

Abstract
The authors developed a mathematical model of arachidonic acid (AA) degradation to prostaglandins (PGs) and leukotrienes (LTs), which are implicated in the processes of inflammation and hypersensitivity to non‐steroidal anti‐inflammatory drugs (NSAIDs). The model focuses on two PGs (PGE2 and PGD2) and one LT (LTC4), their % increases and their ratios. Results are compared with experimental studies obtained from non‐asthmatics (NAs), and asthmatics tolerant (ATA) or intolerant (AIA) to aspirin. Simulations are carried out for predefined model populations NA, ATA and three AIA, based on the differences of two enzymes, PG E synthase and/or LTC4‐synthase in two states, that is, no‐inflammation and inflammation. Their model reveals that the model population with concomitant malfunctions in both enzymes is the most sensitive to NSAIDs, since the duration and the capacity for bronchoconstriction risk are highest after simulated oral dosing of indomethacin. Furthermore, inflammation prolongs the duration of the bronchoconstriction risk in all AIA model populations, and the sensitivity analysis reveals multiple possible scenarios leading to hypersensitivity, especially if inflammatory processes affect the expression of multiple enzymes of the AA metabolic pathway. Their model estimates the expected fold‐changes in enzyme activities and gives valuable information for further targeted transcriptomic/proteomic and metabolomic studies.
Inspec keywords: enzymes, drugs, molecular biophysics, biochemistry, molecular configurations, diseases, pneumodynamics, patient diagnosis
Other keywords: dynamic model, eicosanoid production, nonsteroidal anti‐inflammatory drug‐triggered hypersensitivity, dynamic mathematical model, complex metabolic pathway, arachidonic acid degradation, prostaglandins, leukotrienes, inflammation, NSAID, asthmatics tolerance, nonasthmatic tolerance, asthmatics intolerance, PG E synthase, LTC4‐synthase, enzymes, bronchoconstriction risk, indomethacin, sensitivity analysis, AA metabolic pathway, fold‐changes, targeted transcriptomic studies, proteomics, metabolomics, noninvasive diagnostic tools, aspirin‐exacerbated respiratory disease
1. Introduction
Eicosanoids are chemical compounds with essential functions for maintaining cellular activities in living organisms. They operate as paracrine and autocrine mediators. Therefore eicosanoids are also implicated in many inflammatory processes and diseases. Thus the production and signalling pathways of eicosanoids are the targets of several pharmacological interventions. For all mammalian, arachidonic acid (AA) is the essential precursor of eicosanoids. The metabolism of AA is a complex network of non‐enzymatic and enzyme‐driven steps, comprising a multitude of different enzymes, receptors and additional compounds. These metabolic pathways are also the targets of multiple endogenous as well as exogenous modulators, eventually causing up‐ and down‐regulation of eicosanoid production, accompanied by various physiological effects [1, 2, 3, 4].
On the basis of an enzymatic view, metabolism of AA can be roughly subdivided into the cyclooxygenase (COX) pathway and the lipoxygenase (LOX) pathway revealing prostanoids and leukotrienes (LTs) as metabolites, respectively [5]. Representative bioactive metabolites of the COX‐pathway are prostaglandin (PG) E2 (PGE2) and PG D2 (PGD2), and main metabolites of LOX pathway are cysteinyl‐LTs (cys‐LTs, i.e. LTC4, LTD4, LTE4) [5]. The biological effects of eicosanoids are pleiotropic. Some effects and related diseases of the above mentioned eicosanoids are selected to give a general idea of their pivotal biological relevance. To begin with the LOX‐pathway: cys‐LTs cause bronchoconstriction [6], recruit inflammatory cells [7], increase swelling of the nasal mucosa and increase mucus secretion [5]. They are approximately 30–3000‐times more potent bronchoconstrictors than histamine in healthy humans in‐vivo [8]. Their main systemic sources are leukocytes like eosinophils and basophils, as well as mast cells and epithelial cells [9, 10, 11, 12]. Overproduction of cys‐LTs is a hallmark of the aspirin‐exacerbated respiratory disease (AERD), which is characterised by chronic rhinosinusitis, nasal polyps and severe bronchial asthma because of the hypersensitivity to non‐steroidal anti‐inflammatory drugs (NSAIDs) [13]. The effects of the metabolites of the COX‐pathway are more diverse. For instance, PG D2 (PGD2) induces bronchoconstriction in allergic asthma [14], whereas PG E2 (PGE2) prevents bronchoconstriction [15, 16], protects airways against inflammation, reduces LT production through the inhibition of the 5‐LOX pathway [12, 16, 17, 18, 19], inhibits mast cell degranulation, and promotes normal airway function [20, 21, 22, 23]. Underproduction of PGE2 strongly diminishes the inhibitory effects of PGE2 on the 5‐LOX pathway, thus enhances cys‐LT production [12] and it reduces the inhibitory effect of PGE2 on mast cells [13, 22, 24]. On the basis of the thereon, it was hypothesised that decreased levels of PGE2 might induce bronchoconstriction by underproduction of cAMP [11, 12, 19]. On the contrary, locally increased levels of PGE2, for example, by inhalation, might induce bronchodilation and could potentially protect aspirin‐precipitated attacks of asthma [16, 23, 25]. In human airways PGE2 is produced by many cells including epithelium, fibroblasts, smooth muscle cells, alveolar cells, macrophages, phagocytes and lymphocytes [2, 26]. The COX pathway comprises of two isozymes, COX‐1 and COX‐2. Both pathways are inhibited by NSAIDs, comprising non‐selective and selective COX‐inhibitors. The LOX pathway is most often blocked by LOX‐inhibitors or it is down‐regulated by the action of cys‐LT‐receptor antagonists. These substances are well known and prominent drugs for many diseases like tumour or asthma [27, 28, 29]. NSAIDs play a crucial role in tilting AA metabolism in favour of the LOX pathway causing cys‐LT overproduction [4, 9, 11, 12, 18, 19, 30, 31]. There is increasing evidence that NSAID‐triggered hypersensitivity, like AERD [also known as aspirin induced asthma (AIA)], partly originates from the imbalance of eicosanoids, secreted by activated cells. These imbalances, in concert with others, result in bronchial hyper‐reactivity – a characteristic symptom of asthma.
Patients suffering from AIA demonstrate a characteristic pattern of multiple defects, observed either as hyper‐inflammation or underproduction of anti‐inflammatory mediators, but also detected as overproduction of bronchoconstrictors and pro‐inflammatory mediators [9, 10, 11, 12, 13, 19, 25, 32, 33, 34, 35, 36, 37, 38]. A strict separation of eicosanoid profiles of un‐inflamed and inflamed states had not been completely accomplished by metabolomic studies because of the considerably overlapping metabolomic profiles of these entities. Nevertheless, there have been several promising attempts during the recent years [10, 11, 12, 13, 19, 25, 30, 32, 33, 34, 35, 36, 37, 38].
In one of these studies, Pierzchalska et al. [32] systematically studied and measured eicosanoid production in response to proinflammatory stimuli in different asthmatic patient groups and controls. Large deviations of cellular eicosanoid production were noted between the subgroups of asthmatic patients that are tolerant or intolerant to aspirin as well as between the latter two subgroups and a group of non‐asthmatics (NAs) in their experiments [32]. We performed this theoretical study to find out with the help of mathematical modelling whether the differences in eicosanoid profiles of different patient groups might be predicted from the differences in their enzyme‐expression profiles within the cells responsible for eicosanoid production. We focused on the maximal velocities of the enzymes PG E synthase (PGES) and LT C4‐synthase (LTC4S). The selection of these two parameters was encouraged by experimental findings that different patient groups exhibited different expressions of LTC4S [10, 39], and, moreover, that they differently responded to inflammatory stimuli in terms of PGES up‐regulation [32]. We investigated whether maximal velocities of enzymes PGES and LTC4S characterise and distinguish different patient groups [i.e. NA, asthmatics tolerant to aspirin (ATA) and asthmatics intolerant to aspirin (AIA)] at the level of eicosanoid production. The effect of inflammation on eicosanoid production was also modelled, both at rest in equilibrium conditions without the drug as well as after simulated oral dosing of indomethacin. The impact of the state of inflammation on the model results (concerning absolute and relative values of [PGE2], [PGD2] and [LTC4] as well as the ratios [PGE2]/[LTC4] and [PGE2]/[PGD2]) was analysed especially with regard to sensitivity of different model populations to indomethacin – a typical exemplar of NSAIDs.
2. Materials and methods
2.1. Kinetic scheme and mathematical model
The kinetic scheme of AA metabolism and its interaction with an NSAID, as considered for mathematical modelling, is presented in Fig. 1. In brief, AA with constant influx is first transformed via a LOX pathway into either 5‐ or 15‐hydroperoxyeicosatetraenoic acid (5‐ or 15‐HPETE). Enzymes involved in this step are 5‐LOX for the conversion of AA into 5‐HPETE, and, 15‐LOX for the conversion of AA into 15‐HPETE. 15‐HPETE is further transformed into 15‐HETE by phospholipid‐hydroperoxy‐glutathione‐peroxidase (PHGPx), whereas 5‐HPETE is further transformed either into LT A4 (LTA4) by 5‐LOX or into 5‐HETE by PHGPx. LTA4 is then transformed either by LTC4S into LT C4 (LTC4) or by LTA4 hydrolase (LTA4H) into LT B4 (LTB4), which is exported from the cells via ATP‐binding cassette transporter C4 (ABCC4). Via COX pathway, AA is transformed into PG H2 (PGH2) by two isozymes, COX‐1 and COX‐2, acting in parallel. PGH2 is the precursor of all prostanoids, that is, PGs (PGE2, PGD2 and PGF2α ), prostacyclin (PGI2) and thromboxane (TXB2). The synthases PGES, PGDS, PGFS, PGIS and TXS are the corresponding enzymes of the subsequent catalytic reactions generating the above mentioned prostanoids. An efflux of all modelled eicosanoids is considered. Their degradation rate might be interpreted either by their export or by further metabolisation. The inhibitory effect of PGE2 on 5‐LOX, and of NSAIDs on COX‐1 and COX‐2, plus the auto‐regulation of LTC4S is in Fig. 1 denoted by dashed lines.
Fig. 1.
