

Abstract
HDAC inhibitors show therapeutic promise for skin malignancies; however, the roles of specific HDACs in adult epidermal homeostasis and disease are poorly understood. We find that homozygous epidermal co-deletion of Hdac1 and Hdac2 in adult mouse epidermis causes reduced basal cell proliferation, apoptosis, inappropriate differentiation, and eventual loss of Hdac1/2-null keratinocytes. Hdac1/2 deficient epidermis displays elevated acetylated p53 and increased expression of the senescence gene p16. Loss of p53 partially restores basal proliferation, whereas p16 deletion promotes long-term survival of Hdac1/2-null keratinocytes. In activated GLI2-driven pre-basal cell carcinoma, Hdac1/2 deletion dramatically reduces proliferation and increases apoptosis, and knockout of either p53 or p16 partially rescues both proliferation and basal cell viability. Topical application of the HDAC inhibitor Romidepsin to normal epidermis or GLI2ΔN-driven lesions produces similar defects to genetic Hdac1/2 deletion, and these are partially rescued by loss of p16. These data reveal essential roles for HDAC1/2 in maintaining proliferation and survival of adult epidermal and basal cell carcinoma progenitors and suggest efficacy of therapeutic HDAC1/2 inhibition will depend in part on the mutational status of p53 and p16.
INTRODUCTION
Adult epidermal homeostasis involves both self-renewal and differentiation of stem and progenitor cells. The delicate balance between these two processes is perturbed in common skin conditions including epidermal malignancies and hyperproliferative diseases. Chromatin regulators such as histone deacetylases (HDACs) play critical roles in ensuring faithful inheritance of gene expression programs from mother to daughter cells and in permitting broad-scale, rapid responses to differentiation signals. HDACs function by removing acetyl groups from histone tails, generating compacted chromatin and a repressive transcriptional environment (Kouzarides, 2000). They also deacetylate key transcription factors, altering their activity and/or stability (Higashitsuji et al., 2007, Tang et al., 2008). HDAC inhibitors show promise as a potential treatment for cutaneous diseases including skin cancers. However, the genetic requirements for individual HDAC proteins in adult skin are not fully understood. Delineating these requirements and unraveling the underlying mechanisms will be important in designing more specific and effective therapeutics.
In line with decreased proliferation generally caused by HDAC inhibitors (Duvic, 2015), constitutive co-deletion of Hdac1 or Hdac2 in embryonic epidermis causes complete failure of epidermal stratification, reduced proliferation and increased apoptosis, associated with upregulation of the senescence factor p16/INK4a (p16) and hyperacetylation of the apoptotic regulator p53 (LeBoeuf et al., 2010). By contrast, homozygous epidermal deletion of Hdac1 combined with heterozygous deletion of Hdac2 using a constitutive Keratin 5 (K5)-Cre driver causes hyperproliferation, accelerated differentiation, hair follicle degeneration, and accumulation of c-MYC protein in adult epidermis, and accelerated tumor development in a mouse squamous cell carcinoma (SCC) model (Winter et al., 2013) (Hughes et al., 2014), suggesting that the pro-proliferative functions of elevated c-MYC may override the effects of partial loss of HDAC1/2 in this context. The consequences of double homozygous genetic deletion of Hdac1/2 in vivo have not yet been evaluated either in normal adult epidermis or in skin tumor models.
Basal cell carcinoma (BCC) is the most common skin tumor in Caucasian populations and is linked to sporadic or familial mutations, most commonly in the HH receptor PTCH1 or its coreceptor Smoothened (SMO), that cause constitutive signaling through the Hedgehog (HH) pathway. Although BCC lesions rarely metastasize, they are locally invasive and can cause massive local tissue damage if not treated promptly. Furthermore, familial BCC causing multiple lesions is hard to treat by excision (Atwood et al., 2014). The FDA-approved SMO antagonists Vismodegib and Sonidegib are effective in initial treatment of BCC (Sekulic and Von Hoff, 2016), but lesions recur due to acquisition of secondary downstream mutations in, or downstream of, SMO (Brinkhuizen et al., 2014) (Pricl et al., 2015) (Wang et al., 2014); (Urman et al., 2016); (Atwood et al., 2013) (Sharpe et al., 2015), resulting in a need for additional therapeutics that function downstream of SMO or target parallel pathways important for tumor development.
HH signaling can upregulate HDAC1 which deacetylates GLI1 to enhance its activity in a positive feedback loop, suggesting HDAC inhibitors as potential treatments for HH-mediated malignancies (Canettieri et al., 2010). The HDAC inhibitor Voronistat was found to be mildly effective in reducing growth of mouse BCC cells in vitro and in xenografts, and its effects were enhanced by co-treatment with a small molecule inhibitor of an atypical protein kinase C family member that recruits HDAC1 to GLI1 (Mirza et al., 2017). The latter study utilized cells that were deficient in p53 as well as PTCH1; however, approximately 50% of human BCC do not display P53 mutations (Rady et al., 1992, Soufir et al., 1999, Zhang et al., 2001). Thus, the consequences of homozygous loss of both HDAC1 and HDAC2 in postnatal epidermis in vivo, and the extent to which targeting these factors could be therapeutically useful in BCC and other hyperproliferative skin conditions is not yet entirely clear.
To address these questions, we carried out inducible in vivo epidermal-specific homozygous co-deletion of Hdac1 and Hdac2 in postnatal mouse epidermis and in a mouse model of pre-BCC expressing a mutant, constitutively active form of the SHH pathway downstream transcription factor GLI2 (GLI2ΔN) (Grachtchouk et al., 2011). Our findings reveal crucial roles for HDAC1/2 in maintaining proliferation and survival of adult epidermal progenitor cells. Our data further suggest HDAC inhibitors as promising therapeutics for epidermal hyperproliferative conditions and malignancies, but indicate that their efficacy would be reduced in carcinomas that carry concomitant loss of function mutations in p53 and/or p16.
RESULTS
Short-term inducible deletion of Hdac1/2 causes defective plantar epidermis
As epidermal homeostasis is influenced by secreted signals from cycling hair follicles (Oh and Smart, 1996) and Hdac1 deletion affects postnatal hair follicle differentiation and cycling (Hughes et al., 2014, Winter et al., 2013), we examined the effects of Hdac1/2 deletion in the epidermis by analyzing hairless plantar skin (Figure 1a), which exhibits ubiquitous expression of HDAC1 and HDAC2 (Figure 1b). We used a bi-transgenic K5-reverse tetracycline transactivator (K5-rtTA) tetO-Cre system for doxycycline-inducible deletion of Hdac1 and Hdac2 in K5 promoter-active basal cells (Ramirez et al., 1994, Teta et al., 2012). K5 promoter-active basal cells serve as stem cells for the epidermis: they both self-renew and give rise to suprabasal progeny (Blanpain and Fuchs, 2006). Thus, as the epidermis turns over, which is completed within approximately 8-10 days in adult mice (Potten et al., 1987), the Hdac1/2-deleted basal cells give rise to Hdac1/2-deleted suprabasal and terminally differentiated cells. Hdac1/2 deletion was initiated in K5-rtTA tetO-Cre mice carrying Hdac1fl/fl Hdac2fl/+ (Hdac1cKO Hdac2cHet), Hdac1fl/+ Hdac2fl/fl (Hdac1cHet Hdac2cKO) or Hdac1fl/fl Hdac2fl/fl (Hdac1/2cKO) alleles from postnatal day (P) 20 and plantar skin samples were analyzed at P27. At this time point, HDAC1 and HDAC2 were efficiently deleted in basal and many suprabasal cells in the plantar epidermis of Hdac1cKO Hdac2cHet and Hdac1cHet Hdac2cKO mice, respectively (Figure S1e–h); however epidermal thickness and proliferation were not significantly altered compared with littermate controls lacking K5-rtTA or tetO-Cre (Figure S1a–d; i–l; q,r), and apoptotic basal cells were absent (Figure S1m–p).
