

SUMMARY
Tumor genotype can influence the immune microenvironment, which plays a critical role in cancer development and therapy resistance. However, the immune effects of gain-of-function Trp53 mutations have not been defined in pancreatic cancer. We compare the immune profiles generated by KrasG12D-mutated mouse pancreatic ductal epithelial cells (PDECs) engineered genetically to express the Trp53R172H mutation with their p53 wild-type control. KrasG12D/+;Trp53R172H/+ tumors have a distinct immune profile characterized by an influx of CD11b+Ly6G+ neutrophils and concomitant decreases in CD3+ T cells, CD8+ T cells, and CD4+ T helper 1 cells. Knockdown of CXCL2, a neutrophil chemokine, in the tumor epithelial compartment of CRISPR KrasG12D/+;Trp53R172H/+ PDEC tumors reverses the neutrophil phenotype. Neutrophil depletion of mice bearing CRISPR KrasG12D/+;Trp53R172H/+ tumors augments sensitivity to combined CD40 immunotherapy and chemotherapy. These data link Trp53R172H to the presence of intratumoral neutrophils in pancreatic cancer and suggest that tumor genotypes could inform selection of affected individuals for immunotherapy.
In brief
Siolas et al. demonstrate that the gain-offunction Trp53R172H mutation promotes CD11b+Ly6G+ neutrophil recruitment to KrasG12D pancreatic tumors, which is distinct from tumors with loss-of-function or wild-type Trp53. The presence of neutrophils in the tumor microenvironment promotes resistance to combination CD40 immunotherapy and chemotherapy treatment, suggesting that tumor genotype may guide therapy selection.
Graphical Abstract
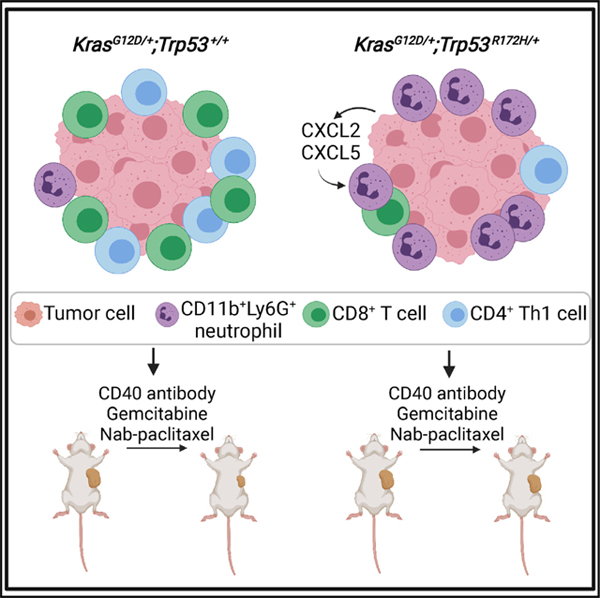
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is an intractable disease with a low survival rate, limited treatment options, and a poor therapy response rate (Raj et al., 2017). This is in part due to the immunosuppressive and fibrotic microenvironment of pancreatic tumors (Murakami et al., 2019). PDAC progression is typified by a multitude of immunosuppressive cells, including regulatory T cells, myeloid-derived suppressor cells (MDSCs), neutrophils, and tumor-associated macrophages (TAMs), leading to immune evasion (Hiraoka et al., 2006; Ho et al., 2020; Lesina et al., 2011; Steele et al., 2015). There is an urgent need to increase our understanding of how tumors create an immunosuppressive microenvironment that leads to tumor growth and therapy resistance (Martinez-Bosch et al., 2018).
Different tumor genetic backgrounds can profoundly influence the composition of the immune microenvironment, as demonstrated in melanoma, breast, and prostate cancers, through distinct cellular mechanisms (Bezzi et al., 2018; Spranger et al., 2015; Wellenstein et al., 2019). For example, activation of β-catenin signaling in melanoma cells leads to T cell exclusion and resistance to checkpoint immunotherapy in genetically engineered mouse models (GEMMs) (Spranger et al., 2015). In prostate cancer GEMMs, loss of the tumor suppressor gene Pten alone or in combination with loss of Trp53, Zbtb7a, or Pml resulted in diverse immune profiles affecting MDSCs, monocytes, and T cells (Bezzi et al., 2018). Although changes to the immune microenvironment over time have been described during development of pancreatic cancer, how distinct immune cell populations correlate to specific tumor genotypes has not been determined (Clark et al., 2007).
In PDAC, KRAS mutations are an initiating genetic event and the most frequent gene alteration, occurring in over 90% of tumors (Raphael et al., 2017). Several labs have demonstrated that KRASG12D mutations in tumor cells remodel the pancreatic immune microenvironment by recruiting immunosuppressive MDSCs and regulatory T cells (Bayne et al., 2012; Mathew et al., 2014; Pylayeva-Gupta et al., 2012; Zhang et al., 2017), but the effects of additional mutations have not been explored.
TP53 is mutated in up to 70% of PDACs, second in mutation frequency only to KRAS (Raphael et al., 2017). Homozygous TP53 deletions, found in less than 2% of tumors, are a much rarer genetic event than p53 mutation in human PDAC (Raphael et al., 2017). Trp53 deletions occurring in the context of oncogenic Kras mutations have been demonstrated experimentally to drive recruitment of CD11b+F480+ macrophages in autochthonous pancreatic tumor models through increased expression of CXCR3and CCR2-associated chemokines and macrophage colony-stimulating factor (M-CSF) (Blagih et al., 2020). However, most alterations affecting TP53 in human PDAC are missense mutations affecting the DNA-binding domain, which may be gain-of-function mutations that bestow neomorphic capabilities or loss-of-function mutations that abrogate its tumor suppressor activity (Pfister and Prives, 2017).
An integrated genomics analysis of 456 human PDACs defined four subtypes of pancreatic cancer (Bailey et al., 2016). In particular, the squamous subtype is correlated with TP53 mutations and has been observed to be enriched for a distinct immune cell-specific gene expression signature (Bailey et al., 2016). A separate study found that the human PDAC squamous subtype was enriched significantly for infiltration of tumor-associated neutrophils, suggesting a connection between TP53 mutations and neutrophils (Chao et al., 2016). In addition, tumors from KrasG12D;Trp53R172H;Cxcr2−/− - GEMM mice, whose intratumoral neutrophil recruitment is abrogated because of deletion of the CXCR2 receptor, had decreased expression of genes associated with the PDAC squamous subtype compared with controls (Chao et al., 2016; Steele et al., 2016). Neutrophil presence (Chao et al., 2016; Steele et al., 2016) and Trp53R172H gain-of-function mutation in tumor cells (Morton et al., 2010; Weissmueller et al., 2014) have been implicated independently in promoting metastasis in mouse models of PDAC. Although these studies implicate the Trp53R172H mutation in mediating immune regulation, the immune effects directly attributable to acquisition of the gain-of-function Trp53R172H mutation in pancreatic ductal epithelial cells have not been characterized.
Here we used CRISPR-Cas9 to engineer a Trp53R172H mutation in primary murine pancreatic ductal epithelial cells (PDECs) derived from genetically engineered KrasG12D mice. Orthotopic implantation of these cell lines into wild-type immunocompetent C57BL/6 mice revealed that CRISPR KrasG12D/+;Trp53R172H/+ PDEC tumors have a distinct immune profile characterized by an increase in intratumoral neutrophils and a concomitant decrease in T cells compared with KrasG12D/+;Trp53+/+ lesions. Correspondingly, CRISPR KrasG12D/+;Trp53R172H/+ PDEC tumor epithelial cells produced elevated levels of two chemokines involved in neutrophil accumulation, CXCL2 and CXCL5, but only knockdown of CXCL2 in CRISPR KrasG12D/+;Trp53R172H/+ PDEC tumors abrogated neutrophil recruitment. Finally, depletion of neutrophils from CRISPR KrasG12D/+;Trp53R172H/+ PDEC tumors augmented sensitivity to CD40 agonist combination chemotherapy and immunotherapy. These results support a role of the intratumoral Trp53R172H mutation in modulating the tumor microenvironment in pancreatic cancer and mediating therapeutic response to immunotherapy.
RESULTS
Introduction of a Trp53R172H gain-of-function mutation into KrasG12D PDECs through conditionally active CRISPR-Cas9
To understand the potential connection between the immune microenvironment and gain-of-function mutant Trp53R172H, we used PDECs obtained from KrasG12D/+;Trp53+/+ GEMMs. Because these can be cultivated ex vivo (Pylayeva-Gupta et al., 2012), they are amenable to genetic targeting, allowing us to focus selectively on Trp53. We targeted the most common TP53 mutation in human pancreatic cancer, R175H (Bailey et al., 2016; Witkiewicz et al., 2015), which is the equivalent of mouse R172H. To create isogenic PDEC lines with a defined Trp53 mutation while minimizing off-target effects, we selected a conditional CRISPR-Cas9 gene editing platform in which a FKBP12-derived destabilizing domain is fused to Cas9 (DD-Cas9), enabling conditional Cas9 expression and temporal control of gene editing in the presence of an FKBP12 small-molecule synthetic ligand, Shield-1 (Senturk et al., 2017). Following genetic sequence verification, CRISPR KrasG12D/+;Trp53+/+ or KrasG12D/+;Trp53R172H/+ cells were implanted orthotopically into the pancreata of wild-type mice, and tumors that formed were harvested 2 weeks later (Figure 1A). Compared with KrasG12D/+;Trp53+/+ tumors, CRISPR KrasG12D/+;Trp53R172H/+ tumors had a higher tumor volume and weight (Figure 1B) and displayed more aggressive histological features, including a complex architecture consisting of poor gland formation, predominantly solid tumor growth, and abundant neutrophilic inflammatory infiltrates (Figure 1C), indicating that the edited Trp53R172H mutation promoted neoplastic progression.
Figure 1. Introduction of a p53 gain-of function mutation in KrasG12D PDECs through conditionally active CRISPR-Cas9.

(A) Experimental design. Representative gross tumors are shown.
(B) Tumor weight. Each dot represents one mouse. n = 6. Error bars, ± SD.
(C) Representative images of tumor sections stained by H&E; scale bars, 100 μm.
(D) mRNA transcript levels as measured by qPCR. The graph shows quantification of the ratio of transcripts for doxorubicin-treated versus untreated cells. Error bars, ± SEM.
(E) Left: representative images of tumor sections stained for CK19, Ki-67, and DAPI by immunofluorescence. Scale bars, 100 μm. Right: Ki-67 quantification per field of view. Each point on the graph represents one mouse; n = 5 (KrasG12D/+; Trp53R172H/+), n = 4 (KrasG12D/+;Trp53+/+). Error bars, ± SD.
