

Abstract
Emergence of antibiotic-resistant Mycobacterium tuberculosis (M. tuberculosis) restricts the availability of drugs for the treatment of tuberculosis, which leads to the increased morbidity and mortality of the disease worldwide. There are many intrinsic and extrinsic factors that have been reported for the resistance mechanism. To overcome such mechanisms, chemically synthesized benzaldehyde thiosemicarbazone derivatives were screened against M. tuberculosis to find potential inhibitor for tuberculosis. Such filtering process resulted in compound 13, compound 21, and compound 20 as the best binding energy compounds against DNA gyrase B, an important protein in the replication process. The ADMET prediction has shown the oral bioavailability of the novel compounds.
1. Introduction
Tuberculosis (TB) is a potentially serious communicable disease caused by the bacillus Mycobacterium tuberculosis (M. tuberculosis) [1]. TB was found with increased mortality, and it spreads from person to person through tiny droplets released into the air via coughs and sneezes [2]. According to the World Health Organization (WHO) report, worldwide, around 10 million people were affected with TB [3]. Here, 56% were men, 32% were women, and children accounted for 12%. Also, 1.2 million deaths were reported in HIV-negative people and 208000 deaths were reported among HIV-positive people. The largest number of new TB cases occurred in the Southeast Asian region with 44% of new cases, followed by the African region with 25% of new cases and Western Pacific with 18%. Eight countries accounted for two-thirds of the new TB cases including India (26%), Indonesia (8.5%), China (8.4%), Philippines (6.0%), Pakistan (5.7%), Nigeria (4.4%), Bangladesh (3.6%), and South Africa (3.6) [3]. M. tuberculosis does not have any particular virulence factor, but it perseveres long-term in the human body without causing any significant damage and transmission, if not, the immune system of the host is compromised. It secrets effector proteins to complicate the immune system, thereby, stimulating its intracellular survival in granulomas throughout the latency period of infection [4]. M. tuberculosis can develop in different conditions such as pulmonary and extrapulmonary TB (pleural, lymphadenitis, skeletal, gastrointestinal, and ocular) [5]. Once M. tuberculosis entered into the lungs, alveolar macrophages engulf the organism, and it was captured by phagosomes. Finally, it was delivered to lysosomes and get degraded. However, in most of the instances, to persist in human alveolar macrophages, the organism inhibits the acidification and phagosomes maturation. Also, to escape from the immune system recognition and hypoxic condition, the organism remains in a “quiescent status” (in nongrowing state but metabolically active) in 90% of the diseased individuals [6]. The innate (antimycobacterial elements, IFNγ and TNFα) as well as adaptive immunity (T cells, Th17, CD4+, and CD8+) controls M. tuberculosis activity in the latent phase of the infection. Hence, it is known as the conditional pathogenic bacterium [7]. If the host immune system is compromised, the organism gets activated and initiate replication. This promotes the diseased macrophage necrosis and, thereby, discharges the intracellular bacteria, which infect novel cells and invade other tissues [8]. Recent reports suggest that TB is also related with many other human complications, namely, autoimmune diseases (sarcoidosis: M. tuberculosis activates toll-like receptors, thereby, promoting pulmonary sarcoidosis [9]; systemic lupus erythematosus: immunosuppressive therapy and several immune abnormalities cause reactivation and diffusion of TB [10]), metabolic syndromes (diabetes mellitus: promotes the proliferation of mycobacterium [11]; atherosclerosis: mycobacterium rushes the development of atherosclerosis [12]), and pulmonary complications (pneumonia: TB infection rises the risk of secondary bacterial infection in children [13]; chronic obstructive pulmonary disease: pulmonary TB may alter the lung architecture [14]; lung cancer: TB is one of the risk factors for the development of lung cancer [15, 16]).
Treatment options available for drug-susceptible TB involve first-line drugs (isoniazid, rifampicin, ethambutol, and pyrazinamide) for six months. The indiscriminate use of the antibiotics leads to the development of resistance. The WHO classifies the resistance as multidrug resistance (MDR), extensive drug resistance (XDR), and total drug resistance (TDR). The rise of resistance to the first-line drugs termed as multidrug resistance (MDR) and resistance to the fluoroquinolones, one of the second-line drugs (capreomycin, kanamycin, and amikacin) tends to the occurrence of XDR. Both resistance to the first and second-line drugs leads to the development of TDR [3]. The U.S Food and Drug Administration (FDA) has developed bedaquiline, a novel drug against MDR M. tuberculosis. Over the 40 years, bedaquiline have been in use to treat TB. Unfortunately, resistance to bedaquiline antibiotic has been reported in recent years [17, 18]. The extrinsic factors associated with antibiotic resistance are social elements of TB in inhabitants and the eminence of prevention and control services of TB. Gygli et al., in 2017, explained the mechanism of intrinsic drug resistance in M. tuberculosis, such as decreased cell wall permeation, secretion of drug inactivating enzymes, mutation in the cell wall efflux system, bacterial drug target modification, and overexpression of drug targets. Here, isoniazid prodrug is activated by catalase or peroxidase (gene: katG), and it functions through enoyl acyl carrier protein reductase (gene: inhA). Rifampicin acts against M. tuberculosis through binding to the RNA polymerase β subunit (gene: rpoB). Ethambutol inhibits arabinosyl transferase (gene: embB) involved in the biosynthesis of cell wall arabinogalactan. Pyrazinamide, a nicotinamide analog, requires pyrazinamidase or nicotinamidase (gene: pncA) to get converted into pyrazinoic acid, an active form. For second-line drugs, fluoroquinolones inhibit the topoisomerase II (DNA gyrase), an essential enzyme involved in the replication process (genes: gyrA and gyrB). The role of kanamycin and amikacin modifies the range of 16S rRNA (gene: rrs) and, thereby, inhibits the protein synthesis. The capreomycin is found to inhibit the translation process in mycobacterium. The target gene tlyA participates in the rRNA ribose specific 2′-O-methylation. Here, the development of resistance to these first-line and second-line drugs is due to the mutations in the drug-targeting genes [19–23]. Such situation necessitates the design and development of novel drug with high antitubercular activity. In the recent study, Volynets et al., in 2019, have experimentally proved the activity of benzaldehyde thiosemicarbazone against M. tuberculosis. Hence, in the present study, we have retrieved the synthesized benzaldehyde thiosemicarbazone derivatives from the literature and screened against one of the main target DNA gyrases to find a potential inhibitor for M. tuberculosis [24] through molecular modelling methods. Our study may give an idea to the researchers who are designing drug against M. tuberculosis in the molecular level.
2. Materials and Methods
2.1. Protein Structure Preparation
The 3-dimensional (3D) structure of the target protein DNA gyrase was obtained from the protein data bank (PDB: 6GAU) [25, 26]. The PDB structure is a homodimer, which has two chains, chain A (DNA gyrase subunit B) and B (DNA gyrase subunit A) (Figure 1). For our virtual screening and molecular docking studies, we have utilized chain A. The sequence length of the subunit is 1179 amino acids. The 3D structure was crystalized by the X-ray diffraction method. The nonamino acid structures cocrystallized with DNA gyrase, phosphoaminophosphonic acid-adenylate ester, magnesium ions, and water molecules were detached from the structure.
Figure 1.