Figure 1 -. Hdac1/2 deletion causes apoptosis and reduced proliferation of plantar epidermis that is partially rescued by co-deletion of p53.
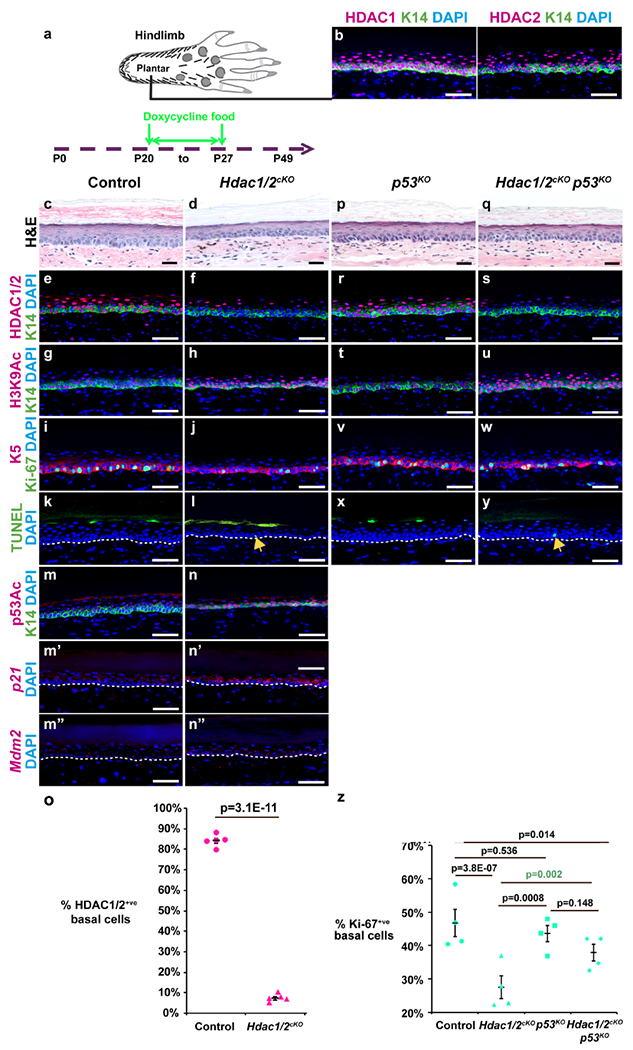
(a) Schematic of plantar foot and timing of doxycycline treatment in panels (c-z); samples in (c-z) were analyzed at P27. (b) IF for HDAC1, HDAC2 and K14 in wild-type P30 plantar skin. (c,d,p,q) Plantar skin histology for the genotypes indicated. (e-j,m,n,r-w,m,n) IF for the indicated proteins and genotypes. (m’-n”) RNAscope with the indicated probes and genotypes. (k,l,x,y) TUNEL staining for the genotypes indicated. Arrows in (l,y) indicate apoptotic basal cells. White dashed lines indicate epidermal-dermal borders. (o) Quantitation of HDAC1/2-positive plantar epidermal basal cells; n=5 Hdac1/2cKO and control mice. (z) Quantitation of Ki67-positive plantar epidermal basal cells in the genotypes indicated. Each data point represents the average percentage from ≥5 20X fields from each of 4 mice of each genotype. P-values calculated with two-tailed Student’s t-test; error bars indicate SEM. Scale bars: 50μm (b,e-n”,r-y) and 25μm (c,d,p,q).
By contrast, in Hdac1/2cKO double knockout plantar epidermis, which displayed efficient removal of both HDAC1 and HDAC2 in the basal cell layer, with less than 10% of basal cells remaining positive for HDAC1/2 by IF after 7 days of induction (Figure 1e,f,o), epidermal thickness (Figure 1c,d) and proliferation (Figure 1i,j,z) were significantly reduced and sporadic TUNEL-positive basal cells were present that were never observed in littermate controls lacking K5-rtTA or tetO-Cre (Figure 1k,l). Furthermore, levels of H3K9Ac, which is normally deacetylated by HDACs (Seto and Yoshida, 2014) were dramatically elevated (Figure 1g,h).
Decreased proliferation of Hdac1/2cKO basal cells is partially rescued by loss of p53
In line with data from embryonic epidermis, we found that acetylated p53 was elevated in postnatal Hdac1/2 deficient plantar epidermis (Figure 1m,n), and p16 mRNA and protein were respectively increased in plantar epidermis and hairy skin epithelial cells lacking HDAC1/2 (Figure S2a,b) after 7 days of induction. Furthermore, mRNA levels for the p53 target genes p21 and Mdm2 were elevated in Hdac1/2 deficient plantar epidermis (Figure 1m’–n”). To test whether the effects of short-term Hdac1/2 deletion were dependent on p53 or p16, we generated Hdac1/2cKO mice that were also null for either p53 (p53KO) (Jacks et al., 1994) or p16 (p16KO) (Sharpless et al., 2001). Loss of p53 alone had no effect on epidermal thickness relative to controls (Figure 1c,p). Absence of p53 in Hdac1/2cKO mice did not restore epidermal thickness to control levels (Figure 1q, compare with c,d) and did not rescue hyperacetylation of H3K9Ac (Figure 1g,h,t,u). Loss of p53 partially restored proliferation (Figure 1i,j,v,w,z) but did not prevent apoptosis (Figure 1k,l,x,y) of Hdac1/2 deleted basal cells. Interestingly, loss of p16 did not significantly rescue epidermal thickness (Figure S2c–f), H3K9 hyper-acetylation (Figure S2k–n), proliferation (Figure S2o–r,w) or apoptosis (Figure S2s–v) of Hdac1/2-deleted epidermis. Together, these data indicate that the immediate effects of HDAC1/2 deletion on proliferation are mediated in part via increased p53 activity.
HDAC1/2 are required for long-term maintenance of postnatal epidermal progenitor cells
5-10% of basal cells remained undeleted 7 days after initiating doxycycline treatment in Hdac1/2cKO mice (Figure 1o). To determine whether undeleted cells could out-compete deleted cells in the longer term, we analyzed plantar epidermis from mice that were induced with doxycycline food for 14 days (from P20 to P34), or 22 days (P20 to P42). At P34, the majority of epidermal basal stem cells still lacked HDAC1/2 and had populated the suprabasal layers with Hdac1/2-deleted progeny (Figure 2c,d). Basal cell proliferation remained reduced at this stage (Figure 2e,f). We noted the appearance of suprabasal cells with enlarged nuclei and premature cornification (Figure 2a,b, arrow), indicating that HDAC1/2 play critical roles in suprabasal as well as basal cells. In line with this, K5 and K14 expression was expanded and cells double positive for K14 and the suprabasal marker K10 or the granular layer marker loricrin were present (Figure 2e–j, arrows). Consistent with aberrant differentiation, we found that acetylated c-Myc was dramatically elevated in Hdac1/2cKO plantar epidermis (Figure 2k,l).
