

Abstract
This study aimed to investigate the clinicopathological significance and prospective molecular mechanism of RUNX family transcription factor 2 (RUNX2) in lung squamous cell carcinoma (LUSC). The authors used immunohistochemistry (IHC), RNA‐seq, and microarray data from multi‐platforms to conduct a comprehensive analysis of the clinicopathological significance and molecular mechanism of RUNX2 in the occurrence and development of LUSC. RUNX2 expression was significantly higher in 16 LUSC tissues than in paired non‐cancerous tissues detected by IHC (P < 0.05). RNA‐seq data from the combination of TCGA and genotype‐tissue expression (GTEx) revealed significantly higher expression of RUNX2 in 502 LUSC samples than in 476 non‐cancer samples. The expression of RUNX2 protein was also significantly higher in pathologic T3‐T4 than in T1‐T2 samples (P = 0.031). The pooled standardised mean difference (SMD) for RUNX2 was 0.87 (95% CI, 0.58–1.16), including 29 microarrays from GEO and one from ArrayExpress. The co‐expression network of RUNX2 revealed complicated connections between RUNX2 and 45 co‐expressed genes, which were significantly clustered in pathways including ECM‐receptor interaction, focal adhesion, protein digestion and absorption, human papillomavirus infection and PI3K‐Akt signalling pathway. Overexpression of RUNX2 plays an essential role in the clinical progression of LUSC.
Inspec keywords: bone, bioinformatics, biological organs, cellular biophysics, cancer, biological tissues, RNA, tumours, adhesion, proteins, lung, biochemistry, genetics, gynaecology, molecular biophysics
Other keywords: RUNX2 expression, RUNX2 protein, RUNX2 upregulation, lung squamous cell carcinoma tissues, RUNX family transcription factor 2
1 Introduction
With ∼228,820 estimated new cases and 135,720 estimated deaths in 2019, lung cancer (LC) is one of the most common malignant tumours and leads all cancer‐related deaths in the world [1, 2 ]. The majority of LC cases are non‐small cell LC (NSCLC), which can be divided into three subtypes: lung adenocarcinoma (LUAD), lung squamous cell carcinoma (LUSC) and large cell lung carcinoma (LCLC). Specifically, LUSC is the second‐most‐common type of NSCLC, with a high incidence of >400,000 new cases worldwide per year [3 ]. Current therapeutic strategies for LUSC are mainly surgery and chemotherapy. Although significant progress has been made in the targeted treatment of LUSC, the survival condition of LUSC patients remains poor [4 ]. Thus, it is of crucial importance to identify a novel molecular indicator to combat LUSC.
RUNX family transcription factor 2 (RUNX2) plays a crucial role in osteoblastic differentiation and skeletal morphogenesis [5 ]. Mounting evidence suggests that RUNX2 is involved in growth, proliferation and metastasis of various human cancers, including LC [6 ]. Li et al. demonstrated that RUNX2 overexpression in NSCLC and related to the poor prognosis of NSCLC patients [7 ]. Herreño et al. [8 ] reported that the expression of RUNX2 notably elevated in NSCLC and was related to the EMT process through direct regulation of E‐CADHERIN, VIMENTIN, TWIST1, and SNAIL1 expression. Tandon et al. [9 ] reported that RUNX2 is overexpressed in LC cells and that RUNX2 mediates epigenetic silencing of BMP‐3B to downregulate the expression of BMP‐3B. The study conducted by Zheng et al. [10 ] revealed that WWOX reduces the invasiveness of LC via suppression of the expression of RUNX2. These findings imply that RUNX2 may serve as a promising anti‐tumour target in LC. However, these studies lacked a comprehensive evaluation of the clinicopathological significance of RUNX2 in LC through multiple methods and investigation of the molecular mechanism of RUNX2 in LC from multiple perspectives. No study has yet explored the role of RUNX2 in LUSC in further depth.
In this work, we intend to investigate the clinicopathological significance and prospective molecular mechanism of RUNX2 in LUSC through a comprehensive analysis of immunohistochemistry (IHC), RNA‐seq and microarray data. We narrated the experiments in the sequential order of IHC, RNA‐seq and microarray data, all of which were used for evaluating RUNX2 expression in LUSC and non‐cancer tissues. RUNX2 expression in LUSC was assessed firstly in the protein level, then in the RNA level, lastly in the integrated level of protein and RNA.
2 Materials and methods
2.1 Immunohistochemistry
A total of 16 LUSC tissues and 16 paired non‐cancerous tissues (3 females, 13 males; average age 60 years) collected during the period of January 2016 to August 2017 by the Department of Pathology, Guigang People's Hospital of Guangxi/the Eighth Affiliated Hospital of Guangxi Medical University (Guangxi, China) were included for IHC. Approvement was acquired from the Ethical Committee of the Guigang People's Hospital of Guangxi/the Eighth Affiliated Hospital of Guangxi Medical University, and each patient signed written informed consent. Antigen retrieval was conducted by boiling tissue sections in sodium citrate buffer (pH 6.0) at 100–120°C for 5 min; endogenous peroxidase activity was blocked with 3% hydrogen peroxide at room temperature for 10 min; sections were then incubated with anti‐RUNX2 antibody [EPR14334] (cat. no. ab192256; 1: 500 dilution; Abcam, Cambridge, MA, USA) overnight at 4°C; followed by 3′,3′‐diaminoben‐zidene staining at room temperature for 5 min. The average score was calculated by randomly selecting ten fields under a light microscope (magnification ×200). The immunoreaction score (IRS) was calculated according to the intensity of staining and the percentage of positive cells. The scores were divided into 0, 1, 2, and 3, which represented negative, weak, medium, and strong, respectively. The positive staining proportion was classified as 0 for <10%, 1 for 11–25%, 2 for 26–50%, 3 for 51–75%, and 4 for 76–100%. The ultimate score was obtained based on the product of the positive proportion and the staining intensity [11 ].
