

Abstract
Constructing interaction network from biomedical texts is a very important and interesting work. The authors take advantage of text mining and reinforcement learning approaches to establish protein interaction network. Considering the high computational efficiency of co‐occurrence‐based interaction extraction approaches and high precision of linguistic patterns approaches, the authors propose an interaction extracting algorithm where they utilise frequently used linguistic patterns to extract the interactions from texts and then find out interactions from extended unprocessed texts under the basic idea of co‐occurrence approach, meanwhile they discount the interaction extracted from extended texts. They put forward a reinforcement learning‐based algorithm to establish a protein interaction network, where nodes represent proteins and edges denote interactions. During the evolutionary process, a node selects another node and the attained reward determines which predicted interaction should be reinforced. The topology of the network is updated by the agent until an optimal network is formed. They used texts downloaded from PubMed to construct a prostate cancer protein interaction network by the proposed methods. The results show that their method brought out pretty good matching rate. Network topology analysis results also demonstrate that the curves of node degree distribution, node degree probability and probability distribution of constructed network accord with those of the scale‐free network well.
Inspec keywords: cancer, proteins, molecular biophysics, learning (artificial intelligence), data mining, text analysis, medical computing, topology, statistical distributions
Other keywords: text mining, reinforcement learning, cooccurrence‐based interaction extraction approach, reinforcement learning‐based algorithm, prostate cancer protein interaction network, matching rate, scale‐free network, probability distribution, node degree probability, node degree distribution, network topology
1 Introduction
As an infamous lethal disease, cancer has caused more than millions of human deaths [1 ]. How to cure cancer has been a concern for a long time. Accordingly, a myriad of valuable biomedical texts on cancer‐related research have been accumulated. By searching PubMed with ‘cancer’ as the keyword, we retrieved more than three million publications [2 ]. Accordingly, how to fully take advantage of the massive amounts of biomedical texts turns out to be a new challenge for us as it is impossible to manually process these materials thoroughly. Text mining, with the goal of finding exciting outcomes hidden in mountains of unstructured texts to obtain new information and knowledge, has now been extensively applied in biomedical research [3, 4 ]. Many have benefited from the convenience of text mining technology to discover novel knowledge to improve the development of biomedical research, especially those pertaining to malignant diseases, such as cancer.
As a promising machine learning approach, reinforcement learning, which can optimise policy in unknown environments, provides a framework to learn directly from the interaction and achieve goals [5 ]. It is suitable for many systematic complex diseases, such as cancer, of which some biological mechanisms have not yet been understood clearly [6, 7 ]. Moreover, the versatility and openness of reinforcement learning ensure that it can make full use of biological knowledge. In our work of protein interaction network construction, multiple sources of biological knowledge can be seamlessly embedded such that the constructed network is more credible than those constructed without biological domain knowledge. Furthermore, since reinforcement learning algorithms are based on the principles of statistics and probability, the scale‐free property of the network, which has been found to be the property of many complex networks in real world, will be preserved during the network construction and evolution.
In this work, we take advantage of the reinforcement learning to build‐up a prostate cancer protein interaction network from texts, where a node represents a protein and an edge denotes an interaction. A node selects another node under the decision of reinforcement learning agent, and then it will get a reward after each attempt. The value of reward determines which interactions will be reinforced. The topology of the interaction network is yielded by the continuing iteration of the agent gradually and the final network is the output of learning result.
2 Text mining in cancer systems biology
Text mining is the process of extracting unknown and understandable information from large amounts of texts, and forming well‐defined knowledge. Text mining is generally composed of four tasks [3 ]: information retrieval, information extraction, knowledge discovery and hypothesis generation.
More and more interests have been shown in text mining to discover new knowledge to promote biomedical research, particularly in some areas of malignant diseases, such as cancer research. By retrieval results from PubMed [2 ] with ‘text mining’ as the keyword, we can see that there has been a significant increase in publication number since 2000, as shown in Fig. 1.
Fig 1.