Figure 2 -. Accelerated differentiation of Hdac1/2 mutant epidermis is associated with increased levels of acetylated c-MYC.
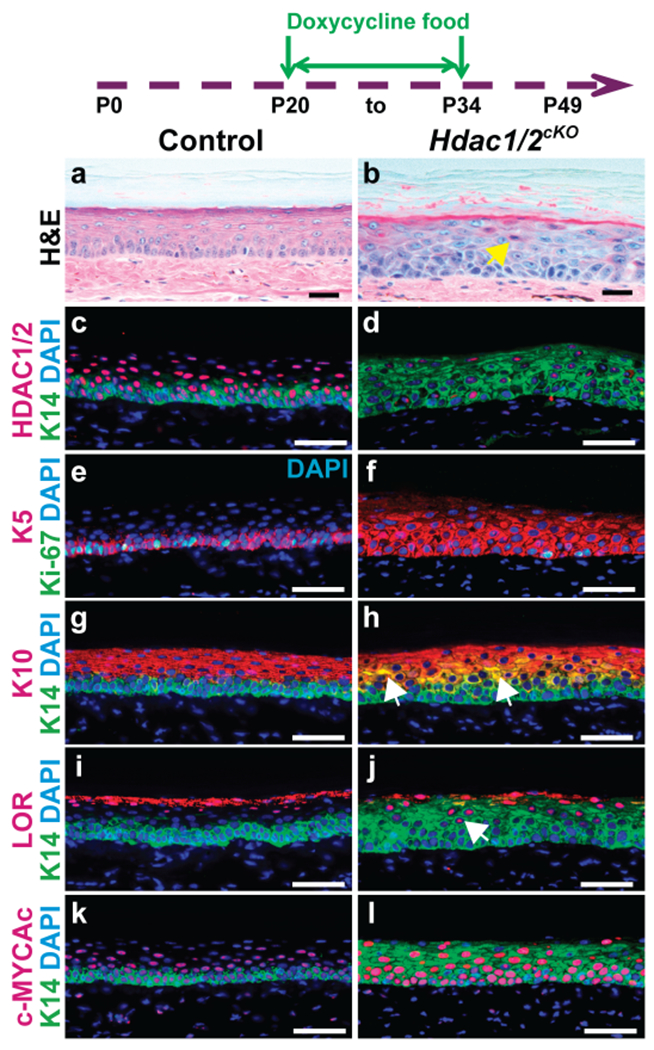
Schematic shows timing of doxycycline treatment; plantar skin samples were analyzed at P34. (a-l) Histology (a,b) and IF for the indicated proteins (c-l) in control and Hdac1/2 mutant skin as indicated; n≥4 mice for each genotype. Scale bars: 25μm (a,b), 50μm (c-l).
By contrast with persistence of HDAC1/2-deleted cells at P34, by P42 the epidermis was largely repopulated by undeleted cells (Figure 3a’,b’,y). The repopulated epidermis was thickened (Figure 3a,b) and hyperproliferative (Figure 3c,d,z) relative to controls. Expression of K14, K5 and K10 was expanded (Figure 3a’,b’,c,d,e,f; Figure S3a–d), and cells double-positive for K14 and loricrin were present (Figure S3c,d, arrow); however, premature cornification was less obvious than at P34 (Figure 3b, compare with Figure 2b), and levels of acetylated c-MYC were similar to controls (Figure S3e,f). Basal cells no longer showed evidence of increased H3K9Ac (Figure 3e,f). Consistent with loss of Hdac1/2-deleted cells, basal cell apoptosis was absent (Figure 3g,h) and levels of p53Ac in Hdac1/2cKO mutant epidermis were similar to controls (Figure S3g,h). CD3+ T-cells were absent in mutant plantar skin at P42 (Figure S4a,b); however, the repopulated skin displayed signs of a persistent inflammatory response, including elevated expression of the inflammatory marker CD26 (dipeptidyl peptidase-4) (Arwert et al., 2012, Novelli et al., 1996) in the epidermis and underlying dermis (Figure S4c,d), and increased numbers of dermal mast cells compared with controls (Figure S4e–g). These observations suggested that hyperplasia and disorganized differentiation were secondary to a damage response caused by loss of HDAC1/2 at earlier stages.
Figure 3 – Long-term maintenance of Hdac1/2-deleted cells requires p16.
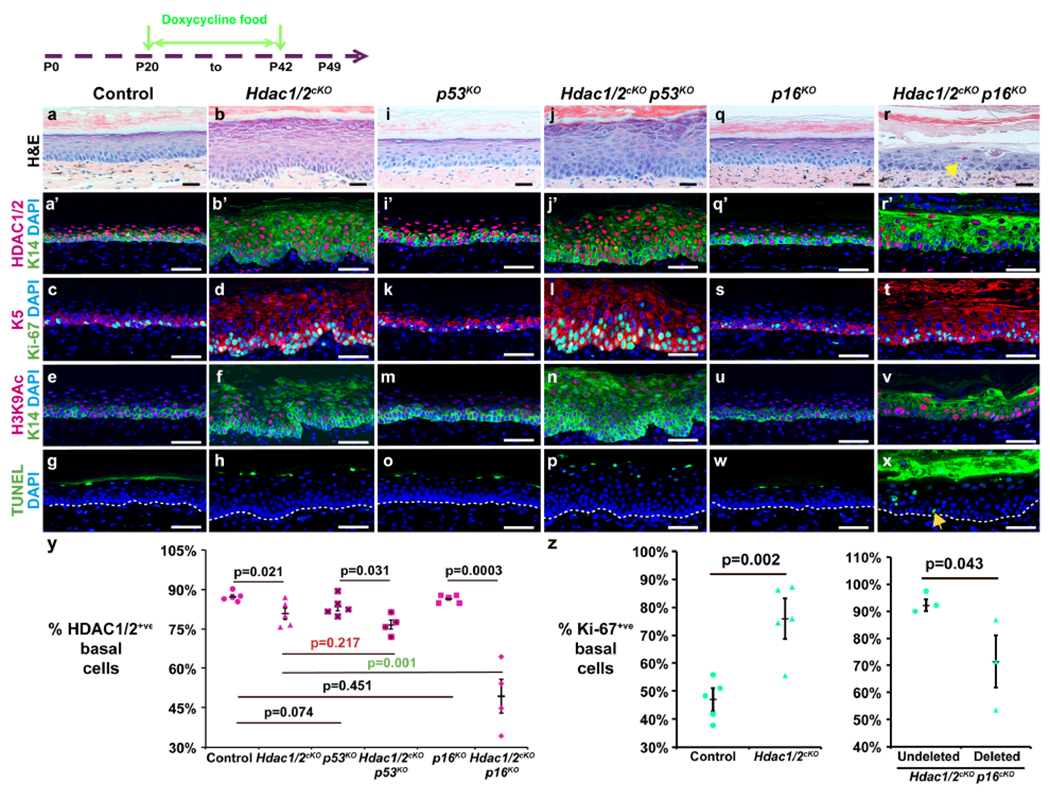
The schematic shows timing of doxycycline treatment; samples were analyzed at P42. (a,b,i,j,q,r) Histology of plantar skin for the genotypes indicated. Arrow in (r) indicates premature cornification. (a’-f, i’-n, q’-v) IF for the indicated proteins and genotypes. (g,h,o,p,w,x) TUNEL staining for the genotypes indicated. Arrow in (x) indicates apoptotic cell. (y) Quantitation of HDAC1/2-positive plantar epidermal basal cells in the genotypes indicated. N≥4 mice of each genotype were analyzed. (z) Left graph: quantitation of Ki-67-positive basal cells in control and Hdctc1/2-mutant plantar epidermis; n=5 mice of each genotype were analyzed. Right graph: quantitation HDAC1/2-positive and HDAC1/2-negative (deleted) basal cells staining positive for Ki-67 in Hdac1/2cKO p16KO plantar epidermis; n=3 mice analyzed. In (y,z), each data point represents the average percentage from ≥5 20X fields from a single mouse. P-values calculated with two-tailed Student’s t-test; error bars indicate SEM. Scale bars: 25μm (a,b,i,j,q,r), 50μm (a’-h, i’-p, q’-x).
