

Abstract
Damage to specific brain circuits can cause specific neuropsychiatric symptoms. Therapeutic stimulation to these same circuits may modulate these symptoms. To determine if these circuits converge, we studied depression severity after brain lesions (n=461, five datasets), transcranial magnetic stimulation (TMS) (n=151, four datasets), and deep brain stimulation (DBS) (n=101, five datasets). Lesions and stimulation sites most associated with depression severity were connected to a similar brain circuit across all 14 datasets (p<0.001). Circuits derived from lesions, DBS, and TMS were similar (p<0.0005), as were circuits derived from patients with major depression versus other diagnoses (p<0.001). Connectivity to this circuit predicted out-of-sample antidepressant efficacy of TMS and DBS sites (p<0.0001). In an independent analysis, 29 lesions and 95 stimulation sites converged on a distinct circuit for motor symptoms of Parkinson’s disease (p<0.05). We conclude that lesions, TMS, and DBS converge on common brain circuitry that may represent improved neurostimulation targets for depression and other disorders.
Introduction
Causal neuroanatomy can be mapped in animal models by precisely modulating different brain circuits in well-controlled experiments1,2. However, it can be challenging to translate these findings into human therapeutics3,4. In humans, mapping of psychiatric symptoms is based primarily on correlation, resulting in a “causality” gap when attempting to translate this information into effective treatments. Causality may be inferred in humans based on the clinical effects of focal brain lesions, transcranial magnetic stimulation (TMS), and deep brain stimulation (DBS)2. These modalities have each been used to link depression symptoms to specific brain circuits based on the location of lesions or stimulation sites that affect depression severity2,5–9. Each result has been proposed as a potential solution to the causality gap between neuroimaging correlates and effective treatments2,10.
It remains unclear whether these three causal sources of information converge on the same circuit or therapeutic target2,11,12. Heterogeneity in lesion location, stimulation site location, neuromodulation modality, patient population, depression symptoms, depression subtypes, and numerous other factors argue against a common neuroanatomical substrate. If these causal sources of information converge on a similar brain circuit despite this heterogeneity, this would have implications for localization and treatment of depression and for bridging the causality gap more generally2. For example, it has been proposed that TMS and DBS sites connected to similar circuits may modulate similar symptoms13, lesions causing a symptom may be connected to the same circuit as brain stimulation targets that relieve that symptom5, and similar symptoms map to similar circuits across different diagnoses6,14. Confirmation of these hypotheses may lead to a transformative framework for targeting brain stimulation treatments2,12.
To address these questions, we analyzed 14 independent datasets of patients with brain lesions, TMS, or DBS. Each dataset included variability in the lesion or stimulation locations and variability in depression symptoms, measured after the lesion or before and after therapeutic brain stimulation. We also extended this approach to three additional datasets of patients with brain lesions or DBS sites associated with motor symptoms of Parkinson’s disease (PD). The brain regions functionally connected to each location were identified using a normative connectome database. This method identifies a polysynaptic brain circuit underlying each location, allowing one to test whether lesions or stimulation sites in different brain regions intersect the same population-derived circuit5. We test 1) whether TMS sites and DBS sites that affect depression are connected to the same brain circuit, 2) whether lesion locations associated with depression and stimulation sites that affect depression are connected to the same brain circuit, 3) whether this circuit is associated with depression severity irrespective of baseline diagnosis, and 4) whether this approach is relevant beyond depression.
Results
Characteristics of included datasets
We identified 14 datasets including 461 lesions (Fig. 1a)15, 151 TMS sites (Fig. 1b)8,16–18, and 101 DBS sites (Fig. 1c)9,19–23 (Supplementary Table 1). Five datasets included patients who were evaluated for depression severity after penetrating brain injury, ischemic stroke, or hemorrhagic stroke. Seven datasets included patients who were treated for primary MDD with either TMS (four datasets) or DBS (three datasets). Finally, two datasets included patients receiving DBS for other disorders (PD or epilepsy), but which measured change in depressive symptoms as potential side effect.
Figure 1: Lesion locations and brain stimulation sites across 14 datasets.
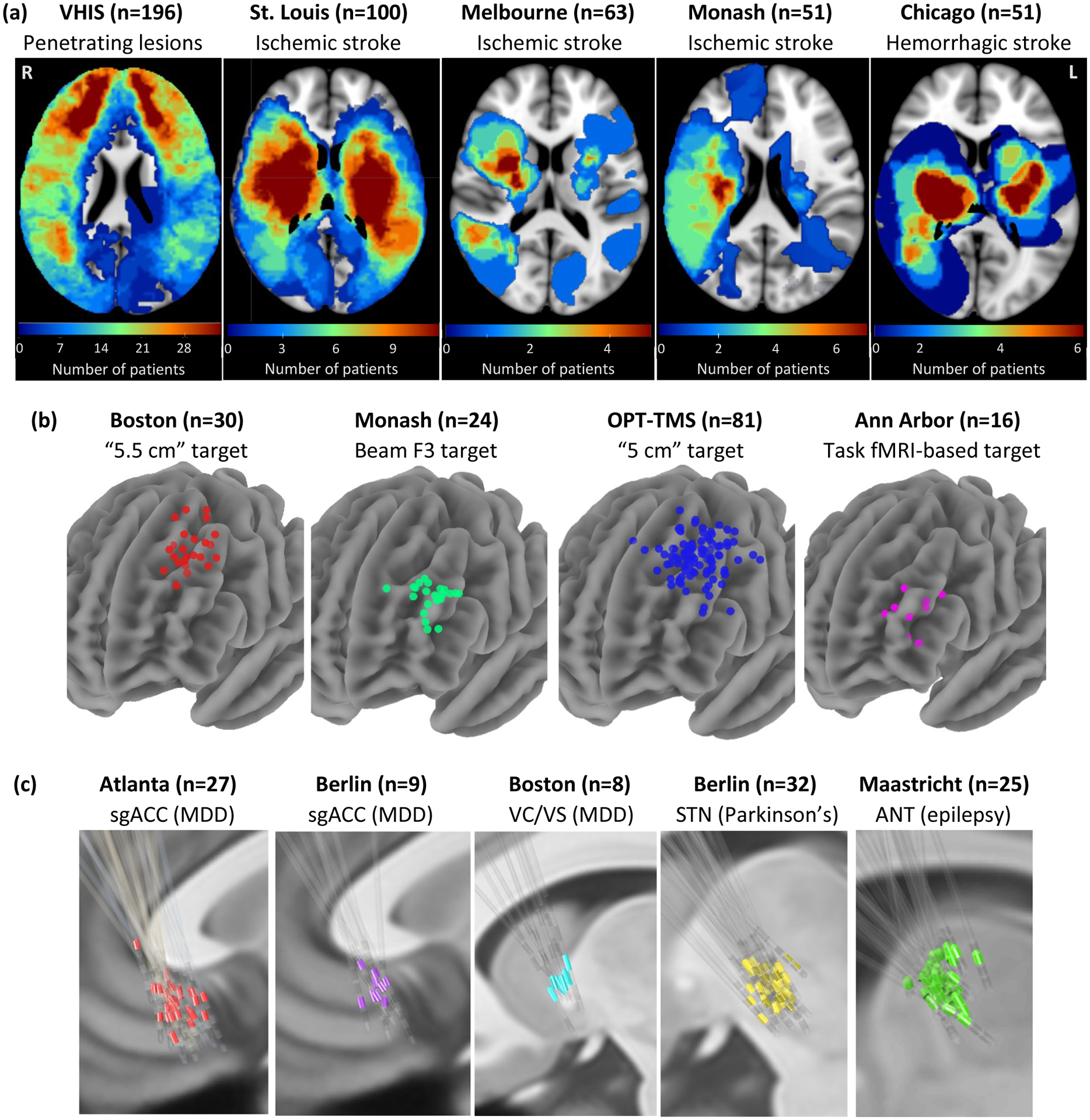
Our analysis included: (a) 461 brain lesions across five datasets and three different diagnoses. (b) 151 TMS sites across four datasets, one diagnosis (major depressive disorder), and four different TMS targets. (c) 101 DBS sites across five datasets, three different diagnoses, and four different DBS targets. VHIS = Vietnam Head Injury Study, OPT-TMS = Optimizing TMS for the Treatment of Depression Study, sgACC = subgenual anterior cingulate cortex, MDD = major depressive disorder, VC/VS = ventral capsule/ventral striatum, STN = subthalamic nucleus, ANT = anterior nucleus of the thalamus.
