetic behavior will be the same as that of the CrEL control and [4] the amount of drug released from particles in the peripheral compartment is negligible. With these assumptions in place, we set the k1,3 term to link the two sub-models and represent the release mechanism of drug from DcNP into the central compartment. The DcNP associated species of G and T was then parameterized with the k4,3 and k3,4 to account for distribution. By fitting the additive sum of compartments 1 and 3 to the observed GT concentrations, we estimated the fraction of drug that is associated or dissociated in plasma and the relevant pharmacokinetic parameters.
Model Simulations and Verification with Experimental Data
For G, the estimated volume of central compartment decreased 4.6 times when administered as DcNP compared to administration in CrEL form(24 mL vs 5.2 mL) which likely reflects a reduced distribution of DcNP-G into tissues. For T, the estimated volume of distribution was slightly less than the physiological plasma volume of a mouse (~0.8 mL) in both DcNP and CrEL groups. It is likely that T association to DcNPs or CrEL micelles limits the distribution of T from the plasma. Thus, the volume parameter was fixed at 0.8 mL to retain physiological context. It is important to note that our dissociated T parameters were derived from an injection of the CrEL control and not completely soluble drug (due to solubility limitations) and the CrEL micelles may limit distribution of T from plasma. The estimated release parameter of G (k1,3) was 9.5-fold lower than T (0.2 h−1 vs 1.9 h−1, Table III) and corresponds to the relative 11.3-fold difference in mean residence times of G and T after DcNP administration (11.3 h vs 1.0 h, Table II). Based on the parameters generated in this model (Table III), a simulation was performed to predict the plasma concentration time course kinetics of dissociated and associated G and T (Fig. 5). The ratio of dissociated over total G AUCs as simulated by the model was 1.5%, which agrees with our maximum fraction dissociated in plasma estimate of 1.6% from Eq. 1. For T, the model simulated ratio of dissociated over associated T AUCs is 24.7% and in close agreement with our boundary estimate using Eq. 1 (26.6%). It is interesting to note that the in vivo association of G is greater than T (~98% vs ~73%), but the opposite is true for in vitro association (9% versus 95%). There is likely an unknown in vivo mechanism that enables the substantial association of G to DcNPs, however further investigation is required. Despite these unknowns, the current results clearly show that DcNPs retain hydrophilic G and hydrophobic T together in plasma (up to 8 h) and may enable the co-delivery of GT to target cancer cells.
Table III.
Model Derived Pharmacokinetic Parameters for Gemcitabine and Paclitaxel When Administered as a Single IV Dose in GT DcNP Dosage Form
| DcNP Dissociated Parameters | Gemcitabine | Paclitaxel |
| V (mL) | 24 ± 12 | 0.8a |
| K0,1 (1/h) | 3.9 ± 0.9 | 7.0 ± 2.0 |
| K1,2 (1/h) | 0.5 ± 0.2 | 0.6 ± 0.1 |
| K2,1 (1/h) | 0.2 ± 0.1 | 4.4 ± 1.0 |
| DcNP Associated Parameters | Gemcitabine | Paclitaxel |
| VDcNP (mL) | 5.2 ± 0.6 | 0.8a |
| K1,3 (1/h) | 0.2 ± 0.02 | 1.9 ± 0.1 |
| K3,4 (1/h) | 0.6 ± 0.1 | 0.7 ± 0.2 |
| K4,3 (1/h) | 1.8 ± 0.5 | 1.6 ± 0.5 |
Volume was fixed to plasma volume assuming a blood volume of ~2 mL for 8 to 9-week-old mice and a hematocrit of 40%
Effect of DcNP Formulation on Gemcitabine and Paclitaxel Tissue Distribution
We next determined the effect of DcNP on preferential GT tissue distribution in mice. Non-specific, off-target accumulation of drugs in healthy tissues can limit the therapeutic potential of GT DcNPs. In addition, accumulation of cytotoxic drug in healthy tissues can pose a safety concern. Therefore, we compared tissue-to-plasma drug concentration ratios at 3 h after IV administration in mice dosed with GT in DcNP or the CrEL control dosage form. The 3 h time point was selected to ensure that both drugs are detectable in both plasma and tissues for animals treated with DcNP or CrEL control. As shown in Fig. 6, DcNPs retain G in the plasma relative to CrEL control 3 h post-injection in all tissues tested (p < 0.05, Student’s T Test). For T, lung and kidney tissue-to-plasma ratios were reduced in DcNP vs CrEL control; while liver and spleen ratios increased. These data indicate that G in DcNPs does not accumulate in off-target organs such as the liver and spleen and, instead, GT in DcNP are better retained in blood and plasma. The effect of DcNPs on T distribution is less dramatic but there does not appear to be a substantial trend towards healthy organ accumulation (Fig. 6). Taken together, these data suggest that G bound to DcNPs does not accumulate in any of the sampled tissues and likely provides drug associated to DcNP a greater opportunity to reach target tumor cells by remaining in the systemic circulation. It is possible that T binding to serum protein, as well as stripping of T from DcNPs, may contribute to the differential tissue distribution of G and T in healthy mice. However, this difference, particularly the T increase in liver and spleen, is minimal. The large reduction of G in the four off-target tissues are significant compared to that of CrEL control formulation and may result in less off-target toxicity.
Fig. 6.