Structure of M. tuberculosis DNA gyrase.
2.2. Active Site Prediction
The active site of the DNA gyrase subunit B was predicted through the DoGSiteScorer [27]. Finding the low molecular weight ligand molecule by the target protein is the source for maintenance of the biological system. Here, active site is crucial for the function of an enzyme [28]. Usually, the active site prediction helps to ensure the protein function, druggability, and family classification. Such prediction is carried out by the DoGSite server, a structure-based prediction method, which functions on the basis of the difference of Gaussian (DoG) method. Most of the computational techniques available are geometry-based, energy-based, evolutionary-based, and combine method predictions. All of these approaches have disadvantages [29]. In contrast, DoGSite uses the pattern recognition method to predict the active sites. This locates the active site regions by sieving the grid of the protein using the DoG filter, and it finds the spherically designed structures called DoG cores in the grid. Finally, these detected DoG cores are gathered to form pockets, where the ligand molecules can fit firmly.
2.3. Ligand Preparation
The synthesized 30 benzaldehyde thiosemicarbazone (Figure 2) derivatives were obtained from the literature (9). The structures were drawn with the help of ChemSketch software and optimized (10) (Table 1).
Figure 2.

Structure of benzaldehyde thiosemicarbazone.
Table 1.
PyRx (AutoDock Vina) virtual screening results for benzaldehyde thiosemicarbazone derivatives against DNA gyrase subunit B.
| S. no. | Derivatives | Structure | Binding affinity (kcal/mol) |
|---|---|---|---|
| 1 | Compound 13 |

|
−8.5 |
| 2 | Compound 21 |

|
−7.8 |
| 3 | Compound 20 |

|
−7.7 |
| 4 | Compound 5 |

|
−6.9 |
| 5 | Compound 7 |
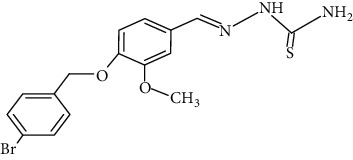
|
−6.9 |
| 6 | Compound 1 |

