

Abstract
Background:
Several monogenic causes for isolated dystonia have been identified, but they collectively account for only a small proportion of cases. Two genome-wide association studies have reported a few potential dystonia risk loci; but conclusions have been limited by small sample sizes, partial coverage of genetic variants, or poor reproducibility.
Objective:
To identify robust genetic variants and loci in a large multi-center cervical dystonia cohort using a genome-wide approach.
Methods:
We performed a genome-wide association study using cervical dystonia samples from the Dystonia Coalition. Logistic and linear regressions, including age, sex and population structure as covariates, were employed to assess variant- and gene-based genetic associations with disease status and age at onset. We also performed a replication study for an identified genome-wide significant signal.
Results:
After quality control, 919 cervical dystonia patients compared with 1,491 controls of European ancestry were included in the analyses. We identified one genome-wide significant variant (rs2219975, chromosome 3, upstream of COL8A1, p-value 3.04×10−8). The association was not replicated in a newly genotyped sample of 473 cervical dystonia cases and 481 controls. Gene-based analysis identified DENND1A to be significantly associated with cervical dystonia (p-value 1.23×10−6). One low-frequency variant was associated with lower age-at-onset (16.4±2.9 years, p-value=3.07×10−8, minor allele frequency=0.01), located within the GABBR2 gene on chromosome 9 (rs147331823).
Conclusion:
The genetic underpinnings of cervical dystonia are complex and likely consist of multiple distinct variants of small effect sizes. Larger sample sizes may be needed to provide sufficient statistical power to address the presumably multi-genic etiology of cervical dystonia.
Keywords: Cervical dystonia, genome-wide association study, GWAS, rare disease, movement disorder
Introduction
The dystonias are a group of disorders characterized by involuntary, repetitive twisting movements, abnormal postures, or other manifestations of excessive muscle contraction.[1, 2] Cervical dystonia (CD) is the most common adult-onset dystonia, and is characterized by involuntary patterned contractions of cervical musculature resulting in abnormal movements or postural changes of the head and neck. CD is rare, and estimates of incidence and prevalence are limited by a paucity of comprehensive epidemiological studies.[3]
Some forms of dystonia are genetically determined, and several genes have been identified so far.[4-6] However, these genes collectively account for only a small fraction of all isolated dystonias. Thus, additional genetic and/or environmental factors probably play an important role. The identification of genes among affected pedigrees has been complicated by the fact that many appear to be dominantly inherited with partial penetrance. Genome-wide association studies (GWAS) agnostically scan markers across the genome in a large population to identify genetic variations associated with a disease trait. GWAS have successfully identified thousands of markers that are associated with a wide spectrum of diseases,[7] and are the most cost-effective approach to discovering any genetic association with a disease. The power of this method to detect genetic variants depends on study sample size and the underlying genetic architecture. Although variants with large effect sizes can sometimes be detected with sample sizes as small as 100, far more variants with smaller effect sizes can be detected when more than 10,000 cases and controls are examined. Currently, only a few very small GWAS have been published for dystonia. One GWAS involving 212 CD cases from the United Kingdom revealed a genetic variant in NALCN gene,[8] but the association was not replicated in another study involving additional 252 CD patients from Spain.[9] A GWAS of 127 cases with musician’s dystonia identified a significant association in a locus of ARSG gene, but it has not yet been independently replicated.[10]
These studies demonstrate that GWAS is a promising approach for the discovery of candidate genes and loci related to the dystonias. However, they are limited by small sample sizes and lack of genome-wide gene-based analysis of low frequency variants. To address these limitations, the present study provides a comprehensive coverage of the genetic susceptibility in idiopathic CD using a large cohort of 919 cases. Additionally, we investigated potential genetic determinants of age-at-onset and tremor among CD cases.