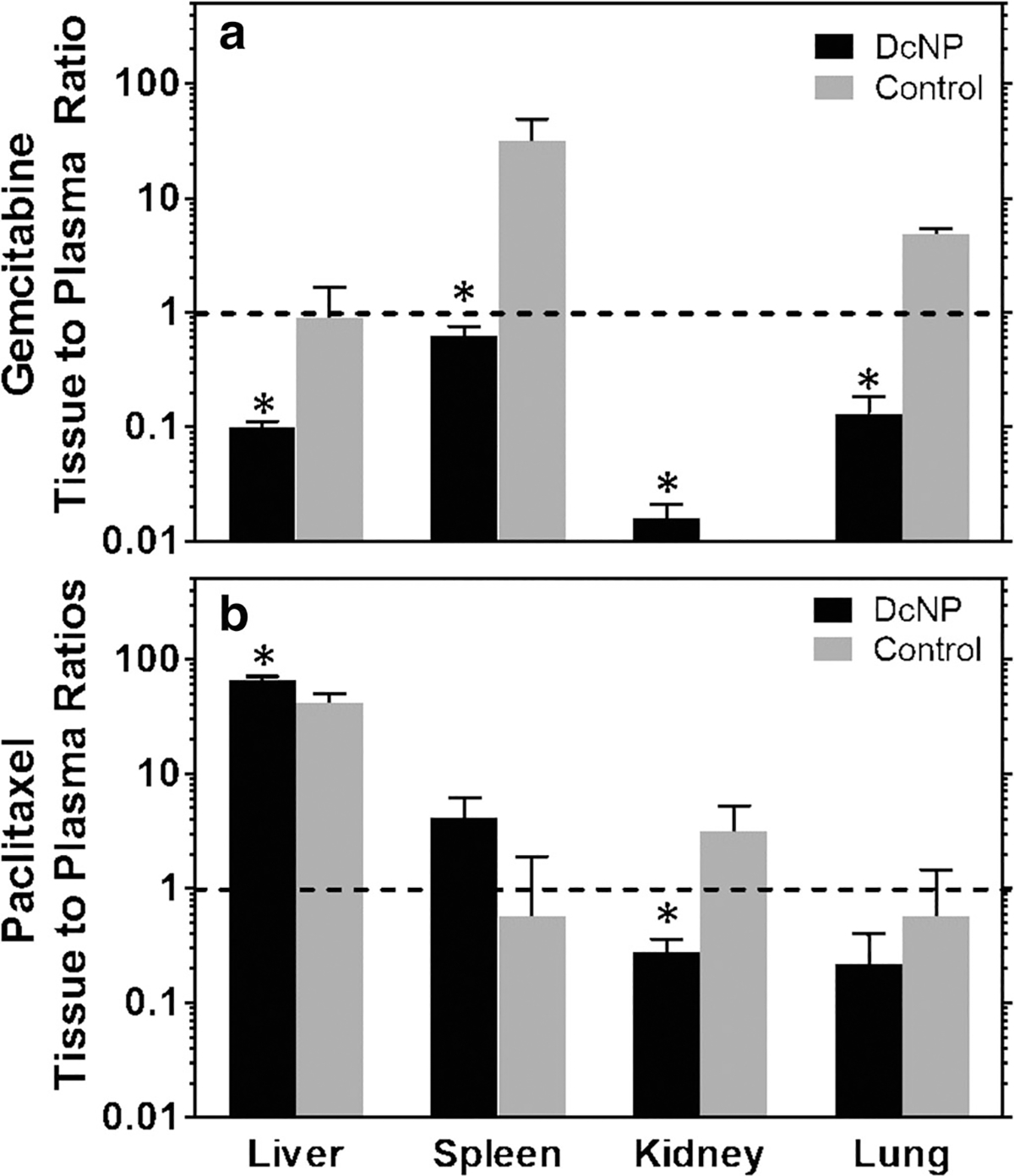
Effects of DcNP on gemcitabine and paclitaxel tissue distribution 3 h after intravenous injection compared to the control suspension. Mice (n = 3) were intravenously administered with GT DcNP or a control dosage form (CrEL suspension) at 50 mg/kg gemcitabine and 5 mg/kg paclitaxel. Gemcitabine and paclitaxel concentrations were measured in the listed tissues 3 h after injection; the respective tissue to plasma ratios for each animal were analyzed and presented as a mean ± SD for each dosage form. Panel (a) represents gemcitabine and panel (b) represents paclitaxel tissue to plasma ratios. The black bars indicate GT DcNP while the gray bars indicate the CrEL control dosage form. *denotes p < .05.
Effect of DcNP Formulation on Gemcitabine and Paclitaxel Localization in Healthy Versus Tumor Bearing Lung Tissue
To determine whether DcNPs enhance GT distribution into target tissues, namely cancer nodules, we next investigated the differential localization of GT in DcNP or CrEL form in healthy versus cancer nodule bearing mice. Since lungs are a common metastatic site for breast cancer disease progression, we utilized a model for metastasis that forms cancer nodules in the lungs. Cancer nodules were established in female BALB/c mice via intravenous inoculation of syngeneic breast cancer cells (4T1). This process has been shown to consistently produce detectable and multiple 4T1 cancer nodules in lungs within 14 days. Once nodules were established and confirmed with IVIS imaging, mice were administered with GT in either DcNP or conventionally solubilized form at equivalent doses (50/5 mg/kg, GT) and euthanized at pre-determined time intervals. We compared the effect of DcNP and CrEL dosage forms on GT distribution at two time points. To capture rapidly cleared free GT (CrEL) in the lungs and plasma, an early time point (1 h) was chosen. After 1 h, GT in both DcNP and CrEL are in their distribution phase after IV dosing. A later time point (24 h) was chosen to capture GT distribution after distribution equilibrium is reached. Nodule-free and nodule-bearing lungs were harvested, and lung tissue drug concentrations were compared in these two groups.
