

Abstract
Background:
Despite guidelines recommending genetic counseling for patients with early-onset renal cell carcinoma (RCC), studies interrogating the spectrum of germline mutations and clinical associations in patients with early-onset RCC are lacking.
Objective:
To define the germline genetic spectrum and clinical associations for patients with early-onset RCC diagnosed at age ≤46 yr who underwent genetic testing.
Design, setting, and participants:
We retrospectively identified patients with early-onset RCC who underwent germline testing at our institution from February 2003 to June 2020.
Outcome measurement and statistical analysis:
The frequency and spectrum of pathogenic/likely pathogenic (P/LP) variants were determined. Clinical characteristics associated with mutation status were analyzed using two-sample comparison (Fisher’s exact or χ2 test).
Results and limitations:
Of 232 patients with early-onset RCC, 50% had non–clear-cell histology, including unclassified RCC (12.1%), chromophobe RCC (9.7%), FH-deficient RCC (7.0%), papillary RCC (6.6%), and translocation-associated RCC (4.3%). Overall, 43.5% had metastatic disease. Germline P/LP variants were identified in 41 patients (17.7%), of which 21 (9.1%) were in an RCC-associated gene and 20 (8.6%) in a non–RCC-associated gene, including 17 (7.3%) in DNA damage repair genes such as BRCA1/2, ATM, and CHEK2. Factors associated with RCC P/LP variants include bilateral/multifocal renal tumors, non–clear-cell histology, and additional extrarenal primary malignancies. In patients with only a solitary clear-cell RCC, the prevalence of P/LP variants in RCC-associated and non–RCC-associated genes was 0% and 9.9%, respectively.
Conclusions:
Patients with early-onset RCC had high frequencies of germline P/LP variants in genes associated with both hereditary RCC and other cancer predispositions. Germline RCC panel testing has the highest yield when patients have clinical phenotypes suggestive of underlying RCC gene mutations. Patients with early-onset RCC should undergo comprehensive assessment of personal and family history to guide appropriate genetic testing.
Patient summary:
In this study of 232 patients with early-onset kidney cancer who underwent genetic testing, we found a high prevalence of mutations in genes that increase the risk of cancer in both kidneys and other organs for patients and their at-risk family members. Our study suggests that patients with early-onset kidney cancer should undergo comprehensive genetic risk assessment.
Keywords: Early-onset renal cell carcinoma, Hereditary renal cell carcinoma syndromes, Germline sequencing, Genetic counseling, Kidney cancer
1. Introduction
Renal cell carcinoma (RCC) is typically diagnosed between the sixth and seventh decade of life, with a median age at diagnosis of 64 yr [1]. Early-onset RCC diagnosed before the age of 46 yr has been associated with hereditary RCC syndromes [2]. There are several well-known hereditary RCC syndromes, accounting for approximately 5–9% of all RCC cases, including von Hippel Lindau (VHL) disease, Birt-Hogg-Dubé (BHD) syndrome, and hereditary leiomyomatosis and RCC (HLRCC) [3]. To date, studies evaluating germline genetics in patients with early-onset RCC have been limited to select populations with disparate age cutoffs. A study of 190 Chinese patients with RCC diagnosed at the age of ≤45 yr found that 9.5% had germline pathogenic/likely pathogenic (P/LP) variants, of which two-thirds were in RCC predisposition genes and one-third were in cancer predisposition genes that have not been linked to RCC risk, such as BRCA1/2 [4]. In the USA, another study of 844 patients with RCC diagnosed at the age of ≤60 yr found germline mutations in 12.8% of patients, but only 3.7% had mutations in RCC predisposition genes [5].
Selection of patients for genetic testing has traditionally been based on phenotypic indicators suggestive of a specific gene mutation, such as tumor pathology, syndromic features, and personal and family history of cancer. For patients who present with only an early-onset solitary RCC tumor, it is challenging to decide how to perform genetic testing. In addition, the incidence of RCC in patients younger than 50 yr has steadily increased from 2.4 to 4.4 per 100 000 over the past two decades [6]. The reasons for this rise are unclear, but it results in a larger pool of patients who can be considered for germline genetic testing.
Beyond the recognition of well-defined hereditary RCC syndromes, the genetic landscape and clinical characteristics of patients with early-onset RCC are not well characterized. Prior large-scale germline sequencing studies of RCC lacked central pathology review to confirm diagnostic accuracy and detailed clinical annotations to provide information on the characteristics of patients with germline mutations [5,7]. Owing to limited and conflicting data on the genetics of patients with early-onset RCC and the rising incidence of RCC among younger patients, we sought to determine the germline variants, associated clinical characteristics, and tumor histology in a large cohort of patients with early-onset RCC who underwent genetic testing.
2. Patients and methods
2.1. Patient selection
We identified patients with early-onset RCC (age at diagnosis ≤46 yr) who underwent genetic testing from two institutional cohorts (Fig. 1). The first cohort was identified from a database of patients seen at the Memorial Sloan Kettering Cancer Center (MSK) Clinical Genetics Service from February 1, 2003 to July 1, 2020 (henceforth referred to as the guideline-based cohort). Germline testing on the basis of personal and family history was performed as available at the time of testing. The second cohort was identified from patients who consented to matched tumor-germline next-generation sequencing under the MSK-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT) protocol from May 1, 2015 to July 1, 2020; data for 47 patients in this group were previously reported [8]. In addition to tumor molecular profiling, these patients also underwent germline analysis of more than 75 cancer susceptibility genes independent of clinical or tumor phenotype (hereafter referred to as the universal testing cohort). Detailed clinical annotations were collected from electronic medical records and clinical genetics visit notes and were manually validated to confirm accuracy. Three-generation family history information was assessed. Patients with a first- or second-degree relative with RCC were classified as having a family history of RCC. This study was approved by the institutional review board at MSK.
