

Abstract
Biomolecular regulatory networks are organised around hubs, which can interact with a large number of targets. These targets compete with each other for access to their common hubs, but whether the effect of this competition would be significant in magnitude and in function is not clear. In this review, the authors discuss recent in vivo studies that analysed the system level retroactive effects induced by target competition in microRNA and mitogen‐activated protein kinase regulatory networks. The results of these studies suggest that downstream targets can regulate the overall state of their upstream regulators, and thus cannot be ignored in analysing biomolecular networks.
Inspec keywords: reviews, RNA, molecular biophysics, enzymes
Other keywords: target‐mediated reverse signalling, mitogen‐activated protein kinase regulatory networks, biomolecular regulatory networks, microRNA regulatory networks, review, in vivo study
1. Introduction
Large‐scale biomolecular regulatory networks are organised around highly connected nodes or hubs (Fig. 1a ) [3, 4]. In analysing such networks, we often rely on a key concept of systems biology (module), which is defined as a small‐scale interaction network with a function that is largely independent of its context [5]. On the basis of the modular decomposition of large networks, it may be possible to predict their dynamics and properties [6]. However, challenge lies in defining individual modules and distinguishing their core components from rest of the network. While conceptually possible, this task can be highly non‐trivial in real systems [7, 8]. To illustrate, let us consider target competition for access to hub of the network.
Fig. 1.
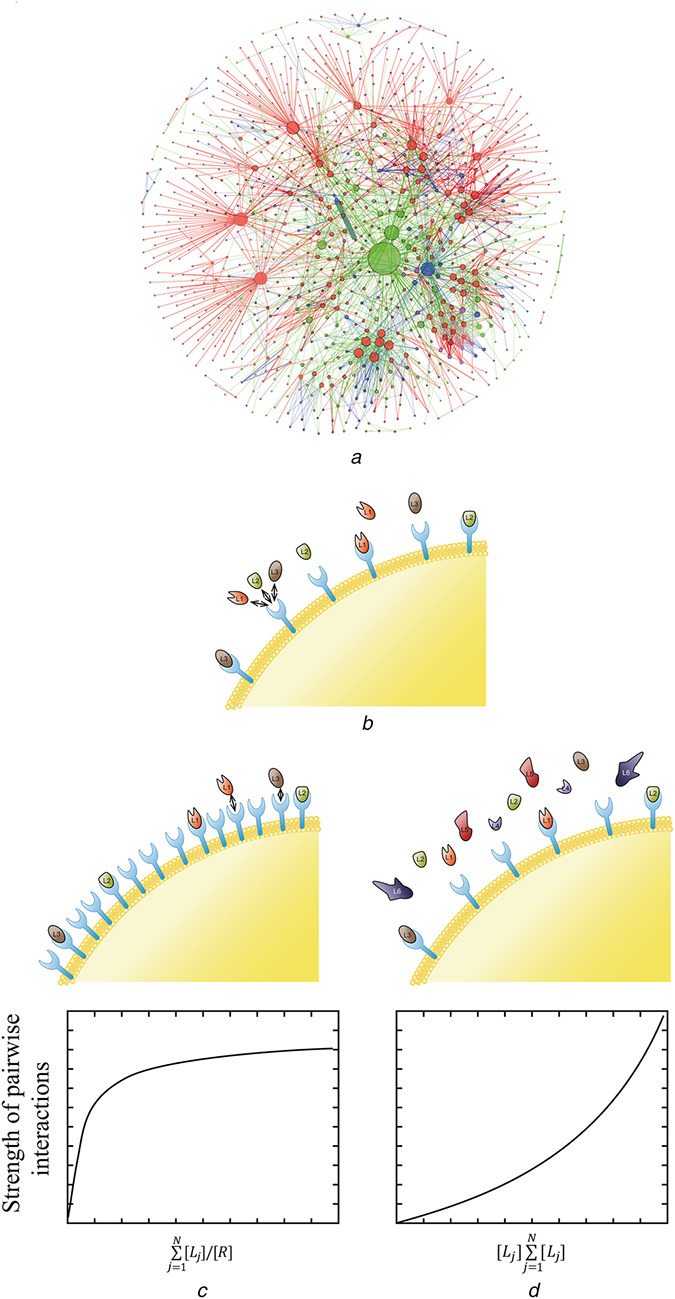
Model for competitive interactions
(a) Diagram of human RNA network. Red, blue, green, and magenta coloured nodes represent miRNA, circRNA, lncRNA, and mRNA, respectively. The size of nodes represents the number of interactions of each node. The thickness of the edge denotes the number of target sites between nodes [1, 2], (b) Simple model of a biomolecular network where one receptor (R) interacts with multiple ligands (Li for i = 1, 2, 3,…, 6), (c), (d) Model identifies two operating regime of the network where the strength of pairwise interaction is weak: when the receptor is in excess (c) and when individual ligand constitutes a small fraction of total ligand concentration (d)
Hubs, by definition, can interact with a large number of targets, creating the possibility of target–target competition. The network level consequences of this target competition can be demonstrated with a simple thermodynamic model, where one generalised receptor interacts with many ligands (Fig. 1b ). This model serves as a simplified description of more complex systems such as one enzyme processing many substrates, one transcription factor binding to multiple loci, or one microRNA (miRNA) downregulating multiple transcripts. According to the model, there are two operating regimes that can result in a situation where competitive effects are negligible, and system can be understood as a combination of independent modules. In the first regime, receptor is present in excess, such that there are enough receptors for all the ligands (Fig. 1c ). In the second regime, individual ligand constitutes only a small fraction of the total ligand pool, such that each ligand cannot influence the overall state of the system (Fig. 1d ). This regime most likely corresponds to the case where one transcription factor binds to multiple regulatory regions in the genome. In this system, addition or removal of a single regulatory region does not affect the ability of the transcription factor to control rest of its target genes.
However, many other operating regimes are also possible where removal of a single ligand can generate a strong perturbation in the overall state of the network. In this scenario, targets can potentially sequester and act as competitive inhibitors of the hub by competing with each other for access of the hub. In addition, targets can also compete with the regulators of the hub, and in this way control its abundance or post‐translational modification status. This example illustrates a more general effect called retroactivity, defined as the ability of a downstream target of a module to induce a change in the internal state of the module [8, 9, 10]. As a physicochemical consequence of biomolecular interactions, retroactive effects from target–target competition are always present. However, it has been proposed that in vivo retroactive effects should be minimised because they reduce modularity of a network, which is viewed as essential for network robustness [11]. Therefore, cells must have evolved mechanisms to insulate hubs from retroactive effects from their downstream targets, allowing modular decomposition of the network [7].
Contrary to the conventional school of thoughts, several recent in vivo studies demonstrated that retroactive effects from target competition can be strong in magnitude and biologically significant, implying that retroactive effects cannot be neglected. Moreover, retroactive effects are detectable regardless of the type of the network, i.e. RNA–RNA, RNA–protein, and protein–protein interaction networks all can exhibit strong retroactive effects. Here, we review these studies of retroactive effects from target competition using two exemplary regulatory networks: post‐transcriptional regulatory network of miRNA and enzymatic signal transduction network of mitogen‐activated protein kinase (MAPK).
2. Target‐dependent inhibition of miRNA activity
miRNAs belong to a family of small non‐coding RNAs that induce post‐transcriptional regulation of gene expression through translational repression and/or degradation of target transcripts (reviewed in [12, 13]). Although miRNAs are only ∼21–24 nucleotides long, they comprise one of the largest gene families in higher eukaryotes [12]. miRNAs are used in a broad range of processes such as cell differentiation, proliferation, metabolism, and apoptosis, and deregulation of miRNAs is often involved in developmental disorders and diseases such as cancer [14, 15, 16].
One interesting characteristic of miRNA‐mediated post‐transcriptional regulation is that miRNAs can interact with a large number of targets in a given cellular context (Fig. 2). In theory, different targets of a given miRNA will inevitably compete with each other for access to the miRNA. In this way, a certain transcript can influence the translation of other transcripts by controlling the effective activity of their common regulating miRNA. However, integrative genomics and bioinformatics studies have shown that on average, vertebrate miRNAs can control ∼50 different transcripts, and some miRNAs are predicted to have up to 800 targets [17, 18, 19]. Although theoretically possible, it is likely that individual targets cannot influence the overall activity of the miRNA, and thus cannot affect the translational status of other mRNA targets of the miRNA. Nonetheless, several studies provided counterexamples and showed that removal of even a single target can affect the internal state of the miRNA regulatory network.
Fig. 2.
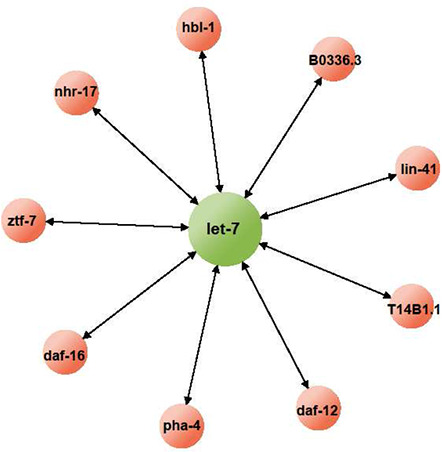
Targets of a miRNA
As the hub of a regulatory network, miRNA is expected to interact with a large number of targets. However, this idea is supported mainly by computational studies and lacks experimental support. Thus far, let‐7, one of the most extensively studied miRNA in C. elegans, is shown to regulate at least nine transcripts. However, it is not clear whether these transcripts are all present at the same time and in the same place. Considering that individual miRNAs can have pleiotropic functions in development and homoeostasis, it is important to identify the number of targets an miRNA can have at a given cellular context in understanding the regulation of the miRNA network.