As shown in Table IV, mice bearing 4T1 cancer nodules had a higher concentration of G (p < 0.01) and T (p < 0.12) in lung tissue 1 h after CrEL administration compared to healthy tissue. Increases of lung concentration reflect increases of plasma concentration of G (p = 0.14) and T (p < 0.01). Comparison of lung-to-plasma ratios 1 h after CrEL administration shows that G (p = 0.24) and T (p = 0.4) distribution are not significantly different between healthy and cancer nodule burdened animals. For cancer nodule burdened mice given DcNPs, no statistical significance was observed in lung, plasma, or lung-to-plasma ratios. At later time points (24 h), free drug rapidly clears from the systemic circulation and only the DcNP group has detectable levels of drug in plasma and lung. The lung-to-plasma ratios of G in cancer nodule bearing mice at 24 h are 5 times greater than in healthy with a p value of 0.04. Although a lung-to-plasma ratio of T cannot be determined at 24 h due to rapid plasma clearance, the concentration of T is 7x greater in cancer nodule bearing mice versus healthy mice (p = 0.008). These data are summarized in Table IV. The results from this experiment show that when administered to healthy mice, DcNPs can sustain drug levels in the lung (a major site of metastasis) for a longer time than the CrEL control formulation. When mice have cancer nodules present, G and T levels in the lung are increased relative to healthy mice. These increases in lung concentrations are disproportionately larger than the elevated concentrations in plasma, suggesting that the particles preferentially target cancer burdened tissue. Furthermore, when we compare the GT concentration ratios in lungs, an 8.8 to 1 ratio is observed, similar to the original drug ratio in formulation. This further supports the idea that particles may selectively deposit and retain in cancer burdened tissue.
Table IV.
Effect of 4T1 Tumors on Gemcitabine and Paclitaxel Localization in Tumor Burdened Lung Tissues After Dosing with GT DcNP or CrEL Control Formulation
| Gemcitabine |
Paclitaxel |
|||||
|---|---|---|---|---|---|---|
| Healthy | 4T1 | p value | Healthy | 4T1 | p value | |
| 1 h after control | ||||||
| Lung (μg/g) | 0.9 ± 0.4 | 6.0 ± 0.8 | <0.01* | 1.0 ± 0.3 | 1.7 ± 0.5 | 0.12 |
| Plasma (μg/mL) | 0.9 ± 0.2 | 2.8 ± 1.4 | 0.14 | 0.3 ± 0.05 | 0.7 ± 0.1 | 0.01* |
| Lung/Plasma Ratio | 0.9 ± 0.3 | 2.2 ± 1.4 | 0.24 | 3.3 ± 1.2 | 2.5 ± 0.8 | 0.40 |
| 1 h after DcNP | ||||||
| Lung (μg/g) | 7.5 ± 1.2 | 12.5 ± 4.9 | 0.21 | 0.6 ± 0.4 | 0.9 ± 0.5 | 0.46 |
| Plasma (μg/mL) | 52.2 ± 64 | 72.6 ± 60.0 | 0.71 | 1.4 ± 0.9 | 2.2 ± 1.1 | 0.39 |
| Lung/Plasma Ratio | 0.1 ± 0.2 | 0.2 ± 0.1 | 0.85 | 0.4 ± 0.01 | 0.4 ± 0.2 | 0.92 |
| 24 h after DcNP | ||||||
| Lung (μg/g) | 0.3 ± 0.1 | 3.5 ± 1.0 | 0.01* | 0.06 ± 0.01 | 0.4 ± 0.1 | 0.03* |
| Plasma (μg/mL) | 12.7 ± 1.5 | 34.2 ± 0.6 | <.01* | BDL | BDL | NA |
| Lung/Plasma Ratio | 0.02 ± .01 | 0.10 ± .03 | 0.04* | NA | NA | NA |
Data in the table are presented as the geometric mean of three biological replicates ± standard deviation. No drug was detected with the CrEL control 24 h after drug administration
denotes p < 0.05
NA denotes a ratio that is not calculable
BDL denotes no detectable drug in the sample
DISCUSSION
A key challenge in the treatment of breast cancer is metastasis and poor tolerability of highly potent, chemotherapeutic drug combinations. Off-target drug distribution and asynchronous concentrations of drug combinations in target tissues (tumors) likely contribute to the high dose requirements for metastatic control and dose-limiting toxicities. To coordinate the anticancer effects of two chemotherapeutic agents, we have successfully co-formulated chemically distinct gemcitabine (G) and paclitaxel (T) in a drug combination nanoparticle (DcNP). When intravenously given to mice laden with metastatic 4T1 breast cancer nodules in the lung, the GT DcNP demonstrated improved tissue selectivity and long-acting exposure of both drugs in metastatic cancer bearing tissues (Table IV).