|
−6.7 |
| 7 | Compound 26 |

|
−6.7 |
| 8 | Compound 6 |

|
−6.7 |
| 9 | Compound 28 |

|
−6.6 |
| 10 | Compound 15 |

|
−6.6 |
| 11 | Compound 9 |

|
−6.5 |
| 12 | Compound 27 |

|
−6.5 |
| 13 | Compound 10 |

|
−6.4 |
| 14 | Compound 25 |

|
−6.3 |
| 15 | Compound 19 |

|
−6.2 |
| 16 | Compound 29 |

|
−6.2 |
| 17 | Compound 24 |

|
−6.2 |
| 18 | Compound 30 |

|
−6.1 |
| 19 | Compound 8 |

|
−6.1 |
| 20 | Compound 11 |

|
−6.1 |
| 21 | Compound 12 |

|
−6.1 |
| 22 | Compound 17 |

|
−6.1 |
| 23 | Compound 22 |

|
−6.0 |
| 24 | Compound 4 |
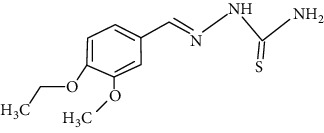
|
−5.9 |
| 25 | Compound 14 |

|
−5.9 |
| 26 | Compound 23 |
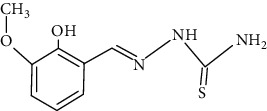
|
−5.9 |
| 27 | Compound 2 |

|
−5.8 |
| 28 | Compound 3 |

|
−5.7 |
| 29 | Compound 18 |

|
−5.7 |
| 30 | Compound 16 |

|
−5.6 |
2.4. Virtual Screening
A total of 30 benzaldehyde thiosemicarbazone derivatives were screened against gyrB using the python prescription virtual screening tool (PyRx) [30], a structure-based virtual screening process, which usually screens compound libraries against protein targets. Using the OpenBabel tool combined with the PyRx server, the ligand molecules were added and subjected to energy minimization with the help of the universal force field (UFF) by the conjugate gradient algorithm. Both protein and ligand structures were converted into PDBQT format. The docking procedure was carried out through the AutoDock Vina of PyRx Tool. From the 30 compounds, best binding energy was selected and passed to the next procedure, docking studies to get final best compounds.
2.5. Molecular Docking Studies
Prediction of binding orientation of ligand with the protein is carried out through the molecular docking studies; in the current study, AutoDock version 4.2.6 was utilized [31]. It works on the basis of the Lamarckian genetic algorithm [32]. The target protein DNA gyrase B 3D structure was added with polar hydrogen atoms using AutoDock Tool (ADT). Both ligand and the protein structures were converted into PDBQT format. The grid was fixed with 80 × 80 x 80 size, which covers the active site of the protein. The desolvation map and grid map was created by the AutoGrid program. The algorithm was set with energy evaluation of 2500000, population of 150, 27000 generations, 0.02 mutation rate, and 0.8 crossover rate. Next, the AutoDock run was simulated, and the binding energy of protein-ligand affinity was checked to identify the best binding affinity. AutoDock produces empirical scoring functions; it is the combination of hydrogen bonding, van der Waals, electrostatic, hydrophobicity, entropy, and desolvation energies. The unit of binding energy is kcal/mol. The higher binding affinity between the complexes reflects the increased intermolecular forces. The lowest binding energy stabilizes the complex. The hydrophobic interactions were also explored using LigPlot+ v.2.2 analysis.
2.6. Absorption, Distribution, Metabolism, Excretion, and Toxicity Prediction
The pharmacokinetic properties such as absorption (A), distribution (D), metabolism (M), excretion (E), and toxicity (T) were predicted through the online server ADMET structure activity relationship (admetSAR). The database has structure and text search options, and also, the database collects, curates, and holds ADMET-related properties data from the available literature. The database includes 210,000 curated data for greater than 96,000 ligand molecules with 45 different ADMET-related properties, species, and proteins. The admetSAR interface is to search particular ligand profile, common name, and analog search. It has 5 quantitative regression models and 22 qualitative classifications with increased prediction accuracy. ADMET prediction is a distinctive interdisciplinary linking between the biologist, medicinal chemist, formulators, toxicologist, and regulators across India. The ADMET prediction of drug candidates has majorly reduced the drug failure in clinical trials [33].
3. Results and Discussion
TB, malaria, and acquired immune deficiency syndrome (AIDS) are the top most fatal infectious disease becoming a worldwide public health threat. The development of TB causes increased morbidity and mortality [34]. Different combinations of drugs have been given in treatment. There are several factors that have been reported for the failure of TB treatment, namely, delay in diagnosis, lack of effective drug administration, decreased accessibility of inexpensive, low toxic and effective drugs, prolong intake of drugs, lack of adherence to drug regimen, and rise of drug-resistant TB strains. Hence, to overcome the resistance mechanism, novel compounds were synthesized against the M. tuberculosis, and molecular modelling studies were carried out to find the potential drug candidate. The DoGSite server predicted the binding site pocket on the DNA gyrase B, which covered the area of 2834.65 Å3, surface 3418.38 Å2, and the drug score is 0.81. The drug score falls between 0 and 1, and the higher drug score reveals the high potential of the predicted pocket on the protein (Figure 3).
Figure 3.