Fig. 1 –
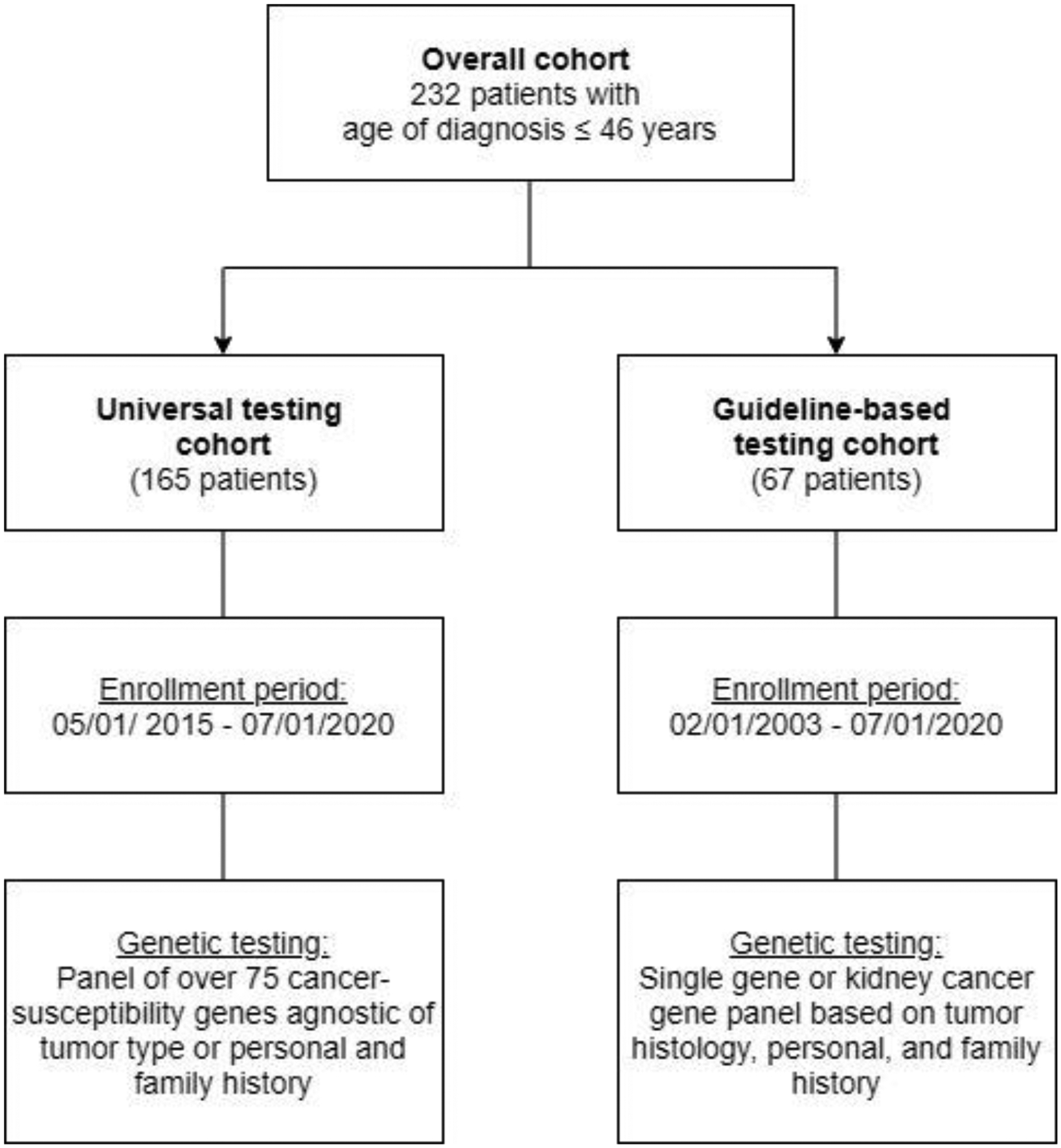
Flow diagram of the study cohort.
2.2. Somatic and germline sequencing and germline variant analysis
Germline DNA was extracted from blood samples and sequenced using MSK-IMPACT as previously described [9] or sent to commercial, New York state–approved laboratories for sequencing. MSK-IMPACT includes more than 75 genes associated with hereditary cancer predisposition, covering all cancer-predisposing genes identified in the American College of Medical Genetics and Genomics (ACMG) guidelines (Supplementary Table 1) [10].
Germline variants were interpreted by a clinical molecular geneticist or molecular pathologist according to the ACMG/Association for Molecular Pathology criteria [11]. Patient with P/LP variants were referred for in-person genetic counseling. P/LP variants were classified at the gene level as high penetrance (relative risk >4), moderate penetrance (relative risk 2–4), low penetrance (relative risk <2), uncertain penetrance, or recessive on the basis of known disease-associated risks. Patients in the universal testing cohort also had kidney tumor sequencing and somatic panel analysis of ≥341 genes using the MSK-IMPACT platform [12]. The Fraction and Allele-Specific Copy Number Estimates from Tumor Sequencing (FACETS) algorithm was used to evaluate loss of heterozygosity (LOH) at the locus of the variants [13]. Hereditary RCC syndrome genes (hereafter referred to as RCC genes) were defined as VHL, FH, FLCN, MET, TSC1/2, BAP1, SDHA/B/C/D, MITF, and PTEN (Supplementary Table 2) [3].