Methods
Data Sources and Study Population
The dystonia subjects studied in this project are from the Dystonia Coalition, which is an ongoing study to facilitate collaborations and to advance the pace of clinical and translational research for isolated dystonia syndromes (i.e. "primary" dystonias).[11-13] The Dystonia Coalition collected DNA samples and detailed clinical information on adult-onset isolated dystonia patients (n=3,119) including several subtypes to identify risk factors and biomarkers to improve prognosis, treatment and prevention. Because different forms of dystonia may have different genetic substrates, we focused our GWAS on the most common subtype, CD. We included all available CD samples of European ancestry at the time of the study. All cases were evaluated and recruited by individuals with expertise in movement disorders using a standardized protocol, which was videotaped for verification.[11] Acquired forms of CD and CD combined with other movement disorders (e.g. parkinsonism) were excluded. We included cases who started with dystonia in the neck region (isolated CD). Cases with minor dystonic symptoms in other body regions were also included, if CD was the main problem (segmental or multifocal dystonias).
Control samples without neurological disorders were obtained from the Emory Cardiovascular Biobank study and the Mental Stress-Induced Ischemia, Mechanisms and Prognosis (MIPS). Blood specimens of participants were collected and used to extract genomic DNA. The Dystonia Coalition study and the present GWAS were approved by the Institutional Research Board committee at Emory University. All participants provided written consent. We obtained demographic variables such as age, sex, age-at-onset from the survey and medical records.
Genotypic Measures and Data Processing for the GWAS
Genomic DNA (gDNA) was extracted from blood samples of study participants by a central laboratory at the Coriell Institute for Medical Research (Camden, New Jersey, USA). Each gDNA sample was quantified using the PicoGreen assay, standardized to 50ng/mL, and processed following the standard Illumina protocol including hybridization, incubation and scanning. We used Illumina’s Multi-Ethnic Genotyping Array (MEGA) platform, which is optimized for genome-wide association studies in multi-ethnic populations. The MEGA chip directly measures 1.7 million genetic markers, including improved genome-wide coverage of non-European ancestry, exonic content of over 400,000 markers, more than 17,000 variants relevant to clinical and pharmacogenetic studies, and an additional 23,000 variants selected for functional, immunological, oncological, ancestry, forensic, and other common and rare diseases.
Genotypes were measured and called using Illumina’s GenomeStudio software. Participants were excluded if they had an overall SNP call rate (ratio of measured SNPs per participant over the total number of SNPs in the dataset) < 95% or mismatch between genotypic and phenotypic measurements (e.g., sex and ethnicity). Individuals related closer than second degree cousins were identified. Only one individual from each related pair was kept in the study. In addition, individual SNPs were excluded from the analyses if they had unknown chromosomal location, were missing for more than 5% of the total participant samples, had Hardy-Weinberg Equilibrium (HWE) p-value less than 10−6 among the controls, or had a minor allele frequency (MAF) less than 0.01 among controls.[14] Base-pair position of each variant was annotated to human genome reference GRCh37/hg19.
Cleaned genotype data of common variants were further pruned using plink software with r2 less than 0.1, and window size and step of 50 and 5 SNPs, respectively. We used ADMIXTURE [15] analysis with the 1000 Genome phase 3 samples and known continental ancestries to assign genotyped individuals to European (>80% European Ancestry). Principal component analyses were performed using flashPCA software.[16] The dosage data of SNPs in the 1000 Genomes Project phase 3 version 5 reference panel were imputed by the Michigan Imputation Server (https://imputationserver.sph.umich.edu/) using minimac.[17] Following imputation, variant level quality control was performed using the EasyQC R package (www.R-project.org), and exclusion metrics included: Hardy-Weinberg equilibrium p-value <10−6, posterior call probability < 0.9, imputation quality <0.3, MAF < 0.05, call rate < 97.5%. Variants were also excluded if they deviated > 10% from their expected allele frequency based on reference data from the Genome Aggregation Database (gnomAD).[18] Among 968 genotyped CD cases with phenotypic data, 13, 14 and 22 samples were excluded due to high missing rate, sex-mismatch, and non-European ancestry, respectively. Following variant level quality control, we obtained 8,053,795 common variants (MAF>5%) in 919 CD cases and 1,491 controls of European ancestry.