For (B), (D), and (E), Student’s t test (two-tailed, unpaired); *p < 0.05, **p < 0.01, ****p < 0.0001.
Next we sought to confirm the functionality of mutant Trp53R172H in CRISPR-Cas9-modified cells by examining the expression of well-established target genes of Trp53R172H. Specifically, we chose to focus on the Ccna, Ccnb1, Ccnb2, and Cdk1 cell cycle control genes, whose expression has been shown to increase after DNA damage through Trp53R172Hdependent activation of nuclear transcription factor Y (NF-Y) (Di Agostino et al., 2006). Exposure of CRISPR-Cas9-modified cells to the DNA-damaging agent doxorubicin was accompanied by higher expression of Ccna, Ccnb1, Ccnb2, and Cdk1 in CRISPR KrasG12D/+;Trp53R172H/+ cells in comparison with KrasG12D/+;Trp53+/+ cells (Figure 1D), consistent with the gain-of-function mode of action attributed to this mutation. In addition, CRISPR KrasG12D/+;Trp53R172H/+ tumors displayed higher proliferative rates compared with KrasG12D/+;Trp53+/+ tumors, as determined by epithelium-associated Ki-67 staining (Figure 1E). Our analyses demonstrate the feasibility of introducing a Trp53R172H gain-of-function mutation using a conditionally active CRISPR-Cas9 system and the utility of this approach for creation of an isogenic model system to pursue Trp53R172H functional studies.
Acquisition of the Trp53R172H gain-of-function mutation promotes neutrophil recruitment
To assess the immune landscape of gain-of-function mutant Trp53R172H tumors, we performed immune profiling of mouse orthotopic tumors using multicolor flow cytometry 2 weeks after implantation. CRISPR KrasG12D/+;Trp53R172H/+ tumors displayed a significantly elevated proportion of tumor-associated neutrophils (CD11b+Ly6G+) in comparison with KrasG12D/+;Trp53+/+ lesions (Figure 2A). This was confirmed by immunohistochemistry (IHC) analysis (Figure S1A) and remains significant when accounting for tumor weight (Figure S1B). The infiltration of neutrophils is in contrast to the lack of observed changes in other cells of myeloid lineage, including TAMs (CD11b+F480+), M1 (F480+MHCII+CD80+CD86+) or M2 (F480+MHCII–CD206+) macrophages (Figures 2B–2D), CD11c+ dendritic cells, or CD11c+major histocompatibility complex (MHC) class II+ classical dendritic cells (Figures S1C and S1D).
Figure 2. Acquisition of p53R172H by PDEC KrasG12D tumors alters their immune profile in vivo.

(A–G) Flow cytometric plots and quantification of (A) neutrophils, (B) TAMs, (C) M1 macrophages, (D) M2 macrophages, (E) CD3+ T cells, (F) T helper cells and CD8+ T cells, and (G) Th1 cells. n = 6–8; error bars, ± SD. Student’s t test (two-tailed, unpaired). **p < 0.01, ****p < 0.0001; n.s., not significant.
(H) Flow cytometry plots (left) and quantification (right) of neutrophils from orthotopically implanted CRISPR KrasG12D/+;Trp53+/+ tumors or CRISPR KrasG12D/+; Trp53−/− tumors. n = 8; error bars, ± SD. Student’s t test (two-tailed, unpaired).
(I) Gene Ontology analysis of the human TCGA PDAC cohort for genes differentially expressed with p < 0.05 in p53 wild-type (n = 63) compared with p53 mutant tumors (n = 58), highlighting the top significantly altered immune related pathways. See also Figures S1–S3.
Examination of the T cell compartment in CRISPR-generated tumors by flow cytometry demonstrated decreases in CD3+ T cells, CD8+ T cells, and Th1 helper cells (CD4+Tbet+) in CRISPR KrasG12D/+;Trp53R172H/+ tumors, but there were no changes in CD4+ T cells (Figures 2E–2G). The observed changes in the T cell compartment are consistent with published reports demonstrating an inverse relationship between neutrophil accumulation and T cell infiltration in pancreatic cancer (Chao et al., 2016; Steele et al., 2015). There were no observed changes in CD4+Foxp3+ regulatory T cells or PD1+ CD8+ T cells (Figures S1E and S1F). All flow cytometry analyses were carried out using three independent sequence-verified CRISPR-Cas9-modified clones from each genotype (Figures S2A–S2H). No significant inter-clonal variations were observed, ruling out the possibility that the observed immune profiles are clone dependent. Furthermore, immunofluorescence detection of neutrophils in pancreatic tumors derived from mouse models of autochthonous PDAC driven by a pancreas-specific G12D mutation in KRAS that carried wildtype p53 (KC model: KrasLSL-G12D/+; Trp53+/+;p48Cre/+) or the pancreas-specific R172H mutation in p53 (KPC model: KrasLSL-G12D/+;Trp53LSL-R172H/+;p48Cre/+) revealed a significantly higher neutrophil density number, as indicated by the higher number of myeloperoxidase (MPO+) cells in KPC tumors compared with KC tumors (Figure S3A). These data suggest that acquisition of the gain-of-function Trp53R172H mutation leads to intratumoral neutrophil accumulation. To examine whether this immune phenotype is specific to the Trp53R172H mutation, we used CRISPR-Cas9 gene editing to create a homozygous p53 deletion in our KrasG12D/+;Trp53+/+ PDECs (Figure S3B). CRISPR KrasG12D/+;Trp53−/− orthotopic tumors did not display an increase in intratumoral neutrophil accumulation compared with CRISPR KrasG12D/+;Trp53+/+ tumors (Figure 2H; Figure S3C), indicating that the Trp53R172H mutation exerts a distinct immune phenotype to the tumor microenvironment.
To further explore this observation, we examined the human pancreatic adenocarcinoma cohort from The Cancer Genome Atlas (TCGA) for genes significantly differentially expressed (p < 0.05) between tumors with missense mutations in the DNA binding domain of TP53 compared with tumors with wild-type TP53. Highly frequent “hotspot” missense mutations located in the p53 DNA binding domain are a key feature of gain-of-function activity (Bargonetti and Prives, 2019). Although multiple metabolic pathways associated with mutant TP53 function (Alvarado-Ortiz et al., 2021; Liu et al., 2019) were highly upregulated by Gene Ontology analysis in the mutated PDAC tumors, neutrophil-related pathways, including neutrophil-mediated immunity, neutrophil activation, and neutrophil degranulation, were also enriched significantly (Figure 2I). Because of the strong association between purported gain-of-function p53 mutations and neutrophils in pancreatic cancer in humans and mice, we focused on the mechanism of neutrophil infiltration in Trp53R172H tumors.
Neutrophil infiltration in CRISPR KrasG12D/+;Trp53R172H/+ tumors is dependent on tumor-cell-derived chemokines
Neutrophil recruitment is mainly mediated by chemokines that have a glutamate-leucine-arginine motif (ELR+ chemokines), which consist primarily of CXCL1, CXCL2, and CXCL5 (Disteldorf et al., 2015). CXCR2, a G-protein-coupled receptor, regulates neutrophil migration through these chemokines (Jaffer and Ma, 2016). Human PDAC tumors display increased expression of CXCL2 and CXCL5 in comparison with normal pancreas (Figure 3A), suggesting that production of these chemokines may be responsible for intratumoral neutrophil accumulation. To assess the relevance of this recruitment mechanism to the infiltration of neutrophils observed in CRISPR KrasG12D/+; Trp53R172H/+ tumors, we analyzed the relative levels of CXCL2 and CXCL5 production by quantitative PCR (qPCR) in tumor epithelial cells (CD45–PDGFR–CD34–EPCAM+ epithelium) and CD45+ intratumoral immune cells isolated by flow cytometry from CRISPR KrasG12D/+;Trp53R172H/+or KrasG12D/+;Trp53+/+ tumors. The epithelial compartment of CRISPR KrasG12D/+; Trp53R172H/+ tumors displayed significantly higher levels of Cxcl2 and Cxcl5 expression than the epithelial compartment of CRISPR KrasG12D/+;Trp53+/+ tumors (Figure 3B). Enhanced production of the CXCL2 and CXCL5 chemokines in the CRISPR KrasG12D/+;Trp53R172H/+ tumors in comparison to KrasG12D/+; Trp53+/+ was confirmed using mouse chemokine magnetic bead multiplex immunoassay (Figure S3D). Focusing on CRISPR KrasG12D/+;Trp53R172H/+ tumors, we found that CXCL5 expression was significantly higher in tumor epithelial cells than in the immune cell compartment, whereas the reverse was true for CXCL2 expression (Figure S3E). High levels of CXCL2 in the microenvironment are not unexpected because neutrophils can express abundant CXCL2 to amplify neutrophil recruitment and activation in an autocrine and/or paracrine manner (Li et al., 2016).
Figure 3. Neutrophil infiltration in CRISPR KrasG12D/+;Trp53R172H/+ tumors is dependent on chemokine secretion from p53 mutant cells.

(A) mRNA transcript levels in PDACs compared with normal adjacent tissue (n = 75 PDACs and n = 55 adjacent normal cells). Each data point indicates an individual tissue sample. Error bars, ± SD.
(B) mRNA expression levels as measured by qPCR. Error bars, ± SEM; n = 3 using pooled samples of 5 tumors each.
(C) mRNA expression levels in vitro measured by qPCR. Error bars, ± SEM; n = 3–4.
(D) Gross histology (top) and graph of tumor weight (bottom, in milligrams) from CRISPR KrasG12D/+;Trp53R172H/+ tumors stably expressing scramble shRNA (shScr) and shRNA specific for CXCL5 (shCXCL5) or CXCL2 (shCXCL2). Error bars, ± SD.
(E–J) Representative flow cytometry plots and quantification of (E) neutrophils, (F) CD3+ T cells, (G) T helper cells, (H) CD8+ T cells, (I) Th1 cells, and (J) regulatory T cells. Each dot represents one mouse. Error bars, ± SD. For (A)–(J), Student’s t test (two-tailed, unpaired); *p < 0.05, **p < 0.01, ***p < 0.001 ****p < 0.0001. See also Figure S3.