Similar “depression circuits” across 14 independent datasets
The location of each lesion or brain stimulation site (Fig. 2a–c, top panels) was mapped to an underlying brain circuit using a large normative connectome database (n=1000) and previously validated methods (Fig. 2a–c, bottom panels)5. The normative connectome was used to estimate connectivity of each lesion or stimulation site to every voxel in the brain. At each voxel, a Pearson r-value was computed for the correlation between depression score and lesion or stimulation site connectivity to that voxel (Fig. 2a–c, right panels), yielding a population-derived “circuit map” for each of the 14 datasets (Supplementary Figure 1).
Figure 2: Identifying depression circuit maps for each cohort.
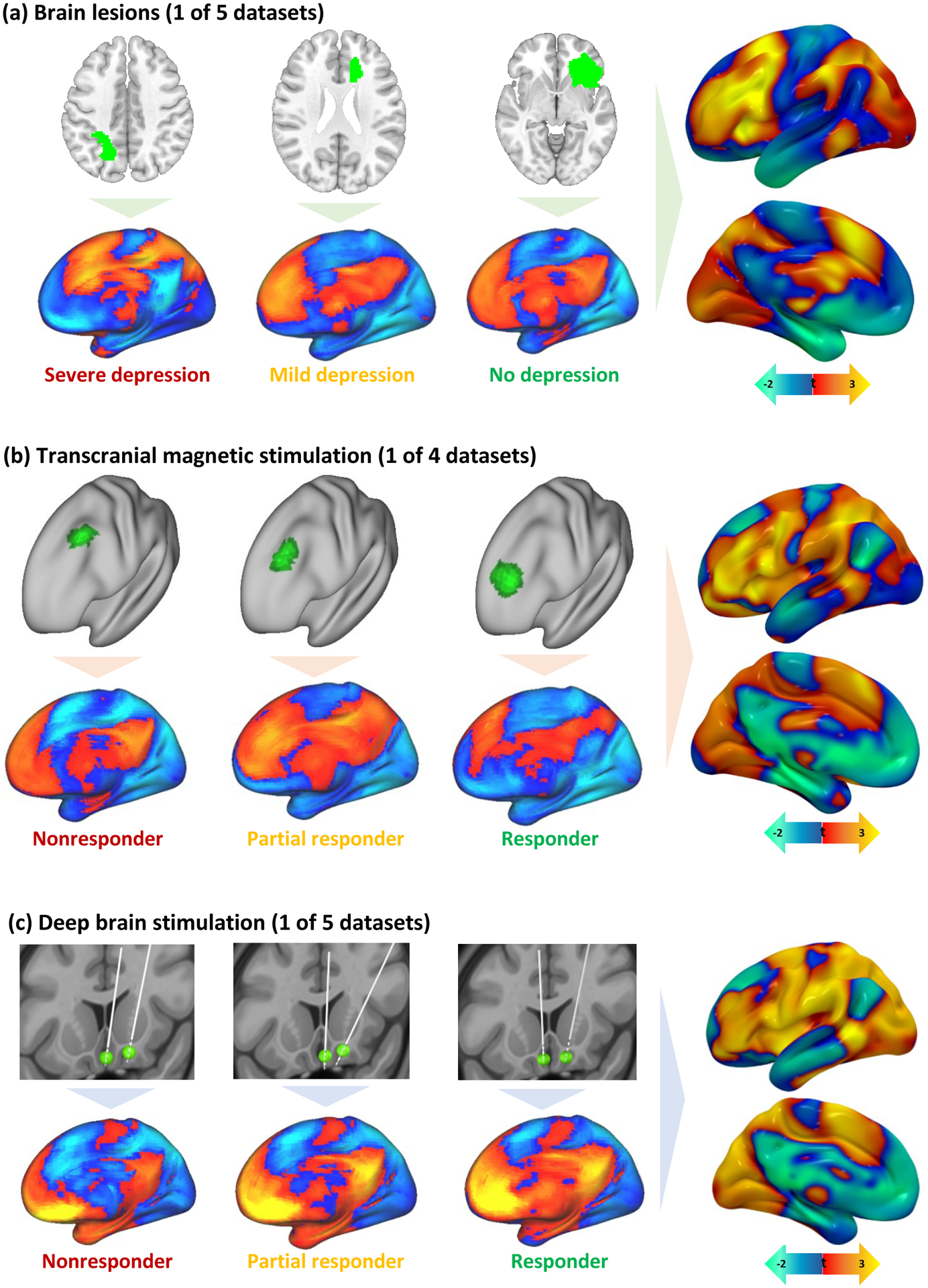
Brain lesions (a), TMS sites (b), and DBS sites (c) were all mapped to a common brain atlas (top row in each panel). Functional connectivity of each lesion location or stimulation site was computed using a normative connectome database (bottom row in each panel). Positive functional connectivity is shown in warm colors and negative functional connectivity in cool colors. Connections most associated with depression score (lesion datasets) or change in depression score (brain stimulation datasets) were identified for each dataset (right column in each panel). The color scale was inverted for TMS datasets because TMS sites that improve depression are thought to be anti-correlated to DBS sites that improve depression or lesion sites associated with lower risk of depression.
Cross-dataset similarity was assessed by computing the spatial correlation between each pair of circuit maps (e.g. dataset 1 vs dataset 2) and by comparing each circuit map to a combined map from the other 13 datasets. Significance was assessed using permutation testing, in which the spatial correlation was recomputed after randomly pairing each patient’s lesion or stimulation site with a different patient’s depression score within the same dataset6. The average pairwise similarity between circuit maps, weighted by sample size, was higher than expected by chance (mean spatial r=0.24, 95%CI 0.19–0.29, p<0.001) (Fig. 3a, S2a) and similar to a weighted mean map generated from the other 13 datasets (mean spatial r=0.45, 95%CI 0.33–0.57, p<0.001). Results were unchanged when using Kendall tau (p<0.001) or Euclidean distance (p=0.0013) instead of Pearson correlation or when including lesion size as a covariate.
Figure 3: Depression circuit maps are similar across 14 datasets (n=713).