Interestingly, the single nanoparticle composed of the unlikely partners—water soluble G and water insoluble T—not only demonstrated long acting tissue selectivity for breast cancer nodules in the lung, but also minimal distribution into healthy organs. The tissue-to-plasma ratios of GT DcNPs, which also produced long acting plasma circulation, did not significantly differ from the control GT administration in mice 3 h after IV injection (Fig. 6). An unexpected finding is that both G and T remain well associated to DcNPs for the duration of the time course study, which may be related to target tissue localization and the long-acting pharmacokinetics of GT enabled by the DcNP platform. In vitro association efficiency suggests that paclitaxel has a greater affinity for DcNPs than does gemcitabine. However, when given in DcNP form, gemcitabine has a greater enhancement in plasma exposure than paclitaxel compared to their respective controls. Pharmacokinetic modeling and simulation was used as a novel tool to distinguish the in vivo associated and dissociated fractions of gemcitabine. This approach may be used to estimate the associated and dissociated fractions of drug over time for other nanoparticle drug delivery systems where isolation of in vivo associated and dissociated drug is experimentally challenging. Although this work focuses specifically on the use of combination G and T, DcNPs represent a potential approach to the synchronized delivery of other combination regimens used in breast cancer treatment such as targeted therapy or hormone therapy.
Combination drug nanoparticles have been previously reported as potential therapies for cancer. However, it remains a challenge to co-formulate chemically dissimilar drugs such as G and T (water soluble and insoluble drugs). To our knowledge, there are only a few published reports that achieve the co-formulation of GT to target breast cancer and each study notes an improved effect of combination particles versus individual GT which highlights the potential for combination particles (23–28). Water soluble G and water insoluble T are brought together by approaches such as chemical conjugation of both drugs to polymers (25,28) or encapsulation in calcium phosphate nanoparticles with a lipid bilayer coating (23). However, chemical conjugation will produce a new chemical entity that requires a long journey of regulatory approval and filing as a new drug. Calcium phosphate precipitation requires multiple filtration steps to remove organic solvents such as THF or chloroform.
In contrast, the distinction of the DcNP process is that no chemical conjugation is required to produce substantial in vivo association of both gemcitabine and paclitaxel. The DcNP process does not require filtration of unassociated drug or co-solvents as described in other reports. Even with a limited AE% of 9% for gemcitabine, a 50-fold increase in gemcitabine plasma exposure is observed when compared to the CrEL control. Further analysis based on a combination of metabolite kinetics and MBPK modeling suggests that water soluble gemcitabine is highly associated with DcNP in vivo. Paclitaxel was found to be highly associated both in vitro and in vivo, although the lower dose (5 mg/kg) limits its duration in plasma. While the exact mechanism leading to the stable association of water soluble gemcitabine to GT DcNPs is beyond the scope of this report, it is worth noting the resemblance with another similarly-manufactured DcNP formulation for HIV antivirals. Lopinavir, ritonavir and tenofovir association to DcNPs resulted in stable, water insoluble drug association (lopinavir and ritonavir) both in-vitro and in-vivo in non-human primates (NHP). On the other hand, water soluble tenofovir exhibited low in-vitro association (10%), but high in vivo association in NHPs (97%) (15). The stable circulation of GT well associated to DcNPs in plasma demonstrates the ability of one carrier to load two anticancer drugs while targeting cancer cells (Fig. 3).
Clinical studies have shown that prolonged infusion rates of G (10 mg/m2/min) confer a survival advantage over standard 30-min infusions (29). Deoxycytidine kinase (dCK), which converts G to its active triphosphate form, has been shown to be rapidly saturated after G infusion (30). As a result, a large fraction of the total dose of G is lost to metabolism by CDA before activation by dCK. Increasing the infusion time of G can allow more drug to be converted to active form and produce a greater pharmacologic effect. As an example, Eckel et al. showed that 24 h infusions of G can have similar pharmacologic effect as a 30 min conventional infusion for advanced pancreatic adenocarcinoma at 1/10th the dose (100 mg/m2 vs 1000 mg/m2) (31).
In this study, we have found that a single dose of GT in DcNPs can increase the apparent plasma half-life of G from 1.6 h to 13.72 h in mice (Fig. 3). No infusion is necessary in this case with GT DcNP administration. Although total drug concentration in plasma does not directly reflect the free fraction of G available for phosphorylation, the persistent circulation of parent drug can increase the opportunity for drug to reach target cells for phosphorylation instead of inactivation as seen with other long-acting nucleoside analogs (14). Thus, extending the plasma circulation of parent G may act similarly as a prolonged infusion and may produce a greater pharmacologic effect. Regarding T, clinical studies have not established a relationship between infusion rate and pharmacologic effect. Instead, conventional Taxol is infused over 3 h to mitigate the toxicity of Cremophor EL, a solubilizing excipient. Minor and major hypersensitivity reactions have been linked to rapid infusion of Cremophor El. When this excipient is not present, such as in albumin bound paclitaxel formulations (Abraxane), infusions can be administered in as little as 30 min without prophylactic medications for hypersensitivity reactions (32). In our study, the use of biocompatible lipid excipients with proven human safety in other dosage formulations (33) enables T to stay suspended in nanoparticle form. T in DcNP can be administered in a single dose (with G) without the need for Cremophor EL.
The pharmacokinetic profiles of nanoparticle delivery systems are often described using total drug concentrations instead of unbound drug concentrations. This is partly due to the complexity of separating bound and unbound fractions of drug from biological matrices (34). Total drug concentrations can provide an adequate description of particle circulation but may confound the prediction of pharmacologic effect. In this report, we offer novel approaches to estimate the fraction of drug that is associated to nanoparticles in vivo. GT association to DcNPs was first estimated by in vitro dialysis under sink conditions (G: 9%, T = 95%). However, this estimate did not correspond with our in vivo results and may be due to the lack of blood components in the dialysis experiment, which can affect drug dissociation.