To examine whether intratumoral neutrophil accumulation is mediated by chemokines generated by tumor epithelial cells, we stably transfected short hairpin RNAs (shRNAs) targeting CXCL2 or CXCL5 or a scramble hairpin control into CRISPR KrasG12D/+;Trp53R172H/+ cells. These hairpins induced more than 80% knockdown efficiency, as assessed by qPCR of in vitro mRNA (Figure 3C). Two weeks after orthotopic implantation, CRISPR KrasG12D/+;Trp53R172H/+ tumors with a CXCL2 or CXCL5 hairpin were smaller in size (Figure 3D), consistent with published reports showing that these chemokines affect tumor growth (Keeley et al., 2010; Zhao et al., 2017). Furthermore, CRISPR KrasG12D/+;Trp53R172H/+ tumors bearing a CXCL2 hairpin showed fewer intratumoral neutrophils than tumors bearing a scramble control hairpin (Figure 3E). Flow cytometry analysis of CRISPR KrasG12D/+;Trp53R172H/+ shCXCL2 tumors demonstrated a greater abundance of CD3+, CD4+, and CD8+ T cells compared to scramble hairpin control tumors (Figures 3F–3H). Further examination of the CD4 compartment in CRISPR KrasG12D/+;Trp53R172H/+shCXCL2 tumors revealed a significant increase in the CD4+Tbet+ Th1 population and a slight decrease in the CD4+Foxp3+ regulatory T cell population (Figures 3I and 3J). In contrast, no significant difference in neutrophil accumulation was detected in CRISPR KrasG12D/+; Trp53R172H/+ tumors stably expressing shCXCL5 (Figure 3E). These results implicate tumor-cell-derived CXCL2 production in mediating the neutrophil immune phenotype of CRISPR KrasG12D/+;Trp53R172H/+ tumors. Analysis of the peripheral blood of mice bearing orthotopic CRISPR KrasG12D/+;Trp53R172H/+ tumors or KrasG12D/+;Trp53+/+ tumors revealed no significant difference in the abundance of neutrophils between the two tumor genotypes, ruling out a potential contribution of systemic neutrophilia (Figure S3F).
Neutrophils confer resistance to CD40 combination immunotherapy and chemotherapy
The combination of agonist CD40 immunotherapy with gemcitabine and nab-paclitaxel chemotherapy is currently in phase II clinical trials for treatment of pancreatic cancer. Treatment with a CD40 antibody activates antigen-presenting cells (APCs) that can elicit an immune response from T cells using tumor antigens released by chemotherapy-mediated cell destruction (Byrne and Vonderheide, 2016). Because neutrophils may interfere with T cell proliferation and function as well as differentiation and maturation of APCs (Li et al., 2019; Schuster et al., 2013), we hypothesized that neutrophils may cause resistance to CD40 combination therapy. We treated mice bearing orthotopic CRISPR KrasG12D/+; Trp53+/+ tumors or CRISPR KrasG12D/+;Trp53R172H/+ tumors with a single dose of gemcitabine and nab-paclitaxel, followed by one dose of mouse CD40 (FGK45) 48 h later, as established in prior publications (Byrne and Vonderheide, 2016). Tumors were harvested 4 weeks after the drug treatment (Figure S4A). CRISPR KrasG12D/+;Trp53R172H/+tumors were more resistant to CD40 combination treatment than CRISPR KrasG12D/+;Trp53+/+ tumors, as measured by tumor weight (Figure S4B).
To determine whether neutrophils are a source of resistance to this drug combination, we used a neutrophil depletion strategy in mice bearing CRISPR KrasG12D/+;Trp53R172H/+ tumors. The anti-Ly6G antibody 1A8 is an established means for successfully depleting Ly6G+ cells in vivo (Boivin et al., 2020; Jamieson et al., 2012; Steele et al., 2016), and we confirmed the efficiency of intratumoral neutrophil depletion in our mouse model using flow cytometry (Figure S4C). CRISPR KrasG12D/+; Trp53R172H/+ tumor-bearing mice were treated continuously with anti-Ly6G or control antibody every 48 h in addition to a single treatment of combination CD40 immunotherapy and gemcitabine and nab-paclitaxel chemotherapy commencing 2 weeks after orthotopic implantation (Figure 4A). CD40 combination treatment was more effective in slowing tumor growth when administered together with anti-Ly6G compared with the control antibody combination (Figures 4B and 4C). Examination of intratumoral lymphocytes from CD40 combination immunotherapy/chemotherapy-treated tumors by flow cytometry 2 weeks after drug treatment revealed significant increases in CD3+, CD4+, and CD8+ cells in neutrophil-depleted mice compared with control mice (Figures 4D–4F). Although there was no difference in PD1+CD8+ T cells, a marker of T cell exhaustion, or in CD4+Foxp3+ regulatory T cells (Figures S4D and S4E), we observed an increase in CD4+Tbet+ Th1 cells (Figure 4G) in tumors treated with CD40 combination immunotherapy and the Ly6G antibody in comparison with mice treated with combination immunotherapy and a control antibody. These results support a model where the gain-of-function Trp53R172H mutation promotes neutrophil infiltration, which, in turn, could confer resistance to immunotherapy combination drug treatment.
Figure 4. Neutrophils confer resistance to CD40 combination immunotherapy and chemotherapy.

(A) Experimental design.
(B and C) Tumor growth curves (B) and waterfall plots (C) for 21 days after the start of the indicated therapy (n = 9–10 mice/group). Each symbol or bar represents the mean of a group (B) or a single mouse (C), respectively, with error bars indicating SEM. Statistical differences were determined by linear mixed-effects modeling with Tukey’s honest significance (HSD) post-test (B) with significance indicated. **p < 0.01, ***p < 0.001.
(D–G) Representative flow cytometry plots and quantification of (D) CD3+ T cells, (E) CD8+ T cells, (F) T helper cells, and (G) Th1 cells. Each dot represents one mouse; n = 8–9. Error bars, ± SD. Student’s t test (two-tailed, unpaired). *p < 0.05, **p < 0.01, ****p < 0.0001. See also Figure S4.
DISCUSSION
Increasing evidence suggests that oncogenic drivers can coordinate with different secondary mutations to create diverse immune landscapes (Bezzi et al., 2018). Loss of p53 expression can cooperate with oncogenic KRAS to promote tumor development through the presence of CD11b+F480+ macrophages (Blagih et al., 2020), and our work indicates a neutrophil-rich predominance when KRASG12D tumors acquire a gain-of-function p53 mutation, suggesting that shifts in the predominant myeloid population may occur based on secondary mutations. In non small cell lung cancer (NSCLC) mouse models, KRasG12D and MYC cooperate to induce CCL9-mediated recruitment of CD206+ macrophages and PD-L1-dependent expulsion of T cells (Kortlever et al., 2017). In contrast, STK11/LKB1 loss cooperates with KRasG12D to promote intratumoral accumulation of neutrophils in NSCLC (Koyama et al., 2016), indicating that secondary mutations may refine the composition of intratumoral immune populations and that different immune profiles may occur within the same cancer type. Although tumor genotype is not currently utilized for selecting individuals for immunotherapy, with the exception of those with high mutational burden being considered for treatment with PD1 therapy (Marabelle et al., 2020), our study adds to the mounting evidence that suggests that affected individuals may be stratified for clinical trials based on their genotype and/or immunophenotype to tailor precision therapy (Bezzi et al., 2018).
There are several possible mechanisms whereby p53 mutation might mediate an increase in ELR+ chemokines. Increased transcription of CXCL5 has been found to be dependent on gain-of function mutant p53 in multiple cancer cell lines, including lung, breast, and melanoma (Yeudall et al., 2012). In addition, there are several studies suggesting that the p53R172H mutation may elevate CXC chemokine production through nuclear factor kB (NF-kB). Mutant Trp53R172H is a well-established activator of NF-kB (Cooks et al., 2013; Weisz et al., 2007), and examination of the KPC mouse model revealed that NF-kB activation can potently increase CXCL5 levels (Chao et al., 2016). In addition, an analysis of the pancreatic cancer TCGA dataset demonstrated that tumors with high CXCR2 ligand expression were enriched significantly in expression of genes associated with inflammatory signaling pathways involving NF-kB (Chao et al., 2016). However, the mechanism by which p53 mutations can influence ELR+ chemokine expression may also depend on tumor tissue type. Most recently, a separate study examining Trp53R172H mutant osteosarcoma cell lines demonstrated an increase in CXCL5-mediated by PLAC8 (ONZIN) overexpression (Zhang et al., 2018). PLAC8 is a small cysteine-rich protein that is strongly expressed in human invasive PDAC and is involved in pancreatic cell growth and progression, but its relationship to neutrophil chemokines has not been defined (Kaistha et al., 2016).
It has been proposed that neutrophils can be pro- or antitumorigenic depending in part on the specific cancer type and stage (Jaillon et al., 2020). Efforts have been made to classify neutrophils into anti-tumor (N1) neutrophils or pro-tumor (N2) neutrophils based on their activation status, cytokine production, and effects on tumor cells (Masucci et al., 2019; Fridlender et al., 2009). N1 neutrophils direct cytotoxic activity against tumor cells through production of hydrogen peroxide and nitric oxide, activate T cell-dependent adaptive immunity, and also limit bacterially driven inflammation (Dmitrieva-Posocco et al., 2019; Finisguerra et al., 2015; Fridlender et al., 2009). In contrast, N2 neutrophils can promote tumor cell proliferation by inhibiting T cell activation through release of reactive oxygen species and arginase 1 and also facilitate metastasis by assisting with formation of a pre-metastatic niche (Coffelt et al., 2016; Gabrilovich et al., 2012; Park et al., 2016). In the context of pancreatic cancer, neutrophil phenotypes are consistent with a pro-tumorigenic role. Human pancreatic cancer has abundant neutrophil infiltration (Chao et al., 2016; Reid et al., 2011), which is associated with poor prognosis (Wang et al., 2016). In addition, a high peripheral blood neutrophil-to-lymphocyte ratio is a negative predictor of overall survival and disease-free survival (Zhou et al., 2018). Neutrophil chemokines are correlated significantly with advanced clinical stage and shorter survival in human PDA (Li et al., 2011; Nywening et al., 2018). In our study, acquisition of neutrophils was associated with tumor growth and reduction of T cell accumulation, also suggestive of a pro-tumorigenic function. Two separate reports have explored the functional role of neutrophils in KPC mouse models of pancreatic cancer, primarily by modulating CXCR2 (Chao et al., 2016; Steele et al., 2016). Disrupting CXCR2 signaling prevented accumulation of neutrophils and induced T cell-dependent suppression of tumor growth in KPC tumors (Chao et al., 2016).
