

Summary
Lung cancer is one of the most aggressive tumors. Targeted therapies stratified by oncogenic drivers has remarkably improved therapeutic outcomes in patients with non-small cell lung cancer (NSCLC) 1. However, such oncogenic drivers are not found in 25-40% of NSCLC cases 2. Here, we identified a novel fusion transcript of CLIP1 and LTK using whole transcriptome sequencing in a multi-institutional genome screening platform (LC-SCRUM-Asia, UMIN000036871). The CLIP1-LTK fusion was present in 0.4% of NSCLCs and was mutually exclusive with other known oncogenic drivers. We show that kinase activity of CLIP1-LTK fusion protein is constitutively activated and has transformation potential. Treatment of Ba/F3 cells expressing CLIP1-LTK with lorlatinib, an ALK inhibitor, inhibited CLIP1-LTK kinase activity, suppressed proliferation, and induced apoptosis. One NSCLC patient harboring the CLIP1-LTK fusion showed dramatic clinical responses to lorlatinib treatment. To our knowledge, this is the first description of LTK alterations with oncogenic activity in cancers. Our results demonstrate that the CLIP1-LTK fusion is a novel target in NSCLC, which could be treated with lorlatinib.
The discovery of oncogenic drivers has uncovered the pathogenesis of non-small cell lung cancer (NSCLC). Comprehensive molecular analyses revealed that somatic driver events can be identified in the receptor tyrosine kinase (RTK)/RAS/RAF pathway in the 76% of lung adenocarcinoma samples. These include somatic mutations (e.g. KRAS, EGFR and BRAF mutations) and fusions (e.g. ROS1, ALK, and RET) 3. Furthermore, transcriptome sequencing-based approach identified additional actionable oncogenic drivers such as CD74-NRG1 fusion in NSCLC 4. Development of corresponding kinase inhibitors has changed treatment strategies and improved survival for patients with targetable oncogenic drivers. Nonetheless, no targeted therapies have been developed for the large proportion of advanced NSCLC patients whose tumors lack such known oncogenic drivers.
In 2013, we established a multi-institutional lung cancer genome screening platform (LC-SCRUM-Asia) to identify lung cancer patients with oncogenic drivers and to clinically develop molecular-targeted therapies. Here, we apply this platform to search for novel oncogenic drivers that could be targeted in patients with currently untreatable NSCLC.
Results
Identification of the CLIP1-LTK fusion
To identify novel oncogenic gene fusion, we initiated whole transcriptome sequencing (WTS) of NSCLC samples negative for known oncogenic drivers in the LC-SCRUM-Asia cohort 5. Of consecutive 75 samples analyzed between October of 2020 and December of 2020 (Extended Data Table 1), we identified an in-frame fusion transcript of CLIP1 on chromosome 12q24 and LTK on chromosome 15q15 in one patient (patient #1) (Fig. 1a). LTK and ALK constitute the ALK/LTK subfamily of receptor tyrosine kinases (RTKs) 6, while CLIP1 is a member of the microtubule plus-end tracking protein family7. A CLIP1-LTK fusion protein is predicted to harbor multiple coiled-coil domains found in CLIP1 upstream of the full LTK kinase domain (Fig. 1b). To confirm that transcripts encoding the fusion protein were present in tumors from patient #1, we used two sets of RT-PCR primers to detect amplicons of the predicted size in tissues from two metastatic sites, the supraclavicular lymph node and liver (Fig. 1c). Sanger sequencing verified the fusion sequence of CLIP1 exon 16 to LTK exon 11 in the expressed transcript (Extended Data Fig. 1). In addition, we performed break-apart fluorescence in situ hybridization (FISH) assay for further validation of the LTK rearrangement in this patient. Hybridization of these probes showed orange (5’)-green (3’) fused signals in non-tumoral cells (Extended Data Fig. 2: left panel). However, 82 % of scored tumor cells showed 1≥ sole green (3’) signals including the kinase domain, which indicates the presence of LTK rearrangement (Extended Data Fig. 2: right panel). Lack of orange signals (5’) in tumor cells may suggest deletion in this chromosomal region.
Fig. 1. Identification of the CLIP1-LTK fusion.
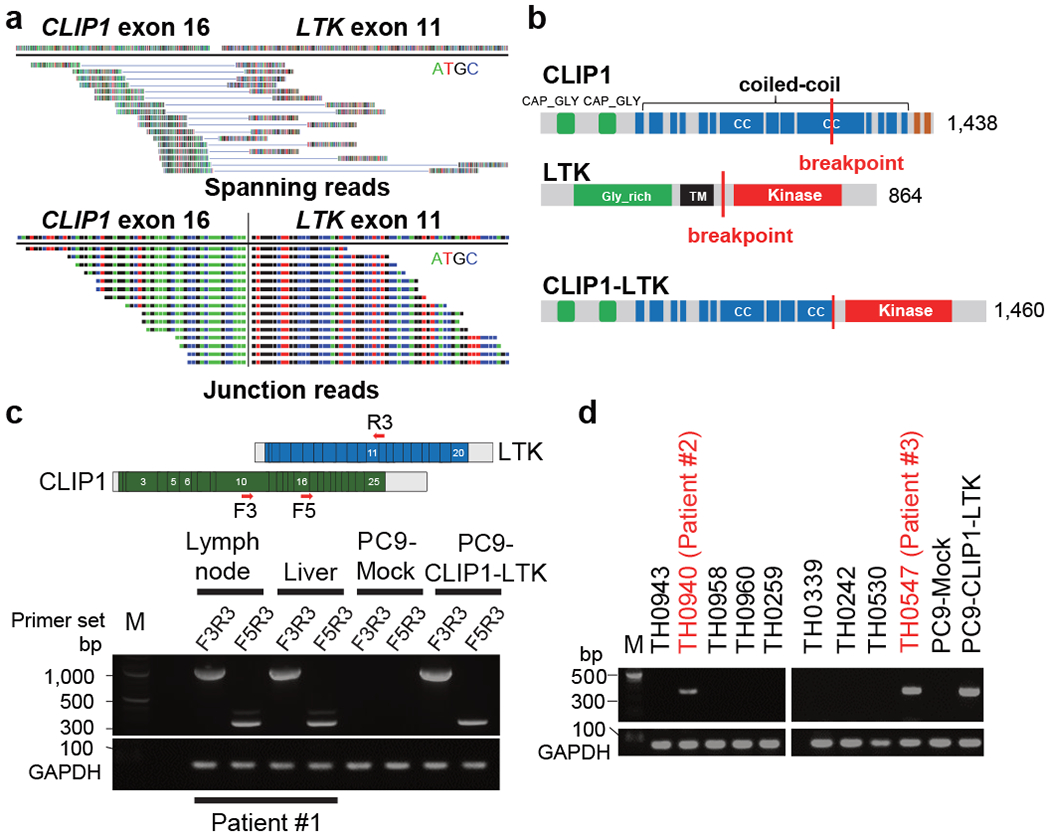
a, Spanning (top) and junction (bottom) reads for the CLIP1-LTK fusion transcript, as detected by WTS. b, Schematic representations of WT CLIP1 (top), WT LTK (middle), and the CLIP1-LTK fusion protein (bottom). CC, coiled-coil; TM, transmembrane. c, Detection by RT-PCR of CLIP1-LTK transcripts in two metastatic sites, the supraclavicular lymph node and liver from Patient #1. Primer locations are indicated at the top. cDNA isolated from PC9 cells expressing the CLIP1-LTK fusion or Mock cells (see Methods) served as respective positive and negative controls. GAPDH transcripts serve as an internal control. M, 100-bp DNA ladder marker. d, Screening of lung cancer specimens for the CLIP1-LTK transcript using the F5 and R3 primer set. Shown are representative panels of RT-PCR screening that contain two positive samples (#2 and #3), as well as GAPDH.
Next, to determine how prevalent the CLIP1-LTK fusion is in lung cancers, we examined 572 tumor samples from patients with various types of lung cancer in LC-SCRUM-Asia, irrespective of oncogenic driver status, using RT-PCR described above. Of 30 small cell lung cancers, all were negative for this fusion. Of 542 NSCLC samples analyzed, two were positive for CLIP1-LTK fusion transcripts (0.4% in NSCLC) (see patient #2 and #3 in Fig. 1e). Overall, the 3 tumors positive for the fusion (the tumors from patient #1-3) were negative for other known oncogenic drivers, suggesting that the CLIP1-LTK fusion is mutually exclusive with other defined oncogene drivers. ALK immunohistochemistry analysis, which was approved as an assay for detecting ALK fusions, was also performed, and all 3 tumors were negative. All 3 tumors with CLIP1-LTK fusion were classified histologically as adenocarcinoma (Extended Data Fig. 3 and Extended Data Table 2).