Multiple in vivo studies demonstrated that phenotypic defects in an miRNA mutant can be rescued by reducing concentration of a single target of the miRNA [20, 21, 22, 23]. For example, in nematode Caenorhabditis elegans (C. elegans), removal of let‐7 miRNA resulted in developmental defects where mutant worms fail to transform into adults from the final larval stage. Computationally, let‐7 is predicted to target over 60 different transcripts [21]. However, the lethality of the let‐7 hypomorphic mutant can be rescued by repressing any one of the four confirmed targets of let‐7 (lin‐41, daf‐12, hbl‐1, and pha‐4) [22, 24]. If the phenotypic defects of let‐7 mutant are due to increased expression of its targets, then these defects should be rescued only if all of the targets are repressed, not when only one is repressed.
Similarly, a study on lin‐4 miRNA also came to a similar conclusion that lin‐4 miRNA induces its regulatory effect by mainly regulating translational status of only one target. According to TargetScan, lin‐4 is predicted to bind and regulate over 20 different transcripts [25]. However, the cell lineage defects observed in lin‐4 mutant embryos show striking similarities as those from increased expression of Lin‐14 protein [23, 26]. Indeed, it has been shown that lin‐4 can bind directly to the 3′ untranslated region (UTR) of lin‐14 mRNA and downregulate its translation [23], but this still cannot explain how Lin‐14 alone can account for lin‐4 mutant phenotypes.
One possible explanation that can account for these observations is that most of the target transcripts of a given miRNA are actually pseudo‐targets, and thus, their function is to regulate activity of the miRNA on its authentic target by acting as competitive inhibitors [21]. Repressing any one of these pseudo‐targets will enhance the effective activity of the miRNA, and this can rescue phenotype of miRNA mutants. Therefore, it has been postulated that the main function of miRNA is to regulate a few main transcripts rather than repressing dozens of targets simultaneously. According to this argument, an miRNA has only one or a few authentic targets and the rest of the targets only function to fine‐tune the miRNA's ability to regulate these authentic targets [21]. Of note, this argument raises an interesting possibility where mRNAs, especially abundant ones, may affect the translational status of other mRNAs in a protein‐coding independent manner by regulating the activity of their common binding miRNAs [21, 27].
These in vivo data using C. elegans clearly demonstrate strong competitive effects where different targets of a given miRNA can influence the ability of the miRNA to downregulate its targets. In the next section, we present evidence of retroactive effects from the target competition where competitive effects propagate upward to influence abundance of the miRNA.
3. Target‐mediated protection of miRNAs
Post‐transcriptional regulation by miRNAs is a dynamic process where miRNAs are constantly produced and degraded (Fig. 3a ). A recent study investigated the fate of an miRNA on regulating its target. The authors showed that miRNA lifetime is closely related to interaction with its targets [28]. According to their analysis, targets promote post‐transcriptional modification on the miRNA, resulting in its decay. Moreover, each miRNA on average can regulate at least two transcripts before turnover [28]. Using C. elegans, Chatterjee and Grosshans [29] showed the opposite effect where targets protect the miRNA from degradation, which is mediated by exoribonuclease 5'‐3' Exoribonuclease 2 (XRN‐2). They found that XRN‐2‐mediated degradation of miRNA can be blocked by introducing additional targets of the miRNA [29]. Since XRN‐2‐mediated degradation requires disassembly of miRNA‐argonaute (Ago) RNA‐induced silencing complex (RISC), the observed effect can be explained by target‐mediated sequestration of the miRNA to RISC, and in this way protecting the miRNA from XRN‐2. Indeed, the authors showed that let‐7(n2853) loss‐of‐function mutation destabilises let‐7 miRNA by impairing binding between let‐7 and its targets, and consequently eliminating the protective effect induced by the targets [30].
Fig. 3.
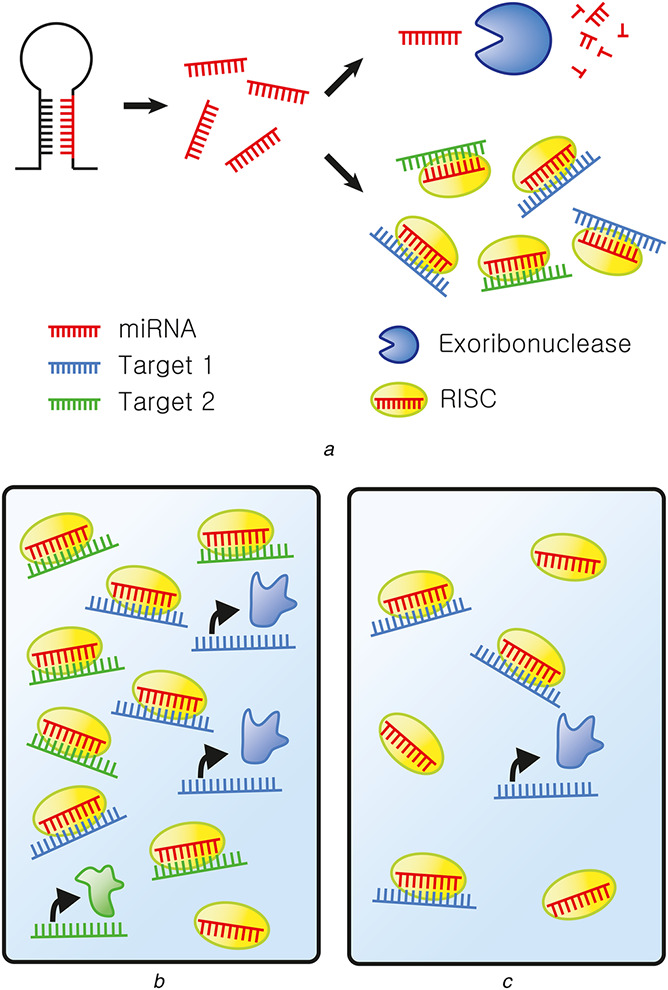
Target competition in the miRNA regulatory network
(a) Post‐transcriptional regulation by miRNAs. miRNAs (red) are generated from their precursor transcripts through successive cleavage by Drosha and Dicer. These single‐strand miRNAs can regulate translation of their multiple target transcripts (green and blue). This requires miRNAs to bind to Ago proteins to form RISC (yellow). When miRNAs are not bound to their targets, they dissociate from RISC and are degraded by exoribonuclease (blue), (b) When the expression of level of miRNA–target is high, effective activity of miRNA is low, allowing translation of proteins encoded by the target transcripts of the miRNA. Furthermore, targets also sequester miRNAs into RISC and protect them from degradation, resulting in higher level of miRNA, (c) However, when the target level is low, the abundance of miRNA is low, but its apparent activity in repressing translation of its target transcripts is high
In addition, the authors demonstrated that the abundance of targets can be important in determining the identity of miRNAs expressed in a given cellular context [30]. During miRNA biogenesis, miRNA precursor transcripts are cleaved subsequently by Drosha and Dicer RNase III type enzymes [13]. This results in an miRNA:miRNA* duplex, but only one of the strands (miRNA) is preferentially loaded to RISC, whereas the other strand (miRNA*) undergoes degradation. According to the current models, strand selection of the miRNA duplex depends on the relative thermodynamic stability of each strand, such that the strand with less stable 5′ end is preferentially loaded to RISC [31, 32]. However, when the stabilities of the two strands are similar, both strands can potentially be loaded onto RISC and regulate their targets. Since the turnover rate of a given miRNA depends on the abundance of its targets, Chatterjee et al. [30] proposed that target abundance may play a role in determining which of the two strands of the miRNA duplex will be chosen. According to their model, the strand with greater number of targets is likely to be present in higher levels because it is protected from degradation through its target interaction. The authors demonstrated this idea by expressing a target of miR‐241* strand in C. elegans, which resulted in accumulation of miR‐241* [30].
Consistent with this notion, a number of miRNAs in both nematodes and Drosophila undergoes ‘arm switching’, meaning miRNA to miRNA* transition [30, 33, 34]. For example, Okamura et al. [34] demonstrated that miRNA* species can be loaded onto RISC and repress translation of their target transcripts. Moreover, this regulatory activity of miRNA* strand provides the driving force for arm switching. Although the mechanism of this arm switching is not clear, it may involve differences in the target pool from one cellular context to another.
To summarise, these studies suggest the two main system level effects of miRNA's ability to interact with a large number of targets: (i) different targets compete with each other and negatively regulate miRNA activity; (ii) targets sequester miRNA to RISC and prevent its interaction with exoribonuclease, which results in increased abundance of the miRNA by protecting it from degradation (Figs. 3b and c ).
4. miRNA sponges and competing endogenous RNA
Target‐mediated control of miRNA activity is also demonstrated in mammalian cell culture systems. Ebert et al. [35] showed that when an artificial target was expressed at sufficiently high levels, it competed with the endogenous targets of the miRNA, resulting in inhibition of the miRNA's ability to repress its endogenous targets. These miRNA decoy targets are named miRNA sponges (also known as target mimicry in plants [36] and competing endogenous RNA (ceRNA) in mammals [37]) and can be used to induce loss‐of‐function phenotype of the target miRNA [35].
The presence of strong competitive effects in culture cells is demonstrated by Arvey et al. [38] who showed that the effectiveness of a given miRNA in downregulating each individual targets depends on the total concentration of its target pool. In other words, an miRNA with lower target abundance is more effective in repressing translation of a single target transcript compared with one with higher target abundance.
