

Abstract
Laser Capture Microdissection (LCM) provides a fast, specific, and versatile method to isolate and enrich cells in mixed populations and/or subcellular structures, for further proteomic study. Furthermore, Mass Spectrometry (MS) can quickly and accurately generate differential protein expression profiles from small amounts of samples. Although cellular protrusions—such as tunneling nanotubes, filopodia, growth cones, invadopodia, etc.—are involved in essential physiological and pathological actions such as phagocytosis or cancer-cell invasion, the study of their protein composition is progressing slowly due to their fragility and transient nature. The method described herein, combining LCM and MS, has been designed to identify the proteome of different cellular protrusions. First, cells are fixed with a novel fixative method to preserve the cellular protrusions, which are isolated by LCM. Next, the extraction of proteins from the enriched sample is optimized to de-crosslink the fixative agent to improve the identification of proteins by MS. The efficient protein recovery and high sample quality of this method enables the protein profiling of these small and diverse subcellular structures.
Keywords: cellular protrusion, laser capture microdissection, proteomics, fixation, DTBP, mass spectrometry
1. Introduction
Laser Capture Microdissection (LCM) is a tool that facilitates, via direct visualization of cells or tissues, the enrichment and isolation of cells of interest in a heterogeneous sample [1]. There are different classes of LCM depending on the type of laser and capture method. Overall, laser capture microdissection is an optical microscope, which can be coupled to fluorescence, with a finely focused laser and a system for capturing dissected samples. The cells of interest can be selected from the heterogeneous sample through a touch screen enabling the selection of regions of interest (ROI) using an interactive pen or the mouse. Each ROI can be drawn freehand or using predefined geometrical shapes. This technique has been successfully applied in the isolation of tumoral cells, neurons affected by Parkinson or Alzheimer diseases, virus-infected cells, etc. [2–4]. Moreover, LCM was shown to be able to separate cellular organelles from specific cells [5], growth cones [6] and invadopodia [7]. One advantage of using LCM to enrich a sample with the cells of interest is the possibility of performing subsequent downstream studies of DNA, RNA or protein composition.
Proteomics, the study of proteins, is one of the most important technologies used to acquire new insights into cellular function since proteins are the primary effectors of all biological activity. However, the feasibility of combining proteomic techniques with LCM depends primarily on the sensitivity of the technique used, as the amount of protein retrieved from the dissected sample can be very small. Traditional proteomic techniques cannot solve this problem since they require a high degree of sample homogeneity to obtain significant results. Nevertheless, mass spectrometry can be applied to identify and quantify proteins from different samples. In microproteomics, much smaller amounts, in the attomole or even zeptomole range, are required to sequence a peptide [8].
Cellular protrusions are involved in several biological functions, such as cell migration, neurite outgrowth, phagocytosis and cell-to-cell communication. More recently, these structures have also been related to cancer-cell invasion, intercellular transmission of misfolded proteins in degenerative diseases and intercellular spread of infectious pathogens. These physiological and pathological processes are conducted by different types of cellular protrusions, such as lamellipodia, pseudopodia, filopodia, growth cone, tunneling nanotubes and others [9–17]. However, there are many unanswered questions related to their mechanism of formation, their structural components or signaling pathways [18]. One of the principal reasons for the lack of knowledge regarding cellular protrusions is the limited techniques that allows to specifically study them. Previous attempts have been made to isolate cellular protrusions by using a Boyden chamber [19–21] or an excimer laser [22]. However, none of these methods are specific, since they do not visualize what they are isolating and they were only able to enrich pseudopodia, a specific subtype of cellular protrusion, but could not specifically isolate different types of cellular protrusions.
Here, we proposed a protocol that combines two techniques to enhance our knowledge of the composition of these structures: LCM, to get an enrichment of cellular protrusions based on morphological features, and MS, to uncover the proteomes of these structures.
The first road block in using LCM to isolate cellular protrusions is their transient nature and structural fragility. Thus, we first needed to identify fixative procedures that would be strong enough to maintain the structural integrity of the cellular protrusions for laser microdissection but also compatible with downstream microproteomic analyses. Fixative solutions with glutaraldehyde are ideal to maintain cellular protrusions, including fragile protrusions such as tunneling nanotubes [23]. Unfortunately, glutaraldehyde fixations, even in low concentrations, create thorough cross-linking that seriously hampers the quality sample for MS analysis [24]. Here, we use dimethyl-3–3’-dithiobispropiomimidate (DTBP), which can be de-crosslinked by using a reducing agent and a temperature-step process [25, 26]. In fact, the currently optimized fixation with DTBP not only preserves cellular protrusions but also allows for the identification of high quality and high quantity proteins, which reduces the number of cellular protrusions needed for MS analysis [27]. The current protocol allows one to specifically select any type of cellular protrusion based on morphological and/or fluorescence markers, thus greatly improving its selectivity compared to previous methods [6, 7, 19–22]. Importantly, we were able to isolate TNTs, a very delicate and specialized cell-to-cell communication conduit, that have been related to several diseases, such as cancer, human immunodeficiency virus or prion infection [28–31].
2. Materials
2.1. Cell culture
CAD (cath-a-differentiated) cell line (Sigma, Acc. # 081000805).
25 cm2 cell culture flasks, 5-, 10-, and 25-mL individually wrapped pipettes, 1-mL aspirating pipettes and 15- and 50-mL conical tubes.
An optical biological inverted microscope, a biological safety cabinet, e.g. 1300 Series Class II, Type A2 Biological safety cabinet (Thermo Scientific, # 1335), a CO2 incubator and an aspiration pump.
CAD cell Culture Medium: Add 50 mL of Fetal Bovine Serum (FBS) (10%) to 450 mL of OPTI-MEM (Gibco/ThF). Filter-sterilize and store at 4 °C for no longer than 4 weeks.
Cell counting: Trypan Blue solution, 0.4% (Sigma-Aldrich, # T8154) and a metallized haemocytometer (Hausser Scientific/Reichert Bright-Line, # 1492)
2.2. Preparation for LCM
As stated above, there are different types of LCM. In our studies, we used a laser microdissection system from Molecular Machines and Industries (MMI CellCut Laser Microdissection, Eching, Germany) controlled by the MMI Cell Tools Software from the same company.
MMI Live cell chamber with membrane and petri dish (Molecular machines and industries, # 50301). This includes a membrane ring for the initial cultivation of cells and a UV-permeable microdissection chamber to isolate the cellular protrusions of interest.
Phosphate buffer saline (PBS) 10X, pH 7.4 and fibronectin solution from bovine plasma (Sigma-Aldrich, # 86088-83-7).
Tweezers, filter tips, pipettes and an aspiration pump.
2.3. Fixation protocol
Protrusion induction: Hydrogen peroxide (H2O2), 30% (Fisher Scientific, # BP2633–500)
Fixation: Paraformaldehyde (PFA), 16%, EM Grade (Electron Microscopy Sciences, # 15710); Dimethyl-3–3’-Dithiobispropiomimidate (DTBP) (ThermoFisher, # 20665); HEPES, 1M (Gibco/ThF, # 15630–080); pH Meter; vortex mixer and chemical hood; microcentrifuge tubes; pipette tips and Pipetman classic™ Starter Kit (Gilson, # F167300).
To evaluate fixation and H2O2 treatment: a cell membrane marker (wheat germ agglutinin tetramethylrhodamine conjugate; ThermoFisher), Ibidi μ-dishes and a fluorescence microscope.