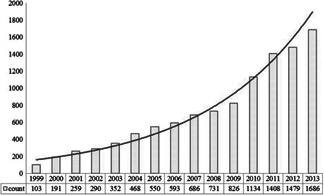
Retrieval results and its trend by using ‘text mining’ from 1999 to 2013
There are many efforts that use text mining to facilitate the development of biology. For instance, the network of genes, genetic diseases and brain areas introduced by Hayasaka et al. [8 ] used extracted associations from the texts as construction basis. The interaction network through literature mining provided by Sharma et al. [9 ] demonstrated that 19 genes were confirmed to be related to prostate cancer. Text mining will facilitate cancer research, by contributing to finding new knowledge for cancer diagnostics, treatment, prevention and patient management so as to reduce the risk of death from cancer. Korhonen et al. [10 ] and Guo et al. [11 ] developed systems for assessment of the causes of cancer by evidence drawn from biomedical literature. Some efforts [12 –13 ] have been made to extract the relationships among cancer, potential factors and treatments by using clinical records so as to facilitate the cancer cure study. As it can be seen, the full utilisation of text mining to enhance cancer systems biology and biomedical research is a new hot topic.
3 Reinforcement learning
Reinforcement learning is based on the idea that the system learns directly from the interaction during the process of approaching the goals. The reinforcement learning framework has five fundamental elements: controller, environment, state, reward and action [14 ], as shown in Fig. 2.
Fig 2.
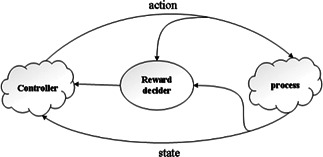
Framework of reinforcement learning
Controller selects an action; the environment responds to the controller, generates new scenes to the agent and then returns a reward
At each time step k, the controller selects an action uk ∈ U from action space U . As a result, the state changes to xk + 1 ∈ X from xk ∈ X in accordance with a transition function probability f : X × U →[0,∞)
| (1) |
The controller attains a reward rk + 1 according to rewarding function
| (2) |
The state‐action value function Qπ : X × U → R of some policy π and state value function Vπ yield the return, a long‐term reward, from a starting state
| (3) |
| (4) |
where γ ∈ [0,1] is a discount rate which shows how far sighted the controller is in considering the rewards and is also a factor for increasing uncertainty on future rewards. The ultimate goal of reinforcement learning is to obtain an optimum strategy through Q value or V value.
In recent years, reinforcement learning has drawn growing attention from researchers, and has been used in many biomedical fields. Farhang et al. [15 ] used reinforcement learning to segment computed tomography images to identify suspected cancer part. Fei et al. [16 ] proposed a multi‐scale fused edge detection algorithm, with a Sarsa (λ )‐based boundary amending to connect gaps in detected edges, to segment neuronal structure from neuronal electron microscopy images. However, few use reinforcement learning method and analysis of protein interaction networks.
4 Interaction network and scale‐free property
4.1 Scale‐free property
There are plenty of important and complex networks all around us. Edros and Renyi [17 ] have assumed that the complex networks are randomly intertwined, which means, in a random network, any two nodes are connected with the probability p (0 < p < 1) that obeys Poisson distribution, as shown in Fig. 3.
Fig 3.
Distribution of node degree that obeys Poisson distribution
Barabási [18 ] has found that in most cases, the node degree of a complex network of real world obeys power rate law distribution rather than Poisson distribution. For a randomly selected node, the probability of its degree being k is , where r and k are constants, as shown in Fig. 4. The network thereby is called scale‐free network. Researchers have concluded that many complex networks are characterised by scale‐free property. Many biological networks, such as protein interaction networks of yeast, Caenorhabditis elegans and Drosophila [18 ], are scale free.
Fig 4.

Distribution of node degree that obeys power rate law distribution
4.2 Biomedical entities and interaction network
Along with the advances in biomedical research, there is a growing recognition that complex biological functions are determined by complex interactions between various basic bio‐units rather than by a single biomolecular. Hartwell et al. [19 ] has proposed that modern biology also requires studying a variety of biomolecular interaction through different levels and forms of networks, such as metabolic networks, gene regulatory networks and protein interaction networks.