HDAC1/2 maintain epidermal progenitor cell fitness by suppressing p16
To determine whether HDAC1/2-deleted cells were lost via apoptosis, decreased proliferation, accelerated differentiation, and/or senescence, we examined whether their ability to compete with undeleted cells was altered by co-deletion of p53 or p16. p53 co-deletion failed to rescue long-term survival of Hdac1/2 deleted cells in Hdac1/2cKO p53KO triple mutant plantar epidermis (Figure 3a–p,y); however, p16 co-deletion resulted in statistically significantly increased retention of HDAC1/2-deleted basal cells (Figure 3a’,b’,q’,r’,y). Epidermal thickness in p16KO mice was similar to controls (Figure 3a,q), but in Hdac1/2cKO p16KO mice was reduced compared with Hdac1/2cKO epidermis at P42 (Figure 3b,r). Triple mutant epidermis had an abnormal, disorganized structure characterized by appearance of enlarged cells, loss of cell-cell adhesion in more superficial epidermal layers, and premature cornification (Figure 3r, arrow). Cells double positive for K14/K10 (Figure S3a,b,i,j, arrow) and K14/loricrin (Figure S3c,d,k,l, arrows) were more prominent in Hdac1/2cKO p16KO compared with Hdac1/2cKO epidermis, and acetylated c-MYC was elevated (Figure S3e,f,m,n). Furthermore, Hdac1/2cKO p16KO triple mutant epidermis displayed higher numbers of nuclei showing elevated H3K9Ac than were observed in Hdac1/2cKO skin at the same stage (Figure 3e,f,u,v).
Interestingly, persistence of HDAC1/2-deleted cells in triple mutant epidermis was not due to rescue of their apoptosis by p16 deletion, as Hdac1/2cKO p16KO triple mutant plantar epidermis at P42 displayed increased apoptosis (Figure 3g,h,w,x, arrow) and elevated acetylated p53 (Figure S3g,h,o,p) compared with control, Hdac1/2cKO or p16KO epidermis at the same stage. Furthermore, analysis of the proliferative status of HDAC1/2-negative cells compared with undeleted cells in the same triple mutant Hdac1/2cKO p16KO samples showed that cells lacking HDAC1/2 were less proliferative than the undeleted cells (Figure 3z). These results suggest that p16 deletion enables survival of Hdac1/2 deleted keratinocytes by preventing their senescence, rather than by rescuing decreased proliferation, accelerated differentiation, or apoptosis.
Taken together, our data indicate that HDAC1/2 play at least three major and distinct roles in postnatal epidermis: (i) deacetylation of p53 to permit progenitor cell proliferation; (ii) deacetylation of c-MYC to prevent premature and disordered differentiation; and (iii) repression p16 to suppress senescence and allow for long-term maintenance of progenitor cells.
Forced GLI2ΔN expression in epidermis initiates BCC-like tumorigenesis
To investigate whether homozygous co-deletion of Hdac1/2 could combat initial stages of epidermal tumorigenesis, we utilized doxycycline-inducible epithelial-specific expression of a mutant, constitutively active form of GLI2, GLI2ΔN in K5-rtTA tetO-GLI2ΔN mice. GLI2ΔN stimulates HH signaling and can promote formation of BCC-like growths (Grachtchouk et al., 2011). Adult K5-rtTA tetO-GLI2ΔN mice placed on high dose (200 μg/ml) doxycycline drinking water displayed invagination of plantar epidermis (Figure S5a,b), efficient expression of MYC-tagged GLI2ΔN (Figure S5c,d), increased proliferation (Figure S5c–e), and expression of the BCC markers K17 and SOX9 (Anderson-Dockter et al., 2012, Vidal et al., 2008) (Figure S5f–i) within 7 days of initiating induction, providing a rapid and consistent system for analyzing molecular mechanisms underlying proliferative responses to GLI2ΔN. Forced expression of GLI2ΔN did not affect HDAC1/2 expression (Figure S5j,k). Interestingly, though, acetylated p53 was slightly increased (Figure S5l,m) and p16 mRNA levels were elevated (Figure S5n,o) in GLI2ΔN-expressing epidermis.
GLI2ΔN-expressing mice died after approximately 10-days of induction with high dose doxycycline water; however, mice treated with a lower dose of doxycycline water (10 μg/ml), survived for at least one month and developed tumors in plantar skin resembling human BCC (Figure S5p,q), including the appearance of highly proliferative cells at the leading edges of tumors (Figure S5r,s), and expression of K17 and SOX9 (Figure S5t–w). Thus, forced expression of Gli2ΔN promotes BCC-like tumorigenesis in the plantar skin of K5-rtTA tetO-GLI2ΔN mice.
Hdac1/2 deletion in pre-BCC-like lesions significantly reduces proliferation and induces apoptosis
Immunofluorescence for HDAC1 or HDAC2 revealed expression of both these proteins in pre-BCC lesions of K5-rtTA tetO-GLI2ΔN mice induced with high dose doxycycline water for 7 days (Figure 4a,b). To test whether deletion of HDAC1 and HDAC2 affected initiation of pre-BCC, we generated K5-rtTA tetO-Gli2ΔN tetO-Cre Hdac1fl/+ Hdac2fl/fl (Gli2ΔNK5-rtTA Hdac1cHet/Hdac2cKO), K5-rtTA tetO-Gli2ΔN tetO-Cre Hdac1fl/fl Hdac2fl/+ (GliNΔNK5-rtTA Hdac1cKO/Hdac2cHet) and K5-rtTA tetO-Gli2ΔN tetO-Cre Hdac1fl/fl Hdacf2fl/fl (Gli2ΔNK5-rtTA Hdac1/2cKO) mice as well as controls lacking tetO-Cre and placed them on high dose doxycycline water. GLI2ΔN expression was apparent by 4 days after initiating doxycycline treatment (Figure 4c,d). HDAC1/2 were mosaically deleted in basal cells at 4 days (Figure 4c,d); by 7 days after initiating treatment, HDAC1/2 were efficiently deleted (Figure 4e,f), p53 and c-MYC were hyperacetylated, and p16 mRNA levels were significantly increased (Figure 4g–i; Figure S7i,j). Subsequent analyses were therefore conducted on tissue from mice at the 7-day time point.