Fixation solution 1: 4% PFA solution, 200 mM HEPES and 1x PBS (see Note 1).
Fixation solution 2: 5 mM DTBP solution. Weight DTBP powder to make up a 50 mM DTBP stock and dissolve in 1x PBS pH 8.6. Calculate the necessary volume of the stock solution to get a final concentration of 5 mM DTBP and add 25 mM HEPES in 1x PBS pH 8.6 (see Note 2).
2.4. Laser capture microdissection
MMI CellCut Laser Microdissection (Molecular Machines and Industries) and the Software (MMI Cell Tools Software; Ver. 4.3.3. Molecular Machines and Industries/ Olympus IX81)
2.5. Protein extraction protocol
Digital dry bath (37 °C; 60°C and 100°C); Sonicator (e.g. Avanti Polar, model G1125P1T_B); refrigerated microcentrifuge
To measure protein: Bovine Serum Albumin Standard; RC DC™ Protein Assay Kit II (Bio-rad, # 5000122); Truview Cuvettes (Biorad, # 1702510); a spectrophotometer, e.g. Smartspec™ Plus Spectrophotometer (Biorad, # 1702525).
Alternatively, samples can be acquired based on the number of cellular protrusions isolated by LCM, rather than protein assay.
Lysis buffer (RIPA): 10 mM Tris-HCl pH 8.0, 1mM EDTA, 0.5 mM EGTA, 2% SDS, 1% Triton X-100, 0.1% Sodium Deoxycholate, 140mM NaCl, 100mM DTT (see Note 3), 1% protease inhibitors (e.g. Halt™ Protease Inhibitor Cocktail, 100X; ThermoFisher, # 78429) and molecular water.
2.6. Mass Spectrometry sample preparation
5X Laemmli buffer: 25 mM Tris-HCl pH 6.8, 25% glycerol, 10% SDS, 0.02% bromophenol blue, 150 mM DTT and molecular water. (see Note 4).
An 8% acrylamide Bis-Tris gel for mass spectrometry: Prepare a 1.25 M Bis-Tris stock and adjust pH to 6.5–6.8. For the resolving gel: calculate the necessary volume of the stock solution to get a final concentration of 350 mM Bis-Tris and add the corresponding volume of 30% acrylamide/bis-acrylamide (37.5:1) to get a final concentration of 8%. Add H20 to make up a final volume of 5 ml. Finally, add 10% ammonium persulfate (APS) and TEMED (10 μl) to polymerize it. For the stacking gel: calculate the necessary volume of the stock solution to get a final concentration of 350 mM Bis-Tris and add the corresponding volume of 30% acrylamide to get a final concentration of 4%. Add H20 to make up a final volume of 1.75 ml. Finally, add 10% APS and TEMED (10 μl) to polymerize it.
20X MOPS Buffer: 1 M 3-(N-morpholino) propane sulfonic acid (MOPS), 1 M Tris Base, 20 mM EDTA, 2% SDS and molecular water (see Note 5).
Running buffer: 20% MOPS Buffer, 0.5 % sodium bisulfite and molecular water (see Note 1).
Fixing solution for gels: methanol, acetic acid and molecular water (40:40:8, respectively) (see Note 1).
Precision Plus Protein™ Dual Color Standards (Bio-Rad, # 161–0374), loading tips.
Powerpac™ Basic Power Supply (Biorad, # 1645050) and Mini-PROTEAN® Tetra cell system (Biorad, # 1658000)
Compact digital rocker (Fisher Scientific, # 11-676-333)
Speed vacuum.
iST Sample Preparation Kit from PreOmics.
2.7. Mass Spectrometers
Thermo Scientific Orbitrap Fusion mass spectrometer or timsTOF Pro mass spectrometer from Bruker.
3. Methods
3.1. Cell culture
The method described here is to get an enrichment of cellular protrusions and subsequently, to extract the proteins and analyze them by MS. Cell density is a critical step when isolating cellular protrusions. It can vary not only with the types of cellular protrusions that need to be isolated but also with the cell types used for the experiments. For example, if you are interested in isolating filopodia, we recommend plating low cell density in order to ensure that filopodia can be identified and selected from single cells. If you are interested in isolating TNTs, then the cells need to be in closer proximity in order to be able to form TNTs and therefore a higher cell density is required. These numbers must be pre-determined for each cell types used, as well as for the types of cellular protrusion of interest.
Pre-warm a 50-ml centrifuge tube with CAD media in the 37 °C water bath. Place 3 ml of media into a 15-ml conical tube and 5 ml of media into a T25 plate. Remove the cells from the liquid nitrogen tank. Thaw the cryotube with CAD cells in the 37 °C water bath. When almost thawed (it is acceptable for some ice crystals to be left), take the tube out of the water bath and move to the tissue culture hood. As freezing media contains DMSO, which is toxic for cells, these steps have to be done quickly.
Add 1 ml of media to the cells (approximately 1,500,000 cells), resuspend them, and transfer them into the prepared 15-ml conical tube. Spin the conical tube in a centrifuge at 1,000Xg for 4 minutes (the cells will collect at the bottom as a pellet). Carefully aspirate and discard the supernatant from the 15 ml conical tube and add 1 ml of media to the cell pellet and re-suspend gently. Then, pull up all of the cell mixture into the pipette and transfer to the prepared T25 plate. Look at cells before placing in the incubator. The cells should be round and shiny.
Next day, check that the cells have adhere to the plate and are healthy, non-differentiated and look at their confluency. Grow cells until they become 80% confluent (usually 2 days).
To split the cells, remove the media and dead cells, wash once with PBS, and add 1 ml of media. Tap the flask to detach the cells (mechanical dissociation) and observe under the microscope to ensure that the majority of the cells are detached. Add 3 ml of fresh media along the walls to scrape out all the cells. Pipette out the total 5 ml of the cultures, by pressing the tip of the pipet on the bottom of the flask to break clumps and check under the microscope to ensure that you have mostly single cells. Add 1 ml of the culture (approximately 1,000,000 cells) to the new flask and take the volume of fresh media needed to make up the new flask to 6 ml.
Alternatively, cells can be grown in a 60 or 10 cm culture dish. In this case, cells can be detached mechanically by pipetting them up and down, rather than tapping the flask.
After 2 days, cells should be 80% confluent and they can be split again. Cells should be split a minimum of 2 times before seeding them in a MMI live chamber dish to ensure cells viability. Once the cells have been split 2 times, they can be counted by mixing a sample of the cell solution and with Trypan blue (dilution 1:1). Count the cells using a haemocytometer or an automated cell counter. Calculate the volume for your desired cell concentration (i.e. 200,000 cells for TNTs) to be seeded in a MMI live chamber dish.
Differentiated CAD cells
In order to obtain axonal and dendritic protrusions, we differentiated CAD cells by serum starvation.
Seed 30,000 CAD cells in a MMI live chamber dish.
To differentiate these cells, they have to be grown in OPTI-MEM without FBS for 10 days.
The rest of the protocol is the same as for CAD cells.
3.2. Preparation of MMI live chamber dish
Optimal conditions for cell attachment will depend on the cell type and should be pre-determined. For CAD cells, we use fibronectin coating but there are other extracellular matrix protein coatings available for different cell lines (see Note 6).
Remove the membrane ring from the 35 mm dish with regular tweezers and transfer it to the adhesive area of the UV-permeable microdissection chamber. Press carefully with your fingers and, once attached, do not move the ring to prevent wrinkles and/or breakages (see Note 7).