2.2 Clinicopathological significance of RUNX2 in LUSC from RNA‐seq data
Level three fragments per kilobase million (FPKM) expression data of RUNX2 in 502 LUSC tissues and 49 non‐cancer tissues, as well as the clinical information, were achieved from the GDC data portal (https://portal.gdc.cancer.gov/ ). FPKM expression value of RUNX2 was further transformed into log2(transcripts per million [TPM] + 0.001) format. We also included 427 normal lung tissues from the genotype‐tissue expression (GTEx) (https://www.gtexportal.org/home/ ) as the complementary non‐cancer controls. The clinicopathological significance of RUNX2 was appraised in a merged cohort of 502 LUSC and 476 non‐cancer lung samples.
Integrated standardised mean difference (SMD) and summarised receiver's operating characteristics (SROCs) curves of RUNX2 expression in LUSC and non‐cancer lung tissues. We searched in Gene Expression Omnibus (GEO) (https://www.ncbi.nim.nih.gov/geo/ ), Sequence Read Archive (SRA) (https://www.sra.org.uk/ ), Oncomine (https://www.oncomine.org/ ) and ArrayExpress (https://www.ebi.ac.uk/arrayexpress/ ) databases to seek microarrays with RUNX2 expression in LUSC and non‐cancer lung tissues reported before 10 May 2020. Studies with more than three LUSC or non‐cancer lung samples containing RUNX2 expression data were included for the calculation of SMD and plotting of SROC. Forest plots of SMD and SROC curves were constructed for pooled RNA‐seq data and microarray data, as stated in previous works [12 ].
2.3 RUNX2 expression in all pan‐squamous cell carcinomas (SCCs)
In order to investigate whether the high expression of RUNX2 is a tumour‐ and site‐specific, or whether its expression in SCC at other sites is similar, we analysed RUNX2 expression status in pan‐SCC. RUNX2 expression profiles in four types of SCCs – including cervical SCC (CESC), oesophageal carcinoma (ESCA), head and neck SCC (HNSC) and LUSC – and corresponding normal tissues were downloaded from firebrowse (http://www.firebrowse.org/) [13 ].
2.4 Prognostic significance of RUNX2 in LC from KM plotter
Information of overall survival (OS) of LC patients and RUNX2 expression in LC were extracted from probe sets including 216994_s_at, 221283_at, 221282_x_at, 236858_s_at, 236859_at and 232231_at. We also extracted the prognosis data of LUSC and log2(TPM + 0.001) transformed expression data of RUNX2 in LUSC from TCGA (https://tcga‐data.nci.nih.gov/docs/publications/tcga/ ). The prognostic influence of RUNX2 on the OS of 1925 LC patients and 502 LUSC patients from the above probe sets and TCGA database were evaluated by Kaplan–Meier survival curves, and HR values were analysed by SPSS 22.0 [14, 15 ]. The cut‐off value for dividing LC or LUSC patients into different survival groups was the median expression value of RUNX2. In order to evaluate the prognosis of RUNX2 in LUSC patients, we also screened the data sets containing LUSC prognosis data in the public database. The median value of RUNX2 expression was taken as the boundary to divide the cases into high and low expression groups. The HR and 95% CI of each dataset were assessed using Stata 12.0. Then the HRs were integrated to evaluate the overall prognostic effect of RUNX2 on LUSC patients.
2.5 Alteration profile of RUNX2 in LUSC
To investigate the transcriptional mechanism of aberrant RUNX2 expression in LUSC, we queried the genetic alteration status of RUNX2 in 511 LUSC patients from the TCGA‐Firehose Legacy dataset in the OncoPrint module of cBioPortal v.3.3.1 (https://www.cbioportal.org/) [16, 17 ]. We also checked indels (insertion and deletions) and synonymous mutations of RUNX2 in LUSC on the Human Gene Mutation Database (HGMD) (http://www.hgmd.org/ ), 1000 Genomes Project (1000GP) (https://www.genome.gov/27528684/1000‐genomes‐project ), National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project (https://evs.gs.washington.edu/EVS/ ), and Database of Cancer Driver InDels (dbCID) (http://bioinfo.ahu.edu.cn:8080/dbCID/ ), since these two types of mutations are also likely to be engaged in cancer development, progression or therapy [18 –20 ].
2.6 Co‐expression network of RUNX2
Co‐expression analysis was conducted on the log2(TPM + 0.001) expression matrix of 502 LUSC tissues and 49 non‐cancer tissues from TCGA using weighted correlation network analysis (WGCNA) package in R software v.3.6.1. Genes in the same module with RUNX2 with a weight value of more than 0.1 were considered as the co‐expressed genes of RUNX2 [21, 22 ]. The co‐expression network of RUNX2 and its co‐expressed genes was built by Cytoskape v.3.7. Functional enrichment analyses of the co‐expressed genes were carried out via the ClusterProfiler package in R software v.3.6.1. Terms with a P ‐value of <0.05 were considered as a significant.