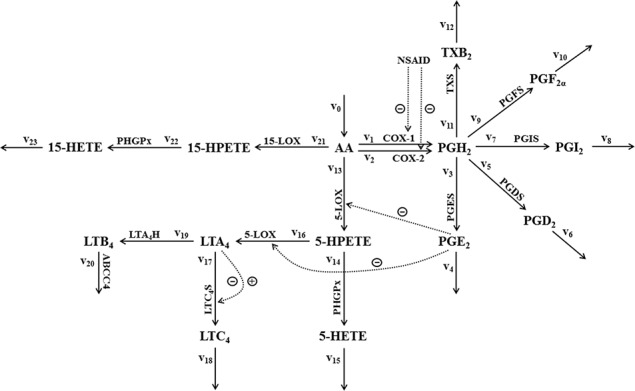
Kinetic scheme of AA metabolism considered for mathematical modelling
Metabolic degradation of AA via COX and LOX pathway yields eicosanoids: PGs, thromboxane (TX), LTs and hydro(pero)xyeicosatetraenoic acids
Full lines with the corresponding enzymes depict the direction of metabolic fluxes, dotted lines depict either inhibitory or activating effects on enzymes
Numeration of the fluxes reflects the reaction rates of the mathematical model
The mathematical model consists of a set of fourteen first‐order non‐linear ordinary differential equations, describing the time evolution of system variables, which are concentrations of fourteen metabolites (M), that is, [AA], [PGH2], [PGE2], [PGD2], [PGI2], [PGF2α], [TXB2], [5‐HPETE], [5‐HETE], [LTA4], [LTC4], [LTB4], [15‐HPETE] and [15‐HETE]. The time derivative of a selected system variable (d[M]/dt) equals the difference between the sum of all incoming and the sum of all outgoing metabolic fluxes, all with respect to numeration and the direction of arrows in the kinetic scheme in Fig. 1
| (1) |
Metabolic fluxes or reaction velocities (vi ) are defined as
for i = 0;
| (2) |
for i = 3, 5, 7, 9, 11, 14, 19, 20, 21, 22 (decoding enzymes PGES, PGDS, PGIS, PGFS, TXS, PHGPx, LTA4H, ABCC4, 15‐LOX and PHGPx, respectively), where v maxi is the maximal reaction velocity, Ki is the Michaelis–Menten constant and S is either PGH2 (for i = 3, 5, 7, 9, 11), AA (for i = 21), 15‐HPETE (for i = 22), 5‐HPETE (for i = 14), LTA4 (for i = 19) or LTB4 (for i = 20)
| (3) |
for i = 1, 2, 13, 16, (decoding enzymes COX‐1, COX‐2 and 5‐LOX(2 ×), respectively), where v maxi is the maximal reaction velocity, Ki is the Michaelis–Menten constant, KIi is the equilibrium dissociation constant of the inhibitor I from an enzyme in case of reversible competitive enzyme inhibition, S is either AA (for i = 1, 2, 13) or 5‐HPETE (for i = 16) and I is either NSAID (for i = 1, 2) or PGE2 (for i = 13, 16)
| (4) |
for i = 17 (decoding enzyme LTC4S), where A, B and C are the corresponding parameters, and S is LTA4
| (5) |
for i = 4, 6, 8, 10, 12, 15, 18, 23, S is either PGE2 (for i = 4), PGD2 (for i = 6), PGI2 (for i = 8), PGF2α (for i = 10), TXB2 (for i = 12), 5‐HETE (for i = 15), LTC4 (for i = 18) or 15‐HETE (for i = 23) and ki is the rate constant.
Equation (2) known as steady‐state Michaelis–Menten kinetics, was used in all enzyme‐driven reaction steps, which corresponded well with this type of enzyme kinetics. In the cases in which the effect of an inhibitor on an enzyme was simulated a reversible competitive inhibition, described by (3), was used. This was the case for the modelling of the inhibitory effect of NSAIDs. Indomethacin is known as a reversible competitive inhibitor of COX‐1 and COX‐2. We considered also the inhibitory effect of PGE2 on 5‐LOX [12, 16, 17, 18, 19] in the model. Since the entire mechanism of this inhibition is not completely elucidated [9, 11, 18, 40], a reversible competitive enzyme inhibition was considered here as in our previous [41, 42, 43] and the other models [44] of eicosanoid production. Equation (4) was used in the case of reaction step 17, in which enzyme LTC4S catalyses conversion of substrate LTA4 into the product LTC4. Kinetics of this particular enzyme and the substrate involves substrate auto‐inhibition [45], which is characterised by a bell‐shaped velocity‐substrate relationship. In other words, this is the enzyme kinetics, in which LTA4 auto‐regulates the velocity of the enzyme LTC4S. Equation (5), known as linear mass‐action law, was used in those cases in which enzymes were either not involved or not identified. This was primarily the case for eicosanoid effluxes except for the efflux of LTB4. Similar approaches in modelling were used also in other models of eicosanoid production [29, 44, 46, 47, 48].
In the dynamic simulations, time dependent drug plasma concentration [NSAID] is modelled with the standard two‐store pharmacokinetic model for oral drug dosing, taking into account absorption and elimination phases
| (6) |
where D is the drug dose, M is the molecular mass of the drug, V is the apparent volume of drug distribution normalised with the fraction of the absorbed drug, and, k a and k e are the first‐order absorption and elimination rate constants, respectively. These pharmacokinetic parameters are presented in Supplementary Table 2 along with other kinetic parameters of the model.
Table 2.
Values of maximal velocities of the enzyme LTC4S (v max17) for: NA model population, aspirin‐tolerant asthmatic model population (ATA) and three different aspirin‐intolerant‐asthmatic model populations (AIA(1), AIA(2), AIA(3))
| Parameter | Enzyme | NA | ATA | AIA(1) | AIA(2) | AIA(3) |
|---|---|---|---|---|---|---|
| v max17, µM s−1 | LTC4S | 0.057 | 0.23 | 0.23 | 1.15 | 1.15 |
For the purpose of sensitivity analysis, the response coefficients, R, defined as the ratio between the relative change of the selected system variable with respect to small alterations of a single parameter value, and the relative change of this particular model parameter, were calculated according to the equation
| (7) |
where X is the model variable and P is the model parameter. For instance, response coefficient −2 means that system variable decreased for 20%, if one parameter value increased for 10%.
Software used for all calculations was Berkeley Madonna 8.0.1 (R. Macey and G. Oster, University of California at Berkeley). The system of fourteen differential equations (explicitly presented in Supplemental information), evolved from (1) by considering all definitions of the reaction velocities (2)–(5) and in dynamic simulations considering also the time‐dependent plasma drug concentration (6), was numerically integrated from the initial stationary state, which was determined separately for each model variable in the absence of NSAID for each particular simulation (see Supplementary Table 5 for the complete set of initial conditions used in the modelling). The numerical integration method with variable integration step was used. The complete set of parameters for a reference state, that is, NA model population in the state of no‐inflammation (NI), is presented in Supplementary Table 2. Some parameter values are subject to changes. These values depend on the model population and the model state and are separately presented in Tables 1, 2, 3, and they are discussed in the following sections.
Table 1.
Values of model parameters that define model states of NI and I in all model populations
| Parameter | Enzyme | NI | I |
|---|---|---|---|
| v max2, µM s−1 | COX‐2 | 0.055 | 0.28 |
| v max13, 16, µM s−1 | 5‐LOX | 2.53 | 86 |
| v max21, µM s−1 | 15‐LOX | 2.5 | 25 |
| v 0, µM s−1 | PLA2 | 0.07 | 0.7 |
Table 3.
Values of maximal velocities of the enzyme PGES (v max3) for: NA model population, aspirin‐tolerant asthmatic model population (ATA) and three different aspirin‐intolerant‐asthmatic model populations (AIA(1), AIA(2), AIA(3)) either in model states NI or inflammation (I)
| Model state | Parameter | Enzyme | NA | ATA | AIA(1) | AIA(2) | AIA(3) |
|---|---|---|---|---|---|---|---|
| NI | v max3, µM s−1 | PGES | 5.0 | 5.0 | 2.5 | 5.0 | 2.5 |
| I | v max3, µM s−1 | PGES | 20 | 20 | 5.0 | 10 | 5.0 |
2.2. Reference points for model simulations
In the process of constructing the model, we first determined the set of parameter values simulating NA model population in state of NI (NA‐NI), which first served as a reference for the definition of the model state of inflammation (I), and also later for the definition of all other model populations. In addition to NA, we defined four hypothetical model populations simulating asthmatic patients. One of them a simulated patient group of ATA and three of them simulated patient group of asthmatics intolerant to aspirin (AIA(1), AIA(2) and AIA(3)). A similar approach was used in our previous, simpler model of eicosanoid production [41, 43, 49]. One of the differences among both models is that in the previous one, the populations were defined differently and the model states of inflammation and NI were not considered for modelling. Since each of the model population in the present study could exist in either state of NI or inflammation we will analyse and compare ten model cases, in total. The following paragraphs will present the parameter estimation procedures for the model state of inflammation (I) and the model populations that are all derived from the reference model state – NA in state of NI (NA‐NI). The complete description of the definition of the reference model state is presented in the Supplementary Information.
Here we first focus on the definition of transition from the model state of NI to inflammation (I) for the model population NA. The modelling of two different states, inflammation and NI, was motivated by the publication of Pierzchalska et al. [32], who investigated the impact of proinflammatory stimuli on the cellular production of eicosanoids and on the expression of enzymes in cells obtained from patient groups NA, ATA and AIA. Their results were obtained from cultured fibroblasts gained from human airways. Fibroblasts were cultured with or without addition of cytomix and the supernatants were analysed by gas chromatography/mass spectrometry. Furthermore, they estimated COX‐1 and COX‐2 expression by RT‐PCR and immunoblotting. The addition of cytomix imitated cytokine‐ and bacterial‐driven inflammation, whereby cytomix used in experiments was a mixture of human recombinant interleukin (IL‐1β), tumour necrosis factor (TNF‐β) and lipopolysaccharide from Pseudomonas aeruginosa [32]. This experiment revealed several‐fold higher concentrations of PGE2 and PGD2 in sample cells obtained from NA after the addition of cytomix. These results are depicted in Figs. 2a and b as grey columns with error bars for NA, where % increases of [PGE2] and [PGD2] are presented for all patient groups (NA, ATA and AIA). In the NA group more than twenty‐five‐fold increase in [PGE2] and approximately four‐fold increase in [PGD2] was observed after the addition of cytomix. Such extensive increases of the model variables [PGE2] and [PGD2] had to be predicted in the transition from the model state of NI to inflammation via changes in particular model parameters. Therefore we first carried out the sensitivity analysis for our reference model state (NA‐NI) to identify the parameters with the highest impact on the model variables. Response coefficients for the variables [PGE2] and [PGD2] as well as of the ratios [PGE2]/[LTC4] and [PGE2]/[PGD2], with respect to small alterations in maximal velocities of the enzymes 5‐LOX (v max13, 16), COX‐1 (v max1), COX‐2 (v max2), PGES (v max3), PGDS (v max5), 15‐LOX (v max21) and LTC4S (v max17) as well as of the inflow of AA (v 0) were calculated according to (7). Maximal velocities of enzymes (v max) were selected for the sensitivity analysis, since they reflect the differences in total concentrations of the enzymes, which in turn depend also on their gene expression. Specifically, v max is defined in all expressions for the enzyme kinetics used here (2)–(4) as a product of total enzyme concentration ([E]tot) and the catalytic rate constant (k cat) (v max = k cat [E]tot). The sensitivity analysis revealed (for details see Supplemental Figures 2, 3, 4, 5):
(i) variables [PGE2] and [PGD2] are both equally sensitive to changes in v 0 and v max of both COX enzymes, whereby in both cases response coefficients for COX‐2 (v max2) are four‐fold smaller than those for COX‐1 (v max1),
(ii) with regards to enzymes in the prostanoid production pathway downstream of PGH2, variables [PGE2] and [PGD2] are sensitive exclusively to changes of the enzyme of their own production pathway (e.g. PGE2 is sensitive exclusively to changes in PGES) and not to the changes of the others,
(iii) variable [PGE2]/[PGD2] is sensitive exclusively (and almost equally with the opposite signs) to changes in parameters of the enzymes PGES (v max3) and PGDS (v max5),
(iv) variable [PGE2]/[LTC4] is not sensitive to any of the v max in the prostanoid production pathway downstream of PGH2 except of v max for PGES (v max3),
(v) increases in parameter values concerning the enzymes COX‐1 (v max1), COX‐2 (v max2), PGES (v max3) and PLA2 (v 0) result in positive response coefficients for [PGE2]/[LTC4] with a maximal value for v max3,
(vi) increases in parameter values concerning the enzymes 5‐LOX (v max13,16), 15‐LOX (v max21) and LTC4S (v max17) result in negative response coefficients for [PGE2]/[LTC4] with the most negative value for v max13, 16,
(vii) in order to simulate several‐fold‐increases of the system variables under study almost the same fold‐changes of parameter values, to which the variables are most sensitive, will be necessary.