To confirm that the membrane ring is properly attached and does not leak, add 1 ml of filtered PBS to the dish and wait at least 5 minutes. If no leaks are observed, proceed with the sample preparation. If PBS is seen on the outside of the membrane ring, discard the ring/chamber and start anew.
Prepare 300 μl of filtered PBS and 24 μl of fibronectin.
Remove the PBS, add the fibronectin solution and make sure that the fibronectin solution covers the entire surface of the ring, since the membrane is hydrophobic. Place the dish in the 37° C/5% CO2 incubator for 20 minutes.
Wash 3 times with filtered PBS and 2 times with CAD media.
Add the desired volume of cells (in our case 200,000 cells for TNT samples) and leave them in the incubator until ready for fixation (see Note 8).
Several protocols can be applied to increase cellular protrusions and should be pre-determined based on your cell types and cellular protrusion of interest. We have included two of them (serum starvation for axons/dendrites or exposure to H2O2 for TNTs) but many other stimuli can be applied depending on your research goals (see Note 9).
3.3. Fixation protocol
As previously stated, cellular protrusions are fragile and must be fixed prior to LCM isolation [26, 27]. However, while a strong fixative like glutaraldehyde is ideal to maintain the integrity of cellular protrusions, it is irreversible and does not allow for efficient protein extraction post cell lysis (Fig. 1A&B). On another hand, we have recently demonstrated that using a cleavable crosslinker like DTBP not only preserved cellular protrusions [26] but it also allows for a greatly improved protein recovery after cell lysis (Fig. 1C). Overall, the use of DTBP along with the alternate protein extraction protocol we have developed [26] allows for the reduction of the overall number of cellular protrusions needed to be isolated by LCM, as it greatly increases the number of spectra, number of proteins and number of total unique proteins identified by MS compared to glutaraldehyde-fixed samples (Fig. 2).
Fig. 1. Effect of fixations, protein extractions and dehydration of samples on protein yield.
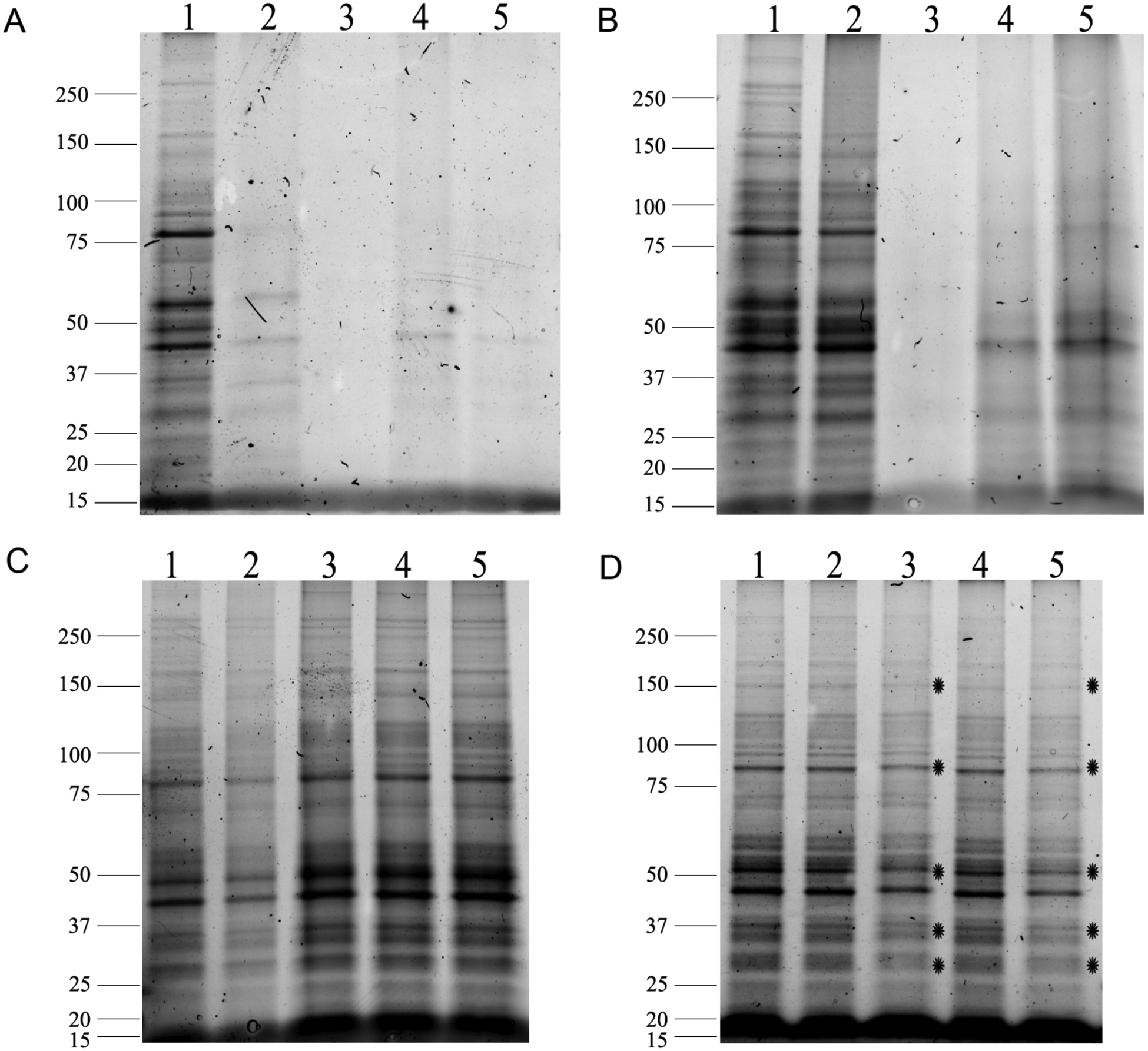
GLU/PFA fixed samples (A) lysed with RIPA buffer (0.01% SDS) on ice or (B) lysed with RIPA buffer (2% SDS) and incubations at 100°C/20 min and 60°C/2h; (1) Unfixed lysates, (2) 4% PFA alone, (3) GLU (0.05%)/PFA, (4) GLU (0.01%)/PFA, (5) GLU (0.005%)/PFA; (C) PFA/DTBP fixed samples were lysed with RIPA buffer (2% SDS and 100 mM DTT) and incubations at 37°C/30 min, 100°C/20 min and 60°C/2h; (1) Unfixed lysates, (2) 4% PFA alone, (3) PFA/3mM DTBP, (4) PFA/5mM DTBP, (5) PFA/10mM DTBP. (D) PFA/5mM DTBP fixed samples extracted and loaded as in gel C; (1) Cells were fixed and lysed the same day; (2) Cells were fixed and kept in PBS for 24h before lysing; (3) Cells were fixed and kept dry for 24h before lysing; (4) Cells were fixed and kept in PBS for 72h before lysing; (5) Cells were fixed and kept dry for 72h before lysing. Stars indicate protein bands with decreased intensity. Gels are representative of 3 independent experiments (reproduced from ref. 26 with permission from Proteomics).
Fig. 2. Effect of type of fixation on protein yield and MS results from 1,000 cells.