Figure 4 -. Hdac1/2 deletion increases p53 acetylation and elevates p16 mRNA levels in GLI2ΔN-expressing plantar epidermis.
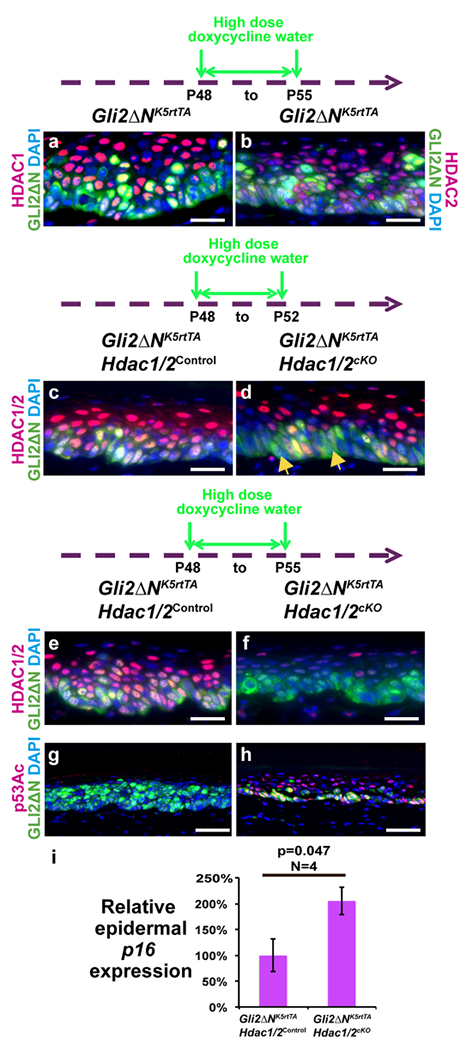
Schematics show the timing of high-dose doxycycline treatment; samples were analyzed at P55 (a,b,e-i) or P52 (c,d). (a,b) IF for Myc-tagged GLI2ΔN, HDAC1 (a) and HDAC2 (b) in induced K5-rtTA tetO-GLI2ΔN plantar epidermis. (c-f) IF for GLI2ΔN and HDAC1/2 in plantar epidermis of the indicated genotypes induced for 4 (c,d) or 7 (e,f) days. (g,h) IF for GLI2ΔN and acetylated p53 in plantar epidermis of the indicated genotypes induced for 7 days. (i) qPCR forp16 mRNA in induced K5-rtTA tetO-GLI2ΔN Hdac1/2cKO plantar epidermis compared with K5-rtTA tetO-GLI2ΔN Hdac1/2control. P-values calculated with two-tailed Student’s t-test; error bars indicate SEM. N≥5 pairs of mutant and control mice in (a-h); n=4 pairs of mutant and control mice in (i). Scale bars: 25μm (a-f), 50μm (g,h).
Homozygous loss of Hdac1 combined with heterozygous loss of Hdac2 in Gli2ΔN-expressing plantar epidermis (Figure S6e,f) produced a slight decrease in epidermal thickening and invagination (Figure S6a,b), a mild but significant decrease in proliferation (Figure S6i,j,q), but no increase in apoptosis (Figure S6m,n), while heterozygous loss of Hdac1 combined with homozygous loss of Hdac2 (Figure S6g,h) had no obvious impact on epidermal thickening and invagination (Figure S6c,d), proliferation (Figure S6k,l,r) or apoptosis (Figure S6o,p), indicating that the functions of HDAC2 in this context are partially redundant with those of HDAC1.
Double homozygous deletion of Hdac1/2 in GLI2ΔN-expressing epidermis (Figure 5g,h) significantly reduced thickening and invagination (Figure 5a,b) and proliferation (Figure 5m,n,y), increased basal cell apoptosis (Figure 5s,t,z), and caused slightly expanded K10 (Figure S7a,b) and loricrin (Figure S7e,f) expression in some regions compared with controls lacking tetO-Cre.
Figure 5 – Hdac1/2 deletion reduces proliferation and increases apoptosis of GLI2ΔN-expressing plantar epidermis via p53- and p16-dependent mechanisms.
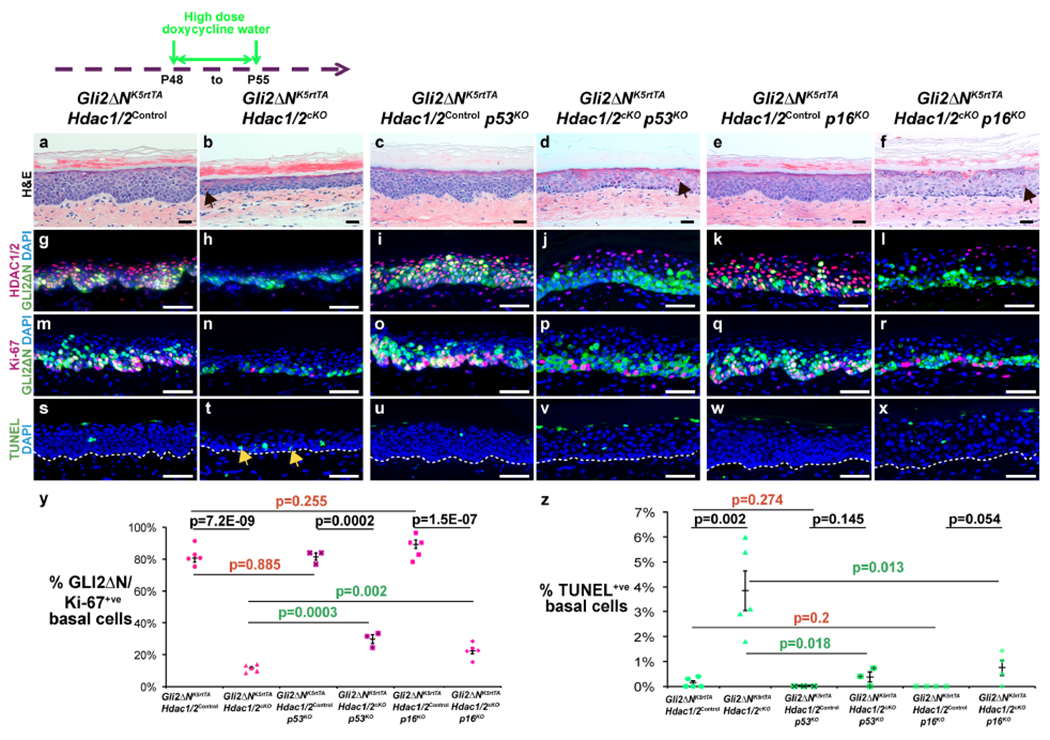
Schematic shows timing of doxycycline treatment; samples were analyzed at P55. (a-f) Histology of plantar epidermis for the indicated genotypes. Arrows in (b,d,f) indicate prematurely cornified cells. (g-r) IF for the indicated proteins and genotypes. (s-x) TUNEL staining for the indicated genotypes. Arrows in (t) indicate apoptotic cells. White dashed lines indicate epidermal-dermal boundaries. (y,z) Quantitation of Ki-67-positive (y) or TUNEL-positive (z) GLI2ΔN-expressing plantar epidermal basal cells for the indicated genotypes; n≥3 mice of each genotype were analyzed. P-values calculated with two-tailed Student’s t-test; error bars indicate SEM. Scale bars: 25μm (a-f), 50μm (g-x).