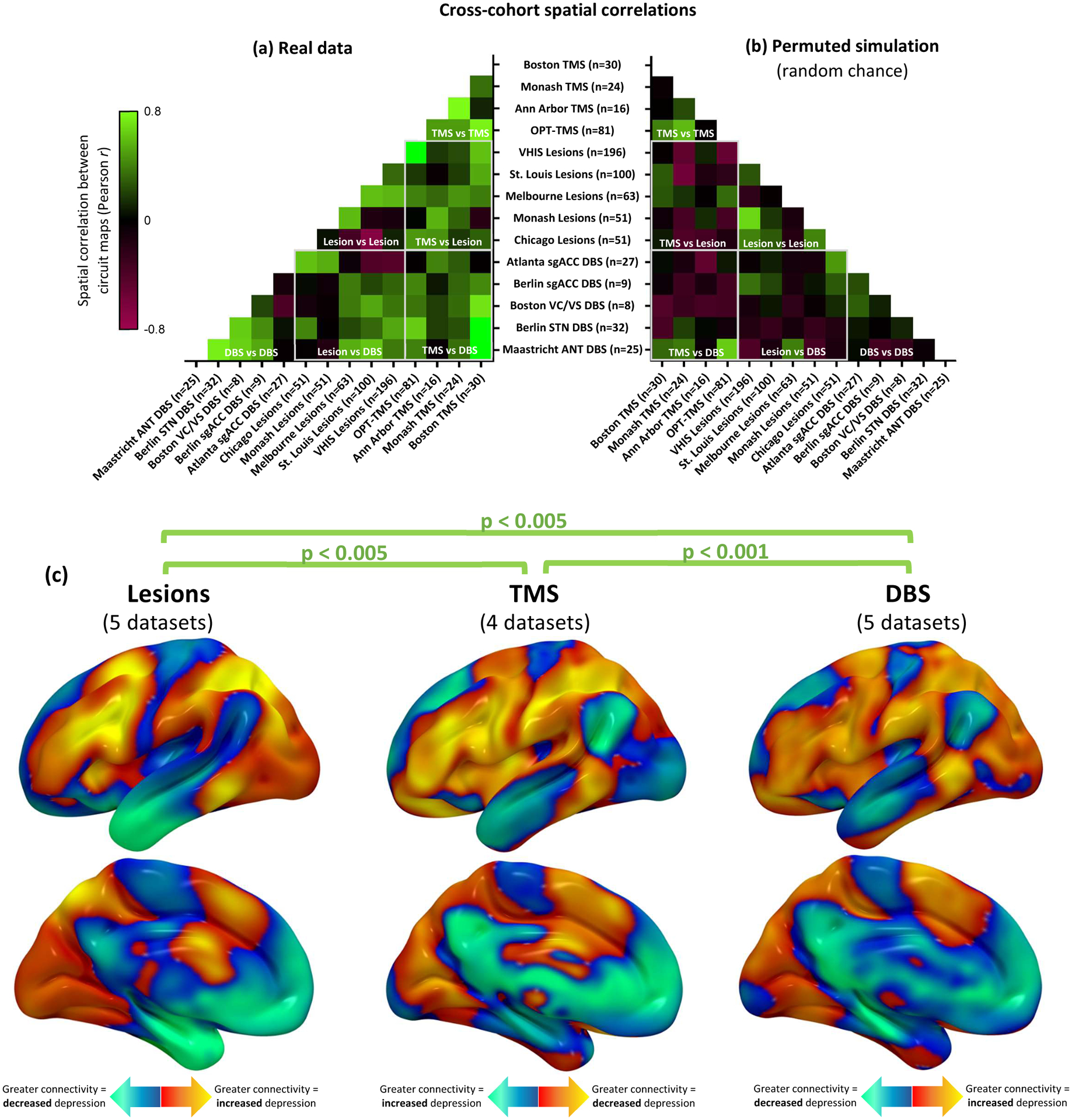
(a) The 14 circuit maps were consistently similar to one another (mean r=0.24, 95%CI 0.19–0.29), as depicted in this cross-correlogram comparing different datasets. Permutation testing confirmed that the weighted mean cross-correlation was significantly stronger than expected by chance (p<0.001, 10,000 permutations) Green colors represent high spatial correlation between circuit maps, black boxes represent neutral correlation, and red boxes represent negative correlation. (b) Representative example of correlation between circuit maps generated from randomly-permuted data. This analysis confirmed that no overall cross-correlation is expected by chance (mean r = 0.00, 95%CI −0.01–0.01). (c) Depression circuit maps were similar between lesion datasets (n=461), TMS datasets (n=151), and DBS datasets (n=101). Permutation testing confirmed that each comparison was significantly stronger than expected by chance (p<0.005, 10,000 permutations). For display purposes, depression circuit maps were averaged (weighted mean) across datasets within each modality. The color scale on TMS circuit maps is inverted to facilitate visual comparison to lesion and DBS circuit maps.
To rule out methodological bias, we conducted a control analysis using patient age instead of depression scores. Age is presumably unrelated to stimulation or lesion location, so we hypothesized that this analysis would yield significantly weaker cross-dataset similarity. Indeed, the 14 control maps did not match one another (mean spatial r= −0.02, 95%CI −0.09–0.05, p=0.86, BF01=1.01) and did not match a map generated from the other 13 datasets (mean spatial r=−0.01, 95%CI −0.14–0.11, BF01=1.001). The control maps did not match the depression circuit maps (mean spatial r= −0.05, 95%CI −0.12–0.02, p=0.93, BF01=1.003). Similarity between control maps was significantly weaker than similarity between depression circuit maps (p=0.0023).
Convergence across brain lesions, TMS, and DBS
To determine whether lesions, TMS, and DBS converge on the same circuit, we grouped the different datasets according to modality. Depression circuit maps derived from brain lesion datasets were similar to circuit maps derived from TMS datasets (mean spatial r=0.28, 95%CI 0.17–0.39, p=0.0025), DBS datasets (mean spatial r=0.19, 95%CI 0.10–0.28, p=0.0037), or both neuromodulation modalities combined (mean spatial r=0.25, 95%CI 0.18–0.32, p<0.001)(Fig. 3, Supplementary Figure 2a). Depression circuit maps derived from TMS were similar to those derived from DBS (mean spatial r=0.25, 95%CI 0.11–0.39, p<0.001) (Fig. 3, Supplementary Figure 2a).
As a control, this analysis was also repeated using patient age instead of depression score. We hypothesized that this analysis would yield significnatly weaker cross-dataset spatial correlation. Age-based circuit maps derived from brain lesions were not similar to those derived from TMS (mean spatial r=−0.04, 95%CI −0.17–0.09, p=0.70, BF01=1.07), DBS (mean spatial r= −0.14, 95%CI −0.26– −0.02, p=0.97, BF01=6.8), or both neuromodulation modalities combined (mean spatial r= −0.07, 95%CI −0.17–0.02, BF01=3.4). Control maps derived from TMS were not similar to those derived from DBS (mean spatial r= 0.01, 95%CI −0.14–0.16, p=0.43, BF01=0.99). In all cases, similarity between control maps was significantly weaker than similarity between depression circuit maps (p=0.0038). Control maps from neuromodulation datasets did not match depression circuit maps from lesion datasets (mean spatial r= −0.11, 95%CI −0.19– −0.03, BF01=16.9). Control maps from lesion datasets also did not significantly match depression circuit maps from neuromodulation datasets (mean spatial r= 0.07, 95%CI −0.02–0.17), although Bayesian analysis indicates moderate evidence for a correlation (BF01=0.29) (Fig. 4a).
Figure 4: Depression circuit maps are similar across lesions, neuromodulation, and diagnoses.
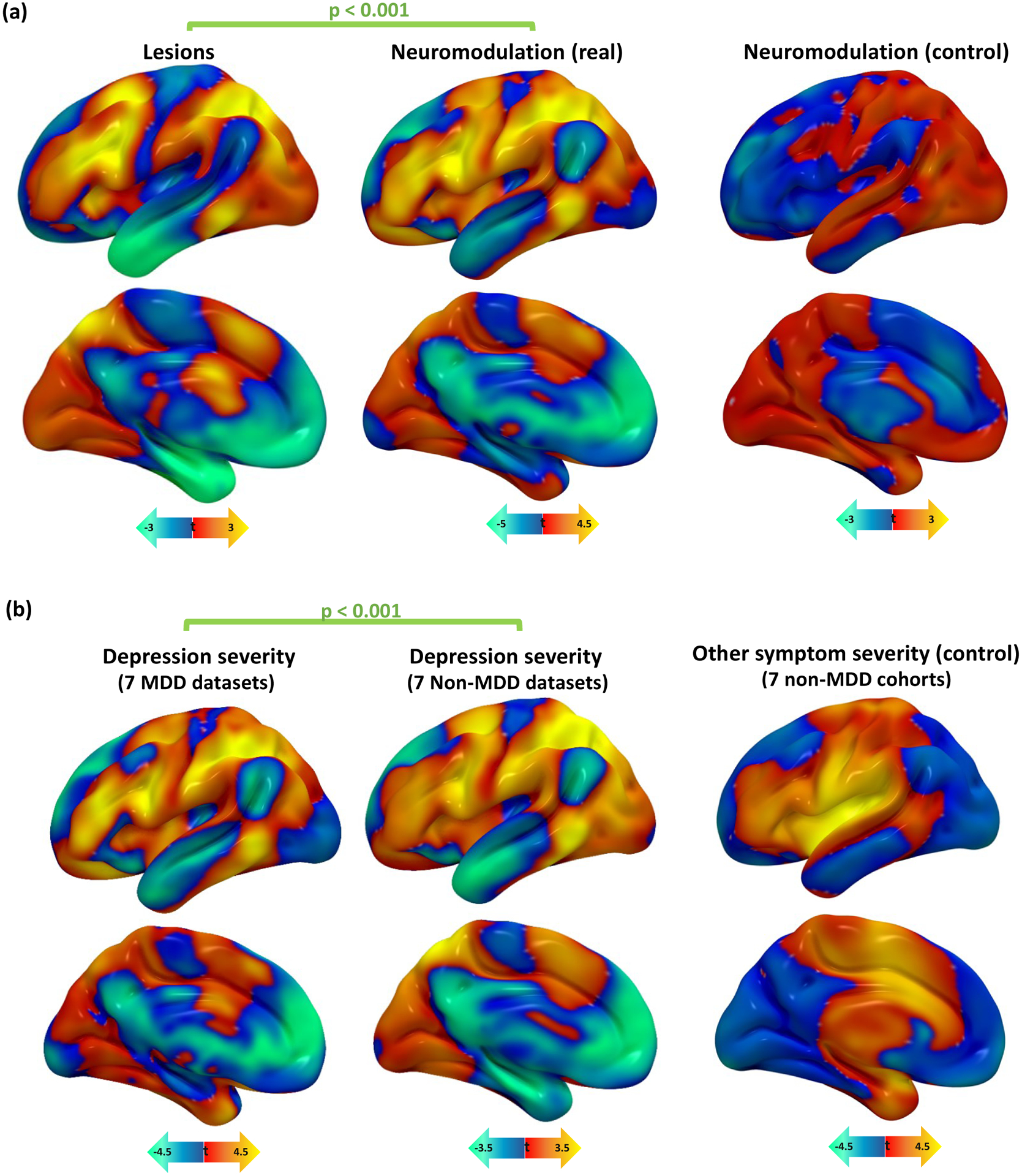
(a) Depression circuit maps were similar between lesion datasets and neuromodulation datasets (mean r=0.25, 95%CI 0.16–0.34). Permutation testing confirmed that this similarity was stronger than expected by chance (p<0.001, 10,000 permutations). In a control analysis, there was no similarity between depression circuit maps from lesion datasets and age-based circuit maps from neuromodulation datasets (r= −0.11, 95%CI −0.21 – −0.01, p=0.93).
