

Abstract
Aberrancy in cell cycle progression is one of the fundamental mechanisms underlying tumorigenesis, making regulators of the cell cycle machinery rational anti-cancer therapeutic targets. Growing body of evidence indicates that the cell cycle regulatory pathway integrates into other hallmarks of cancer, including metabolism remodeling and immune escaping. Thus, therapies against cell cycle machinery components can not only repress the division of cancer cells, but also reverse cancer metabolism and restore cancer immune surveillance. Apart from the ongoing effects on developing small molecule inhibitors (SMIs) of cell cycle machinery, PROTACs have recently been used for targeting these oncogenic proteins related to cell cycle progression. Here, we discuss the rationale of cell cycle targeting therapies, particularly PROTACs, to more efficiently retard tumorigenesis.
Keywords: Cell cycle, cancer, metabolism, cancer immune, PROTAC, degradation
Cell cycle machinery and its function in cancer
After the identification of first cell cycle regulator gene cdc28 (the analog of CDK1 in yeast) in early 1970s [1], the long history of cell cycle study reveals the mechanisms of how cell cycle functions as the fundamental event in cell biology [2,3]. Cell cycle represents a series of strictly-controlled events that lead to the faithful duplication of the genome DNA and the reproduction of two daughter cells [2,3]. Briefly, cell cycle could be divided into four major phases: gap 1 (G1), DNA synthesis (S), gap 2 (G2) and mitosis (M) (Figure 1). The fidelity and integrity of DNA replication, and the timely and accurate separation of sister chromatids, are quality controlled by various cell cycle checkpoints, including the G1/S checkpoint which could be relieved by the sequential phosphorylation of Rb by Cyclin D-CDK4/6 and Cyclin E-CDK2 to dissociate E2F [4,5]; the G2/M checkpoint where the Cyclin B-CDK1 kinase activity is precisely controlled by the Aurora A/PLK1/CDC25 and Chk1/WEE1 signaling [6]; and the spindle assembly checkpoint (SAC) where the mitotic checkpoint complex (MCC) inhibits APCCdc20 E3 ligase activity to disable the degradation of Cyclin B and Securin to halt the mitotic progression [7] (Figure 1). The fine-tuned and orderly progression of cell cycle is controlled by sequential destruction of Cyclins by the anaphase promoting complex/cyclosome (APC/C, also called APC) [8] and Skp1-Cullin 1-F-box (SCF) E3 ligase complexes [9–12], and subsequent activation of the cell cycle machinery, including Cyclin D-CDK4/6 in G1 phase, Cyclin E-CDK2 at the end of G1 phase, Cyclin A-CDK2 at the end of S phase, and Cyclin B-CDK1 at the end of G2 phase [13,14], respectively (Figure 1). A comprehensive map of human proteomics at the single cell level with precise annotation of individual cell in the cell cycle using the fluorescence ubiquitination cell cycle indicator (FUCCI) technology [15], further validate the timely regulation of most cycling proteins at the post-translational level, including phosphorylated by cell cycle-controlling kinases, and degradation by SCF or APC/C E3 ligases.
Figure 1. Cell cycle machinery and checkpoints.

The proceeding of the cell cycle depends on subsequential degradation of Cyclins and other key cell cycle regulators by the APC/C and SCF E3 ligase complexes, to ensure the faithful completion of DNA replication and orderly cell division. In G1 phase, Cyclin D-CDK4/6 and Cyclin E-CDK2 phosphorylate Rb protein, leading to the relief from G1/S checkpoint by triggering E2F release from Rb and the repressive complex for the transcription of downstream targets, to prepare for the synthesis of protein and DNA. After the replication of genome DNA in S phase, cells in G2 phase pass through the G2/M checkpoint, where errors during DNA synthesis activate the DNA damage pathway to repress Cyclin B-CDK1 kinase activity through the ATR-Chk1-WEE1/CDC25C signaling axis. During mitosis, the spindle assembly checkpoint is governed by MCC-mediated repression of APCCdc20, which is responsible for the degradation of Securin to activate Separase and the degradation of Cyclin B to activate CDK1. Among these cell cycle controlling E3 ligases, APCCdh1 controls the ubiquitination and protein stability of Cdc20 and Skp2, thus ensuring the ordered activation and inactivation of APC/C and SCF in different cell cycle phases.
In keeping with the key role for the cell cycle machinery in determining the faithful duplication of the genome DNA, genetic variation or signaling transduction interruption of cell cycle regulators and/or cell cycle checkpoints cause improper re-entry into the cell cycle and aberrant cell divisions, which is a major hallmark for cancer [16,17]. For example, overexpression of Cyclin D/E, PLK1, Aurora A/B, Skp2, and Cdc20 have been frequently detected in various types of human cancers, and these oncoproteins serve as therapeutic targets in clinic with several inhibitors been approved, such as CDK4/6 inhibitors (palbociclib [18], ribociclib [19] and abemaciclib), or in clinical trials, such as PLK1 inhibitors (rigosertib and volasertib, NCT01928537 or NCT01721876, phase III/IV), Aurora A inhibitor (alisertib, NCT02530619, phase III) and WEE1 inhibitor [20] (MK-1775, phase II), Chk1/2 (LY2606368, NCT02203513, phase II). Growing body of evidence suggests that the cell cycle machinery integrates into other hallmarks of cancer, including metabolism remodeling and immune escaping. Therefore, therapies against the cell cycle regulatory proteins not only repress cancer cell division by blocking cell cycle progression, but also reverse cancer cell metabolism or restore cancer immune surveillance, thus providing tremendous effects for combating tumors in vivo. Recently, the emerging proteolysis targeting chimera (PROTAC) technology has been adopted to target the oncogenic component of the cell cycle machinery for ubiquitination-mediated proteasomal degradation, with advantages over traditional small molecule inhibitors (SMIs), which might be a promising approach for cell cycle-focused cancer therapy in future endeavors.
Cell cycle and cancer metabolism
Compared to most normal tissue cells, cancer cells are characterized with a distinct metabolic phenotype known as the Warburg effect, including aerobic glycolysis and reduced oxidative phosphorylation (OXPHOS), which provides biosynthetic intermediates to fuel cancer cell proliferation and division at elevated levels [21]. The cell cycle-coupled metabolism oscillation has been firstly observed in yeasts, where yeasts appear to prefer oxidative metabolism at the G1 cell cycle phase, and then shift to rely more on glycolysis at the reductive/building (R/B) phase [22]. DNA replication occurs exclusively at the R/B phase, and the reductant NADPH at this phase ensures a low frequency of spontaneous DNA mutation rate [23]. However, it still remains largely elusive whether and how cell metabolism is regulated by the cell cycle machinery in mammalian cells. Moreover, the metabolic cycle in yeasts is based on the transcriptional profile for cells at different cell cycle phases in a bulk RNA sequencing analysis [22]. In mammalian cells, the cycling proteins whose expression levels fluctuate during cell cycle, are mainly regulated at the post-translational level rather than at the transcriptional level, based on a comprehensive proteomics mapping of human cells at the single cell level with precise annotation of cell cycle phases for individual cell [15]. Collectively, multiple metabolic enzymes and/or their upstream regulators are regulated by cell cycle machinery, either through CDKs-mediated phosphorylation [24–28], and/or through SCF [29,30] and APC/C [29,31–33] E3 ligase-mediated ubiquitination and subsequent proteasomal degradation (Figure 2).
Figure 2. Regulation of cell metabolism by the cell cycle machinery at the post-translational level.

In G1 phase, Cyclin D-CDK4/6 phosphorylates and inactivates the glycolytic enzymes, PFK and PKM2, to rewire glycolysis into more reliance on the pentose phosphate pathway (PPP) and serine pathway, or phosphorylates and inactivates the master regulators of mitochondrial biogenesis, NRF1 and PGC1α, to repress oxidative phosphorylation (OXPHOS), or indirectly represses metabolic-related proteins, including VDAC/HK2 and PPARα On the other hand, APCCdh1 ubiquitinates and degrades multiple metabolic enzymes or regulators, such as PFKBP3, GLS1 and IDH3β, to repress glycolysis and glutaminolysis. In late G1 and S phases, Cyclin E-CDK2 and Cyclin A-CDK2 phosphorylate the TCA cycle enzyme IDH1/2, which are further ubiquitinated by SCFSkp2 to render more glycolysis in S phase. In M phase, Cyclin B-CDK1 phosphorylates and inactivates various mitochondrial proteins to repress OXPHOS.