Fig. 2.

Percentage increases (% increase) of PGs in the transition from NI to I
a [PGE2]
b [PGD2]
Model simulations (open columns) and experimentally induced values upon exposure to cytomix, mimicking inflammation (grey columns with error bars)
Experimental data are obtained from [32] by digitalisation of their Fig. 2
Fig. 3.
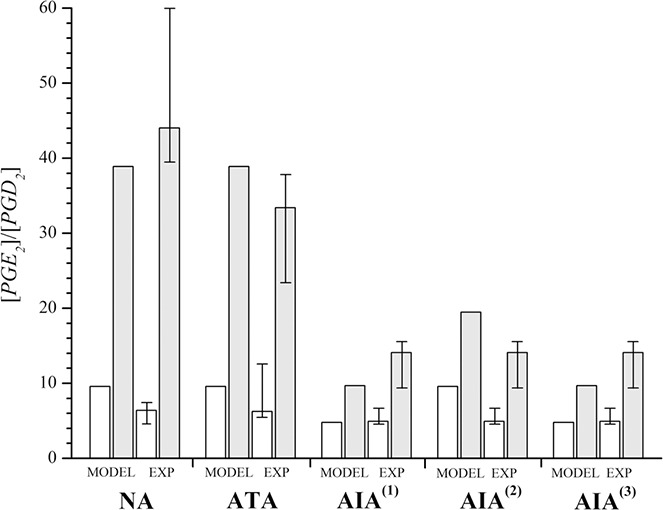
[PGE2]/[PGD2] ratio in the model states (MODEL) of NI (open columns) and I (grey columns) compared to the experimental setup (EXP) without cytomix (open columns with error bars) and with cytomix (grey columns with error bars)
Experimental data are obtained from [32] by digitalisation of their Fig. 3 (without indomethacin)
Fig. 4.
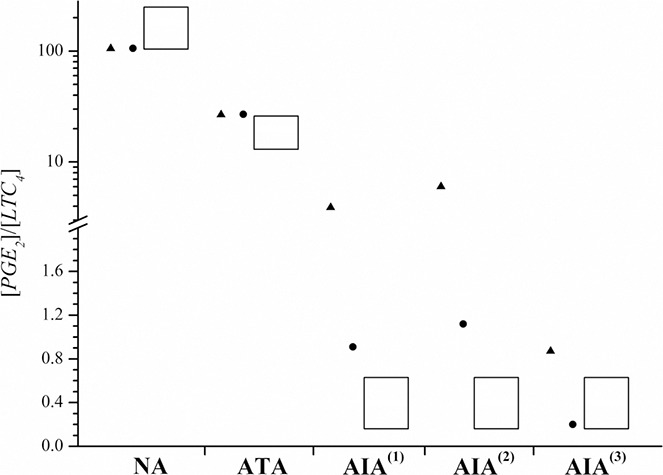
[PGE2]/[LTC4] ratio simulated by the model in the states of NI (black up‐pointing triangle) and I (black circle)
Measured values (Rf) presented with columns are from [19]
Note: the scale on the ordinate is broken, whereby the upper part has logarithmic scale and the lower part has linear one
Logarithmic scaling was chosen for displaying all results
Fig. 5.
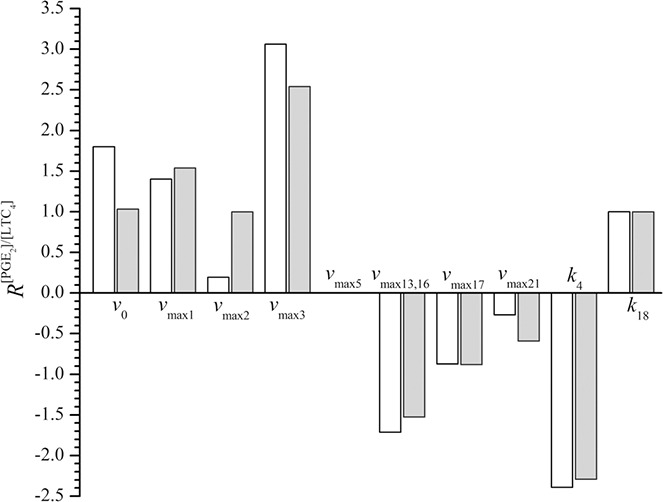
Response coefficients (R) of the model variable [PGE2]/[LTC4] in model population AIA(3) with respect to variations in parameter values vmaxi (i = 0, 1, 2, 3, 5, 13(16), 17, 21) and ki (i = 4, 18) by 10% in both model states NI (open columns) and I (grey columns)
Furthermore, published results of experimental studies considering the expressions of enzymes involved in the pathway of eicosanoid production under inflammatory conditions demonstrated:
(i) increased expression of COX‐2 in cultured human fibroblasts after addition of cytomix, mimicking inflammation [32],
(ii) increased expression of 15‐LOX in chronic asthma [50], in mild atopic asthma after allergen challenge [51], and as a result of different interleukins actions in various types of cells [51, 52, 53, 54, 55],
(iii) increased activation and gene expression of 5‐LOX in response to action of various cytokines produced under inflammatory conditions [56, 57, 58],
(iv) substantially increased expression of X phospholipase A2 (sPLA2‐X) in acute and chronic mouse asthma models [59],
(v) higher fold‐increase in microsomal PGES expression in control than in asthmatic mice in response to IL‐1β stimulation [60].
In order to simulate the transition of our reference model state (NA‐NI) into the state of inflammation (NA‐I), and, moreover, to be in a position to compare our model predictions to measured cytomix‐provoked results [32] as well as the others [19], explicit constraints and postulates had to be considered:
(i) based on measured data for NA [32], [PGE2] has to increase approximately 15–35‐fold in inflammation (see Fig. 2a ); NA – grey column with error bars),
(ii) based on measured data for NA [32], [PGD2] has to increase approximately 3–8‐fold in inflammation (see Fig. 2b ); NA – grey column with error bars),
(iii) based on measured data for NA [32], [PGE2]/[PGD2] ratio has to increase approximately four‐fold in inflammation (see Fig. 3; compare the values of open and grey columns with error bars for NA),
(iv) based on measured data for NA [19], the [PGE2]/[LTC4] ratio has to be between 100 and 200 (see Fig. 4; column for NA),
(v) postulate of the model is that the [PGE2]/[LTC4] ratio has to be conserved in the transition from NI to inflammation in the model population NA.
Note that the latter postulate serves purely as a forecast, if this conservation holds also for the other model populations or not. Based on these five constraints, we determined the values of five model parameters for the state of inflammation in the model population NA. The model parameters were v max of those enzymes, for which experimental evidence was found concerning their up‐ or down‐regulation under different experimental inflammatory conditions as described above [32, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60]. These enzymes and their corresponding parameters given in parentheses are: PLA2 (v 0), COX‐2 (v max2), 5‐LOX (v max13,16), 15‐LOX (v max21) and PGES (v max3). Parameter estimation advanced step‐by‐step with constant rechecking if the results fulfil the proposed constraints. First, v max3 was increased four‐fold, which increased the [PGE2]/[PGD2] ratio in inflammation for four‐fold with respect to NI. Then, v 0 and v max2 were arbitrarily increased for ten‐fold and five‐fold, respectively, which resulted in an increase of [PGE2] and [PGD2] as well as in a decrease of [LTC4], and therefore in a drastic increase of [PGE2]/[LTC4]. Finally, by keeping all other parameters, that is, v 0, v max2 and v max3 fixed, the v max13, 16 and v max21, were determined such that:
(i) absolute concentration of PGD2 [PGD2] increased four‐fold. Note: this is a typical % increase for NA group, determined in experiment [32] (see Fig. 2b ); NA – grey column with error bars),
(ii) absolute concentration of PGE2 [PGE2] increased four‐times as much as that of PGD2; that is, 16‐fold. Note: this is the lowest margin of % increase for NA group, determined in experiment [32] (see Fig. 2a ); NA – grey column with error bars),
(iii) the ratio [PGE2]/[LTC4] was conserved in the transition from NI to inflammation.
Thirty‐four‐fold and ten‐fold larger values of parameters v max13, 16 and v max21, respectively, reasonably fulfilled all above mentioned constraints. In this way, we have defined a set of parameter values that encodes the model state of inflammation (see Table 1), which will be applied to all other model populations. Parameter value of v max3 was intentionally left out, since changes in this parameter value will be considered also in the definition of the model populations.
From this point on, four different sets of parameters for four hypothetical asthmatic model populations were proposed comprising ATA and three types of asthmatics intolerant to aspirin specified as AIA(1), AIA(2) and AIA(3). The AIA model populations differ in their model parameter of maximal enzyme velocities of PGES and LTC4S. Using this approach, we intended to find out, which of the predicted model populations agrees most consistently with measured results from patient groups. In return, we reveal information which parameters of our model populations might reproduce most likely findings of the published patient groups. Up‐ and/or down‐regulations of PGES and LTC4S were either measured [10, 39] or hypothesised [32] in experiments related to NSAID‐triggered hypersensitivity and inflammation in humans. PGES up‐regulation was found also in mouse asthma models [60]. In this respect, v max of PGES and LTC4S are evidently implicated in the occurrence of NSAID‐triggered hypersensitivity. Our model states NA and ATA differ among each other only in the value of maximal velocity for LTC4S. On the other hand, model‐predicted AIA populations differ from ATA in the maximal velocity of either PGES or LTC4S or in maximal velocities of both. Model population AIA(1) differs from model population ATA only in one parameter value, that is, v max of PGES, whereas model population AIA(3) differs from model population ATA in two parameters, that is, v max of PGES and LTC4S. Model population AIA(2) in state of NI is characterised by the same v max of PGES as model population ATA. Model populations NA, ATA and AIA have been introduced already in our previous model [41, 43, 49]. That model was simpler, as it did not consider the conversion of AA to PGH2 and its further conversions to other prostanoids (particularly not to PGE2). Therefore our previous model did not enable the explicit simulation of the differences in v max of PGES. It also did not consider conversions of AA to 5‐ and 15‐HETE. In the previous model, we defined model populations with differences in v max of COX‐1, COX‐2 or LTC4S. COX‐2 is known as an inducible enzyme in inflammation [32, 61]. Pierzchalska et al. [32] reported no statistically significant differences in the expression of COX‐2, when comparing the patient ATA and AIA or NAs after cytomix‐induced inflammation. Therefore we considered in our present model the differences in v max of COX‐2 for the characterisation of the model states of NI and of inflammation. By assuming diminished v max of LTC4S our previous model [41, 43, 49] described AIA patients most consistently. Based thereon, we considered the same assumptions for the definition of model populations based on LTC4S, applying v max of LTC4S and fold‐ratios as used in [41, 43, 49]. Accordingly, the measured five‐fold higher expression of LTC4S in patient group AIA compared to ATA [10, 39] is reflected in our recent model by a five‐fold higher value of v max of LTC4S in the model populations AIA(2) and AIA(3) compared to ATA. In addition, measured four‐fold lower expression of LTC4S in group NA compared to patient group ATA [10, 39] is reflected by a four‐fold lower value of v max of LTC4S in the model population NA compared to model population ATA. The explicit parameter values are presented in Table 2.