Transforming potential of CLIP1-LTK
As coiled-coil domains reportedly facilitate protein dimerization 8, we asked whether the CLIP1-LTK protein formed dimers that might constitutively activate LTK kinase. To do so, we transiently transfected NIH3T3 cells with either CLIP1-LTK, wild-type (WT) LTK or Mock control expression constructs and probed immunoblots of cell extracts using an antibody which detects LTK phosphorylated at tyrosine 672. We detected robust phosphorylation in CLIP1-LTK-expressing cells but much weaker phosphorylation when cells were transfected with WT LTK (Fig. 2a). Next, we asked whether CLIP1-LTK protein could transform mouse lines commonly used to analyze transforming potential, namely NIH3T3 fibroblasts 9 and the Ba/F3 pro-B line 10. To do so, we transduced both lines with MIGR1 retroviral constructs to allow expression of human CLIP1, LTK, CLIP-LTK as well as GFP, or Mock virus. As an additional control we transduced cells with CLIP1-LTK harboring a K1140M mutation (CLIP1-LTK-K1140M), which corresponds to EML4-ALK K589M and renders the fusion protein kinase-dead 11. GFP positive cells were sorted and immunoblot analysis of cell extracts with a phospho-LTK antibody revealed a strong band in NIH3T3 or Ba/F3 cells expressing CLIP1-LTK (NIH3T3-CLIP1-LTK or Ba/F3-CLIP1-LTK) but no visible bands in Mock cells or in cells expressing LTK (NIH3T3-LTK or Ba/F3-LTK) or CLIP1-LTK-K1140M (NIH3T3-CLIP1-LTK-K1140M or Ba/F3-CLIP1-LTK-K1140M) (Extended Data Fig. 4). These results confirm that CLIP1-LTK is constitutively activated in stably transduced cell lines. The flow cytometry analysis of Mock-transduced NIH3T3 cells, NIH3T3-LTK, and NIH3T3-CLIP1-LTK revealed that LTK, but not CLIP1-LTK, was expressed on the cell surface (Extended Data Fig. 5a), while both LTK and CLIP-LTK were detected in the cytoplasm (Extended Data Fig. 5b). These results suggest that CLIP1-LTK is located in the cytoplasm as it lacks the transmembrane domain of LTK.
Fig. 2. Transforming activity of the CLIP1-LTK fusion.
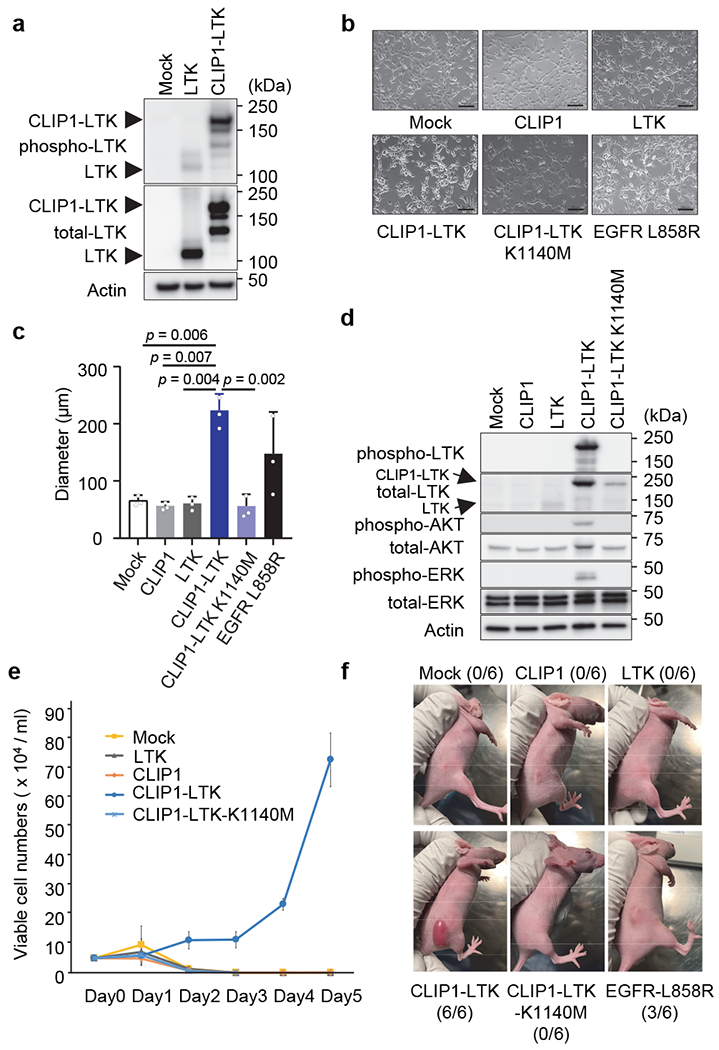
a, Constitutive tyrosine kinase activity of CLIP1-LTK fusion protein. NIH3T3 cells were transiently transfected with empty vector or WT LTK or CLIP1-LTK expression plasmids, and 48 hours later cell extracts were immunoblotted with anti-phospho LTK antibodies. b, Light microscopy images of indicated stably-transduced NIH3T3 cells. Cells were plated in 10 cm plates at 2 x 105 cells/ml and cultured for 1-2 days until cells reached 50-60 % confluency. Bars = 100 μm. c, A soft agar colony formation assay of indicated NIH3T3 stably-transduced cells. Colony diameters were measured and average values ± standard deviations from three independent experiments are shown. d, Activation of AKT and ERK downstream of CLIP1-LTK. Ba/F3 cells transduced with either Mock, CLIP1, LTK, CLIP1-LTK, or CLIP1-LTK-K1140M were cultured at 5 x 105 cells/ml in the absence of 5% WEHI medium for 12 hours. Cell extracts were then subjected to western blotting analysis with indicated antibodies. e, IL-3-independent growth of Ba/F3-CLIP1-LTK cells. Prior to plating, Ba/F3 cells transduced with indicated constructs and growing in 5% WEHI medium were washed and resuspended in RPMI medium at 5 x 104 cells/ml. Living cells were counted daily and shown as an average ± standard deviation from three independent experiments. f, CLIP1-LTK has tumor transforming activity. NIH3T3 cells transduced with transduced with either Mock, CLIP1, LTK, CLIP1-LTK, CLIP1-LTK-K1140M, or EGFR-L858R were injected into the flanks of nude mice and observed for tumor growth. Representative pictures taken at day 10 following injection are shown. The numbers of tumors induced in the injected animals are shown in parentheses.
Further analysis indicated that NIH3T3 cells expressing CLIP1 (NIH3T3-CLIP1) or NIH3T3-LTK cells as well as Mock-transduced cells were contact inhibited and showed normal cobblestone-like morphology, while NIH3T3-CLIP1-LTK cells showed a round-shape morphology (Fig. 2b) and lack of contact-inhibition (Extended Data Fig. 6a). Interestingly, that analysis also showed that NIH3T3-CLIP1-LTK-K1140M cells showed phenotypically resembled Mock-transduced cells and normal contact inhibition. Furthermore, analysis of colony formation in soft agar revealed significantly larger NIH3T3-CLIP1-LTK colonies compared to either Mock-transduced or NIH3T3-CLIP1 or NIH3T3-LTK cells after two weeks of growth (Extended Data Fig. 6b). In fact, NIH3T3-CLIP1-LTK colonies were larger in diameter than positive control NIH3T3 cells expressing EGFR-L858R, which has transforming activity 12 (Fig. 2c). Finally, as expected, NIH3T3-CLIP1-LTK-K1140M cells did not exhibit anchorage-independent growth (Fig. 2c).
Ba/F3 cells require IL-3 for growth, but expression of oncogenic kinases renders them IL-3 independent 10,13,14. Indeed, AKT and ERK remained activated in Ba/F3-CLIP1-LTK cells but not in Ba/F3 cells transduced with either Mock, CLIP1, LTK, or CLIP1-LTK-K1140M cells in the absence of WEHI medium as a source of IL-3 (Fig. 2d). Consistent with this observation, unlike other Ba/F3 stable cells, Ba/F3-CLIP1-LTK cells showed IL-3-independent growth (Fig. 2e), suggesting that CLIP1-LTK harbors transforming activity.
To further confirm the transforming activity of CLIP1-LTK in vivo, we subcutaneously injected NIH3T3 cells transduced with either Mock, CLIP1, LTK, CLIP1-LTK, or CLIP1-LTK-K1140M cells into the flank of nude mice. Tumors were detected only in nude mice injected with NIH3T3-CLIP1-LTK cells (Fig. 2f). Interestingly, no tumor was observed in mice injected with NIH3T3-CLIP1-LTK-K1140M. Taken together, these results support the conclusion that CLIP1-LTK fusion induces oncogenic transformation dependent on its kinase activity.
Lorlatinib inhibits CLIP1-LTK activity
As LTK and ALK share nearly 80% identity in their respective kinase domains, we hypothesized that compounds developed as ALK tyrosine kinase inhibitors would inhibit constitutive kinase activity of the CLIP1-LTK fusion protein. To test this hypothesis, we treated NIH3T3 cells transiently transfected with a CLIP1-LTK expression construct with varying concentrations of 7 FDA-approved or investigational tyrosine kinase inhibitors known to inhibit ALK. We then prepared immunoblots from cell extracts and probed each with a phospho-LTK antibody. Among inhibitors tested, lorlatinib was most potent and nearly completely blocked CLIP1-LTK phosphorylation at 10 nM (Fig. 3a). Lorlatinib is a third-generation ALK/ROS1 inhibitor with IC50 of <0.07 nM, based on a biochemical kinase assay using WT ALK 15. Consistent with these observations, in a cell viability assay using Ba/F3-CLIP1-LTK cells, the lorlatinib IC50 was 1.1 nM, while values obtained using other ALK inhibitors were in the 10-20 nM range (Fig. 3b). In contrast, osimertinib was not as effective as ALK inhibitors and showed an IC50 value of >100 nM. Lorlatinib did not inhibit cell viability when Ba/F3-CLIP1-LTK cells were cultured in the presence of WEHI medium containing IL-3, excluding the possibility of nonspecific toxicity (Extended Data Fig. 7). Moreover, lorlatinib treatment inhibited anchorage-independent growth of NIH3T3-CLIP1-LTK cells in soft agar (Extended Data Fig. 8), suggesting that lorlatinib may be effective for tumors induced by CLIP1-LTK.
Fig. 3. Lorlatinib inhibits CLIP1-LTK kinase activity and suppresses tumor growth.