2.3. Statistical analysis
Patients were grouped into three categories: no P/LP variant identified, P/LP variant(s) identified in a non-RCC gene, and P/LP variant(s) identified in an RCC-associated gene. Patients who had P/LP variants in both RCC and non-RCC genes were only included in the RCC category for the primary analysis. Our primary analysis entailed a two-sample comparison (Fisher’s exact or χ2 test) of clinical and pathologic factors between (1) patients without P/LP variants and patients with P/LP variants in RCC genes and (2) patients without P/LP variants and patients with P/LP variants in non-RCC genes. All analyses were conducted using R v3.6.0 (R Foundation for Statistical Computing, Austria, Vienna). Statistical significance was set at p < 0.05.
3. Results
3.1. Cohort characteristics
A total of 232 patients with early-onset RCC were identified; 165 (71.1%) had universal genetic testing and 67 (28.9%) underwent guideline-based testing. Clinical characteristics are summarized in Table 1. The median age of onset was 38 yr (range 21–46). Of the 232 patients, 26 (11%) had bilateral/multifocal renal tumors, of whom six (23%) had discordant RCC histologic subtypes. The most common histologic subtypes were clear-cell RCC (50%), unclassified RCC (12%), and chromophobe RCC (10%). A total of 51 patients (22%) had multiple primary malignancies, including 25 (11%) with a different primary malignancy before their RCC diagnosis and 26 (11%) who developed additional cancer after their RCC diagnosis. The most common additional malignancies were thyroid cancer (n = 8), colorectal cancer (n = 5), sarcoma (n = 5), prostate cancer (n = 5), and lymphoma (n = 5). Thirty-five patients (15%) had a family history of RCC. Forty-eight patients (21%) had metastatic disease at the time of diagnosis. The median follow up was 3.4 yr (range 0–39). A total of 53 patients (23%) developed metachronous metastasis and 42 patients (18%) died of an RCC-related cause.
Table 1 –
Demographic and histologic data and clinical outcomes for patients with early-onset RCC in the overall, universal testing, and guideline-based cohorts
| Characteristic | Overall cohort (n = 232) | Universal testing cohort (n = 165) | Guideline-based cohort (n = 67) |
|---|---|---|---|
| Male, n (%) | 141 (60.8) | 106 (64.2) | 35 (52.2) |
| Median age at initial diagnosis, yr (range) | 38 (21–46) | 40 (21–46) | 38 (24–46) |
| Race, n (%) | |||
| White | 172 (74.1) | 119 (72.1) | 53 (79.1) |
| Black | 19 (8.2) | 15 (9.1) | 4 (6.0) |
| Asian | 15 (6.5) | 11 (6.7) | 4 (6.0) |
| Other/unknown | 26 (11.2) | 20 (12.1) | 6 (9.0) |
| Extrarenal primary malignancies, n (%) | 51 (21.9) | 33 (20.0) | 18 (26.9) |
| Before RCC diagnosis | 25 (10.8) | 14 (8.5) | 11 (16.4) |
| After RCC diagnosis | 26 (11.2) | 19 (11.5) | 7 (10.4) |
| Bilateral/multifocal renal tumors, n (%) | 26 (11.2) | 15 (9.1) | 11 (16.4) |
| Family history of RCC, n (%) | 35 (15.1) | 20 (12.1) | 15 (22.4) |
| RCC histology per renal tumor, n (%) | N = 257 tumors | N = 176 tumors | N = 81 tumors |
| Clear-cell RCC | 129 (50.2) | 86 (48.9) | 43 (53.1) |
| Unclassified RCC | 31 (12.1) | 24 (13.6) | 7 (8.6) |
| Chromophobe RCC | 25 (9.7) | 16 (9.1) | 9 (11.1) |
| FH-deficient RCC | 18 (7.0) | 12 (6.8) | 6 (7.4) |
| Papillary RCC | 17 (6.6) | 8 (4.5) | 9 (11.1) |
| Translocation association RCC | 11 (4.3) | 11 (6.3) | 0 |
| Angiomyolipoma | 7 (2.7) | 4 (2.3) | 3 (3.7) |
| SDH-deficient RCC | 2 (0.8) | 2 (1.1) | 0 |
| Other | 12 (4.7) | 9 (5.1) | 3 (3.7) |
| Unknown | 5 (1.9) | 4 (2.3) | 1 (1.2) |
| Metastatic disease at diagnosis, n (%) | 48 (20.7) | 35 (21.2) | 13 (19.4) |
RCC = renal cell carcinoma.