(b) Depression circuit maps were similar between MDD patients and non-MDD patients (mean r=0.26, 95%CI 0.16–0.36, p<0.001). Permutation testing confirmed that this similarity was stronger than expected by chance (p<0.001, 10,000 permutations). In a control analysis, there was no similarity between depression circuit maps from MDD datasets and “other symptom severity” circuit maps in non-MDD datasets (r= −0.03, 95%CI −0.12–0.06, p=0.77).
MDD = major depressive disorder.
Finally, we assessed whether within-modality similarity of our depression circuit maps was stronger than between-modality similarity. We compared each depression circuit map to a combined map generated from the remaining datasets within a modality (e.g. TMS dataset 1 vs three other TMS datasets) or between different modalities (e.g. TMS dataset 1 vs nine DBS/lesion datasets). Within-modality similarity (spatial r=0.46) was idental to between-modality similarity (spatial r=0.46). We also repeated this analysis using pairwise comparisons between circuit maps, which yielded a similar result (spatial r=0.24 vs r=0.25, respectively).
The circuit is transdiagnostic, but specific to depression
We compared depression circuit maps derived from datasets of patients with MDD (7 datasets, n=199) to those derived from datasets of patients with other diagnoses such as stroke, penetrating head trauma, PD, and epilepsy (7 datasets, n=518). Depression circuit maps derived from MDD datasets were similar to depression circuit maps derived from patients without MDD (mean spatial r=0.26, 95%CI 0.19–0.33, p<0.001) (Fig. 4b, Supplementary Figure 2).
To assess whether this result was driven by overall clinical severity/disability rather than depression, this analysis was repeated using the severity of the primary presenting symptom in non-MDD datasets. This control analysis included stroke severity, PD motor improvement, or seizure frequency improvement. Control circuit maps from non-MDD datasets failed to match depression circuit maps from MDD datasets (mean spatial r= −0.03, 95%CI −0.09–0.03, BF01=1.04), and this spatial cross-correlation was significantly weaker than the cross-correlation between the depression circuit maps used in our primary analysis (p<0.001) (Fig. 4b).
To assess specificity to depression versus other cognitive or emotional symptoms, we generated control circuit maps using 34 other cognitive/emotional scores, which were available in our two largest datasets (VHIS and St. Louis). Our leave-one-dataset-out depression circuit map (generated from the other 13 datasets) was more similar to the VHIS depression circuit map than to the 28 control circuit maps (r=0.54 versus r<0.35) (Supplementary Figure 3a). Our leave-one-dataset-out depression circuit map was also more similar to the St. Louis depression circuit map than to the 6 control circuit maps (r=0.39 versus r<0.23) (Supplementary Figure 3b). Across both datasets, the leave-one-dataset-out maps were significantly more similar to the depression circuit maps than to the other circuit maps (p=0.0032).
Combining all datasets and explaining clinical variance
We generated a combined depression circuit map by taking the mean of all 14 circuit maps, weighted by the sample size of each dataset (Fig. 5a). Peak regions in this combined map include the intraparietal sulcus, dorsolateral prefrontal cortex, inferior frontal gyrus, ventromedial prefrontal cortex, and subgenual cingulate cortex (Supplementary Table 2). Compared to a consensus brain network parcellation24, our circuit was most similar to the dorsal attention network and frontoparietal control network, and was most anti-correlated to the default mode network and limbic network (Supplementary Figure 4).
Figure 5: Combining all circuit maps and predicting clinical variance.
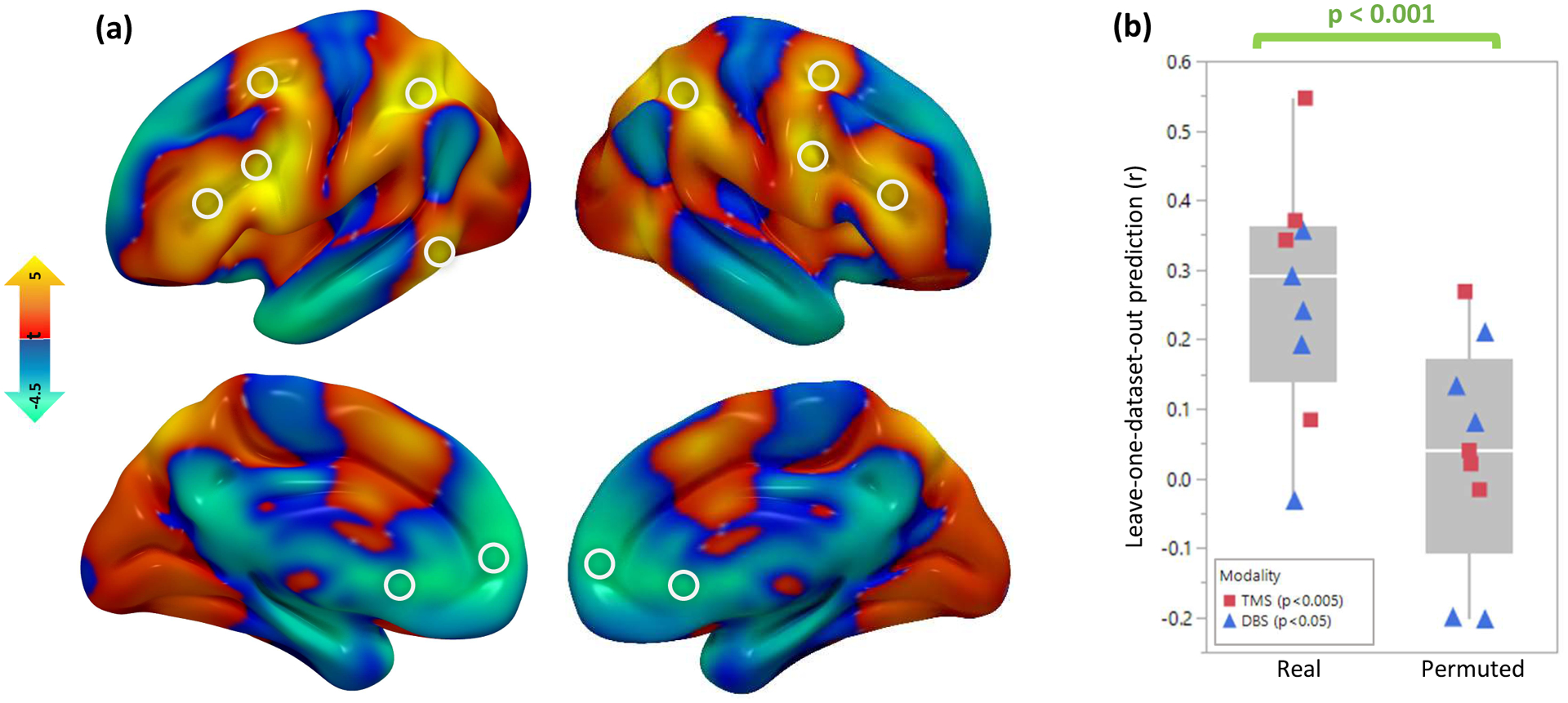
(a) A combined “depression circuit” was generated from all 14 datasets. Peaks in this circuit are depicted with white circles. Positive peaks included the dorsolateral prefrontal cortex, frontal eye fields, inferior frontal gyrus, intraparietal sulcus, and extrastriate visual cortex. Negative peaks included the subgenual cingulate cortex and ventromedial prefrontal cortex. Peaks are listed in Supplementary Table 2. (b) Across the 9 neuromodulation cohorts (n=252), antidepressant efficacy was predicted by stimulation site connectivity to a circuit generated from the remaining 13 cohorts (mean r=0.22). Permutation testing confirmed that this similarity was stronger than expected by chance (p<0.001, 10,000 permutations). This was true for both TMS (n=151, r=0.24, p=0.0034 with 10,000 permutations) and DBS (n=101, r=0.21, p=0.033 with 10,000 permutations). Line = median, box limits = interquartile range, whiskers = outliers, points = individual correlation value for each neuromodulation dataset.