In G1 phase, Cyclin D-CDK4/6 plays a major role for the cell cycle progression and cell metabolism, and the protein level of Cyclin D is controlled by the CRL4AMBRA E3 ubiquitin ligase [34–36]. On one hand, Cyclin D1 represses OXPHOS, either through direct phosphorylation of nuclear respiratory factor 1 (NRF-1) [24] and peroxisome proliferator-activated receptor-γ coactivator-1α (PGC1α) [25] by Cyclin D-CDK4, or through direct translocation onto the outer membrane of mitochondria to bind with voltage-dependent anion channel (VDAC) [25] and HK2 [37] (Figure 2). Besides, Cyclin D1 also represses fatty acid oxidation through inhibiting peroxisome proliferator-activated receptor α (PPARα)-mediated transcriptome in a CDK-independent manner [38] (Figure 2). On the other hand, Cyclin D-CDK6 phosphorylates and inactivates two glycolytic enzymes, 6-phosphofructokinase (PFK1) and pyruvate kinase M2 (PKM2), thus rendering the tetramers form of these two enzymes to a less-active dimmer formation, which subsequently leads to the accumulation of glycolytic intermediates and rewiring of cancer cell metabolism towards more reliance to the pentose phosphate pathway (PPP) and serine pathway [26] (Figure 2). Furthermore, the enzyme 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3 (PFKFB3), which generates fructose-2,6-bisphosphate (F2,6P(2)) to activate the glycolytic enzyme PFK1 [39], is sequentially ubiquitinated by APCCdh1 in G1 phase [29,31,32] and by SCFβ-TRCP in S phase [29] (Figure 2). Apart from the glycolytic enzyme, the first enzyme in glutaminolysis, Glutaminase 1 (GLS1) is also ubiquitinated and destructed by APCCdh1 in G1 phases [29,32], thus GLS1 is accumulated in S phase to enhance glutaminolysis and sustain DNA replication (Figure 2).
In late G1 and S phase, Cyclin E-CDK2 and Cyclin A-CDK2 phosphorylate IDH1/2, the rate-limiting enzyme in the tricarboxylic acid (TCA) cycle, to facilitate their ubiquitination by the SCFSkp2 E3 ligase in S phase and subsequent degradation by 26S proteasome, resulting in cancer cell metabolism shift from OXPHOS at G1 phase to glycolysis at S phase [30] (Figure 2). Different from IDH1/2, IDH3β, a regulatory subunit of IDH3 hetero-tetramer enzyme, is ubiquitinated by the APCCdh1 E3 ligase in early G1 phase [33] (Figure 2). During the G2/M transition, the Cyclin B1-CDK1 kinase translocates into mitochondria to phosphorylate mitochondrial proteins, such as the electron transport chain (ETC) Complex I [27] and SIRT3 to impact cellular respiration [28] (Figure 2), suggesting that the physiological CDK1 inhibitor, such as DUX4 [40], might perturb cancer cell metabolism. In addition to these enzymes, other master regulators of cancer metabolism, such as Akt, can also be regulated by the cell cycle machinery at the post-translational level [41,42]. Akt is a key mediator for the Warburg effect and a critical metabolism remodeling factor in cancer, by promoting the uptake of glucose and amino acid, fostering glycolysis, and rewiring glucose metabolism to lipid synthesis [43]. Akt itself is regulated in a cell cycle-dependent manner, including the phosphorylation by Cyclin A-CDK1 [41] and the ubiquitination by SCFSkp2 [42]. On the other way around, Skp2 can also be phosphorylated by Akt, which renders the cytosol translocation of Skp2 and also disturbs the degradation of Skp2 by APCCdh1 E3 ligase complex [44].
These cell cycle-dependent phosphorylation/inhibition and destruction of metabolic enzymes provide multiple-disciplinary regulatory mechanisms for the shift of metabolic preference and the oscillation of metabolic intermediates, which is essential for the appropriate syntheses of DNA and proteins for the duplicated daughter cells. Perturbation of these regulator pathway lead to oxidative stress, DNA mutation and metabolism remodeling that eventually elicit uncontrollable cell proliferation and tumorigenesis [26,30]. For instance, T-cell acute lymphoblastic leukemia (T-ALL) cells express the dominant Cyclin D3 isoform, and Cyclin D3-CDK6 over-activation represses glycolysis by inhibiting PFK1 and PKM2, thus pushing more flux to PPP and serine pathway to produce reductive NADPH and glutathione, which favors the proliferation and drug-resistance of T-ALL cells [26]. To this end, this metabolic remodeling caused by Cyclin D3-CDK6 could be efficiently reversed by CDK4/6 inhibitors, thus making T-ALL cells susceptible to oxidative stress and apoptosis [26]. Moreover, prostate cancer (PrCa) expresses relatively high level of Skp2, which is reversely correlated with IDH1 protein level [30]. This character renders PrCa cells to rely more on glycolysis that facilitates the accumulation of glycolysis and PPP intermediates for the synthesis of amino acid and nucleotide to feed the division of daughter cells [30]. In keeping with this notion, the glycolysis preference in PrCa cells presents as a vulnerability that could be reversed by the Skp2 inhibitor, SKPin C1, to efficiently repress prostatic tumorigenesis in vitro and in vivo [30].
Although the oncogenic traits of several metabolic enzymes have been well defined, there is still lack of efficient approach to target them to combat tumorigenesis. Hence, defining the upstream regulators, especially those related to cell cycle mechanism, can achieve the goal of killing two birds (cell cycle and cancer metabolism) with one stone. To this end, the revealing of regulatory mechanism(s) between cell cycle and cancer metabolism not only provide novel insights for understanding the pathogenesis of cancer, but also provide the rational to target the cell cycle machinery to reverse cancer cell metabolism for better treatment of cancer.
Cell cycle and cancer immune surveillance
The cancer immune surveillance theory is formulated about half a century ago by Burnet and Thomas, promoting the development of the most favorable therapeutic approach nowadays, namely cancer immunotherapy [45]. Transcriptome by bulk-RNA-seq [46] and scRNA-seq [47,48] about the immune checkpoint blockade (ICB)-response versus ICB-resistant tumors coherently show that cell-cycle related genes, including the key G2/M checkpoint and E2F target genes are enriched in the ICB-resistant groups [47]. Furthermore, genetic amplifications of Cyclin D1 and CDK4 are reversely correlated with ICB response in human solid tumors [49], while a preference of deletions and mutations in Cdkn2a/b [50], upstream inhibitory regulator of CDK4/6 occurs in non-T cell-inflamed pancreatic cancers, compared with CD8+ T cell-inflamed cancers. The malfunction of the Rb tumor suppressor protein, which functions largely by repressing the transcriptional activity of E2F, is one of the most common features of most human cancer types. As such, CDK4/6 inhibitors have been applied for the treatment of cancers in part by restoring Rb tumor suppressive function through suppressing CDK4/6-mediated phosphorylation of Rb [18,19]. Later on, an unexpected cancer immunity regulatory effect has been observed during the preclinical investigation of these CDK4/6 inhibitors in breast cancer (BRCA), lung cancer, pancreatic cancer, and melanoma, in which multiple mechanisms have been defined [51–53] (Figure 3).
Figure 3. CDK4/6 inhibitor at the crosstalk of cell cycle and immune surveillance.