Integration of various interactions and functional relations network has been applied to complex diseases such as cancer. Generally speaking, in a biomedical network, a node represents a biomedical entity and an edge represents a relation between entities. However, as a biological problem, cancer is a sophisticated disease [20 –23 ], and many interactions remain unclear, leading to incompleteness of interaction networks. On the other hand, many high‐quality interaction network databases provide a wealth of up‐to‐date information that is useful to understand complex human diseases. However, as various approaches arbitrarily simplify complex biomedical systems by representing biomedical entities with nodes and representing biomedical interactions with edges, much information on biomedical entities and their relations will be omitted, causing understanding limitations.
4.3 Protein interaction network
Interactions between proteins are the basis of many biological functions. The topology of the interaction network is also very important. It has been found that the protein interaction network is scale free, which indicates that node degree distribution obeys power rate law distribution [24, 25 ]. There are many protein interaction databases, such as HPRD [26 ], IntAct [27 ] and STRING [28 ]. Generally speaking, we can use three different methods to construct protein interaction networks. The first one is to utilise text mining technology, the second one is to perform prediction based on known homologous facts and the third one is to take advantage of high throughput experiment.
Each of these methods has its merits and drawbacks. The text mining‐based approach obtains available data from published literature. Thereby, the gathered data are usually of high quality but are lacking in systematic view because the negative data, which are in fact very necessary to system analysis, are generally unavailable; moreover, the data in a single text are usually likely to cause the halo effect. Although the method which carry out prediction from homologous data is frequently utilised because of its convenience of implementation, it suffers from noise and deviation as the data are not attained directly. The method that is based on the experiment usually guarantees high reliability. However, as the experiment cost is exorbitant, it is impossible to apply the method to building up large‐scale interaction networks. Therefore under the restrictions of budget, it is very important to integrate diverse data sources and methods by making full use of the data model to build networks with maximal confidence for better and deeper understanding of the disease mechanisms.
5 Modelling with reinforcement learning
5.1 Advantage of using reinforcement learning
In this work, we propose an approach to construct biomolecular interaction networks using reinforcement learning algorithm where nodes represent the biomolecules and edges represent biomolecular interactions. A biological molecule selects a biological molecule to have interaction with. The attained reward value determines which interactions are enhanced. The agent keeps selecting repeatedly, and researches a decision finally. The whole process is iterative until in the end the network is formed by the dynamics of the system [29, 30 ].
Using reinforcement learning method has several advantages. First, reinforcement learning method could ensure the scale‐free property of the network constructed. As revealed by the Barabasi‐Albert model (BA model) for network dynamic evolution which was proposed by Barabasi and Albert [18 ], growth and preferential attachment are two fundamental reasons that lead the node degree distribution of scale‐free network to obey power rate law distribution, where growth means the number of nodes in the network increases over time and preferential attachment means a node with higher connection degree has more chances to receive new links. Since reinforcement learning algorithms are based on the principles of statistics and probability, the algorithms keep the characteristics of growth and preferential attachment during the network evolution and therefore the constructed network is able to follow the scale‐free property. Meanwhile, compared with most stochastic approaches, reinforcement learning algorithms are able to better exploit existing knowledge and then converge to optimal faster. Second, reinforcement learning ensures network stable and optimal in an unknown environment. Using reinforcement learning method, the agent repeatedly tries to select node and rewards returned by the agent and determine which interactions will be enhanced. The structure of the network will be established as the learning optimal result of the agent. The scale‐free property of the network will be maintained. Third, cancer is a system of a complex disease, a few mechanisms of which remain unclear. As reinforcement learning is good at dealing with the learning problem in unknown and random environment, the constructed interaction network based on reinforcement learning can be best guaranteed to converge to a stable and optimal one. Besides, the network construction approach that is based on reinforcement learning method could make use of the openness of reinforcement learning, so that during the process of establishing the network, the approach could be seamlessly combined with biological knowledge and biological data from multiple sources to enhance biological facts, therefore improving the confidence of the network and ensuring the basic biological characteristics of the network.