In the biologic milieu and limited blood volume, our in vivo data indicates an association of G to DcNPs much greater than the 9% found in vitro. We reconcile these differences by using a non-compartmental approach to estimate the maximum time-averaged fraction of dissociated drug that can be present in vivo (Eq. 1, fdiss. max). This approach was first utilized with anti-HIV DcNPs and we have further extended its application toward an estimate for the fraction of drug available for metabolism when administered as a DcNP (Eq. 2, fdiss. G→dFdU). For G, the estimate for fdiss. G→dFdU was greater than fdiss. max suggesting that metabolism for DcNP-G can occur in tissues that are not part of the central compartment (such as lean tissue). It is possible that DcNPs distribute into peripheral tissue where local dissociation and metabolism of G can occur. Since this loss of parent drug does not re-enter the systemic circulation, Eq. 1 may underpredict the dissociated fraction. Alternatively, Eq. 2 captures the transit of dFdU from peripheral tissue back into the central compartment and account for peripheral metabolism but both fdiss. max and fdiss. G→dFdU indicate that G is highly associated to DcNPs. Regarding T association to DcNPs, the clearance pathway for dissociated T is in the liver, which resides in the central compartment. Under these conditions, fdiss. max can provide an estimate of the dissociated fraction without needing to account for loss of parent drug in peripheral tissues; it shows that T is also highly associated to DcNPs in vivo. Taken together, both estimates show that G and T mostly circulate in vivo as associated forms.
As an extension to those non-compartmental estimates, we next adapted a mechanism-based pharmacokinetic model (MBPK) to derive a dynamic simulation of both G and T association to DcNPs in plasma (11). Results from this MBPK model showed that both hydrophilic G and hydrophobic T are well associated in vivo and correlate closely to our non-compartmental estimates. These early results demonstrate a novel application of pharmacokinetic modeling to understand the species of GT that circulate in vivo. In future studies, we plan to incorporate metabolite markers for both G and T to further expand the concept of accounting for peripheral clearance. Use of non-compartmental estimates and further expansion of this MBPK model to include metabolite markers can provide a quantitative tool to characterize novel nanoparticle formulations in development.
Our next finding was that GT DcNPs do not appear to accumulate in healthy organs. Depending on the composition and size of nanoparticle formulations, the liver and spleen can sequester as much as 99% of other types of nanoparticles (35). This is mainly due to the fenestrations in liver and spleen microvasculature and direct interactions of traditional nanoparticles with endocytic cells. Although the removal of particulates constitutes an essential component of the immune system, the premature clearance of particles prior to their interaction with target cells poses a barrier to effective nanoparticle delivery. Lipid-based particles such as liposomes are often associated with liver and spleen uptake (36). For example, when large (378 nm) and small liposomes (113 nm) were intravenously administered in mice, 93% of large liposomes and 67% of small liposomes were recovered in the liver and spleen after 4 h (37). The hepatic uptake of particles is also observed with non-lipid nanoparticle delivery systems. Yeo et al. intravenously administered albumin bound paclitaxel (Abraxane, 30 mg/kg) in mice and found that after 3 h, liver concentrations of paclitaxel were 100-fold greater than plasma concentrations. In the same study, an F127 stabilized nanocrystal form of paclitaxel was intravenously injected (30 mg/kg) in mice and after 3 h, liver concentrations were 50-fold greater than plasma (38). Compared to these liposome and nanoparticle drug delivery systems, the GT in DcNP form have remarkably different biodistribution characteristics. The biodistribution properties of GT DcNPs may be related to the discoid-like morphology and absence of a membrane structure (as opposed to enclosed, spherical liposomes).
GT DcNPs are a combination particle that contains multiple active drugs to overcome drug resistance unlike single drug particles. Both water soluble G and water insoluble T are stabilized together by two lipid excipients, without the need for a membrane structure such as liposomes. Electron microscopic analysis of GT DcNP product reveals that they contain neither membrane structures nor spherical enclosures typically observed with liposomes. After IV injection, GT DcNPs do not appear to distribute into or accumulate in the liver or spleen. For G administered as DcNPs, the tissue-to-plasma ratios show that DcNPs significantly limit tissue distribution compared to the control formulation (<1/10th across all tissues) (Fig. 6a). This effect on G disposition is particularly promising as hepatic distribution of free drug is associated with hepatic abnormalities and dysfunction (39). For T, there is no clear trend on tissue distribution of drug when administered as a DcNP compared to the conventionally solubilized form. T is known to interact readily with albumin, while G does not, and this drug specific property may affect T disposition (40). Regardless of how T can dissociate from combination nanoparticles, the overall accumulation of both G and T in off-target organs is minimal compared to previous reports and may lead to improved safety.