Deletion of p53 or p16 partially rescues basal proliferation and apoptosis in Hdac1/2 deficient GLI2ΔN-expressing plantar epidermis
To determine whether the effects of Hdac1/2 deletion in pre-BCC were mediated by p53 or p16, we generated Gli2ΔNK5-rtTA Hdac1/2control mice lacking tetO-Cre, and Gli2ΔNk5-rtTA Hdac1/2cKO mice that either carried null mutations in p53 or p16 or were wild-type for p53 and p16. In the absence of Hdac1/2 deletion, loss of p53 or p16 did not affect GLI2ΔN-mediated epidermal thickening and invagination (Figure 5a,c,e), HDAC1/2 expression (Figure 5g,i,k), proliferation (Figure 5m,o,q,y), or apoptosis (Figure 5s,u,w,z). However, concomitant loss of either p53 or p16 partially rescued the decreased thickness (Figure 5b,d,f), decreased proliferation (Figure 5n,p,r,y), and increased apoptosis (Fig 5 t,v,x,z) caused by Hdac1/2 deletion in GLI2ΔN-expressing epidermis. Premature cornification was observed in GLI2ΔN-expressing plantar epidermis deleted for Hdac1/2, with or without concomitant loss of p53 or p16 (arrows in b,d,f). Interestingly, co-deletion of p16 further exacerbated expanded expression of K10 (Figure S7a–d, arrow) and loricrin (Figure S7e–h, arrows), enlargement of suprabasal nuclei (Figure 5b,f), and elevated acetylated c-MYC levels (Figure S7i–l) caused by Hdac1/2 deletion in GLI2ΔN-expressing epidermis. Thus, both p53 and p16 are required to maintain proliferation and prevent apoptosis in pre-BCC.
Romidepsin reduces proliferation and induces apoptosis in normal and GLI2ΔN-expressing plantar epidermis
Our data from genetic deletion experiments suggested that small molecule HDAC1/2 inhibitors might be effective in treating BCC and other hyperproliferative skin conditions. As mouse plantar epidermis is too thick to permit easy access of topically applied chemical inhibitors, we tested this by topically applying Romidepsin or vehicle-only to dorsal paw skin of P50 control or GLI2ΔN-expressing mice that had been induced with high dose doxycycline water from P46. After 4 consecutive days of inhibitor application to dorsal paw skin, Romidepsin caused histological changes (Figure 6a,b,a’,b’), including epidermal damage, hair follicle defects, and the presence of an inflammatory infiltrate in the dermis (Figure S8a,b). Romidepsin treatment resulted in increased H3K9 acetylation (Figure 6c,d,c’,d’), significantly reduced proliferation (Figure 6e,f,q,e’,f’,q’), and basal cell apoptosis (Figure 6g,h,g’,h’, arrows) of both normal and GLI2ΔN-expressing epidermis compared with vehicle treated skin. Notably, the epidermal thickening and invaginations observed in GLI2ΔN-expressing dorsal paw epidermis were diminished following Romidepsin treatment (Figure 6a’,b’). In parallel, topical Romidepsin altered the patterns of K10 and loricrin expression (Figure S8c–f,k–n), and caused hyperacetylation of both c-MYC (Figure S8g,h,o,p) and p53 (Figure S8i,j,q,r). These phenotypes were in line with the effects of genetic deletion of Hdac1/2 in normal and GLI2ΔN-expressing interfollicular epidermis. By contrast with the short-term effects of genetic loss of Hdac1/2, and despite reduced proliferation, epidermal thickness was increased, rather than decreased by topical Romidepsin (Figure 6a,b). This was likely due to a more pronounced effect on c-MYC hyperacetylation in suprabasal cells caused by topical application of inhibitor, which can be expected to penetrate suprabasal layers before affecting Hdac1/2 basal cells, compared with K5 promoter-driven deletion which first removes Hdac1/2 in the basal layer. In line with this, forced expression of c-Myc in suprabasal epidermis causes hyperplasia (Waikel et al., 1999). Together with our observation of differentiation defects in Hdac1/2-deleted suprabasal cells (Figure 2), these data indicate that HDAC1/2 play key roles in suprabasal as well as basal cells. These results suggest topical Romidepsin as a promising therapeutic for BCC and other hyperproliferative skin conditions in human patients.
Figure 6 – p16 deletion partially rescues reduced IFE and pre-BCC proliferation and increased apoptosis caused by topical Romidepsin.
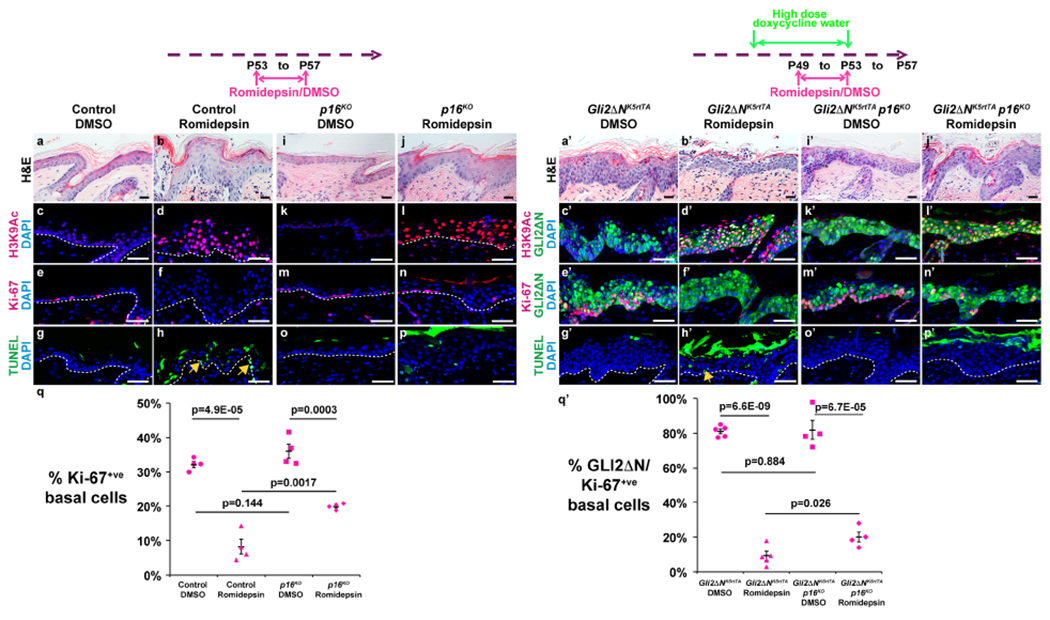
Schematics show time lines for topical application of Romidepsin or vehicle to dorsal paw skin starting at P53 (both diagrams) and high-dose oral doxycycline treatment starting at P49 (right diagram). All samples were analyzed at P57. (a,b,i,j,a’,b’,i’,j’) Histology of dorsal paw skin of the indicated genotypes following treatment with vehicle (DMSO) or Romidepsin as indicated. (c-f, k-n, c’-f’, k’-n’) IF for the indicated proteins in dorsal paw skin of the indicated genotypes following vehicle or Romidepsin treatment. (g,h,o,p,g’,h’,o’,p’) TUNEL assay for the genotypes indicated following vehicle or Romidepsin treatment. Arrows in (h,h’) indicate apoptotic cells. (q) Quantitation of Ki-67-positive IFE basal cells in control or p16-null dorsal paw IFE treated with vehicle or Romidepsin. (q’) Quantitation of Ki-67-positive basal cells in GLI2ΔN-expressing dorsal paw IFE with or without p16-deletion following treatment with vehicle or Romidepsin; n=4 mice of each genotype analyzed. P-values calculated with two-tailed Student’s t-test; error bars indicate SEM. Scale bars: 25μm (a,b,i,j,a’,b’,i′,j’), 50μm (c-h, k-p, c’-h’, k’-p’).