Association Analysis
Genome-wide association analysis of CD status was adjusted for age, sex, and genotype-derived principal components in a logistic regression model. Imputed genetic variants were tested for association with CD assuming an additive genetic model. Among 919 CD patients with available phenotypes, genome-wide association analyses of age-at-onset, tremor and affected area (segmental vs. focal) were adjusted for age, sex, and genotype-derived principal components in linear or logistic regression models. We summarized the GWAS results using FUMA (http://fuma.ctglab.nl/), which is a platform that can be used to annotate, prioritize, visualize and interpret GWAS results.[19] Genome-wide significant SNPs (p < 5 ×10−8) were grouped into genomic loci if they are dependent on each other at r2 > 0.1 or physically close (distance < 500kb). Lead SNPs were defined within each locus if they are independent (r2 < 0.1). FUMA uses GWAS summary statistics to compute gene-based P-values using the MAGMA tool.[19] The gene-based P-value is computed for protein-coding genes by mapping SNPs to genes if SNPs are located within the genes. The Bonferroni correction was used to correct for multiple testing based on total number of tested protein coding genes. The 1000 Genomes phase 3 was used as a reference panel to calculate LD across SNPs and genes. All other statistical analyses were performed in the R statistical environment version 3.2.5 (http://www.r-project.org/)).
Replication Study
We investigated 473 independent CD patients that originated from the Dystonia Coalition (n=270) or the German Dystonia Registry (DysTract, http://dystract.cio-marburg.de/en/, n=203) using a similar protocol for enrollment. We also included 481 healthy controls from Northern Germany (Luebeck area). Variant rs2219975 was genotyped using a melting curve analysis on the LightCycler or by Sanger sequencing.
Results
After quality control, the analysis included 919 cases of CD and 1,491 controls, all of European ancestry. The PCA plots showed that the genetic ancestry of the study sample was consistent with the HapMap panel, and the genetic background was similar between cases and controls (Supplementary Figure 1A and 1B). Characteristics of these groups are summarized in Table 1. In brief, the CD subjects had a younger average age of 60.7±11.4 years, compared to controls (64.8±11.5 years). More CD participants were females than in the control group (75.7 % vs. 48.0%). Among 919 CD subjects, the average age at onset was 45.6±13.5 years.
Table 1:
Dystonia patients and controls of European ancestry using for the GWAS stage.
| Variables | Cases (N=919) Mean±SD or count (%) |
Controls (N=1491) Mean±SD or count (%) |
|---|---|---|
| Age (years) | 60.7±11.4 | 64.8±11.5 |
| Female | 696 (75.7%) | 716 (48.0%) |
| Age at Onset (years) | 45.6±13.5 | N/A |
| Focal | 769 (83.7%) | N/A |
| Segmental | 150 (16.3%) | N/A |
| Tremor | 616 (67.0%) | N/A |
There was moderate global inflation (inflation factor of 1.07) of over 8 million common variants. Using the 1000 Genome Project-based imputation panel, we identified one genome-wide significant common (MAF>5%) SNP from a locus on chromosome 3 (Figure 1 and Table 2). The lead common SNP (rs2219975) of the chromosome 3 locus had a MAF of 0.06 in controls and 0.10 in CD cases. The minor allele C was associated with higher risk of CD (OR of 2.1, p-value=3.04×10−8). Two variants (chr3:99071470 and chr3:99072830, GRCh37/hg19) with lower MAF (~1%) and high LD in the chromosome 3 locus were also associated with CD with p-values of 6.74×10−9 and 8.49×10−9, and ORs of 46 and 50, respectively (see summary statistics of these two low-frequency variants including minor allele counts in Supplementary Table 1).
Figure 1.
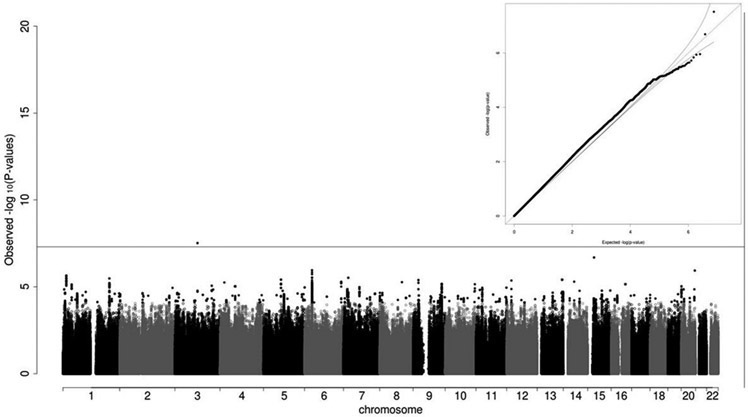
Summary of the GWAS of cervical dystonia. Manhattan plot showed a significant locus on chromosome 3. The horizontal line represented the genome-wide significant threshold of 5×10−8. The quantile-quantile plot showed overall distribution of p-values in the GWAS.