In a leave-one-dataset-out analysis, we assessed whether connectivity of the stimulaton site to our depression circuit could predict depression outcomes after TMS and DBS. In each neuromodulation dataset, each patient’s stimulation site connectivity profile was compared to a circuit map generated from the remaining 13 datasets using spatial correlations. Across all neuromodulation datasets, connectivity to our circuit predicted the efficacy of treatment targets (weighted mean r=0.22, 95%CI 0.11–0.33 p<0.001) (Fig. 5b). The leave-one-dataset-out circuit independently predicted clinical variance in TMS datasets (weighted mean r=0.24, p=0.0034) and DBS datasets (weighted mean r=0.21, p=0.033).
Comparison to prior established methods
We hypothesized that our mapping and targeting approach would outperform established methods for both causal brain mapping and neuromodulation targeting. First, we repeated the primary analysis using voxel-lesion symptom mapping (VLSM), a tool that is widely used to localize behaviors using lesions25. Similar approaches have also been applied to TMS16 and DBS26. VLSM failed to detect significant similarity across all 14 datasets (mean spatial r= −0.03, p=0.91, BF01=1.001).
Next, we compared our approach to existing approaches for connectivity-based neuromodulation targeting. For each TMS and DBS site, we computed connectivity to the subgenual cingulate cortex, which has been shown to predict TMS response8,18 and has been used as a DBS target27. Indeed, antidepressant efficacy of each stimulation site was correlated with its connectivity to the subgenual cingulate (weighted mean r= −0.13, 95%CI −0.24– −0.02, p=0.039). Connectivity to our leave-one-dataset-out depression circuit predicted outcomes (weighted mean r=0.22, 95%CI 0.11–0.33, p<0.001) significantly better than connectivity to the subgenual cingulate (p=0.012).
Generalizability of the method beyond depression
To demonstrate that this approach can generalize to other neuropsychiatric disorders, we also repeated the analysis using previously-published data on motor symptoms of PD, the most common clinical indication for DBS. This included 29 case reports of lesion-induced parkinsonism28, 95 patients (two datasets) who received DBS for PD28, and one TMS site (primary motor cortex, hand knob) which has demonstrated efficacy for PD in a meta-analysis of 10 randomized trials29.
The PD circuit derived from lesions was similar to the PD circuit derived from DBS (p=0.01) (Supplementary Figure 5). Connectivity to the motor cortex TMS target predicted change in PD motor symptoms with DBS (p=0.02) and risk of parkinsonism after a brain lesion (p=0.0005) (Supplementary Figure 5). In a leave-one-dataset-out analysis, the PD circuit predicted motor improvement with DBS (r=0.26, p=0.01).
To confirm specificity, we used the PD circuit as a control for depression and vice versa. Connectivity to the PD circuit was independently predictive of motor improvement (p=0.0003) after controlling for connectivity to the depression circuit. Connectivity to the depression circuit was independently predictive of mood improvement (p=0.02) after controlling for connectivity to the PD circuit. By itself, the depression circuit did not significantly predict motor improvement with DBS (r= −0.06, p=0.58, BF01=3.4). The PD circuit also did not significantly predict depression improvement with TMS and DBS (r=0.06, p=0.32), although Bayesian analysis indicates moderate evidence for a correlation (BF01=0.29).
Discussion
Across 14 independent datasets, we found that mapping depression based on brain lesions, TMS sites, and DBS sites converged on a common neuroanatomical substrate. This convergence was robust despite many sources of heterogeneity that should bias us against a common substrate, including different lesion distributions, lesion etiologies, stimulation targets, stimulation modalities, and neuropsychiatric diagnoses. Our convergent circuit includes regions previously implicated in depression such as the subgenual cingulate, ventromedial prefrontal cortex, and dorsolateral prefrontal cortex30–34. However, our different datasets converged on a common brain circuit or brain network, not an individual brain region. The circuit was consistent with prior work on large-scale brain networks in depression, as it is similar to the dorsal attention network and the frontoparietal control network, and anti-correlated with the default mode network and limbic network35. This neuroanatomical convergence has several important implications.
First, TMS sites and DBS sites that modulate depression were connected to a similar circuit. To our knowledge, this is the strongest evidence to date that invasive and noninvasive brain stimulation are targeting the same circuit to treat the same symptom12,13. Given recent negative trials of DBS20,36 and TMS37 for depression, our circuit may serve as a refined therapeutic target to improve neuromodulation outcomes in future trials. More broadly, this finding supports the use of circuit mapping to define neuromodulation targets6,8,9 and translate therapy between stimulation modalities for various neuropsychiatric disorders13. Furthermore, our findings support the notion that high-frequency TMS and high-frequency DBS modulate brain circuits in opposite directions13, as the TMS and DBS maps were inverted with respect to each other.
Second, lesion locations associated with depression and stimulation sites that modulate depression were connected to a similar circuit. This finding generalized to Parkinson’s disease – lesion locations associated with parkinsonism and stimulation sites that modulate parkinsonism were connected to a similar circuit, which was distinct from our depression circuit. To our knowledge, this is the strongest evidence to date showing that lesions causing a symptom can identify therapeutic targets for symptom relief. Given that lesion network mapping has been used to map a broad range of neuropsychiatric symptoms, from amnesia to criminality5, our approach may have therapeutic implications well beyond depression and Parkinson disease.
Third, we identified similar depression circuits in patients with MDD, penetrating brain injury, stroke, epilepsy, and PD. This suggests that depression symptoms map to a similar neuroanatomical substrate independent of whether the symptoms are caused by a primary psychiatric disorder, a structural brain lesion, or a side effect of DBS. This finding is consistent with the recent Research Domain Criteria initiative, which seeks to establish transdiagnostic constructs for psychiatric symptom severity38. Our findings were also specific to depression relative to other neuropsychiatric symptoms, but further work is needed to conclusively confirm specificity.
Fourth, our findings were consistent across 14 independent datasets. Most prior studies in depression have focused on a single dataset30–34, although larger studies are beginning to appear14. Meta-analyses often find poor consistency in neuroimaging correlates of depression33,34. To our knowledge, our consistency across 14 datasets, including a leave-one-dataset-out analysis, is one of the strongest demonstrations of result consistency for a psychiatric condition. Furthermore, the results survived rigorous permutation-based statistical testing, a highly conservative approach that prevents Type I error due to multiple comparisons or a biased analysis.
Fifth, it is worth highlighting our focus on “causal” sources of information such as lesions and brain stimulation. This resolves some of the interpretive ambiguity associated with neuroimaging correlates of depressive symptoms or antidepressant efficacy of non-anatomically-targeted treatments39. By combining brain lesions and brain stimulation, this study moves us towards the goal of “mapping causal circuitry in human depression2,” potentially facilitating more direct translation to targeted therapeutics.
Finally, our parsimonious mapping and targeting model outperformed established approaches for both lesion-based brain mapping and connectivity-based neuromodulation targeting. Our approach identified relationships that were not apparent using VLSM, illustrating the potential of brain connectivity to detect trends beyond what is possible using anatomical location alone. Our approach also explained more clinical variance than subgenual connectivity, which is widely used to target neuromodulation40–44.