Because of the lack of response to single-agent immunotherapy in individuals with pancreatic cancer, combinatorial strategies are being actively explored in clinical trials (Siolas et al., 2020). Combination of the CD40 agonist antibody APX005M and chemotherapy (with or without nivolumab PD1 therapy) showed an encouraging overall response rate of 54% in a phase Ib trial of individuals with metastatic pancreatic cancer and is currently under evaluation in an ongoing randomized phase II study (Vonderheide, 2020). Given that neutrophils suppress T cell proliferation and function, the ability to therapeutically target neutrophils is appealing and may open the door to improved efficacy of T cell-based therapeutics (Chao et al., 2016). There are two therapeutic agents targeting the CXCR2 chemokine axis currently in separate clinical trials, but neither has been explored with CD40 therapy (Bilusic et al., 2019; Evans, 2019). Our findings indicate that the gain-of-function mutant Trp53R172H can cause accumulation of tumor-associated neutrophils, whose targeting may be used to augment the therapeutic efficacy of combination CD40 immunotherapy in pancreatic cancer. Furthermore, this contributes to the growing body of work showing that different tumor genotypes can have distinct immune profiles within pancreatic cancer, suggesting immunophenotype should be considered in therapy selection.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dafna Bar-Sagi (Dafna.Bar-sagi@nyulangone.org).
Materials availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and code availability
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table.
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE Antibodies | SOURCE | IDENTIFIER |
|---|---|---|
| APC/Cy7 anti-mouse CD3 (17A2) | BioLegend | Cat# 100221; RRID:AB_2057374 |
| BV-421 CD45 (30-F11) | BioLegend | Cat# 103133; RRID:AB_10899570 |
| PE anti-mouse F4/80 (BM8) | BioLegend | Cat# 123109; RRID:AB_893498 |
| PE anti-mouse Foxp3 (FJK-16 s) | Invitrogen | Cat# 12-5773-80; RRID:AB_465935 |
| APC/Cy7 anti-mouse CD11b (M1/70) | BioLegend | Cat# 101225; RRID:AB_830641 |
| PE anti-mouse CD80 (16-10A1) | BioLegend | Cat# 104733; RRID:AB_2563112 |
| PerCP/Cy5.5 anti-mouse CTLA4 (UC10-4B9) | BioLegend | Cat# 106315; RRID:AB_2564473 |
| APC/Cy7 anti-mouse MHCII I-A/I-E (M5/114.15.2) | BioLegend | Cat# 107627; RRID:AB_1659252 |
| PE anti-mouse Gr1 (RB6-8C5) | BioLegend | Cat# 108407; RRID:AB_313372 |
| APC anti-mouse Ly6G (1A8) | BioLegend | Cat# 127613; RRID:AB_1877163 |
| APC anti-mouse CD206 (C068C2) | BioLegend | Cat# 141707; RRID:AB_10896057 |
| Mouse BD Fc Block (2.4G2) | BD pharmingen | Cat# 553142; RRID:AB_394657 |
| PE mouse anti-mouse T-Bet (4B10) | BD pharmingen | Cat# 561265; RRID:AB_10565980 |
| APC anti-mouse CD4 (GK1.5) | BioLegend | Cat# 100411; RRID:AB_312696 |
| PE/Cy7 CD8 (53-6.7) | BioLegend | Cat# 100721; RRID:AB_312760 |
| PerCP/Cy5.5 anti-mouse CD8 (53-6.7) | BioLegend | Cat# 100733; RRID:AB_2075239 |
| PerCP/Cy5.5 anti-mouse CD86 (GL-1) | BioLegend | Cat# 105027; RRID:AB_893420 |
| PerCP/Cy5.5 Ep-CAM (G8.8) | BioLegend | Cat# 107627; RRID:AB_1659252 |
| PerCP/Cy5.5 anti-mouse PD1 (29F.1A12) | BioLegend | Cat# 135207; RRID:AB_10550092 |
| Pe/Cy7 anti-mouse IFN-g (XMG1.2) | BioLegend | Cat# 505825; RRID:AB_1595591 |
| PE anti-mouse CD31 (390) | BioLegend | Cat# 102407; RRID:AB_312902 |
| Pe/Cy7 anti-mouse PDGFRa (APA5) | BioLegend | Cat# 135911; RRID:AB_2715973 |
| PE anti-mouse CD11c (N418) | BioLegend | Cat# 117307; RRID:AB_313776 |
| PE IgG1k isotype control | Invitrogen | Cat# 12-4301-81; RRID:AB_470046 |
| Pe/Cy7 Rat IgG1 isotype control | BioLegend | Cat# 400415; RRID:AB_326521 |
| InVivo rat IgG2a mAb (2A3) isotype control | BioXCell | Cat# BE0089; RRID:AB_1107769 |
| InVivoMAb monoclonal anti-mouse Ly6G (1A8) | BioXCell | Cat# BE0075-1; RRID:AB_1107721 |
| InVivoMAb CD40 rat anti-mouse IgG2a mAb (FGK45) | BioXCell | Cat# BE0016-2; RRID:AB_1107647 |
| InVivoMAb anti-rat Kappa Immunoglobulin Light Chain | BioXCell | Cat# BE0122; RRID:AB_10951292 |
| APC anti-mouse CD34 (HM34) | BioLegend | Cat #128611; RRID:AB_10553895 |
| MPO antibody | Santa Cruz | Cat# sc-390109; RRID:AB_2892996 |
| Neutrophil Marker (NIMP-R14) antibody | Santa Cruz | Cat# sc-59338; RRID:AB_2167795 |
| P53 (1C12) Mouse antibody | Cell Signaling | Cat# 2524S; RRID:AB_331743 |
| Anti-Vinculin mouse antibody | SigmaAldrich | Cat# V9264; RRID:AB_10603627 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| STBL3 | ThermoFisher | Cat# C737303 |
|
| ||
| Chemicals, Peptides, and Recombinant Proteins | ||
|
| ||
| Nab-paclitaxel | Celgene | Cat# 68817-134-50 |
| Gemcitabine | Hospira | Cat# 0409-0181-01 |
| Albumin | Sigma Aldrich | Cat# A5843 |
| Xtremegene 9 | Roche | Cat# 06 365 779 001 |
| Polybrene | Sigma Aldrich | Cat# TR-1003-G |
| Hygromycin | Sigma Aldrich | Cat# 400053 |
| G418 | Sigma Aldrich | Cat# 345812 |
| PfIFI | New England Biolabs | Cat# R0595S |
| BsmBI | New England Biolabs | Cat#R0739S |
| Shield-1 | Cheminpharma | Cat# S1-001 |
| Lipofectamine 3000 | Life Technologies | Cat# L3000008 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Surveyor DNA mutation Kit | IDT | Cat# 706020 |
| TOPO TA Cloning Kit | Thermo-Fisher | Cat# K4575J10 |
| SYBR Green PCR Master Mix | Thermo-Fisher | Cat# 4309155 |
| Quantitect Reverse Transcription Kit | QIAGEN | Cat# 205311 |
| Zombie Green Fixable Viability Kit | Biolegend | Cat# 423111 |
| RNeasy Mini Kit | QIAGEN | Cat# 157029548 |
| Milliplex Mouse Cytokine Magnetic Bead Panel | Millipore Sigma | Cat# MCYTOMAG-70K-03 |
| KAPA Mouse Genotyping Kit | Roche | Cat# 07961804001 |
| Foxp3/Transcription Factor Staining Buffer Set | Invitrogen | Cat# 00-5523-00 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| HEK293T | ATCC | Cat# ATCC® CRL-3216 |
| KrasG12D/+;Trp53+/+PDEC | Pylayeva-Gupta et al., 2012 | N/A |
| KrasG12D/+;Trp53−/− PDEC | This manuscript | N/A |
| KrasG12D/+;Trp53R172H/+PDEC | This manuscript | N/A |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| C57Bl6/J mice females (6-10weeks) | Charles River Laboratories | CR: 632 |
|
| ||
| Oligonucleotides | ||
| p53 sgRNA, CGG AGC AGC GCT CAT GGT GG | This manuscript | N/A |
|
| ||
| p53 HDR template, CAC CTC CAG CTG GGA GCC GTG TCC GCG CCA TGG CCA TCT ACA AGA AGT CAC AGC ACA TGA CGG AAG TCG TGA GAC ACT GTC CCC ACC ATG AGC GCT GCT CCG ATG GTG ATG GTA AGC CCT CAA CAC CGC CTG TGG GGT TAG GAC TGG CAG | This manuscript | N/A |
| p53 Forward PCR primer, TGGGA CAGCCAAGTCTGTTA | This manuscript | N/A |
| p53 Reverse PCR primer CCTGCT GTCTCCAGACTCCT | This manuscript | N/A |
| shScramble CCGGTCCTAAGGTTA AGTCGCCCTCGCTCGAGCGAGGG CGACTTAACCTTAGGTTTT | Sarbassov et al., 2005 | Addgene Cat # 1864 |
| CXCL2 forward PCR primer ATGC CTGAAGACCCTGCCAAG | Roy et al., 2012 | N/A |
| CXCL2 reverse PCR primer GGTC AGTTAGCCTTGCCTTTG | Roy et al., 2012 | N/A |
| CXCL5 forward PCR primer ACAGT GCCCTACGGTGGAAGT | Roy et al., 2012 | N/A |
| CXCL5 reverse PCR primer CGAGT GCATTCCGCTTAGCTT | Roy et al., 2012 | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| pLKO.1 neo | Gift from Sheila Stewart | Addgene Cat# 13425 |
| psPAX2 | Gift from Didier Trono | Addgene Cat# 12260 |
| pMD2G | Gift from Didier Trono | Addgene Cat# 12259 |
| pLKO.1 hygro | Gift from Bob Weinberg | Addgene Cat# 24150 |
| LentiCRISPRv2 hygro | Stringer et al., 2019 | Addgene Cat# 98291 |
| conditional EDCPV | Gift from R. Sordella, Cold Spring Harbor Laboratory | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/; RRID:SCR_001935 |
| FlowJo version 10.2 | FlowJo | https://www.flowjo.com/solutions/flowjo/downloads; RRID:SCR_008520 |
| R version 3.4.3 | RStudio | http://www.R-project.org; RRID:SCR_000432 |
| Prism Version 7 | GraphPad | https://www.graphpad.com/scientific-software/prism/; RRID:SCR_002798 |
| Vevo Lab | FUJIFILM-VisualSonics | https://www.visualsonics.com/product/software/vevo-lab |
|
| ||
| Other | ||
|
| ||
| Whole-Tissue Gene Expression Data of Pancreatic Ductal Adenocarcinoma | Badea et al., 2008 | GSE15471 |
| Expression data from Mayo Clinic Pancreatic Tumor and Normal samples | Pei et al., 2009 | GSE16515 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
The LSL-KrasG12D/+;Trp53+/+;p48Cre/+ and LSL-KrasG12D/+;Trp53LSL-R172H/+;p48Cre/+ mouse strains were previously described (Pylayeva-Gupta et al., 2012). The Institutional Animal Care and Use Committee at the New York University (NYU) School of Medicine approved all animal care and procedures. All mice were housed under conditions in line with NYU Institutional Animal Care and Use Committee guidelines. Mice were housed from 3–5 per cage and were kept in a 12-h day/night cycle with light from 6:30 until 18:30. For orthotopic mouse models, 7to 9-week-old wild-type (WT) C57BL/6 (stock 027) mice were purchased from The Charles River Laboratories. All mice were from a C57BL/6 genetic background. Female mice were used for orthotopic injections of CRISPR KrasG12D/+;Trp53R172H/+ PDEC, KrasG12D/+;Trp53−/− PDEC, and KrasG12D/+;Trp53+/+ PDEC cell lines using the method described in Das et al. (2020). For chemotherapy treatment, gemcitabine, pharmaceutical grade suspension at 38 mg/ml, was diluted to 12 mg/ml in PBS and administered at 120 mg/kg via intraperitoneal (i.p.) injection (Byrne and Vonderheide, 2016). Nab-paclitaxel (Abraxane, Celgene) pharmaceutical grade powder was resuspended at 12 mg/ml in PBS and administered at 120 mg/kg i.p., or an equivalent molar dose of human albumin (Sigma) was administered i.p. as control (Byrne and Vonderheide, 2016). Mice were injected on day 14 after orthotopic implantation. Chemotherapy was purchased through the NYU Langone Health pharmacy. Mice received 100 μg of either agonist CD40 rat anti-mouse IgG2a mAb (clone FGK45, endotoxin-free) or the isotype control IgG2a mAb (clone 2A3) on day 16 after orthotopic surgery. Neutrophils were depleted by i.p. injection of 200 μg Ly6G+ Ab (clone 1A8) and 200 μg mouse IgG2a anti-rat antibody (clone Mar 18.5) or isotype control mAb (clone 2A3) on day 12, repeated every 48 h. All antibodies were purchased from BioXCell. Mice were euthanized by carbon-dioxide-induced narcosis for flow cytometry analysis and tumor size assessment.