A recent work by Poliseno et al. [27] further showed that miRNA activity can be influenced by removal of a single target. They found that the levels of Phosphatase and tensin homolog (PTEN) mRNA and protein are reduced upon removal of Phosphatase and tensin homolog pseudogene 1 (PTENP1), a pseudogene of PTEN. Pseudogenes cannot code for functional proteins, but they share the 5′ and 3′ UTR sequences with their protein‐coding relatives, and thus are regulated by the same group of miRNAs [39]. The authors showed that the negative effect of PTENP1 on PTEN required Dicer, an important regulator in the miRNA biogenesis, and similar effect can be induced by expressing just the 3′ UTR of PTENP1 [27]. These results suggest that the effect of PTENP1 on PTEN is mediated by miRNAs, providing a possible biological function of a pseudogene as a regulator of miRNA activity to control the expression of its protein‐coding relatives [27]. The authors further argued that regulation of miRNA activity by acting as competitive inhibitors is a general biological function of pseudogenes because V‐Ki‐ras2 Kirsten rat sarcoma viral oncogene homolog (KRAS) and its pseudogene KRAS1P also showed similar correlation in their abundance as PTEN and PTENP1 did. In addition, miRNA binding sites are well conserved in multiple gene–pseudogene pairs, which make pseudogenes perfect decoys of miRNAs that target the corresponding protein‐coding genes [27].
A powerful example of a ceRNA in the miRNA regulatory network is provided by circular RNAs (circRNAs). CircRNAs are a large class of non‐coding RNAs that are formed by non‐canonical splicing where a splicing donor site is joined to an upstream acceptor site, resulting in an RNA with circular structure [40, 41]. Although circRNAs were first identified over 20 years ago, biological function of this new class of non‐coding RNA was obscure. Recently, it was shown that Cerebellar degeneration related protein 1 (CDR1) antisense (CDR1as) circRNA contains over 70 binding sites for miR‐7, enabling it to act as an effective ceRNA to competitively inhibit miR‐7 activity [42, 43]. The circular structure of CDR1as further enhanced its effectiveness as a ceRNA because it lacks the extremities, and thus is resistant to exonucleolytic RNA decay [42].
In addition to CDR1as, another circRNA called Sry has been identified and has been shown to contain 16 putative binding sites for miR‐138. Further investigation revealed that Sry can act as a sponge for miR‐138, generalising the biological function of circRNA as ceRNA for miRNAs [42, 43]. More recently, Liu et al. [44] constructed and analysed a biochemical network of circRNAs–miRNAs–mRNAs. Using bioinformatics approach to predict possible inter‐connections between circRNAs and mRNAs, the authors found that circRNA–CER and mRNA–matrix metalloproteinase 13 (MMP13) both contain miR‐136 binding sites. Consistent with the target competition model, suppressing expression of CER resulted in decreased MMP13 level [44], which is indicative of increased miR‐136 activity. Hence, this demonstrates that a circRNA (CER) is able to regulate not only an miRNA (miR‐136), but also an mRNA (MMP13) expression by functioning as a decoy for their common interacting miRNA.
Subsequent studies reported other types of circRNAs and non‐coding RNAs that can act as ceRNAs [44, 45, 46]. Combined, these studies demonstrate the generality of target‐mediated control of miRNA activity and suggest that one possible biological function of non‐coding RNAs is to regulate the activity of their binding miRNAs.
5. Computational approaches for system level analysis of the miRNA network
Until recently, experimental studies on the regulatory function of ceRNAs have been concentrated on identification and validation of binding interaction between a ceRNA with its corresponding miRNA. Accordingly, initial computational interest in ceRNA–miRNA regulation has been focused on genome‐wide search for ceRNAs and matching them with regulating miRNAs [47]. However, accumulating evidence reveal complexities in ceRNA–miRNA networks where ceRNAs can also act as hubs for multiple miRNAs, leading to miRNA–miRNA crosstalk mediated by their common binding ceRNA. For instance, Hansen et al. [42, 48] found that CDR1as contains nearly perfect binding site for miR‐671, which can induce endonucleolytic cleavage of CDR1as. In other words, by regulating the abundance of CDR1as, miR‐671 can indirectly control the activity of miR‐7, which is inhibited by CDR1as. In addition, despite the effort to generalise the biological function of circRNAs as ceRNAs, accumulating evidences indicate that most circRNAs are not expressed at high enough levels to be effective in sequestering miRNAs [47, 49].
As the biological significance and complexity of ceRNA–miRNA interaction grew, computational studies began to construct and analyse mathematical models for miRNA–target interaction networks. For example, Ala et al. [50] performed mass‐action kinetic modelling analysis to simulate a simple miRNA network with only two mRNAs and one miRNA by employing dynamic properties of each element and measuring catalytic activity of the miRNA. They demonstrated that simple titration mechanism exhibits ultra‐sensitive responses to small changes in the miRNA or mRNA concentration near the threshold concentration, which could be determined once quantitative information on the kinetic parameters become available [50].
Recently, a number of studies used computational approach to analyse miRNA networks in the context of various cancer models such as colorectal cancer [51], ovarian cancer [52], and breast cancer (BRCA) [53]. However, most of the current effort still utilises qualitative modelling based on expression correlations [50, 53, 54] and relies on target predictions from several web‐based software such as TargetScan, miRanda, and TargetProfiler [25, 55, 56]. For example, Paci et al. [53] examined expression correlations between 10,492 mRNAs, 311 miRNAs, and 833 long non‐coding RNAs (lncRNAs) from BRCA profiled The Cancer Genome Atlas (TCGA) database. By applying multivariate analysis, the authors identified mRNA/lncRNA pairs where lncRNA may behave as miRNA sponge, which they denote as ‘pure sponge modules’ [53]. In this example, the actual expression levels of miRNAs and their targets are poorly considered. This is largely contributed by the lack of quantitative information on the biochemical reactions of the miRNA network. In modelling a regulatory network, in addition to the qualitative factors such as seed match or expression correlation, quantitative variables such as binding affinities, concentrations, and degrees of catalytic reaction can exert significant effect on the model outcome.
Despite these limitations, a number of computational studies formulated mathematical models to analyse the system level effects of ceRNAs on the internal state of the miRNA regulatory networks. Using kinetics modelling consisting of ordinary differential equations, Figliuzzi et al. [57, 58] analysed a network composed of one miRNA regulating N target transcripts. First, they analysed the network at steady state and found that cross‐regulation between ceRNAs via their common regulating miRNA can occur, but is rare [58]. For ceRNAs to have strong retroactive effects, miRNA–ceRNA complex must decay partially, in other words, miRNAs cannot induce target cleavage catalytically [58]. However, when the authors analysed transient response to small perturbations in the system component, they found that ceRNA crosstalk can occur even in the absence of miRNA decay [57]. In addition, large perturbations can lead to non‐linear response with strong crosstalk between ceRNAs [57].
These computational effort led to the development of new experimental approaches that allowed quantitative assessment of miRNA networks [59, 60]. Denzler et al. [59] introduced artificial miR‐122 targets with one, three, or mutated miR‐122 binding sites. Using these constructs, they quantitatively analysed the stoichiometric relationship between miRNA and its targets. However, they could not detect any ceRNA effects when miR‐122 target was overexpressed. This is most likely because the level of miR‐122 targets was too low to induce competitive effect [59]. Yuan et al. [60] used similar approach to quantitate miRNA and target interaction by introducing artificial targets of miR‐21 to cells. In their paper, retroactive effects were clearly detected when medium and high levels of artificial miR‐21 target were introduced [60]. These quantitative measurements call for re‐examination for the conditions necessary to observe retroactive effects, which provide the driving force for the formulation of more realistic models. This cooperation between modelling and experimental effort will assist us in understanding and predicting cellular phenotypes modulated by perturbation in miRNA networks.
6. Competition during the biogenesis of miRNAs
Although studies discussed above only consider competition at the level of miRNA–target interaction, competition is rather a common feature in the miRNA biogenesis pathway. Throughout the multiple steps of the miRNA biogenesis, different transcripts can compete with miRNA precursor transcripts and inhibit the generation of mature miRNAs.
Several studies have shown that transfection of small hairpin RNAs (shRNAs) can lead to upregulation, rather than downregulation, of a large number of genes [61, 62, 63]. Since the structure of shRNAs closely resembles that of precursor‐miRNAs (pre‐miRNAs), this off target effect can be readily explained by competition between shRNAs and pre‐miRNAs for machineries of miRNA biogenesis. Consequently, this competition will lead to decreased level of endogenous miRNAs, which in turn results in upregulation of genes that are repressed by these miRNAs. One likely bottleneck of miRNA biogenesis is exportin, which transports pre‐miRNAs from nucleus to cytoplasm where they are further processed by Dicer. Indeed, it has been shown that both pre‐miRNAs and shRNAs can bind to Exportin‐5 and co‐transfection of shRNAs with Exportin‐5 results in increased level of mature shRNAs, suggesting that Exportin‐5 is likely to be saturated in cells [64, 65]. Notably, a recent study showed that Exportin‐5 is dispensable during miRNA biogenesis and can be replaced by other factors [66]. This raises an interesting question, whether multiple exportins can compete with each other for access to pre‐miRNAs. The mechanism of pre‐miRNA export to cytoplasm needs further investigation in the future.