Our analysis may seem circular or biased given that the TMS and DBS sites for MDD were chosen because they were already known to be part of a “depression circuit.” However, our depression circuit was derived from the variance across stimulation sites within each target, not simply the location of the intended target. For example, the left prefrontal cortex appears as part of our depression circuit not because it was targeted with TMS, but because different TMS sites across the left prefrontal cortex produced different effects on depression, different DBS sites produced different effects on depression symptoms depending on their connectivity to the left prefrontal cortex, and different lesion locations were associated with different amounts of depression depending on their connectivity to left prefrontal cortex. It is also worth noting that this concern is not relevant for lesions, which were randomly distributed throughout the brain, yet identified a depression circuit that was very similar to the circuit identified from TMS or DBS sites.
There are several limitations. First, this analysis was retrospective, taking advantage of existing datasets with heterogeneous populations and outcome metrics, limiting the amount of variance that can be explained. Prospective validation is required to confirm whether targeting our circuit results in improved antidepressant response. Second, most datasets only included a single depression score without subscales, which may also limit the amount of variance that can be explained. Given that different symptom clusters respond to stimulation of different circuits with TMS6, future work with more detailed phenotyping may enable further subclassification. Third, we used a normative functional connectome for all circuit mapping, as prior work suggests that using a disease-matched connectome makes little difference for either depression or Parkinson’s disease6,8. However, this analysis could be repeated using connectomes that are age, gender, and disease matched to each dataset. Similarly, this analysis could be repeated using measures of structural white matter connectivity or individualized functional connectivity9,18,45. Individualized connectivity may explain additional variance, but adds additional noise to the analysis46. Individualized neurostimulation-induced electric field modeling may also be valuable, but prior work has shown it to yield similar functional connectivity estimates to our simplified model47.
In conclusion, these results support the existence of at least one neuroanatomical substrate for depression symptoms. More broadly, by combining lesion locations, noninvasive stimulation sites, and invasive stimulation sites, we introduce a method for identifying a convergent neuroanatomical substrate for neurological and psychiatric symptoms. Future work should seek to prospectively determine whether this convergent substrate provides an improved target for neuromodulation therapies.
Methods
Characteristics of included datasets
We sought out multiple datasets that included MRI or CT of focal brain lesions and stimulation sites. Lesions and stimulation sites showed incidentally variable locations in different patients. Localization methods are described in the supplement. All depression datasets included continuous scores on a validated depression metric. All PD datasets included either a clear case description of lesion-induced parkinsonism or continuous scores on the Unified Parkinson’s Disease Rating Scale (UPDRS). In each dataset, participants provided informed consent to data collection or the institutional review board approved retrospective analysis of symptom and imaging data.
Patients with missing data were excluded from the analysis. To avoid bias due to unequal variances, unequal sample sizes, or inconsistent severity cut-offs for different datasets, each dataset was analyzed independently. Study characteristics are summarized in Supplementary Table 1.
No statistical methods were used to pre-determine sample size, but our sample sizes are larger than the largest prior studies of lesions7, TMS sites6, or DBS sites21 in depression.
Generation of circuit maps
A normative human connectome database was used to compute mean resting-state functional connectivity of each patient’s lesion or stimulation site based on 1000 healthy subjects, as previously described5–7. This yielded a whole-brain connectivity map of each patient’s lesion or stimulation site (Figure 2).
In the TMS and DBS datasets with depression outcomes, these connectivity maps were compared with change in depression score using partial Pearson correlation at each voxel, controlling for pre-treatment depression severity. In the lesion datasets with depression outcomes, connectivity maps were compared with overall depression scores using Pearson correlation at each voxel. For each dataset, this analysis yielded a whole-brain “circuit map” of connections correlated with antidepressant efficacy (for TMS and DBS) or depression severity (for lesions). TMS-based circuit maps were multiplied by −1 because TMS sites that improve depression are thought to be anti-correlated to DBS sites that improve depression13 or lesion sites associated with lower risk of depression5,7. Inverting the circuit maps for TMS also facilitates visual comparison across all three modalities (see figure 2).
In the PD DBS datasets, patient-specific connectivity maps were compared with change in UPDRS score. The connectivity of lesions causing parkinsonism was estimated using a one-sample t-test at each voxel. For each dataset, this yielded a whole-brain circuit map of connections associated with parkinsonism. In the absence of individualized TMS sites, we generated a group-mean region of interest at the M1 hand knob (MNI coordinates [−40, −20, 62]), which has been shown to be the most effective TMS target for Parkinson’s disease29.
We generated control circuit maps using two different approaches. For all datasets, control maps were generated using patient age, which is presumably unrelated to stimulation site or lesion location, rather than depression scores. For all non-MDD datasets, additional control maps were generated using severity of the primary presenting symptom, including NIH Stroke Scale (stroke patients), Neurobehavioral Rating Scale (penetrating brain injury patients), UPDRS (Parkinson’s Disease patients), and seizure frequency (epilepsy patients).
Computational and statistical methods
All computational/statistical analyses were conducted using customized MATLAB scripts, except as otherwise specified. All correlation coefficients were Fisher-transformed before further analysis. To facilitate comparison across datasets with different sample sizes, voxel-wise Fisher z-values were converted to t-values. All parametric p-values were computed using a two-tailed hypothesis test. Similarity between different maps was assessed using spatial correlations.
To confirm similarity across different datasets, we computed the mean spatial cross-correlation between the circuit maps in each analysis. Because the datasets were collected in highly heterogeneous settings, they could not be assumed to have identical distributions. To address this, statistical significance was addressed using a non-parametric multi-level block permutation testing approach. In this permutation test, the mean spatial correlation was re-computed 25,000 times in simulated data. The null distribution of this permutation test was defined by randomly re-assigning each patient’s connectivity map with a different patient’s clinical variables within the same dataset. A p-value was defined as the percentage of randomly-permuted results that were stronger than the real result, as in prior work6.
For null findings, the resulting t-values were used to compute Bayes factors, which were used to compare likelihood of the null hypothesis to the likelihood of the alternative hypothesis48. In the case of spatial correlations, the null hypothesis was that there is no similarity between the two maps in question. Thus, for the purpose of calculating Bayes factors, stronger positive correlations were considered to support the alternative hypothesis, while weaker positive correlations and negative correlations were considered to support the null hypothesis49.
Combining and comparing circuit maps
The 14 circuit maps were then categorized to assess for similarity between different modalities or diagnoses. Categories included TMS, DBS, neuromodulation (TMS and DBS combined), lesions, MDD (all modalities), and non-MDD (all modalities). MDD and non-MDD datasets were defined according to the inclusion criteria of the original study. We hypothesized that (1) TMS, DBS, and lesion datasets would yield similar circuits, (2) lesions and neuromodulation would yield similar circuits, and (3) MDD and non-MDD patients would yield similar circuits. To statistically compare different categories, we computed the mean spatial cross-correlation of all circuit maps in one category with all circuit maps in the other category. Significance was assessed using permutation testing as above.
To visualize the map for each category, circuit maps from different datasets were combined into a mean circuit map across all voxels, weighted by the sample size of each dataset. This weighted mean approach was chosen over a combined linear model because it maintains independence between datasets, thus reducing the statistical penalty associated with combining heterogeneous datasets50.
Each dataset’s circuit map was also compared with a “leave-one-dataset-out” circuit map generated by taking the weighted mean of the other 13 circuit maps. This yielded a leave-one-dataset-out spatial correlation for each dataset. The weighted mean of these spatial correlations was considered to represent the overall similarity between each circuit map and the remaining circuit maps. This value was assessed for significance using permutation testing as above.
Assessing specificity to depression
To confirm that the results were not driven by overall clinical severity, we repeated the analysis using the control circuit maps generated from severity of non-depressive symptoms in non-MDD datasets. Using the same statistical methods described above, we hypothesized that (1) the control circuit maps would not be significantly similar between different datasets, modalities, or diagnoses, and (2) the control circuit maps would not significantly match the depression circuit maps. We also hypothesized that the spatial cross-correlation between depression circuit maps would be significantly stronger than the spatial cross-correlation between control circuit maps using a paired t-test.