Binding pocket of DNA gyrase B.
Virtual screening of 30 benzaldehyde thiosemicarbazone derivatives against DNA gyrase B reveals the high binding affinity of compound 13 with the binding energy of −8.5 kcal/mol (Table 1), followed by compounds 20 and 21 with the binding energies of −7.7 kcal/mol and −7.8 kcal/mol. Here, both compounds 20 and 21 have more or less similar binding energies. Other compounds, namely, 1, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 17, 19, 22, 24, 25, 26, 27, 28, 29, and 30 have their binding energies ranging from −6.0 to −6.9 kcal/mol. Also, the binding energies of compounds 2, 3, 4, 14, 16, 18, and 23 range from −5.6 to −5.9 kcal/mol. From Table 1, 10 best binding energy compounds were selected and passed through the molecular docking simulation studies using the AutoDock Tool.
Molecular docking studies of 10 best compounds against DNA gyrase B result in compound 13 as the best compound with the binding energy of −8.2 kcal/mol, which has formed four amino acid interactions with the bond distances of ILE493: 2.0 Å, SER413: 1.9 Å, LYS409: 2.8 Å, and SER12: 2.6 and 2.1 Å, and 11 hydrophobic interactions, namely, LYS409, SER413, ALA416, GLY296, TYR297, ARG492, ALA491, SER298, HIS514, GLU352, and PHE551 (Figure 4). Next, compound 21 is with −7.5 kcal/mol of binding energy, 3 hydrogen bond interactions such as GLY612 (2.1 Å), ARG634 (1.8 Å), and PRO567 (2.0 Å), and 12 hydrophobic interactions of MET616, LYS611, LEU613, PRO566, ALA533, TYR610, GLN538, SER541, ALA531, ALA564, LEU563, and LEU529 (Figure 5). Here, compounds 20 and 21 have only slight variations in their binding energies (Table 2). Compound 20 has the binding energy of −6.9 kcal/mol. The amino acids, namely, GLY612 (2.1 Å), ARG634 (1.8 Å), and PRO567 (2.0 Å) have been found in the hydrogen bond interaction. In hydrophobic interaction, the amino acids MET616, LYS611, LEU613, PRO566, ALA533, TYR610, GLN538, SER541, ALA531, ALA564, LEU563, and LEU529 have been found in the interaction (Figure 6). The higher the hydrophobic interaction, the lower the hydrogen bond interaction and vice versa. Our findings are similar to that of the experimental studies carried out by Volynets et al. [24]. The in vitro studies on benzaldehyde thiosemicarbazone derivatives against M. tuberculosis results in the minimal inhibitory concentration (MIC) of 0.68 μM for compound 13 and 0.74 μM for compound 20. In our findings, compound 21 is the second most high binding energy compound. Also, compounds 20 and 21 slightly vary in their binding energies.
Figure 4.
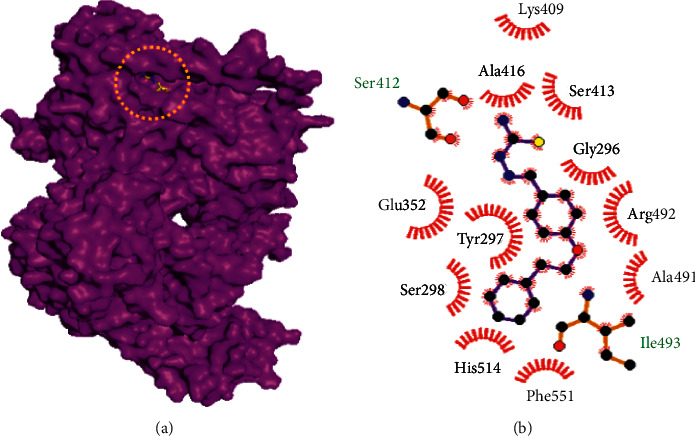
Binding mode of DNA gyrase B with the benzaldehyde thiosemicarbazone derivative compound 13. (a) Binding mode of compound 13 in the active site of DNA gyrase B. (b) Hydrophobic interactions of DNA gyrase B and compound 13.
Figure 5.