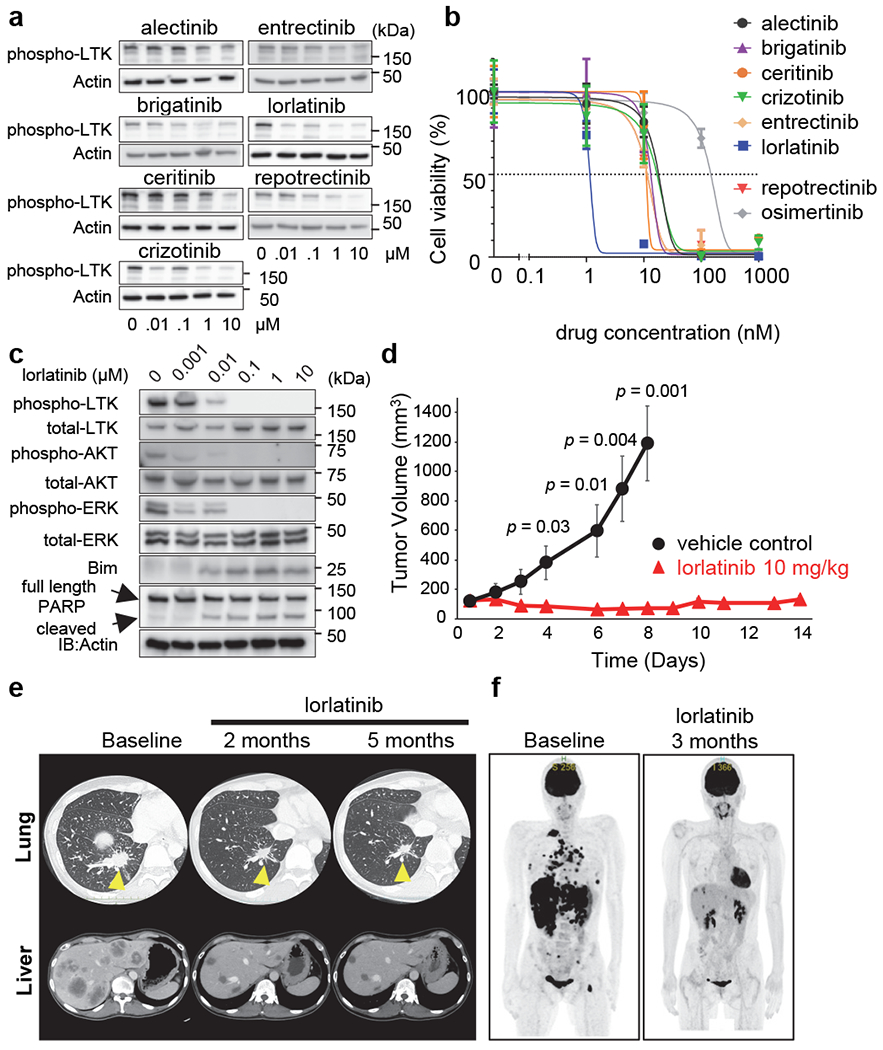
a, Screening for CLIP1-LTK inhibitors. NIH3T3 cells were transiently transfected with pCMV3-CLIP1-LTK, cultured 24 hours, and then treated 6 hours with indicated TKIs. Cell extracts were subjected to western blotting analysis (using antibodies against phospho-LTK and Actin). b, Viability assays of inhibitor-treated cells. Ba/F3-CLIP-LTK cells were treated 48 hours with increasing concentrations of indicated chemicals and viability was measured using the Cell Counting Kit-8. Results are shown as an average ± standard deviation from three independent experiments. c, Inhibition of CLIP-LTK kinase activity and downstream signaling by lorlatinib. Ba/F3-CLIP-LTK cells (4 x 105 cells/well in 6 well plates) were treated 24 hours with increasing lorlatinib concentrations. Cell extracts were then subjected to western blotting analysis with indicated antibodies. d, Inhibition of tumor growth by lorlatinib in vivo. NIH3T3-CLIP-LTK cells (3 x 106 cells) were transplanted into the flanks of nude mice. When the mean tumor volume reached 100-200 mm3, mice were randomized into two groups and treated with either lorlatinib (10 mg/kg once daily) or vehicle control by oral gavage for 14 days and 8 days, respectively. Tumor volumes are shown as an average ± standard error (n=6 for each group). e, Time course CT scan images of right lung (upper panels) and liver (lower panels) in Patient #1, who had been treated with lorlatinib orally at 100 mg daily. f, Time course PET scan images of the whole body.
A previous report indicated that LTK activates AKT and ERK 16. Thus, we asked whether inhibition of CLIP1-LTK kinase activity by lorlatinib would impact downstream signaling responsible for cell proliferation or apoptosis. To do so, we treated Ba/F3-CLIP-LTK cells for 24 hours with various concentrations of lorlatinib and then prepared cell lysates for immunoblotting with indicated antibodies (Fig. 3c). That analysis revealed decreased phosphorylation of CLIP1-LTK, AKT, and ERK relative to untreated controls, as well as induction of a non-phosphorylated, stabilized form of Bim and cleavage of full-length PARP (also indicative of apoptosis) at lorlatinib concentrations as low as 10 nM. These results suggest that activation of AKT and ERK downstream of CLIP1-LTK enhances proliferation and cell survival, while inhibition of CLIP1-LTK kinase activity induces apoptosis.
To further evaluate the activity of lorlatinib against CLIP1-LTK expressing tumor, we subcutaneously injected NIH3T3-CLIP1-LTK cells into the flanks of nude mice and treated them with lorlatinib or vehicle control for 2 weeks when tumors reach a volume of approximately 100 mm3. Consistent with cell viability assay, lorlatinib treatment significantly inhibited tumor growth (Fig. 3d).
CLIP1-LTK as a therapeutic target
Patient #1 described above (Extended Data Table 2) received first-line therapy with 4 cycles of carboplatin, pemetrexed and pembrolizumab, resulting in tumor shrinkage. However, after subsequent maintenance therapy with pemetrexed plus pembrolizumab, computed tomography (CT) images showed rapid disease progression with enlargement of both the primary tumor and multiple metastatic tumors, including liver metastases (Fig. 3e). WTS and Sanger sequencing of the patient’s tumor cDNA revealed the CLIP1-LTK fusion (Fig. 1a, c and Extended Data Fig. 1), and the LTK rearrangement was confirmed by break-apart FISH (Extended Data Fig. 2). The patient, who was advised of potential benefits and risks of lorlatinib treatment and informed of our preclinical findings, then provided consent for treatment with LTK-targeted therapy. After the off-label use was approved by National Cancer Center Hospital East review board, lorlatinib was started at a regular dosage of 100 mg once daily every day. The patient’s two- and five-month follow-up CT images showed rapid and dramatic shrinkage of the primary tumor and multiple metastatic tumors (Fig. 3e). In addition, as shown by Positron Emission Tomography (PET) images of whole body, all the primary and metastatic tumors at baseline responded dramatically to lorlatinib treatment (Fig. 3f). These results indicate that NSCLC patients with the CLIP1-LTK fusion may be candidates for lorlatinib therapy.
Discussion
In this study, we identified the CLIP1-LTK fusion in NSCLC patients without known oncogenic drivers and demonstrated that CLIP1-LTK kinase is constitutively activated and has transforming capacity that can be suppressed by lorlatinib treatment. Clinically, we report that a NSCLC patient harboring the CLIP1-LTK fusion showed a robust positive response to lorlatinib.
To identify novel oncogenic gene fusions, we used an enriched cohort of pan-negative for oncogenic drivers determined by molecular analyses in LC-SCRUM-Asia. In addition, we used an RNA-based targeted approach, which is more accurate than DNA sequencing but occasionally limited by RNA quality and quantity, particularly when samples are derived from formalin-fixed, paraffin-embedded tissues 17. However, in the LC-SCRUM-Asia, fresh-frozen tissues have been submitted for molecular analyses and derived RNA samples were of sufficient quantity and quality, being factors that likely contributed to successful identification of the CLIP1-LTK fusion transcript as a novel oncogenic driver in NSCLC.
In our Japanese cohort, 0.4% of NSCLC specimens harbored the CLIP1-LTK fusion. Oncogenic fusions such as ALK, ROS1, RET are rare fractions, less than 5% in NSCLC, and their prevalence was observed to be similar in any racial/ethnic cohort 18. Therefore, it is predicted that there are more than 7,500 cases with NSCLC harboring CLIP1-LTK fusion worldwide. However, due to the limited number of patients who exhibit this fusion, it is not yet clear whether NSCLC marked by CLIP1-LTK fusion will emerge as showing unique clinical characteristics. An ongoing retrospective screening for the CLIP1-LTK fusion using LC-SCRUM-Asia cohort (n=1,500) will provide more detailed information on prevalence and clinicopathologic features. Moreover, additional effort is required to determine the prevalence of the fusion in other racial or ethnic groups.
To our knowledge, this is the first report of functional oncogenic alterations in LTK in cancers. Although the non-recurrent UACA-LTK fusion was identified in a thyroid carcinoma by The Cancer Genome Atlas Research project 19, it seems to be an out-of-frame fusion and its oncogenic potential has been unknown 20. However, previous reports suggested that LTK hyperactivation can induce transformation6,21. In addition, WT LTK is expressed in brain, placenta, and hematopoietic cells such as those of B cell lineages 6, while CLIP1 is ubiquitously expressed 22. Transcription of the CLIP1-LTK fusion is driven by the CLIP1 promoter, and thus CLIP1-LTK protein could be expressed in a variety of cell types, including alveolar cells. It is also predicted that fusion to CLIP1 constitutively activates LTK due to dimerization through CLIP1 coiled-coil domains. This idea is supported by observations that CLIP1 is also a fusion partner of the RTK genes ALK, RET, and ROS1 in multiple cancers such as Spitz tumors, renal cell carcinoma, and NSCLC 23–26.