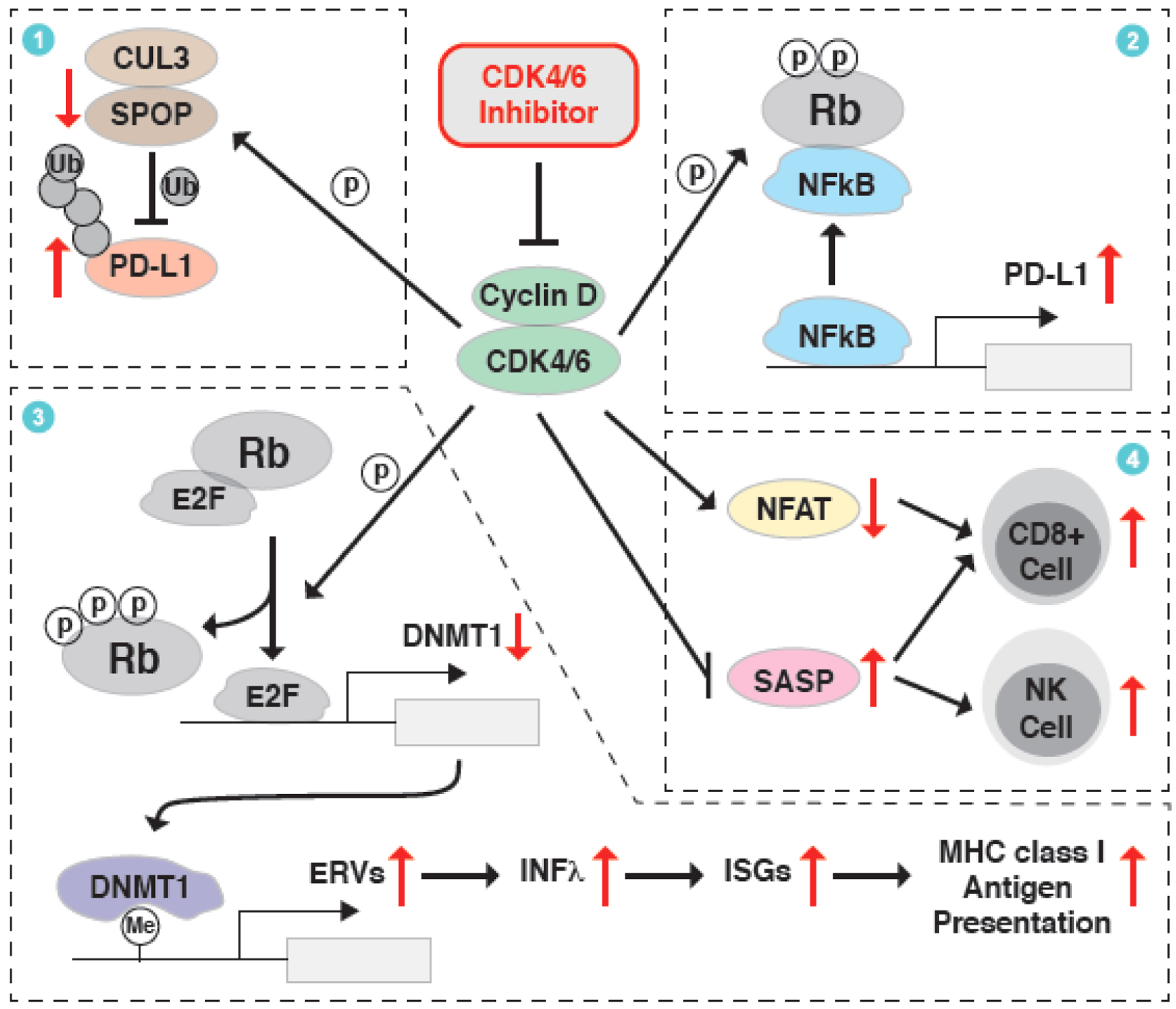
An unexpected immune-regulatory function of CDK4/6 inhibitor has been observed in clinic and various underlying mechanisms have been revealed to contribute to this phenotype. First, Cyclin D-CDK4/6 kinase can phosphorylate and stabilize SPOP, thus facilitating CUL3SPOP E3 ligase-mediated ubiquitination and proteasomal degradation of PD-L1, which could be blocked by the CDK4/6 inhibitor. Second, Cyclin D-CDK4/6 phosphorylates Rb, promotes its binding to NFκB/p65 and thus inactivates the transcription of PD-L1, while CDK4/6 inhibition triggers the transcription of PD-L1. Third, Cyclin D-CDK4/6 phosphorylates Rb to active E2F-mediated transcription of DNMT1, an epigenetic transcription repressor of endogenous retroviral elements (ERVs). This process could be antagonized by CDK4/6 inhibitor to trigger the IFNλ/ISGs/MHC signaling axis and the presentation of tumor antigens. Lastly, inhibition of CDK4/6 stimulates ASAP and represses NFAT-mediated transcriptome, which eventually promotes the infiltration of CD8+ T cells and/or NK cells for the killing of cancer cells.
A major mechanism underlying the immunity boosting effect of CDK4/6 inhibitors is due to a transcriptional and post-translational regulation on the immune checkpoint protein PD-L1 [54,55]. On one hand, the immune checkpoint protein PD-L1 is ubiquitinated by CUL3SPOP E3 ligase, while the stability of the protein stability of SPOP is controlled by the Cyclin D-CDK4-mediated phosphorylation and APCCdh1 E3 ligase-mediated ubiquitination, resulting in a cell-cycle dependent fluctuation of PD-L1 protein, which peaked in M phase [54] (Figure 3). In keeping with this model, CDK4/6 inhibitor palbociclib causes the reduction in SPOP protein abundance and subsequent accumulation of PD-L1 protein, which subsequently enhances the effect of anti-PD-1 immunotherapy in syngeneic mouse models [54] (Table 1). On the other hand, CDK4/6 phosphorylates Rb on the S249/T252 residuals to trigger the binding between Rb and p65, thus repressing NFκB-mediated transcription of PD-L1 and causing immune escape of cancer cells, which could be efficiently reversed by CDK4/6 inhibitors [55] (Figure 3).
Table 1.
Combination therapy of CDK inhibitors and immune checkpoint blockades (ICBs).
| Combination therapy | Cancer type/model | Outcomes | Mechanisms |
|---|---|---|---|
| CDK4/6i (Trilaciclib/Palboci clib) + PD-1 mAb | Colon cancer (CT26 and MC38) | Palbociclib enhances tumor regression and improves overall survival rates in CT26 (survivor: 8/12 in combination therapy group vs. 3/14 in PD-1 mAb group) and MC38 (6/12 vs. 3/15) syngeneic mouse models that treated with PD-1 mAb [54]. The combination therapy (Trilaciclib/Palbociclib + PD-1 mAb) nearly eliminates tumors in CT26 and MC38 syngeneic mouse models [59]. |
PD-L1 protein stability is controlled by CUL-3SPOP-mediated ubiquitination and subsequent proteasomal degradation [54]. Cyclin D-CDK4 phosphorylates SPOP, and protects it from APCCdh1-mediated ubiquitination and subsequent proteasomal degradation [54]. CDK4/6i enhances T cell activation, through the de-repression of Nuclear Factor of Activated T cell (NFAT) family proteins and their target genes [59]. |
| CDK4/6i (Abemaciclib) + PD-L1 mAb | Breast cancer (MMTV-rtTA/tetO-HER2); Colon cancer (CT26) | The combination therapy regresses to the growth of MMTV-rtTA/tetO-HER2 tumor (~70% reduction by day 13, no resume growth by day 35) and CT26 mouse model (4/4 combination therapy group in vs 2/4 in PD-L1 mAb group) [53]. Continuous combination therapy leads to complete tumor rejection and immunological memory [56]. |
CDK4/6 inhibition increases endogenous retroviral elements (ERVs) expression by repressing DNMT1 transcription, thus increasing intracellular double-stranded RNA levels, which in turn stimulates IFNλ production and hence enhances tumor antigen presentation [53]. CDK4/6 inhibition suppresses the proliferation of Treg cells [53]. CDK4/6 inhibition increase NFAT, and synergizes with PD-L1 blockade to enhance adaptive and innate immune activation [56]. |
| MEKi (Trametinib) + CDK4/6i (Palbociclib) + PD-1 mAb | KRAS-driven Pancreas cancer (KPCflox GEMM and KPCmut transplant) | The combination therapy produces an over 5-fold increase in the median survival of the KPCflox GEMM and KPCmut organoid transplant tumors, compared to PD-1 alone treatment [58]. | MEK and CDK4/6 inhibitors induce a senescence-associated secretory phenotype (SASP), which further stimulates the accumulation of CD8+ T cells into tumors, thus sensitizing tumors to PD-1 antibody [58]. |
| CDK4/6i (Trilaciclib) + 5-FU/Oxaliplatin + PD-L1 mAb | Colon cancer (CT26 and MC38) | CDK4/6i enhances anti-tumor response and overall survival compared with chemotherapy and ICI combinations alone [60]. | CDK4/6i modulates the proliferation and composition of T-cell subsets in the tumor microenvironment and increases effector function [60]. |
| CDK7i (YKL-5-124) + PD-1 mAb | Small cell lung cancer (SCLC) | The combination therapy offers significant survival benefit in 4 highly aggressive SCLC murine models (Rb1L/Lp53L/Lp130L/L (RPP) GEMM; RPP orthotopic, Rb1−/−p53−/− (RP); RPP-MYC orthotopic) [62]. | CDK7 inhibition triggers immune response signaling and induces pro-inflammatory cytokines and chemokines production, thus provoking a robust immune surveillance program elicited by T cells [62]. |
The other mechanism of CDK4/6 inhibitors’ immunity enhancing effect is owing to the improved tumor antigens present and immune cell infiltration [53]. For instance, CDK4/6 inhibitor activates the expression of endogenous retroviral elements (ERVs) by repressing the transcription of DNA methyltransferase 1 (DNMT1), which triggers a transcriptome characterized by increasing interferon λ (IFN λ) and MHC class I/II antigens [53,56] (Figure 3). These tumor-derived antigens recruit more immune cell infiltration into tumor site to repress tumor growth and facilitate the clearance of tumor cells, thus improving the effect of ICB, such as PD-L1 antibody, in a MMTV-rtTA/tetO-HER2 BRCA mouse model [53] (Table 1). Likewise, CDK4/6 inhibitor, in combination with MEK inhibitor, suppresses the tumorigenesis of KRAS-driven lung cancer [57] and pancreatic ductal adenocarcinoma [58], in part through inducing tumor necrosis factor-α (TNFα) and intercellular adhesion molecule-1 (ICAM-1) to trigger an immunomodulatory senescence-associated secretory phenotype (SASP) (Figure 3). SASP-mediated endothelial cell activation prompts the infiltration of CD8+ T cells into tumors, thus sensitizing them to PD-1/PD-L1 blockades [58] (Table 1). Besides, CDK4/6 inhibitor-induced SASP further elicits natural killer (NK) cells-mediated immune surveillance program [57], and activates endothelial cell to favor the infiltration of CD8+ T cells into tumors [58], thus sensitizing tumors to ICB such as PD-1 antibody treatment (Figure 3, Table 1). In keeping with these findings, CDK4/6 inhibition can antagonize the transcriptome associated with T cell exclusion and immune evasion, which are frequently observed in ICB-resistant melanoma patients [48]. Besides, CDK4/6 inhibitor also enhances T-cell activation and augments antitumor immunity through de-repressing the activity of nuclear factor of activated T-cells (NFAT) [59]. Recently, CDK4/6 inhibitor has also been reported to enhance the anti-tumor effect for chemotherapy/ICB combination regimen (oxaliplatin/5-FU together with PD-L1 antibody) in syngeneic mouse models [60] (Table 1). To be noted, in the clinical trial for trilaciclib in combination with etoposide/carboplatin for small cell lung cancer (SCLC, NCT02499770, phase Ib/IIa), trilaciclib can preserve peripheral lymphocyte and enhance T-cell activation, suggesting an effect of CDK4/6 inhibitor in improving antitumor immunity in SCLC patient during chemotherapy [60].