If exportin is the only limiting component among miRNA biogenesis machineries, then transfection of small‐interfering RNAs (siRNAs) will provide more effective tool for gene knockdown because processing of these RNAs do not require nuclear export. However, transfecting cells with siRNAs also leads to similar upregulation of genes as siRNAs still compete with endogenous miRNAs for RISC components [67]. In support of this, it has been shown that a major bottleneck in miRNA‐mediated gene regulation occurs at the level of Ago loading during RISC assembly [68]. Thus, exogenous RNAs can compete with endogenous miRNA precursors and miRNAs themselves in multiple layers of the miRNA biogenesis pathway. In contrast, it has been shown that an exogenous miRNA does not compete with other miRNAs or shRNAs [63]. However, this result is likely to be misleading since the authors used weak polymerase‐II promoter to express the miRNA, i.e. the expression level is too low to saturate the miRNA machineries. Indeed, it has been shown that exogenous miRNAs driven by stronger polymerase‐III promoter competed with endogenous miRNAs [62]. Therefore, to better understand biogenesis of miRNAs and to design more effective small RNAs for RNA interference, it will be necessary to monitor saturation of each of the miRNA biogenesis factors.
Another evidence of competition among miRNA biogenesis factors is presented by knockout studies. Kim et al. [66] compared the effects of miRNA biogenesis factor knockout on the expression of mature miRNAs. While Drosha knockout dramatically reduced the expression of almost all miRNA species, a number of 5′ miRNAs is still generated in the absence of Dicer [66]. The authors postulated that these miRNAs might be generated by Ago2 which can directly bind to pre‐miRNAs and can potentially generate miRNAs in a Dicer‐independent manner [69, 70]. These studies suggest that Dicer and Ago proteins may compete for access to pre‐miRNAs and call for future examination for possible consequences of this competition. More importantly, they indicate that competitive effects are not restricted between two RNA species, but can also occur between proteins for access to their common binding RNAs.
Thus far, we have discussed studies that indicate in vivo evidence of retroactive effects in miRNA regulatory networks. These studies clearly demonstrate that competition among different target transcripts of a given miRNA can influence activity of the miRNA. Furthermore, these competitive effects can propagate upstream to affect abundance of the miRNA. In addition, competition is not restricted to miRNA–targets, but is present in almost every step of miRNA biogenesis pathway. In the following sections, we will use MAPK signalling pathway as an example to illustrate that retroactive effects are also present and biologically significant in protein–protein interaction networks.
7. Competitive interaction in MAPK signalling pathway
Another example of a biomolecular regulatory network where different components compete for access to hubs is an enzymatic network formed by MAPK and its multiple substrates. MAPK signalling pathway is a three‐tiered cascade of phosphorylation–dephosphorylation cycles (reviewed in [71, 72]). An input to the pathway can be provided by a cell surface receptor; its output is the activation level of MAPK, a kinase at the bottom of the cascade. MAPK is activated when it is phosphorylated by an MAPK kinase; this process is reversed by an MAPK phosphatase. Active MAPK controls cellular processes by phosphorylating its multiple substrates.
Previous structural studies of MAPK revealed that MAPK contains two substrate interaction sites; the D recruitment site (DRS) and the F recruitment site [73, 74]. Biochemical studies suggested that regulators of MAPK as well as most of MAPK substrates interact with the DRS [75]. Thus, similar to miRNA–target transcripts, MAPK substrates can compete with each other, as well as with the regulators of MAPK phosphorylation for binding to MAPK (Fig. 4a ). The possibility of substrate competition was further solidified by a recent experiment by Futran and colleagues who attempted to directly determine MAPK and substrate interaction site. Using a photoactivatable non‐canonical amino acid, p‐azidophenylalanine (AzF), the authors photo‐cross‐linked Capicua (Cic; a substrate of MAPK) bound to MAPK and identified MAPK–Cic interaction residues using mass spectrometry [76]. They successfully showed that this interaction site is mapped to the DRS, providing the molecular details for MAPK–substrate interaction [76].
Fig. 4.
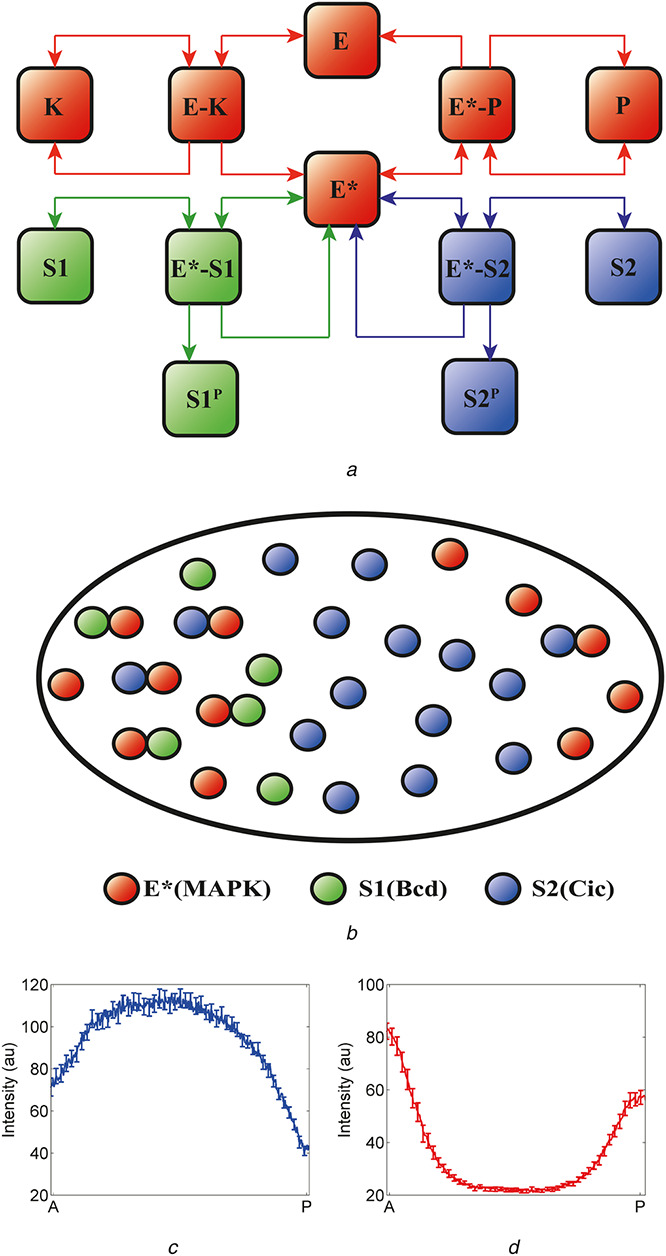
Substrate competition in the terminal patterning of the Drosophila embryo
(a) Substrate competition model: an enzyme (E) is activated when phosphorylated by a kinase (K). Activated enzyme (E*) then phosphorylates two substrates (S1 and S2) and is deactivated when dephosphorylated by a phosphatase (P). The two substrates compete both with each other and with the phosphatase for binding to the activated enzyme, (b) Schematic representation of the proposed substrate competition in the early Drosophila embryo. An enzyme is activated at the poles of the embryo (E*, red). At the anterior pole, there are both localised and uniform substrates, S1 (localised, green) and S2 (uniform, blue); at the posterior pole, there is only S2. In the context of the embryo, E* is phosphorylated MAPK, S1 models the combined effects of uniformly distributed substrates (such as Cic) and S2 models anteriorly localised substrates (such as Bcd), (c), (d) Non‐uniform spatial distribution of substrates leads to asymmetric pattern of phosphorylated MAPK and nuclear Cic. At the anterior pole, higher level of substrates provides effective protection of phosphorylated MAPK from the phosphatase, resulting in higher level of phosphorylated MAPK (d). At the same time, the level of Cic is also higher at the anterior pole due to the anteriorly localised substrates which act as competitive inhibitors of MAPK‐mediated downregulation of Cic (c)
Although theoretically possible, MAPK signalling system was believed to be modular and retroactive effects from substrate competition would be minimal. Recently, however, studies demonstrated the presence and significance of MAPK substrate competition using the terminal patterning system of the Drosophila embryo. First, we will present evidence indicating substrate‐mediated control of MAPK activity.
8. Substrates dependent inhibition of MAPK enzymatic activity
In early fly embryo, locally activated MAPK specifies the future terminal structures of the larva by phosphorylating uniformly expressed transcriptional repressors such as Cic and Groucho (Gro) [77, 78, 79]. In addition to Cic and Gro, MAPK also phosphorylates two anteriorly localised transcription factors Bicoid (Bcd) and Hunchback (Hb) [80, 81]. This results in a non‐uniform spatial distribution of MAPK substrates where MAPK can interact with two different populations of substrates in two distinct locations within a single embryo (Fig. 4b ).
Studies on miRNA networks suggest that targets can act as competitive inhibitors of miRNA activity. Similar negative effect is induced by the substrates of MAPK in the early embryo. Kim and colleagues showed that MAPK signalling activity is weaker in the anterior region of the embryo due to higher levels of MAPK substrates in this region. They found that the spatial pattern of Cic exhibited striking asymmetry at the poles, with the level of Cic significantly higher at the anterior pole (Fig. 4c ) [81]. Since cic mRNA is expressed uniformly throughout the embryo and Cic is downregulated in response to its phosphorylation by MAPK [82], this asymmetry in Cic protein expression pattern can be explained by weaker enzymatic activity of MAPK in the anterior region, where the total concentration of MAPK substrate is high. Indeed, it has been shown that MAPK enzymatic activity, measured by Cic downregulation, can be enhanced by removal of other MAPK substrates such as Bcd and Hb [83].