To assess specificity to depression, we then generated symptom-specific circuit maps based on other cognitive/emotional scales, which were available in our two largest datasets. In the VHIS dataset (n=196), we generated 28 circuit maps based on the Mini Mental State Examination and each of the 27 symptoms measured by the neurobehavioral rating scale. In the St. Louis dataset (n=100), we generated six circuit maps based on the Boston Naming Test, animal naming test (verbal fluency), Hopkins Verbal Learning Test (learning/memory), Brief Visuospatial Memory Test (visual memory), clock draw test (visuospatial skills), and spatial span test (attention). In each dataset, we used spatial correlations to compare the symptom-specific maps to a leave-one-dataset-out depression map generated from the other 13 datasets. We hypothesized that the leave-one-dataset-out depression maps would be more similar to each dataset’s depression map than to its other symptom-specific maps.
To test for significance, we regenerated these cognitive/emotional circuit maps 25,000 times after randomly permuting each patient’s clinical outcomes with a different patient’s neuroimaging results. We again used spatial correlation to compare each of these maps to a leave-one-dataset-out depression map. We averaged the resulting Fisher-transformed spatial correlations, yielding a null distribution of 25,000 spatial correlation values expected by random chance. We computed a p-value as the percentage of these values that exceeded the weighted mean correlation between the leave-one-dataset-out map and each dataset’s depression circuit map.
Explaining clinical variance
For each neuromodulation dataset, treatment-induced change in depression score was predicted using a leave-one-dataset-out map constructed from the other 13 datasets. Within each dataset, spatial correlations were computed between each patient’s stimulation site connectivity profile and the leave-one-dataset-out map. This yielded a metric representing the similarity between the patient’s stimulation site connectivity and the “ideal” stimulation site connectivity. In each dataset, this similarity metric was compared with improvement in depression score using partial Pearson correlation, controlling for baseline depression severity. Across all datasets, these correlations were combined into a single weighted mean value representing the degree to which our circuit predicted neuromodulation outcomes across all datasets. Significance was assessed using permutation testing as above.
Finally, a combined depression circuit map was generated based on the weighted mean of all 14 datasets. Peaks in this circuit map were identified using the FSL “cluster” algorithm with a detection threshold of p<0.00005 and minimum cluster extent of 100 mm3, consistent with conservative statistical guidelines51.
Comparison to prior established methods
We hypothesized that our model would be superior to existing methods for both causal brain mapping and neuromodulation targeting. First, we compared our causal mapping approach to voxel-lesion symptom mapping (VLSM), a tool that can identify lesion locations or stimulation sites associated with a particular behavioral outcome (without considering connectivity)25. Next, we compared our connectivity-based targeting approach to the current consensus approach, which identifies optimal TMS targets based on subgenual cingulate connectivity8,18.
Using VLSM, we assessed whether particular lesion locations and stimulation sites were associated with depression, irrespective of their connectivity. At each voxel, we used a t-test to compare depression severity between patients whose lesions or stimulation sites overlapped with that voxel versus patients whose lesions or stimulation sites did not overlap with that voxel. This yielded a whole-brain map of lesion locations or stimulation sites associated with depression severity.
We then attempted to explain clinical variance using stimulation site connectivity to the subgenual cingulate. Within each dataset, we computed mean connectivity of each patient’s stimulation site to the subgenual cingulate, following the methods described in Weigand et al8. In each dataset, subgenual connectivity was compared with improvement in depression score using partial Pearson correlation, controlling for baseline depression severity. Across all datasets, these correlations were combined into a single weighted mean value representing the degree to which our circuit predicted neuromodulation outcomes across all datasets. The predictive value of subgenual connectivity was compared with the predictive value of our depression circuit using a Z-test for dependent correlations within each dataset.
Data availability statement
This paper used de-identified data from 14 different datasets collected by 14 different teams of investigators at various institutions across four different countries. Each dataset is available upon reasonable request from each respective team of investigators. Data sharing will be subject to the policies and procedures of the institution where each dataset was collected as well as the laws of the country where each dataset was collected.
Code availability statement
All custom MATLAB code used in this study is available upon reasonable request from the corresponding author.
Supplementary Material
Acknowledgements
We thank all research participants, funding bodies, allied health staff, and other research staff that made this work possible. The present work was supported by the Sidney R. Baer Foundation (SHS, JLP, MDF), the Brain & Behavior Research Foundation (SHS), and the National Institute of Mental Health (Grant No. K23MH121657 to SHS; Grant Nos. R01MH113929 and R01MH115949 to MDF). The funders were not directly involved in the conceptualization, design, data collection, analysis, decision to publish, or preparation of the manuscript.
Competing Interests/Disclosures
SHS serves as a clinical consultant for Kaizen Brain Center. SHS and MDF have jointly received investigator-initiated research support from Neuronetics. None of these organizations were involved in the present work.
SHS and MDF each own independent intellectual property on the use of brain network mapping to target neuromodulation. The present work did not utilize any of this intellectual property.
The authors report no other conflicts of interest related to the present work.
References
- 1.Czéh B, Fuchs E, Wiborg O & Simon M Animal models of major depression and their clinical implications. Progress in Neuro-Psychopharmacology and Biological Psychiatry 64, 293–310, doi: 10.1016/j.pnpbp.2015.04.004 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Etkin A Mapping Causal Circuitry in Human Depression. Biological psychiatry 86, 732–733, doi: 10.1016/j.biopsych.2019.09.009 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Nestler EJ & Hyman SE Animal models of neuropsychiatric disorders. Nat Neurosci 13, 1161–1169, doi: 10.1038/nn.2647 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Monteggia LM, Heimer H & Nestler EJ Meeting Report: Can We Make Animal Models of Human Mental Illness? Biol Psychiatry 84, 542–545, doi: 10.1016/j.biopsych.2018.02.010 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fox MD Mapping Symptoms to Brain Networks with the Human Connectome. N Engl J Med 379, 2237–2245, doi: 10.1056/NEJMra1706158 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Siddiqi SH et al. Distinct symptom-specific treatment targets for circuit-based neuromodulation. American Journal of Psychiatry (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Padmanabhan JL et al. A human depression circuit derived from focal brain lesions. Biological Psychiatry, doi: 10.1016/j.biopsych.2019.07.023 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weigand A et al. Prospective Validation That Subgenual Connectivity Predicts Antidepressant Efficacy of Transcranial Magnetic Stimulation Sites. Biol Psychiatry 84, 28–37, doi: 10.1016/j.biopsych.2017.10.028 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Riva-Posse P et al. Defining critical white matter pathways mediating successful subcallosal cingulate deep brain stimulation for treatment-resistant depression. Biol Psychiatry 76, 963–969, doi: 10.1016/j.biopsych.2014.03.029 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Etkin A Addressing the Causality Gap in Human Psychiatric Neuroscience. JAMA Psychiatry 75, 3–4, doi: 10.1001/jamapsychiatry.2017.3610 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Ressler KJ & Mayberg HS Targeting abnormal neural circuits in mood and anxiety disorders: from the laboratory to the clinic. Nature neuroscience 10, 1116–1124, doi: 10.1038/nn1944 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matthews PM & Hampshire A Clinical Concepts Emerging from fMRI Functional Connectomics. Neuron 91, 511–528, doi: 10.1016/j.neuron.2016.07.031 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Fox MD et al. Resting-state networks link invasive and noninvasive brain stimulation across diverse psychiatric and neurological diseases. Proc Natl Acad Sci U S A 111, E4367–4375, doi: 10.1073/pnas.1405003111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Drysdale AT et al. Resting-state connectivity biomarkers define neurophysiological subtypes of depression. Nat Med, doi: 10.1038/nm.4246 10.1038/nm.4246. (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koenigs M et al. Focal brain damage protects against post-traumatic stress disorder in combat veterans. Nat Neurosci 11, 232–237, doi: 10.1038/nn2032 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johnson KA et al. Prefrontal rTMS for treating depression: location and intensity results from the OPT-TMS multi-site clinical trial. Brain Stimul 6, 108–117, doi: 10.1016/j.brs.2012.02.003 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taylor SF et al. Changes in brain connectivity during a sham-controlled, transcranial magnetic stimulation trial for depression. J Affect Disord 232, 143–151, doi: 10.1016/j.jad.2018.02.019 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cash RFH et al. Subgenual Functional Connectivity Predicts Antidepressant Treatment Response to Transcranial Magnetic Stimulation: Independent Validation and Evaluation of Personalization. Biol Psychiatry 86, e5–e7, doi: 10.1016/j.biopsych.2018.12.002 (2019). [DOI] [PubMed] [Google Scholar]