Although CDK4/6 inhibitors induce the infiltration of immune cells into tumor and the activation of CD8+ T cells, these inhibitors also display inhibitory effect on the proliferation of immune cells, including T-cell [59]. Hence, mechanistic dissection of the cell cycle regulatory effect and immunomodulatory effect of CDK4/6 might inspire the identification of novel therapeutic target(s) downstream of CDK4/6 to avoid unwanted toxicity to normal immune cells. During senescence, the ribosome biogenesis defects lead to the accumulation of ribosomal proteins RPS14 and RPL22 to inhibit CDK4/6 [13,61], which might modulate immune surveillance. Apart from CDK4/6, CDK7 inhibitor (YKL-5-124) also provokes a robust T cell-based immune surveillance program in SCLC through activating IFNγ pathway, thus enhances the anti-tumor effect of ICB [62] (Table 1). Furthermore, G2/M cell cycle arrest with CDK1 inhibitor RO-3306 can abolish irradiation-induced antitumor effect through suppressing micronuclei formation and DNA damage-induced inflammatory signaling [63]. These studies altogether indicate that therapeutic approaches targeting cell cycle regulators, especially CDK4/6, might achieve the goal of “one stone, two birds” in anti-cancer treatment by repressing cancer cell division and restoring cancer immune surveillance.
Targeting cell cycle regulators with PROTAC
Cell cycle machinery proteins represent a major type of cancer therapeutic targets, and several kinase inhibitor drugs are now in clinical use or in preclinical/clinical trials, including the inhibitors for CDKs, PLK, WEE1, Aurora A, and Chk1/2. Similar as other traditional SMIs, these inhibitors for cell cycle machinery have potential shortcomings, including: 1) off-target inhibitory effect on close family members of target protein, such as the inhibitory effect of CDK2i on CDK1 [64]; 2) acquired drug resistance due to mutants on target or upstream regulators, such as RB mutation [65], CDK6 amplification [66], and loss of PTEN [67] or FAT1 [68] during the establishment of CDK4/6 inhibitor resistance [69]; 3) minimal effect on kinase-independent role of CDK targets, such as for CDK6 [70–72]. These shortages of inhibitor could be overcome by the emerging PROTAC technology, which provide a new way to target those so-called undruggable targets, such as transcriptional factors and scaffold proteins [73]. PROTAC is a hetero-bifunctional molecule that recruits endogenous E3 ligase, such as cereblon (CRBN) and von Hippel-Lindau (VHL), to degrade disease-causing proteins of interest (POIs), including those oncogenic players in cell cycle machinery, such as CDKs [74–80], WEE1 [81], Aurora A [82] and Cdc20 [83] (Box 1).
Box 1. PROTAC.
Proteolysis targeting chimeras (PROTAC) is an emerging and promising approach to target disease-causing proteins for ubiquitination-mediated proteasomal degradation. By hijacking the endogenous E3 ubiquitin ligase for subsequent degradation by the 26S proteasome, PROTAC represents a major type of targeted protein degradation (TPD) techniques, which also includes lysosome targeting chimeras (LYTAC), autophagy targeting chimeras (AUTAC), and autophagosome tethering compound (ATTEC) [103].
PROTAC molecule consists of three functional moieties: a ligand for recruiting endogenous E3 ubiquitin ligase, a warhead for recognizing proteins of interest (POIs), and a linker region to achieve best efficiency and specificity. The E3 ligase cereblon (CRBN) and von Hippel-Lindau (VHL) are the best-defined E3 ligases for PROTAC design, with immunomodulatory imide drugs (IMiDs, such as pomalidomide and lenalidomide) and the chemical synthesized VHL ligands (such as VHL ligand-1 and VHL-032) as ligands, respectively. PROTAC recruits the close proximity between E3 ligand and POI, facilitating the transfer of poly-ubiquitin chain onto POI by the multi-subunit E3 ligases, CRL4CRBN or CRL2VHL E3 ligase. To date, two PROTACs, ARV-110 and ARV-471 are in clinical trials for the treatment of metastatic castration resistant PrCa (mCRPC) and ER+/HER2− metastatic BRCA, respectively.
As an ultimate effecter of the G1/S checkpoint, E2F1 play an oncogenic role by governing the transcription of the downstream targets [84]. However, no SMI of E2F1 has been developed yet. Recently, a DNA oligomer-based PROTAC platform has been developed to degrade oncogenic transcriptional factors, including E2F1 [85] (Figure 4). CDK4/6 kinases are upstream regulators for Rb/E2F1, and CDK4/6 inhibitors are conventionally used to block E2F1 transcriptional function. By linking CDK4/6 inhibitor palbociclib to pomalidomide to recruit the CRBN E3 ligase, CDK4/6 PROTACs have been developed to degrade either CDK4/6 (BSJ-02-162 [76], BSJ-03-204 [76], pal-pom [86]) or CDK4 only (BSJ-03-132) [76] (Figure 4). In addition, several CDK6 specific degraders (BSJ-03-123 [77], CP-10 [78], YX-2-107 [72], Degrader 6 [87]) have also been developed based on abemaciclib or palbociclib, which are effective in inhibiting the proliferation of hematopoietic cancer cells [72,77,78] (Figure 4). Compared with SMIs, these CDK6 specific degraders have advantages in overcoming the resistance to CDK6 inhibitor [78] or in inhibiting the kinase-independent function of CDK6 [72].
Figure 4. PROTACs present new avenues to degrade oncogenic proteins in the cell cycle machinery.