2.7 Statistical analysis
The statistical analyses of RNA‐seq and IHC data were conducted by using SPSS 22.0. RUNX2 expression was presented in the form of a mean (M ) ± standard deviation (SD). Differential expression of RUNX2 between LUSC and non‐cancer tissues from IHC and RNA‐seq data was assessed by independent sample t‐tests and paired sample t‐tests, respectively. The relationship between RUNX2 expression and clinicopathological parameters with two subgroups was examined by independent samples’ t‐tests. When the clinicopathological parameters contained three or more subgroups, the Kruskal–Wallis test was performed to examine the significance of RUNX2 differential expression. Violin plots in each dataset were drawn by GraphPad Prism 8.0. According to the maximum Youden index and the corresponding cut‐off values of the receiver operating characteristics (ROCs), RUNX2 expression was input into SPSS (version 23.0, IBM Corp., Armonk, NY, USA) to compute true positive (TP), true negative (TN), false positive (FP), and false negative (FN) counts. The SROC curve was plotted by an INLA package of R software (V3.6.1) based on TP, TN, FP and FN. ROC curves and Kaplan–Meier survival curves were constructed to appraise the distinguishing ability and prognostic value of RUNX2 in LUSC. Hazard ratio (HR) and log‐rank p values for survival analysis were examined. P values <0.05 were regarded to be statistically significant.
3 Results
3.1 Overexpression of RUNX2 protein in LUSC
According to the results from IHC, remarkably stronger immunostaining of RUNX2 was observed in LUSC tissues than in adjacent lung tissues (Fig. 1 ).
Fig 1.
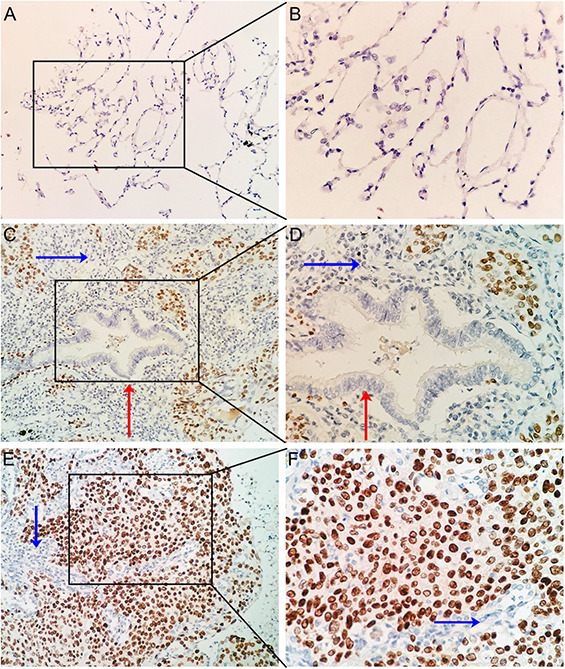
IHC staining of RUNX2 in LUSC and non‐cancer lung tissues
(a) IHC staining of normal alveoli tissues at 10 × 20
(b) IHC staining of normal alveoli tissues at 10 × 40
(c) IHC staining of LUSC with bronchus tissues at 10 × 20
(d) IHC staining of LUSC with bronchus tissues at 10 × 40
(e) IHC staining of LUSC tissues at 10 × 20
(f) IHC staining of LUSC tissues at 10 × 40
RUNX2 expression was pronouncedly higher in LUSC tissues than in the adjacent normal bronchial mucosa or alveoli (Figs. 2 a and b ) (P = 0.049, P < 0.001). RUNX2 also exhibited a supreme ability in separating LUSC tissues from normal alveoli tissues (Fig. 2 d ) (AUC = 0.900, P < 0.001).
Fig 2.
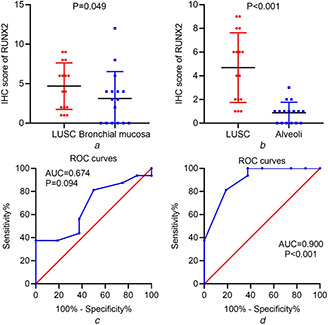
Differential expression of RUNX2 in LUSC and normal lung tissues from IHC data
(a) Scatter plot of IHC scores of RUNX2 in LUSC and normal bronchial mucosa tissues
(b) Scatter plot of IHC scores of RUNX2 in LUSC and normal alveoli tissues
(c) ROC curves of RUNX2 expression in distinguishing LUSC and normal bronchial mucosa tissues
(d) ROC curves of RUNX2 expression in distinguishing LUSC and normal alveoli tissues
3.2 RUNX2 expression from integrated RNA‐seq and microarray data
RNA‐seq data from the combination of TCGA and GTEx showed significantly higher expression of RUNX2 in 502 LUSC samples than in 476 non‐cancer samples (Fig. 3 ). Overexpression of RUNX2 could discriminate LUSC from non‐cancer lung tissues well (AUC = 0.724) (Fig. 4 ). Moreover, RUNX2 expression was closely related to the T stage of LUSC from the TCGA cohort. Patients with advanced T stage (T3‐4) presented higher RUNX2 expression (9.796 ± 1.092) than patients with early T stage (T1‐2) (9.518 ± 1.127) (P = 0.031) (Fig. 5 a ). With regard to microarrays, a total of 30 microarray datasets – 29 from the GEO database and one from ArrayExpress – were included in the present study (Fig. S1). It can be noted from a panel of violin plots and ROC curves that RUNX2 showed overexpression in LUSC tissues, and RUNX2 performed well in differentiating LUSC from non‐cancer lung tissues in most microarray datasets (Figs. 3 and 4 ). The forest plot of SMD indicated the overall upregulation of RUNX2 in LUSC tissues (SMD = 0.87, 95%CI = 0.58–1.16) (Fig. S2). SROC curves and forest plots of the positive and negative likelihood ratio (PLR and NLR), sensitivity, (SEN) specificity (SPE) and diagnostic odds ratio (DOR) supported the power of RUNX2 in distinguishing LUSC from non‐cancer tissues (AUC = 0.80, overall PLR = 2.85, NLR = 0.44, DOR = 7.58, SEN = 0.70, SPE = 0.83) (Fig. S3).