Table 2.
Summary of common genetic variants (MAF>0.05) of genome-wide significant locus associated with cervical dystonia.
| SNP ID | rs number | CHR | POS | REF | ALT | AF_Control | AF_CD | OR | T Statistic | p-value |
|---|---|---|---|---|---|---|---|---|---|---|
| 3:99070019 | rs2219975 | 3 | 99070019 | T | C | 0.06 | 0.10 | 2.123 | 5.539 | 3.04×10−8 |
CHR: chromosome number; POS: base-pair position in GRCh37/hg19; REF: reference allele; ALT: alternative (effective) allele; AF: allele frequency of alternative allele; OR: odds ratio.
The regional plot (Supplementary Figure 2) showed all tested SNPs in the neighboring regions of the lead SNP and additional association signals with CD within these regions. The CD-associated locus is located in intergenic region, and is about 300 kb upstream of COL8A1.
Genotyping of the lead SNP rs2219975 in the replication samples found the minor C allele less frequently in cases (overall: 6.4%, DC subgroup: 7.4%; DysTract subgroup: 5.2%) compared to controls (9.0%).
Complementary to single variant association test, gene-based statistical methods can analyze multiple genetic markers simultaneously to determine their joint association with a disease trait. Using the MAGMA gene-based analysis method available in the online tool, functional mapping and annotation (FUMA),[19] the DENND1A was significantly associated with CD after correction for multiple testing. We also identified another 4 genes (Supplementary Table 2; ATP11A, ST7-OT4, PPP1R16B, SLC39A12) with nominal p-values less than 10−4. None of the previously reported genes potentially related to adult-onset isolated dystonia [20] (i.e., ANO3, CIZ1, COL6A3, GNAL, THAP1, TOR1A) were significantly associated with CD in the gene-based analysis. We also examined the genetic associations of individual SNPs with CD in regions harboring six previously reported genes related to dystonias, including ANO3, CIZ1, COL6A3, GNAL, THAP1 and TOR1A. None of the SNPs in these regions were significantly associated with CD after multiple testing correction. Gene expression quantitative trait locus (eQTL) analysis showed that eQTLs from the GWAS of CD were the most enriched for brain tissue as well as specific sub-regions of the brain (e.g., cerebellum, cerebellar hemisphere, spinal cord cervical, hippocampus), although not statistically significant after correcting for multiple testing.
There were no common variants significantly associated with age at onset (ranging 8 to 77 years) among 919 CD patients adjusted for sex and population structure. Interestingly, one low frequency SNP on chromosome 9:101222141 (rs147331823, MAF ~0.01) showed strong association with age at onset (p-value=3.07×10−9) as shown in Supplementary Figure 3. The minor allele G was associated with a younger age at onset of 16.4±2.9 years.
The current analyses failed to replicate the CD-associated SNPs in the NALCN region on chromosome 13 previously reported.[8] None of these reported SNPs (reported p-values range from 9.76×10−7 to 2.54×10−6) had nominal p-values less than 0.05. We also searched the NALCN region including ±200 kb flanking regions. Among all imputed SNPs in the region, none had a p-value lower than 10−3.