3.2. Frequency and spectrum of germline mutations
All patients in the universal testing cohort had at least 76 cancer-associated genes sequenced. The patients in the guideline-based cohort had a median of eight genes sequenced (range 1–20). In the overall cohort, 45 P/LP variants in 22 genes were identified in 41 patients (17.7%). Twenty-one patients (9.1%) had P/LP variants in RCC genes, nine (3.9%) had P/LP variants in moderate- or high-penetrance non-RCC genes, and 11 (4.7%) had P/LP variants in low-, uncertain-, or recessive-penetrance non-RCC genes (Supplementary Table 3, Fig. 2A). In the universal testing cohort, 12 patients (7.3%) had P/LP variants in RCC genes and 20 (12.1%) had P/LP variants in non-RCC genes. In the guideline-based cohort, nine patients (13.4%) had P/LP variants in RCC genes and no patient had P/LP variants in non-RCC genes (Fig. 2B). Two patients had two P/LP variants each (FH/RECQL4 and CHEK2/FANCA). One patient with both early-onset high-risk prostate cancer and FH-deficient RCC carried three P/LP variants (FH/BRCA2/FANCA). The most frequently mutated RCC genes were FH (n = 12; 5.2%), VHL (n = 4; 1.7%), and SDHB (n = 2; 0.9%). The most common moderate- or high-penetrance non-RCC genes were ATM (n = 2; 0.9%), BRCA1 (n = 2; 0.9%), and CHEK2 (n = 2; 0.9%).
Fig. 2 –
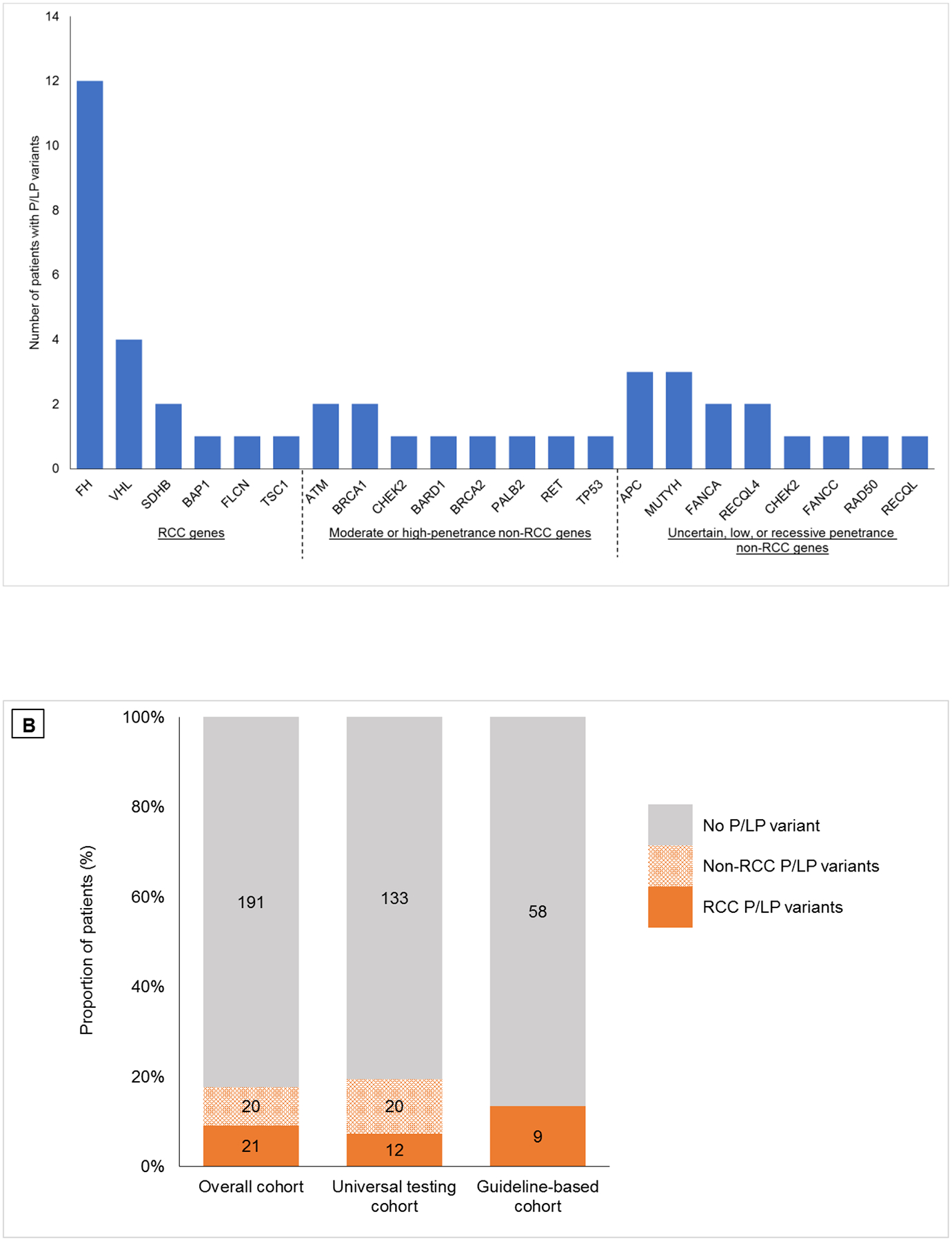
(A) Spectrum of germline pathogenic/likely pathogenic (P/LP) variants found in the overall study cohort. (B) Frequency of germline P/LP variants in cancer susceptibility genes in patients with early-onset renal cell carcinoma (RCC) in the overall cohort, the universal testing cohort who underwent Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT) sequencing of 76 or more genes, and the guideline-based cohort who underwent germline sequencing on the basis of personal and family history.
3.3. Clinical phenotypes of germline P/LP variant carriers
Compared to patients with no P/LP variants, clinical characteristics associated with patients carrying RCC P/LP variants included bilateral/multifocal renal tumors, non–clear-cell RCC histology, and an additional primary malignancy before RCC diagnosis (Table 2). As expected, these factors were not associated with the presence of non-RCC P/LP variants. Family history of RCC and metastatic disease at the time of diagnosis were not associated with the presence of a P/LP variant in RCC or non-RCC genes.