Tumor tissues from 3 patients described here harboring the CLIP1-LTK fusion did not exhibit other known oncogenic drivers. Thus, it is likely that in these cases, CLIP1-LTK is a sole driver of lung tumorigenesis. Although no LTK-specific inhibitors are currently available, we evaluated known ALK inhibitors, as most inhibit LTK kinase activity in vitro. Among inhibitors tested, lorlatinib was most potent, with an IC50 of 1.1 nM in Ba/F3-CLIP1-LTK cells, a value similar to that seen in Ba/F3 cells expressing EML4-ALK27. Successful treatment of patient #1 with loratinib justifies immediate investigation using a larger cohort of lung cancer patients with the CLIP1-LTK fusion. Indeed, a large-scale prospective CLIP1-LTK screening using LC-SCRUM-Asia platform (n=5,000) is planned to answer this question. Furthermore, our study provides a rationale for development of more specific LTK inhibitors.
In summary, we identified the CLIP1-LTK fusion as a novel oncogenic driver in NSCLC, and the fusion protein can be targeted by lorlatinib. Urgent clinical development of molecular targeted agents as well as routine screening and clinical validation for this new oncogenic driver are now warranted.
METHODS
Multi-institutional lung cancer genome screening platform (LC-SCRUM-Asia)
As of May 2021, more than 13,000 lung cancer patients were enrolled from a total of 307 institutions across Japan. For the molecular screening, DNA and RNA were extracted from fresh and frozen tissues and/or from formalin-fixed paraffin-embedded (FFPE) samples. Between February of 2013 and March of 2015, RT-PCR and FISH analysis revealed ALK, ROS1, and RET fusions. From March 2015, a targeted next-generation sequencing (NGS) system (Oncomine Comprehensive Assay, Thermo Fisher Scientific, MA, USA) has been used for the molecular screening, and from September 2019, a multi-gene quantitative PCR (qPCR) assay (Amoy 9-in-1 kit, AmoyDx, Xiamen, China) has been also implemented. The research protocol (#2018-010) was approved by the Research Ethics Committees at National Cancer Center and all institutions participating in LC-SCRUM-Asia, and written informed consent was provided from each subject. This research protocol includes whole-transcriptome sequencing described below.
Whole-transcriptome sequencing (WTS)
Based on results of the molecular screening in LC-SCRUM-Asia, we selected consecutive non-squamous NSCLC samples lacking previously defined oncogenic drivers (namely, EGFR, ALK, ROS1, BRAF, MET, NTRK, HER2, RET, NRG1, and KRAS) described in the National Comprehensive Cancer Network (NCCN) guidelines. cDNA libraries were prepared from residual RNA samples (20-100 ng) using the TruSeq RNA Exome system (Illumina) according to the manufacturer’s protocol. Libraries were subjected to paired-end sequencing of 75-bp reads using the NextSeq 500/550 High Output Kit v2.5 on the NextSeq 550 system (Illumina). Alignment of sequencing reads and detection of fusion transcripts were performed using the STAR-fusion pipeline (v1.9.0) 28, and detected fusion transcripts were annotated using the CTAT database (Release v0.2.0: https://github.com/FusionAnnotator/CTAT_HumanFusionLib/releases/tag/v0.2.0).
RT-PCR and Sanger sequencing
Total RNA (20-200 ng) was reverse-transcribed to cDNA using Superscript™ VILO™ MasterMix (Invitrogen). cDNA was subjected to PCR amplification using HotStarTaq DNA polymerase (QIAGEN). PCR reactions were performed under the following conditions: 95 °C for 15 min, 40 cycles of 94 °C for 1 min, 55 °C for 1min, and 72 °C for 1min, followed by final extension for 10 min at 72 °C. PCR products were directly sequenced in both directions using an ABI PRISM 3500×I Genetic Analyzer (Applied Biosystems). Sequences of primers used for PCR in this study are listed in Supplementary Table 1. The study protocols (2016-288 and 2018-404) for RT-PCR and Sanger sequencing were approved by the Research Ethics Committee at National Cancer Center.
Fluorescence in situ hybridization
Fluorescence in situ hybridization (FISH) analysis was performed with 4 μm-sectioned FFPE samples. LTK BA Probe Set (LTKBA20-GROR, Empire Genomics) which were designed to flank the LTK gene, were used as probes, following manufacturer’s protocol.
Reagents
NIH3T3 and Phoenix-AMPHO cells were purchased from ATCC. Ba/F3, WEHI, and BOSC23 cells were kindly provided by Dr. Daniel G. Tenen (Harvard Medical School). PC9 cells were kindly provided by Dr. Pasi Jänne (Dana Farber Cancer Institute). Crizotinib, ceritinib, alectinib, brigatinib, lorlatinib, entrectinib, and reptrectinib were purchased from Selleck.
CLIP1-LTK constructs used for functional analysis
Empty pCMV3 vectors as well as those harboring CLIP1 and LTK were purchased from SinoBiological. CLIP1-LTK fusion cDNA was constructed using the In-fusion HD Cloning Plus kit (Takara). The kinase-dead CLIP1-LTK-K1140M expression plasmid was generated using the QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent). CLIP1, LTK, CLIP1-LTK, and CLIP1-LTK-KD cDNA were also subcloned into the MIGR1 retroviral vector 29 (gift from Dr. Warren Pear: Addgene plasmid # 27490; http://n2t.net/addgene:27490 ; RRID:Addgene_27490) using the In-fusion HD Cloning Plus kit. MIGR1 vector harboring EGFR-L858R was previously described 14. The integrity of all vector constructs was confirmed by Sanger sequencing. Oligonucleotides used for cloning are listed in Supplementary Table 1.
Viral transduction
BOSC23 or Phoenix-AMPHO cells were transfected with empty MIGR1 vectors or those harboring CLIP1, LTK, CLIP1-LTK, CLIP1-LTK-K1140M, or EGFR L858R constructs to produce retrovirus particles. Retrovirus-containing supernatants were harvested and concentrated with a Retro-X™ concentrator (Takara). Ba/F3 cells were transduced using the RetroNectin-bound Virus Infection Method (Takara). NIH3T3 and PC9 cells were transduced in the presence of 8 μg/ml polybrene. Transduced cells were selected by GFP sorting. Ba/F3 stably-transduced cells were maintained in RPMI supplemented with 10% FBS, 5% WEHI conditional medium (as a source of IL-3), and 100 units/ml penicillin and 100 μg/ml streptomycin (P/S). NIH3T3 and PC9 stably-transduced cells were maintained in DMEM supplemented with 10% FBS and P/S.
Ba/F3 transformation Assay
Ba/F3 stably-transduced cells in RPMI 1640 supplemented with 10% FBS plus P/S were plated in triplicate at a density of 5 x 104 cells/ml in 12-well plates. Cell viability and changes in cell number were determined using trypan blue staining.
Western blotting
Cells were lysed in SDS sample buffer and immediately boiled for 5 minutes. Lysates were subjected to SDS polyacrylamide gel electrophoresis and blotted on to PVDF membranes (Millipore). The antibodies and dilutions used are listed in Supplementary Table 2. Images were captured using ImageQuant LAS 4000 (GE Healthcare) and analyzed using ImageJ software (ver. 1.53). All images were assembled and figures were generated using Microsoft PowerPoint 2016 software (ver. 2108).
Cell viability assay
Ba/F3 cells (10,000 cells per well) were seeded in 96-well plates, treated with inhibitors of interest for 48 hours, and then viability was evaluated using the Cell Counting Kit-8 (Fujifilm). Data were captured using Spectra Max Paradigm (Molecular Devices) with SoftMax Pro software (ver.7.10). Absorbance was read at 450 nm. IC50 value was determined with a nonlinear regression model (four parameter) using GraphPad Prism software (ver. 9.0.2).
Soft agar colony formation assay
Anchorage-independent growth was assayed as described 30. A total of 10,000 cells/well was plated in 6 well plates, and colonies were counted or measured after 14 days. Cell images were captured using BZ-II Viewer software (ver. 2.10) and analyzed using BZ-II Analyzer software (ver. 1.42).
Flow cytometry
NIH3T3 cells transduced with transduced with either Mock, LTK, or CLIP1-LTK were stained with primary antibody specific for LTK (ab129155, Abcam, 1:200 dilution) for 30 min at 4°C, followed by washing and incubation with anti-rabbit antibody conjugated to Alexa Fluor 488 (A-21206, Invitrogen, 1:100 dilution) for 30 min at 4°C, and then acquired on FACSCanto™ II (BD Biosciences). Data were captured using FACSDiva software (ver. 9.0) and analyzed using FlowJo software (ver. 10.7.1).
Immunofluorescence microscopy
Cells were seeded at 1 x 105 cells on glass coverslip and fixed with 4% paraformaldehyde and permeabilized with 0.5% Triton X-100 in PBS, followed by incubation with anti-DDDDK-tag antibody conjugated to Alexa Fluor 594 (M185-A59, MBL, 1:200 dilution). Samples were mounted on slides with DAPI-containing ProLong Diamond Antifade Mountant (Thermo Fisher Scientific) and images were acquired with a Biorevo BZ-9000 fluorescence microscope (Keyence).
Xenograft experiments
All animal experiments were approved by the Institutional Animal Care and Use Committee of National Cancer Center (K20-009). The mice were housed on a 12:12 light/dark cycle, and temperature was kept at 24°C (23-25°C) and humidity at 49% (40-60%). To establish tumor xenografts, NIH3T3 cells transduced with Mock, CLIP1, LTK, CLIP1-LTK, CLIP1-LTK-K1140M, or EGFR L858R (3 x 106 cells) were transplanted into the flanks of athymic nude mice (BALB/cAJcl-Foxn1nu, CLEA Japan). To evaluate the efficacy of lorlatinib, nude mice bearing NIH3T3-CLIP1-LTK tumors were used. Lorlatinib was formulated in 2% DMSO + 30% polyethylene glycol 300 in H2O. When the mean tumor volume reached 100-200 mm3, mice were randomized into two groups and treated with either lorlatinib (10 mg/kg once daily) or vehicle control by oral gavage. Tumor volumes (6 tumors per each group) were calculated by the following fomula: 1/2(length × width2) 31.