Discussion
The results from this study are derived from the largest GWAS for any type of dystonia to date. CD is considered a rare disorder, and this study was made possible only by pooling samples through a multicenter collaborative effort. However, the sample size of this study (919 cases and 1491 controls) is considerably smaller than other GWAS for more common disorders such as Parkinson’s disease and the finding of a genome-wide significant SNP on chromosome 3 could not be confirmed in a small replication sample. There might be several reasons for the lack of replication: (1) rs2219975 has a rather low frequency (~7%) compared to other genetic risk variants and thus, larger sample sizes are needed to detect and replicate an association; (2) The frequency of this variant highly depends on genetic ancestry (e.g., 0.5481 in Africans and 0.07303 in non-Finnish Europeans, https://gnomad.broadinstitute.org/variant/3-99070019-T-C?dataset=gnomad_r2_1). Small differences in the origin of cases and controls may lead to false positive associations in a GWAS; (3) The individual effect size of genetic risk variants might be low given the known etiological heterogeneity of CD. The sample size was sufficient for genome-wide discovery of CD-related genes with large effects (e.g., at alpha level of 5×10−8, OR 1.5 and 1.68 for MAF of 0.2 and 0.1, respectively), but insufficient for genetic associations with small effects or very low risk allele frequency.
In the present GWAS of CD, we could not confirm a previously reported association of CD in NALCN from an independent GWAS. We also were unable to replicate the association of dystonia with the ARSG gene, although the latter may reflect the different type of dystonia studied (e.g., task specific dystonia).[10]
The locus on chromosome 3 is located upstream of COL8A1. The COL8A1 gene encodes one of the two alpha chains of type VIII collagen. Defects in COL8A1 are associated with corneal dystrophy and age-related macular degeneration. It has not been associated with any type of dystonia, but it is interesting to note that dystonia has been reported to be potentially caused by biallelic mutations in COL6A3, another member of the collagen family.[20, 21]
Gene-based analysis identified DENND1A on chromosome 9 to be significantly associated with CD. DENND1A encodes DENN domain containing protein 1A, a member of the connecdenn family. It serves as a guanine nucleotide exchange factor that is expressed in brain and plays a role in vesicle function. It has not yet been associated with dystonia, although pathogenic variants in different guanine nucleotide exchange factors encoded by GNAL and GNAO1 have been linked to adult onset focal and segmental dystonias and a childhood-onset movement disorder that combines chorea and dystonia[4], respectively. SLC39A12 (solute carrier family 39 member 12) encodes a zinc transporter and belongs to a subfamily of proteins that share structural characteristics of zinc transporters. Mutations in SLC39A14, another member of the same ZIP transporter family, have been linked to hypermanganesemia associated with infantile-onset dystonia and parkinsonism-dystonia.[22, 23]
Although we did not identify any genome-wide significant common variant associated with age at onset for CD, a low-frequency variant (MAF ~1%) was associated with a decrease of age at onset (16.4±2.9 years, p-value=3.07×10−8), located within the GABBR2 gene on chromosome 9. This gene encodes a subunit of GABA-B receptors, which play an important role in neuronal excitability. This gene has not been previously associated with dystonia, although GABA receptor activators such as baclofen and benzodiazepines can be effective in treating some types of dystonia. Since the strong association was based on nine carriers of the minor allele, future association study is warranted to validate the finding.
In conclusion, we have conducted the largest GWAS of any type of dystonia so far assembled, and attempted to address heterogeneity by relying on a standardized exam and by focusing on a single dystonia subtype, CD. Despite these measures, GWAS did not reveal any robust locus, indicating a presumably rather small impact of an individual common variant at the population level. Future genetic studies of dystonias require the whole scientific community to join effort to reveal the complex genetic architecture of CD and other dystonias.
Supplementary Material
Funding agencies:
YVS, CL, QH and HAJ are partially supported by the National Institute of Health grant NS096455 and the Dystonia Medical Research Foundation. This work was also supported in part by grants to the Dystonia Coalition, a part of the Rare Diseases Clinical Research Network of the Office of Rare Diseases Research at the National Center for Advancing Translational Sciences (Grant numbers NS065701, TR001456, and NS116025). KL, IRK, and CK are supported by the DFG (FOR2488). The DysTract study has been supported by the Federal Ministry of Education and Sciences (BMBF) in Germany (01GM1514B, to KL and CK). UK, MK, and CK were supported by the DFG (TR-SFB134).