Substrate‐dependent inhibition of MAPK activity is also demonstrated by E26 transformation‐specific family proteins Pointed (Pnt) and Yan. Throughout the development of fly oocyte and embryo, MAPK regulates gene expression by phosphorylating Pnt, a transcriptional activator, and Yan, a transcriptional repressor [84]. MAPK‐mediated phosphorylation of Pnt results in enhancement of its transcriptional activity while phosphorylated Yan undergoes degradation [85, 86]. Therefore, it was believed that Pnt and Yan show mutually exclusive expression patterns in regions where MAPK signalling is active. However, closer examination of Pnt and Yan expression revealed that in numerous tissues, two proteins are co‐expressed despite the presence of active MAPK [87]. This co‐expression could be understood using the competition model where Pnt competitively inhibits MAPK‐mediated phosphorylation of Yan [87]. Of note, MAPK substrates discussed in these studies are all transcription factors that regulate gene expression throughout embryonic development. Hence, these studies demonstrate that the context‐dependent network topology may affect the transcriptional response in developing tissues.
The competitive effects by the MAPK substrates are not only restricted to transcription factors. Using Drosophila imaginal discs, Kim et al. [88] showed that p90 ribosomal S6 kinase (RSK), a downstream effector kinase of MAPK, can negatively regulate MAPK signalling activity. Although the authors did not attribute their observations as competitive effects induced by RSK, their results are strikingly similar to those induced by the MAPK substrates in the terminal patterning of the early embryo. In both cases, activity of downstream substrates was dispensable for their inhibitory effect on MAPK enzymatic activity [81, 83, 88]. Therefore, negative regulation of MAPK signalling by RSK in wing and eye discs of Drosophila can be attributed as another in vivo example of competitive effects.
Similar to the target transcripts of miRNAs discussed earlier, multiple substrates of MAPK can affect the activity of MAPK studies discussed below show that this competitive effect can propagate upward to control the abundance of active MAPK by competing with MAPK phosphatase.
9. Substrate‐dependent protection of phosphorylated MAPK
Activation/phosphorylation status of MAPK is regulated by the phosphorylation–dephosphorylation cycle. In the nematode miRNA network, target transcripts can positively regulate the abundance of miRNA by protecting it from degradation by exoribonuclease [30]. Similar positive retroactive effect mediated by the substrates of MAPK is also observed in the terminal patterning system of the Drosophila embryo.
Kim et al. [89] demonstrated that individual MAPK substrates can control the phosphorylation status of MAPK in vivo. The authors showed that removal of any one of the four known nuclear substrates of MAPK led to a significant decrease in the level of phosphorylated MAPK. Furthermore, removal of multiple substrates has a cumulative effect, as removal of two substrates together led to a stronger reduction in the level of phosphorylated MAPK compared with the effect induced by removing them individually [89]. Conversely, overexpression of the substrates resulted in increased phosphorylation of MAPK [89]. More importantly, when Yan, an MAPK substrate that is not present in this developmental context, is introduced, it had similar protective effect on MAPK phosphorylation [89]. Thus, phosphorylation status of MAPK is positively regulated by the substrates of MAPK (Fig. 4d ).
As a step toward understanding the mechanism behind this substrate‐dependent control of MAPK phosphorylation, the authors expressed different Cic variants to determine conditions necessary for Cic to positively regulate phosphorylated MAPK. They found that endogenous function of Cic as a transcriptional repressor is dispensable, while nuclear localisation of Cic and its physical association with MAPK are important for its regulation on MAPK phosphorylation [89]. Using a combination of mass‐action modelling and quantitative imaging in multiple mutant embryos, authors showed that MAPK substrates sequester phosphorylated MAPK to the nucleus and protect it from the action of cytoplasmic phosphatase [89]. Note that this sequestration of phosphorylated MAPK is similar to transcript‐mediated sequestration of miRNAs to miRNA–RISC complex and subsequent protection of miRNAs from exoribonuclease.
Moving beyond Drosophila, substrate‐mediated control of MAPK subcellular localisation is also demonstrated in budding yeasts. Blackwell et al. [90] showed that the nuclear accumulation of Fus3 MAPK is reduced in the absence of Dig1 or Dig2, two nuclear substrates of Fus3. Similarly, overexpression of Ste12, another nuclear substrate of Fus3, increased the nuclear accumulation of Fus3 [90]. Although this paper did not show whether substrates can regulate the phosphorylation of Fus3, it clearly demonstrated that substrates of MAPK are acting as nuclear tethers and controlling the subcellular localisation of MAPK. Finally, in an effort to identify MAPK substrates in C. elegans germ‐line development, Arur et al. [91] found that 10 of 30 predicted targets of MAPK can alter the spatial pattern of MAPK phosphorylation. Removal of any one of these ten substrates resulted in altered pattern of phosphorylated MAPK [91]. Although the molecular mechanism is yet to be determined, this work also supports that removal of MAPK substrate can influence phosphorylation status of MAPK.
Overall, these studies clearly demonstrate that MAPK substrates can control the phosphorylation status of MAPK and its subcellular localisation. In the following section, we discuss recent studies that used computational and experimental approaches to elucidate the significance of the competitive effects.
10. Biological significance of MAPK–substrate competition
Studies on target competition in miRNA networks provide a valuable insight regarding a possible function of MAPK–substrate competition. Although numerous biochemical experiments suggest that Bcd and Hb are direct substrates of MAPK in early fly embryo [80, 81], the biological significance of their phosphorylation by MAPK is unclear. In fact, it has been shown that phosphorylation of Bcd by MAPK is dispensable for Bcd's ability to pattern the anterior region of the embryo [80]. On the basis of the studies of competitive effect in the miRNA network, we can speculate that phosphorylation of Bcd and Hb is just a by‐product of the spatial regulation of MAPK signalling activity by these two substrates. Similar to the miRNA network where multiple targets compete to regulate miRNA's ability to act on its authentic target [21], phosphorylation of Bcd and Hb might function to regulate MAPK signalling activity in determining the spatial pattern of transcriptional repressors that are downregulated by MAPK such as Cic and Gro.
Studies on the MAPK–substrate competition in termini of early Drosophila embryo provide new insights on interpretation of MAPK signalling. As discussed in the previous section, Kim and colleagues demonstrated that MAPK substrates all induce positive effects on the phosphorylation level of MAPK. Consequently, this protective effect resulted in higher level of phosphorylated MAPK in the anterior region because there are more substrates of MAPK present in this region. At the same time, these substrates also act as inhibitors of MAPK activity, resulting in weaker apparent MAPK activity on a given substrate in the anterior region of the embryo [81, 89] (Figs. 4c and d ). Therefore, these results suggest that MAPK phosphorylation level may not reflect its activity, and one needs to be cautious in interpreting MAPK phosphorylation data.
Ahmed and co‐workers used single‐cell approach to quantitatively assess subcellular localisation, phosphorylation status, and enzymatic activity of MAPK in fibroblasts. They found that there is a time lag between activation of MAPK and its apparent enzymatic activity. Using a combination of biochemical experiments and kinetic modelling of systems of ordinary differential equations, the authors showed that this discrepancy could be resolved by considering competitive effects induced by the substrates [92]. According to their model, the initial interaction between MAPK with its substrates depletes the pool of free active MAPK, and at the same time protects phosphorylated MAPK from phosphatase. Consequently, despite the high level of phosphorylated MAPK, its apparent activity is weak [92]. Consistent with this, studies using synthetic circuit analyses also showed that the competitive binding between upstream regulator (phosphatase) and downstream target (substrate) for a common hub (MAPK) can result in a significant delay in system response time [93, 94].
In addition to delaying response time of the system, substrate competition can generate complex system behaviour such as oscillation. Motivated by the fact that substrates can induce retroactive effects on MAPK signalling status and activity, Liu and colleagues proposed a kinetics model with two modules: dual phosphorylation and substrate interaction [95]. Previous studies have shown that MAPK signalling can exhibit oscillatory behaviour due to the negative feedback regulation by Sprouty [96, 97, 98]. Liu's model suggested an alternative mechanism where retroactive effects from the substrate interaction module can influence the dual phosphorylation module, generating periodic oscillations in MAPK phosphorylation [99]. In contrast to previous models where activated MAPK turns on a negative regulator of its upstream kinase, the current model suggests oscillatory behaviour can be achieved with constant MAPK activity [98]. Moreover, Rowland et al. proposed a kinetics model with ordinary differential equations and analysed effects induced by substrates [99]. Their analysis showed that individual substrates can influence the overall state of the system. Interestingly, they proposed that complex crosstalk among different kinase signalling pathways are possible as they often share a common phosphatase [99]. These mathematical studies demonstrated that substrate competition can be responsible for complex system behaviour and cannot be ignored in understanding cellular signalling pathways [95, 99, 100]. To fully understand the regulatory effects of the substrate competition, these model predictions should be validated through experiments in the future.
A remaining question is whether an animal such as Drosophila can utilise the retroactive effects in MAPK signalling pathway to govern biological processes. A work by Kim et al. [83] showed that this is indeed the case; competitive effects are important in positioning gene expression boundaries in developing embryos. In early Drosophila embryo, multi‐dimensional gene expression patterns can arise through crosstalk among multiple signalling cues. Using the two‐dimensional pattern of zerknűllt (zen) as an experimental system, the authors demonstrate how information from three maternal patterning systems (Bcd, Dorsal, and MAPK) are integrated through an enzymatic network composed MAPK and its substrates [83]. Specifically, Bcd acts as an anteriorly localised MAPK substrates to antagonise the signalling activity of MAPK in this region by competing with other MAPK substrates [81, 83]. This interaction results in repression of zen at the anterior pole, while it is expressed at the posterior pole where Bcd is not present. The authors supported this mechanism using a mass‐action kinetics model analysed at steady state and validated the model predictions using a number of mutant fly embryos [83].