Table 2 –
Association of patient clinicopathologic characteristics with germline P/LP variants in RCC and non-RCC genes
| Characteristic | No P/LP VTs (n = 191) |
RCC VTs (n = 21) |
Non-RCC VTs (n = 20) |
p value versus no VTs | |
|---|---|---|---|---|---|
| RCC VTs | Non-RCC VTs | ||||
| Family history of RCC | 0.1 | 0.3 | |||
| No | 163 (85) | 15 (71) | 19 (95) | ||
| Yes | 28 (15) | 6 (29) | 1 (5) | ||
| Clear-cell histology | 0.003 | 0.8 | |||
| No | 84 (45) | 17 (81) | 7 (39) | ||
| Yes | 104 (55) | 4 (19) | 11 (61) | ||
| Unknown | 3 | 0 | 2 | ||
| Bilateral/multifocal renal tumor at Dx | 0.001 | >0.9 | |||
| No | 178 (93) | 14 (67) | 19 (95) | ||
| Yes | 13 (7) | 7 (33) | 1 (5) | ||
| Pre-RCC extrarenal primary malignancy | 0.01 | 0.7 | |||
| No | 175 (92) | 15(71) | 18 (90) | ||
| Yes | 16 (8) | 6 (29) | 2 (10) | ||
| Disease extent at initial RCC Dx | 0.1 | 0.08 | |||
| Localized | 156 (82) | 14 (67) | 13 (65) | ||
| Metastatic | 34 (18) | 7 (33) | 7 (35) | ||
| Unknown | 1 | ||||
Dx = diagnosis; P/LP = pathogenic/likely pathogenic; RCC = renal cell carcinoma; VTs = variants.
To understand how the presence of these predictive clinicopathologic factors modifies the probability of finding RCC P/LP variants, we stratified patients according to the presence of each factor (Fig. 3). The frequencies of RCC P/LP variants in the groups with bilateral/multifocal renal tumors, an additional primary malignancy before RCC diagnosis, or non–clear-cell histology were 33.3%, 25.0%, and 15.7%, respectively. We found that patients with a solitary clear-cell RCC and no other syndromic phenotype have no probability of carrying an RCC P/LP variant. However, the absence of clinical phenotypes of RCC P/LP variants does not exclude the possibility of other germline P/LP variants. The frequency of germline P/LP variants in non-RCC genes for patients with only a solitary clear-cell RCC was 9.9%. Six of 11 (54.5%) moderate-/high-penetrance non-RCC P/LP variants were unexpected on the basis of patients’ personal or family history of cancer.
Fig. 3 –
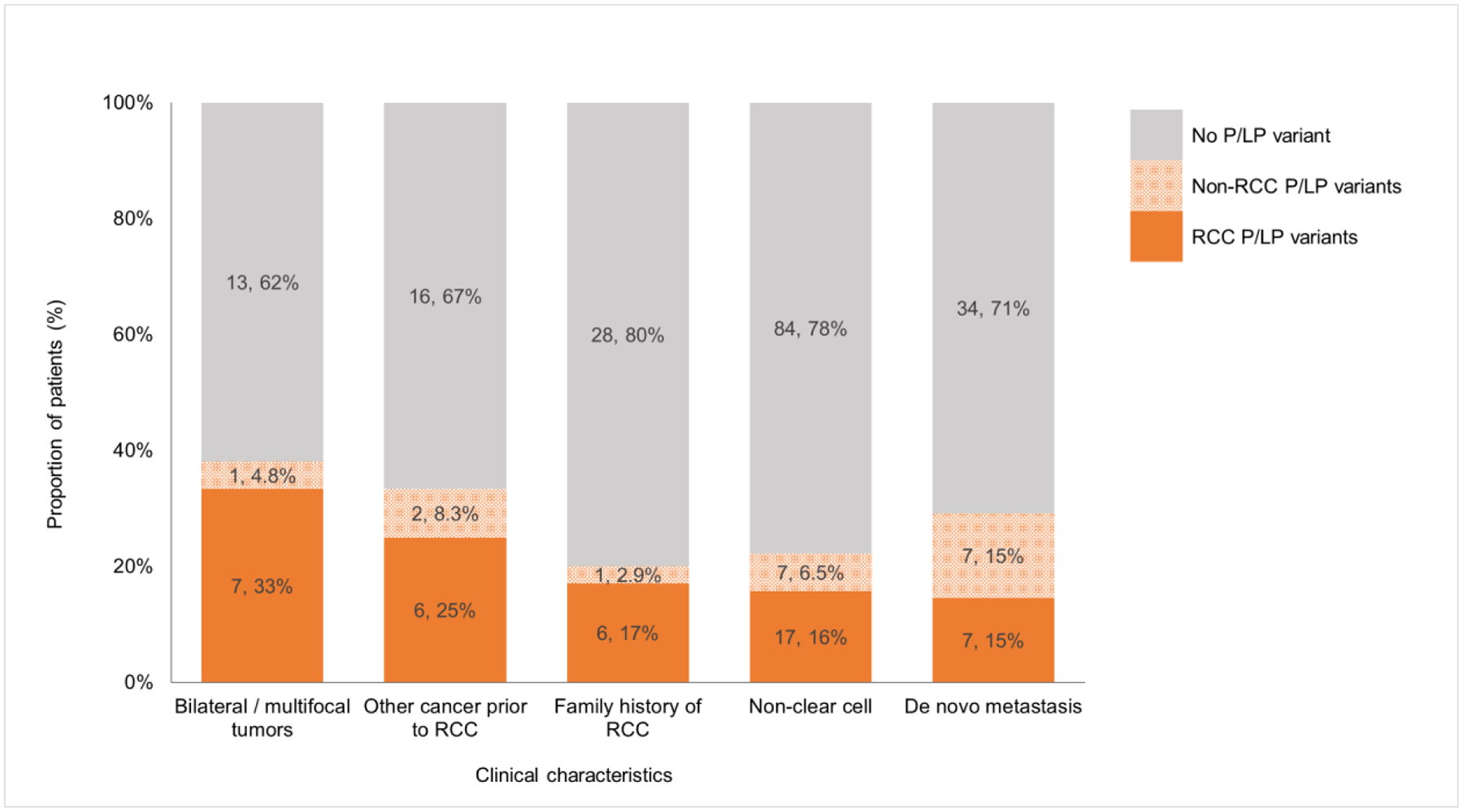
Distribution of germline pathogenic/likely pathogenic (P/LP) variants in genes with and without association to hereditary renal cell carcinoma (RCC) syndromes in patients with early-onset RCC and specific clinical characteristics.