Cell Lines and Stable Expression of Hairpins
Isolation, culture, and adenoviral infection of pancreatic ductal epithelial cells (PDECs) was carried out as previously described (Pylayeva-Gupta et al., 2012). CRISPR-generated cell lines are described in the detailed Methods. HEK293T cells were purchased from the ATCC and maintained in DMEM with 10% FBS and penicillin-streptomycin at 37°C. Scramble control shRNA (Sarbassov et al., 2005) and shRNAs against CXCL2 and CXCL5 were cloned into the lentiviral pLKO.1 neo (gift from Sheila Stewart) and pLKO.1 hygro (gift from Bob Weinberg) vectors, respectively, obtained from Addgene. Lentiviral particles were generated by transfecting HEK293T cells using Xtremegene 9 with the pLKO.1 vector, the packaging construct (psPAX2, gift from Didier Trono), and the envelope plasmid (pMD2G, gift from Didier Trono). Supernatants containing viral particles were collected over a period of 48 h and stored at 4°C. Following final collection, supernatants were filtered through a 0.45-μm-pore-size syringe filter and concentrated using 100-MWCO Amicon Ultra centrifugal filters (Millipore). A multiplicity of infection (MOI) of 10 was used for lentiviral infection of PDEC cells in the presence of 10 μg/ml Polybrene (Chemicon), and infected cells were selected using 150 μg/ml hygromycin (Sigma) or 400 μg/ml G418 (Sigma). All data representative of 3 independent clones from 3 independent experiments
METHOD DETAILS
CRISPR/Cas9 Editing of Trp53
A sgRNA was designed to target the desired Trp53 mutation using CRISPR Design. The sgRNA sequence 5’-CGG AGC GCT CAT GGT GG-3’ was cloned into LentiCRISPRv2 (Stringer et al., 2019) from Addgene according to a published method (Sanjana et al., 2014) and infected into PDEC cells as described above. sgRNA efficiency was verified via a Surveyor nuclease assay as described (Senturk et al., 2017). sgRNA was also cloned into the conditional EDCPV vector (gift from R. Sordella) as detailed in Senturk et al. (2017). Virus packaging was achieved by transiently co-transfecting HEK293T cells in 10-cm culture dish with 3 μg of the p53sgRNAEDCPV, 6 μg of the packaging plasmid psPAX2, and 3 μg of the envelope plasmid pMD2.G (Addgene) using 30 μl of Lipofectamine 3000 reagent (Life Technologies). Viral particles of 10 ml were collected after 48 h of transfection by clarifying the supernatant through 0.45-μm filter membrane (GE Healthcare). Virus transduction was optimized in order to achieve low-MOI transduction. A single-strand DNA homology-directed repair (HDR) template was created with sequence 5’-CAC CTC CAG CTG GGA GCC GTG TCC GCG CCA TGG CCA TCT ACA AGA AGT CAC AGC ACA TGA CGG AAG TCG TGA GAC ACT GTC CCC ACC ATG AGC GCT CCG ATG GTG ATG GTA AGC CCT CAA CAC CGC CTG TGG GGT TAG GAC TGG CAG-3’. A silent mutation (G> A) was placed in the 5’ protospacer adjacent motif (PAM) region and another silent mutation was designed for the 3’ PAM region (C> T) adjacent to the target to prevent multiple recombination events. This resulted in the creation of a unique restriction enzyme cloning site, PflFI (New England Biolabs), which would appear in target cells upon successful HDR, in conjunction with loss of the BsmBI restriction enzyme site (New England Biolabs). A 10-cm dish of PDEC cells was transfected with single-strand DNA HDR template using 25 μL of Lipofectamine 3000 (Life Technologies). Shield-1, obtained from Cheminpharma, was solubilized in pure ethanol and added to the culture medium at a concentration of 250 nM (final), and the cells were cultured for 24 h. After 24 h, Venus-positive single cells were sorted into five 96-well plates using a BD FACS ARIA II sorter at the NYU Cytometry and Cell Sorting Facility and allowed to grow for 10 days. Genomic DNA was extracted and amplified as per manufacturer’s directions using KAPA Hot Start Genotyping kit (Sigma) and the primers 5’-TGG GAC AGC CAA GTC TGT TA-3’ and 5’-TAA GGA TAG GTC GGC GGT TC-3’. Restriction enzyme digestion was performed separately for BsmBI and PflFI to screen for successful HDR. Clones that were not digested by BsmBI, but were successfully digested by PflFI, were sent to Genewiz for Sanger sequencing. To determine heterozygosity, genomic DNA was amplified using the primers above and Phusion High-Fidelity DNA Polymerase (New England Biolabs). Amplicons were cloned using a TOPO TA Cloning Kit (Thermo Fisher) as per manufacturer’s instructions and transformed into STBL3 chemically competent cells. Twenty bacterial colonies containing plasmids were selected for DNA isolation and PCR. PCR products were used for direct Sanger sequencing (Genewiz). In addition, all CRISPR KrasG12D/+;Trp53R172H/+ and KrasG12D/+;Trp53+/+ PDEC cell lines used in this paper were analyzed by RNA sequencing. RNA was isolated using an RNeasy kit (QIAGEN) and genomic DNA was removed using an RNase-free DNase kit (QIAGEN). RNA sequencing was performed by NYU Langone’s Genome Technology Center using an Illumina HiSeq2500 instrument. Three cell lines were created for each genotype. KrasG12D/+;Trp53+/+ cell lines were infected and single cell sorted as above, but were not treated with Shield-1 and therefore did not undergo DNA editing. KrasG12D/+;Trp53−/− PDEC cell lines were generated by treating with Shield-1 as above, but without the addition of a HDR template.
Flow Cytometry
Single-cell suspensions were prepared from pancreata as described (Pylayeva-Gupta et al., 2016), and tumor-infiltrating lymphocytes were isolated as described (Das et al., 2020). All samples were acquired on an LSR II instrument (BD Bioscience) at the NYU Cytometry and Cell Sorting Facility and analyzed with FlowJo version 10.2 (TreeStar, Inc.). Cell sorting using a BD FACS ARIA II sorter was performed to isolate Ep-CAM+ cells and CD45+ cells, and > 95% purity of sorted cells was achieved. All data representative of 3 independent clones from 3 independent experiments
Tissue Collection, Immunohistochemistry, Immunofluorescence, and Scoring
Mouse pancreata were fixed overnight in 10% buffered formalin (Fisher Scientific) and embedded in paraffin as described earlier (Pylayeva-Gupta et al., 2012). For histology analysis, excised tumors were processed for routine hematoxylin and eosin staining. For immunehistochemistry, methods were as in (Das et al., 2020), with rat anti-Ly6G (1:200, Santa Cruz) used as the primary antibody. Slides were examined on a Nikon Eclipse 80i microscope. Immunofluorescence staining using Ki-67 or MPO, along with CK19, was performed at the NYU Experimental Pathology Research Laboratory. Scoring of tumor sections for individual markers was performed by counting 10 fields using Omero imaging software and images were analyzed to measure stained area using ImageJ software (Schneider et al., 2012).
RNA Extraction and QPCR
For assessment of cell cycle genes, cultured PDEC cells were treated with 0.75 μM of doxorubicin for 36 hr and then subjected to RNA extraction with a RNeasy Kit (QIAGEN) as per the manufacturer’s instructions. Total RNA (1 μg) was reverse-transcribed using the Quantitect Reverse Transcription Kit (QIAGEN). Subsequently, specific transcripts were amplified with SYBR Green PCR Master Mix (USB) using a Stratagene Mx 3005P thermocycler. Primer sequences for Cxcl2 and Cxcl5 are in key resources table (Roy et al., 2012). Where fold expression is specified, the comparative CT method was used to quantify gene expression. Expression was normalized to that of GAPDH. For RNA isolation from tumors, pancreata processed to single-cell suspension were stained for flow cytometry. CD45–CD34–PDGFRa–Ep-CAM+ cells or CD45+ immune cells were FACS sorted using a 100-μm nozzle into the lysing reagent RLT, and total RNA was extracted as per the manufacturer’s instructions (RNeasy Mini Kit, QIAGEN). To check knockdown in PDEC cells, 105 cells were lysed in 350 μL RLT reagent and total RNA was extracted as per the manufacturer’s instructions (RNeasy Mini Kit, QIAGEN). QPCR was performed as described above.
Bioinformatics Analysis of Public Datasets
130 human PDAC tumor (n = 75) and Adj Norm (n = 55) mRNA expression profiles generated on the same array (Affymetrix GeneChip Human Genome U133 Plus 2.0) were downloaded from GEO (GEO: GSE15471 (Badea et al., 2008), GSE16515 (Pei et al., 2009)) and processed as previously described (Gadaleta et al., 2011). CXCL cytokines plots and an unpaired Student’s t test were generated in GraphPad Prism (GraphPad Software, CA, USA; https://www.graphpad.com:443/). TCGA data (Pancreatic Adenocarcinoma, TCGA Firehose Legacy) were analyzed in cBioPortal (https://www.cbioportal.org/). PDAC tumors with very low tumor cell content (< 33%) or labeled as non-PDAC, were removed and n = 58 tumors with hotspot missense mutations in TP53 DNA binding domain and n = 63 tumors without mutations or homozygous deletions were compared using cBioPortal’s Groups analysis function. Genes with significantly higher expression (q value < 0.05) in p53 mutated versus WT tumors were imported into Enrichr (https://maayanlab.cloud/Enrichr/), and assessed for enrichment in Gene Ontology Biological Process 2018 gene sets.