In metastatic breast cancer, solid tumors and metastatic nodules induce major changes to their surrounding microenvironment. These changes can limit the effectiveness of nanoparticle delivery systems by reducing penetration into solid tumors (41). Various approaches have been investigated to overcome these limitations such as active targeting and tumor priming with limited clinical success (41). In our study, we found that intravenously administered DcNPs can produce greater concentrations in tumor burdened pulmonary tissue compared to healthy pulmonary tissue. This enhancement of drug accumulation in tumor-bearing lungs is likely due to increased distribution of DcNP from plasma to peripheral tumor fenestrations within tumor foci. The small size (60–70 nm) and prolonged circulation (48 h) in plasma of DcNPs may allow particles to penetrate the fenestrations of tumor foci, which typically range from 0.3 to 4.7 μm in size (42,43). Other nanoparticle systems such as liposomes or polymeric particles have been reported to leverage the leaky neovasculature around tumors to enhance drug permeation and retention (commonly referred to as the EPR effect). In these scenarios, new blood vessels formed to support rapid tumor growth are leaky due to poorly developed endothelial cells lining the vessels. This allow for the passive distribution, diffusion or penetration of nano-sized particles into solid tumors. In the current work, metastatic 4T1 cancer cells present as lung cancer nodules in already highly perfused capillary beds. Under these conditions, it is not clear whether neovasculature has formed or the role of the EPR effect on the observed tumor tissue targeting by GT DcNPs. Nevertheless, our data demonstrates the selective deposition of GT DcNPs in tumor burdened tissue. When we calculate the concentration ratios of G and T in tumor burdened lungs, we yield a ratio similar to the administered dose (8.8:1 versus 10:1, G/T). This suggests that intact DcNPs are depositing in tumor bearing lungs without having a large fraction of the dose sequestered in the reticuloendothelial system, reflected in the liver and spleen. The observation that untargeted DcNPs can have a tumor specific deposition in pulmonary tissue is a promising feature of DcNPs. In future studies, we will investigate the role of the EPR effect in the selective accumulation of GT DcNPs in cancer burdened tissue. While one could seek to improve the dispositional advantage of GT DcNPs with the use of active targeting ligands (such as those targeted to EGFR, integrin or other MBC markers), such studies are beyond the scope of this report but are under consideration for future studies.
We have recently evaluated the therapeutic effects of this GT DcNP composition on 4 T1 breast cancer metastasis to lungs in mice (44). A single GT (20/2 mg/kg) dose in DcNP form nearly eliminated breast cancer colonization in the lungs, while this effect was not achievable by a CrEL drug combination at a 5-fold higher dose (i.e., 100/10 mg/kg GT). Dose-response curves of cancer nodule inhibition and systemic toxicity through body weight loss demonstrated a therapeutic index of about 15.8. These results may be related to the preferential distribution and long acting pharmacokinetic properties contributed by stable association of GT to DcNPs in vivo.
CONCLUSION
In summary, we have developed stable drug combination nanoparticles composed of water soluble gemcitabine and water insoluble paclitaxel. GT DcNP stabilization is enabled by lipid excipient composition and a novel but simple process that does not require complex free drug removal. By doing so, this highly potent combination of chemotherapy has been transformed from a short-acting regimen to a long-acting regimen. The development and application of a mechanism based pharmacokinetic model elucidates the time course of associated and dissociated fractions of GT in vivo. This validated model indicates that GT remains associated to DcNP in vivo and displays an enhanced distribution toward cancer burdened tissue over healthy tissue, which may improve the therapeutic effect of this combination. In a recent report, these novel GT DcNPs were evaluated as a treatment using a highly aggressive 4T1 model of breast cancer metastasis. The DcNP platform is able to incorporate multiple drugs and allow water soluble and insoluble chemotherapeutic agents to form a single nano-dosage form. Thus, it may be used for other cancer drug combinations, either in clinical use or in development, with higher potencies. In addition, it may be possible to incorporate targeting ligands in the DcNP to provide additional cancer cell selectivity and preferential distribution of the chosen drug combinations. The long-acting and cancer tissue selective drug combination kinetics provided by this DcNP platform technology may lead to a meaningful impact on the development of targeted, combination treatment of metastatic breast cancer.
ACKNOWLEDGMENTS AND DISCLOSURES.
We thank Stanley Riddell laboratory in Fred Hutchinson Cancer Research Center for providing 4T1-luc and Matthew Hartman for editorial assistance. This work was supported in part by NIH grant UM1 AI120176, R61 AI149665, U01 AI148055 and J Yu was supported in part by a NIH pharmacology training grant T32 GM00750.
FINANCIAL SUPPORT
This work was supported in part by NIH grants UM1 AI120176, R61 AI149665, U01 AI1448055 and T32 GM007750 Pharmacological training for J Yu.
ABBREVIATIONS
- DcNP
Drug combination nanoparticle
- CrEL
Cremophor El suspension