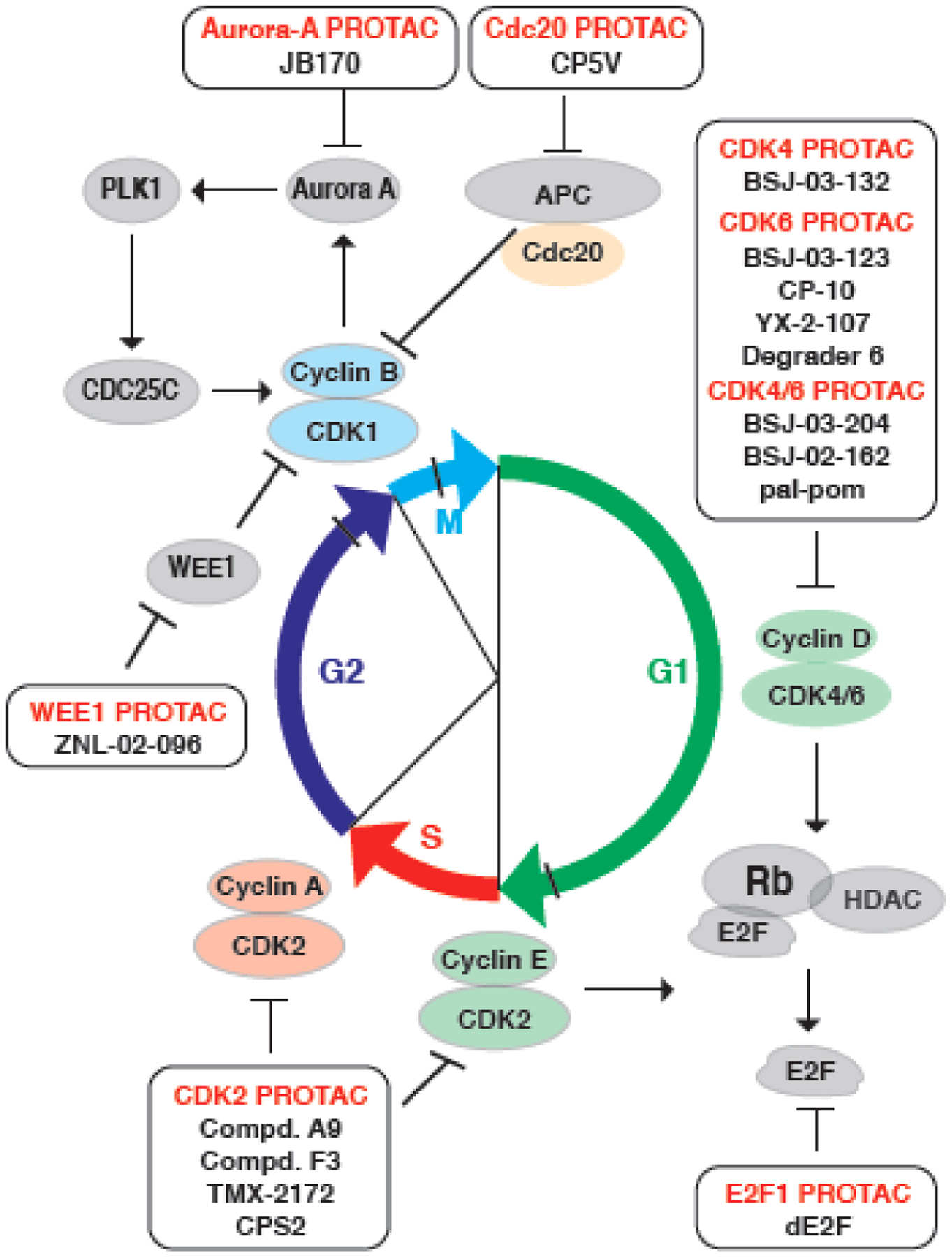
PROTACs against cell cycle controlling kinases, including CDK2/4/6, WEE1, Aurora A, as well as E2F1 and Cdc20, have been developed recently as potent therapeutics to combat cancer. By targeting upstream regulators of the G2/M checkpoint, WEE1-PROTAC (ZNL-20-096), Aurora A-PROTAC (JB170) and Cdc20-PROTAC (CP5V) block the mitosis entry and progression. Based on the CDK4/6 inhibitors, PROTACs have been developed to either target CDK4/6 (BSJ-03-024, BSJ-02-162, and pal-pom), or CDK4 only (BSJ-03-132), or CDK6 only (BSJ-03-123, CP-10, YX-2-107, and Degrader 6), which elicit potent degradation efficiency and anti-proliferation effect in cancer cells. For the S-phase related CDK2, several PROTACs (Compd. A9, Compd. F3, TMC-2172, and CPS2) have been developed to specifically degrade CDK2, sparing its close analog CDK1 and other CDKs. Downstream of the G1/S checkpoint, the effecter transcription factor E2F1 can also be targeted by dE2F, a DNA-based PROTAC for E2F. Compared with SMIs, PROTACs have several superior advantages, including the ability to distinguish close family members (such as CDK4 vs. CDK6, or CDK2 vs. CDK1), the capability of blocking the kinase activity-independent function of CDKs, as well as the potential to overcome drug resistance to inhibitor.
Although many CDK inhibitors have been approved to be used in clinical practice or in clinical trial, a common shortage of these CDK inhibitors is an off-target inhibitory effect on the close family member of POI. For example, CDK2 controls the G1/S checkpoint and the entry of cell cycle S phase, making it a promising drug target for cancer [88], and CDK2 inhibitor, FN-1501 (NCT03690154, phase I) and PF-07104091 (NCT04553133, phase I/II), are now in clinical trials for AML and lung cancer, respectively [89]. However, it is extremely challenging to develop a specific inhibitor solely for CDK2, in part due to a similar ATP binding pocket is shared by CDK1 and CDK2, while inhibition of CDK1 might introduce unwanted toxicity [64]. To this end, several CDK2 degraders have been developed based on CDK2 inhibitors (FN-1501 and TMX-2039) to degrade CDK2 only (CPS2 [90] and Compound A9 [74]), CDK2/9 (Compound F3 [74]), or CDK2/5, respectively (TMX-2172 [75]) (Figure 4). Compared with CDK2 inhibitors, these CDK2 degraders have better anti-proliferative effects for PC3 PrCa cells [74], OVCAR8 ovarian cancer cells [75], AML cell lines and primary patient cells [90]. Usually, inhibitors cannot phenocopy the biological consequence of genetic knockdown or knockout, indicating a kinase-independent role of kinase [91], and PROTACs can overcome this shortcoming of inhibitor and mime the effects of genetic ablation [92]. For example, CDK2 inhibitor is not sufficient to drive AML cell differentiation as genetic ablation of CDK2 using short hairpin [93], and CDK2 PROTAC efficient promote AML cell differentiation in a CDK2-dependent manner [90].
In G2/M checkpoint, WEE1 phosphorylates CDK1 to prevent mitotic entry [6], making it a reasonable therapeutic target for cancer, and WEE1 inhibitor (adavosertib/AZD1775) shows promising result in the phase II clinical trial for uterine serous carcinoma [20]. By converting adavosertib into a PROTAC, the WEE1 degrader (ZNL-02-096) has roughly 10-fold potency in blocking mitotic entry through a robust effect in degrading WEE1, and effectively inhibits the proliferation of ovarian cancer cells in vitro [81] (Figure 4). Additionally, Aurora-A kinase is essential for the rapid recruitment of G2/M checkpoint protein to kinetochores in early M phase, and Aurora-A is amplified or overexpressed in tumors [6]. Aurora-A PROTAC (JB170) has been developed based on the Aurora-A inhibitor alisertib, and it induces S-phase defect and apoptosis in MV4-11 cancer cells [82] (Figure 4). Given the critical of Cdc20 in initiation of anaphase and exist from mitosis [7], Cdc20 plays a oncogenic role for tumorigenesis, which inspires the development of specific inhibitor, such as Apcin [94,95]. By conjugation of VHL ligand onto Apcin, a Cdc20 degrader (CP5V) has been developed for the destruction of Cdc20, and it blocks the mitosis and represses the proliferation of MCF-7 BRCA cells, leading to tumor regression in a 4T1 BRCA xenograft mouse model [83] (Figure 4). Likewise, CDK9 degraders (Compound 3 [79] and Compound 11c [80]) have also been developed based on the CDK5/9 inhibitor aminopyrazole or the CDK9 inhibitor flavopiridol, to selectively degrade CDK9 and inhibit the proliferation of CDK9-overexpressed cancer cells.
Taken together, PROTACs against cell cycle machinery proteins and upstream regulators could provide the advantage of relatively more specificity, genetic ablation-mimic function which cannot be achieved by inhibitor, and the potential to overcome drug resistance due to mutations frequently observed in drug target proteins. Although no PROTAC for cell cycle machinery proteins has been proceeded into clinical trials yet, in part due to the potential weakness in permeability, oral availability and stability, PROTAC is still one of the most promising and attractive approaches to target the cell cycle machinery for cancer therapy. Nevertheless, the potential limits of PROTACs should be considered during the development of drugs for cell cycle machinery components and regulators. First, compared with canonical SMIs, PROTACs have relatively higher molecule weight (usually over 600 Da) that possible compromises their permeability and oral bioavailability. Although several orally bioavailable PROTACs, such as AR PROTAC (ARV-110, NCT03888612, phase I/II), ER PROTAC (ARV-471, NCT04072952, phase I/II) and CDK2/4/6 PROTAC [96] have been developed for clinical and preclinical study, the in vivo efficiency is still under investigation. Recently, several molecule glues have been proved to recruit the CUL4-DDB1 E3 ligase onto the Cyclin K-CDK12 kinase complex, subsequently triggering the ubiquitination and proteasomal degradation of Cyclin K [97–99]. Thus, molecule glue likely represents a complementary way of targeted protein degradation (TPD), possibly with better pharmacodynamics and pharmacokinetics properties [97–99]. Second, most present PROTACs are developed based on well-defined inhibitors for the POI, thus rendering them easy to inherit potential off-target effect and drug resistance derived from the parental inhibitors. Last but not the least, due to its catalytic manner of action, PROTACs might possess stronger toxicity to normal cell/tissues, for which the recently-developed controllable [100,101] and targeted-delivery [102] PROTAC platforms may provide a solution.