Statistics and Reproducibility
Group size was based on previous experience. No statistical method was used to predetermine sample size. Unless otherwise noted, each experiment was repeated three or more times with similar results. Statistical analysis was conducted on data from three or more biologically independent experimental replicates. Student’s t-test (two-tailed) was used to determine statistical significance.
Extended Data
Extended Data Fig. 1: Electropherogram showing Sanger sequencing of the CLIP1-LTK fusion transcript.
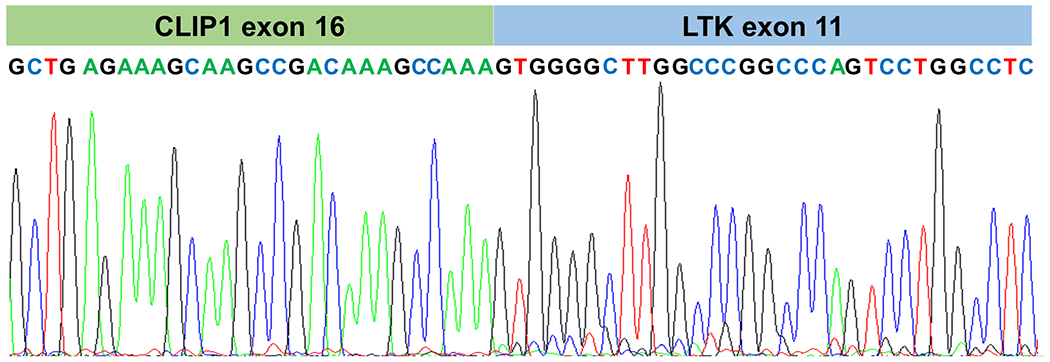
cDNAs were generated from RNAs isolated from patient tumors and amplified by RT-PCR with CLIP1-LTK F3 and R3 primers. PCR products were directly sequenced using respective primers.
Extended Data Fig. 2: LTK break-apart FISH assay.
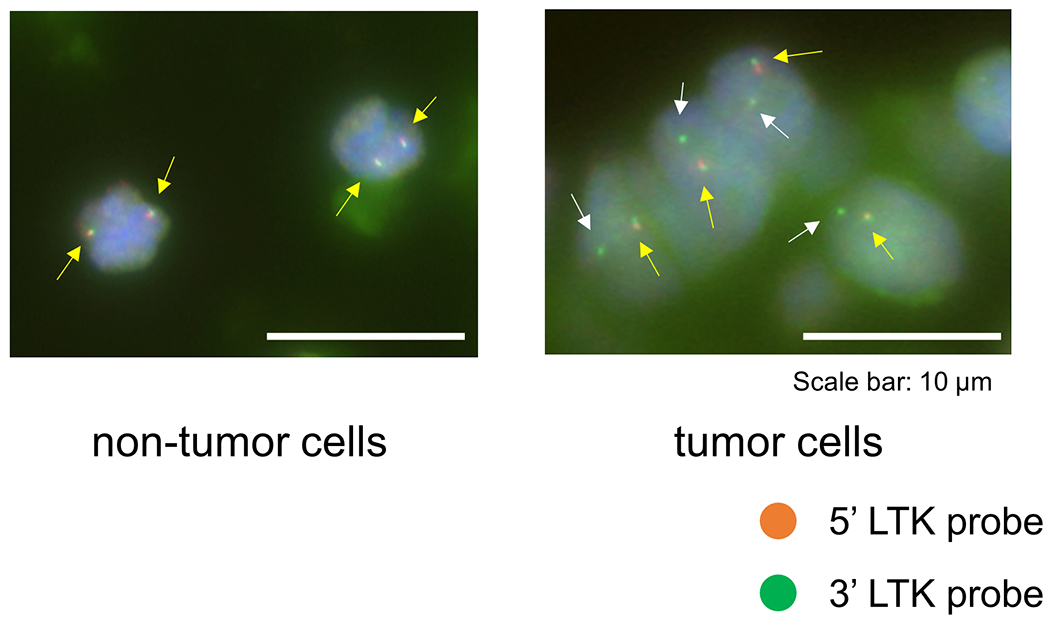
Non-tumor cells from Patient #1 showed orange (5’)-green (3’) fused signals (yellow arrows), while 82% (41/50) of scored tumor cells showed one fused (yellow arrows) and at least one green (3’) signals (white arrows), which indicates the presence of LTK rearrangement. Bars = 10 μm.
Extended Data Fig. 3: Histological findings of tumors positive for the CLIP1-LTK fusion.
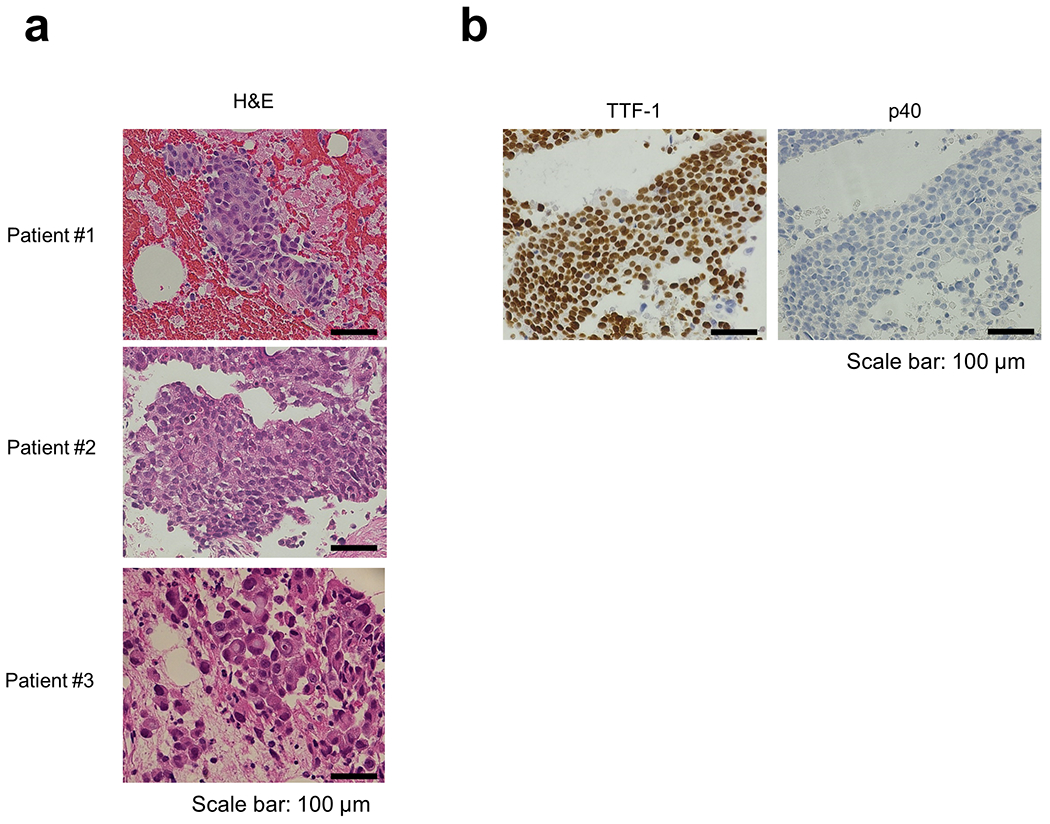
a, Hematoxylin and eosin (H&E) stained images. Samples from Patient #1 and #3 were diagnosed with adenocarcinoma morphologically. Samples from Patient #2 was diagnosed with NSCLC. Bars = 100 μm. b, Immunohistochemical analysis of samples from Patient #2. TTF-1 positive (upper panel) and p40 negative (lower panel) staining supported the diagnosis of NSCLC favor adenocarcinoma. Bars = 100 μm.
Extended Data Fig. 4: Analysis of Ba/F3 (a) and NIH3T3 (b) cells stably transduced using MIGR1 IRES-GFP vectors harboring indicated constructs.
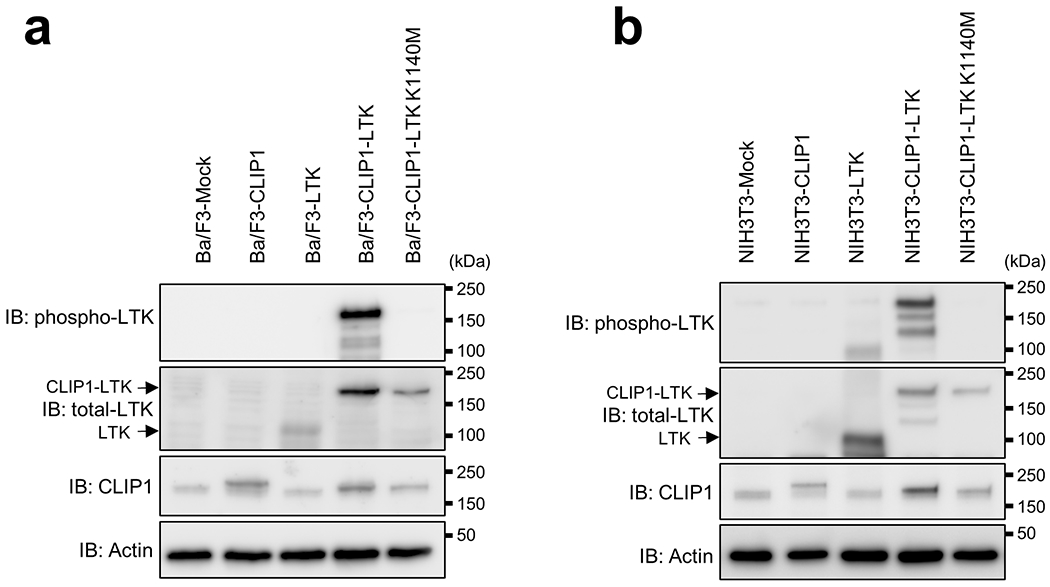
GFP-positive cells were sorted and expanded and then cell extracts were immunoblotted with antibodies indicated at the left side of each graph.