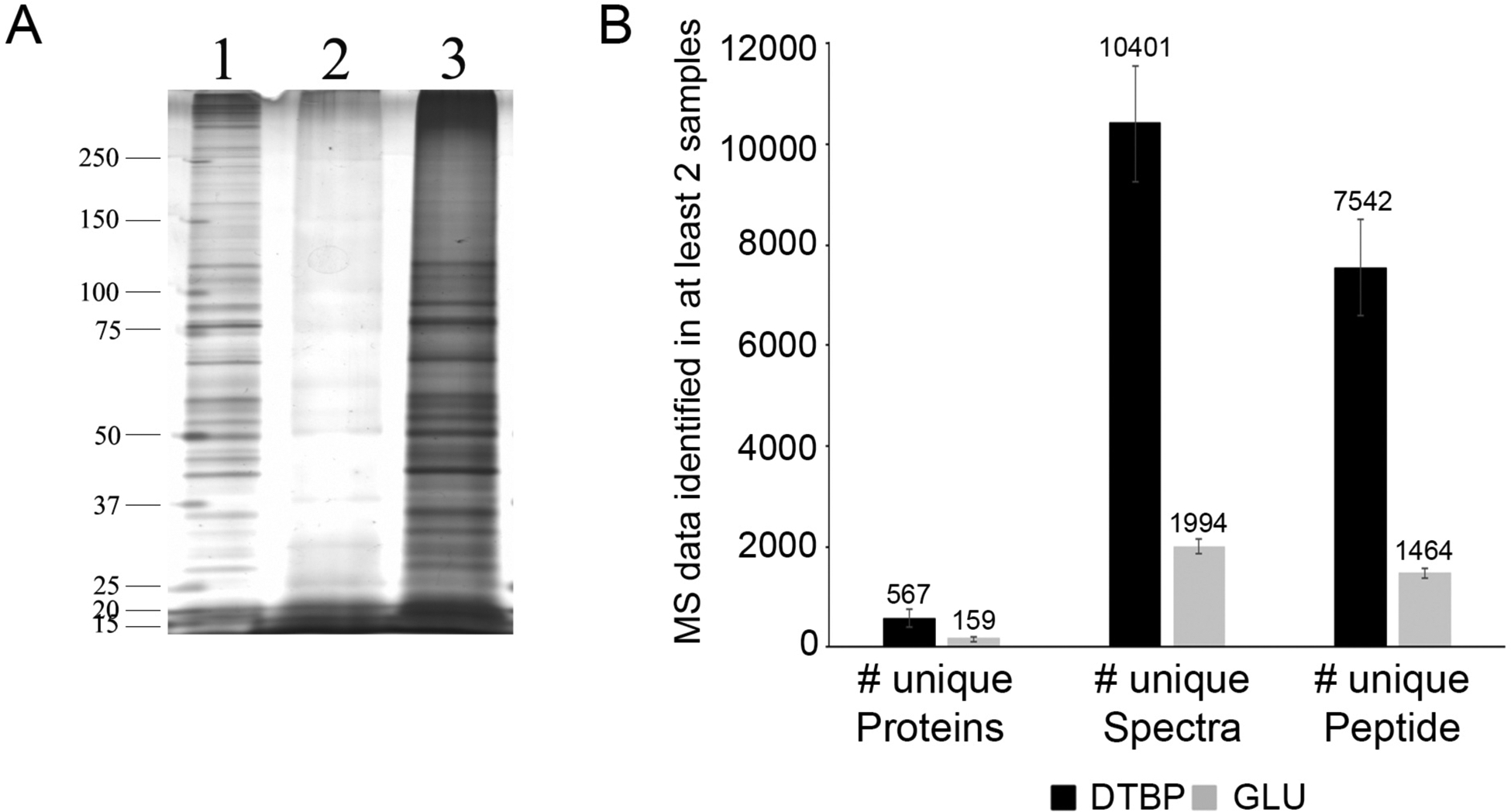
(A) Silver-stained representative gel from (1) Unfixed cells (0.1 μg of protein); (2) 1,000 GLU/PFA fixed cells; (3) 1,000 PFA/DTBP fixed cells; (B) Average and Standard Deviation of unique proteins, unique spectra and unique peptides of samples from 1,000 PFA/DTBP or GLU/PFA fixed cells (reproduced from ref. 26 with permission from Proteomics).
To increase the number of cellular protrusions, a concentration of 100 μM of H2O2 in warm CAD media is added to the cells for 5 minutes (see Note 10).
Add 100 μl for 500 μl total volume or 200 μl for 1 ml total volume of the fixation solution 1 (4% PFA solution) directly to the cells in their culture media and wait 5 minutes.
Aspirate and add 1 ml of fixation solution 1 for 15 minutes (see Note 11). Change fixative solutions by tilting the dish, allowing and carefully aspirating the solution near the wall, without disturbing cells or they might detach, and cellular protrusions could break. Take the same considerations for steps 4 and 5.
Aspirate and add 1 ml of fixation solution 2 (5 mM DTBP solution) for 15 minutes.
Aspirate and add 1 ml of filtered PBS (see Note 12).
We recommend pre-establishing the best conditions (time and concentration), for induction of cellular protrusions with H2O2 for different cell types. Similarly, different concentrations of DTBP might be required to optimize the maintenance of cellular protrusions for different cell types. Trials should be carried out with a cell membrane marker (e.g. wheat germ agglutinin tetramethylrhodamine conjugate (WGA-Rhod, 1:200 in PBS) in Ibidi μ-dishes (Ibidi) and check for cellular protrusion stability under a fluorescence microscope [26]. The cells should be washed twice with PBS after fixation and labeled for 10–20 min in the dark at room temperature with WGA-Rhod.
3.4. Laser Capture Microdissection (LCM)
LCM parameters have to be set-up to improve the isolation of different cellular protrusions from the cells of interest. Here, are some key steps to take into account when using the LCM.
Mount the microdissection chamber dish onto the stage of the LCM microscope and open the MMI Cell Tool program.
The CellCut mode of the software allows for the calibration of the slide geometry of the dish that keeps the plane of focus steady as the laser cuts around the protrusions. With the ×4 objective, focus the dish and set the proper plane tilt of the dish. Then, scan the dish to visualize the entire cell-covered area and help determining what area to cut.
The MMI cell tools software also allows the manipulation of the laser parameters (speed, focus and power) to get a precise cut of the desired protrusions. The settings for LCM have to be adjusted for each sample with the objective required to cut the cellular protrusions of interest. Different objectives may be used depending on the cellular protrusions of interest (see Fig. 3).
The laser speed should be slowed down to avoid detaching the cells or disturbing the cellular protrusions during the process of cutting. This will need to be determined for each sample depending on the types of isolated cellular protrusions. For example, for growth cones or filopodia, which are strongly attached to the substratum, the cut velocity can be higher (e.g. 35 μm/s). On the other hand, for more fragile structures like TNTs, the cut velocity should be reduced (e.g. 10 μm/s).
Laser focus is a way to adjust the position of the laser beam in the Z-direction within the sample, determining the thickness of the cut. It should neither be too low to detach the membrane as it is cutting, nor too high to not cut the membrane at all. This will be determined by calibrating the laser focus according to the plane tilt of the dish. Thus, the laser focus should be adjusted for each experiment as it may vary from dish to dish and sample to sample.
The power needed to cut the sample is proportional to the sample thickness. It is represented in percentage of UV light transmitted by the laser. The laser power should be set to the minimal value sufficient to cut through the membrane and we suggest having at least 2 laser cut repetitions (i.e., the laser will cut the same area twice). This is critical to ensure that the membrane is properly cut from the rest of the samples, since these are the samples of interest that will need to stay attached to the chamber after removal of the ring (in our case, 75–85% of laser power was enough).