The effects of topical Romidepsin depend in part on intact p16 function
To test whether loss of p16 could impair the effectiveness of chemical HDAC inhibition in BCC, we compared the effects of topical Romidepsin on normal dorsal paw skin and GLI2ΔN-induced pre-BCC lesions in mice with homozygous p16 deletion and control littermates wild-type for p16. Loss of p16 had no effect on morphology (Figure 6a,i,a’,i’), H3K9Ac levels (Figure 6c,k,c’,k’), proliferation (Figure 6e,m,q,e’,m’,q’) or apoptosis (Figure 6g,o,g’,o’) of vehicle-treated normal or GLI2’N-expressing epidermis. By contrast, whilep16 deletion in Romidepsin-treated epidermis did not alter H3K9Ac levels caused by topical Romidepsin (Figure 6d,l,d’,l’), it restored proliferation from approximately 25% to approximately 60% of the levels seen in vehicle-treated normal epidermis, which was highly statistically significant (Figure 6e,f,m,n,q), and also decreased basal cell apoptosis resulting from topical Romidepsin treatment (Figure 6g,h,o,p). Similarly, loss of p16 in Romidepsin-treated pre-BCC lesions restored epithelial invaginations (Figure 6b’,j’), increased proliferation from less than 10% to approximately 25% of the levels seen in vehicle-treated lesions (Figure 6e’,f’,m’,n’,q’), and prevented basal cell apoptosis (Figure 6g’,h’,o’,p’). Thus, the efficacy of Romidepsin in blocking proliferation and inducing apoptosis in both normal skin and pre-BCC-like lesions was significantly diminished in the absence of p16.
DISCUSSION
We show here that HDAC1 and HDAC2 function in postnatal epidermis to maintain proliferation in part by deacetylating p53, and permit long-term cell survival by repressingp16. Interestingly, while we found that inducible homozygous deletion of Hdac1 together with heterozygous loss of Hdac2 did not alter proliferation in postnatal plantar epidermis in the short term, previous studies have described hair follicle defects and epidermal hyperproliferation in hairy skin of mice with constitutive homozygous loss of Hdac1, which were exacerbated by heterozygous Hdac2 deletion (Hughes et al., 2014) (Winter et al., 2013). Thus, the influence of degenerating hair follicles, and the effects of longer-term compound homozygous/heterozygous deletion of Hdac1 and Hdac2, reveal non-redundant functions of these regulators in adult epidermis.
Similar to its effects in normal plantar epidermis, homozygous Hdac1/2 deletion caused decreased proliferation, increased apoptosis and premature differentiation in GLI2ΔN-expressing pre-BCC lesions. Co-deletion of p53 partially rescued both proliferation and apoptosis, providing an explanation for the relatively mild effects of the HDAC inhibitor Voronistat observed in mice xenografted with Ptch1−/− P53−/− BCC cells (Mirza et al., 2017). While not as common as P53 mutations, loss of heterozygosity for P16 occurs in approximately 4% of human BCC (Soufir et al., 1999). Interestingly, we observed partial rescue of proliferation and decreased apoptosis when p16 was co-deleted with Hdac1/2. These data indicate that in hyperproliferative lesions HDAC1/2-mediated suppression of the functions of both p16 and p53 is important to maintain proliferation and prevent apoptosis.
To test directly whether HDAC inhibition could reduce proliferation and increase apoptosis of GLI2ΔN-expressing pre-BCC lesions, we used topical application of the Class I HDAC inhibitor Romidepsin. Topical Romidepsin was effective at increasing H3K9 acetylation, increasing apoptosis, and inducing premature differentiation, and decreased proliferation to less than 10% of the levels observed in vehicle-treated lesions. Homozygous deletion of p16 restored proliferation of Romidepsin-treated lesions to approximately 25% of the levels seen in vehicle-treated skin.
Taken together, our data suggest that topical HDAC1/2 inhibition is likely to prove effective in reducing proliferation of pre-BCC lesions that lack P53 or P16 mutations. Collectively, approximately 46% of human BCC lack such mutations, suggesting potential susceptibility of a large fraction of BCC to Romidepsin treatment, and highlighting the importance of genotyping BCC for P53 and P16 before initiating therapy.
The experiments described here focused on the effects of HDAC1/2 deletion or inhibition on early stages of BCC initiation rather than in established tumors. Importantly, however, we observed similar effects of homozygous Hdac1/2 deletion in normal hairless skin, where HH signaling is generally not active, and in pre-BCC lesions caused by GLI2ΔN-induced constitutive HH signaling. These results demonstrate that removal of HDAC1/2 function has significant consequences unrelated to the HH pathway. This conclusion is supported by partial rescue of Hdac1/2 mutant phenotypes upon co-deletion of p53 or p16, which are not canonical HH targets. Thus, our data reveal general effects of HDAC inhibition on epidermal proliferation, suggesting that this approach could be useful in treating established BCC lesions, and hyperproliferative skin conditions unrelated to BCC. The effects of topical inhibition on normal skin also highlight the potential for side effects in normal tissues; indeed, systemic Romidepsin is associated with thrombocytopenia (Bishton et al., 2011) and electrocardiographic changes (Sager et al., 2015). In light of this, topical application limited to lesional skin may be an attractive therapeutic option.
MATERIALS AND METHODS
Mice
The mouse lines used and methods for genotyping, induction, and topical Romidepsin application are detailed in Supplementary Data. All animal experiments were performed with approved animal protocols according to institutional guidelines established by the IACUC committees of the University of Pennsylvania and the Icahn School of Medicine at Mount Sinai.
Histology, immunostaining and TUNEL assays
Tissues dissected from euthanized mice were fixed in 4% Paraformaldehyde/PBS (Affymetrix/USB) overnight at 4 °C, and then paraffin embedded and sectioned at 5μm. Detailed experimental procedures, antibodies and statistical analysis methods are provided in Supplementary Data.
Real-time PCR
Reverse-transcription was performed using a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems), and cDNA was subjected to real time PCR using the StepOnePlus system and SYBR Green Kit (Applied Biosystems). Detailed methods and primers are provided in Supplementary Text.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Adam Glick for K5-rtTA mice; Eric N. Olson for Hdac1fl/fl and Hdac2fl/fl mice; Steven Prouty for histology; and Mingang Xu, Katherine M. Szigety, and Deborah J. Moran for helpful discussions. This work was supported by RO1AR063146 (SEM) and the Penn Skin Biology and Diseases Resource-based Center P30AR069589.
Abbreviations:
- HDAC
histone deacetylase
- p16
p16/INK4a
- K5
keratin 5
- rtTA
reverse tetracycline transactivator
- SCC
squamous cell carcinoma
- BCC
basal cell carcinoma
- SMO
Smoothened
- HH
hedgehog
- P
postnatal day
- IF
immunofluorescence
- SEM
standard error of the mean
- IFE
interfollicular epidermis
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST
The authors declare that they have no conflicts of interest.