5.2 Protein interaction extraction
Biomedical interactions, such as gene–gene interactions, protein–protein interactions and other interactions in genome‐wide associations as well as other extensive relationships provide us with useful scaffolds for further integrative analysis and study. There are many interaction extraction approaches, such as co‐occurrence‐based text mining approach, linguistic patterns approach, machine learning‐based approach. The co‐occurrence method, which assumes that there is some relationship if the two biological entities appear in a text concurrently, is commonly used to find out interactions between biological entities. Co‐occurrence method can be easily implemented, but will bring about a higher false positive rate. In fact, most biomedical texts use some kinds of fixed grammar patterns to describe the interaction of two different biological entities. From the perspective of English grammar, the function term and related grammar structure have changed few for a long time. For instance, the following pattern examples are most frequently used:
Pattern example 1: function term‐{of}‐entity‐and‐ entity
Pattern example 2: noun form of function term‐{of}‐entity‐and‐ entity
Pattern example 3: entity‐{in}‐entity‐noun form of function term
Pattern example 4: entity‐attribute‐{function term}‐with‐ {of}‐ entity
Pattern example 5: noun form of function term‐{ preposition }‐ entity‐{by}‐ entity
The interaction extraction approach that is based on co‐occurrence is inclined to include false positive interactions while the one that is based on pattern matching tend to miss interactions in spite of high precision rate. Hereby, we propose an interaction identification approach which first uses pattern matching approach to identify the interactions that match the pattern rule and then takes advantage of co‐occurrence‐based approach to deal with the unmatched texts by extending the text with the assumption that co‐occurrence terms are possible to have some relationship where we also discount the interaction weight to demonstrate that the interaction is extracted from extended texts rather than original sentences. The protein interaction extracting using extended pattern matching with discount is shown in Algorithm 1 (see Fig. 5 ).
Fig 5.

Algorithm 1: Interaction extracting using extended pattern matching with discount
5.3 Reinforced network constructing model
In our proposed model, a node i selects a node to interact with under the decision of reinforcement learning agent, and it will attain a decision. The node will get a reward after each attempt. The value of reward determines which interactions will be reinforced. The structure of the interaction network is the result of continuing iteration of the agent. In this way, both the evolution of the interaction network and the evolution of the individual protein are taken into consideration.
Node i randomly selects other nodes to establish an interaction with the probability that is assigned to other nodes. Each node has the policy to choose nodes and get reinforced in each iteration. Each node maintains a weight vector 〈 w i 1, …, w in 〉. As each time newly added nodes connect existing node i with a given probability, any node i is selected by weight vector w i (t ) at time t. We will introduce the reinforcement learning model for constructing protein interaction network, as well as the fundamental elements of reinforcement learning, reward, actions and states, will be discussed.
The construction of a network can be modelled as a Markov decision process model, and can obtain optimal strategies by reinforcement learning algorithms. A node chooses other nodes as interaction node according to a certain probability. There are many methods to obtain the probability. In this work, we obtain the access probability by computing the rate of occurrence of node i choosing node j to all occurrence of node i
| (5) |
Meanwhile, texts from different sources have different levels of authority and credibility. When evaluating the strength of the interaction between biological entities, it is necessary to take into consideration the influence of text data. We utilise the factor of the text to account for the significance of it and occurrence to denote the intensity of it. We choose impact factor (IF) value as a weight factor. Generally speaking, if the journal has a larger IF, the internal research texts tend to have higher weight. The ultimate strength of the interaction is the multiplication of the significance value and the intensity value of the text. The weight can be calculated as
| (6) |
We can view the probability of selecting a node as the weight assigned by other nodes, which is used to measure the probability of interaction between two nodes. Thus, in an n ‐node network, any node i, with a weight vector, the access probability of node j is . Finally, we obtain a probability matrix, through which node i can obtain the probability of interacting with node j. At each stage, there is a probability matrix which represents the probability of interaction. The network topology changes with the procedure of matrix updating. During each iteration, each node has access policy to choose other nodes, and the knowledge will be reinforced in the form of updating access probability matrix which records the probability of interaction. The matrix is to be updated along with the evolution of the network topology. When the matrix keeps stable, the evolution can be considered as being completed, and the topology of the network is formed.
In the process of the network construction, the actions of an agent can be described as determining whether there is an interaction between the current node and another node, as shown in Table 1.
Table 1.
Available actions
| # | Action |
|---|---|
| 0 | pending as unable to determine whether there is an interaction between two nodes |
| 1 | no interaction between two nodes |
| 2 | interaction between two nodes |
Agent in the current state chooses an action, and enters the next state. Hereby, the state can be regarded as the static image of the external environment in a given state after taking some action. In protein interaction networks, the state at a given time is the description of all proteins and interactions. From the perspective of graph theory, it is a description of all nodes and edges between nodes, as well as a corresponding transition probability matrix.