Concluding Remarks
Aberrancies in cell cycle-regulatory proteins have been defined in virtually all types of human cancers, based on which multiple therapies have been developed either in clinical practice or in clinical trial at present. Growing evidence shows that these therapies not only restrict cancer cell division, but also trigger unexpected effects, including reversing cancer metabolism remodeling and restoring cancer immune surveillance. For example, CDK inhibitors, including CDK4/6 and CDK2, have potential metabolism regulatory effects, either by restoring the metabolism from PPP and serine pathway back to glycolysis [26], or by antagonizing the Warburg effect [30]. However, it warrants further investigation whether the metabolic phenotypes observed after manipulation of the cell cycle machinery is a direct effect of change in specific metabolic enzyme(s) or an indirect effect caused by disruption of cell cycle progression due to the genetically or pharmaceutically ablation of cell cycle regulators (see Outstanding Questions). Moreover, directly perturbation of cellular metabolism disrupts cell cycle progression vice versa, thus making it complicate whether cellular metabolism remodeling is the cause or result of cell cycle disruption.
Outstanding Questions Box.
Whether the metabolic phenotypes observed after manipulation of cell cycle machinery is a direct effect of specific protein target or indirect effect caused by disruption of cell cycle progression?
Aberrant cell cycle leads to metabolism remodeling, and perturbation of cell metabolism also disrupts cell cycle progression vice versa. Which is the causing event and how does their interplay dictate the tumorigenesis process?
Whether inhibition of other cell cycle machinery proteins also causes immuno-modulatory effect as CDK4/6 inhibitors do?
How to dissect the immuno-modulatory effect and cell cycle regulatory effect of CDK4/6 inhibitors, which guides us to better define therapeutic target(s) for efficiently triggering anti-cancer immunity without adverse effect(s) on the proliferation of normal immune cells?
Whether PROTACs against cell cycle machinery proteins are capable of retarding tumorigenesis in vivo, due to the fact that the efficacy of PROTACs is potentially limited by their cellular permeability, oral bioavailability and stability?
Whether PROTACs against cell cycle machinery proteins will also face drug resistance, which can be likely derived from a complementary elevation of a close family member of the target protein with similar biological function?
Nowadays, CDK4/6 inhibitors triggers rampant anti-tumor immunity by either promoting the presentation tumor antigen derived from the DMNT1/ERVs/ISGs axis [53], or recruiting the infiltration of immune cells into tumor and activation of T cells [56][57–60], or transcriptionally and post-translational regulation of immune checkpoint protein(s) [54,55]. Thus, the combination therapies of CDK4/6 inhibitors with ICBs show promising outcomes in several types of human cancer patients [53,54,56,58–60]. However, due to the potential toxicity of CDK4/6 inhibitors on the proliferation of normal immune cells, it is necessary to dissect the critical function of CDK4/6 in regulating cell cycle progression and anti-cancer immunity, thus allow us to further develop a targeted therapy to only trigger cancer immune surveillance without inhibition on the proliferation of normal immune cells. Moreover, it warrants further in-depth investigation on the possible function of other cell cycle regulatory protein(s), besides CDK4/6, on cell metabolism and/or immune surveillance. These new knowledge will provide more therapeutic targets for cell cycle-focused cancer treatments to better retard tumorigenesis.
Due to the presence of multiple close family members for CDKs and the high similarity of inhibitor binding pocket in these kinases, it is extremely challenging to develop specific inhibitors that spare other close family CDK members. The emerging PROTAC technology provides a viable option, and PRTOACs that uniquely degrade CDK2 [74,90], CDK4 [76], or CDK6 [72,77,78,87] have been developed with low or no effect on other CDK family members. Likewise, PROTACs have superior effects in repressing the potential kinase-independent effect of CDKs [72] and in overcoming potential drug resistance [78], making it a promising way to dissect the biological functions of different CDKs, and to specifically target a unique protein along the cell cycle machinery to achieve novel anti-cancer therapeutic option. However, it awaits for further determination the effect of PROTACs against cell cycle machinery proteins in regressing tumor in vivo, and more effects are warranted to improve the permeability and bioavailability of PROTACs, as well as to develop PROTACs for those undruggable targets in the cell cycle machinery.
Highlights.
Cell cycle machinery regulates cancer metabolism primarily at the post-translational level, either through phosphorylation of metabolic enzymes or upstream regulators by CDKs, or through ubiquitination of these proteins by APC/C or SCF E3 ligases.
CDK inhibitors restore cancer immune surveillance, in part by triggering the tumor antigens presentation or stimulating the infiltration of immune cells into tumor, thus providing synergetic effects with immune checkpoint blockades (ICBs).
PROTACs provide a new cancer therapeutic approach to target cell cycle machinery components with oncogenic roles for proteasomal degradation, which holds advantages over traditional small molecular inhibitors (SIMs) in many aspects includes distinguishing protein target with its close family member(s), overcoming potential drug resistance, or repressing the kinase-independent function of protein targets.
Acknowledgements:
We apologize for not citing all relevant reports owing to space limitations. This work was supported in part by funding from NIH (R35CA253027 to W.W.).
Glossary box
- Cell cycle checkpoints
a control mechanism that halt cell cycle progress to ensure the accurate replication of genomic DNA and faithful separation of the sister chromatids into daughter cells.
- PLK1
polo-like kinase 1, also known as serine/threonine-protein kinase 13 (STPK13).
- SAC
spindle assembly checkpoint, also known as the mitotic checkpoint, is a cell cycle checkpoint during mitosis or meiosis that ensures the proper transition from metaphase to anaphase and accurate separation of the duplicated chromatids.
- APC/C
anaphase promoting complex/cyclosome, a multisubunit E3 ubiquitin ligase complex that targets cell cycle regulatory proteins for degradation by the 26S proteasome during exit from the mitosis and in the G1 phase of the cell cycle. Among APC/C, the Cdh1 and Cdc20 subunit determines the specificity of ubiquitination substrate.
- SCF
Skp1-Cullin 1-F-box, a multiple protein E3 ubiquitin ligase complex that composes of the scarfold Cullin 1, the E2-recruiting subunit Rbx1, the adaptor protein Skp1 that binds to the F-box motif, and the substrate-binding F-box protein.
- FUCCI
an technique for visualizing of cell cycle progression in living cells, in which G1 phase cells express RFP-Cdt1 with red fluorescence and S/G2/M phases cells with green GFP-geminin.
- PROTAC
proteolysis targeting chimera, a hetero-bifunctional small molecule composed of a ligand to recruit E3 ubiquitin ligase, a moiety to recognize protein of interest (POI), and a linker region, which bridges the transfering of poly-ubiquitin chain from the E3 ubiquitin ligase onto POI for the ubiquitination and subsequent proteasomal degradation of POI.
- OXPHOS
oxidative phosphorylation, a process taking place in mitochondria, which transfers electrons from the reductive NADH or FADH2 to oxygen by a series of electron carriers to produce ATP.
- Warburg effect
a metabolism phenotype of cancer cells, which is characterized by favoring glycolysis rather than TCA cycle for the carbohydrate metabolism.
- PPP
pentose phosphate pathway, also known as hexose monophosphate shunt, a metabolic pathway parallel to glycolysis that uses glucose-6-p to generate reductive DNAPDH and ribose-5-phosphate as a precursor for nucleotide synthesis.
- Glutaminolysis
a metabolic process occurs in mitochondia, in which glutamine is lysed to glutamate to feed the TCA cycle.
- TCA cycle
tricarboxylic acid, also known as the Krebs cycle or the citric acid cycle, a metabolic process that occurs in mitochondia, through which Acetyl-CoA is lysed to CO2 in an enzymatic chain reaction, to produce ATP and NADH.
- ICB
immune checkpoint blockade, also known as immune checkpoint inhibitor or ICI, which represents the antibodies bind with immune checkpoint proteins to prevent their interactions with the corresponding receptor/ligand. The representive ICBs include PD-1 monoclonal antibody (mAb), PD-L1 mAb or CTLA-4 mAb.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: W.W. is a co-founder and consultant for the ReKindle Therapeutics. Other authors declare no competing financial interests.