Fig 3.
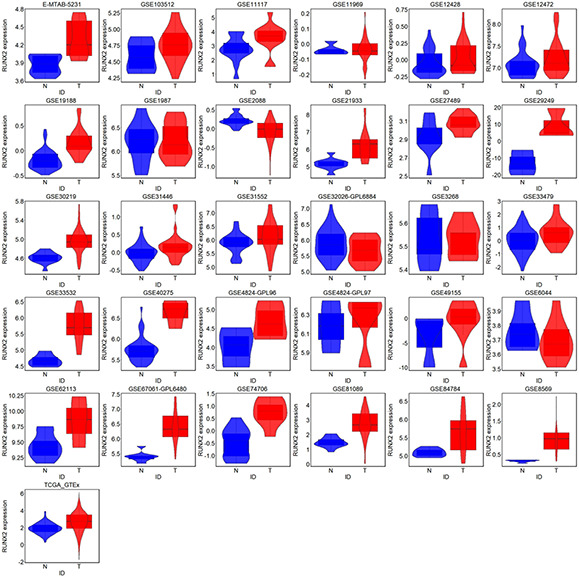
Violin plot for RUNX2 expression in LUSC and non‐cancer lung tissues from all RNA‐seq and microarray datasets. LUSC and non‐cancer tissues were marked by blue and red, respectively. T is for tumour and N is for non‐cancer
Fig 4.
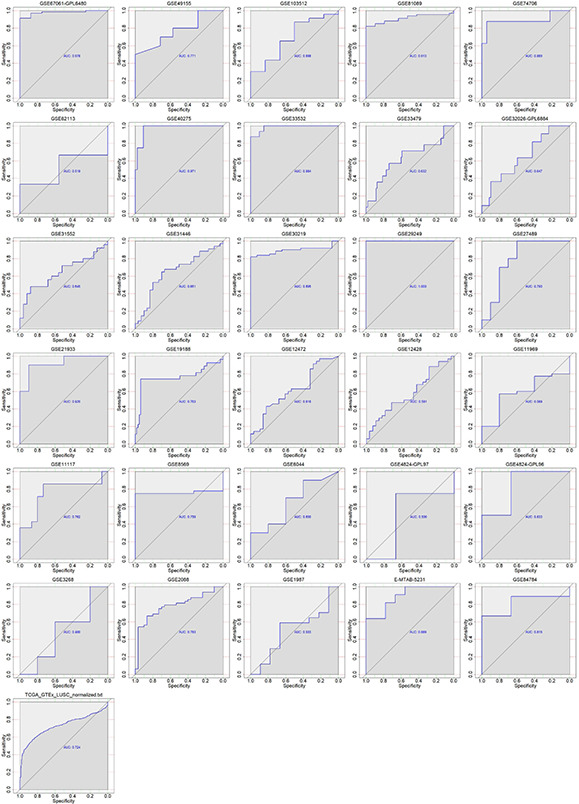
ROC curves of the ability of RUNX2 to discriminate LUSC from non‐cancer tissues in all RNA‐seq and microarray datasets. AUC: area under curve
Fig 5.
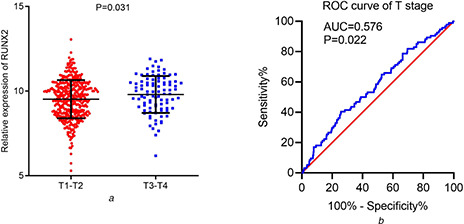
Relationship between RUNX2 and T stage of LUSC patients from TCGA cohorts
(a) Scatter plot of differential RUNX2 expression between early and advanced T stages
(b) ROC curves of the ability of RUNX2 to separate patients with different T stages
3.3 Overexpression of RUNX2 in pan‐SCC tissues
As shown in Fig. 6, RUNX2 demonstrated overexpression in all types of SCCs, including CESC, ESCA, HNSC, and LUSC. Particularly, the log2 normalised RUNX2 expression contrasted sharply between CESC, ESCA, LUSC, and respective normal lung tissues. We also found that the RUNX2 expression level was not significantly different between distinct SCC tissues based on the result of firebrowse (Fig. 6 ).
Fig 6.
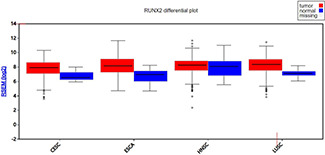
RUNX2 expression profiles in pan‐SCCs from firebrowse. CESC: cervical SCC; ESCA: oesophageal carcinoma; HNSC: head and neck SCC; LUSC: lung SCC; RSEM: RNA‐Seq by expectation‐maximisation
3.4 Impact of RUNX2 on the prognosis of LC
Kaplan–Meier survival curves for 221283_at, 221282_x_at, and 216994_s_at probe sets indicated that LC patients with higher RUNX2 expression had poorer survival outcomes than those with lower RUNX2 expression (P < 0.05) (Figs. 7 a –c ). The medium survival months of LC patients in 221283_at probe set with high or low RUNX2 expression were 64.1 and 76, respectively. LC patients in 221282_x_at probe set with high or low RUNX2 expression had medium survival months of 57 and 101.47, respectively. LC patients in the 216994_s_at probe set with high or low RUNX2 expression had medium survival months of 56 and 88.7, respectively. There was no obvious difference between the survival outcomes of LUSC patients from RNA‐seq data with high or low RUNX2 expression (data not shown). The flowchart of screening for the prognostic role of RUNX2 in LUSC is shown in Fig. S4, and 16 datasets were finally included. The integrated HR is 1.13 (95%CI: 0.96–1.32) (Fig. S5), indicating that increased expression of RUNX2 mRNA had a trend to act as a risk factor for the poor survival of LUSC.