Financial Disclosures
Pinky Agarwal has received honoraria from Sunovion, Acadia, Amneal, Accorda, ADAMA, Kyowa Kirin, and Supernus for speaking. She has also received consulting fees from ADAMAS CALA Health, and Sunovion. Research support has been received from US WorldMeds, Centogene, Biogen, Merck, Sun Pharma, Advanced Research Company Limited (SPARC) Anexxon Inc, CHDI Foundation, Roche.
T. Bäumer has received grant support from DFG FOR 2698. He has also received honoraria or consulting fees from Ipsen Pharma, Allergan, Merz Pharmaceuticals
B.D. Berman has received research grant support from the Parkinson’s Foundation, Dana Foundation, NIH (NIH/NCATS Colorado CTSI Grant Number KL2 TR001080), Dystonia Coalition (receives the majority of its support through NIH grant NS065701 from the Office of Rare Diseases Research in the National Center for Advancing Translational Science and National Institute of Neurological Disorders and Stroke), and from Mary Rossick Kern and Jerome H. Kern. He also serves on the medical advisory boards for the Benign Essential Blepharospasm Research Foundation and the National Spasmodic Torticollis Association.
F. Borngräber has received grant support from Clinician Scientist Program, BIH, Charité-Universitätsmedizin Berlin and Max-Delbrück Centre.
M. Borsche has received honoraria from Ipsen Pharma.
A. Brashear has active or recent grant support from the NIH Revance, Allergan, Merz, Ipsen. She has served as a consultant or on advisory boards for Revance, Ipsen, Allergan and Merz. She is a board member for the American Board of Psychiatry and Neurology, California Institute of Regenerative Medicine, McKnight Brain Research Foundation, Latinx Physicians of California. She is Founder and Board Member of Care Directions.
C. Cruchaga has received research support from: Biogen, EISAI, and Alector. He has consulted or is a member of the advisory board of Vivid genetics, Halia Therapeutics and Takeda and GSK.
A.J. Espay has received grant support from the NIH and the Michael J Fox Foundation; personal compensation as a consultant/scientific advisory board member for Abbvie, Neuroderm, Neurocrine, Amneal, Acadia, Acorda, Kyowa Kirin, Sunovion, Lundbeck, and USWorldMeds; honoraria from Acadia, Sunovion, USWorldMeds; and publishing royalties from Lippincott Williams & Wilkins, Cambridge University Press, and Springer.
S. Factor has Honoraria from Lundbeck, Teva, Sunovion, Biogen, Acadia, Neuroderm, Acorda, CereSpir; Grants from Ipsen, Medtronics, Boston Scientific, Teva, US World Meds, Sunovion Therapeutics, Vaccinex, Voyager, Jazz Pharmaceuticals, Lilly, CHDI Foundation, Michael J. Fox Foundation, NIH; Royalties from Demos, Blackwell Futura, Springer for textbooks, Uptodate; and other support from Bracket Global LLC, CNS Ratings LLC.
S. Fox has received Clinic support from the Edmond J Safra Foundation for Parkinson Research; National Parkinson Foundation and the Toronto Western and General Foundation. Salary from UHN Dept of Medicine Practice Plan. Research Funding from Michael J Fox Foundation for Parkinson Research, NIH (Dystonia Coalition); CIHR; Parkinson Canada. Honoraria from the International Parkinson and Movement Disorder Society and American Academy of Neurology. Site PI for Clinical Trials for Biotie, Cynapsus, Eisai, Revance. Consultancy/Speaker fees from Acadia; Atuka; Sunovion; Teva; Paladin; Royalties from Oxford University Press.
M. Gelderblom has received grant support from Merz Pharmaceuticals, Allergan, DFG, Werner Otto Stiftung, Ostseeförderung. He has served on Advisory Boards for Merz Pharmaceuticals and Allergan, and has received honoraria from Merz Pharmaceuticals, Allergan and Ipsen.