Binding mode of DNA gyrase B with the benzaldehyde thiosemicarbazone derivative compound 21. (a) Binding mode of compound 21 in the active site of DNA gyrase B. (b) Hydrophobic interactions of DNA gyrase B and compound 21.
Table 2.
AutoDock results for benzaldehyde thiosemicarbazone derivatives against DNA gyrase subunit B.
| S. no: | Compounds | Binding energy (kcal/mol) | Amino acid interactions and distances (Å) | Hydrophobic interactions |
|---|---|---|---|---|
| 1 | Compound 13 | −8.2 | ILE493 (2.0 Å), SER413 (1.9 Å), LYS409 (2.8 Å), and SER12 (2.6 and 2.1 Å) | LYS409, SER413, ALA416, GLY296, TYR297, ARG492, ALA491, SER298, HIS514, GLU352, and PHE551 |
| 2 | Compound 21 | −7.5 | GLY612 (2.1 Å), ARG634 (1.8 Å), and PRO567 (2.0 Å) | MET616, LYS611, LEU613, PRO566, ALA533, TYR610, GLN538, SER541, ALA531, ALA564, LEU563, and LEU529 |
| 3 | Compound 20 | −6.9 | ALA564 (1.9 Å) and ALA531 (2.0 Å) | TYR610, LEU613, ALA533, GLY612, PRO567, LEU568, PRO566, ASP532, LYS611, LEU529, LEU563, and ALA531 |
| 4 | Levofloxacin | −7.0 | ASN309 (2.5 Å) and ARG40 (2.0 Å) | LEU200, THR307, PHE304, GLU196, MET197, ASP639, TRP47, ASP640, ARG193, ASN309, ILE308, and HIS44 |
| 5 | Rifampicin | −6.7 | THR250 (2.3 Å), ASP259 (2.1 Å and 2.2 Å), and LYS262 (2.0 Å) | VAL301, ARG550, ASP348, HIS514, GLU317, HIS311, GLU312, PRO554, ASN558, and GLU557 |
| 6 | Isoniazid | −5.0 | SER1027 (2.7 Å), ALA531 (2.0 Å), LEU529 (2.1 Å), and ALA564 (2.2 Å) | ALA564, MET530, ARG634, LEU563, SER541, GLY537, GLU1023, and SER1027 |
Figure 6.