Using the proper objective (depending on the size of the cellular protrusions of interest) [27], you can select the ROIs by manually drawing a line with a digital pen, directly on the computer screen, around the protrusions of interest (Fig. 3 shows different types of cellular protrusions that can be isolated by LCM).
We recommend selecting 10–100 ROIs at a time, prior to cutting, but these can be changed according to need or user preferences.
Between cutting sessions, replace the PBS with fresh PBS to remove any dead cells or debris and store the dish at 4 °C until further use.
The dish can be used for 4 to 5 days approximately. Thus, as many ROIs should be isolated as possible per day and a new dish need to be prepared if more ROIs are required after 5 days post-fixation (see Note 13).
To isolate the ROIs cut, remove the PBS, wash the dish gently with filtered PBS, aspirate the PBS and carefully lift the ring from the microdissection chamber with tweezers. All of the desired ROIs should be left on the sticky membrane of the microdissection chamber, while the rest of the cells will be removed along with the ring. It is imperative to go back to the LCM and look at the isolated ROIs left on the microdissection chamber to ensure that the expected ROIs are present and that no whole cells or other cellular debris are present. If a cell is found, it must be obliterated with the laser.
Once the ROIs have been examined and validated, the cellular protrusions can be lifted from the microdissection chamber with a micropipette by taking a small volume of lysis buffer (i.e., 5 μl) and pipetting up and down all over the microdissection chamber where the cuts are located (see Note 14). Avoid making bubbles. The cuts should be transferred to a 1.5 ml centrifuge tube, and the process can be repeated to ensure that all cuts have been lifted by observing the dish under the microscope. The final volume should be small (i.e., 15 μl total for all of your samples).
For the whole cell lysate sample, cut the membrane from the ring that was lifted away from the dissection chamber in step 11 above with a scalpel and put it in 1.5 ml centrifuge tube with lysis buffer (i.e., 40 μl). Make sure to push the membrane at the bottom of the tube and that it is immersed in lysis buffer (see Note 15).
Fig. 3. Cellular protrusions isolated by Laser Capture Microdissection.
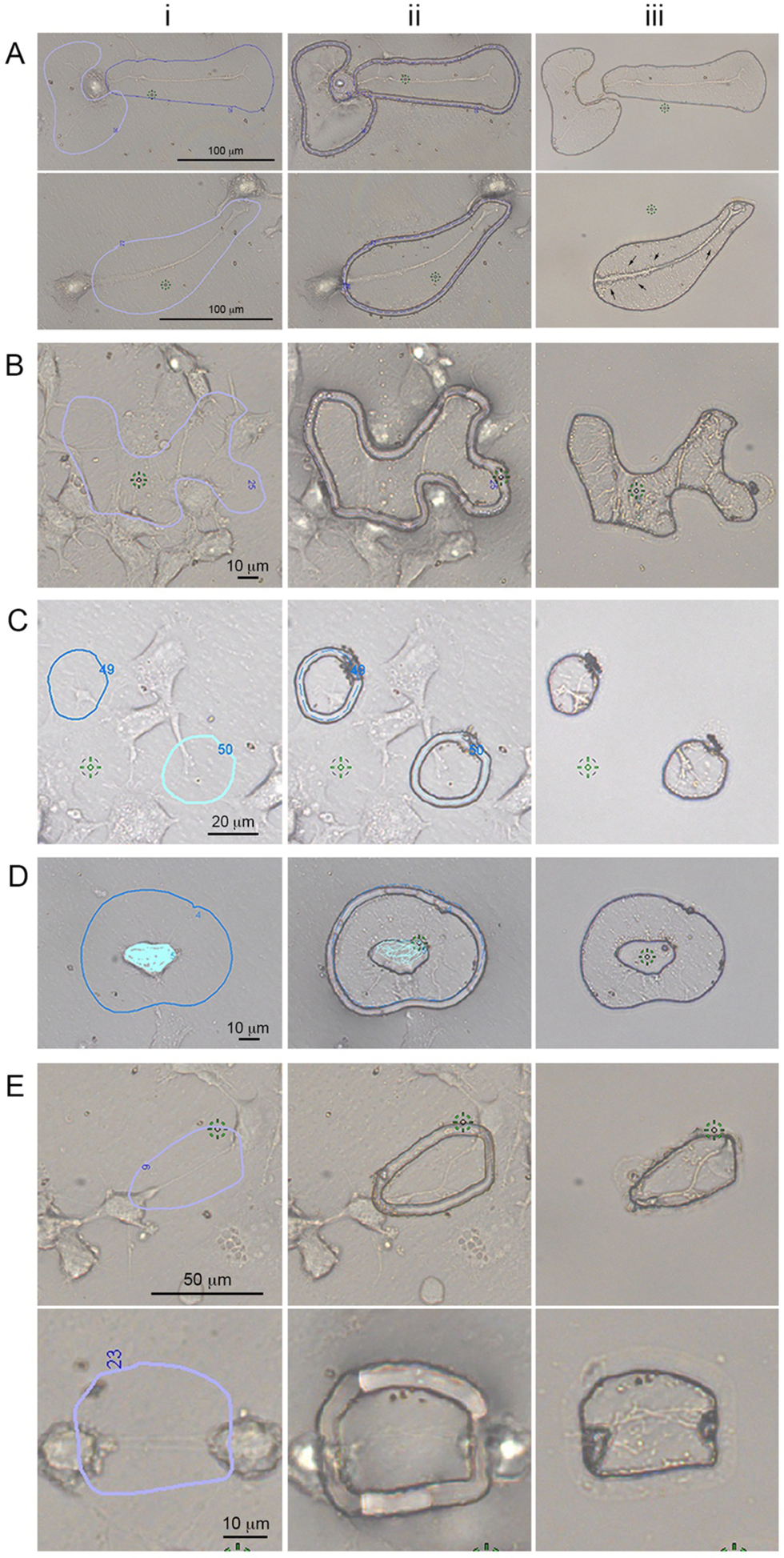
LCM, which uses a finely focused laser to cut a region of interest (ROI), is ideal to isolate cellular protrusions. Cells were plated on MMI live cell chambers, fixed, and imaged at ×20 (A), ×40 (B–D) or ×60 (E) magnifications. Various types of cellular protrusions are shown. For all cases, (i) ROIs are drawn around the protrusions of interest; (ii) are representative images of cellular protrusions after the laser cut, and (iii) images of the desired isolated cellular protrusions after removal of the LCM membrane containing the “unwanted” cells. Examples of different types of protrusions are shown: (A) axons and dendrites from dCADs are shown (scale bar = 100 μm). Small dendritic filopodia can be observed (black arrows); (B) in order to increase the number of cellular protrusions, CADs cells were treated for 5 min with 100 μM H2O2 prior to fixation. Various types of cellular protrusions are shown (scale bar = 10 μm). Individual subtypes of cellular protrusions can be specifically isolated such as (C) GCs (scale bar = 20 μm), (D) filopodia (scale bar = 10 μm), and (E) TNTs (scale bar top = 50 μm; bottom = 10 μm). TNTs do not touch the substratum and tension is visible within these structures (Ei). As expected, after being cut, the structures collapsed onto the LCM membrane (Eii,iii), clearly demonstrating that these protrusions were TNTs, and not attached filopodia or other types of protrusions (reproduced from ref. 27 with permission from International Journal of Molecular Science).