Our previous model [41, 43, 49] could not consider differences in v max of PGES. On the other hand, our present model considers differences in v max of PGES twice; first, in the definitions of the model states inflammation and NI, and, second, in the definition of the model populations. The latter consideration was again motivated by observations of Pierzchalska et al. They reported the discrepancies in the production of PGE2 in the cultured samples of bronchial fibroblasts, obtained from NA, ATA and AIA patient groups, which were analysed under two conditions – with or without addition of cytomix in vitro. They concluded [32]:
(i) measured [PGE2]/[PGD2] ratio in experimental groups NA and ATA was approximately four‐fold larger in case of added cytomix than without cytomix,
(ii) measured [PGE2]/[PGD2] ratio in AIA patient group was approximately two‐fold larger in case of added cytomix than without cytomix,
(iii) measured [PGE2]/[PGD2] ratio was in patient groups NA and ATA not significantly different neither in case of added cytomix nor in case without cytomix,
(iv) measured [PGE2] increased in patient group ATA approximately two‐fold more compared with AIA after addition of cytomix to sample cells.
Pierzchalska et al. suggested that cytomix‐caused changes in the [PGE2]/[PGD2] ratio in different patient groups arise from the different impact of cytokines on the expression of PGES in cells, which were taken from each particular patient group. A different expression of PGES, however, has not been explicitly confirmed by their experiments, since they did not measure it. According to these experimental findings and in relation to the previously defined reference model state – NA‐NI, we implemented the following v max of PGES for our model populations for each model state, inflammation or NI:
(i) in NI, in ATA and AIA(2) v max3 is the same as in NA,
(ii) for ATA (and in NA), v max3 is four‐fold higher in inflammation than in NI,
(iii) for AIA(2), v max3 is two‐fold higher in inflammation than in NI,
(iv) in inflammation, in AIA(1) and AIA(3) v max3 is four‐fold lower than in ATA,
(v) for AIA(1) and AIA(3), v max3 is two‐fold lower in NI than in inflammation.
Explicit values of v max3 derived from these postulates are presented in Table 3.
2.3. Aims of the investigation
For each model population, that is, NA, ATA and all three AIA, the state of NI and of inflammation (I) was investigated. The absolute concentrations of the main system variables [PGE2], [PGD2] and [LTC4] were determined. In addition, their percentage increases (% increases) in the transition from the state of NI to inflammation were analysed and compared to the data measured on patients. Moreover, the [PGE2]/[LTC4] ratio and [PGE2]/[PGD2] ratio were predicted in the basal state (i.e. at rest without drug) and after simulated model‐based oral dosing of indomethacin. The latter resulted in a dynamical response of the system to the drug. In this concern, we tested the simulated model populations with respect to sensitivity to NSADs. We investigated whether our model results, based on the proposed model populations, agree with the published eicosanoid profiles from asthmatic patient groups. In addition, we aimed to find out whether or not our model might give some rationale for bio‐pharmacological‐based explanations of the differences observed among patient groups with respect to the metabolomic level of eicosanoid production from the proteomic point of view. Pure prediction of our dynamic model of eicosanoid production depicts the simulation of oral drug dosing for each of our model population, either in model state of NI or in a state of inflammation. A rough estimation for the bronchoconstriction which does not provide any information of the airway smooth muscle reactivity of a patient group or an individual patient will here be predicted purely on the basis of calculated the ratio [PGE2]/[LTC4] after oral indomethacin dosing. Finally, the sensitivity analysis of the model population AIA(3), which exhibited the most sensitive response to simulated oral indomethacin dosing, was worked out in terms of response coefficient (R) for the model variable [PGE2]/[LTC4].
Throughout Section 3, the results of our model are compared with two independently performed and published experiments, which are closely related with NSAID‐triggered hypersensitivity in humans [19, 32]. First, both experimental references accounted for the proper definition of our model's reference state, and partially also for the definition of the model populations. Later, their results served as a strong reference when comparing our model results with those obtained from asthmatic patient groups. Furthermore, these publications served as references for adequacy testing of our predicted kinetic scheme, the methods of mathematical modelling and the consistency of our proposed asthmatic model populations in the model states NI and inflammation.
3. Results and discussion
3.1. Experimental results from patients against results from model populations
Cultured cells, obtained from different patient groups, pre‐incubated for 18 h with cytomix, mimicking inflammation, dramatically increased the release of both types of PGs, PGE2 and PGD2 [32]. As presented in Fig. 2a , significant differences in % increases of [PGE2] were observed among the patient groups after exposure to cytomix in vitro [32]. However, no significant differences were observed in % increase of [PGD2] among these groups [32] (see Fig. 2b ). The in vitro addition of cytomix resulted in the approximately 2500% (26‐fold), approximately 1000% (11‐fold) and 500% (6‐fold) increase in [PGE2] in patient groups NA, ATA and AIA, respectively [32]. Our model populations, analysed at rest (i.e. without simulated drug dosing) revealed the following increases of [PGE2] in the transition from our model state of NI to inflammation: 1500% (16‐fold) for NA and ATA, and 750% (8.5‐fold) for all three AIA model populations (see Fig. 2a ). Measured % increases of [PGD2] ranged between 200% (three‐fold) and 400% (five‐fold), whereas model predicted values were approximately 300% (four‐fold) and were the same for all model populations (see Fig. 2b ). According to their results, Pierzchalska et al. [32] hypothesised that cytomix, mimicking inflammation, had no effect on the expression of PGDS but had varying effects on the expression of PGES in different patient groups. Our model results agree reasonably well with these measured % increases of [32] which speaks in favour of the consistency of our definitions of the model states and accounts for the definition of populations based on the differences in PGES.
Pierzchalska et al. did not measure increases in LTC4 but our model predicted them: The % increases of [LTC4] and [LTB4] were both equal and were approximately 1500% (16‐fold) for ATA and NA, approximately 3000% (31‐fold) for AIA(1) and AIA(3), and, approximately 4000% (41‐fold) for AIA(2). We compared these model results to those measured after ovalbumin‐triggered inflammation in BAL fluid obtained from mouse asthma models of Henderson et al. [59]. They measured increases of [cys‐LT] by approximately 1100% (12‐fold), [PGE2] by approximately 300% (4‐fold), [PGD2] by approximately 100% (2‐fold) and [LTB4] by approximately 300% (4‐fold) [59]. A general comparison of results from the asthma mouse model and from our model revealed that our model populations AIA in a state of inflammation consistently predicted approximately four‐fold higher increases for all studied eicosanoids as reported by Henderson et al. [59] for the mouse asthma model. This outcome indicates and speaks in favour of the validity of the definition of our model states of NI and inflammation.
The next comparison focused on the [PGE2]/[PGD2] ratio, which was measured by Pierzchalska et al. [32]. They analysed cellular samples of patient groups AIA, ATA and NA, which were exposed to cytomix in vitro. They revealed significantly smaller increases of [PGE2]/[PGD2] ratio in patient group AIA than in that of patient groups ATA and NA (see Fig. 3 for reproduced data). On the basis of their results they concluded that the activity of PGES in cells of patients suffering from AIA is lower than in those of ATA and NA. They also hypothesised that cytomix, mimicking inflammation in vitro, would up‐regulate PGES in cells of the AIA patient group to a smaller extent than those from NA and ATA patient groups. Our model‐predicted increases of [PGE2]/[PGD2] in transition from the model state of NI to that of inflammation were in all our AIA model populations obviously lower than in NA and ATA. These findings of our model are the consequence of smaller fold‐differences in v max for PGES in AIA than in ATA and NA model populations, when comparing the values in the model states of NI and inflammation (see Table 3 for comparison). The results of our model predictions for ATA and all AIA model populations revealed good agreement with measured results of [32]. For AIA(1) and AIA(3), the agreement was better than for AIA(2), since AIA(2) was characterised by the same v max of PGES (v max3) as the model population ATA in state of NI. On the other hand, the increase in v max3 from NI to inflammation was two‐fold in all of our AIA model populations. Model simulations thus revealed that the fold‐changes of v max for PGES (v max3) are almost directly reflected in the fold‐changes of the ratio [PGE2]/[PGD2].
Good agreement between our model results and the experimental results [19] was achieved also when looking at our model‐predicted ratio [PGE2]/[LTC4] and measured Rf, defined as [PGE2]/[cys‐LT] in [19]. Our results are outlined in Fig. 4. The columns in Fig. 4 represent the intervals of measured Rf obtained from patients [19], whereas dots (black circle) and triangles (black up‐pointing triangle) represent the results of mathematical model in states of inflammation (I) and NI, respectively. Note that results of mathematical model for ATA of both model states (i.e. NI and I) fall fully within the measured intervals of [19]. This is due to the fact that parameter values for the model population NA in the model state of inflammation (NA‐I) were deduced from the reference model population (NA‐NI) and the values were chosen so that the [PGE2]/[LTC4] ratio did not change in the transition from NI to inflammation. It turned out that model population ATA also possesses the same property as NA. On the other hand, [PGE2]/[LTC4] ratios for all AIA model populations were significantly smaller in states of inflammation than in states of NI (see Fig. 4 and Table 4 for exact model‐predicted values).
Table 4.