Ultrasound Imaging
Image acquisition and analysis were performed at the NYU Preclinical Imaging Laboratory. Mice were anesthetized with 2% isoflurane and placed on a physiological stage interfaced to a Vevo 3100 (Visualsonics, Fujifilm) high frequency ultrasound scanner. The stage monitored the mouse’s core temperature, respiration rate and heart rate. The abdominal area was wet shaved, ultrasound gel applied and an MXD550D transducer lowered until a transverse section of the tumor was centered in the field of view. The transducer was connected to a stepper motor and a 3D volume was acquired using a 200 micron slice spacing. The volumetric data was then imported to the Vevo LAB (Visualsonics, Fujifilm) analysis software. The tumor was segmented by drawing contours around the periphery of the tumor for each slice and the volume calculated by the volume measurement tool.
Immunoblotting
Cultured PDEC cells were treated with 0.6 μM of doxorubicin for 24 hr. Cells were lysed in sample buffer, denatured at 95°C, and resolved on polyacrylamide gels. Cells were probed for p53 with monoclonal anti-mouse p53 antibody. Vinculin was detected as a loading control. Secondary antibodies were purchased from LiCOR IRDye 800CW and 700CW.
Extracellular Cytokine Measurements
Orthotopic tumors were minced with a razor blade and incubated in RPMI media for one hour at 37°C. The solution was centrifuged for 2 min at 2000 RPM in a microcentrifuge. The supernatant was then assayed according to the manufacturer’s instructions using a MILLIPLEX Mouse Cytokine/Chemokine Magnetic Bead Panel, in a Luminex 200 (Luminex Corporation, Austin, TX, USA) machine.
QUANTIFICATION AND STATISTICAL ANALYSIS
At least 5 mice were included in each group, unless noted and the experiments were repeated a minimum 2 times. Group means were compared with Student t tests. Significance of variations between two groups was determined by an unpaired Student t test (two-tailed). Statistical analyses were performed using GraphPad Prism software (version 7.0d), and data are presented as mean ± SD p < 0.05 was considered statistically significant. Statistical details and p values of experiments can be found in the corresponding figure legends.
Supplementary Material
Highlights.
Gain-of-function Trp53R172H promotes neutrophil recruitment to pancreatic tumors
Neutrophils in KrasG12D/+;Trp53R172H/+ tumors are due to tumor-cell-derived chemokines
Neutrophils confer resistance to CD40 combination immunotherapy and chemotherapy
ACKNOWLEDGMENTS
We thank L.J. Taylor for help with manuscript preparation. We thank the NYU Cytometry and Cell Sorting Laboratory Facility, Preclinical Imaging Laboratory, and Experimental Pathology Research Laboratory, which are partially supported by Perlmutter Cancer Center grant P30CA016087 and NIBIB grant NIH P41 EB017183. This work was funded by NIH/NCI CA210263 (to D.B.S.) and by the Lustgarten Foundation Pancreatic Cancer Dream Team (SU2C-AACR-DT14-14 to D.B.-S.). D.S. is supported by NIH K08CA241341. E.A.V. was supported by a Canadian Institutes of Health research fellowship (146792). E.K. was supported by NIH F30 CA243205. The graphical abstract was created with BioRender.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109578.
DECLARATION OF INTERESTS
D.S. is a consultant for Ciox Health and a shareholder in Mirimus. She served on a scientific advisory board for MabImmune and received royalties from Cold Spring Harbor Laboratory.
REFERENCES
- Alvarado-Ortiz E, de la Cruz-Ló pez KG, Becerril-Rico J, Sarabia-Sá nchez MA, Ortiz-Sá nchez E, and García-Carrancá A. (2021). Mutant p53 Gain-of-Function: Role in Cancer Development, Progression, and Therapeutic Approaches. Front. Cell Dev. Biol 8, 607670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badea L, Herlea V, Dima SO, Dumitrascu T, and Popescu I. (2008). Com- bined gene expression analysis of whole-tissue and microdissected pancreatic ductal adenocarcinoma identifies genes specifically overexpressed in tu- mor epithelia. Hepatogastroenterology 55, 2016–2027. [PubMed] [Google Scholar]
- Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, Miller DK, Christ AN, Bruxner TJ, Quinn MC, et al. ; Australian Pancreatic Cancer Genome Initiative (2016). Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 531, 47–52. [DOI] [PubMed] [Google Scholar]
- Bargonetti J, and Prives C. (2019). Gain-of-function mutant p53: history and speculation. J. Mol. Cell Biol 11, 605–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, and Vonderheide RH (2012). Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell 21, 822–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezzi M, Seitzer N, Ishikawa T, Reschke M, Chen M, Wang G, Mitchell C, Ng C, Katon J, Lunardi A, et al. (2018). Diverse genetic-driven immune landscapes dictate tumor progression through distinct mechanisms. Nat. Med 24, 165–175. [DOI] [PubMed] [Google Scholar]
- Bilusic M, Heery CR, Collins JM, Donahue RN, Palena C, Madan RA, Karzai F, Marté JL, Strauss J, Gatti-Mays ME, et al. (2019). Phase I trial of HuMax-IL8 (BMS-986253), an anti-IL-8 monoclonal antibody, in patients with metastatic or unresectable solid tumors. J. Immunother. Cancer 7, 240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blagih J, Zani F, Chakravarty P, Hennequart M, Pilley S, Hobor S, Hock AK, Walton JB, Morton JP, Gronroos E, et al. (2020). Cancer-Specific Loss of p53 Leads to a Modulation of Myeloid and T Cell Responses. Cell Rep. 30, 481–496.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boivin G, Faget J, Ancey P-B, Gkasti A, Mussard J, Engblom C, Pfirschke C, Contat C, Pascual J, Vazquez J, et al. (2020). Durable and controlled depletion of neutrophils in mice. Nat. Commun 11, 2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne KT, and Vonderheide RH (2016). CD40 Stimulation Obviates Innate Sensors and Drives T Cell Immunity in Cancer. Cell Rep. 15, 2719–2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao T, Furth EE, and Vonderheide RH (2016). CXCR2-Dependent Accumulation of Tumor-Associated Neutrophils Regulates T-cell Immunity in Pancreatic Ductal Adenocarcinoma. Cancer Immunol. Res 4, 968–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark CE, Hingorani SR, Mick R, Combs C, Tuveson DA, and Vonderheide RH (2007). Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 67, 9518–9527. [DOI] [PubMed] [Google Scholar]
- Coffelt SB, Wellenstein MD, and de Visser KE (2016). Neutrophils in can- cer: neutral no more. Nat. Rev. Cancer 16, 431–446. [DOI] [PubMed] [Google Scholar]
- Cooks T, Pateras IS, Tarcic O, Solomon H, Schetter AJ, Wilder S, Lozano G, Pikarsky E, Forshew T, Rosenfeld N, et al. (2013). Mutant p53 prolongs NF-kB activation and promotes chronic inflammation and inflammation- associated colorectal cancer. Cancer Cell 23, 634–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Shapiro B, Vucic EA, Vogt S, and Bar-Sagi D. (2020). Tumor Cell-Derived IL1b Promotes Desmoplasia and Immune Suppression in Pancreatic Cancer. Cancer Res. 80, 1088–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Agostino S, Strano S, Emiliozzi V, Zerbini V, Mottolese M, Sacchi A, Blandino G, and Piaggio G. (2006). Gain of function of mutant p53: the mutant p53/NF-Y protein complex reveals an aberrant transcriptional mechanism of cell cycle regulation. Cancer Cell 10, 191–202. [DOI] [PubMed] [Google Scholar]
- Disteldorf EM, Krebs CF, Paust HJ, Turner JE, Nouailles G, Tittel A, Meyer-Schwesinger C, Stege G, Brix S, Velden J, et al. (2015). CXCL5 drives neutrophil recruitment in TH17-mediated GN. J. Am. Soc. Nephrol 26, 55–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dmitrieva-Posocco O, Dzutsev A, Posocco DF, Hou V, Yuan W, Thovarai V, Mufazalov IA, Gunzer M, Shilovskiy IP, Khaitov MR, et al. (2019). Cell-Type-Specific Responses to Interleukin-1 Control Microbial Invasion and Tumor-Elicited Inflammation in Colorectal Cancer. Immunity 50, 166–180.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans J. (2019). Phase Ib/II Study of MEDI4736 Evaluated in Different Combinations in Metastatic Pancreatic Ductal Carcinoma. https://clinicaltrials.gov/ct2/show/NCT02583477.