In this work, we introduce an algorithm, in which each node has to choose a node to form interaction, and the change of the transfer probability matrix interference the topology of the network. A matrix w can be used for action selection. The final attained matrix w can be seen as the topology of the network, and the updating process can be regarded as the construction process of the network.
6 Experiment and analysis
Prostate cancer is one of the most common cancers worldwide. In 2008, it was the top one cancer killer for men according to United States statistics [1 ]. In recent years, with ageing population and changes in lifestyle, a significant growth trend in the incidence of prostate cancer has become one of the main malignant neoplasms of men. On the other hand, there have accumulated large amounts of research work concerning prostate cancer. We obtained more than 110 000 texts retrieving from PubMed by ‘prostate cancer’, and the number is keeping increasing. As we can see, prostate cancer is an important biomedical research.
6.1 Workflow and data acquisition
Protein interaction text mining refers to automatically digging out protein interactions from biomedical texts. Generally, the process includes text data acquisition, protein named entity recognition, relationship extraction and network establishment.
As an online literature database, PubMed contains over 22 million texts, including life science, behavioural science, chemistry, biology and other fields, which provide researchers with a wealth of resources to carry out biological text mining research. We develop a tool, which is based on E‐utilities [31 ] provided by PubMed as an application programming interface, to obtain 80 841 texts from PubMed with the keyword ‘prostate cancer’. The downloaded texts are used for subsequent processing.
There are many protein term identification approaches, including dictionary‐based approach, machine learning approaches, rule‐based approaches and some combined approaches [32 ]. Among various approaches, the dictionary‐based approach generally ensures highest identification precision. In this work, to avoid mistakes caused by incorrect protein terms identification, we utilise dictionary‐based approach to find out protein name from texts. We set‐up a protein name list composed by protein names downloaded from HPRD, IntAct and STRING, and then used it to obtain protein names by string comparing and matching. After that, we extracted interactions using the approach based on Fig. 5 and constructed network using Algorithm 2 (see Fig. 6 ).
Fig 6.

Algorithm 2: Protein interaction network construction with reinforced learning approach
6.2 Results and analysis
In our experiment, we set window size as 1 and discount rate as 0.5. We obtained 4544 effective protein–protein interactions through the downloaded 80 841 abstracts texts using the proposed approach. The edge numbers under different thresholds of node degree are as shown in Table 2. We check the interaction in HPRD, IntAct and STRING. Table 2 shows the different edges, number of unmatched edges and overall matching rates under different edge weight thresholds.
Table 2.
Different edges, number of unmatched edges and overall matching rates under different edge weight thresholds
| Threshold | Edges | Unmatched edges | Matching rate, % |
|---|---|---|---|
| 4.5 | 416 | 71 | 82.93 |
| 4 | 494 | 97 | 80.36 |
| 3.5 | 608 | 136 | 77.63 |
| 3 | 791 | 176 | 77.74 |
| 2.5 | 908 | 181 | 80.07 |
| 2 | 1327 | 207 | 84.40 |
We can obtain more than 77% of the matching rate by the proposed method with different thresholds. It can be seen that higher thresholds bright out significant improvement in matching rate. It can be also noted that when the threshold value is set to 2, the matching rates go up. We conclude that there are some reasons. First, in the interaction network, edges are assigned with different weights, which is a feature of edge. In our work, we care more about whether the edge in the network has a corresponding interaction between proteins. The matching rates of different thresholds indicate how many predicted protein interactions are consistent with the data of HPRD, IntAct and STRING. Therefore the overall matching rate rather than the matching rates under different threshold is more important. Second, setting lower threshold value will include additional edges on the basis of the edges of higher thresholds. Since the overall performance of the proposed method is good, and moreover, as the source data from texts are relatively reliable, a great portion part of included addition edges are true. As a result, the matching rating hovers around 80%. Third, when we set the threshold value as 2, the majority of attained interactions occur in a sentence, which satisfies the assumption of co‐occurrence‐based algorithm: terms co‐concurrently in a sentence are very much likely to have relationship, causing matching rate to rise. Besides, in some cases, some newly studied interactions, though probably being pending ones, are more likely to be discussed in academic literature than those already known and proved interactions, causing the occurrence times of newly studied interactions to be greater than those of proved ones. As stated above, since the occurrence time is used to calculate the edge weight, it could give rise to the fluctuation of matching rate. The average matching rate of our approach, 80.52%, is higher than that of the approach by Chun et al. [33 ], 78.5% of precision rate; and our approach is almost even with the approach of Giles and Wren [34 ] which brought out 80% precision.