Extended Data Fig. 5: Cellular localization of LTK and CLIP1-LTK.
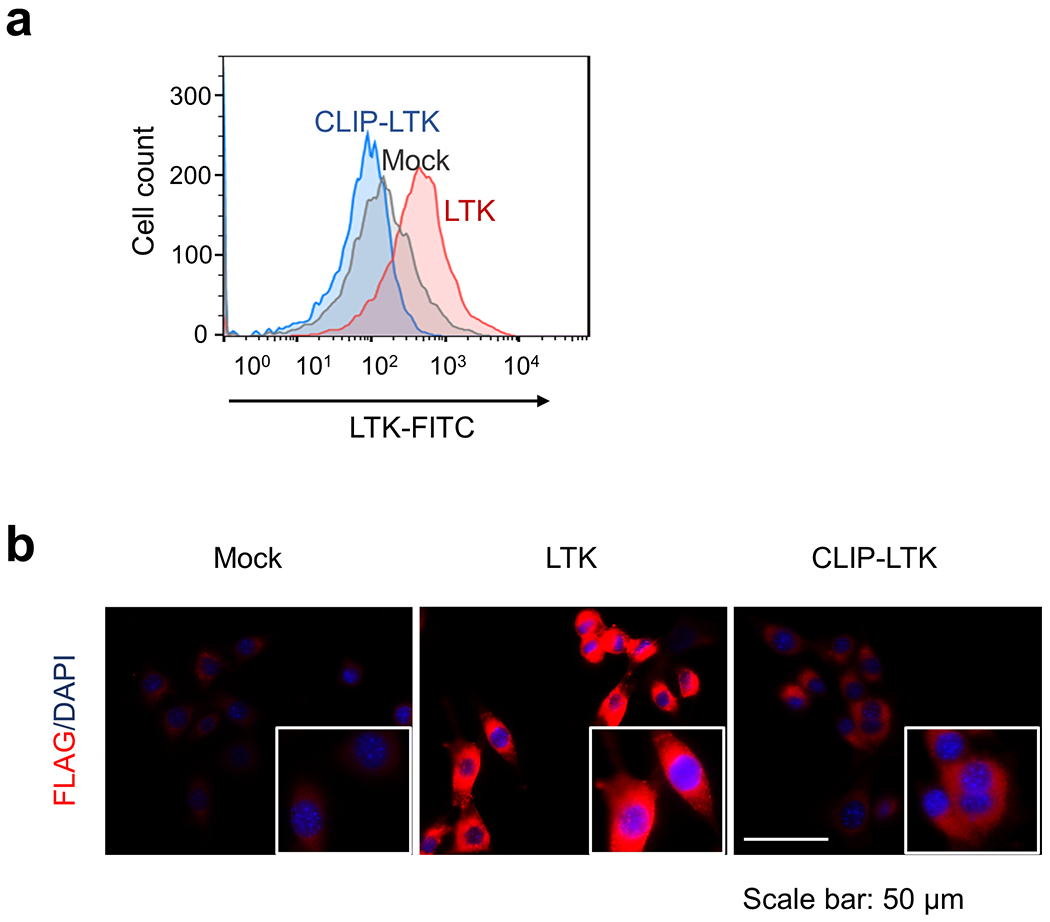
a, NIH3T3-Mock NIH3T3-LTK, or NIH3T3-CLIP1-LTK cells were stained with primary antibody specific for LTK, conjugated with Alexa Fluor 488, and subjected to flow cytometry analysis. Viable cells were gated as shown in Supplementary Information Figure 2. b, NIH3T3-Mock NIH3T3-LTK, or NIH3T3-CLIP1-LTK cells were fixed, permeabilized, and stained with anti-DDDDK-tag antibody conjugated to Alexa Fluor 594. Cells were subjected to immunofluorescence analysis.
Extended Data Fig. 6: Light microscopy images of indicated stably-transduced NIH3T3 cells.
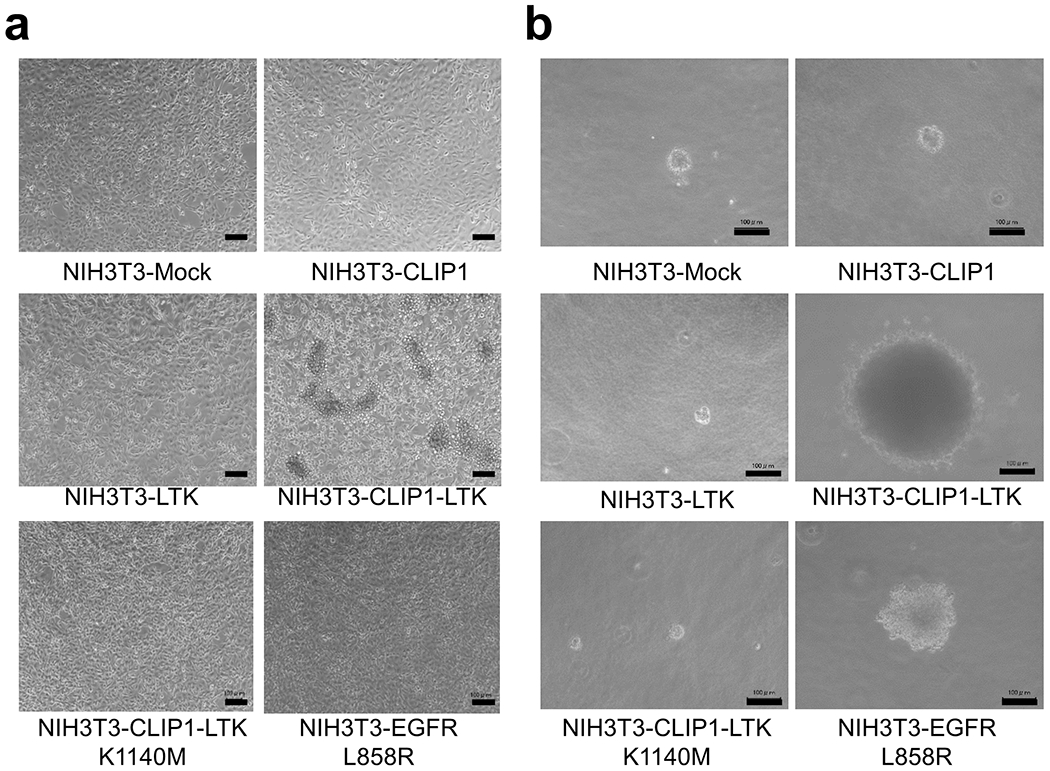
a, Cells were plated in 10 cm plates at 2 x 105 cells/ml and cultured in DMEM supplemented with 10% FBS and P/S for 2-3 days until cells reached 100% confluency. Bars = 100 μm. b, Cells were plated in 6-well plates at 1 x 104 cells/ml and cultured in a soft agar medium for 14 days. Bars = 100 μm.
Extended Data Fig. 7: Analysis of viability of Ba/F3-CLIP1-LTK cells.
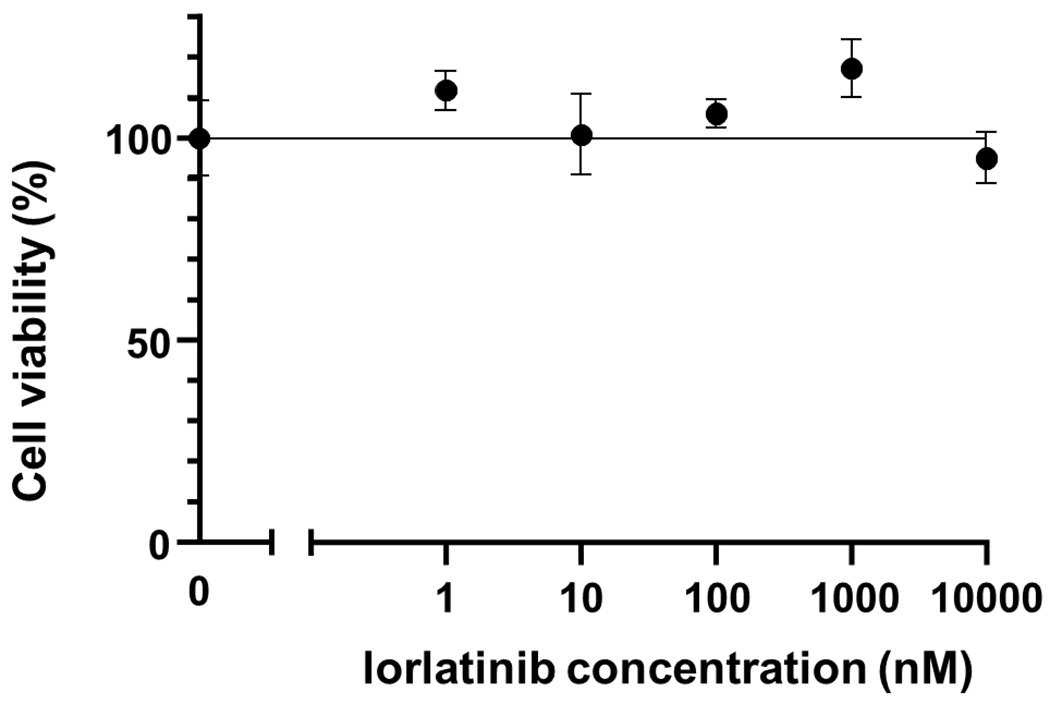
Ba/F3-CLIP1-LTK cells were treated with various concentrations of lorlatinib in the presence of 5% WEHI medium as a source of IL-3. Results are shown as an average ± standard deviation from three independent experiments.