- Finisguerra V, Di Conza G, Di Matteo M, Serneels J, Costa S, Thompson AA, Wauters E, Walmsley S, Prenen H, Granot Z, et al. (2015). MET is required for the recruitment of anti-tumoural neutrophils. Nature 522, 349–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, Worthen GS, and Albelda SM (2009). Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 16, 183–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrilovich DI, Ostrand-Rosenberg S, and Bronte V. (2012). Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol 12, 253–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadaleta E, Cutts RJ, Kelly GP, Crnogorac-Jurcevic T, Kocher HM, Lemoine NR, and Chelala C. (2011). A global insight into a cancer transcriptional space using pancreatic data: importance, findings and flaws. Nucleic Acids Res. 39, 7900–7907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiraoka N, Onozato K, Kosuge T, and Hirohashi S. (2006). Prevalence of FOXP3+ regulatory T cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions. Clin. Cancer Res 12, 5423–5434. [DOI] [PubMed] [Google Scholar]
- Ho WJ, Jaffee EM, and Zheng L. (2020). The tumour microenvironment in pancreatic cancer clinical challenges and opportunities. Nat. Rev. Clin. Oncol 17, 527–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffer T, and Ma D. (2016). The emerging role of chemokine receptor CXCR2 in cancer progression. Transl. Cancer Res 5, S616–S628. [Google Scholar]
- Jaillon S, Ponzetta A, Di Mitri D, Santoni A, Bonecchi R, and Mantovani A. (2020). Neutrophil diversity and plasticity in tumour progression and therapy. Nat. Rev. Cancer 20, 485–503. [DOI] [PubMed] [Google Scholar]
- Jamieson T, Clarke M, Steele CW, Samuel MS, Neumann J, Jung A, Huels D, Olson MF, Das S, Nibbs RJB, and Sansom OJ (2012). Inhibition of CXCR2 profoundly suppresses inflammation-driven and spontaneous tumorigenesis. J. Clin. Invest 122, 3127–3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaistha BP, Lorenz H, Schmidt H, Sipos B, Pawlak M, Gierke B, Kreider R, Lankat-Buttgereit B, Sauer M, Fiedler L, et al. (2016). PLAC8 Localizes to the Inner Plasma Membrane of Pancreatic Cancer Cells and Reg- ulates Cell Growth and Disease Progression through Critical Cell-Cycle Regulatory Pathways. Cancer Res. 76, 96–107. [DOI] [PubMed] [Google Scholar]
- Keeley EC, Mehrad B, and Strieter RM (2010). CXC chemokines in cancer angiogenesis and metastases. Adv. Cancer Res 106, 91–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortlever RM, Sodir NM, Wilson CH, Burkhart DL, Pellegrinet L, Brown Swigart L, Littlewood TD, and Evan GI (2017). Myc Cooperates with Ras by Programming Inflammation and Immune Suppression. Cell 171, 1301–1315.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama S, Akbay EA, Li YY, Aref AR, Skoulidis F, Herter-Sprie GS, Buczkowski KA, Liu Y, Awad MM, Denning WL, et al. (2016). STK11/ LKB1 Deficiency Promotes Neutrophil Recruitment and Proinflammatory Cytokine Production to Suppress T-cell Activity in the Lung Tumor Microenvironment. Cancer Res. 76, 999–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesina M, Kurkowski MU, Ludes K, Rose-John S, Treiber M, Klöppel G, Yoshimura A, Reindl W, Sipos B, Akira S, et al. (2011). Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell 19, 456–469. [DOI] [PubMed] [Google Scholar]
- Li A, King J, Moro A, Sugi MD, Dawson DW, Kaplan J, Li G, Lu X, Strieter RM, Burdick M, et al. (2011). Overexpression of CXCL5 is associated with poor survival in patients with pancreatic cancer. Am. J. Pathol 178, 1340–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JL, Lim CH, Tay FW, Goh CC, Devi S, Malleret B, Lee B, Bako- cevic N, Chong SZ, Evrard M, et al. (2016). Neutrophils Self-Regulate Immune Complex-Mediated Cutaneous Inflammation through CXCL2. J. Invest. Dermatol 136, 416–424. [DOI] [PubMed] [Google Scholar]
- Li Y, Wang W, Yang F, Xu Y, Feng C, and Zhao Y. (2019). The regulatory roles of neutrophils in adaptive immunity. Cell Commun. Signal 17, 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Zhang C, Hu W, and Feng Z. (2019). Tumor suppressor p53 and metabolism. J. Mol. Cell Biol 11, 284–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marabelle A, Fakih M, Lopez J, Shah M, Shapira-Frommer R, Naka- gawa K, Chung HC, Kindler HL, Lopez-Martin JA, Miller WH Jr., et al. (2020). Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 21, 1353–1365. [DOI] [PubMed] [Google Scholar]
- Martinez-Bosch N, Vinaixa J, and Navarro P. (2018). Immune Evasion in Pancreatic Cancer: From Mechanisms to Therapy. Cancers (Basel) 10, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masucci MT, Minopoli M, and Carriero MV (2019). Tumor Associated Neutrophils. Their Role in Tumorigenesis, Metastasis, Prognosis and Therapy. Front. Oncol 9, 1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew E, Collins MA, Fernandez-Barrena MG, Holtz AM, Yan W, Hogan JO, Tata Z, Allen BL, Fernandez-Zapico ME, and di Magliano MP (2014). The transcription factor GLI1 modulates the inflammatory response during pancreatic tissue remodeling. J. Biol. Chem 289, 27727–27743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton JP, Timpson P, Karim SA, Ridgway RA, Athineos D, Doyle B, Jamieson NB, Oien KA, Lowy AM, Brunton VG, et al. (2010). Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc. Natl. Acad. Sci. USA 107, 246–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami T, Hiroshima Y, Matsuyama R, Homma Y, Hoffman RM, and Endo I. (2019). Role of the tumor microenvironment in pancreatic cancer. Ann. Gastroenterol. Surg 3, 130–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nywening TM, Belt BA, Cullinan DR, Panni RZ, Han BJ, Sanford DE, Jacobs RC, Ye J, Patel AA, Gillanders WE, et al. (2018). Targeting both tumour-associated CXCR2+ neutrophils and CCR2+ macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut 67, 1112–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Wysocki RW, Amoozgar Z, Maiorino L, Fein MR, Jorns J, Schott AF, Kinugasa-Katayama Y, Lee Y, Won NH, et al. (2016). Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci. Transl. Med 8, 361ra138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei H, Li L, Fridley BL, Jenkins GD, Kalari KR, Lingle W, Petersen G, Lou Z, and Wang L. (2009). FKBP51 affects cancer cell response to chemotherapy by negatively regulating Akt. Cancer Cell 16, 259–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfister NT, and Prives C. (2017). Transcriptional Regulation by Wild-Type and Cancer-Related Mutant Forms of p53. Cold Spring Harb. Perspect. Med 7, a026054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, and Bar-Sagi D. (2012). Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell 21, 836–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pylayeva-Gupta Y, Das S, Handler JS, Hajdu CH, Coffre M, Koralov SB, and Bar-Sagi D. (2016). IL35-Producing B Cells Promote the Development of Pancreatic Neoplasia. Cancer Discov. 6, 247–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj N, Klimstra DS, Horvat N, Zhang L, Chou JF, Capanu M, Basturk O, Do RKG, Allen PJ, and Reidy-Lagunes D. (2017). O6-Methylguanine DNA Methyltransferase Status Does Not Predict Response or Resistance to Alkylating Agents in Well-Differentiated Pancreatic Neuroendocrine Tumors. Pancreas 46, 758–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raphael BJ, Hruban RH, Aguirre AJ, Moffitt RA, Yeh JJ, Stewart C, Robertson AG, Cherniack AD, Gupta M, Getz G, et al. ; Cancer Genome Atlas Research Network. Electronic address: andrew_aguirre@dfci.harvard.edu; Cancer Genome Atlas Research Network (2017). Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 32, 185–203.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid MD, Basturk O, Thirabanjasak D, Hruban RH, Klimstra DS, Bagci P, Altinel D, and Adsay V. (2011). Tumor-infiltrating neutrophils in pancreatic neoplasia. Mod. Pathol 24, 1612–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy M, Richard J-F, Dumas A, and Valliè res L. (2012). CXCL1 can be regulated by IL-6 and promotes granulocyte adhesion to brain capillaries during bacterial toxin exposure and encephalomyelitis. J. Neuroinflammation 9, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F. (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, and Sabatini DM (2005). Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to Image J: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster S, Hurrell B, and Tacchini-Cottier F. (2013). Crosstalk between neutrophils and dendritic cells: a context-dependent process. J. Leukoc. Biol 94, 671–675. [DOI] [PubMed] [Google Scholar]
- Senturk S, Shirole NH, Nowak DG, Corbo V, Pal D, Vaughan A, Tuveson DA, Trotman LC, Kinney JB, and Sordella R. (2017). Rapid and tunable method to temporally control gene editing based on conditional Cas9 stabilization. Nat. Commun 8, 14370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siolas D, Morrissey C, and Oberstein PE (2020). The Achilles’ Heel of Pancreatic Cancer: Targeting pancreatic cancer’s unique immunologic characteristics and metabolic dependencies in clinical trials. J. Pancreatol 3, 121–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spranger S, Bao R, and Gajewski TF (2015). Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 523, 231–235. [DOI] [PubMed] [Google Scholar]
- Steele CW, Karim SA, Foth M, Rishi L, Leach JD, Porter RJ, Nixon C, Jeffry Evans TR, Carter CR, Nibbs RJ, et al. (2015). CXCR2 inhibition suppresses acute and chronic pancreatic inflammation. J. Pathol 237, 85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele CW, Karim SA, Leach JDG, Bailey P, Upstill-Goddard R, Rishi L, Foth M, Bryson S, McDaid K, Wilson Z, et al. (2016). CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell 29, 832–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stringer BW, Day BW, D’Souza RCJ, Jamieson PR, Ensbey KS, Bruce ZC, Lim YC, Goasdoué K, Offenhä user C, Akgu€l S, et al. (2019). A reference collection of patient-derived cell line and xenograft models of proneural, classical and mesenchymal glioblastoma. Sci. Rep 9, 4902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonderheide RH (2020). CD40 Agonist Antibodies in Cancer Immuno- therapy. Annu. Rev. Med 71, 47–58. [DOI] [PubMed] [Google Scholar]
- Wang WQ, Liu L, Xu HX, Wu CT, Xiang JF, Xu J, Liu C, Long J, Ni QX, and Yu XJ (2016). Infiltrating immune cells and gene mutations in pancreatic ductal adenocarcinoma. Br. J. Surg 103, 1189–1199. [DOI] [PubMed] [Google Scholar]
- Weissmueller S, Manchado E, Saborowski M, Morris JP 4th, Wagen- blast E, Davis CA, Moon SH, Pfister NT, Tschaharganeh DF, Kitzing T, et al. (2014). Mutant p53 drives pancreatic cancer metastasis through cell- autonomous PDGF receptor b signaling. Cell 157, 382–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisz L, Damalas A, Liontos M, Karakaidos P, Fontemaggi G, Maor- Aloni R, Kalis M, Levrero M, Strano S, Gorgoulis VG, et al. (2007). Mutant p53 enhances nuclear factor kappaB activation by tumor necrosis factor alpha in cancer cells. Cancer Res. 67, 2396–2401. [DOI] [PubMed] [Google Scholar]
- Wellenstein MD, Coffelt SB, Duits DEM, van Miltenburg MH, Slagter M, de Rink I, Henneman L, Kas SM, Prekovic S, Hau CS, et al. (2019). Loss of p53 triggers WNT-dependent systemic inflammation to drive breast cancer metastasis. Nature 572, 538–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witkiewicz AK, McMillan EA, Balaji U, Baek G, Lin W-C, Mansour J, Mollaee M, Wagner K-U, Koduru P, Yopp A, et al. (2015). Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic tar- gets. Nat. Commun 6, 6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeudall WA, Vaughan CA, Miyazaki H, Ramamoorthy M, Choi MY, Chapman CG, Wang H, Black E, Bulysheva AA, Deb SP, et al. (2012). Gain-of-function mutant p53 upregulates CXC chemokines and enhances cell migration. Carcinogenesis 33, 442–451. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Velez-Delgado A, Mathew E, Li D, Mendez FM, Flannagan K, Rhim AD, Simeone DM, Beatty GL, and Pasca di Magliano M. (2017). Myeloid cells are required for PD-1/PD-L1 checkpoint activation and the establishment of an immunosuppressive environment in pancreatic cancer. Gut 66, 124–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Hu Q, Li G, Li L, Liang S, Zhang Y, Liu J, Fan Z, Li L, Zhou B, et al. (2018). ONZIN Upregulation by Mutant p53 Contributes to Osteosarcoma Metastasis Through the CXCL5-MAPK Signaling Pathway. Cell. Physiol. Biochem 48, 1099–1111. [DOI] [PubMed] [Google Scholar]
- Zhao J, Ou B, Han D, Wang P, Zong Y, Zhu C, Liu D, Zheng M, Sun J, Feng H, and Lu A. (2017). Tumor-derived CXCL5 promotes human colorectal cancer metastasis through activation of the ERK/Elk-1/Snail and AKT/ GSK3b/b-catenin pathways. Mol. Cancer 16, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Wei Q, Fan J, Cheng S, Ding W, and Hua Z. (2018). Prognostic role of the neutrophil-to-lymphocyte ratio in pancreatic cancer: A meta-analysis containing 8252 patients. Clin. Chim. Acta 479, 181–189. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table.