In summary, these works show multiple roles of downstream substrates of MAPK in regulating properties of their upstream regulator. The substrates compete with each other and act as inhibitors of MAPK enzymatic activity. The substrates also can positively regulate MAPK phosphorylation by protecting phosphorylated MAPK from the action of phosphatase. Furthermore, localisation of the substrates can affect the subcellular localisation of phosphorylated MAPK in response to external stimuli. Finally, these effects can propagate through MAPK patterning network and affect expression of genes that are regulated by MAPK signalling system.
11. Conclusions and outlook
In this review, we present recent studies on the target competition in biomolecular networks where one regulator interacts with a large number of targets. Although a number of studies raise concerns regarding the competition model [47, 101, 102], multiple in vivo experimental evidences clearly demonstrate that competitive effects are both strong in magnitude and biologically significant.
The competitive effects in miRNA and MAPK networks are strikingly similar (Fig. 5). In both cases, targets can negatively control the activity of their regulator, and at the same time increase the abundance of the regulator by protecting it from either degradation (for miRNA) or dephosphorylation (for MAPK). Interestingly, individual targets can regulate the activity and abundance of its upstream regulator in both networks despite the large number of predicted targets. Finally, these competitive effects are functionally significant in tuning the activity of the regulator toward its authentic target.
Fig. 5.
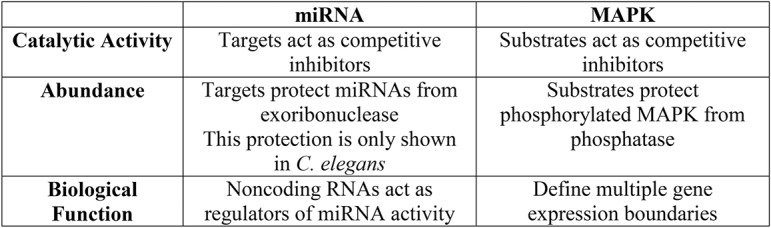
Summary of competitive effects in miRNA and MAPK regulatory networks
Results from these studies raise important questions when understanding results of overexpression experiments. For example, as discussed earlier, transfecting cells with shRNAs or siRNAs induce competition over miRNA processing machineries, which result in upregulation of genes that are controlled by endogenous miRNAs. This situation where a reporter protein or exogenous molecules skew the state of the system and induce competition with endogenous system components is rather a common phenomenon. It has been shown that exogenous expression of Rho GTPases compete with endogenous Rho proteins for binding to Rho guanine nucleotide dissociation inhibitors (RhoGDIs). Since RhoGDIs are present in limited amount and RhoGDI controls the homoeostasis of Rho proteins, this work raises concerns on previous interpretation of overexpression experiments [103]. In addition, in Xenopus egg extract, Kopito and Elbaum [104] observed that introducing one important cargo resulted in nuclear export of another, suggesting that capacity of nuclear accumulation is limited and different cargoes compete with each other for importin.
Given the generality of competitive effects in numerous signalling systems, we expect that target competition is an important regulatory strategy in biomolecular networks and quantitative and computational studies will provide valuable insights toward system level understanding of these networks.
12. Acknowledgments
The authors Yongjin Jang and Min A Kim both contributed equally to this work and are both first co‐authors. This work was supported by KAIST research allowance (G04160001) and the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Korean government Ministry of Science, AQ3ICT & Future Planning (2016R1C1B2009886).
13 References
- 1. Li, J.H. , Liu, S. , Zhou, H. , et al.: ‘Starbase V2.0: decoding MiRNA‐CeRNA, MiRNA‐NcRNA and Protein‐RNA interaction networks from large‐scale Clip‐Seq data’, Nucl. Acids Res., 2014, 42, (Database issue), pp. D92–D97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yang, J.H. , Li, J.H. , Shao, P. , et al.: ‘Starbase: a database for exploring MicroRNA‐MRNA interaction maps from argonaute Clip‐Seq and Degradome‐Seq data’, Nucl. Acids Res., 2011, 39, (Database issue), pp. D202–D209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Albert, R. : ‘Scale‐free networks in cell biology’, J. Cell Sci., 2005, 118, (Pt 21), pp. 4947–4957 [DOI] [PubMed] [Google Scholar]
- 4. Zhu, X. , Gerstein, M. , Snyder, M. : ‘Getting connected: analysis and principles of biological networks’, Genes Dev., 2007, 21, (9), pp. 1010–1024 [DOI] [PubMed] [Google Scholar]
- 5. Hartwell, L.H. , Hopfield, J.J. , Leibler, S. , et al.: ‘From molecular to modular cell biology’, Nature, 1999, 402, (6761 Suppl), pp. C47–C52 [DOI] [PubMed] [Google Scholar]
- 6. Lauffenburger, D.A. : ‘Cell signaling pathways as control modules: complexity for simplicity?’, Proc. Natl. Acad. Sci. USA, 2000, 97, (10), pp. 5031–5033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Del Vecchio, D. , Ninfa, A.J. , Sontag, E.D. : ‘Modular cell biology: retroactivity and insulation’, Mol. Syst. Biol., 2008, 4, p. 161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Saez‐Rodriguez, J. , Gayer, S. , Ginkel, M. , et al.: ‘Automatic decomposition of kinetic models of signaling networks minimizing the retroactivity among modules’, Bioinformatics, 2008, 24, (16), pp. i213–i219 [DOI] [PubMed] [Google Scholar]
- 9. Sauro, H.M. : ‘Modularity defined’, Mol. Syst. Biol., 2008, 4, p. 166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang, L. , Sontag, E.D. : ‘On the number of steady states in a multiple futile cycle’, J. Math. Biol., 2008, 57, (1), pp. 29–52 [DOI] [PubMed] [Google Scholar]
- 11. Wagner, G.P. , Pavlicev, M. , Cheverud, J.M. : ‘The road to modularity’, Nat. Rev. Genet., 2007, 8, (12), pp. 921–931 [DOI] [PubMed] [Google Scholar]
- 12. Bartel, D.P. : ‘MicroRNAs: genomics, biogenesis, mechanism, and function’, Cell, 2004, 116, (2), pp. 281–297 [DOI] [PubMed] [Google Scholar]
- 13. Ha, M. , Kim, V.N. : ‘Regulation of microRNA biogenesis’, Nat. Rev. Mol. Cell Biol., 2014, 15, (8), pp. 509–524 [DOI] [PubMed] [Google Scholar]
- 14. Gammell, P. : ‘MicroRNAs: recently discovered key regulators of proliferation and apoptosis in animal cells: identification of miRNAs regulating growth and survival’, Cytotechnology, 2007, 53, (1–3), pp. 55–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lu, J. , Getz, G. , Miska, E.A. , et al.: ‘MicroRNA expression profiles classify human cancers’, Nature, 2005, 435, (7043), pp. 834–838 [DOI] [PubMed] [Google Scholar]
- 16. Kloosterman, W.P. , Plasterk, R.H. : ‘The diverse functions of microRNAs in animal development and disease’, Dev. Cell, 2006, 11, (4), pp. 441–450 [DOI] [PubMed] [Google Scholar]
- 17. Krek, A. , Grun, D. , Poy, M.N. , et al.: ‘Combinatorial microRNA target predictions’, Nat. Genet., 2005, 37, (5), pp. 495–500 [DOI] [PubMed] [Google Scholar]
- 18. Rajewsky, N. : ‘MicroRNA target predictions in animals’, Nat. Genet., 2006, 38, (6), pp. S8–13 [DOI] [PubMed] [Google Scholar]
- 19. Cotterell, J. , Sharpe, J. : ‘An atlas of gene regulatory networks reveals multiple three‐gene mechanisms for interpreting morphogen gradients’, Mol. Syst. Biol., 2011, 6, p. 425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Johnston, R.J. , Hobert, O. : ‘A microRNA controlling left/right neuronal asymmetry in Caenorhabditis elegans ’, Nature, 2003, 426, (6968), pp. 845–849 [DOI] [PubMed] [Google Scholar]
- 21. Seitz, H. : ‘Redefining microRNA targets’, Curr. Biol., 2009, 19, (10), pp. 870–873 [DOI] [PubMed] [Google Scholar]
- 22. Slack, F.J. , Basson, M. , Liu, Z. , et al.: ‘The Lin‐41 Rbcc gene acts in the C. Elegans heterochronic pathway between the Let‐7 regulatory RNA and the Lin‐29 transcription factor’, Mol. Cell, 2000, 5, (4), pp. 659–669 [DOI] [PubMed] [Google Scholar]
- 23. Wightman, B. , Ha, I. , Ruvkun, G. : ‘Posttranscriptional regulation of the heterochronic gene Lin‐14 by Lin‐4 mediates temporal pattern formation in C. Elegans’, Cell, 1993, 75, (5), pp. 855–862 [DOI] [PubMed] [Google Scholar]
- 24. Grosshans, H. , Johnson, T. , Reinert, K.L. , et al.: ‘The temporal patterning microRNA Let‐7 regulates several transcription factors at the larval to adult transition in C. Elegans’, Dev. Cell, 2005, 8, (3), pp. 321–330 [DOI] [PubMed] [Google Scholar]
- 25. Lewis, B.P. , Burge, C.B. , Bartel, D.P. : ‘Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets’, Cell, 2005, 120, (1), pp. 15–20 [DOI] [PubMed] [Google Scholar]
- 26. Ambros, V. , Horvitz, H.R. : ‘The Lin‐14 locus of Caenorhabditis elegans controls the time of expression of specific postembryonic developmental events’, Genes Dev., 1987, 1, (4), pp. 398–414 [DOI] [PubMed] [Google Scholar]