Values of [PGE2]/[LTC4] ratio at rest and minimal values after dosing of 25 mg indomethacin (∼at t = 4 h) for all model populations in states of NI and I
| Condition | Model state | NA | ATA | AIA(1) | AIA(2) | AIA(3) |
|---|---|---|---|---|---|---|
| basal (without drug) | NI | 110.4 | 28.0 | 4.1 | 6.2 | 0.91 |
| I | 110.4 | 28.0 | 0.94 | 1.2 | 0.21 | |
| minimum after indomethacin (25 mg) | NI | 1.1 | 0.28 | 0.045 | 0.063 | 0.010 |
| I | 7.0 | 1.8 | 0.044 | 0.073 | 0.0099 |
Published Rf for AIA patient group was around or below 1.0, ranging from 0.16 to 0.63 [19]. In our model state of NI (black up‐pointing triangle), the model predicted values were slightly larger than 1.0 for AIA(1) and AIA(2) (4.1 and 6.2, respectively) and lower than one for AIA(3) (0.91). In the model state of inflammation (black circle), the values were: 0.94, 1.2 and 0.21 for AIA(1), AIA(2) and AIA(3), respectively. These results were in good agreement with reported intervals of 0.16–0.63 [19]. According to this criterion, our model population AIA(3) described a patient group suffering from AIA slightly better than model populations AIA(1) and AIA(2), although, both of them roughly matched the properties of experimentally revealed results from AIA patients. Based on our model results for AIA(3) (i.e. model population with altered PGES and LTC4S maximal velocities with respect to ATA), these results suggest that differences in maximal velocity of PGES obviously largely contribute to a lower [PGE2]/[LTC4] ratio. This property could not be recognised solely from the data concerning [PGE2] and [PGD2] as described above (see Figs. 2a and b ). The results of the model simulations thus suggest that differences in maximal velocities of each particular enzyme, that is, LTC4S or PGES, might lead to NSAID‐triggered hypersensitivity. On the basis of our model, the worst case of hypersensitivity is expected, if maximal velocities of both enzymes, PGES and LTC4S, are affected (with a decrease in PGES and an increase in LTC4S maximal velocity) at the same time. The same property was revealed by the sensitivity analysis and by the dynamic simulation of eicosanoid production after indomethacin dosing, which is presented in the following sections.
3.2. Sensitivity analysis
Sensitivity analysis of the model variable [PGE2]/[LTC4] for the model population AIA(3) was carried out. This was performed with respect to small alterations in maximal velocities of the enzymes 5‐LOX (v max13, 16), COX‐1 (v max1), COX‐2 (v max2), PGES (v max3), PGDS (v max5), 15‐LOX (v max21) and LTC4S (v max17), the inflow of AA (v 0), plus the parameter values concerning PGE2 and LTC4 elimination rates (k 4 and k 18, respectively), as described in Sections 2.1 and 2.2. The response coefficients (R) are presented in Fig. 5. In both states of NI and inflammation, the [PGE2]/[LTC4] ratios were most sensitive to changes in the parameter v max of PGES (v max3). Almost the same but negative response coefficient was because of changes in parameter value k 4, which describes the PGE2 elimination rate. Response coefficients for 5‐LOX and 15‐LOX were negative, whereas those for COX‐1 and COX‐2, which indirectly increase [PGE2] levels in the model, were positive. Unexpectedly, the [PGE2]/[LTC4] ratio was somewhat less sensitive to changes in v max of LTC4S (v max17) than in v max of 5‐LOX (v max13,16). The opposite would be expected, since 5‐LOX is more upstream of the production of LTC4 than LTC4S. This is probably because of the kinetics of LTC4S used for modelling, which is not simple Michaelis–Menten kinetics but implements the enzyme kinetics with the substrate auto‐inhibition [see (4)]. In the COX pathway, the sensitivity of [PGE2]/[LTC4] to changes in v max of PGES (v max3), which is less upstream of the PGE2 production, was larger than the sensitivity to changes in v max of COX‐1 (v max1) and COX‐2 (v max2), which are more upstream (see Fig. 5 for comparison). The system was also almost equally sensitive (albeit with different signs) to changes in the parameters implicating inflow and elimination rates of LTC4, that is, v max17 and k 18, respectively, plus inflow and elimination rates of PGE2, that is, v max3 and k 4, respectively. The same effects on the production of either LTC4 or PGE2 could thus be achieved either by enhancing the inflow rates or reducing the corresponding elimination rates for the same factor. Changes in v max of PGDS (v max5) or in v max of any other of the second level enzymes within the COX pathway, except PGES, did not have any effect on the [PGE2]/[LTC4] ratio, neither in a state of inflammation nor in the state of NI. Fig. 5 presents the results of modifications for PGDS, solely. Furthermore, there were no major differences in the sensitivity analysis comparing the model states of NI and of inflammation for each particular model population, apart from one exception. When focusing on v max of COX‐2 (v max2), the sensitivity was much lower in state of NI than in that of inflammation. Finally, it is worth noting that although response coefficients are rather high, our model is robust to small perturbations. This means that small changes of parameter values do not cause the transitions from one model population to another. For such changes substantial changes of parameter values, that is, fold‐increases/decreases, are necessary.
3.3. Dynamic simulations
The effect of NSAIDs on [PGE2], [PGD2] and [LTC4] as well as on their ratios was studied with our dynamic model simulations using indomethacin. All parameters and initial conditions for the dynamic simulations are presented in the Supplementary Information. Supplementary Table 2 summarises all kinetic and pharmacokinetic parameters for the reference model state (NA‐NI), and Supplementary Table 5 summarises initial conditions for simulation of all model populations in both model states. For the simulation of the time evolution of the system variables from their initial concentrations the time‐dependent NSAID plasma drug concentration, described by (6), was applied. Equation (6) was derived from a standard two‐store pharmacokinetic model with absorption and elimination phases. With the use of pharmacokinetic parameters: k a = 1.0·10−4 s−1, k e = 9.5·10−5 s−1 and V = 45 L this model consistently described measured indomethacin plasma concentration after oral dosing in [62]. Indomethacin was used for simulations since Pierzchalska et al. [32] also used this drug in their experiments. The time‐evolution of all above mentioned absolute concentrations is presented in Fig. 6 for our model population AIA(3) in state of inflammation after simulated oral dosing of 25 mg indomethacin. This is a typical oral therapeutic dose. The dose of 25 mg indomethacin resulted in maximal blood plasma concentration of indomethacin yielding 0.59 µmol/L [NSAID] approximately 3 h after simulated application time (at t = 0).
Fig. 6.
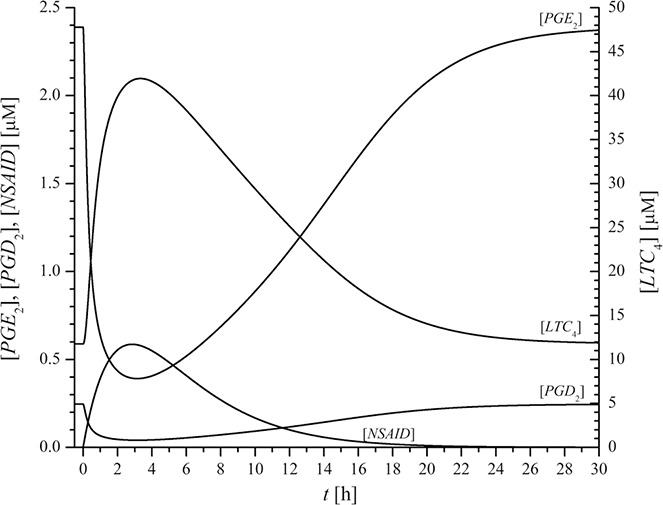
Time evolution of the absolute concentrations [PGE2], [PGD2] and [LTC4] as well as of plasma indomethacin concentration [NSAID] in model population AIA(3) in state of inflammation after simulated oral dosing of 25 mg indomethacin
NSAID dosing causes decreased synthesis of PGD2 and PGE2 [12, 63]. The dynamic simulations of NSAID dosing revealed decreased values of PGD2 and PGE2, accompanied by increased synthesis of LTC4. This indicates a tilt of the AA metabolic pathway in favour of the LTC4 production because of NSAIDs. For indomethacin, the extremes of [PGE2], [PGD2] and [LTC4] were achieved approximately 3–4 h after the moment of dosage, which almost coincided with the maximal indomethacin plasma concentration. The recovery of [PGE2], [PGD2] and [LTC4] back to their initial values was slow and lasted up to 24 h (see Fig. 6).
The ratios of [PGE2]/[PGD2] (full line) and [PGE2]/[LTC4] (dashed line) after indomethacin dosing are presented in Fig. 7a for AIA(3) model states of NI and of inflammation (I). Evidently, indomethacin revealed no effect on the [PGE2]/[PGD2] ratio. The same was observed for all other cases. These results are in complete accordance with the experiment of Pierzchalska et al., where no significant differences in [PGE2]/[PGD2] ratio were found after addition of 10 µmol/L indomethacin to samples with and without cytomix [32]. In our model, maximal plasma concentration of indomethacin 10 µmol/L was achieved with the dosing of 430 mg indomethacin. Even this high dose of indomethacin did not change the [PGE2]/[PGD2] ratio of any of the model populations. On the other hand, after simulated indomethacin dosing the [PGE2]/[LTC4] ratio dropped significantly, for approximately 20‐fold in a state of inflammation and approximately 90‐fold in state of NI. The time interval of extremely low [PGE2]/[LTC4] ratios (e.g. values lower than 0.16, the minimal values measured in AIA patients without drug) represents the duration of the largest risk for bronchoconstriction in our model simulation. Note, it is assumed that extended time interval in combination with the drop to the lowest absolute values of the [PGE2]/[LTC4] ratio goes along with increasing risk of bronchoconstriction. Among all AIA model populations simulated with the dosing of 25 mg indomethacin, AIA(3) exhibited the broadest time interval and the lowest [PGE2]/[LTC4] ratio (i.e. below 0.1 for ∼ 12 h) (see Fig. 7b ). Thus, model population AIA(3) was recognised as the most sensitive among the predicted three AIA model populations. For NA and ATA model populations, the minimal [PGE2]/[LTC4] ratios were much higher (several orders of magnitude) after 25 mg of indomethacin than those for AIA model populations (see Table 4 for the exact values). Furthermore, our dynamic model simulations with indomethacin dosing revealed smaller minimal [PGE2]/[LTC4] ratios for NA and ATA in state of NI than in state inflammation, whereas in AIA model populations no significant differences were calculated. However, our dynamic simulations revealed that those time intervals indicating risk of bronchoconstriction (with [PGE2]/[LTC4] ratios below 0.16) were in all AIA model populations significantly more extended in inflammation than in NI (see Fig. 7b ) for comparison). Note: the ordinate is drawn in the logarithmic scale, in order to present all model populations in a single diagram; therefore minima look apparently narrower.
Fig. 7.

Simulated time dependencies of the
a [PGE2]/[LTC4] (full line) and [PGE2]/[PGD2] (dashed line) ratios for the model population AIA(3) in states of NI and I for indomethacin dose 25 mg
b [PGE2]/[LTC4] ratios for all model populations in both model states, NI (dashed lines) and I (full lines), for indomethacin dose 25 mg
Note: Scale of ordinate in Fig. 7b is logarithmic and thus minima look apparently narrower
Drug is in all cases applied at time 0
The results of our model thus indicate that the state of inflammation of the underlying model population prolongs the duration of the risk for bronchoconstriction. This was demonstrated for all three investigated AIA model populations. Among them, model population AIA(3), which is characterised by concomitantly increased v max of LTC4S and decreased v max of PGES, exhibited the highest sensitivity to NSAIDs. It demonstrated the lowest [PGE2]/[LTC4] ratio and the longest duration of extremely low ratio (below 0.1). PGE2 is attributed to bronchodilation [15, 16] and LTC4 to bronchoconstriction [6]. Therefore a rough estimation of bronchoconstriction risk might be defined and predicted from their ratio. However, this value does not provide any information to the patient's individual bronchial hyper‐reactivity and hypersensitivity. With this in mind, our model population AIA(3) demonstrated the highest and the most long‐lasting risk for bronchoconstriction. In addition, our model results also suggest that differences in v max of either enzyme, LTC4S or PGES, might also lead to NSAID hypersensitivity, as demonstrated for model populations AIA(1) and AIA(2), albeit, their capacity and duration of bronchoconstriction risk were weaker and shorter than in AIA(3).