References
- 1.Hartwell LH et al. (1973) Genetic Control of the Cell Division Cycle in Yeast: V. Genetic Analysis of cdc Mutants. Genetics 74, 267–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nurse P (2000) A long twentieth century of the cell cycle and beyond. Cell 100, 71–78. [DOI] [PubMed] [Google Scholar]
- 3.Nasmyth K (2001) A prize for proliferation. Cell 107, 689–701. [DOI] [PubMed] [Google Scholar]
- 4.Bertoli C et al. (2013) Control of cell cycle transcription during G1 and S phases. Nat Rev Mol Cell Biol 14, 518–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guardavaccaro D and Pagano M (2006) Stabilizers and destabilizers controlling cell cycle oscillators. Mol Cell 22, 1–4. [DOI] [PubMed] [Google Scholar]
- 6.O’Connell MJ et al. (2000) The G2-phase DNA-damage checkpoint. Trends Cell Biol 10, 296–303. [DOI] [PubMed] [Google Scholar]
- 7.Lara-Gonzalez P et al. (2012) The spindle assembly checkpoint. Curr Biol 22, R966–980. [DOI] [PubMed] [Google Scholar]
- 8.Peters JM (2006) The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat Rev Mol Cell Biol 7, 644–656. [DOI] [PubMed] [Google Scholar]
- 9.Wang Z et al. (2014) Roles of F-box proteins in cancer. Nat Rev Cancer 14, 233–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu J et al. (2020) Targeting SCF E3 Ligases for Cancer Therapies. Adv Exp Med Biol 1217, 123–146. [DOI] [PubMed] [Google Scholar]
- 11.Frescas D and Pagano M (2008) Deregulated proteolysis by the F-box proteins SKP2 and beta-TrCP: tipping the scales of cancer. Nat Rev Cancer 8, 438–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Skaar JR et al. (2013) Mechanisms and function of substrate recruitment by F-box proteins. Nat Rev Mol Cell Biol 14, 369–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bury M et al. (2021) New Insights into CDK Regulators: Novel Opportunities for Cancer Therapy. Trends Cell Biol 31, 331–344. [DOI] [PubMed] [Google Scholar]
- 14.Swaffer MP et al. (2016) CDK Substrate Phosphorylation and Ordering the Cell Cycle. Cell 167, 1750–1761 e1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mahdessian D et al. (2021) Spatiotemporal dissection of the cell cycle with single-cell proteogenomics. Nature 590, 649–654. [DOI] [PubMed] [Google Scholar]
- 16.Malumbres M and Barbacid M (2009) Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer 9, 153–166. [DOI] [PubMed] [Google Scholar]
- 17.Kastan MB and Bartek J (2004) Cell-cycle checkpoints and cancer. Nature 432, 316–323. [DOI] [PubMed] [Google Scholar]
- 18.Beaver JA et al. (2015) FDA Approval: Palbociclib for the Treatment of Postmenopausal Patients with Estrogen Receptor-Positive, HER2-Negative Metastatic Breast Cancer. Clin Cancer Res 21, 4760–4766. [DOI] [PubMed] [Google Scholar]
- 19.Tripathy D et al. (2017) Ribociclib (LEE011): Mechanism of Action and Clinical Impact of This Selective Cyclin-Dependent Kinase 4/6 Inhibitor in Various Solid Tumors. Clin Cancer Res 23, 3251–3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu JF et al. (2021) Phase II Study of the WEE1 Inhibitor Adavosertib in Recurrent Uterine Serous Carcinoma. J Clin Oncol 39, 1531–1539. [DOI] [PubMed] [Google Scholar]
- 21.Hanahan D and Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- 22.Tu BP et al. (2005) Logic of the yeast metabolic cycle: temporal compartmentalization of cellular processes. Science 310, 1152–1158. [DOI] [PubMed] [Google Scholar]
- 23.Chen Z et al. (2007) Restriction of DNA replication to the reductive phase of the metabolic cycle protects genome integrity. Science 316, 1916–1919. [DOI] [PubMed] [Google Scholar]
- 24.Wang C et al. (2006) Cyclin D1 repression of nuclear respiratory factor 1 integrates nuclear DNA synthesis and mitochondrial function. Proc Natl Acad Sci U S A 103, 11567–11572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bhalla K et al. (2014) Cyclin D1 represses gluconeogenesis via inhibition of the transcriptional coactivator PGC1alpha. Diabetes 63, 3266–3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang H et al. (2017) The metabolic function of cyclin D3-CDK6 kinase in cancer cell survival. Nature 546, 426–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Z et al. (2014) Cyclin B1/Cdk1 coordinates mitochondrial respiration for cell-cycle G2/M progression. Dev Cell 29, 217–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu R et al. (2015) CDK1-Mediated SIRT3 Activation Enhances Mitochondrial Function and Tumor Radioresistance. Mol Cancer Ther 14, 2090–2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Colombo SL et al. (2010) Anaphase-promoting complex/cyclosome-Cdh1 coordinates glycolysis and glutaminolysis with transition to S phase in human T lymphocytes. Proc Natl Acad Sci U S A 107, 18868–18873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu J et al. (2021) Skp2 dictates cell cycle-dependent metabolic oscillation between glycolysis and TCA cycle. Cell Res 31, 80–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Herrero-Mendez A et al. (2009) The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat Cell Biol 11, 747–752. [DOI] [PubMed] [Google Scholar]
- 32.Colombo SL et al. (2011) Molecular basis for the differential use of glucose and glutamine in cell proliferation as revealed by synchronized HeLa cells. Proc Natl Acad Sci U S A 108, 21069–21074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu Q et al. (2019) APC/C-CDH1-Regulated IDH3beta Coordinates with the Cell Cycle to Promote Cell Proliferation. Cancer Res 79, 3281–3293. [DOI] [PubMed] [Google Scholar]
- 34.Simoneschi D et al. (2021) CRL4(AMBRA1) is a master regulator of D-type cyclins. Nature 592, 789–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maiani E et al. (2021) AMBRA1 regulates cyclin D to guard S-phase entry and genomic integrity. Nature 592, 799–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chaikovsky AC et al. (2021) The AMBRA1 E3 ligase adaptor regulates the stability of cyclin D. Nature 592, 794–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Caillot M et al. (2020) Cyclin D1 targets hexokinase 2 to control aerobic glycolysis in myeloma cells. Oncogenesis 9, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kamarajugadda S et al. (2016) Cyclin D1 represses peroxisome proliferator-activated receptor alpha and inhibits fatty acid oxidation. Oncotarget 7, 47674–47686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yalcin A et al. (2009) Nuclear targeting of 6-phosphofructo-2-kinase (PFKFB3) increases proliferation via cyclin-dependent kinases. J Biol Chem 284, 24223–24232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bury M et al. (2019) NFE2L3 Controls Colon Cancer Cell Growth through Regulation of DUX4, a CDK1 Inhibitor. Cell Rep 29, 1469–1481 e1469. [DOI] [PubMed] [Google Scholar]
- 41.Liu P et al. (2014) Cell-cycle-regulated activation of Akt kinase by phosphorylation at its carboxyl terminus. Nature 508, 541–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chan CH et al. (2012) The Skp2-SCF E3 ligase regulates Akt ubiquitination, glycolysis, herceptin sensitivity, and tumorigenesis. Cell 149, 1098–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhu J and Thompson CB (2019) Metabolic regulation of cell growth and proliferation. Nat Rev Mol Cell Biol 20, 436–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gao D et al. (2009) Phosphorylation by Akt1 promotes cytoplasmic localization of Skp2 and impairs APCCdh1-mediated Skp2 destruction. Nat Cell Biol 11, 397–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ribatti D (2017) The concept of immune surveillance against tumors. The first theories. Oncotarget 8, 7175–7180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Galvani E et al. (2020) Stroma remodeling and reduced cell division define durable response to PD-1 blockade in melanoma. Nat Commun 11, 853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peng J et al. (2019) Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res 29, 725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jerby-Arnon L et al. (2018) A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 175, 984–997 e924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miao D et al. (2018) Genomic correlates of response to immune checkpoint blockade in microsatellite-stable solid tumors. Nat Genet 50, 1271–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li J et al. (2018) Tumor Cell-Intrinsic Factors Underlie Heterogeneity of Immune Cell Infiltration and Response to Immunotherapy. Immunity 49, 178–193 e177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li J and Stanger BZ (2020) Cell Cycle Regulation Meets Tumor Immunosuppression. Trends Immunol 41, 859–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goel S et al. (2018) CDK4/6 Inhibition in Cancer: Beyond Cell Cycle Arrest. Trends Cell Biol 28, 911–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Goel S et al. (2017) CDK4/6 inhibition triggers anti-tumour immunity. Nature 548, 471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang J et al. (2018) Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature 553, 91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jin X et al. (2019) Phosphorylated RB Promotes Cancer Immunity by Inhibiting NF-kappaB Activation and PD-L1 Expression. Mol Cell 73, 22–35 e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schaer DA et al. (2018) The CDK4/6 Inhibitor Abemaciclib Induces a T Cell Inflamed Tumor Microenvironment and Enhances the Efficacy of PD-L1 Checkpoint Blockade. Cell Rep 22, 2978–2994. [DOI] [PubMed] [Google Scholar]