Extended Data Fig. 8: Lorlatinib suppresses anchorage-independent growth of NIH3T3-CLIP1-LTK cell colonies.
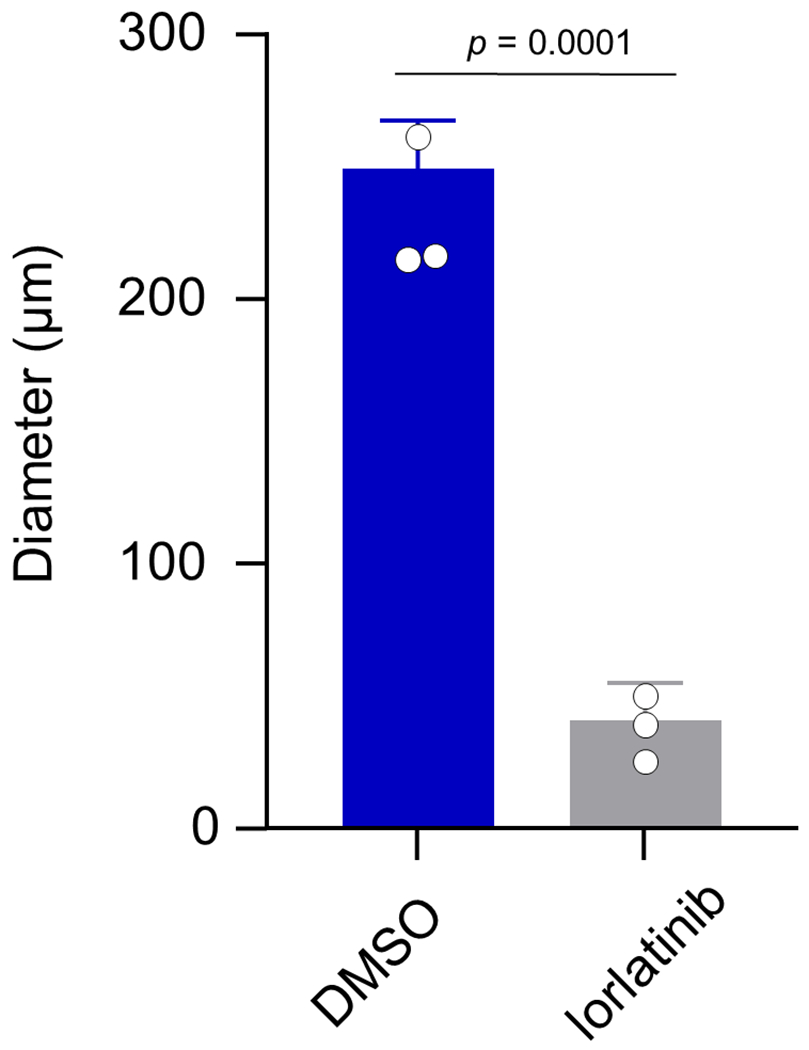
Colony diameters were measured in lorlatinib- versus vehicle (DMSO)-treated cells and shown as average ± standard deviation from three independent experiments.
Extended Data Table 1:
Characteristics of patients subjected to WTS (n=75)
| Patient characteristics (n=75) | |
|---|---|
| Median age (range) | 60 (34-84) |
|
| |
| Sex | |
| male | 54 (72%) |
| female | 21 (28%) |
|
| |
| Histology | |
| adenocarcinoma | 67 (89%) |
| others | 8 (11%) |
|
| |
| Smoking | |
| never | 15 (20%) |
| former | 48 (64%) |
| current | 12 (16%) |
| Median pack-year (range) | 35 (0-132) |
|
| |
| Stage | |
| III | 14 (19%) |
| IV | 52 (69%) |
| recurrence | 9 (12%) |
Extended Data Table 2.
Characteristics of lung cancer patients with the CLIP1-LTK fusion
| Patient | #1 | #2 | #3 |
|---|---|---|---|
| Age/Sex | 44/Female | 69/Male | 58/Male |
| TNM classification | T4N3M1C | T3N3M0 | T4N3M1C |
| Stage | IVB | IIIC | IVB |
| Histology | adenocarcinoma | adenocarcinoma | adenocarcinoma |
| Known driver | negative | negative | negative |
| Smoking (pack-year) | ex-smoker (1) | ex-smoker (44) | ex-smoker (28) |
| Metastasis/recurrence site | brain, lung, liver | none | bone |
| 1st line treatment | CBDCA+PEM+pembrolizumab | CDDP+VNR+TRT | CBDCA+PEM+pembrolizumab |
| (best response) | (PR) | (SD) | (PR) |
| Radiation therapy | brain | thorax | none |
| Co-occurring gene alterations | TP53 p.Tyr205Ter | TP53 p.Ser240Gly | TP53 p.Met160_Ala161delinslleSer, STK11 p.Asp194Tyr |
| Tumor mutation burden(mutations/Mb) | 1.69 | 5.9 | N/A |
CBDCA, carboplatin; PEM, pemetrexed; CDDP, cisplatin; VNR, vinorelbine; TRT, thoracic radiotherapy; PR, partial response; SD, stable disease; N/A, not available
Supplementary Material
Acknowledgements
We thank Ms. Yuri Murata and PREMIA Inc. for administrative assistance to manage clinical samples, molecular screening and clinico-genomic database in LC-SCRUM-Asia. We also thank Dr. Tomoko Yamamori Morita for assistance to flow cytometry analyses. This work was supported by the Princess Takamatsu Cancer Research Fund 18-250 (S.S.K), JSPS KAKENHI Grant Number JP20K17215 (H.I.), JP16K21746 (S.S.K.), and JP21K15541 (K.T.), the National Cancer Center Research and Development Fund 31-A-5 (A.O.), 31-A-6 (S.S.K), and 28-A-6 (K.G.), and AMED Grant Number JP21ck0106289 (K.G.), JP21ck0106294 (K.Y.), JP21ck0106483 (K.No.), JP21ck0106568 (K.G.), JP20ck0106411 (S.Ma.), JP20ck0106449 (I.O.), JP20ck0106450 (S.N.), JP20ak0101050 (Katsuya Tsuchihara), JP18Ik0201056 (A.O.), JP18kk0205004 (Hitoshi Nakagama), JP17Ack0106148 (K.G.), JP17Ack0106147 (Seiji Yano), and JP16ck0106041 (K.G.). Molecular screening in LC-SCRUM-Asia was supported by Amgen, Astellas, AstraZeneca, Boehringer Ingelheim, Bristol-Myers Squibb, Chugai, Daiichi sankyo, Eisai, Janssen, Kyowa kirin, Merck, MEDICAL & BIOLOGICAL LABORATORIES, MSD, Novartis, Ono, Pfizer, Sumitomo Dainippon, Taiho, and Takeda. Whole-transcriptome sequencing was supported by Merus Inc.
Declaration of interests
H.I. reports research support from Amgen and personal fees (honoraria) from Ono. S.Ma. reports research support from Chugai, Novartis, Eli Lilly, Merck and MSD, and personal fees (honoraria) from AstraZeneca, Chugai, Novartis, Pfizer and Eli Lilly. Y.S. reports research support from MSD, and personal fees (honoraria) from Ono, Pfizer, Chugai, Novartis, Bristol-Myers Squibb, AstraZeneca, and Taiho Pharmaceutical Co. T.F. reports research support from Pfizer, Chgai, Novartis, and AstraZeneca. K.Y. reports research support from AstraZeneca, Eli Lilly, Pfizer, Daiichi sankyo, Abbvie, Taiho, Bayer, Takeda, MSD, and personal fees (honoraria) from AstraZeneca, Bristol-Myers Squibb, Chugai, Daiichi sankyo, Janssen, Eli Lilly, Taiho, Novartis, Kyowa kirin, Boehringer Ingelheim. T.K. reports research support from Pfizer, Chugai, Takeda, Novartis, Turning Point Therapeutics, and AstraZeneca, and personal fees (honoraria) from Pfizer, Chugai, Takeda, Novartis, and AstraZeneca. K.Ni. reports research support from Pfizer, and personal fees (honoraria) from Pfizer, Chugai, Takeda, Novartis, and AstraZeneca. A.N. reports personal fees (honoraria) from Chgai and Novartis. S.Kuy. reports personal fees (honoraria) from Pfizer, Chugai, and AstraZeneca. N.F. reports personal fees (honoraria) from Pfizer, Chugai, Novartis, and AstraZeneca. I.O. reports research support from Chugai, Takeda, and Novartis, and personal fees (honoraria) from Pfizer, Chugai, Novartis, and AstraZeneca. K.T. reports personal fees (honoraria) from Chugai, Novartis, and AstraZeneca. H.D. reports research support from Pfizer, Chugai, and Takeda, and personal fees from Chugai. A.Y. reports personal fees (honoraria) from Takeda. K.K. reports personal fees (honoraria) from Pfizer, Chugai, Novartis, and AstraZeneca. Y.Z. reports research support from AstraZeneca, and personal fees (honoraria) from Pfizer, Chugai, Takeda, and AstraZeneca. K.No. reports research support from Chugai and Takeda, and personal fees (honoraria) from Pfizer, Chugai, Takeda, Novartis, and AstraZeneca. T.S. reports personal fees (honoraria) from Chugai and AstraZeneca. G.I. reports research support from Takeda, and personal fees (honoraria) from Pfizer, Chugai, Takeda, Novartis, and AstraZeneca. S.N. reports research support from Pfizer, Chugai, Takeda, and AstraZeneca, and personal fees (honoraria) from Pfizer, Chugai, Takeda, Novartis, and AstraZeneca. A.O. reports personal fees (honoraria) from Chugai. S.S.K. reports research support from Boehringer Ingelheim, MiNA Therapeutics, and Taiho Therapeutics, as well as personal fees (honoraria) from Boehringer Ingelheim, Bristol Meyers Squibb, AstraZeneca, Chugai Pharmaceutical, and Takeda Pharmaceuticals, all outside of the submitted work. K.G. reports research support from Boehringer Ingelheim, Bristol-Myers Squibb, Chugai, Daiichi sankyo, Eisai, Eli Lilly, Guardant Health, Janssen, Kyowa Kirin, Life Technologies Japan, MSD, Novartis, Ono, Otsuka, Pfizer, Taiho, Takeda, and personal fees (honoraria) from Bristol-Myers Squibb, Chugai, Daiichi sankyo, Eisai, Eli Lilly, Haihe Biopharma, Ignyta, Janssen, KISSEI, Kyowa Kirin, LOXO Oncology, MEDICAL & BIOLOGICAL LABORATORIES, Merck Biopharma, Merus, MSD, Ono, Pfizer, Sumitomo Dainippon Pharma, Shanghai Haihe, Sysmex Corporation, Taiho, Takeda, and Xcoo. No other conflict of interest is reported.