3.4. Explanatory impact of the model
The results of our mathematical model correspond to the following experimental patient‐based observations and might explain the reported imbalances of eicosanoids in NSAID‐triggered hypersensitivity:
(i) deficiency in the production of PGE2 in cells obtained from patient group suffering from AIA after addition of cytomix [32],
(ii) increased [PGE2]/[PGD2] ratio after addition of cytomix to cells obtained from NA, ATA and AIA patient groups [32]
(iii) significantly lower increase of [PGE2]/[PGD2] ratio after addition of cytomix to cells obtained from AIA than in those obtained from ATA and NA patient groups [32],
(iv) no significant impact of NSAID on [PGE2]/[PGD2] ratio in any of the patient groups [32],
(v) order of 1000‐fold lower [PGE2]/[LTC4] ratio in AIA than in NA patient groups after NSAID dosing [19],
(vi) the ratio [PGE2]/[LTC4] around 1.0 in AIA patient group and approximately 100‐times larger value in NA group in the basal state (without drug) [19],
(vii) higher basal concentrations of LTC4 in AIA than in ATA and NA patient groups [4, 9, 18, 30, 31].
Our model predicts and gives some hints on imbalances of different eicosanoid ratios, which arise from differences in the maximal velocities of particular enzymes. This endorses the idea that inflammatory stimuli might have a different impact on the protein levels and/or their expressions in the cells obtained from different patient groups. The model predicts which enzymes might be affected and what fold‐changes might occur because of the inflammatory stimuli. However, it has to be noted that the fold‐changes of parameter values which are implicated in the model are only estimates. These estimates might offer preliminary results on the expected orders of magnitude, additionally, they might give some clues as to the parameter values which are more and those which are less affected by inflammation. The experiment of [32] leads to the suggestion that PGES and COX‐2 were affected by inflammation; thereby inflammation was simulated in vitro by cytomix, a mixture of proinflammatory agents. Changes in COX‐2 were experimentally confirmed, whereas the changes in PGES were only hypothesised by this ex vivo/in vitro approach [32]. On the basis of our reference model state (i.e. model population NA‐NI), only the changes of two parameter values, that is, the v max of PGES and COX‐2, could not reflect the results of experimentally induced inflammation, gained from samples of the NA patient group. The reported approximately 20‐fold increase of [PGE2] in patient group NA after cytomix was in our model achieved by a series of fold‐changes of parameter values. An influx of AA was increased, two parameters of the LOX pathway, the v max of 5‐LOX and 15‐LOX, were increased in addition in order to obtain an agreement with the results obtained experimentally for the NA patient group in [19, 32]. Moreover, specific fold‐changes for v max of PGES were applied for the definition of model populations ATA and three AIA as well as for the simulation of the transition from state of NI to inflammation in these model populations (see Tables 2 and 3). Fold‐changes in parameter values used in our model are deduced from the metabolomic studies of eicosanoid profiles [19, 32] and could be used as a starting point for further proteomic or transcriptomic studies.
In addition, we analysed which parameter values of the system variables have the highest impact on the [PGE2]/[LTC4] ratio, known as a sensitive indicator of NSAID‐triggered hypersensitivity [19]. Our study revealed parameters implicated in the PGE2 production and elimination pathway. Therefore patient group AIA might differ from ATA and NA in this particular property. It was hypothesised in [32], that cytomix increased PGES expression in the AIA patient group in a smaller extent than in ATA and NA patient groups. Another possible explanation might be that inflammatory processes differently affected the mechanism of conversion of PGE2 into biologically inactive metabolite(s) of PGE2 – PGE‐M, or, that inflammatory processes might affect both mechanisms. Recent results obtained from exhaled breathe condensates, quantified by highly sensitive gas chromatography/mass spectrometry, revealed higher levels of PGE‐M in AIA than in ATA, and NA patient groups [64, 65]. However, the fact that the [PGE2]/[LTC4] ratio, which became a benchmark for distinguishing AIA from ATA in recent years [11, 12, 19, 33], is up to 1000‐fold lower in patient group AIA than in patient group NA. This in turn might then only be explained by the assumption that the process of PGE2 degradation is faster in AIA than in NA and ATA, which up to date, was not proved.
Sensitivity analysis of our model system revealed that elevation of the parameters v max3 and k 4 (implicated in the production and degradation of PGE2, respectively) by the same factor affects neither the [PGE2]/[LTC4] nor the [PGE2]/[PGD2] ratios (see Fig. 5). In addition, the overall flux directed to the metabolite of PGE2 would increase. These results of our model system might thus explain low levels of PGE2 and high levels of PGE‐M at the same time in patients suffering from AIA.
According to the results of our presented model the worst case of NSAID‐triggered hypersensitivity would be expected from concomitantly increased expression of LTC4S and decreased expression of PGES, although, many other combinations are also possible. This would be the case, if parameters with positive response coefficients with respect to [PGE2]/[LTC4] ratio (e.g. v max of enzymes PLA2, COX‐1, COX‐2 and PGES plus the rate of LTC4 degradation) are down regulated, and/or, if parameters with negative response coefficients (e.g. v max of enzymes 5‐LOX, 15‐LOX and LTC4S, plus the rate of PGE2 degradation), are up regulated at the same time. Various combinations of increased/decreased parameter values provide rationales for a variety of clinical symptoms like bronchoconstriction, nasal polyps, rhinosinusitis, urticaria, conjunctivitis and so on, as well as their acuteness and strength, ranging from acute to chronic and from mild to severe. For a more complete description of such diseases by modelling, additional experimental and theoretical studies will have to be performed, integrating metabolomic and proteomic studies. Our current mathematical modelling approach is one of the first systematical attempts in this regard.
Dynamic model simulations after oral indomethacin dosing endorsed the results of sensitivity analysis. They both reveal the model population AIA(3) as the most sensitive model population with respect to the duration and capacity of the risk for bronchoconstriction. AIA(3) is characterised by concomitantly increased v max of LTC4S and decreased v max of PGES. Moreover, our dynamic simulations revealed that in all AIA model populations the state of inflammation prolonged the duration of bronchoconstriction risk. This property is again most evident in the model population AIA(3). The latter simulations might help to reveal differences in NSAID‐triggered hypersensitivity of different patient groups. Accordingly, this might facilitate estimations on the outcome of oral drug dosing in different patient populations, depending on the anamnesis of their inflammatory state. Future proteomic and transcriptomic studies, combined with metabolomic studies, will contribute to improvements of the models such as ours, and, at least in our opinion, might be of high relevance in discovering more insights on the intrinsic factors involved in AERD.
4. Conclusions
In spite of accumulating experimental and clinical results the molecular and cellular mechanisms, which induce and initiate processes in AERD, are still not completely elucidated and understood. In the past, experimental and clinical research dealing with AERD was primarily focused on a single level, either on concerning the gene, molecular, cellular or tissue/organ level. Theoretical analyses and predictions attained from mathematical models, such as ours, integrate the accumulated knowledge of these different levels of research. Therefore the mathematical model analysis might serve as a suitable tool aiding to figure out and test new hypotheses, check molecular mechanisms, analyse experimental results, guide novel experiments as well as give ideas for new methods and tools for diagnosing and treatment diseases, such as AERD in that case, on a multiscale level.
Our dynamic mathematical model represents a step forward towards multiscale models as described for the lung [46, 66]. In such multi‐cellular models airway smooth muscle cells will be coupled with cells involved in inflammation via inflammatory mediators acting on airway smooth muscle cells by contraction/relaxation mechanisms [67, 68]. These multiscale models might elucidate the ‘functional signature’ of AERD [33], that is, the underlying functional and molecular abnormalities, which result from complex cellular alterations. Those complex alterations are expressed in terms of inter‐ and intra‐cellular signalling of metabolism because of over‐ and under‐expression of genes, gene products, consider possible input stimuli which finally integrate and reveal a unique message in response to external and internal events, for example, an inflammatory disease [11].
Ambitious efforts will be to extentd our recent model to facilitate meaningful estimations for an individual risk of bronchoconstriction regarding asthmatic patients, supposed to be intolerant to aspirin, ideally with regard to the type and dose of the NSAID as well as his/her acute original condition of inflammation and bronchial hyper‐responsiveness.
5. Acknowledgment
The financial support of Slovenian Research Agency (ARRS) under the contract No. P1‐0055 was gratefully acknowledged.