- AUC
Area under the curve
- C0
Concentration at time 0
- T1/2
Half-life
- Dose/AUC
Apparent clearance
- Vss
Volume of distribution at steady state
- MRT
Mean residence time
- AUMC
Area under the moment curve
- GT
Gemcitabine and paclitaxel combination
- G
Gemcitabine
- T
Paclitaxel
- MBPK
Mechanism-based pharmacokinetic model
- K
rate constant
- dFdU
2′,2′-difluoro-deoxyuridine
- CDA
Cytidine deaminase
- dCK
deoxycytidine kinase
REFERENCES
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30. [DOI] [PubMed] [Google Scholar]
- 2.Wang R, Zhu Y, Liu X, Liao X, He J, Niu L. The Clinicopathological features and survival outcomes of patients with different metastatic sites in stage IV breast cancer. BMC Cancer. 2019;19(1):1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rahman AM, Yusuf SW, Ewer MS. Anthracycline-induced cardiotoxicity and the cardiac-sparing effect of liposomal formulation. Int J Nanomedicine. 2007;2(4):567–83. [PMC free article] [PubMed] [Google Scholar]
- 4.Albain KS, Nag SM, Calderillo-Ruiz G, Jordaan JP, Llombart AC, Pluzanska A, et al. Gemcitabine plus paclitaxel versus paclitaxel monotherapy in patients with metastatic breast cancer and prior anthracycline treatment. J Clin Oncol. 2008;26(24):3950–7. [DOI] [PubMed] [Google Scholar]
- 5.Grunewald R, Kantarjian H, Keating MJ, Abbruzzese J, Tarassoff P, Plunkett W. Pharmacologically directed design of the dose rate and schedule of 2′,2′-difluorodeoxycytidine (gemcitabine) administration in leukemia. Cancer Res. 1990;50(21):6823–6. [PubMed] [Google Scholar]
- 6.Luu T, Chow W, Lim D, Koczywas M, Frankel P, Cristea M, et al. Phase I trial of fixed-dose rate gemcitabine in combination with bortezomib in advanced solid tumors. Anticancer Res. 2010;30(1): 167–74. [PubMed] [Google Scholar]
- 7.Gianni L, Kearns CM, Giani A, Capri G, Vigano L, Lacatelli A, et al. Nonlinear pharmacokinetics and metabolism of paclitaxel and its pharmacokinetic/pharmacodynamic relationships in humans. J Clin Oncol. 1995;13(1):180–90. [DOI] [PubMed] [Google Scholar]
- 8.Smith RE, Brown AM, Mamounas EP, Anderson SJ, Lembersky BC, Atkins JH, et al. Randomized trial of 3-hour versus 24-hour infusion of high-dose paclitaxel in patients with metastatic or locally advanced breast cancer: National Surgical Adjuvant Breast and bowel project protocol B-26. J Clin Oncol. 1999;17(11):3403–11. [DOI] [PubMed] [Google Scholar]
- 9.Ait-Oudhia S, Mager DE, Straubinger RM. Application of pharmacokinetic and pharmacodynamic analysis to the development of liposomal formulations for oncology. Pharmaceutics. 2014;6(1): 137–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Freeling JP, Koehn J, Shu C, Sun J, Ho RJ. Anti-HIV drug-combination nanoparticles enhance plasma drug exposure duration as well as triple-drug combination levels in cells within lymph nodes and blood in primates. AIDS Res Hum Retrovir. 2015;31(1):107–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kraft JC, McConnachie LA, Koehn J, Kinman L, Sun J, Collier AC, et al. Mechanism-based pharmacokinetic (MBPK) models describe the complex plasma kinetics of three antiretrovirals delivered by a long-acting anti-HIV drug combination nanoparticle formulation. J Control Release. 2018;275:229–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perazzolo S, Shireman LM, Koehn J, McConnachie LA, Kraft JC, Shen DD, et al. Three HIV drugs, Atazanavir, ritonavir, and Tenofovir, Coformulated in drug-combination nanoparticles exhibit long-acting and lymphocyte-targeting properties in nonhuman Primates. J Pharm Sci. 2018;107(12):3153–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koehn J, Iwamoto JF, Kraft JC, McConnachie LA, Collier AC, Ho RJY. Extended cell and plasma drug levels after one dose of a three-in-one nanosuspension containing lopinavir, efavirenz, and tenofovir in nonhuman primates. AIDS. 2018;32(17):2463–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McConnachie LA, Kinman LM, Koehn J, Kraft JC, Lane S, Lee W, et al. Long-acting profile of 4 drugs in 1 anti-HIV nanosuspension in nonhuman Primates for 5 weeks after a single subcutaneous injection. J Pharm Sci. 2018;107(7):1787–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Perazzolo S, Shireman LM, McConnachie LA, Koehn J, Kinman L, Lee W, et al. Integration of computational and experimental approaches to elucidate mechanisms of first-pass lymphatic drug sequestration and long-acting pharmacokinetics of the injectable triple-HIV drug combination TLC-ART 101. J Pharm Sci. 2020;109:1789–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Veltkamp SA, Pluim D, van Tellingen O, Beijnen JH, Schellens JHM. Extensive metabolism and hepatic accumulation of gemcitabine after multiple oral and intravenous administration in mice. Drug Metab Dispos. 2008;36(8):1606–15. [DOI] [PubMed] [Google Scholar]
- 17.Kiani A, Köhne C-H, Franz T, Passauer J, Haufe T, Gross P, et al. Pharmacokinetics of gemcitabine in a patient with end-stage renal disease: effective clearance of its main metabolite by standard hemodialysis treatment. Cancer Chemother Pharmacol. 2003;51(3): 266–70. [DOI] [PubMed] [Google Scholar]
- 18.Shipley LA, Brown TJ, Cornpropst JD, Hamilton M, Daniels WD, Culp HW. Metabolism and disposition of gemcitabine, and oncolytic deoxycytidine analog, in mice, rats, and dogs. Drug Metab Dispos. 1992;20(6):849–55. [PubMed] [Google Scholar]