Data availability
The WTS data that support the findings of this study are not publicly available and restrictions apply to the availability of these data. Such WTS data are available through to the corresponding authors (Susumu S. Kobayashi: sukobaya@east.ncc.go.jp or Koichi Goto: kgoto@east.ncc.go.jp) for academic non-commercial research purposes upon reasonable request, and subject to review of a project proposal that will be evaluated by a LC-SCRUM-Asia data access committee, entering into an appropriate data access agreement and subject to any applicable ethical approvals. The presence of LTK fusion was explored in various types of cancer using publicly available data generated by TCGA consortium (https://gdc.cancer.gov), accessed through cBioPortal (https://www.cbioportal.org/datasets).
References
- 1.Konig D, Savic Prince S & Rothschild SI Targeted Therapy in Advanced and Metastatic Non-Small Cell Lung Cancer. An Update on Treatment of the Most Important Actionable Oncogenic Driver Alterations. Cancers (Basel) 13, doi: 10.3390/cancers13040804 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saito M et al. Gene aberrations for precision medicine against lung adenocarcinoma. Cancer Sci 107, 713–720, doi: 10.1111/cas.12941 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cancer Genome Atlas Research, N. Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–550, doi: 10.1038/nature13385 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fernandez-Cuesta L et al. CD74-NRG1 fusions in lung adenocarcinoma. Cancer Discov 4, 415–422, doi: 10.1158/2159-8290.CD-13-0633 (2014). [DOI] [PubMed] [Google Scholar]
- 5.National Comprehensive Cancer Network. Non-Small Cell Lung Cancer (Version 4.2021), <https://www.nccn.org/professionals/physician_gls/pdf/nscl.pdf> (2021).
- 6.Roll JD & Reuther GW ALK-activating homologous mutations in LTK induce cellular transformation. PLoS One 7, e31733, doi: 10.1371/journal.pone.0031733 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scheel J et al. Purification and analysis of authentic CLIP-170 and recombinant fragments. J Biol Chem 274, 25883–25891, doi: 10.1074/jbc.274.36.25883 (1999). [DOI] [PubMed] [Google Scholar]
- 8.Grigoryan G & Keating AE Structural specificity in coiled-coil interactions. Curr Opin Struct Biol 18, 477–483, doi: 10.1016/j.sbi.2008.04.008 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Varmus HE The molecular genetics of cellular oncogenes. Annu Rev Genet 18, 553–612, doi: 10.1146/annurev.ge.18.120184.003005 (1984). [DOI] [PubMed] [Google Scholar]
- 10.Warmuth M, Kim S, Gu XJ, Xia G & Adrián F Ba/F3 cells and their use in kinase drug discovery. Curr Opin Oncol 19, 55–60, doi: 10.1097/CCO.0b013e328011a25f (2007). [DOI] [PubMed] [Google Scholar]
- 11.Soda M et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 448, 561–566, doi: 10.1038/nature05945 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Greulich H et al. Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants. PLoS Med 2, e313, doi: 10.1371/journal.pmed.0020313 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kobayashi S et al. An alternative inhibitor overcomes resistance caused by a mutation of the epidermal growth factor receptor. Cancer Res 65, 7096–7101, doi: 10.1158/0008-5472.CAN-05-1346 (2005). [DOI] [PubMed] [Google Scholar]
- 14.Yasuda H et al. Structural, biochemical, and clinical characterization of epidermal growth factor receptor (EGFR) exon 20 insertion mutations in lung cancer. Sci Transl Med 5, 216ra177, doi: 10.1126/scitranslmed.3007205 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zou HY et al. PF-06463922, an ALK/ROS1 Inhibitor, Overcomes Resistance to First and Second Generation ALK Inhibitors in Preclinical Models. Cancer Cell 28, 70–81, doi: 10.1016/j.ccell.2015.05.010 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamada S et al. Expression of a chimeric CSF1R-LTK mediates ligand-dependent neurite outgrowth. Neuroreport 19, 1733–1738, doi: 10.1097/WNR.0b013e3283186bf8 (2008). [DOI] [PubMed] [Google Scholar]
- 17.Bruno R & Fontanini G Next Generation Sequencing for Gene Fusion Analysis in Lung Cancer: A Literature Review. Diagnostics (Basel) 10, doi: 10.3390/diagnostics10080521 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kohno T et al. Beyond ALK-RET, ROS1 and other oncogene fusions in lung cancer. Transl Lung Cancer Res 4, 156–164, doi: 10.3978/j.issn.2218-6751.2014.11.11 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cancer Genome Atlas Research, N. Integrated genomic characterization of papillary thyroid carcinoma. Cell 159, 676–690, doi: 10.1016/j.cell.2014.09.050 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoshihara K et al. The landscape and therapeutic relevance of cancer-associated transcript fusions. Oncogene 34, 4845–4854, doi: 10.1038/onc.2014.406 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Muller-Tidow C et al. High-throughput analysis of genome-wide receptor tyrosine kinase expression in human cancers identifies potential novel drug targets. Clin Cancer Res 10, 1241–1249, doi: 10.1158/1078-0432.ccr-0954-03 (2004). [DOI] [PubMed] [Google Scholar]
- 22.Carvalho P, Gupta ML Jr., Hoyt MA & Pellman D Cell cycle control of kinesin-mediated transport of Bik1 (CLIP-170) regulates microtubule stability and dynein activation. Dev Cell 6, 815–829, doi: 10.1016/j.devcel.2004.05.001 (2004). [DOI] [PubMed] [Google Scholar]
- 23.Drilon A et al. Cabozantinib in patients with advanced RET-rearranged non-small-cell lung cancer: an open-label, single-centre, phase 2, single-arm trial. Lancet Oncol 17, 1653–1660, doi: 10.1016/S1470-2045(16)30562-9 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuroda N et al. ALK rearranged renal cell carcinoma (ALK-RCC): a multi-institutional study of twelve cases with identification of novel partner genes CLIP1, KIF5B and KIAA1217. Mod Pathol 33, 2564–2579, doi: 10.1038/s41379-020-0578-0 (2020). [DOI] [PubMed] [Google Scholar]
- 25.Pinsolle J et al. A Rare Fusion of CLIP1 and ALK in a Case of Non-Small-Cell Lung Cancer With Neuroendocrine Features. Clin Lung Cancer 20, e535–e540, doi: 10.1016/j.cllc.2019.05.001 (2019). [DOI] [PubMed] [Google Scholar]
- 26.Yeh I et al. Clinical, histopathologic, and genomic features of Spitz tumors with ALK fusions. Am J Surg Pathol 39, 581–591, doi: 10.1097/pas.0000000000000387 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gainor JF et al. Molecular Mechanisms of Resistance to First- and Second-Generation ALK Inhibitors in ALK-Rearranged Lung Cancer. Cancer Discov 6, 1118–1133, doi: 10.1158/2159-8290.CD-16-0596 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haas BJ et al. Accuracy assessment of fusion transcript detection via read-mapping and de novo fusion transcript assembly-based methods. Genome Biol 20, 213, doi: 10.1186/s13059-019-1842-9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pear WS et al. Efficient and rapid induction of a chronic myelogenous leukemia-like myeloproliferative disease in mice receiving P210 bcr/abl-transduced bone marrow. Blood 92, 3780–3792 (1998). [PubMed] [Google Scholar]
- 30.Borowicz S et al. The soft agar colony formation assay. J Vis Exp, e51998, doi: 10.3791/51998 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tomayko MM & Reynolds CP Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother Pharmacol 24, 148–154, doi: 10.1007/bf00300234 (1989). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The WTS data that support the findings of this study are not publicly available and restrictions apply to the availability of these data. Such WTS data are available through to the corresponding authors (Susumu S. Kobayashi: sukobaya@east.ncc.go.jp or Koichi Goto: kgoto@east.ncc.go.jp) for academic non-commercial research purposes upon reasonable request, and subject to review of a project proposal that will be evaluated by a LC-SCRUM-Asia data access committee, entering into an appropriate data access agreement and subject to any applicable ethical approvals. The presence of LTK fusion was explored in various types of cancer using publicly available data generated by TCGA consortium (https://gdc.cancer.gov), accessed through cBioPortal (https://www.cbioportal.org/datasets).