Binding mode of DNA gyrase B with the benzaldehyde thiosemicarbazone derivative compound 20. (a) Binding mode of compound 20 in the active site of DNA gyrase B. (b) Hydrophobic interactions of DNA gyrase B and compound 20.
Similar computational studies have been reported in the screening of antimicrobial compounds. Pertersen et al. [35] have used the virtual screening techniques to identify novel inhibitor for M. tuberculosis 3-dehydroquinate. Similarly, another study has employed pharmacophore-based virtual screening to identify the pyrazolo [1,5-a] pyrimidine derivatives against InhA of M. tuberculosis [36, 37].
Here, the first-line antimicrobial drugs rifampicin and isoniazid have lower energies −6.7 kcal/mol and −5.0 kcal/mol than the benzaldehyde thiosemicarbazone derivatives compounds 13, 20, and 21, respectively. The second-line drug levofloxacin is −7.0 kcal/mol.
From the virtual screening and molecular docking studies, it is confirmed that compound 13 could serve as a potential inhibitor for M. tuberculosis. Further preclinical studies have to be conducted to confirm the antimicrobial activity of the compound.
3.1. ADMET Prediction
ADMET prediction of benzaldehyde thiosemicarbazone derivatives using admetSAR server results in intestinal absorption capacity of the compounds 13, 21, and 20. These compounds also act as a nonsubstrate and noninhibitor for P-glycoprotein. Compound 13 serves as an inhibitor for renal organic cation transporter and other two compounds 20 and 21 as a noninhibitor. In the metabolism, for cytochrome P450 2C9, 2D6, and 3A4, the compounds 13, 21, and 20 possess nonsubstrate property and for cytochrome P450 1A2, 2C9, 2D6, 2C19, and 3A4, it results in noninhibitory activity. Toxicity prediction reveals the non-AMES toxicity and noncarcinogenic activity. Overall, the ADMET prediction further confirms the bioavailability of the compounds (Table 3).
Table 3.
ADMET prediction for lead compounds.
| S. no. | Models | Compound 13 | Compound 21 | Compound 20 |
|---|---|---|---|---|
| Absorption | ||||
| 1 | Blood-brain barrier | BBB+ | BBB+ | BBB+ |
| 2 | Human intestinal absorption | HIA+ | HIA+ | HIA+ |
| 3 | Caco-2 permeability | Caco-2 | Caco-2 | Caco-2 |
| 4 | P-Glycoprotein substrate | Nonsubstrate | Nonsubstrate | Nonsubstrate |
| 5 | P-Glycoprotein inhibitor | Noninhibitor | Noninhibitor | Noninhibitor |
| 6 | Renal organic cation transporter | Inhibitor | Noninhibitor | Noninhibitor |
|
| ||||
| Metabolism | ||||
| 7 | CYP450 2C9 substrate | Nonsubstrate | Nonsubstrate | Nonsubstrate |
| 8 | CYP450 2D6 substrate | Nonsubstrate | Nonsubstrate | Nonsubstrate |
| 9 | CYP450 3A4 substrate | Nonsubstrate | Nonsubstrate | Nonsubstrate |
| 10 | CYP450 1A2 inhibitor | Inhibitor | Inhibitor | Inhibitor |
| 11 | CYP450 2C9 inhibitor | Inhibitor | Inhibitor | Inhibitor |
| 12 | CYP450 2D6 inhibitor | Noninhibitor | Noninhibitor | Noninhibitor |
| 13 | CYP450 2C19 inhibitor | Noninhibitor | Inhibitor | Inhibitor |
| 14 | CYP450 3A4 inhibitor | Noninhibitor | Noninhibitor | Noninhibitor |
|
| ||||
| Toxicity | ||||
| 15 | AMES toxicity | Non-AMES toxic | Non-AMES toxic | Non-AMES toxic |
| 16 | Carcinogens | Noncarcinogens | Noncarcinogens | Noncarcinogens |
4. Conclusion
The molecular modelling studies on 30 benzaldehyde thiosemicarbazone derivatives reveals the best binding energy of compound 13, compound 21, and compound 20 against DNA gyrase B of M. tuberculosis. To further confirm the activity of these compounds, preclinical studies have to be conducted.
Acknowledgments
MK, JMA, and SKM would like to thank the management of PMCHRI for providing the necessary facilities to carry out this research work.
Data Availability
The data used to support the findings of this study are available from the corresponding author upon request.
Conflicts of Interest
The authors declare that there are no conflicts of interest.
Authors' Contributions
AK., ABB., VA., PKA., JMA., MK., and SKM conceptualized the study and wrote the original draft of the article. AK., ABB., MK., JMA., and SKM developed methodology, performed formal analysis and investigation. AK., ABB., VA., PKA., JMA., MK., VPV., GR., and SKM wrote and reviewed the article. VPV., GR., and SKM acquired fund and collected resources. SKM supervised the study.
References
- 1.Pai M., Behr M. A., Dowdy D., et al. Tuberculosis. Nature Reviews Disease Primers . 2016;2:p. 16076. doi: 10.1038/nrdp.2016.76. [DOI] [PubMed] [Google Scholar]
- 2.Murray C. J. L., Collaborators G. T. The global burden of tuberculosis: results from the Global Burden of Disease Study 2015. The Lancet Infectious Diseases . 2018;18:261–284. doi: 10.1016/S1473-3099(17)30703-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.WHO. Global Tuberculosis Report 2019 . Geneva, Switzerland: WHO; 2019. https://apps.who.int/iris/bitstream/handle/10665/336069/9789240013131-eng.pdf . [Google Scholar]