3.4. Tumor sequencing analysis
To determine the potential pathogenic role of germline variants in RCC tumors, we evaluated the spectrum of somatic mutations and LOH in the tumors at the loci of interest in 34 germline P/LP variant carriers from the universal testing cohort who had sequencing of matched tumor and normal tissues (Fig. 4). Of ten patients with germline RCC P/LP variants, all had LOH or a somatic second alteration in the synonymous allele in their renal tumors. By contrast, of 24 patients with germline non-RCC P/LP variants, only one (4.2%) with a BRCA1 P/LP variant had LOH in the tumor, suggesting that P/LP variants in non-RCC genes, including DNA damage repair (DDR) genes, are unlikely to drive the development of RCC.
Fig. 4 –
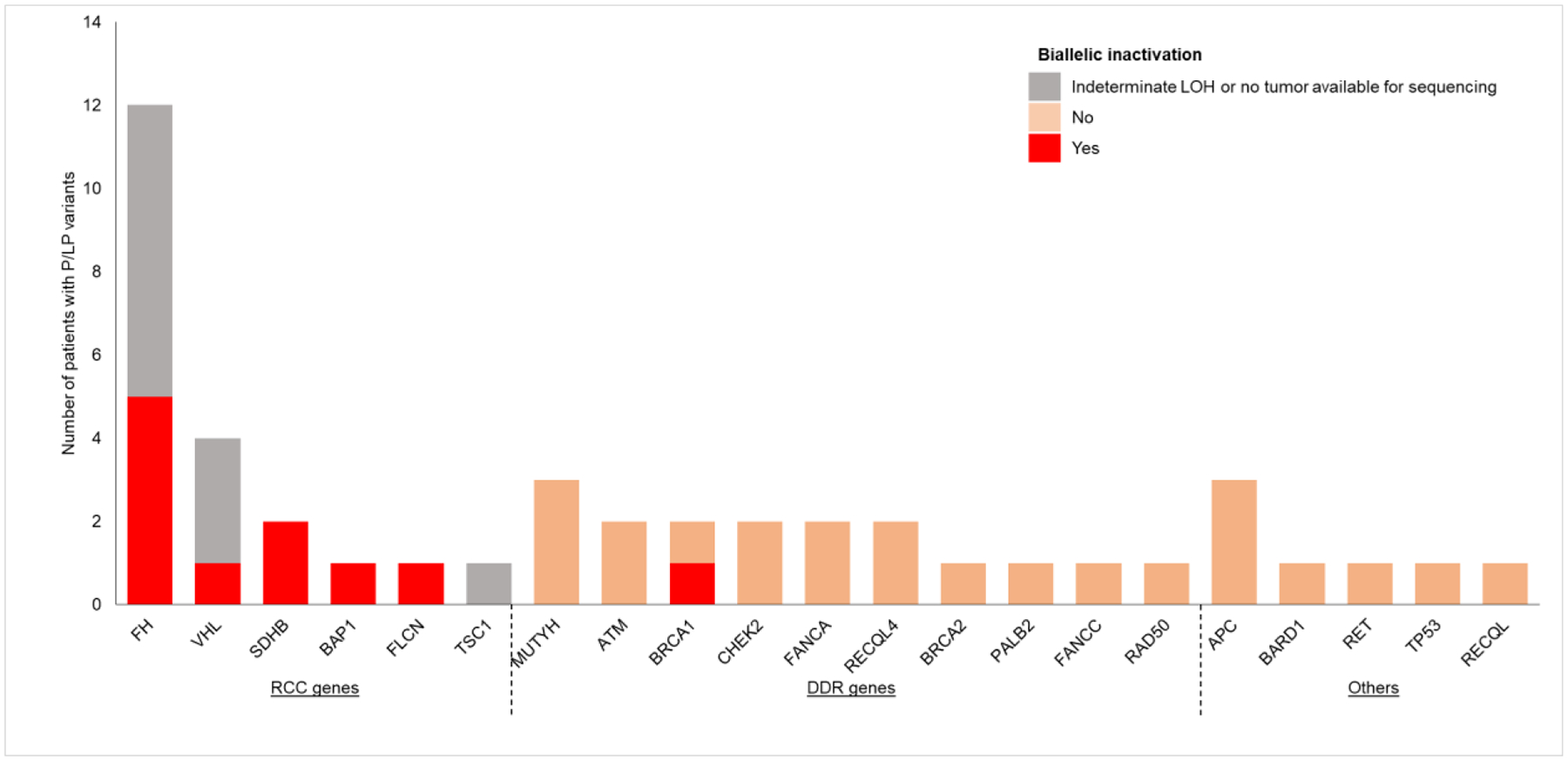
Germline pathogenic/likely pathogenic (P/LP) variants and biallelic inactivation in the renal tumor sequenced. Biallelic inactivation was evaluated by inferring loss of heterozygosity (LOH) or the presence of a somatic mutation in the same gene in the tumor. Renal cell carcinoma (RCC) genes = genes associated with hereditary RCC syndromes. DNA damage repair (DDR) genes = genes with canonical function in DNA damage repair.