Fig 7.
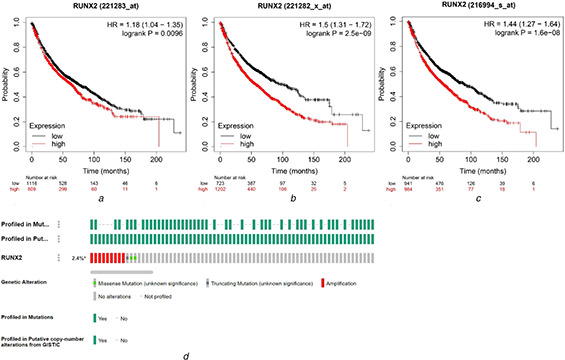
Prognostic influence and alteration status of RUNX2 in LUSC
(a) Kaplan–Meier survival curves of RUNX2 in 1925 LC patients from 221283_at probe set
(b) Kaplan–Meier survival curves of RUNX2 in 1925 LC patients from 221282_x_at probe set
(c) Kaplan–Meier survival curves of RUNX2 in 1925 LC patients from 216994_s_at probe set
(d) Alteration profile of RUNX2 in LUSC from cBioPortal
3.5 Genetic alteration status of RUNX2 in LUSC
The alteration frequency of RUNX2 in 511 LUSC patients from the TCGA‐Firehose Legacy dataset was 2.4%. The 12 LUSC cases with RUNX2 alterations were comprised of nine cases of amplification, one case of truncating mutation and two cases of a missense mutation (Fig. 7 d ). Hence, the predominant type of alterations for RUNX2 in LUSC was amplification. Unfortunately, we have not found any indels (insertion and deletions) and synonymous mutations of RUNX2 in LUSC, according to the HGMD, 1000GP, NHLBI Exome Sequencing Project, and dbCID databases.
3.6 Co‐expression network of RUNX2
WGCNA analysis suggested that a total of 45 genes in green module with a weight value of <0.1 were identified as the co‐expressed genes of RUNX2 in LUSC. The interactions between RUNX2 and co‐expressed genes were illustrated as a network (Fig. 8 ). Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses for the 45 genes indicated that they were mainly enriched in biological processes such as extracellular matrix organisation, extracellular structure organisation, collagen fibril organisation, collagen metabolic process and cell‐substrate adhesion and pathways including ECM‐receptor interaction, focal adhesion, protein digestion and absorption, human papillomavirus infection and PI3K‐Akt signalling pathway (P < 0.05) (Fig. 9, Table 1 ).
Fig 8.
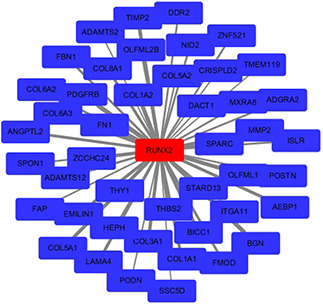
Co‐expression network of RUNX2 in LUSC. RUNX2 and the co‐expressed genes of it were marked in red and blue, respectively. The width of the links between genes represented the value of weights
Fig 9.
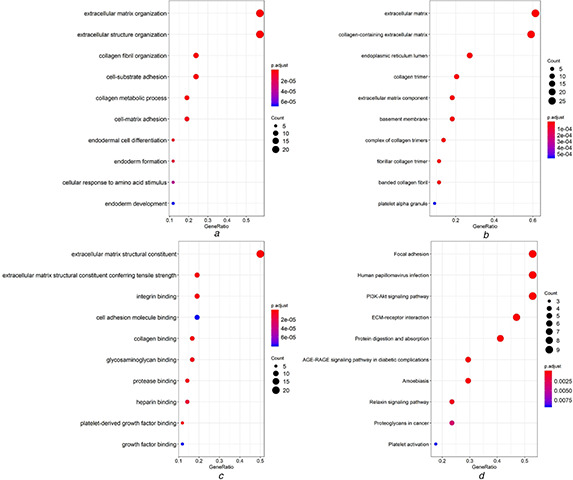
Gene ontology (GO) and Kyoto Encyclopaedia of Genes and Genomes (KEGG) pathway analysis for genes co‐expressed with RUNX2
(a) Dot plot for significant terms of biological functions
(b) Dot plot for significant terms of cellular component
(c) Dot plot for significant terms of molecular functions
(d) Dot plot for significant terms of KEGG pathways
Table 1.