On the other hand, there are a huge number of interactions in STRING; and there are also more than 41 327 interactions in HPRD, and more than 477 526 interactions in IntAct. In our experiment, the learned protein interaction number is very small compared with them. Therefore if STRING, HPRD or IntAct is used as reference, recall rate seems to be low. Actually there are some reasons. The literature authors attach great importance to the credibility of the data rather than the amount, which explains the relatively small amount of the data. What is more, as many interaction data have not been published, we cannot attain them from text. Besides, the recall rate of text mining‐based approach is limited by the text volume. If we obtain texts as source data, we are very much likely to obtain more interactions.
We also analysed the node degree distribution, node degree probability and probability distribution of constructed network. We can see from Fig. 7 that node degrees of most degree are among 1–10. The node degree distribution curve of our method generally accords with the curve of scale‐free network. In Fig. 8, we can see that the node degree probability density of our method also generally fits the node degree probability density of scale‐free network.
Fig 7.
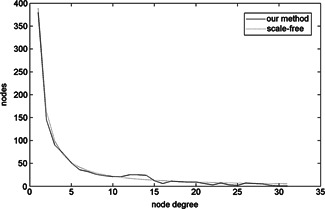
Node degree distribution of constructed protein interaction network and scale‐free network
Fig 8.
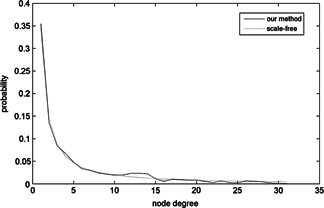
Node degree probability density of constructed protein interaction network and scale‐free network
We utilised the tools provided by Clauset et al. [35, 36 ] to check the likelihood of fitting the power‐law distribution, as shown in Fig. 9. We can see that the probability distribution of the network constructed by our method can fit the probability distribution of the scale‐free network well.
Fig 9.
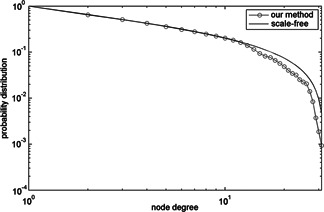
Node degree probability distribution of constructed protein interaction network and scale‐free network
7 Conclusion
With in‐depth research on biology, people have gradually realised that complex biological functions and biological phenomena, are the basic units of the complex interactions between various elements. Building interaction networks and understanding life functions with systems biology view is receiving increasing recognition. In this work, we, within reinforcement learning framework, introduce an algorithm for the establishment of interaction networks. The nodes are considered as proteins, and the edges are viewed as interactions. During the process of network evolution, a node selects which other node to be interacted with. Iterative evolution forms an optimal interaction network.
As prostate cancer is one of the most highly malignant neoplasms, we use the proposed approach to build‐up a prostate cancer protein interaction network, based on the texts attained from PubMed. Network topology analysis of the results shows that node degree distribution of established network is consistent with scale‐free properties. Meanwhile, the matching of edge in protein databases only indicates that the two proteins are interacted. There is still much work on analysing the network, pathways to reveal its role in prostate cancer.
8 Acknowledgments
This work was supported by National Natural Science Foundation of China (61303108, 61472262, 31470821, 61103045), High School Natural Foundation of Jiangsu (13KJB520020), Key Laboratory of Symbolic Computation and Knowledge Engineering of Ministry of Education, Jilin University(93K172014K04), Suzhou Industrial Application of Basic Research Program Part (SYG201422). This work was also partially supported by Collaborative Innovation Center of Novel Software Technology and Industrialization.