- 4.Groschel M. I., Sayes F., Simeone R., Majlessi L., Brosch R. ESX secretion systems: mycobacterial evolution to counter host immunity. Nature Reviews Microbiology . 2016;14(11):677–691. doi: 10.1038/nrmicro.2016.131. [DOI] [PubMed] [Google Scholar]
- 5.Shah M., Chida N. Extrapulmonary tuberculosis. In: Grosset J. H., Chaisson R. E., editors. Handbook of Tuberculosis . Cham, Switzerland: Springer International Publishing; 2017. pp. 91–118. [DOI] [Google Scholar]
- 6.Bhavanam S., Rayat G. R., Keelan M., Kunimoto D., Drews S. J. Understanding the pathophysiology of the human TB lung granuloma using in vitro granuloma models. Future Microbiology . 2016;11:1073–1089. doi: 10.2217/fmb-2016-0005. [DOI] [PubMed] [Google Scholar]
- 7.Cambier C. J., Falkow S., Ramakrishnan L. Host evasion andexploitation schemes of Mycobacterium tuberculosis. Cell . 2014;159:1497–1509. doi: 10.1016/j.cell.2014.11.024. [DOI] [PubMed] [Google Scholar]
- 8.Dutta N. K., Karakousis P. C. Latent tuberculosis infection: myths, models, and molecular mechanisms. Microbiology and Molecular Biology Reviews . 2014;78:343–371. doi: 10.1128/mmbr.00010-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gabrilovich M. I., Walrath J., van Lunteren J., et al. Disordered toll-like receptor 2 responses in the pathogenesis of pulmonary sarcoidosis. Clinical and Experimental Immunology . 2013;173:512–522. doi: 10.1111/cei.12138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yun J. E., Lee S. W., Kim T. H., et al. The incidence and clinical characteristics of Mycobacterium tuberculosis infection among systemic lupus erythematosus and rheumatoid arthritis patients in Korea. Clinical & Experimental Rheumatology . 2002;20:127–132. [PubMed] [Google Scholar]
- 11.Kumar N. P., George P. J., Kumaran P., Dolla C. K., Nutman T. B., Babu S. Diminished systemic and antigen-specific type 1, type 17, and other proinflammatory cytokines in diabetic and prediabetic individuals with latent Mycobacterium tuberculosis infection. The Journal of Infectious Diseases . 2014;210:1670–1678. doi: 10.1093/infdis/jiu329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wick C., Onestingel E., Demetz E., Dietrich H., Wick G. Oral tolerization with mycobacterial heat shock protein 65 reduces chronic experimental atherosclerosis in aged mice. Gerontology . 2017;64:36–48. doi: 10.1159/000480436. [DOI] [PubMed] [Google Scholar]
- 13.Oliwa J. N., Karumbi J. M., Marais B. J., Madhi S. A., Graham S. M. Tuberculosis as a cause or comorbidity of childhood pneumonia in tuberculosis-endemic areas: a systematic review. Lancet Respiratory Medicine . 2015;3:235–243. doi: 10.1016/S2213-2600(15)00028-4. [DOI] [PubMed] [Google Scholar]
- 14.Jordan T. S., Spencer E. M., Davies P. Tuberculosis, bronchiectasis and chronic airflow obstruction. Respirology . 2010;15:623–628. doi: 10.1111/j.1440-1843.2010.01749.x. [DOI] [PubMed] [Google Scholar]
- 15.Shin D. M., Yang C. S., Lee J. Y., et al. Mycobacterium tuberculosis lipoprotein-induced association of TLR2 with protein kinase C zeta in lipid rafts contributes to reactive oxygen species-dependent inflammatory signalling in macrophages. Cellular Microbiology . 2008;10:1893–1905. doi: 10.1111/j.1462-5822.2008.01179.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bell L., Noursadeghi M. Pathogenesis of HIV-1 and Mycobacterium tuberculosis co-infection. Nature Reviews Microbiology . 2018;16:80–90. doi: 10.1038/nrmicro.2017.128. [DOI] [PubMed] [Google Scholar]
- 17.Andries K., Villelas C., Coeck N., et al. Acquired resistance of Mycobacterium tuberculosisto bedaquiline. PLoS One . 2014;9 doi: 10.1371/journal.pone.0102135.e102135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoffmann H., Kohl T. A., Hofmann-Thiel S., et al. Delamanid and bedaquiline resistance inMycobacterium tuberculosis ancestral Beijing genotype causing XDR-TB in a tibetian refugee. American Journal of Respiratory and Critical Care Medicine . 2016;193:337–340. doi: 10.1164/rccm.201502-0372le. [DOI] [PubMed] [Google Scholar]