6 References
- 1. Bergström, B. : ‘Prostaglandines: members of a new hormonal system’, Science, 1967, 157, pp. 323–290 (doi: 10.1126/science.157.3787.382) [DOI] [PubMed] [Google Scholar]
- 2. Funk, C.D. : ‘Prostaglandins and leukotrienes: advances in eicosanoid biology’, Science, 2001, 294, (5548), pp. 1871–1875 (doi: 10.1126/science.294.5548.1871) [DOI] [PubMed] [Google Scholar]
- 3. Pompea, C. , Procopio, J. , Curi, R. : ‘Fatty acids and immune system’, Barz J. Pharm. Sci., 1999, 35, pp. 166–194 [Google Scholar]
- 4. Vane, J. , Botting, R. : ‘Biological properties of cyclooxygenase products’, in Cunningham, F. (ed.): ‘Lipid mediators’, (Academic Press, 1994), pp. 61–97 (doi: 10.1016/B978-0-12-198875-3.50009-8) [DOI] [Google Scholar]
- 5. Harizi, H. , Corcuff, J.B. , Gualde, N. : ‘Arachidonic‐acid‐derived eicosanoids: roles in biology and immunopathology’, Trends in Molecular Medicine, 2008, 14, (10), pp. 461–469 (doi: 10.1016/j.molmed.2008.08.005) [DOI] [PubMed] [Google Scholar]
- 6. Leff, A.R. : ‘Role of leukotrienes in bronchial hyperresponsiveness and cellular responses in airways’, Thorax, 2000, 55, (Suppl 2), pp. S32–37 (doi: 10.1136/thorax.55.suppl_2.S32) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vargaftig, B.B. , Singer, M. : ‘Leukotrienes mediate murine bronchopulmonary hyperreactivity, inflammation, and part of mucosal metaplasia and tissue injury induced by recombinant murine interleukin‐13’, Am. J. Respir Cell Mol. Biol., 2003, 28, (4), pp. 410–419 (doi: 10.1165/rcmb.2002-0032OC) [DOI] [PubMed] [Google Scholar]
- 8. Drazen, J.M. : ‘Leukotrienes as mediators of airway obstruction’, Am. J. Respir. Crit. Care Med., 1998, 158, (5 Pt 3), pp. S193–200 (doi: 10.1164/ajrccm.158.supplement_2.13tac180) [DOI] [PubMed] [Google Scholar]
- 9. Stevenson, D. , Szczeklik, A. : ‘Clinical and pathologic perspectives on aspirin sensitivity and asthma’, J. Allergy Clin. Immunol., 2006, 118, (4), pp. 773–786 (doi: 10.1016/j.jaci.2006.07.024) [DOI] [PubMed] [Google Scholar]
- 10. Szczeklik, A. , Stevenson, D. : ‘Aspirin‐induced asthma: advances in pathogenesis and management’, J. Allergy Clin. Immunol., 1999, 104, (1), pp. 5–13 (doi: 10.1016/S0091-6749(99)70106-5) [DOI] [PubMed] [Google Scholar]
- 11. Schäfer, D. : ‘Testing and typing of eicosanoid‐patterns’, J. Physiol. Pharmacol., 2006, 57, (Suppl 12), pp. 47–64 [PubMed] [Google Scholar]
- 12. Schäfer, D. , Lindenthal, U. , Wagner, M. , Bolcskei, P.L. , Baenkler, H.W. : ‘Effect of prostaglandin E2 on eicosanoid release by human bronchial biopsy specimens from normal and inflamed mucosa’, Thorax, 1996, 51, (9), pp. 919–923 (doi: 10.1136/thx.51.9.919) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Szczeklik, A. , Sanak, M. : ‘The broken balance in aspirin hypersensitivity’, Eur. J. Pharmacol., 2006, 533, (1–3), pp. 145–155 (doi: 10.1016/j.ejphar.2005.12.053) [DOI] [PubMed] [Google Scholar]
- 14. Matsuoka, T. , Hirata, M. , Tanaka, H. , et al.: ‘Prostaglandin D2 as a mediator of allergic asthma’, Science, 2000, 287, (5460), pp. 2013–2017 (doi: 10.1126/science.287.5460.2013) [DOI] [PubMed] [Google Scholar]
- 15. Hartney, J.M. , Coggins, K.G. , Tilley, S.L. , et al.: ‘Prostaglandin E2 protects lower airways against bronchoconstriction’, Am. J. Physiol. Lung Cell Mol. Physiol., 2006, 290, (1), pp. L105–113 (doi: 10.1152/ajplung.00221.2005) [DOI] [PubMed] [Google Scholar]
- 16. Sestini, P. , Armetti, L. , Gambaro, G. , et al.: ‘Inhaled Pge2 prevents aspirin‐induced bronchoconstriction and urinary Lte4 excretion in aspirin‐sensitive asthma’, Am. J. Respir. Crit. Care Med., 1996, 153, (2), pp. 572–575 (doi: 10.1164/ajrccm.153.2.8564100) [DOI] [PubMed] [Google Scholar]
- 17. Christman, B.W. , Christman, J.W. , Dworski, R. , Blair, I.A. , Prakash, C. : ‘Prostaglandin E2 limits arachidonic acid availability and inhibits leukotriene B4 synthesis in rat alveolar macrophages by a nonphospholipase A2 mechanism’, J. Immunol., 1993, 151, (4), pp. 2096–2104 [PubMed] [Google Scholar]
- 18. Harizi, H. , Juzan, M. , Moreau, J.‐F. , Gualde, N. : ‘Prostaglandins inhibit 5‐lipoxygenase‐activating protein expression and leukotriene B4 production from dendritic cells via an il‐10‐dependent mechanism’, J. Immunol., 2003, 170, (1), pp. 139–146 (doi: 10.4049/jimmunol.170.1.139) [DOI] [PubMed] [Google Scholar]
- 19. Schäfer, D. , Schmid, M. , Göde, U.C. , Baenkler, H.W. : ‘Dynamics of eicosanoids in peripheral blood cells during bronchial provocation in aspirin‐intolerant asthmatics’, Eur. Respir. J., 1999, 13, (3), pp. 638–646 (doi: 10.1183/09031936.99.13363899) [DOI] [PubMed] [Google Scholar]
- 20. Gryglewski, R.J. , Szczeklik, A. , Wandzilak, M. : ‘The effect of six prostaglandins, prostacyclin and iloprost on generation of superoxide anions by human polymorphonuclear leukocytes stimulated by zymosan or formyl‐methionyl‐leucyl‐phenylalanine’, Biochem. Pharmacol., 1987, 36, (24), pp. 4209–4213 (doi: 10.1016/0006-2952(87)90660-5) [DOI] [PubMed] [Google Scholar]
- 21. Minakuchi, R. , Wacholtz, M.C. , Davis, L.S. , Lipsky, P.E. : ‘Delineation of the mechanism of inhibition of human T cell activation by Pge2’, J. Immunol., 1990, 145, (8), pp. 2616–2625 [PubMed] [Google Scholar]
- 22. Kay, L. , Yeo, W. , Peachell, P. : ‘Prostaglandin E2 activates Ep2 receptors to inhibit human lung mast cell degranulation’, Br. J. Pharmacol., 2006, 147, (7), pp. 707–713 (doi: 10.1038/sj.bjp.0706664) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pavord, I.D. , Tattersfield, A.E. : ‘Bronchoprotective role for endogenous prostaglandin E2’, Lancet, 1995, 345, (8947), pp. 436–438 (doi: 10.1016/S0140-6736(95)90409-3) [DOI] [PubMed] [Google Scholar]
- 24. Nguyen, M. , Solle, M. , Audoly, L.P. , et al.: ‘Receptors and signaling mechanisms required for prostaglandin E2‐mediated regulation of mast cell degranulation and Il‐6 production’, J. Immunol., 2002, 169, (8), pp. 4586–4593 (doi: 10.4049/jimmunol.169.8.4586) [DOI] [PubMed] [Google Scholar]
- 25. Szczeklik, A. , Mastalerz, L. , Nizankowska, E. , Cmiel, A. : ‘Protective and bronchodilator effects of prostaglandin E and salbutamol in aspirin‐induced asthma’, Am. J. Respir. Crit Care Med., 1996, 153, (2), pp. 567–571 (doi: 10.1164/ajrccm.153.2.8564099) [DOI] [PubMed] [Google Scholar]
- 26. van Overveld, F.J. , Jorens, P.G. , De Backer, W.A. , Rampart, M. , Bossaert, L. , Vermeire, P.A. : ‘Release of arachidonic acid metabolites from isolated human alveolar type Ii cells’, Prostaglandins, 1992, 44, (2), pp. 101–110 (doi: 10.1016/0090-6980(92)90071-Z) [DOI] [PubMed] [Google Scholar]
- 27. Meade, E.A. , Smith, W.L. , DeWitt, D.L. : ‘Differential inhibition of prostaglandin endoperoxide synthase (cyclooxygenase) isozymes by aspirin and other non‐steroidal anti‐inflammatory drugs’, J. Biol. Chem., 1993, 268, (9), pp. 6610–6614 [PubMed] [Google Scholar]
- 28. So, O.‐Y. , Scarafia, L.E. , Mak, A.Y. , Callan, O.H. , Swinney, D.C. : ‘The dynamics of prostaglandin H synthases. studies with prostaglandin H synthase 2 Y355f unmask mechanisms of time‐dependent inhibition and allosteric activation’, J. Biol. Chem., 1998, 273, (10), pp. 5801–5807 (doi: 10.1074/jbc.273.10.5801) [DOI] [PubMed] [Google Scholar]
- 29. Goltsov, A. , Maryashkin, A. , Swat, M. , et al.: ‘Kinetic modelling of nsaid action on cox‐1: focus on in vitro/in vivo aspects and drug combinations’, Eur. J. Pharm. Sci., 2009, 36, (1), pp. 122–136 (doi: 10.1016/j.ejps.2008.10.015) [DOI] [PubMed] [Google Scholar]
- 30. Daffern, P.J. , Muilenburg, D. , Hugli, T.E. , Stevenson, D.D. : ‘Association of urinary leukotriene E4 excretion during aspirin challenges with severity of respiratory responses’, J. Allergy Clin. Immunol., 1999, 104, (3 Pt 1), pp. 559–564 (doi: 10.1016/S0091-6749(99)70324-6) [DOI] [PubMed] [Google Scholar]
- 31. Dahlen, B. , Nizankowska, E. , Szczeklik, A. , et al.: ‘Benefits from adding the 5‐lipoxygenase inhibitor zileuton to conventional therapy in aspirin‐intolerant asthmatics’, Am. J. Respir. Crit. Care Med., 1998, 157, (4 Pt 1), pp. 1187–1194 (doi: 10.1164/ajrccm.157.4.9707089) [DOI] [PubMed] [Google Scholar]
- 32. Pierzchalska, M. , Szabo, Z. , Sanak, M. , Soja, J. , Szczeklik, A. : ‘Deficient prostaglandin E2 production by bronchial fibroblasts of asthmatic patients, with special reference to aspirin‐induced asthma’, J. Allergy Clin. Immunol., 2003, 111, (5), pp. 1041–1048 (doi: 10.1067/mai.2003.1491) [DOI] [PubMed] [Google Scholar]
- 33. Schäfer, D. , Baenkler, H.W. : ‘Functional eicosanoid test and typing (Fet) of peripheral blood cells in eicosanoids related diseases’, J. Physiol. Pharmacol., 2005, 56, (Suppl 5), pp. 103–118 [PubMed] [Google Scholar]
- 34. Szczeklik, A. : ‘Prostaglandin E2 and aspirin‐induced asthma’, Lancet, 1995, 345, (8956), p. 1056 (doi: 10.1016/S0140-6736(95)90799-8) [DOI] [PubMed] [Google Scholar]
- 35. Szczeklik, A. , Nizankowska, E. , Bochenek, G. , Nagraba, K. , Mejza, F. , Swierczynska, M. : ‘Safety of a specific cox‐2 inhibitor in aspirin‐induced asthma’, Clin. Exp. Allergy, 2001, 31, (2), pp. 219–225 (doi: 10.1046/j.1365-2222.2001.01075.x) [DOI] [PubMed] [Google Scholar]
- 36. Szczeklik, A. , Sanak, M. : ‘The role of cox‐1 and cox‐2 in asthma pathogenesis and its significance in the use of selective inhibitors’, Clin. Exp. Allergy, 2002, 32, (3), pp. 339–342 (doi: 10.1046/j.1365-2222.2002.01333.x) [DOI] [PubMed] [Google Scholar]