- 19.Horn A et al. Connectivity Predicts deep brain stimulation outcome in Parkinson disease. Ann Neurol 82, 67–78, doi: 10.1002/ana.24974 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dougherty DD et al. A Randomized Sham-Controlled Trial of Deep Brain Stimulation of the Ventral Capsule/Ventral Striatum for Chronic Treatment-Resistant Depression. Biol Psychiatry 78, 240–248, doi: 10.1016/j.biopsych.2014.11.023 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Irmen F et al. Left prefrontal impact links subthalamic stimulation with depressive symptoms. Ann Neurol, doi: 10.1002/ana.25734 (2020). [DOI] [PubMed] [Google Scholar]
- 22.Merkl A et al. Antidepressant effects after short-term and chronic stimulation of the subgenual cingulate gyrus in treatment-resistant depression. Exp Neurol 249, 160–168, doi: 10.1016/j.expneurol.2013.08.017 (2013). [DOI] [PubMed] [Google Scholar]
- 23.Schaper FLWVJ et al. Deep Brain Stimulation in Epilepsy: A Role for Modulation of the Mammillothalamic Tract in Seizure Control? Neurosurgery 87, 602–610, doi: 10.1093/neuros/nyaa141 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yeo BT et al. The organization of the human cerebral cortex estimated by intrinsic functional connectivity. J Neurophysiol 106, 1125–1165, doi: 10.1152/jn.00338.2011 10.1152/jn.00338.2011. Epub 2011 Jun 8. (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bates E et al. Voxel-based lesion–symptom mapping. Nature Neuroscience 6, 448–450, doi: 10.1038/nn1050 (2003). [DOI] [PubMed] [Google Scholar]
- 26.Gourisankar A et al. Mapping movement, mood, motivation and mentation in the subthalamic nucleus. R Soc Open Sci 5, 171177, doi: 10.1098/rsos.171177 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Choi KS, Riva-Posse P, Gross RE & Mayberg HS Mapping the “Depression Switch” During Intraoperative Testing of Subcallosal Cingulate Deep Brain Stimulation. JAMA Neurol 72, 1252–1260, doi: 10.1001/jamaneurol.2015.2564 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Joutsa J, Horn A, Hsu J & Fox MD Localizing parkinsonism based on focal brain lesions. Brain 141, 2445–2456, doi: 10.1093/brain/awy161 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang C et al. Repetitive transcranial magnetic stimulation therapy for motor recovery in Parkinson’s disease: A Meta-analysis. Brain Behav 8, e01132, doi: 10.1002/brb3.1132 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.James GA et al. Exploratory structural equation modeling of resting-state fMRI: applicability of group models to individual subjects. NeuroImage 45, 778–787, doi: 10.1016/j.neuroimage.2008.12.049 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mayberg HS et al. Reciprocal limbic-cortical function and negative mood: converging PET findings in depression and normal sadness. Am J Psychiatry 156, 675–682, doi: 10.1176/ajp.156.5.675 (1999). [DOI] [PubMed] [Google Scholar]
- 32.Drevets WC et al. Subgenual prefrontal cortex abnormalities in mood disorders. Nature 386, 824–827, doi: 10.1038/386824a0 (1997). [DOI] [PubMed] [Google Scholar]
- 33.Müller VI et al. Altered Brain Activity in Unipolar Depression Revisited: Meta-analyses of Neuroimaging Studies. JAMA Psychiatry 74, 47–55, doi: 10.1001/jamapsychiatry.2016.2783 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gray JP, Müller VI, Eickhoff SB & Fox PT Multimodal Abnormalities of Brain Structure and Function in Major Depressive Disorder: A Meta-Analysis of Neuroimaging Studies. Am J Psychiatry 177, 422–434, doi: 10.1176/appi.ajp.2019.19050560 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Williams LM Defining biotypes for depression and anxiety based on large-scale circuit dysfunction: a theoretical review of the evidence and future directions for clinical translation. Depress Anxiety 34, 9–24, doi: 10.1002/da.22556 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holtzheimer PE et al. Subcallosal cingulate deep brain stimulation for treatment-resistant depression: a multisite, randomised, sham-controlled trial. Lancet Psychiatry 4, 839–849, doi: 10.1016/S2215-0366(17)30371-1 (2017). [DOI] [PubMed] [Google Scholar]
- 37.Yesavage JA et al. Effect of Repetitive Transcranial Magnetic Stimulation on Treatment-Resistant Major Depression in US Veterans: A Randomized Clinical Trial. JAMA Psychiatry 75, 884–893, doi: 10.1001/jamapsychiatry.2018.1483 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kozak MJ & Cuthbert BN The NIMH Research Domain Criteria Initiative: Background, Issues, and Pragmatics. Psychophysiology 53, 286–297, doi: 10.1111/psyp.12518 (2016). [DOI] [PubMed] [Google Scholar]
- 39.Poldrack RA Inferring mental states from neuroimaging data: from reverse inference to large-scale decoding. Neuron 72, 692–697, doi: 10.1016/j.neuron.2011.11.001 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cole Eleanor J., Ph.D. , et al. Stanford Accelerated Intelligent Neuromodulation Therapy for Treatment-Resistant Depression. American Journal of Psychiatry 0, appi.ajp.2019.19070720, doi: 10.1176/appi.ajp.2019.19070720 (2020). [DOI] [PubMed] [Google Scholar]
- 41.Blumberger DM et al. Effectiveness of theta burst versus high-frequency repetitive transcranial magnetic stimulation in patients with depression (THREE-D): a randomised non-inferiority trial. Lancet 391, 1683–1692, doi: 10.1016/S0140-6736(18)30295-2 (2018). [DOI] [PubMed] [Google Scholar]
- 42.Cash RFH et al. Using Brain Imaging to Improve Spatial Targeting of Transcranial Magnetic Stimulation for Depression. Biol Psychiatry, doi: 10.1016/j.biopsych.2020.05.033 (2020). [DOI] [PubMed] [Google Scholar]
- 43.Cash RFH, Cocchi L, Lv J, Fitzgerald PB & Zalesky A Functional Magnetic Resonance Imaging–Guided Personalization of Transcranial Magnetic Stimulation Treatment for Depression. JAMA Psychiatry, doi: 10.1001/jamapsychiatry.2020.3794 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Siddiqi SH, Weigand A, Pascual-Leone A & Fox MD Identification of personalized TMS targets based on subgenual cingulate connectivity: an independent replication. Biological Psychiatry (2021 (In Press)). [DOI] [PubMed] [Google Scholar]
- 45.Riva-Posse P et al. A connectomic approach for subcallosal cingulate deep brain stimulation surgery: prospective targeting in treatment-resistant depression. Molecular psychiatry 23, 843–849, doi: 10.1038/mp.2017.59 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fox MD, Liu H & Pascual-Leone A Identification of reproducible individualized targets for treatment of depression with TMS based on intrinsic connectivity. Neuroimage 66, 151–160, doi: 10.1016/j.neuroimage.2012.10.082 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Opitz A, Fox MD, Craddock RC, Colcombe S & Milham MP An integrated framework for targeting functional networks via transcranial magnetic stimulation. Neuroimage 127, 86–96, doi: 10.1016/j.neuroimage.2015.11.040 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rouder JN & Morey RD Default Bayes factors for model selection in regression. Multivariate Behavioral Research 47, 877–903 (2012). [DOI] [PubMed] [Google Scholar]
- 49.Wagenmakers E-J, Verhagen J & Ly A How to quantify the evidence for the absence of a correlation. Behavior Research Methods 48, 413–426, doi: 10.3758/s13428-015-0593-0 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Turner JA et al. A multi-site resting state fMRI study on the amplitude of low frequency fluctuations in schizophrenia. Front Neurosci 7, 137, doi: 10.3389/fnins.2013.00137 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Slotnick SD Cluster success: fMRI inferences for spatial extent have acceptable false-positive rates. Cogn Neurosci 8, 150–155, doi: 10.1080/17588928.2017.1319350 (2017). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This paper used de-identified data from 14 different datasets collected by 14 different teams of investigators at various institutions across four different countries. Each dataset is available upon reasonable request from each respective team of investigators. Data sharing will be subject to the policies and procedures of the institution where each dataset was collected as well as the laws of the country where each dataset was collected.