- 19.Lempens P., Meehan C. J., Vandelannoote K., et al. Isoniazid resistance levels of Mycobacterium tuberculosis can largely be predicted by high-confidence resistance-conferring mutations. Scientific Reports . 2018;8:p. 3246. doi: 10.1038/s41598-018-21378-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma P., Luo T., Ge L., et al. Compensatory effects of M. tuberculosis rpoB mutations outside the rifampicin resistance-determining region. Emerging Microbes & Infections . 2021;10(1):743–752. doi: 10.1080/22221751.2021.1908096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li K., Yang Z., Gu J., Luo M., Deng J., Chen Y. Characterization of pncA mutations and prediction of PZA resistance in Mycobacterium tuberculosis clinical isolates from chongqing, China. Frontiers in Microbiology . 2021;11 doi: 10.3389/fmicb.2020.594171.594171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chaoui I., Oudghiri A., El Mzibri M. Characterization of gyrA and gyrB mutations associated with fluoroquinolone resistance in Mycobacterium tuberculosis isolates from Morocco. Journal of Global Antimicrobial Resistance . 2018;12:171–174. doi: 10.1016/j.jgar.2017.10.003. [DOI] [PubMed] [Google Scholar]
- 23.Sowajassatakul A., Prammananan T., Chaiprasert A., Phunpruch S. Molecular characterization of amikacin, kanamycin and capreomycin resistance in M/XDR-TB strains isolated in Thailand. BMC Microbiology . 2014;14:p. 165. doi: 10.1186/1471-2180-14-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Volynets G. P., Tukalo M. A., Bdzhola V. G., et al. Benzaldehyde thiosemicarbazone derivatives against replicating and nonreplicating Mycobacterium tuberculosis. Journal of Antibiotics . 2019;72:218–224. doi: 10.1038/s41429-019-0140-9. [DOI] [PubMed] [Google Scholar]
- 25.Burley S. K., Berman H. M., Kleywegt G. J., Markley J. L., Nakamura H., Velankar S. Protein data bank (PDB): the single global macromolecular structure archive. Methods in Molecular Biology . 2017;1607:627–641. doi: 10.1007/978-1-4939-7000-1_26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Petrella S., Capton E., Raynal B., et al. Overall structures of Mycobacterium tuberculosis DNA gyrase reveal the role of a corynebacteriales GyrB-specific insert in ATPase activity. Structure . 2019;27:579–589. doi: 10.1016/j.str.2019.01.004. [DOI] [PubMed] [Google Scholar]
- 27.Volkamer A., Griewel A., Grombacher T., Rarey M. Analyzing the topology of active sites: on the prediction of pockets and sub-pockets. Journal of Chemical Information and Modeling . 2010;50(11):2041–2052. doi: 10.1021/ci100241y. [DOI] [PubMed] [Google Scholar]
- 28.Shulman-Peleg A., Nussinov R., Wolfson H. Recognition offunctional sites in protein structures. Journal of Molecular Biology . 2004;339:607–633. doi: 10.1016/j.jmb.2004.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nayal M., Honig B. On the nature of cavities on protein surfaces: application to the identification of drug-binding sites. Proteins . 2006;63:892–906. doi: 10.1002/prot.20897. [DOI] [PubMed] [Google Scholar]
- 30.Dallakyan S., Olson A. J. Small-molecule library screening by docking with PyRx. Methods in Molecular Biology . 2015;1263:243–250. doi: 10.1007/978-1-4939-2269-7_19. [DOI] [PubMed] [Google Scholar]
- 31.Morris G. M., Huey R., Lindstrom W., et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. Journal of Computational Chemistry . 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fuhrmann J., Rurainski A., Lenhof H. P., Neumann D. A new Lamarckian genetic algorithm for flexible ligand-receptor docking. Journal of Computational Chemistry . 2010;31(9):1911–1918. doi: 10.1002/jcc.21478. [DOI] [PubMed] [Google Scholar]
- 33.Tsaioun K., Blaauboer B. J., Hartung T. Evidence-based absorption, distribution, metabolism, excretion (ADME) and its interplay with alternative toxicity methods. Altex . 2016;33(4):343–358. doi: 10.14573/altex.1610101. [DOI] [PubMed] [Google Scholar]
- 34.Lohrasbi V., Talebi M., Bialvaei A. Z., et al. Trends in the discovery of new drugs for Mycobacterium tuberculosis therapy with a glance at resistance. Tuberculosis . 2018;109:17–27. doi: 10.1016/j.tube.2017.12.002. [DOI] [PubMed] [Google Scholar]
- 35.Petersen G. O., Saxena S., Renuka J., et al. Structure-based virtual screening as a tool for the identification of novel inhibitors against Mycobacterium tuberculosis 3-dehydroquinate dehydratase. Journal of Molecular Graphics and Modelling . 2015;60:124–131. doi: 10.1016/j.jmgm.2015.05.001. [DOI] [PubMed] [Google Scholar]
- 36.Modi P., Patel S., Chhabria M. T. Identification of some novel pyrazolo [1,5-a] pyrimidine derivatives as InhA inhibitors through pharmacophore-based virtual screening and molecular docking. Journal of Biomolecular Structure and Dynamics . 2019;37(7):1736–1749. doi: 10.1080/07391102.2018.1465852. [DOI] [PubMed] [Google Scholar]
- 37.Gygli S. M., Borrell S., Trauner A., Gagneux S. Antimicrobial resistance in Mycobacterium tuberculosis: mechanistic and evolutionary perspectives. FEMS Microbiology Reviews . 2017;41(3):354–373. doi: 10.1093/femsre/fux011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data used to support the findings of this study are available from the corresponding author upon request.