- 57.Ruscetti M et al. (2018) NK cell-mediated cytotoxicity contributes to tumor control by a cytostatic drug combination. Science 362, 1416–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ruscetti M et al. (2020) Senescence-Induced Vascular Remodeling Creates Therapeutic Vulnerabilities in Pancreas Cancer. Cell 181, 424–441 e421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Deng J et al. (2018) CDK4/6 Inhibition Augments Antitumor Immunity by Enhancing T-cell Activation. Cancer Discov 8, 216–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lai AY et al. (2020) CDK4/6 inhibition enhances antitumor efficacy of chemotherapy and immune checkpoint inhibitor combinations in preclinical models and enhances T-cell activation in patients with SCLC receiving chemotherapy. J Immunother Cancer 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lessard F et al. (2018) Senescence-associated ribosome biogenesis defects contributes to cell cycle arrest through the Rb pathway. Nat Cell Biol 20, 789–799. [DOI] [PubMed] [Google Scholar]
- 62.Zhang H et al. (2020) CDK7 Inhibition Potentiates Genome Instability Triggering Anti-tumor Immunity in Small Cell Lung Cancer. Cancer Cell 37, 37–54 e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen J et al. (2020) Cell Cycle Checkpoints Cooperate to Suppress DNA- and RNA-Associated Molecular Pattern Recognition and Anti-Tumor Immune Responses. Cell Rep 32, 108080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wood DJ et al. (2019) Differences in the Conformational Energy Landscape of CDK1 and CDK2 Suggest a Mechanism for Achieving Selective CDK Inhibition. Cell Chem Biol 26, 121–130 e125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Condorelli R et al. (2018) Polyclonal RB1 mutations and acquired resistance to CDK 4/6 inhibitors in patients with metastatic breast cancer. Ann Oncol 29, 640–645. [DOI] [PubMed] [Google Scholar]
- 66.Yang C et al. (2017) Acquired CDK6 amplification promotes breast cancer resistance to CDK4/6 inhibitors and loss of ER signaling and dependence. Oncogene 36, 2255–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Costa C et al. (2020) PTEN Loss Mediates Clinical Cross-Resistance to CDK4/6 and PI3Kalpha Inhibitors in Breast Cancer. Cancer Discov 10, 72–85. [DOI] [PubMed] [Google Scholar]
- 68.Li Z et al. (2018) Loss of the FAT1 Tumor Suppressor Promotes Resistance to CDK4/6 Inhibitors via the Hippo Pathway. Cancer Cell 34, 893–905 e898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li Z et al. (2020) Mechanisms of CDK4/6 Inhibitor Resistance in Luminal Breast Cancer. Front Pharmacol 11, 580251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kollmann K et al. (2013) A kinase-independent function of CDK6 links the cell cycle to tumor angiogenesis. Cancer Cell 24, 167–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Scheicher R et al. (2015) CDK6 as a key regulator of hematopoietic and leukemic stem cell activation. Blood 125, 90–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.De Dominici M et al. (2020) Selective inhibition of Ph-positive ALL cell growth through kinase-dependent and -independent effects by CDK6-specific PROTACs. Blood 135, 1560–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liu J et al. (2020) PROTACs: A novel strategy for cancer therapy. Semin Cancer Biol 67, 171–179. [DOI] [PubMed] [Google Scholar]
- 74.Zhou F et al. (2020) Development of selective mono or dual PROTAC degrader probe of CDK isoforms. Eur J Med Chem 187, 111952. [DOI] [PubMed] [Google Scholar]
- 75.Teng M et al. (2020) Development of CDK2 and CDK5 Dual Degrader TMX-2172. Angew Chem Int Ed Engl 59, 13865–13870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jiang B et al. (2019) Development of Dual and Selective Degraders of Cyclin-Dependent Kinases 4 and 6. Angew Chem Int Ed Engl 58, 6321–6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Brand M et al. (2019) Homolog-Selective Degradation as a Strategy to Probe the Function of CDK6 in AML. Cell chemical biology 26, 300–306 e309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Su S et al. (2019) Potent and Preferential Degradation of CDK6 via Proteolysis Targeting Chimera Degraders. J Med Chem 62, 7575–7582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Robb CM et al. (2017) Chemically induced degradation of CDK9 by a proteolysis targeting chimera (PROTAC). Chem Commun (Camb) 53, 7577–7580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bian J et al. (2018) Discovery of Wogonin-based PROTACs against CDK9 and capable of achieving antitumor activity. Bioorg Chem 81, 373–381. [DOI] [PubMed] [Google Scholar]
- 81.Li Z et al. (2020) Development and Characterization of a Wee1 Kinase Degrader. Cell Chem Biol 27, 57–65 e59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Adhikari B et al. (2020) PROTAC-mediated degradation reveals a non-catalytic function of AURORA-A kinase. Nat Chem Biol 16, 1179–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chi JJ et al. (2019) A novel strategy to block mitotic progression for targeted therapy. EBioMedicine 49, 40–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kent LN and Leone G (2019) The broken cycle: E2F dysfunction in cancer. Nat Rev Cancer 19, 326–338. [DOI] [PubMed] [Google Scholar]
- 85.Liu J et al. (2021) TF-PROTACs enable targeted degradation of transcription factors. J Am Chem Soc 143, 8902–8910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhao B and Burgess K (2019) PROTACs suppression of CDK4/6, crucial kinases for cell cycle regulation in cancer. Chem Commun (Camb) 55, 2704–2707. [DOI] [PubMed] [Google Scholar]
- 87.Rana S et al. (2019) Selective degradation of CDK6 by a palbociclib based PROTAC. Bioorg Med Chem Lett 29, 1375–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tadesse S et al. (2020) Targeting CDK2 in cancer: challenges and opportunities for therapy. Drug Discov Today 25, 406–413. [DOI] [PubMed] [Google Scholar]
- 89.Wang Y et al. (2018) Discovery of 4-((7H-Pyrrolo[2,3-d]pyrimidin-4-yl)amino)-N-(4-((4-methylpiperazin-1-yl)methyl)p henyl)-1H-pyrazole-3-carboxamide (FN-1501), an FLT3- and CDK-Kinase Inhibitor with Potentially High Efficiency against Acute Myelocytic Leukemia. J Med Chem 61, 1499–1518. [DOI] [PubMed] [Google Scholar]
- 90.Wang L et al. (2021) Discovery of a first-in-class CDK2 selective degrader for AML differentiation therapy. Nat Chem Biol 17, 567–575. [DOI] [PubMed] [Google Scholar]
- 91.Fujimoto T et al. (2007) Cdk6 blocks myeloid differentiation by interfering with Runx1 DNA binding and Runx1-C/EBPalpha interaction. EMBO J 26, 2361–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cromm PM et al. (2018) Addressing Kinase-Independent Functions of Fak via PROTAC-Mediated Degradation. J Am Chem Soc 140, 17019–17026. [DOI] [PubMed] [Google Scholar]
- 93.Ying M et al. (2018) Ubiquitin-dependent degradation of CDK2 drives the therapeutic differentiation of AML by targeting PRDX2. Blood 131, 2698–2711. [DOI] [PubMed] [Google Scholar]
- 94.Verma R et al. (2004) Ubistatins inhibit proteasome-dependent degradation by binding the ubiquitin chain. Science 306, 117–120. [DOI] [PubMed] [Google Scholar]
- 95.Sackton KL et al. (2014) Synergistic blockade of mitotic exit by two chemical inhibitors of the APC/C. Nature 514, 646–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wei M et al. (2021) First orally bioavailable prodrug of proteolysis targeting chimera (PROTAC) degrades cyclin-dependent kinases 2/4/6 in vivo. Eur J Med Chem 209, 112903. [DOI] [PubMed] [Google Scholar]
- 97.Slabicki M et al. (2020) The CDK inhibitor CR8 acts as a molecular glue degrader that depletes cyclin K. Nature 585, 293–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mayor-Ruiz C et al. (2020) Rational discovery of molecular glue degraders via scalable chemical profiling. Nat Chem Biol 16, 1199–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lv L et al. (2020) Discovery of a molecular glue promoting CDK12-DDB1 interaction to trigger cyclin K degradation. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Liu J et al. (2020) Light-induced control of protein destruction by opto-PROTAC. Sci Adv 6, eaay5154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Reynders M et al. (2020) PHOTACs enable optical control of protein degradation. Sci Adv 6, eaay5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Liu J et al. (2021) Cancer Selective Target Degradation by Folate-Caged PROTACs. J Am Chem Soc 143, 7380–7387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ding Y et al. (2020) Emerging New Concepts of Degrader Technologies. Trends Pharmacol Sci 41, 464–474. [DOI] [PMC free article] [PubMed] [Google Scholar]