Significant KEGG enriched terms of 45 RUNX2 co‐expression genes
| Term | Count | P‐ value | Genes |
|---|---|---|---|
| hsa04512: ECM–receptor interaction | 8 | 3.36 × 10−12 | ITGA11/THBS2/LAMA4/FN1/COL6A3/COL6A2/COL1A2/COL1A1 |
| hsa04510: focal adhesion | 9 | 6.00 × 10−11 | ITGA11/THBS2/PDGFRB/LAMA4/FN1/COL6A3/COL6A2/COL1A2/COL1A1 |
| hsa04974: protein digestion and absorption | 7 | 4.56 × 10−10 | COL6A3/COL6A2/COL5A2/COL5A1/COL3A1/COL1A2/COL1A1 |
| hsa05165: human papillomavirus infection | 9 | 5.43 × 10−09 | ITGA11/THBS2/PDGFRB/LAMA4/FN1/COL6A3/COL6A2/COL1A2/COL1A1 |
| hsa04151: PI3K‐Akt signalling pathway | 9 | 1.01 × 10−08 | ITGA11/THBS2/PDGFRB/LAMA4/FN1/COL6A3/COL6A2/COL1A2/COL1A1 |
| hsa04933: AGE‐RAGE signalling pathway in diabetic complications | 5 | 1.49 × 10−06 | MMP2/FN1/COL3A1/COL1A2/COL1A1 |
| hsa05146: amoebiasis | 5 | 1.64 × 10−06 | LAMA4/FN1/COL3A1/COL1A2/COL1A1 |
| hsa04926: relaxin signalling pathway | 4 | 1.28 × 10−04 | MMP2/COL3A1/COL1A2/COL1A1 |
| hsa05205: proteoglycans in cancer | 4 | 7.54 × 10−04 | MMP2/FN1/COL1A2/COL1A1 |
| hsa04611: platelet activation | 3 | 2.08 × 10−03 | COL3A1/COL1A2/COL1A1 |
4 Discussion
Benefiting from the rapid development of high‐throughput sequencing and microarray technologies, we are the first group to investigate the clinicopathological significance and molecular mechanism of RUNX2 in LUSC via integration of IHC, RNA‐seq and microarray data from multi‐platforms.
In this study, results from RNA‐seq, microarrays and IHC data unanimously supported the overexpression of RUNX2 in LUSC and the excellent ability of RUNX2 to differentiate LUSC from non‐cancer tissues. Furthermore, the upregulation of RUNX2 in LUSC was significantly related to the advanced T stage of LUSC, indicating that RUNX2 might be involved in the malignant clinical progression of LUSC. Considering that RUNX2 might be associated with malignant tumours derived from squamous epithelium, we explored RUNX2 expression status in pan‐SCC. The result indicated that the expression level of RUNX2 in other SCCs is consistent with that in LUSC. We could speculate that RUNX2 may play a consistent role in the development of SCC included LUSC. Previous studies have also discovered the close relationship between RUNX2 and malignant phenotypes of other malignancies, such as breast cancer, colorectal cancer, and cervical cancer. Li et al. [23 ] reported that knockdown of CCAT1 suppresses cell growth and epithelial to mesenchymal transition (EMT) of cervical cancer cells through inhibiting RUNX2 expression. In the study conducted by Jiang et al. [24 ], RUNX2 expression was remarkably elevated in recurrent CRC tumours and was intimately related to TMN stages, metastasis, and prognosis of CRC patients. Si et al. [25 ] demonstrated that SET7/9 collaborated with activated RUNX2 to promote the development of breast cancer. Prior research in LC revealed RUNX2 overexpression and the promotive effect of RUNX2 on the aggressiveness of NSCLC [7, 8, 26 ]. Most of the NSCLC cases studied were LUAD, and RUNX2 has not been investigated in LUSC. Li et al. [7 ] verified the overexpression and clinical significance of RUNX2 in NSCLC by IHC, RT‐Qpcr, and Western blot, however, there is a lack of research on the potential mechanism of RUNX2 in NSCLC including LUSC. Herreño et al. [8 ] also demonstrated the overexpression of RUNX2 in NSCLC; however, their research on the transcriptional regulation mechanism of RUNX2 mainly focuses on lung adenocarcinoma. The two above studies suffered from the limitations of a small sample size and lacked independent research on the expression status, clinical significance, and underlying mechanisms of RUNX2 in LUSC. This study is a comprehensive study using multiple approaches and methods to uncover the role and mechanism of RUNX2 in LUSC. RUNX2 overexpression in LUSC was validated using a large number of cases (1159 LUSC and 1072 non‐tumour lung samples) derived from TCGA‐GTEx and public gene microarrays, thereby assuring the accuracy and reliability of our research. Moreover, we further explored the molecular mechanism underlying RUNX2 in LUSC through WGCNA analysis and functional enrichment analysis of co‐expressed genes. Considering the above points, RUNX2 possesses great potential as a therapeutic and diagnostic target for LUSC.
We checked the clinical information of LUSC in the tissues used from in‐house IHC, TCGA and gene microarrays. Unfortunately, only GSE8569 microarray contains details of grade in LUSC tissues in all the included gene microarrays; then, we explored the correlation of RUNX2 expression with a grade of LUSC. We found that in the GSE8569 chip, the worse the LUSC grade, the lower the expression level of RUNX2 (Fig. S6). Unfortunately, this is only a single‐chip study result, which requires us to collect more LUSC patient grade information in future studies to better evaluate the relationship between the expression of RUNX2 and LUSC grade.
After confirming the overexpression and oncogenic role of RUNX2 in LUSC, we further explored the molecular mechanism underlying RUNX2 in LUSC through WGCNA analysis and functional enrichment analysis of co‐expressed genes. The co‐expression network of RUNX2 revealed complicated connections between RUNX2 and 45 co‐expressed genes, which were prominently clustered in pathways including ECM‐receptor interaction, focal adhesion, protein digestion, and absorption, human papillomavirus infection and PI3K‐Akt signalling pathway. Among the significantly enriched pathways, ECM‐receptor interaction, focal adhesion, and PI3K‐Akt signalling pathway played indispensable roles in the growth, invasion, and metastasis of human cancers [27 –29 ]. Pathways such as ECM‐receptor interaction and focal adhesion played crucial roles in EMT, and RUNX2 has been reported to participate in EMT to boost the migratory ability of NSCLC [6, 30 –33 ]. Other researchers have pointed out that RUNX2 may interact with genes including BMP‐3B, miR‐218, WWOX and miR‐196b to regulate the development of LC [9, 10, 26, 34 ]. Therefore, we hypothesised that RUNX2 might interact with the co‐expressed genes in the above pathways or be controlled by upstream molecules to influence the occurrence and progression of LUSC.