3.5. Protein extraction
The optimization of the protein extraction protocol was performed to maximize the de-crosslinking of the fixative agents while keeping the lysate as concentrated as possible. In addition to the experimental samples, the following controls should also be considered: (i) a positive whole cell lysate control to compare with the proteome of the protrusion sample; (ii) a negative control to determine the background proteins that are released to the media and can be found on the membrane, such as exosomes. In this case, the same number of cuts that were isolated for cellular protrusions should be isolated in empty areas surrounding cells. Finally, when setting up conditions, silver staining (e.g. ProteoSilver™ Silver Stain Kit, Sigma-Aldrich, PROTSIL1–1KT) might be used to determine the quality of the proteins present in the isolated samples even in those with low amounts of proteins, such as the cellular protrusion samples (see Note 16).
Take the isolated LCM samples from step 12&13 in section 3.4 and thaw them if they were stored at −80°C (see Note 17).
Sonicate for 5 minutes and do a quick spin to bring all samples down.
Leave 20 minutes on ice. Vortex and do a quick spin to bring all samples down.
Heat at 37 °C for 30 minutes (see Note 18). Vortex and do a quick spin to bring all samples down.
Heat at 100 °C for 20 minutes. Vortex and do a quick spin to bring all samples down.
Leave at 60 °C for 2 hours. Vortex and do a quick spin to bring all samples down.
Sonicate for 5 minutes and do a quick spin to bring all samples down.
The lysates are now ready for 1) RCDC protein assay if the protein concentration needs to be measured (see Note 19); 2) Add Laemmli buffer to run samples in a Bis-Tris gel if a set number of cuts were obtained (see Note 20); or 3) directly proceed to step 1 of the iST Sample Preparation Kit from PreOmics.
3.6. Sample preparation for mass spectrometry using limited gel
Use a 7.5% Mini-Protean TGX gels (BioRad) or alternatively, you can polymerize an 8% acrylamide Bis-Tris gel (see Note 21).
Calculate the protein concentration for each sample (for example, for the orbitrap LC-MS/MS we ran 2 and 5 μg of protein) or load a pre-determined number of cuts (see Note 20).
Add the corresponding volume of 5X Laemmli buffer containing 150 mM DTT, vortex and spin the samples.
Boil (100 °C) the samples for 5 minutes.
When the samples are cool, vortex and spin again.
Load the samples on the acrylamide gel.
Run the gel at 30 mAmp, or about 5–10 min, down to ~1 cm inside the gel (enough to visualize the MW marker ladder separation) (see Note 22).
Stop running the gel and fix it for 60 minutes in the fixing solution (Water:Methanol:Acetic acid = 40:40:8).
Inside a biosafety cabinet, excised each lane carefully from the gel (see Note 23).
Put the sliced gel piece in a 1.5 ml centrifuge tube filled with 1% acetic acid in molecular biology grade water. The samples can be stored in this solution at 4° C until analyzed by MS.
Standard MS sample preparation should be performed (see Note 24).
Briefly, for in-gel digestion, dice the samples into 1mm × 1mm squares, rinse multiple times with 50 mM ammonium bicarbonate and reduce with 5 mM DTT, 50 mM ammonium bicarbonate at 55°C for 30 min.
Remove the residual solvent and perform alkylation using 10 mM propionamide in 50 mM ammonium bicarbonate for 30 min at room temperature.
Rinse the gel pieces with 50% acetonitrile, 50 mM ammonium bicarbonate and place in a speed vacuum for 5 min.
Digest with trypsin/LysC (Promega, Madison, WI, USA) overnight at 37°C.
Peptide extraction: Spin down the tubes and collect the solvent, which contain the peptides. Add 60% acetonitrile, 39.9% water, 0.1% formic acid and incubated for 10–15 min. The peptide pools can be dried in a speed vacuum.
Reconstitute the digested peptide pools and injected onto a 100 micron I.D. C18 reversed phase analytical column (Dr. Maisch, Ammerbuch-Entringen, Germany; 2.4 μM Reprosil-Pur) 25–50 cm in length. The UPLC was a Waters M class, operated at 300 nL/min using a linear gradient from 4% mobile phase B to 35% B. Mobile phase A consisted of 0.2% formic acid, 5% DMSO and water; mobile phase B was 0.2% formic acid, 5% DMSO, acetonitrile.
For data collected using the Orbitrap Fusion mass spectrometer, it was set to acquire data in a data dependent fashion selecting and fragmenting by collision-induced dissociation the most intense precursor ions optimized to maximize duty cycle. An exclusion window of 60 seconds was used to improve proteomic depth and multiple charge states of the same ion were not sampled.
For data collected using the timsTOF Pro mass spectrometer in PASEF mode, the mobility range used was 0.7 1/K0 to 1.5 1/K0 and the ramp time 100ms. The effective duty cycle was 120Hz, where the dynamic exclusion setting was set to 0.4 min.
3.7. Sample preparation for mass spectrometry using iST Sample Preparation Kit from PreOmics
As an alternative to using limited gels, the samples can be directly prepared using the iST Sample Preparation Kit from PreOmics. This allows for a fast, and complete sample preparation that minimize possible background contamination. The kit can be used with samples as small as 1 ug of protein. In our case, we were able to use it for small cellular protrusions, like filopodia. The kit includes lysis, digest and peptide purification protocols (see Note 25).
Follow the manufacturer’s instructions.
The peptide pool is obtained at the end, ready to be run for MS.
3.8. Data Analysis
MS data analysis will vary depending on the mass spectrometer and software used. In our case, the MS data were analyzed using Preview and Byonic v2.6.49 (Protein Metrics) as well as custom tools for data analysis developed in MatLab at Stanford University. Peak selection was handled automatically within Byonic.
MS/MS data were searched against a UniProtKB FASTA database containing 16,972 reviewed Mus musculus entries (various dates). Propionamidation (+71.037114 @ C) was set as a fixed modification, Deamination (+0.984016 @ N) and Acetylation (+42.010565 @ K) were set as a common1 modifications, Oxidation (+15.994915 @ M) was set as common2 modification, and Acetylation (+42.010565 @ Protein N-term) and Methylation (+14.01565 @ K, R) were set as rare1 modifications. Byonic was set to allow a maximum of two common modifications and one rare modification and to allow a maximum of two missed cleavages. MS/MS spectra were matched with a tolerance of 12 ppm on precursor mass and 0.4 Da on fragment mass.
Common contaminants were filtered automatically by Byonic and include: TRYP_PIG, ALBU_BOVIN, ALBU_HUMAN, CASB_BOVIN, CASK_BOVIN, CAS1_BOVIN, CO3_HUMAN, HBA_HUMAN, HBB_HUMAN, K1M1_SHEEP, K2C1_HUMAN, K22E_HUMAN, K1C10_HUMAN, K1C15_SHEEP, K1C9_HUMAN, KRHB1_HUMAN, KRHB3_HUMAN, KRHB5_HUMAN, KRHB6_HUMAN, TRFE_HUMAN.
Using Byonic, the proteome was searched with a reverse-decoy strategy and all data filtered and presented at a 1% false discovery rate. Byonic calculates a Byonic score that is an indicator of the correctness of our peptide-spectrum matches (PSM). Byonic scores reflect the absolute quality of the PSM. Byonic scores range from 0 to 1000, with 300 being a good score, 400 a very good score, and scores over 500 reflecting near perfect matches. For our data, all filtered protein identification hits have an FDR rate lower or equal to 2.5%, a Byonic score greater than 250 and a log probability greater than 3.