4. Discussion
We reported genetic testing results for 232 patients with early-onset RCC diagnosed at age ≤46 yr at a large referral cancer center. Germline P/LP variants were identified in 18% of these patients, with half in RCC genes and half in cancer-susceptibility genes, including 7.3% in DDR genes, not previously associated with RCC syndromes. The high frequency of germline mutations in patients with early-onset RCC supports both the National Comprehensive Cancer Network and American Urological Association guideline recommendations regarding genetic counseling for patients with early-onset RCC [14,15]. The approach to genetic testing for patients with early-onset RCC is more nuanced and requires a risk-stratified approach.
Clinicopathologic factors associated with RCC P/LP variants include bilateral/multifocal renal tumors, non–clear-cell histology, and extrarenal primary malignancies before RCC diagnosis. We found that the frequency of P/LP variants in RCC genes is extremely low for patients with solitary clear-cell RCC. This is not surprising given that the cancer syndromes associated with the clear-cell subtype, mainly VHL, BHD, and to a lesser extent tuberous sclerosis complex, tend to have high penetrance for multiple other clinical features such as the central nervous system and cutaneous lesions [3,16,17]. Therefore, there may be low clinical utility in testing patients with solitary clear-cell RCC and no other syndromic features suggestive of underlying hereditary RCC genes. However, there were patients in our cohort with a strong family history of clear-cell RCC (at least two first- or second-degree relatives) and no identified germline mutations. This underscores the need for further research into environmental and multigenic inheritance mechanisms implicated in RCC development and the rising incidence of RCC among younger patients [6].
A question then arises regarding what should be done when a complete genetic risk assessment cannot be performed, such as when tumor histology or family history is unknown, when patients only have solitary clear-cell RCC tumors, or if the personal and family history are discordant with known RCC syndromes. In these scenarios, when patients have discordant phenotypes, gene panel testing should be expanded to include suspected genotypes beyond RCC genes. For example, one patient in our cohort with early-onset RCC and early-onset localized prostate cancer was found to have P/LP variants in both FH and BRCA2. Several studies indicate that clinical assessment dependent on family history of cancer and clinical phenotype is an incomplete strategy for identifying individuals whose cancers are associated with an actionable germline mutation across different tumor types [18–20]. Thus, in the absence of syndromic phenotype or family history of cancer to guide the selection of a genetic test, an inclusive testing approach for RCC genes and genes associated with cancer predisposition can be considered. Alternatively, testing can be deferred until additional clinical manifestation suggestive of an underlying germline mutation arises.
The role of genitourinary pathologists in the management of patients with RCC cannot be overstated. Accurate classification of renal tumors has profound clinical impact on prognostication, clinical management, and identification of patients with hereditary RCC syndromes. When recognized, syndromic histologic subtypes are more sensitive in detecting patients with underlying germline mutations than personal and family history combined. A total of 11/12 (91.7%) patients with FH mutations had FH-deficient RCC, whereas only 6/12 (50%) had personal clinical features or a family history suggestive of HLRCC. Likewise, both patients with SDHB mutations had SDH-deficient renal tumors, but only one had a history of pheochromocytoma indicative of a hereditary RCC syndrome. Both FH- and SDH-deficient RCC have high metastatic potential, but a prior study has shown that SDH-deficient RCC may be misdiagnosed as oncocytoma if immunohistochemical staining is not performed [21]. These results underscore the importance of updating the RCC classification as more histologic and molecular entities are recognized and of incorporating tumor classification into genetic risk assessment and germline testing.
There are several limitations to our study. Two separate testing approaches were employed: one cohort underwent phenotype-agnostic universal testing, while the other underwent guideline-based testing. This difference was reflected in the lower rate of mutations in non-RCC genes observed in the guideline-based cohort compared to the universal testing cohort. The guideline-based cohort was enrolled over a 17-yr period during which knowledge of hereditary genetics in kidney cancer has evolved, with new genes implicated in hereditary RCC discovered. Therefore, the frequency of germline mutations in this cohort may be underestimated. Our overall cohort had a high proportion of patients with advanced RCC. This may account for the higher prevalence of FH and SDH mutations associated with aggressive disease compared to prior germline sequencing studies in RCC [5,7]. We previously reported that the overall frequency of germline mutations was also higher among patients with advanced RCC [8]. Furthermore, the frequency of germline mutations in our study may not be representative of all patients with early-onset RCC, as patients included in this study probably had a personal or family history beyond early-onset RCC that prompted genetic testing. Owing to the limited number of RCC P/LP variants, multivariable analyses of the data were not performed to adjust for potential confounders.