H.A. Jinnah has active or recent grant support from the US government (National Institutes of Health), private philanthropic organizations (Cure Dystonia Now), and industry (Cavion Therapeutics, Retrophin Inc., Revance Therapeutics). Dr. Jinnah has also served on advisory boards or as a consultant for Abide Therapeutics, Allergan Inc., CoA Therapeutics, Retrophin Inc, and Revance Therapeutics. He has received honoraria or stipends for lectures or administrative work from the International Parkinson’s Disease and Movement Disorders Society. Dr. Jinnah serves on the Scientific Advisory Boards for several private foundations including the Benign Essential Blepharospasm Research Foundation, Cure Dystonia Now, the Dystonia Medical Research Foundation, the Tourette Association of American, and Tyler's Hope for a Cure. He also is Director of the Dystonia Coalition, which receives the majority of its support through NIH grants (NS116015, NS067501, and TR001456 from the National Institutes of Neurological Disorders and Stroke and the Office of Rare Diseases Research at the National Center for Advancing Translational Sciences). The Dystonia Coalition has received additional material or administrative support from industry sponsors (Allergan Inc. and Merz Pharmaceuticals) as well as private foundations (The American Dystonia Society, Beat Dystonia, The Benign Essential Blepharospasm Foundation, Cure Dystonia Now, Dystonia Europe, Dystonia Inc., Dystonia Ireland, The Dystonia Medical Research Foundation, The Foundation for Dystonia Research, The National Spasmodic Dysphonia Association, and The National Spasmodic Torticollis Association).
M. Kasten has received grant support from TCRC-134, FOR 2488.
C. Klein has received grant support from The Hermann and Lilly Schilling Foundation; the German Research Foundation; the BMBF; the German Research Foundation; the European Community; intramural funds from the University of Luebeck. She has served as medical advisor to Centogene. She has also received honoraria from the Else Kroener Fresenius Foundation.
I.R. König has received grant support from the German Research Foundation, BMBF, German Cancer Aid.
U.M. Kramer has received grant support from the German Research Foundation
A. Kuhn has received honoraria or served on advisory boards for Medtronic und Boston Scientific.
M.S. LeDoux serves on the speaker’s bureaus for Acadia, Teva, Acorda and Adamas, and editorial boards of Neurology and Tremor and Other Hyperkinetic Movements; has received research support from the National Institutes of Health, Dystonia Medical Research Foundation, Department of Defense, Michael J. Fox Foundation, Revance, and PharmaTwoB; and receives royalty payments from Elsevier for editing Animal Models of Movement Disorders and Movement Disorders: Genetics and Models.
S. Loens has nothing to disclose.
K. Lohmann has received grant support from German Research Foundation; Federal Ministry of Education and Science (BMBF); Movement Disorder Society; Damp-Stiftung.
A. Maurer has nothing to disclose.
W. Ondo, MD has received honorarium for speaking bureau from TEVA, ACADIA, Acorda, Neurocrine, UCBPharma, USWorldMeds, and Lundbeck. He has received research grants from Biogen, Lundbeck, Sun, Restless Legs Syndrome Foundation, Parkinson’s Study Group, Lilly, and Revance. He receives royalties from the books Restless Legs Syndrome, Movement Disorders in Clinical Practice, and UpToDate.
M. Neis has nothing to disclose.
T. Odorfer has received honoraria or travel grants from Allergan, TEVA and Merz.
E. Roze received research support from Merz-Pharma, Orkyn, Aguettant, Elivie, Ipsen, Fondation Desmarest, AMADYS, Agence Nationale de la Recherche, and Fonds de Dotation Brou de Laurière; has served on scientific advisory boards for Orkyn, Aguettant, Merz-Pharma and has received speech honoraria from Orkyn, Aguettant, Merz-Pharma, Medday-Pharma, Everpharma, International Parkinson and Movement disorders Society; received travel grant from Vitalair, Aguettant, Merz-Pharma, Ipsen, Merck, Orkyn, Elivie, Dystonia medical Research Foundation, International Parkinson and Movement disorders Society, European Academy of Neurology, International Association of Parkinsonism and Related Disorders.
J. Volkman has received grant support from Boston Scientific, Medtronic, BMBF, Deutsche Forschungsgemeinschaft. He has received honoraria from Boston Scientific, Medtronic, BMBF, Deutsche Forschungsgemeinschaft. He has served as a consultant or on an advisory board for Boston Scientific, Newronika, and Medtronic.