- 37. Szczeklik, A. , Stevenson, D. : ‘Aspirin‐induced asthma: advances in pathogenesis, diagnosis, and management’, J. Allergy Clin. Immunol., 2003, 111, (5), pp. 913–921; quiz 922 (doi: 10.1067/mai.2003.1487) [DOI] [PubMed] [Google Scholar]
- 38. Gosepath, J. , Schäfer, D. , Mann, W.J. : ‘Aspirin sensitivity: long term follow‐up after up to 3 years of adaptive desensitization using a maintenance dose of 100 Mg of aspirin a day’, Laryngorhinootologie, 2002, 81, (10), pp. 732–738 (doi: 10.1055/s-2002-35002) [DOI] [PubMed] [Google Scholar]
- 39. Cowburn, A.S. , Sladek, K. , Soja, J. , et al.: ‘Overexpression of leukotriene C4 synthase in bronchial biopsies from patients with aspirin‐intolerant asthma’, J. Clin. Invest., 1998, 101, (4), pp. 834–846 (doi: 10.1172/JCI620) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Maxis, K. , Delalandre, A. , Martel‐Pelletier, J. , Pelletier, J.P. , Duval, N. , Lajeunesse, D. : ‘The shunt from the cyclooxygenase to lipoxygenase pathway in human osteoarthritic subchondral osteoblasts is linked with a variable expression of the 5‐lipoxygenase‐activating protein’, Arthritis Res. Ther., 2006, 8, (6), p. R181 (doi: 10.1186/ar2092) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dobovisek, A. , Fajmut, A. , Brumen, M. : ‘Role of expression of prostaglandin synthases 1 and 2 and leukotriene C4 synthase in aspirin‐intolerant asthma: a theoretical study’, J. Pharmacokinet. Pharmacodyn., 2011, 38, (2), pp. 261–278 (doi: 10.1007/s10928-011-9192-6) [DOI] [PubMed] [Google Scholar]
- 42. Dobovisek, A. , Fajmut, A. , Brumen, M. : ‘Strategy for nsaid administration to aspirin‐intolerant asthmatics in combination with Pge(2) analogue: a theoretical approach’, Med. Biol. Eng.Comput., 2012, 50, (1), pp. 33–42 (doi: 10.1007/s11517-011-0844-x) [DOI] [PubMed] [Google Scholar]
- 43. Fajmut, A. , Dobovisek, A. , Brumen, M. : ‘Mathematical modeling in aspirin‐induced asthma: theory and clinical applications’, in Bislimi, A.H. , Tolka, L.C. (Eds.): ‘Asthma: Causes, Complications and Treatment’ (Nova Science Publishers, Inc., 2012) [Google Scholar]
- 44. Yang, K. , Ma, W. , Liang, H. , Ouyang, Q. , Tang, C. , Lai, L. : ‘Dynamic simulations on the arachidonic acid metabolic network’, PLoS Comput. Biol., 2007, 3, (3), p. e55 (doi: 10.1371/journal.pcbi.0030055) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gupta, N. , Gresser, M.J. , Ford‐Hutchinson, A.W. : ‘Kinetic mechanism of glutathione conjugation to leukotriene A4 by leukotriene C4 synthase’, Biochim. Biophys. Acta, 1998, 1391, (2), pp. 157–168 (doi: 10.1016/S0005-2760(98)00002-2) [DOI] [PubMed] [Google Scholar]
- 46. Demin, O. , Karelina, T. , Svetlichniy, D. , et al.: ‘Systems pharmacology models can be used to understand complex pharmacokinetic‐pharmacodynamic behavior: an example using 5‐lipoxygenase inhibitors’, CPT Pharmacometrics Syst. Pharmacol., 2013, 2, p. e74 (doi: 10.1038/psp.2013.49) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Goltsov, A. , Lebedeva, G. , Humphery‐Smith, I. , Goltsov, G. , Demin, O. , Goryanin, I. : ‘In silico screening of nonsteroidal anti‐inflammatory drugs and their combined action on prostaglandin H synthase‐1’, Pharmaceuticals, 2010, 3, (7), pp. 2059–2081 (doi: 10.3390/ph3072059) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Karelina, T.A. , Zhudenkov, K.V. , Demin, O.O. , et al.: ‘Regulation of leukotriene and 5oxoete synthesis and the effect of 5‐lipoxygenase inhibitors: a mathematical modeling approach’, BMC Syst. Biol., 2012, 6, p. 1410 (doi: 10.1186/1752-0509-6-141) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dobovisek, A. , Fajmut, A. , Brumen, M. : ‘Strategy for nsaid administration to aspirin‐intolerant asthmatics in combination with Pge2 analogue: a theoretical approach’, Med. Biol. Eng. Comput., 2012, 50, (1), pp. 33–42 (doi: 10.1007/s11517-011-0844-x) [DOI] [PubMed] [Google Scholar]
- 50. Profita, M. , Sala, A. , Riccobono, L. , et al.: ‘15‐Lipoxygenase expression and 15(S)‐hydroxyeicoisatetraenoic acid release and reincorporation in induced sputum of asthmatic subjects’, J. Allergy Clin. Immunol., 2000, 105, (4), pp. 711–716 (doi: 10.1067/mai.2000.105122) [DOI] [PubMed] [Google Scholar]
- 51. Chu, H.W. , Balzar, S. , Westcott, J.Y. , et al.: ‘Expression and activation of 15‐lipoxygenase pathway in severe asthma: relationship to eosinophilic phenotype and collagen deposition’, Clin. Exp. Allergy, 2002, 32, (11), pp. 1558–1565 (doi: 10.1046/j.1365-2222.2002.01477.x) [DOI] [PubMed] [Google Scholar]
- 52. Brinckmann, R. , Topp, M.S. , Zalan, I. , et al.: ‘Regulation of 15‐lipoxygenase expression in lung epithelial cells by interleukin‐4’, Biochem. J., 1996, 318, (Pt 1), pp. 305–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Conrad, D.J. , Kuhn, H. , Mulkins, M. , Highland, E. , Sigal, E. : ‘Specific inflammatory cytokines regulate the expression of human monocyte 15‐lipoxygenase’, Proc. Natl. Acad. Sci. USA, 1992, 89, (1), pp. 217–221 (doi: 10.1073/pnas.89.1.217) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gulliksson, M. , Brunnstrom, A. , Johannesson, M. , et al.: ‘Expression of 15‐lipoxygenase type‐1 in human mast cells’, Biochim. Biophys. Acta, 2007, 1771, (9), pp. 1156–1165 (doi: 10.1016/j.bbalip.2007.06.001) [DOI] [PubMed] [Google Scholar]
- 55. Nassar, G.M. , Morrow, J.D. , Roberts, L.J. 2nd , Lakkis, F.G. , Badr, K.F. : ‘Induction of 15‐lipoxygenase by interleukin‐13 in human blood monocytes’, J. Biol. Chem., 1994, 269, (44), pp. 27631–27634 [PubMed] [Google Scholar]
- 56. De Caterina, R. , Zampolli, A. : ‘From asthma to atherosclerosis – 5‐lipoxygenase, leukotrienes, and inflammation’, N. Engl. J. Med., 2004, 350, (1), pp. 4–7 (doi: 10.1056/NEJMp038190) [DOI] [PubMed] [Google Scholar]
- 57. Chu, S.J. , Tang, L.O. , Watney, E. , Chi, E.Y. , Henderson, Jr. W.R. : ‘In situ amplification of 5‐lipoxygenase and 5‐lipoxygenase‐activating protein in allergic airway inflammation and inhibition by leukotriene blockade’, J. Immunol., 2000, 165, (8), pp. 4640–4648 (doi: 10.4049/jimmunol.165.8.4640) [DOI] [PubMed] [Google Scholar]
- 58. Murakami, M. , Austen, K.F. , Bingham, C.O. 3rd , Friend, D.S. , Penrose, J.F. , Arm, J.P. : ‘Interleukin‐3 regulates development of the 5‐lipoxygenase/leukotriene C4 synthase pathway in mouse mast cells’, J. Biol. Chem., 1995, 270, (39), pp. 22653–22656 (doi: 10.1074/jbc.270.39.22653) [DOI] [PubMed] [Google Scholar]
- 59. Henderson, W.R. Jr. , Chi, E.Y. , Bollinger, J.G. , et al.: ‘Importance of group X‐secreted phospholipase A2 in allergen‐induced airway inflammation and remodeling in a mouse asthma model’, J. Exp. Med., 2007, 204, (4), pp. 865–877 (doi: 10.1084/jem.20070029) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Stumm, C.L. , Wettlaufer, S.H. , Jancar, S. , Peters‐Golden, M. : ‘Airway remodeling in murine asthma correlates with a defect in Pge2 synthesis by lung fibroblasts’, Am. J. Physiol. Lung Cell Mol. Physiol., 2011, 301, (5), pp. L636–644 (doi: 10.1152/ajplung.00158.2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Porreca, E. , Reale, M. , Di Febbo, C. , et al.: ‘Down‐regulation of cyclooxygenase‐2 (cox‐2) by interleukin‐1 receptor antagonist in human monocytes’, Immunology, 1996, 89, (3), pp. 424–429 (doi: 10.1046/j.1365-2567.1996.d01-753.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Li, D.M. , Lu, W.L. , Wang, X.Q. , et al.: ‘Pharmacokinetics of indomethacin, a metabolite of acemetacin, following a single dose and multiple doses administered as acemetacin sustained‐release tablets in healthy male volunteers’, J. Health Sci., 2005, 51, (3), pp. 308–316 (doi: 10.1248/jhs.51.308) [DOI] [Google Scholar]
- 63. Vane, J. , Botting, R. : ‘Inflammation and the mechanism of action of anti‐inflammatory drugs’, FASEB J., 1987, 1, (2), pp. 89–96 [PubMed] [Google Scholar]
- 64. Antczak, A. , Montuschi, P. , Kharitonov, S. , Gorski, P. , Barnes, P.J. : ‘Increased exhaled cysteinyl‐leukotrienes and 8‐isoprostane in aspirin‐induced asthma’, Am. J. Respir. Crit. Care Med., 2002, 166, (3), pp. 301–306 (doi: 10.1164/rccm.2101021) [DOI] [PubMed] [Google Scholar]
- 65. Sanak, M. , Gielicz, A. , Bochenek, G. , Kaszuba, M. , Nizankowska‐Mogilnicka, E. , Szczeklik, A. : ‘Targeted eicosanoid lipidomics of exhaled breath condensate provide a distinct pattern in the aspirin‐intolerant asthma phenotype’, J. Allergy Clin. Immunol., 2011, 127, (5), pp. 1141–1147 e1142 (doi: 10.1016/j.jaci.2010.12.1108) [DOI] [PubMed] [Google Scholar]
- 66. Politi, A.Z. , Donovan, G.M. , Tawhai, M.H. , et al.: ‘A multiscale, spatially distributed model of asthmatic airway hyper‐responsiveness’, J. Theor. Biol., 2010, 266, (4), pp. 614–624 (doi: 10.1016/j.jtbi.2010.07.032) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mbikou, P. , Fajmut, A. , Brumen, M. , Roux, E. : ‘Theoretical and experimental investigation of calcium‐contraction coupling in airway smooth muscle’, Cell Biochem. Biophys., 2006, 46, (3), pp. 233–252 (doi: 10.1385/CBB:46:3:233) [DOI] [PubMed] [Google Scholar]
- 68. Mbikou, P. , Fajmut, A. , Brumen, M. , Roux, E. : ‘Contribution of rho kinase to the early phase of the calcium‐contraction coupling in airway smooth muscle’, Exp. Physiol., 2011, 96, (2), pp. 240–258 (doi: 10.1113/expphysiol.2010.054635) [DOI] [PubMed] [Google Scholar]