Despite the novel findings in this work, there were some limitations. Co‐expressed genes of RUNX2 in this study were found via WGCNA analysis; experiments such as RNA Binding Protein Immunoprecipitation should be carried out to validate the interactions between RUNX2 and co‐expressed genes. The protein and gene expression levels of RUNX2 could be further confirmed using Western blot and qRT‐PCR analyses. Further, in vitro and in vivo experiments are also expected to verify the catalytic functions of RUNX2 in biological events of LUSC.
5 Conclusion
In summary, RUNX2 was overexpressed in LUSC and correlated with the clinical progression of LUSC. The novelties of the current study lie in the following aspects. Firstly, multiple datasets with sufficient samples were integrated for evaluating the clinicopathological significance of RUNX2 in LUSC. Secondly, we combined experimental pathology with computational pathology for joint verification of RUNX2 overexpression in LUSC. Thirdly, the molecular mechanism of RUNX2 in LUSC was investigated through analysing alteration status and co‐expression network. The findings of the study are anticipated to provide insights into the potential use of RUNX2 as a biomarker for LUSC.
6 Acknowledgments
This study was supported by the Guigang Scientific Research and Technological Development Plan (no. Guikegong1701008), Innovation Project of Guangxi Graduate Education (YCBZ2020045), Guangxi Degree and Postgraduate Education Reform and Development Research Projects, China (JGY2019050). Da‐Ping Yang and Hui‐Ping Lu are contributed equally to this work.
7 References
- 1. Siegel R.L. Miller K.D. Jemal A.: ‘Cancer statistics, 2020 ’, CA Cancer J. Clin., 2020, 70, (1 ), pp. 7 –30 [DOI] [PubMed] [Google Scholar]
- 2. Li W. Li X. Gao L.N. et al.: ‘Integrated analysis of the functions and prognostic values of RNA binding proteins in lung squamous cell carcinoma ’, Front. Genet., 2020, 11, p. 185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang X. Liao X. Huang K. et al.: ‘Clustered microRNAs hsa‐miR‐221–3p/hsa‐miR‐222‐3p and their targeted genes might be prognostic predictors for hepatocellular carcinoma ’, J. Cancer, 2019, 10, (11 ), pp. 2520 –2533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhang N. Wang H. Xie Q. et al.: ‘Identification of potential diagnostic and therapeutic target genes for lung squamous cell carcinoma ’, Oncol. Lett., 2019, 18, (1 ), pp. 169 –180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zuo Z. Ye F. Liu Z. et al.: ‘MicroRNA‐153 inhibits cell proliferation, migration, invasion and epithelial‐mesenchymal transition in breast cancer via direct targeting of RUNX2 ’, Exp. Ther. Med., 2019, 17, (6 ), pp. 4693 –4702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Huang J. Chang S. Lu Y. et al.: ‘Enhanced osteopontin splicing regulated by RUNX2 is HDAC‐dependent and induces invasive phenotypes in NSCLC cells ’, Cancer Cell Int., 2019, 19, p. 306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li H. Zhou R.J. Zhang G.Q. et al.: ‘Clinical significance of RUNX2 expression in patients with non‐small cell lung cancer: a 5‐year follow‐up study ’, Tumour Biol., 2013, 34, (3 ), pp. 1807 –1812 [DOI] [PubMed] [Google Scholar]
- 8. Herreño A.M. Ramírez A.C. Chaparro V.P. et al.: ‘Role of RUNX2 transcription factor in epithelial mesenchymal transition in non‐small cell lung cancer lung cancer: epigenetic control of the RUNX2 P1 promoter ’, Tumour Biol., 2019, 41, (5 ), p. 1010428319851014 [DOI] [PubMed] [Google Scholar]
- 9. Tandon M. Gokul K. Ali S.A. et al.: ‘Runx2 mediates epigenetic silencing of the bone morphogenetic protein‐3B (BMP‐3B/GDF10) in lung cancer cells ’, Mol. Cancer, 2012, 11, p. 27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zheng Q.W. Zhou Y.L. You Q.J. et al.: ‘WWOX inhibits the invasion of lung cancer cells by downregulating RUNX2 ’, Cancer Gene Ther., 2016, 23, (12 ), pp. 433 –438 [DOI] [PubMed] [Google Scholar]
- 11. Liang L. Zhao K. Zhu J.H. et al.: ‘Comprehensive evaluation of FKBP10 expression and its prognostic potential in gastric cancer ’, Oncol. Rep., 2019, 42, (2 ), pp. 615 –628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gao L. Xiong D.D. He R.Q. et al.: ‘MIR22HG as a tumor suppressive lncRNA In HCC: a comprehensive analysis integrating RT‐qPCR, mRNA‐seq, and microarrays ’, Onco. Targets Ther., 2019, 12, pp. 9827 –9848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li C. Qin F. Hong H. et al.: ‘Identification of flap endonuclease 1 as a potential core gene in hepatocellular carcinoma by integrated bioinformatics analysis ’, PeerJ, 2019, 7, p. e7619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Győrffy B. Surowiak P. Budczies J. et al.: ‘Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non‐small‐cell lung cancer ’, PLoS One, 2013, 8, (12), p. e82241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nagy Á. Lánczky