9 References
- 1. Ahmedin J. D.V.M. Freddie B. Melissa M.C. et al.: ‘Global cancer statistics ’, CA Cancer J. Clin., 2011, 61, (2 ), pp. 69 –90 (doi: 10.3322/caac.20107 ) [DOI] [PubMed] [Google Scholar]
- 2. Pubmed , http://www.ncbi.nlm.nih.gov/pubmed/, accessed November 2014
- 3. Fei Z. Patumcharoenpol P. Cheng Z. et al.: ‘Biomedical text mining and its applications in cancer research ’, J. Biomed. Inf., 2013, 46, (2 ), pp. 200 –211 (doi: 10.1016/j.jbi.2012.10.007 ) [DOI] [PubMed] [Google Scholar]
- 4. Yen‐Ching C. Tzong‐Han T. Wen‐Lian H.: ‘New challenges for biological text‐mining in the next decade ’, J. Comput. Sci. Technol., 2010, 25, pp. 169 –179 [Google Scholar]
- 5. van Hasselt H.: ‘Reinforcement learning: state of the art ’ (Springer, Berlin, 2007. ), pp. 207 –251 [Google Scholar]
- 6. Jorrit H. Frank B. Hans W.: ‘Cancer: a systems biology disease ’, Biosystems, 2006, 83, (2–3 ), pp. 81 –90 (doi: 10.1016/j.biosystems.2005.05.014 ) [DOI] [PubMed] [Google Scholar]
- 7. Gui‐Min Q. Xing‐Ming Z.: ‘A survey on computational approaches to identifying disease biomarkers based on molecular networks ’, J. Theor. Biol., 2014, 362, pp. 9 –16 (doi: 10.1016/j.jtbi.2014.06.007 ) [DOI] [PubMed] [Google Scholar]
- 8. Hayasaka S. Hugenschmidt E. Laurienti J.: ‘A network of genes, genetic disorders, and brain areas ’, PLoS One, 2011, 6, pp. 1 –10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sharma P. Senthilkumar D. Brahmachari V. et al.: ‘Mining literature for a comprehensive pathway analysis: a case study for retrieval of homocysteine related genes for genetic and epigenetic studies ’, Lipids Health Dis., 2006, 5, (1 ), pp. 1 –19 (doi: 10.1186/1476-511X-5-1 ) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Korhonen A. Silins I. Sun L. et al.: ‘The first step in the development of text mining technology for cancer risk assessment: identifying and organizing scientific evidence in risk assessment literature ’, BMC Bioinformatics, 2009, 10, (303 ), pp. 1 –19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Guo Y. Korhonen A. Liakata M. et al.: ‘A comparison and user‐based evaluation of models of textual information structure in the context of cancer risk assessment ’, BMC Bioinformatics, 2011, 12, (69 ), pp. 1 –18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. You M. Zhao W. Li Z. et al.: ‘MAPLSC: a novel multi‐class classifier for medical diagnosis ’, Int. J. Data Mining Bioinf., 2011, 5, pp. 383 –401 (doi: 10.1504/IJDMB.2011.041555 ) [DOI] [PubMed] [Google Scholar]
- 13. Ben A. Zweigenbaum P.: ‘Automatic extraction of semantic relations between medical entities: a rule based approach ’, J. Biomed. Semant., 2011, 2, (5 ), pp. 1 –11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sutton R.S. Barto A.G. et al.: ‘Reinforcement learning ’ (MIT Press, 1998. ), pp. 100 –116 [Google Scholar]
- 15. Farhang S. Hamid T. Magdy S.: ‘A reinforcement learning framework for medical image segmentation ’. Proc. of IEEE Int. Joint Conf. on Neural Networks, 2006, pp. 511 –517
- 16. Fei Z. Quan L. Yuchen F. Bairong S.: ‘Segmentation of neuronal structures using SARSA (λ)‐based boundary amendment with reinforced gradient‐descent curve shape fitting ’, PLoS One, 2014, 9, (3 ), pp. 1 –19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Erdos P. Renyi A. Erdos P. et al.: On random graphs[J]. Publicationes Mathematicae Debrecen, 1959, 6, pp. 290 –297 [Google Scholar]
- 18. Barabási A.L.: ‘Scale‐free networks: a decade and beyond ’, Science, 2009, 325, pp. 412 –413 (doi: 10.1126/science.1173299 ) [DOI] [PubMed] [Google Scholar]