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE Antibodies | SOURCE | IDENTIFIER |
|---|---|---|
| APC/Cy7 anti-mouse CD3 (17A2) | BioLegend | Cat# 100221; RRID:AB_2057374 |
| BV-421 CD45 (30-F11) | BioLegend | Cat# 103133; RRID:AB_10899570 |
| PE anti-mouse F4/80 (BM8) | BioLegend | Cat# 123109; RRID:AB_893498 |
| PE anti-mouse Foxp3 (FJK-16 s) | Invitrogen | Cat# 12-5773-80; RRID:AB_465935 |
| APC/Cy7 anti-mouse CD11b (M1/70) | BioLegend | Cat# 101225; RRID:AB_830641 |
| PE anti-mouse CD80 (16-10A1) | BioLegend | Cat# 104733; RRID:AB_2563112 |
| PerCP/Cy5.5 anti-mouse CTLA4 (UC10-4B9) | BioLegend | Cat# 106315; RRID:AB_2564473 |
| APC/Cy7 anti-mouse MHCII I-A/I-E (M5/114.15.2) | BioLegend | Cat# 107627; RRID:AB_1659252 |
| PE anti-mouse Gr1 (RB6-8C5) | BioLegend | Cat# 108407; RRID:AB_313372 |
| APC anti-mouse Ly6G (1A8) | BioLegend | Cat# 127613; RRID:AB_1877163 |
| APC anti-mouse CD206 (C068C2) | BioLegend | Cat# 141707; RRID:AB_10896057 |
| Mouse BD Fc Block (2.4G2) | BD pharmingen | Cat# 553142; RRID:AB_394657 |
| PE mouse anti-mouse T-Bet (4B10) | BD pharmingen | Cat# 561265; RRID:AB_10565980 |
| APC anti-mouse CD4 (GK1.5) | BioLegend | Cat# 100411; RRID:AB_312696 |
| PE/Cy7 CD8 (53-6.7) | BioLegend | Cat# 100721; RRID:AB_312760 |
| PerCP/Cy5.5 anti-mouse CD8 (53-6.7) | BioLegend | Cat# 100733; RRID:AB_2075239 |
| PerCP/Cy5.5 anti-mouse CD86 (GL-1) | BioLegend | Cat# 105027; RRID:AB_893420 |
| PerCP/Cy5.5 Ep-CAM (G8.8) | BioLegend | Cat# 107627; RRID:AB_1659252 |
| PerCP/Cy5.5 anti-mouse PD1 (29F.1A12) | BioLegend | Cat# 135207; RRID:AB_10550092 |
| Pe/Cy7 anti-mouse IFN-g (XMG1.2) | BioLegend | Cat# 505825; RRID:AB_1595591 |
| PE anti-mouse CD31 (390) | BioLegend | Cat# 102407; RRID:AB_312902 |
| Pe/Cy7 anti-mouse PDGFRa (APA5) | BioLegend | Cat# 135911; RRID:AB_2715973 |
| PE anti-mouse CD11c (N418) | BioLegend | Cat# 117307; RRID:AB_313776 |
| PE IgG1k isotype control | Invitrogen | Cat# 12-4301-81; RRID:AB_470046 |
| Pe/Cy7 Rat IgG1 isotype control | BioLegend | Cat# 400415; RRID:AB_326521 |
| InVivo rat IgG2a mAb (2A3) isotype control | BioXCell | Cat# BE0089; RRID:AB_1107769 |
| InVivoMAb monoclonal anti-mouse Ly6G (1A8) | BioXCell | Cat# BE0075-1; RRID:AB_1107721 |
| InVivoMAb CD40 rat anti-mouse IgG2a mAb (FGK45) | BioXCell | Cat# BE0016-2; RRID:AB_1107647 |
| InVivoMAb anti-rat Kappa Immunoglobulin Light Chain | BioXCell | Cat# BE0122; RRID:AB_10951292 |
| APC anti-mouse CD34 (HM34) | BioLegend | Cat #128611; RRID:AB_10553895 |
| MPO antibody | Santa Cruz | Cat# sc-390109; RRID:AB_2892996 |
| Neutrophil Marker (NIMP-R14) antibody | Santa Cruz | Cat# sc-59338; RRID:AB_2167795 |
| P53 (1C12) Mouse antibody | Cell Signaling | Cat# 2524S; RRID:AB_331743 |
| Anti-Vinculin mouse antibody | SigmaAldrich | Cat# V9264; RRID:AB_10603627 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| STBL3 | ThermoFisher | Cat# C737303 |
|
| ||
| Chemicals, Peptides, and Recombinant Proteins | ||
|
| ||
| Nab-paclitaxel | Celgene | Cat# 68817-134-50 |
| Gemcitabine | Hospira | Cat# 0409-0181-01 |
| Albumin | Sigma Aldrich | Cat# A5843 |
| Xtremegene 9 | Roche | Cat# 06 365 779 001 |
| Polybrene | Sigma Aldrich | Cat# TR-1003-G |
| Hygromycin | Sigma Aldrich | Cat# 400053 |
| G418 | Sigma Aldrich | Cat# 345812 |
| PfIFI | New England Biolabs | Cat# R0595S |
| BsmBI | New England Biolabs | Cat#R0739S |
| Shield-1 | Cheminpharma | Cat# S1-001 |
| Lipofectamine 3000 | Life Technologies | Cat# L3000008 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Surveyor DNA mutation Kit | IDT | Cat# 706020 |
| TOPO TA Cloning Kit | Thermo-Fisher | Cat# K4575J10 |
| SYBR Green PCR Master Mix | Thermo-Fisher | Cat# 4309155 |
| Quantitect Reverse Transcription Kit | QIAGEN | Cat# 205311 |
| Zombie Green Fixable Viability Kit | Biolegend | Cat# 423111 |
| RNeasy Mini Kit | QIAGEN | Cat# 157029548 |
| Milliplex Mouse Cytokine Magnetic Bead Panel | Millipore Sigma | Cat# MCYTOMAG-70K-03 |
| KAPA Mouse Genotyping Kit | Roche | Cat# 07961804001 |
| Foxp3/Transcription Factor Staining Buffer Set | Invitrogen | Cat# 00-5523-00 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| HEK293T | ATCC | Cat# ATCC® CRL-3216 |
| KrasG12D/+;Trp53+/+PDEC | Pylayeva-Gupta et al., 2012 | N/A |
| KrasG12D/+;Trp53−/− PDEC | This manuscript | N/A |
| KrasG12D/+;Trp53R172H/+PDEC | This manuscript | N/A |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| C57Bl6/J mice females (6-10weeks) | Charles River Laboratories | CR: 632 |
|
| ||
| Oligonucleotides | ||
| p53 sgRNA, CGG AGC AGC GCT CAT GGT GG | This manuscript | N/A |
|
| ||
| p53 HDR template, CAC CTC CAG CTG GGA GCC GTG TCC GCG CCA TGG CCA TCT ACA AGA AGT CAC AGC ACA TGA CGG AAG TCG TGA GAC ACT GTC CCC ACC ATG AGC GCT GCT CCG ATG GTG ATG GTA AGC CCT CAA CAC CGC CTG TGG GGT TAG GAC TGG CAG | This manuscript | N/A |
| p53 Forward PCR primer, TGGGA CAGCCAAGTCTGTTA | This manuscript | N/A |
| p53 Reverse PCR primer CCTGCT GTCTCCAGACTCCT | This manuscript | N/A |
| shScramble CCGGTCCTAAGGTTA AGTCGCCCTCGCTCGAGCGAGGG CGACTTAACCTTAGGTTTT | Sarbassov et al., 2005 | Addgene Cat # 1864 |
| CXCL2 forward PCR primer ATGC CTGAAGACCCTGCCAAG | Roy et al., 2012 | N/A |
| CXCL2 reverse PCR primer GGTC AGTTAGCCTTGCCTTTG | Roy et al., 2012 | N/A |
| CXCL5 forward PCR primer ACAGT GCCCTACGGTGGAAGT | Roy et al., 2012 | N/A |
| CXCL5 reverse PCR primer CGAGT GCATTCCGCTTAGCTT | Roy et al., 2012 | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| pLKO.1 neo | Gift from Sheila Stewart | Addgene Cat# 13425 |
| psPAX2 | Gift from Didier Trono | Addgene Cat# 12260 |
| pMD2G | Gift from Didier Trono | Addgene Cat# 12259 |
| pLKO.1 hygro | Gift from Bob Weinberg | Addgene Cat# 24150 |
| LentiCRISPRv2 hygro | Stringer et al., 2019 | Addgene Cat# 98291 |
| conditional EDCPV | Gift from R. Sordella, Cold Spring Harbor Laboratory | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/; RRID:SCR_001935 |
| FlowJo version 10.2 | FlowJo | https://www.flowjo.com/solutions/flowjo/downloads; RRID:SCR_008520 |
| R version 3.4.3 | RStudio | http://www.R-project.org; RRID:SCR_000432 |
| Prism Version 7 | GraphPad | https://www.graphpad.com/scientific-software/prism/; RRID:SCR_002798 |
| Vevo Lab | FUJIFILM-VisualSonics | https://www.visualsonics.com/product/software/vevo-lab |
|
| ||
| Other | ||
|
| ||
| Whole-Tissue Gene Expression Data of Pancreatic Ductal Adenocarcinoma | Badea et al., 2008 | GSE15471 |
| Expression data from Mayo Clinic Pancreatic Tumor and Normal samples | Pei et al., 2009 | GSE16515 |