A. Menyhárt O. et al.: ‘Validation of miRNA prognostic power in hepatocellular carcinoma using expression data of independent datasets ’, Sci. Rep., 2018, 8, (1 ), p. 9227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Buechner P. Hinderer M. Unberath P. et al.: ‘Requirements analysis and specification for a molecular tumor board platform based on cBioPortal ’, Diagnostics (Basel), 2020, 10, (2 ), p. 93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jiao X.D. He X. Qin B.D. et al.: ‘The prognostic value of tumor mutation burden in EGFR‐mutant advanced lung adenocarcinoma, an analysis based on cBioPortal data base ’, J. Thorac. Dis., 2019, 11, (11 ), pp. 4507 –4515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Folkman L. Yang Y. Li Z. et al.: ‘DDIG‐in: detecting disease‐causing genetic variations due to frameshifting indels and nonsense mutations employing sequence and structural properties at nucleotide and protein levels ’, Bioinformatics, 2015, 31, (10 ), pp. 1599 –1606 [DOI] [PubMed] [Google Scholar]
- 19. Cheng N. Li M. Zhao L. et al.: ‘Comparison and integration of computational methods for deleterious synonymous mutation prediction ’, Brief. Bioinform., 2020, 21, (3 ), pp. 970 –981 [DOI] [PubMed] [Google Scholar]
- 20. Yue Z. Chu X. Xia J.: ‘PredCID: prediction of driver frameshift indels in human cancer ’, Brief. Bioinform., 2020, 26, p. bbaa119 [DOI] [PubMed] [Google Scholar]
- 21. Langfelder P. Horvath S.: ‘WGCNA: an R package for weighted correlation network analysis ’, BMC Bioinformatics, 2008, 9, p. 559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jiang C. Cao S. Li N. et al.: ‘PD‐1 and PD‐L1 correlated gene expression profiles and their association with clinical outcomes of breast cancer ’, Cancer Cell Int., 2019, 19, p. 233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li R. Liu J. Qi J.: ‘Knockdown of long non‐coding RNA CCAT1 suppresses proliferation and EMT of human cervical cancer cell lines by down‐regulating Runx2 ’, Exp. Mol. Pathol., 2020, 113, p. 104380 [DOI] [PubMed] [Google Scholar]
- 24. Jiang L. Yu X. Ma X. et al.: ‘Identification of transcription factor‐miRNA‐lncRNA feed‐forward loops in breast cancer subtypes ’, Comput. Biol. Chem., 2019, 78, pp. 1 –7 [DOI] [PubMed] [Google Scholar]
- 25. Si W. Zhou J. Zhao Y. et al.: ‘SET7/9 promotes multiple malignant processes in breast cancer development via RUNX2 activation and is negatively regulated by TRIM21 ’, Cell Death Dis., 2020, 11, (2 ), p. 151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xie J. Yu F. Li D. et al.: ‘MicroRNA‐218 regulates cisplatin (DPP) chemosensitivity in non‐small cell lung cancer by targeting RUNX2 ’, Tumour Biol., 2016, 37, (1 ), pp. 1197 –1204 [DOI] [PubMed] [Google Scholar]
- 27. Cheng H. Jiang X. Zhang Q. et al.: ‘Naringin inhibits colorectal cancer cell growth by repressing the PI3 K/AKT/mTOR signaling pathway ’, Exp. Ther. Med., 2020, 19, (6 ), pp. 3798 –3804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang K. Wang J. Wang J. et al.: ‘LKB1 deficiency promotes proliferation and invasion of glioblastoma through activation of mTOR and focal adhesion kinase signaling pathways ’, Am. J. Cancer Res., 2019, 9, (8 ), pp. 1650 –1663 [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang H.J. Tao J. Sheng L. et al.: ‘Twist2 promotes kidney cancer cell proliferation and invasion by regulating ITGA6 and CD44 expression in the ECM‐receptor interaction pathway ’, Onco. Targets Ther., 2016, 9, pp. 1801 –1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Herreño A.M. Ramírez A.C. Chaparro V.P. et al.: ‘Role of RUNX2 transcription factor in epithelial mesenchymal transition in non‐small cell lung cancer lung cancer: epigenetic control of the RUNX2 P1 promoter ’, Tumour Biol., 2019, 41, (5 ), p. 1010428319851014 [DOI] [PubMed] [Google Scholar]
- 31. Brandão‐Costa R.M. Helal‐Neto E. Vieira A.M. et al.: ‘Extracellular matrix derived from high metastatic human breast cancer triggers epithelial‐mesenchymal transition in epithelial breast cancer cells through αvβ3 integrin ’, Int. J. Mol. Sci., 2020, 21, (8 ), p. 2995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ji M. Li W. He G. et al.: ‘Zinc‐α2‐glycoprotein 1 promotes EMT in colorectal cancer by filamin A mediated focal adhesion pathway ’, J. Cancer, 2019, 10, (22 ), pp. 5557 –5566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hsu Y.L. Huang M.S. Yang C.J. et al.: ‘Lung tumor‐associated osteoblast‐derived bone morphogenetic protein‐2 increased epithelial‐to‐mesenchymal transition of cancer by Runx2/snail signaling pathway ’, J. Biol. Chem., 2011, 286, (43 ), pp. 37335 –37346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bai X. Meng L. Sun H. et al.: ‘MicroRNA‐196b inhibits cell growth and metastasis of lung cancer cells by targeting Runx2 ’, Cell. Physiol. Biochem., 2017, 43, (2 ), pp. 757 –767 [DOI] [PubMed] [Google Scholar]