- 27. Poliseno, L. , Salmena, L. , Zhang, J. , et al.: ‘A coding‐independent function of gene and pseudogene mRNAs regulates tumour biology’, Nature, 2010, 465, (7301), pp. 1033–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Baccarini, A. , Chauhan, H. , Gardner, T.J. , et al.: ‘Kinetic analysis reveals the fate of a microRNA following target regulation in mammalian cells’, Curr. Biol., 2011, 21, (5), pp. 369–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chatterjee, S. , Grosshans, H. : ‘Active turnover modulates mature microRNA activity in Caenorhabditis elegans ’, Nature, 2009, 461, (7263), pp. 546–549 [DOI] [PubMed] [Google Scholar]
- 30. Chatterjee, S. , Fasler, M. , Bussing, I. , et al.: ‘Target‐mediated protection of endogenous microRNAs in C. elegans’, Dev. Cell, 2011, 20, (3), pp. 388–396 [DOI] [PubMed] [Google Scholar]
- 31. Khvorova, A. , Reynolds, A. , Jayasena, S.D. : ‘Functional siRNAs and miRNAs exhibit strand bias’, Cell, 2003, 115, (2), pp. 209–216 [DOI] [PubMed] [Google Scholar]
- 32. Schwarz, D.S. , Hutvagner, G. , Du, T. , et al.: ‘Asymmetry in the assembly of the RNAi enzyme complex’, Cell, 2003, 115, (2), pp. 199–208 [DOI] [PubMed] [Google Scholar]
- 33. de Wit, E. , Linsen, S.E. , Cuppen, E. , et al.: ‘Repertoire and evolution of miRNA genes in four divergent nematode species’, Genome Res., 2009, 19, (11), pp. 2064–2074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Okamura, K. , Phillips, M.D. , Tyler, D.M. , et al.: ‘The regulatory activity of microRNA* species has substantial influence on microRNA and 3′ UTR evolution’, Nat. Struct. Mol. Biol., 2008, 15, (4), pp. 354–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ebert, M.S. , Neilson, J.R. , Sharp, P.A. : ‘MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells’, Nat. Methods, 2007, 4, (9), pp. 721–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Franco‐Zorrilla, J.M. , Valli, A. , Todesco, M. , et al.: ‘Target mimicry provides a new mechanism for regulation of microRNA activity’, Nat. Genet., 2007, 39, (8), pp. 1033–1037 [DOI] [PubMed] [Google Scholar]
- 37. Salmena, L. , Poliseno, L. , Tay, Y. , et al.: ‘A ceRNA hypothesis: the rosetta stone of a hidden RNA language?’, Cell, 2011, 146, (3), pp. 353–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Arvey, A. , Larsson, E. , Sander, C. , et al.: ‘Target MRNA abundance dilutes microRNA and siRNA activity’, Mol. Syst. Biol., 2010, 6, p. 363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. D'Errico, I. , Gadaleta, G. , Saccone, C. : ‘Pseudogenes in metazoa: origin and features’, Brief Funct. Genomics Proteomics, 2004, 3, (2), pp. 157–167 [DOI] [PubMed] [Google Scholar]
- 40. Capel, B. , Swain, A. , Nicolis, S. , et al.: ‘Circular transcripts of the testis‐determining gene sry in adult mouse testis’, Cell, 1993, 73, (5), pp. 1019–1030 [DOI] [PubMed] [Google Scholar]
- 41. Nigro, J.M. , Cho, K.R. , Fearon, E.R. , et al.: ‘Scrambled exons’, Cell, 1991, 64, (3), pp. 607–613 [DOI] [PubMed] [Google Scholar]
- 42. Hansen, T.B. , Jensen, T.I. , Clausen, B.H. , et al.: ‘Natural RNA circles function as efficient microRNA sponges’, Nature, 2013, 495, (7441), pp. 384–388 [DOI] [PubMed] [Google Scholar]
- 43. Memczak, S. , Jens, M. , Elefsinioti, A. , et al.: ‘Circular RNAs are a large class of animal RNAs with regulatory potency’, Nature, 2013, 495, (7441), pp. 333–338 [DOI] [PubMed] [Google Scholar]
- 44. Liu, Q. , Zhang, X. , Hu, X. , et al.: ‘Circular RNA related to the chondrocyte Ecm regulates Mmp13 expression by functioning as a Mir‐136 ‘Sponge’ in human cartilage degradation’, Sci. Rep., 2016, 6, p. 22572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Li, F. , Zhang, L. , Li, W. , et al.: ‘Circular RNA itch has inhibitory effect on Escc by suppressing the Wnt/Beta‐catenin pathway’, Oncotarget, 2015, 6, (8), pp. 6001–6013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Xie, H. , Ren, X. , Xin, S. , et al.: ‘Emerging roles of Circrna_001569 targeting Mir‐145 in the proliferation and invasion of colorectal cancer’, Oncotarget, 2016, 7, (18), pp. 26680–26691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Guo, J.U. , Agarwal, V. , Guo, H. , et al.: ‘Expanded identification and characterization of mammalian circular RNAs’, Genome Biol., 2014, 15, (7), p. 409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hansen, T.B. , Wiklund, E.D. , Bramsen, J.B. , et al.: ‘MiRNA‐dependent gene silencing involving Ago2‐mediated cleavage of a circular antisense RNA’, EMBO J., 2011, 30, (21), pp. 4414–4422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bosson, A.D. , Zamudio, J.R. , Sharp, P.A. : ‘Endogenous miRNA and target concentrations determine susceptibility to potential ceRNA competition’, Mol. Cell, 2014, 56, (3), pp. 347–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ala, U. , Karreth, F.A. , Bosia, C. , et al.: ‘Integrated transcriptional and competitive endogenous RNA networks are cross‐regulated in permissive molecular environments’, Proc. Natl. Acad. Sci. USA, 2013, 110, (18), pp. 7154–7159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Amirkhah, R. , Farazmand, A. , Gupta, S.K. , et al.: ‘Naive Bayes classifier predicts functional microRNA target interactions in colorectal cancer’, Mol. Biosyst., 2015, 11, (8), pp. 2126–2134 [DOI] [PubMed] [Google Scholar]
- 52. Zhou, M. , Wang, X. , Shi, H. , et al.: ‘Characterization of long non‐coding RNA‐associated ceRNA network to reveal potential prognostic lncRNA biomarkers in human ovarian cancer’, Oncotarget, 2016, 7, (11), pp. 12598–12611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Paci, P. , Colombo, T. , Farina, L. : ‘Computational analysis identifies a sponge interaction network between long non‐coding RNAs and messenger RNAs in human breast cancer’, BMC Syst. Biol., 2014, 8, p. 83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bosia, C. , Pagnani, A. , Zecchina, R. : ‘Modelling competing endogenous RNA networks’, PLoS One, 2013, 8, (6), p. e66609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. John, B. , Enright, A.J. , Aravin, A. , et al.: ‘Human microRNA targets’, PLoS Biol., 2004, 2, (11), p. e363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Oulas, A. , Karathanasis, N. , Louloupi, A. , et al.: ‘A new microRNA target prediction tool identifies a novel interaction of a putative MiRNA with Ccnd2’, RNA Biol., 2012, 9, (9), pp. 1196–1207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Figliuzzi, M. , De Martino, A. , Marinari, E. : ‘RNA‐based regulation: dynamics and response to perturbations of competing RNAs’, Biophys. J., 2014, 107, (4), pp. 1011–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Figliuzzi, M. , Marinari, E. , De Martino, A. : ‘MicroRNAs as a selective channel of communication between competing RNAs: a steady‐state theory’, Biophys. J., 2013, 104, (5), pp. 1203–1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Denzler, R. , Agarwal, V. , Stefano, J. , et al.: ‘Assessing the ceRNA hypothesis with quantitative measurements of miRNA and target abundance’, Mol. Cell, 2014, 54, (5), pp. 766–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yuan, Y. , Liu, B. , Xie, P. , et al.: ‘Model‐guided quantitative analysis of microRNA‐mediated regulation on competing endogenous RNAs using a synthetic gene circuit’, Proc. Natl. Acad. Sci. USA, 2015, 112, (10), pp. 3158–3163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Grimm, D. , Streetz, K.L. , Jopling, C.L. , et al.: ‘Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways’, Nature, 2006, 441, (7092), pp. 537–541 [DOI] [PubMed] [Google Scholar]