5. Conclusions
In conclusion, early-onset RCC represents a diverse spectrum of histologic subtypes with varying malignant potential. Approximately 18% of patients with early-onset RCC harbored a germline P/LP variant, half of which are associated with hereditary RCC syndromes. Germline testing for RCC gene mutations has the highest yield when patients have other clinicopathologic features associated with hereditary RCC syndromes, such as bilateral/multifocal renal tumors, non–clear-cell histology, and additional extrarenal primary malignancies. In the setting of solitary clear-cell RCC and no additional distinguishing personal or family history after comprehensive assessment, genetic testing can be deferred and clinicians should follow up if additional clinical manifestations suggestive of an underlying germline mutation arise.
This work was previously presented at the virtual online 21st Annual Meeting of the Society of Urologic Oncology, December 3–5, 2020, and at the virtual online 2021 Genitourinary Cancers Symposium, February 11–13, 2021.
Supplementary Material
Funding/Support and role of the sponsor:
This study was funded in part by a National Institutes of Health/National Cancer Institute grant to Memorial Sloan Kettering Cancer Center (P30 CA008748), a Ruth L Kirschstein Research Service Award (T32CA082088), the J. Randall and Kathleen L. MacDonald Kidney Cancer Research Fund, the Weiss Family Kidney Research Fund, the Robert and Kate Niehaus Center for Inherited Cancer Genomics at Memorial Sloan Kettering Cancer Center, and a Harold Amos Faculty Development Award. The sponsors played no direct role in the study.
Financial disclosures:
Maria I. Carlo certifies that all conflicts of interest, including specific financial interests and relationships and affiliations relevant to the subject matter or materials discussed in the manuscript (eg, employment/affiliation, grants or funding, consultancies, honoraria, stock ownership or options, expert testimony, royalties, or patents filed, received, or pending), are the following: None.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin 2020;70:7–30. [DOI] [PubMed] [Google Scholar]
- [2].Shuch B, Vourganti S, Ricketts CJ, et al. Defining early-onset kidney cancer: implications for germline and somatic mutation testing and clinical management. J Clin Oncol 2014;32:431–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Carlo MI, Hakimi AA, Stewart GD, et al. Familial kidney cancer: implications of new syndromes and molecular insights. Eur Urol 2019;76:754–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wu J, Wang H, Ricketts CJ, et al. Germline mutations of renal cancer predisposition genes and clinical relevance in Chinese patients with sporadic, early-onset disease. Cancer 2019;125:1060–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Hartman TR, Demidova EV, Lesh RW, et al. Prevalence of pathogenic variants in DNA damage response and repair genes in patients undergoing cancer risk assessment and reporting a personal history of early-onset renal cancer. Sci Rep 2020;10:13518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Howlader N, Noone AM, Krapcho M, et al. SEER cancer statistics review, 1975–2017. Bethesda, MD: National Cancer Institute; 2020. https://seer.cancer.gov/archive/csr/1975_2017/ [Google Scholar]
- [7].Nguyen KA, Syed JS, Espenschied CR, et al. Advances in the diagnosis of hereditary kidney cancer: initial results of a multigene panel test. Cancer 2017;123:4363–71. [DOI] [PubMed] [Google Scholar]
- [8].Carlo MI, Mukherjee S, Mandelker D, et al. Prevalence of germline mutations in cancer susceptibility genes in patients with advanced renal cell carcinoma. JAMA Oncol 2018;4:1228–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Cheng DT, Prasad M, Chekaluk Y, et al. Comprehensive detection of germline variants by MSK-IMPACT, a clinical diagnostic platform for solid tumor molecular oncology and concurrent cancer predisposition testing. BMC Med Genomics 2017;10:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kalia SS, Adelman K, Bale SJ, et al. Corrigendum: Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med 2017;19:484. [DOI] [PubMed] [Google Scholar]
- [11].Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Cheng DT, Mitchell TN, Zehir A, et al. Memorial Sloan Kettering-integrated mutation profiling of actionable cancer targets (MSK-IMPACT): a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn 2015;17:251–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Shen R, Seshan VE. Facets: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res 2016;44:e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Motzer RJ, Jonasch E, Boyle S, et al. NCCN guidelines insights: kidney cancer, version 1.2021. J Natl Compr Cancer Netw 2020;18:1160–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Campbell S, Uzzo RG, Allaf ME, et al. Renal mass and localized renal cancer: AUA guideline. J Urol 2017;198:520–29. [DOI] [PubMed] [Google Scholar]
- [16].Lonser RR, Glenn GM, Walther M, et al. von Hippel-Lindau disease. Lancet 2003;361:2059–67. [DOI] [PubMed] [Google Scholar]
- [17].Schmidt LS, Linehan WM. Molecular genetics and clinical features of Birt-Hogg-Dube syndrome. Nat Rev Urol 2015;12:558–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Mandelker D, Zhang L, Kemel Y, et al. Mutation detection in patients with advanced cancer by universal sequencing of cancer-related genes in tumor and normal DNA vs guideline-based germline testing. JAMA 2017;318:825–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zhang J, Walsh MF, Wu G, et al. Germline mutations in predisposition genes in pediatric cancer. N Engl J Med 2015;373:2336–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Pritchard CC, Mateo J, Walsh MF, et al. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N Engl J Med 2016;375:443–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Gupta S, Swanson AA, Chen YB, et al. Incidence of succinate dehydrogenase and fumarate hydratase-deficient renal cell carcinoma based on immunohistochemical screening with SDHA/SDHB and FH/2SC. Hum Pathol 2019;91:114–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.