S. Zittel has received grant support from the European Union and Werner Otto Foundation and honoraria from Merz Pharmaceuticals and Brainlab AG.
Footnotes
Relevant conflicts of interest/financial disclosures: None
References
- 1.Albanese A, et al. , Phenomenology and classification of dystonia: A consensus update. Mov Disord, 2013. 28: p. 863–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jinnah HA, The Dystonias. Continuum (Minneap Minn), 2019. 25(4): p. 976–1000. [DOI] [PubMed] [Google Scholar]
- 3.Defazio G, Berardelli A, and Hallett M, Do primary adult-onset focal dystonias share aetiological factors? Brain, 2007. 130(Pt 5): p. 1183–93. [DOI] [PubMed] [Google Scholar]
- 4.Jinnah HA and Sun YV, Dystonia genes and their biological pathways. Neurobiol Dis, 2019. 129: p. 159–168. [DOI] [PubMed] [Google Scholar]
- 5.Balint B and Bhatia KP, Isolated and combined dystonia syndromes - an update on new genes and their phenotypes. Eur J Neurol, 2015. 22(4): p. 610–7. [DOI] [PubMed] [Google Scholar]
- 6.Balint B, et al. , Dystonia. Nat Rev Dis Primers, 2018. 4(1): p. 25. [DOI] [PubMed] [Google Scholar]
- 7.Welter D, et al. , The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res, 2014. 42(Database issue): p. D1001–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mok KY, et al. , Genomewide association study in cervical dystonia demonstrates possible association with sodium leak channel. Mov Disord, 2014. 29(2): p. 245–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gomez-Garre P, et al. , Lack of validation of variants associated with cervical dystonia risk: a GWAS replication study. Mov Disord, 2014. 29(14): p. 1825–8. [DOI] [PubMed] [Google Scholar]
- 10.Lohmann K, et al. , Genome-wide association study in musician's dystonia: a risk variant at the arylsulfatase G locus? Mov Disord, 2014. 29(7): p. 921–7. [DOI] [PubMed] [Google Scholar]
- 11.Yan L, et al. , Secured web-based video repository for multicenter studies. Parkinsonism Relat Disord, 2015. 21(4): p. 366–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.LeDoux MS, et al. , Clinical and genetic features of cervical dystonia in a large multicenter cohort. Neurol Genet, 2016. 2(3): p. e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kilic-Berkmen G, et al. , The Dystonia Coalition: A Multicenter Network for Clinical and Translational Studies. Frontiers in Neurology, 2021. 12: p. 415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anderson CA, et al. , Data quality control in genetic case-control association studies. Nat Protoc, 2010. 5(9): p. 1564–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alexander DH, Novembre J, and Lange K, Fast model-based estimation of ancestry in unrelated individuals. Genome Res, 2009. 19(9): p. 1655–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsai TH, et al. , LC-MS/MS-based serum proteomics for identification of candidate biomarkers for hepatocellular carcinoma. Proteomics, 2015. 15(13): p. 2369–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Das S, et al. , Next-generation genotype imputation service and methods. Nat Genet, 2016. 48(10): p. 1284–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karczewski KJ, et al. , The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 2020. 581(7809): p. 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Watanabe K, et al. , Functional mapping and annotation of genetic associations with FUMA. Nature communications, 2017. 8(1): p. 1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jinnah HA and Sun YV, Dystonia Genes And Their Biological Pathways. Neurobiology of Disease, 2019. [DOI] [PubMed] [Google Scholar]
- 21.Lohmann K, et al. , The role of mutations in COL6A3 in isolated dystonia. J Neurol, 2016. 263(4): p. 730–4. [DOI] [PubMed] [Google Scholar]
- 22.Tuschl K, et al. , Mutations in SLC39A14 disrupt manganese homeostasis and cause childhood-onset parkinsonism-dystonia. Nat Commun, 2016. 7: p. 11601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Juneja M, et al. , A novel mutation in SLC39A14 causing hypermanganesemia associated with infantile onset dystonia. J Gene Med, 2018. 20(4): p. e3012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.