- 19. Hartwell L. Hopfield J. Leibler S. et al.: ‘From molecular to modular cell biology ’, Nature, 1999, 402, pp. 47 –52 (doi: 10.1038/35011540 ) [DOI] [PubMed] [Google Scholar]
- 20. Albert B. Natali G. Joseph L. et al.: ‘Network medicine: a network‐based approach to human disease ’, Nature, 2011, 12, (1 ), pp. 56 –68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Arunachalam V. Ulrich S. Raphaele F. et al.: ‘A directed protein interaction network for investigating intracellular signal transduction ’, Sci. Signal., 2011, 4, (189 ), pp. 1 –8 [DOI] [PubMed] [Google Scholar]
- 22. Marc V. Michael C. Albert B.: ‘Interactome networks and human disease ’, Cell, 2011, 144, (6 ), pp. 986 –998 (doi: 10.1016/j.cell.2011.02.016 ) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xuebing W. Rui J. Michael Z. et al.: ‘Network based global inference of human disease genes ’, Mol. Syst. Biol., 2008, 4, (189 ), pp. 1 –11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jeong H. Mason S. Barabási L. et al.: ‘Lethality and centrality in protein networks ’, Nature, 2001, 411, (6833 ), pp. 41 –42 (doi: 10.1038/35075138 ) [DOI] [PubMed] [Google Scholar]
- 25. Wagner A.: ‘The yeast protein interaction network evolves rapidly and contains few redundant duplicate genes ’, Mol. Biol. Evol., 2001, 18, (7 ), pp. 1283 –1292 (doi: 10.1093/oxfordjournals.molbev.a003913 ) [DOI] [PubMed] [Google Scholar]
- 26. Suraj P. Navarro J. Ramars A. et al.: ‘Development of human protein reference database as an initial platform for approaching systems biology in humans ’, Genome Res., 2003, 13, (10 ), pp. 2363 –2371 (doi: 10.1101/gr.1680803 ) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kerrien S. Faruque Y. Aranda B. et al.: ‘IntAct‐open source resource for molecular interaction data ’, Nucleic Acids Res., 2007, 35, (Suppl 1 ), pp. 561 –565 (doi: 10.1093/nar/gkl958 ) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lars J. Michael K. Manuel S. et al.: ‘STRING 8‐a global view on proteins and their functional interactions in 630 organisms ’, Nucleic Acids Res., 2009, 37, (Suppl 1 ), pp. 412 –416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Richard D.: ‘Random graph dynamics ’ (Cambridge University Press, 2007. ), pp. 20 –28 [Google Scholar]
- 30. Alison W.: ‘A dynamic model of network formation ’, Games Econ. Behav., 2001, 34, pp. 331 –341 (doi: 10.1006/game.2000.0803 ) [DOI] [Google Scholar]
- 31. E‐utilities : http://www.ncbi.nlm.nih.gov/books/NBK1058/, accessed December 2014
- 32. Fei Z. Bai‐rong S.: ‘Combined SVM‐CRFs for biological named entity recognition with maximal bidirectional squeezing ’, PLoS One, 2012, 7, (6 ), pp. e39230 (doi: 10.1371/journal.pone.0039230 ) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chun H.W. Tsuruoka Y. Kim J.D. et al.: ‘Automatic recognition of topic‐classified relations between prostate cancer and genes using MEDLINE abstracts ’, BMC Bioinformatics, 2006, 7, (3 ), pp. 1 –9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Giles C.B. Wren J.D.: ‘Large‐scale directional relationship extraction and resolution ’, BMC Bioinformatics, 2008, 9, (9 ), pp. 1 –13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Virkar Y. Clauset A.: ‘Power‐law distributions in binned empirical data ’, Ann. Appl. Stat., 2014, 8, (1 ), pp. 89 –119 (doi: 10.1214/13-AOAS710 ) [DOI] [Google Scholar]
- 36. Clauset A. Shalizi C.R. Newman M.E.J.: ‘Power‐law distributions in empirical data ’, SIAM Rev., 2009, 51, (4 ), pp. 661 –703 (doi: 10.1137/070710111 ) [DOI] [Google Scholar]