- 19.Tao K, Fang M, Alroy J, Sahagian GG. Imagable 4T1 model for the study of late stage breast cancer. BMC Cancer. 2008;8:228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Veltkamp SA, Beijnen JH, Schellens JH. Prolonged versus standard gemcitabine infusion: translation of molecular pharmacology to new treatment strategy. Oncologist. 2008;13(3):261–76. [DOI] [PubMed] [Google Scholar]
- 21.Reid JM, Qu W, Safgren SL, Ames MM, Krailo MD, Seibel NL, et al. Phase I trial and pharmacokinetics of gemcitabine in children with advanced solid tumors. J Clin Oncol. 2004;22(12):2445–51. [DOI] [PubMed] [Google Scholar]
- 22.de Jonge ME, Huitema AD, Schellens JH, Rodenhuis S, Beijnen JH. Population pharmacokinetics of orally administered paclitaxel formulated in Cremophor EL. Br J Clin Pharmacol. 2005;59(3): 325–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang J, Zhang P, Zou Q, Li X, Fu J, Luo Y, et al. Co-delivery of gemcitabine and paclitaxel in crgd-modified long circulating nanoparticles with asymmetric lipid layers for breast cancer treatment. Molecules. 2018;23(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dong S, Guo Y, Duan Y, Li Z, Wang C, Niu L, et al. Co-delivery of paclitaxel and gemcitabine by methoxy poly(ethylene glycol)-poly(-lactide-coglycolide)-polypeptide nanoparticles for effective breast cancer therapy. Anti-Cancer Drugs. 2018;29(7):637–45. [DOI] [PubMed] [Google Scholar]
- 25.Noh I, Kim HO, Choi J, Choi Y, Lee DK, Huh YM, et al. Co-delivery of paclitaxel and gemcitabine via CD44-targeting nanocarriers as a prodrug with synergistic antitumor activity against human biliary cancer. Biomaterials. 2015;53:763–74. [DOI] [PubMed] [Google Scholar]
- 26.Meng H, Wang M, Liu H, Liu X, Situ A, Wu B, et al. Use of a lipid-coated mesoporous silica nanoparticle platform for synergistic gemcitabine and paclitaxel delivery to human pancreatic cancer in mice. ACS Nano. 2015;9(4):3540–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lei M, Sha S, Wang X, Wang J, Du X, Miao H, et al. Co-delivery of paclitaxel and gemcitabine via a self-assembling nanoparticle for targeted treatment of breast cancer. RSC Adv. 2019;9(10):5512–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aryal S, Hu C-MJ, Zhang L. Combinatorial drug conjugation enables nanoparticle dual-drug delivery. Small. 2010;6(13):1442–8. [DOI] [PubMed] [Google Scholar]
- 29.Tempero M, Plunkett W, Ruiz Van Haperen V, Hainsworth J, Hochster H, Lenzi R, et al. Randomized phase II comparison of dose-intense gemcitabine: thirty-minute infusion and fixed dose rate infusion in patients with pancreatic adenocarcinoma. J Clin Oncol 2003;21(18):3402–3408. [DOI] [PubMed] [Google Scholar]
- 30.Grunewald R, Abbruzzese JL, Tarassoff P, Plunkett W. Saturation of 2′, 2′-difluorodeoxycytidine 5′-triphosphate accumulation by mononuclear cells during a phase I trial of gemcitabine. Cancer Chemother Pharmacol. 1991;27(4):258–62. [DOI] [PubMed] [Google Scholar]
- 31.Eckel F, Schmelz R, Erdmann J, Mayr M, Lersch C. Phase II trial of a 24-hour infusion of gemcitabine in previously untreated patients with advanced pancreatic adenocarcinoma. Cancer Investig. 2003;21(5):690–4. [DOI] [PubMed] [Google Scholar]
- 32.Gradishar WJ. Albumin-bound paclitaxel: a next-generation taxane. Expert Opin Pharmacother. 2006;7(8):1041–53. [DOI] [PubMed] [Google Scholar]
- 33.Bulbake U, Doppalapudi S, Kommineni N, Khan W. Liposomal formulations in clinical use: an updated review. Pharmaceutics. 2017;9(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Skoczen S, McNeil SE, Stern ST. Stable isotope method to measure drug release from nanomedicines. J Control Release. 2015;220(Pt A):169–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang YN, Poon W, Tavares AJ, McGilvray ID, Chan WCW. Nanoparticle-liver interactions: cellular uptake and hepatobiliary elimination. J Control Release. 2016;240:332–48. [DOI] [PubMed] [Google Scholar]
- 36.Allen TM, Everest JM. Effect of liposome size and drug release properties on pharmacokinetics of encapsulated drug in rats. J Pharmacol Exp Ther. 1983;226(2):539–44. [PubMed] [Google Scholar]
- 37.Hu Q, van Rooijen N, Liu D. Effect of macrophage elimination using liposome-encapsulated dichloromethylene diphosphonate on tissue distribution of liposomes. Journal of Liposome Research. 1996;6(4):681–98. [Google Scholar]
- 38.Park J, Park JE, Hedrick VE, Wood KV, Bonham C, Lee W, et al. A comparative in vivo study of albumin-coated paclitaxel nanocrystals and abraxane. Small. 2018;14(16):1703670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.King PD, Perry MC. Hepatotoxicity of chemotherapy. Oncologist. 2001;6(2):162–76. [DOI] [PubMed] [Google Scholar]
- 40.Paal K, Muller J, Hegedus L. High affinity binding of paclitaxel to human serum albumin. Eur J Biochem. 2001;268(7):2187–91. [DOI] [PubMed] [Google Scholar]
- 41.Jain RK. Delivery of molecular and cellular medicine to solid tumors. Adv Drug Deliv Rev. 2001;46(1–3):149–68. [DOI] [PubMed] [Google Scholar]
- 42.Nagy JA, Chang SH, Dvorak AM, Dvorak HF. Why are tumour blood vessels abnormal and why is it important to know? Br J Cancer. 2009;100(6):865–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hashizume H, Baluk P, Morikawa S, McLean JW, Thurston G, Roberge S, et al. Openings between defective endothelial cells explain tumor vessel leakiness. Am J Pathol. 2000;156(4):1363–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mu Q, Yu J, Griffin JI, Wu Y, Zhu L, McConnachie LA, et al. Novel drug combination nanoparticles exhibit enhanced plasma exposure and dose-responsive effects on eliminating breast cancer lung metastasis. PLoS One. 2020;15(3):e0228557. [DOI] [PMC free article] [PubMed] [Google Scholar]