To further discriminate non-significant to statistically significant proteins, we also recommend running a statistical power analysis [32].
Once the statistically significant proteins are determined, a spectral counting method known as normalized spectral abundance factor (NSAF) [33] can be used to determine the relative abundance of proteins in the samples [33, 34, 27], and to verify the reproducibility, between replicates, of the quantitative data through a correlation analysis [34].
Once the statistically significant proteins are determined, along with their relative abundance, a number of analyses can be performed to look at functional and/or localization differences [27]. The type of analyses will depend on the experimental set-up and research questions and should be determined by each investigator.
A number of programs can be used to do these analyses including R, JMP, SSPS, or Matlab for statistical analysis; the R package SuperExactTest for the statistical analysis and visualization of multi-set intersections [35]; DAVID [36, 37], Proteomaps [38], Panther [39], or GOrilla [40, 41] for gene ontology enrichment analysis, and COMPLEAT for a subcellular localization enrichment analysis [27].
4. Notes
All fixation solutions must be made fresh, just prior to using them. Do not store the solutions.
DTBP needs to be stored at 4° C but equilibrated to room temperature prior to use (30 minutes). The stock solution of 50 mM will turn cloudy in approximately 5 minutes. Use the stock before cloudiness occurs otherwise, discard it. Reaction pH is critical, basic conditions (pH 8–10) favor mechanism of action of crosslinking by DTBP. It must be made fresh. Do not store the solution.
RIPA buffer (without DTT and protease inhibitors) can be stored at 4°C. However, DTT and protease inhibitors should be added freshly before use.
5X Laemmli buffer can be stored for several months at − 20 °C but DTT should be added freshly before use.
Can be stored at room temperature for extended time but protected from light. It might start to turn yellow overtime but this color change does not affect its use.
Cells must adhere well to the membrane in order to isolate them and/or cellular protrusions by LCM. However, whether you use fibronectin, poly-lysine, collagen or any other extracellular matrix protein coatings you need to make sure that it does not hinder the formation of the cellular protrusion of interest.
MMI suggests that you seed the cells in the ring and later move it to the UV-permeable microdissection chamber. However, the most important issue when using a MMI live chamber is to ensure that there is no leakage once the ring is attached to the microdissection chamber. If the ring leaks and wets the bottom of the membrane prior to putting inside the microdissection chamber, it will not properly attach to the adhesive area of the chamber. As a result, the LCM isolated cut will not stick to it and will not be collected after removal of the ring. For this reason, we always attach the ring to the microdissection chamber first and only after asserting that it does not leak, we seed the cells. It is important to minimize bubbles and wrinkles when setting the ring in the microdissection chamber.
Do not disturb the cells by moving the dish after seeding. Leave the cells in the incubator so that they can attach to the membrane and make protrusions. The time of adhesion and formation of cellular protrusions depends on both the cell types used and the types of cellular protrusions of interest. In our case, we incubate CAD cells for 2 to 3 hours, when looking for filopodia and/or TNTs but waited up to 10 days with media change every 2–3 days for axons and dendrites in order to allow for cell differentiation upon serum starvation. The time must be pre-determined by each investigator based on their cell/protrusion types.
Many different ways to induce cell protrusions have been published. Test different substances or physical conditions (i.e. hypoxia) that can work with your cell type to increase the number of cellular protrusions you are interested in. Remember that for each treatment, whole cell control samples should be acquired.
The proper stimuli and or concentrations should be pre-determined by the investigator based on the cell type used and cellular protrusions of interest. For instance, prolong H2O2 exposure and/or high concentration can be harmful to the cells and could result in the cells detaching from the membrane. In addition, to avoid disturbing the cells, care should be taken when adding and aspirating solutions.
Time of crosslinking is critical. Proceed to the next step immediately to avoid over-crosslinking.
All the procedures with the LCM must be done with filtered PBS in the dish, to avoid protein degradation [26].
Because time degrades proteins, longer periods of cutting must be avoided (see Fig. 1D).
If your cuts are limited to a specific area, it helps if you outline with a marker on the bottom of the dissection chamber the desired area.
Both protrusion and membrane samples can be stored at − 80°C for several weeks.
Silver stain is not compatible with some types of MS.
Since the sample are in small volumes, the caps of the centrifuge tubes should be tightly closed and surrounded with parafilm to avoid evaporation and drying of the samples.
DTBP-crosslinked proteins can be cleaved by reducing the disulfide bond of the spacer arm with 100–150 mM DTT at 37 °C for 30 minutes (See reagent setup), thus freeing the isolated proteins from the cross-linked product.
RCDC is compatible with detergents (such as SDS) and reducing agents (such as DTT). Bradford and BCA are compatible with reducing agents and detergents, respectively. Make a standard curve with BSA and use as blank the protein extraction supernatant of a membrane from a prepared MMI live chamber dish but never seeded with cells. Fibronectin is a protein and for that reason, it is likely that fibronectin coating increases protein results.
RCDC can be less reliable for very small samples of isolated cellular protrusions and it takes away a large amount of the isolated samples. In order to use all of the LCM isolated cellular protrusions for MS analysis, we recommend determining what the total number of cuts required are and avoid using the RCDC. The number of cuts required will vary depending on the cellular protrusion types and should be pre-determined by each investigator. When using the orbitrap LC-MS/MS we ran 2 and 5 μg of total protein or 3,000 to 6,000 cuts, depending on the types of cellular protrusions isolated. When using the timsTOF Pro mass spectrometer, the number of cuts was reduced due to the greater sensitivity of this mass spectrometer compared to the Orbitrap.
To avoid keratin contamination all these steps and the required solutions should be done in a chemical hood and with molecular grade water.
This “limited gel” allows for the separation of the proteins by size in preparation for MS and each lane can be run as one sample by MS.
It is easier to cut the samples if they are loaded between stained ladders.
Because of the minute sample size, only very sensitive mass spectrometers should be used. We recommend using either the Thermo Scientific Orbitrap Fusion mass spectrometer or the timsTOF Pro mass spectrometer from Bruker.
The iST sample preparation takes about 2 and half hours and includes all required steps, such as lysis to denature, reduce and alkylate proteins, digestion using a LysC and Trypsin mix and peptide purification, which includes removal of hydrophilic contaminants.
ACKNOWLEDGEMENT
This work was supported by a 2014 CSUPERB New Investigator Grant and the National Institute of General Medical Sciences of the National Institutes of Health under Award Number SC2GM111144 awarded to KG.