- 62. Keck, K. , Volper, E.M. , Spengler, R.M. , et al.: ‘Rational design leads to more potent RNA interference against hepatitis B virus: factors effecting silencing efficiency’, Mol. Ther., 2009, 17, (3), pp. 538–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Castanotto, D. , Sakurai, K. , Lingeman, R. , et al.: ‘Combinatorial delivery of small interfering RNAs reduces RNAi efficacy by selective incorporation into RISC’, Nucl. Acids Res., 2007, 35, (15), pp. 5154–5164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yi, R. , Doehle, B.P. , Qin, Y. , et al.: ‘Overexpression of exportin 5 enhances RNA interference mediated by short hairpin RNAs and microRNAs’, RNA, 2005, 11, (2), pp. 220–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yi, R. , Qin, Y. , Macara, I.G. , et al.: ‘Exportin‐5 mediates the nuclear export of pre‐microRNAs and short hairpin RNAs’, Genes Dev., 2003, 17, (24), pp. 3011–3016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kim, Y.K. , Kim, B. , Kim, V.N. : ‘Re‐evaluation of the roles of drosha, export in 5, and dicer in microRNA biogenesis’, Proc. Natl. Acad. Sci. USA, 2016, 113, (13), pp. E1881–E1889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Khan, A.A. , Betel, D. , Miller, M.L. , et al.: ‘Transfection of small RNAs globally perturbs gene regulation by endogenous microRNAs’, Nat. Biotechnol., 2009, 27, (6), pp. 549–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hausser, J. , Syed, A.P. , Selevsek, N. , et al.: ‘Timescales and bottlenecks in miRNA‐dependent gene regulation’, Mol. Syst. Biol., 2013, 9, p. 711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Yang, J.S. , Maurin, T. , Robine, N. , et al.: ‘Conserved vertebrate Mir‐451 provides a platform for dicer‐independent, Ago2‐mediated microRNA biogenesis’, Proc. Natl. Acad. Sci. USA, 2010, 107, (34), pp. 15163–15168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Liu, X. , Jin, D.Y. , McManus, M.T. , et al.: ‘Precursor microRNA‐programmed silencing complex assembly pathways in mammals’, Mol. Cell, 2012, 46, (4), pp. 507–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Futran, A.S. , Link, A.J. , Seger, R. , et al.: ‘Erk as a model for systems biology of enzyme kinetics in cells’, Curr. Biol., 2013, 23, (21), pp. R972–R979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Shaul, Y.D. , Seger, R. : ‘The Mek/Erk cascade: from signaling specificity to diverse functions’, Biochim. Biophys. Acta, 2007, 1773, (8), pp. 1213–1226 [DOI] [PubMed] [Google Scholar]
- 73. Lee, T. , Hoofnagle, A.N. , Kabuyama, Y. , et al.: ‘Docking Motif interactions in map kinases revealed by hydrogen exchange mass spectrometry’, Mol. Cell, 2004, 14, (1), pp. 43–55 [DOI] [PubMed] [Google Scholar]
- 74. Tanoue, T. , Adachi, M. , Moriguchi, T. , et al.: ‘A conserved docking motif in map kinases common to substrates, activators and regulators’, Nat. Cell Biol., 2000, 2, (2), pp. 110–116 [DOI] [PubMed] [Google Scholar]
- 75. Bardwell, A.J. , Abdollahi, M. , Bardwell, L. : ‘Docking sites on mitogen‐activated protein kinase (MAPK) kinases, MAPK phosphatases and the Elk‐1 transcription factor compete for MAPK binding and are crucial for enzymic activity’, Biochem. J., 2003, 370, (Pt 3), pp. 1077–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Futran, A.S. , Kyin, S. , Shvartsman, S.Y. , et al.: ‘Mapping the binding interface of Erk and transcriptional repressor Capicua using photocrosslinking’, Proc. Natl. Acad. Sci. USA, 2015, 112, (28), pp. 8590–8595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Li, W.X. : ‘Functions and mechanisms of receptor tyrosine kinase torso signaling: lessons from drosophila embryonic terminal development’, Dev. Dyn., 2005, 232, (3), pp. 656–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Fores, M. , Ajuria, L. , Samper, N. , et al.: ‘Origins of context‐dependent gene repression by Capicua’, PLoS Genet., 2015, 11, (1), p. e1004902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Cinnamon, E. , Helman, A. , Ben‐Haroush Schyr, R. , et al.: ‘Multiple Rtk pathways downregulate Groucho‐mediated repression in drosophila embryogenesis’, Development, 2008, 135, (5), pp. 829–837 [DOI] [PubMed] [Google Scholar]
- 80. Janody, F. , Sturny, R. , Catala, F. , et al.: ‘Phosphorylation of bicoid on map‐kinase sites: contribution to its interaction with the torso pathway’, Development, 2000, 127, (2), pp. 279–289 [DOI] [PubMed] [Google Scholar]
- 81. Kim, Y. , Coppey, M. , Grossman, R. , et al.: ‘MAPK substrate competition integrates patterning signals in the drosophila embryo’, Curr. Biol., 2010, 20, (5), pp. 446–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Jimenez, G. , Guichet, A. , Ephrussi, A. , et al.: ‘Relief of gene repression by Torso Rtk signaling: role of Capicua in drosophila terminal and dorsoventral patterning’, Genes Dev., 2000, 14, (2), pp. 224–231 [PMC free article] [PubMed] [Google Scholar]
- 83. Kim, Y. , Andreu, M.J. , Lim, B. , et al.: ‘Gene regulation by MAPK substrate competition’, Dev. Cell, 2011, 20, (6), pp. 880–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Sopko, R. , Perrimon, N. : ‘Receptor tyrosine kinases in drosophila development’, Cold Spring Harb. Perspect. Biol., 2013, 5, (6), pp. 1–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. O'Neill, E.M. , Rebay, I. , Tjian, R. , et al.: ‘The activities of two Ets‐related transcription factors required for drosophila eye development are modulated by the Ras/MAPK pathway’, Cell, 1994, 78, (1), pp. 137–147 [DOI] [PubMed] [Google Scholar]
- 86. Rebay, I. , Rubin, G.M. : ‘Yan functions as a general inhibitor of differentiation and is negatively regulated by activation of the Ras1/MAPK pathway’, Cell, 1995, 81, (6), pp. 857–866 [DOI] [PubMed] [Google Scholar]
- 87. Boisclair Lachance, J.F. , Pelaez, N. , Cassidy, J.J. , et al.: ‘A comparative study of pointed and Yan expression reveals new complexity to the transcriptional networks downstream of receptor tyrosine kinase signaling’, Dev. Biol., 2014, 385, (2), pp. 263–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kim, M. , Lee, J.H. , Koh, H. , et al.: ‘Inhibition of Erk‐map kinase signaling by Rsk during drosophila development’, EMBO J., 2006, 25, (13), pp. 3056–3067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Kim, Y. , Paroush, Z. , Nairz, K. , et al.: ‘Substrate‐dependent control of MAPK phosphorylation in vivo’, Mol. Syst. Biol., 2011, 7, p. 467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Blackwell, E. , Kim, H.J. , Stone, D.E. : ‘The pheromone‐induced nuclear accumulation of the Fus3 MAPK in yeast depends on its phosphorylation state and on Dig1 and Dig2’, BMC Cell Biol., 2007, 8, p. 44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Arur, S. , Ohmachi, M. , Nayak, S. , et al.: ‘Multiple Erk substrates execute single biological processes in Caenorhabditis elegans germ‐line development’, Proc. Natl. Acad. Sci. USA, 2009, 106, (12), pp. 4776–4781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Ahmed, S. , Grant, K.G. , Edwards, L.E. , et al.: ‘Data‐driven modeling reconciles kinetics of Erk phosphorylation, localization, and activity states’, Mol. Syst. Biol., 2014, 10, p. 718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Jayanthi, S. , Nilgiriwala, K.S. , Del Vecchio, D. : ‘Retroactivity controls the temporal dynamics of gene transcription’, ACS Synth. Biol., 2013, 2, (8), pp. 431–441 [DOI] [PubMed] [Google Scholar]
- 94. Mishra, D. , Rivera, P.M. , Lin, A. , et al.: ‘A load driver device for engineering modularity in biological networks’, Nat. Biotechnol., 2014, 32, (12), pp. 1268–1275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Liu, P. , Kevrekidis, I.G. , Shvartsman, S.Y. : ‘Substrate‐dependent control of Erk phosphorylation can lead to oscillations’, Biophys. J., 2011, 101, (11), pp. 2572–2581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Kholodenko, B.N. : ‘Negative feedback and ultrasensitivity can bring about oscillations in the mitogen‐activated protein kinase cascades’, Eur. J. Biochem., 2000, 267, (6), pp. 1583–1588 [DOI] [PubMed] [Google Scholar]
- 97. Shankaran, H. , Ippolito, D.L. , Chrisler, W.B. , et al.: ‘Rapid and sustained nuclear‐cytoplasmic Erk oscillations induced by Epidermal growth factor’, Mol. Syst. Biol., 2009, 5, p. 332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Avraham, R. , Yarden, Y. : ‘Feedback regulation of Egfr signalling: decision making by early and delayed loops’, Nat. Rev. Mol. Cell Biol., 2011, 12, (2), pp. 104–117 [DOI] [PubMed] [Google Scholar]
- 99. Rowland, M.A. , Fontana, W. , Deeds, E.J. : ‘Crosstalk and competition in signaling networks’, Biophys. J., 2012, 103, (11), pp. 2389–2398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Schauble, S. , Stavrum, A.K. , Puntervoll, P. , et al.: ‘Effect of substrate competition in kinetic models of metabolic networks’, FEBS Lett., 2013, 587, (17), pp. 2818–2824 [DOI] [PubMed] [Google Scholar]
- 101. Thomson, D.W. , Dinger, M.E. : ‘Endogenous microRNA sponges: evidence and controversy’, Nat. Rev. Genet., 2016, 17, (5), pp. 272–283 [DOI] [PubMed] [Google Scholar]
- 102. Thomson, D.W. , Pillman, K.A. , Anderson, M.L. , et al.: ‘Assessing the gene regulatory properties of argonaute‐bound small RNAs of diverse genomic origin’, Nucl. Acids Res., 2015, 43, (1), pp. 470–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Boulter, E. , Garcia‐Mata, R. , Guilluy, C. , et al.: ‘Regulation of Rho Gtpase crosstalk, degradation and activity by Rhogdi1’, Nat. Cell Biol., 2010, 12, (5), pp. 477–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Kopito, R.B. , Elbaum, M. : ‘Nucleocytoplasmic transport: a thermodynamic mechanism’, HFSP J., 2009, 3, (2), pp. 130–141 [DOI] [PMC free article] [PubMed] [Google Scholar]