REFERENCES
- 1.Hanson JC, Tangrea MA, Kim S, et al. (2011) Emmert-Buck, Expression microdissection adapted to commercial laser dissection instruments. Nat Protoc 6:457–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grünewald A, Rygiel KA, Hepplewhite PD, et al. (2016) Mitochondrial DNA Depletion in Respiratory Chain-Deficient Parkinson Disease Neurons. Ann Neurol 79:366–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Drummond ES, Nayak S, Ueberheide B, Wisniewski T (2015) Proteomic analysis of neurons microdissected from formalin-fixed, paraffin-embedded Alzheimer’s disease brain tissue. Sci Rep 5:15456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nawandar DM, Wang A, Makielski K, et al. (2015) Differentiation-Dependent KLF4 Expression Promotes Lytic Epstein-Barr Virus Infection in Epithelial Cells. PLoS Pathog 11: e1005195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pflugradt R, Schmidt U, Landenberger B, et al. (2011) A novel and effective separation method for single mitochondria analysis. Mitochondrion 11:308–314. [DOI] [PubMed] [Google Scholar]
- 6.Zivraj KH, Tung YC, Piper M (2010) Subcellular profiling reveals distinct and developmentally regulated repertoire of growth cone mRNAs. J Neurosci 30:15464–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ezzoukhry Z, Henriet E, Cordelières FP (2018) Combining laser capture microdissection and proteomics reveals an active translation machinery controlling invadosome formation. Nat Commun 9:2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gutstein HB, Morris JS, Annangudi SP, Sweedler JV (2008) Microproteomics: analysis of protein diversity in small samples. Mass Spectrom Rev 27:316–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gambade A, Zreika S, Guéguinou M, et al. (2016) Activation of TRPV2 and BKCa channels by the LL-37 enantiomers stimulates calcium entry and migration of cancer cells. Oncotarget doi: 10.18632/oncotarget.8122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Costanzo M, Abounit S, Marzo L, et al. (2013) Transfer of polyglutamine aggregates in neuronal cells occurs in tunneling nanotubes. J Cell Sci 126:3678–3685. [DOI] [PubMed] [Google Scholar]
- 11.Möller J, Lühmann T, Chabria M, et al. (2013) Macrophages lift off surface-bound bacteria using a filopodium-lamellipodium hook-and-shovel mechanism. Sci Rep 3:2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dent EW, Gupton SL, Gertler FB (2011) The growth cone cytoskeleton in axon outgrowth and guidance. Cold Spring Harb Perspect Biol 3:pii: a001800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Audenhove I, Denert M, Boucherie C, et al. (2016) Fascin Rigidity and L-plastin Flexibility Cooperate in Cancer Cell Invadopodia and Filopodia. J Biol Chem 291:9148–9160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gousset K, Schiff E, Langevin C, et al. (2009) Prions hijack tunnelling nanotubes for intercellular spread. Nat Cell Biol 11:328–336. [DOI] [PubMed] [Google Scholar]
- 15.Eugenin EA, Gaskill PJ, Berman JW (2009), Tunneling nanotubes (TNT) are induced by HIV-infection of macrophages: a potential mechanism for intercellular HIV trafficking. Cell Immunol 254:142–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thayanithy V, Dickson EL, Steer C, et al. (2014) Tumor-stromal cross talk: direct cell-to-cell transfer of oncogenic microRNAs via tunneling nanotubes. Transl Res 164:359–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brayford S, Bryce NS, Schevzov G, et al. (2016) Tropomyosin Promotes Lamellipodial Persistence by Collaborating with Arp2/3 at the Leading Edge. Curr Biol pii: S0960–9822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sherer NM, Mothes W (2008) Cytonemes and tunneling nanotubules in cell-cell communication and viral pathogenesis. Trends Cell Biol 18:414–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thomsen R, Lade Nielsen A (2011) A Boyden chamber-based method for characterization of astrocyte protrusion localized RNA and protein. Glia 59:1782–1792. [DOI] [PubMed] [Google Scholar]
- 20.Kadiu I, Gendelman HE (2011) Human Immunodeficiency Virus type 1 Endocytic Trafficking through Macrophage Bridging Conduits Facilitates Spread of Infection. J Neuroimmune Pharmacol 6:658–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mili S, Moissoglu K, Macara IG (2008) Genome-Wide Screen Identifies Localized RNAs Anchored At Cell Protrusions Through Microtubules And APC. Nature 453:115–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mimae T, Ito A (2015) New challenges in pseudopodial proteomics by a laser-assisted cell etching technique. Biochim Biophys Acta 1854:538–546. [DOI] [PubMed] [Google Scholar]
- 23.Gousset K, Marzo L, Commere P, Zurzolo C (2013) Myo10 is a key regulator of TNT formation in neuronal cells. J Cell Sci 126:4424–4435. [DOI] [PubMed] [Google Scholar]
- 24.Eltoum I, Fredenburgh J, Myers RB, Grizzle WE (2001) Introduction to the Theory and Practice of Fixation of Tissues. J Histotech 24:173–190. [Google Scholar]
- 25.Gosselin MA, Guo W, Lee RJ (2001) Efficient Gene Transfer Using Reversibly Cross-Linked Low Molecular Weight Polyethylenimine. Bioconjug Chem 12:989–994. [DOI] [PubMed] [Google Scholar]
- 26.Gordon A, Kannan SK, Gousset K (2018) A Novel Cell Fixation Method that Greatly Enhances Protein Identification in Microproteomic Studies Using Laser Capture Microdissection and Mass Spectrometry. Proteomics 18:e1700294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gousset K, Gordon A, Kumar Kannan S, Tovar (2019) A novel Microproteomic Approach Using Laser Capture Microdissection to Study Cellular Protrusions. Int J Mol Sci 20:pii: E1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Osswald M, Jung E, Sahm F, et al. (2015) Brain tumour cells interconnect to a functional and resistant network. Nature 528:93–98. [DOI] [PubMed] [Google Scholar]
- 29.Desir S, Dickson EL, Vogel RI, et al. (2016) Tunneling nanotube formation is stimulated by hypoxia in ovarian cancer cells. Oncotarget doi: 10.18632/oncotarget.9504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hashimoto M, Bhuyan F, Hiyoshi M, et al. (2016) Potential Role of the Formation of Tunneling Nanotubes in HIV-1 Spread in Macrophages. J Immunol 196:1832–41. [DOI] [PubMed] [Google Scholar]
- 31.Victoria GS, Arkhipenko A, Zhu S, et al. (2016) Astrocyte-to-neuron intercellular prion transfer is mediated by cell-cell contact. Sci Rep 6:20762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Levin Y (2011) The role of statistical power analysis in quantitative proteomics. Proteomics. 11:2565–7. doi: 10.1002/pmic.201100033. [DOI] [PubMed] [Google Scholar]
- 33.Arike L, Peil L (2014) Spectral counting label-free proteomics. Methods Mol. Biol 1156: 213–222. [DOI] [PubMed] [Google Scholar]
- 34.McIlwain S, Mathews M, Bereman MS, Rubel EW, MacCoss MJ, Noble WS (2012) Estimating relative abundances of proteins from shotgun proteomics data. BMC Bioinformatics. 13:308. doi: 10.1186/1471-2105-13-308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang M, Zhao Y, Zhang B (2015) Efficient test and visualization of multiset intersections. Sci. Rep 5:16923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang DW, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources. Nature Protoc. 4:44–57. [DOI] [PubMed] [Google Scholar]
- 37.Huang DW, Sherman BT, Lempicki RA (2009) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res.37:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liebermeister W, Noor E, Flamholz A, Davidi D, Bernhardt J, and Milo R (2014) Visual account of protein investment in cellular functions. Proc Natl Acad Sci U S A. 111: 8488–8493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mi H, Muruganujan A, Ebert D, Huang X, Thomas PD. (2019) PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 47: D419–D426. doi: 10.1093/nar/gky1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z (2009) “GOrilla: A Tool For Discovery And Visualization of Enriched GO Terms in Ranked Gene Lists”, BMC Bioinformatics 10:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eden E, Lipson D, Yogev S, Yakhini Z (2007) “Discovering Motifs in Ranked Lists of DNA sequences”, PLoS Computational Biology, 3:e39. [DOI] [PMC free article] [PubMed] [Google Scholar]