DATA AVAILABILITY STATEMENT
The authors declare that the main data supporting the findings of this study are available within the article and its Supplemental Information files. All correspondence and material requests should be addressed to Sarah E. Millar. This study includes no data deposited in external repositories.
REFERENCES
- Anderson-Dockter H, Clark T, Iwamoto S, Lu M, Fiore D, Falanga JK, et al. Diagnostic utility of cytokeratin 17 immunostaining in morpheaform basal cell carcinoma and for facilitating the detection of tumor cells at the surgical margins. Dermatologic surgery : official publication for American Society for Dermatologic Surgery [et al] 2012;38(8): 1357–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arwert EN, Mentink RA, Driskell RR, Hoste E, Goldie SJ, Quist S, et al. Upregulation of CD26 expression in epithelial cells and stromal cells during wound-induced skin tumour formation. Oncogene 2012;31(8):992–1000. [DOI] [PubMed] [Google Scholar]
- Atwood SX, Li M, Lee A, Tang JY, Oro AE. GLI activation by atypical protein kinase C iota/lambda regulates the growth of basal cell carcinomas. Nature 2013;494(7438):484–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood SX, Whitson RJ, Oro AE. Advanced treatment for basal cell carcinomas. Cold Spring Harb Perspect Med 2014;4(7):a013581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishton MJ, Harrison SJ, Martin BP, McLaughlin N, James C, Josefsson EC, et al. Deciphering the molecular and biologic processes that mediate histone deacetylase inhibitor-induced thrombocytopenia. Blood 2011;117(13):3658–68. [DOI] [PubMed] [Google Scholar]
- Blanpain C, Fuchs E. Epidermal stem cells of the skin. Annu Rev Cell Dev Biol 2006;22:339–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkhuizen T, Reinders MG, van Geel M, Hendriksen AJ, Paulussen AD, Winnepenninckx VJ, et al. Acquired resistance to the Hedgehog pathway inhibitor vismodegib due to smoothened mutations in treatment of locally advanced basal cell carcinoma. J Am Acad Dermatol 2014;71(5):1005–8. [DOI] [PubMed] [Google Scholar]
- Canettieri G, Di Marcotullio L, Greco A, Coni S, Antonucci L, Infante P, et al. Histone deacetylase and Cullin3-REN(KCTD11) ubiquitin ligase interplay regulates Hedgehog signalling through Gli acetylation. Nature cell biology 2010;12(2):132–42. [DOI] [PubMed] [Google Scholar]
- Duvic M Histone Deacetylase Inhibitors for Cutaneous T-Cell Lymphoma. Dermatol Clin 2015;33(4):757–64. [DOI] [PubMed] [Google Scholar]
- Grachtchouk M, Pero J, Yang SH, Ermilov AN, Michael LE, Wang A, et al. Basal cell carcinomas in mice arise from hair follicle stem cells and multiple epithelial progenitor populations. The Journal of clinical investigation 2011;121(5):1768–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashitsuji H, Masuda T, Liu Y, Itoh K, Fujita J. Enhanced deacetylation of p53 by the anti-apoptotic protein HSCO in association with histone deacetylase 1. J Biol Chem 2007;282(18):13716–25. [DOI] [PubMed] [Google Scholar]
- Hughes MW, Jiang TX, Lin SJ, Leung Y, Kobielak K, Widelitz RB, et al. Disrupted ectodermal organ morphogenesis in mice with a conditional histone deacetylase 1, 2 deletion in the epidermis. J Invest Dermatol 2014;134(1):24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, et al. Tumor spectrum analysis in p53-mutant mice. Current biology : CB 1994;4(1):1–7. [DOI] [PubMed] [Google Scholar]
- Kouzarides T Acetylation: a regulatory modification to rival phosphorylation? The EMBO journal 2000;19(6):1176–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBoeuf M, Terrell A, Trivedi S, Sinha S, Epstein JA, Olson EN, et al. Hdac1 and Hdac2 act redundantly to control p63 and p53 functions in epidermal progenitor cells. Dev Cell 2010;19(6):807–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirza AN, Fry MA, Urman NM, Atwood SX, Roffey J, Ott GR, et al. Combined inhibition of atypical PKC and histone deacetylase 1 is cooperative in basal cell carcinoma treatment. JCI insight 2017;2(21). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novelli M, Savoia P, Fierro MT, Verrone A, Quaglino P, Bernengo MG. Keratinocytes express dipeptidyl-peptidase IV (CD26) in benign and malignant skin diseases. The British journal of dermatology 1996;134(6):1052–6. [PubMed] [Google Scholar]
- Oh HS, Smart RC. An estrogen receptor pathway regulates the telogen-anagen hair follicle transition and influences epidermal cell proliferation. Proc Natl Acad Sci U S A 1996;93(22):12525–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potten CS, Saffhill R, Maibach HI. Measurement of the transit time for cells through the epidermis and stratum corneum of the mouse and guinea-pig. Cell Tissue Kinet 1987;20(5):461–72. [DOI] [PubMed] [Google Scholar]
- Pricl S, Cortelazzi B, Dal Col V, Marson D, Laurini E, Fermeglia M, et al. Smoothened (SMO) receptor mutations dictate resistance to vismodegib in basal cell carcinoma. Mol Oncol 2015;9(2):389–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rady P, Scinicariello F, Wagner RF Jr., Tyring SK. p53 mutations in basal cell carcinomas. Cancer research 1992;52(13):3804–6. [PubMed] [Google Scholar]
- Ramirez A, Bravo A, Jorcano JL, Vidal M. Sequences 5′ of the bovine keratin 5 gene direct tissue- and cell-type-specific expression of a lacZ gene in the adult and during development. Differentiation 1994;58(1):53–64. [DOI] [PubMed] [Google Scholar]
- Sager PT, Balser B, Wolfson J, Nichols J, Pilot R, Jones S, et al. Electrocardiographic effects of class 1 selective histone deacetylase inhibitor romidepsin. Cancer Med 2015;4(8):1178–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekulic A, Von Hoff D. Hedgehog Pathway Inhibition. Cell 2016;164(5):831. [DOI] [PubMed] [Google Scholar]
- Seto E, Yoshida M. Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harbor perspectives in biology 2014;6(4):a018713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpe HJ, Pau G, Dijkgraaf GJ, Basset-Seguin N, Modrusan Z, Januario T, et al. Genomic analysis of smoothened inhibitor resistance in basal cell carcinoma. Cancer Cell 2015;27(3):327–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpless NE, Bardeesy N, Lee KH, Carrasco D, Castrillon DH, Aguirre AJ, et al. Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature 2001;413(6851):86–91. [DOI] [PubMed] [Google Scholar]
- Soufir N, Moles JP, Vilmer C, Moch C, Verola O, Rivet J, et al. P16 UV mutations in human skin epithelial tumors. Oncogene 1999;18(39):5477–81. [DOI] [PubMed] [Google Scholar]
- Tang Y, Zhao W, Chen Y, Zhao Y, Gu W. Acetylation is indispensable for p53 activation. Cell 2008;133(4):612–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teta M, Choi YS, Okegbe T, Wong G, Tam OH, Chong MM, et al. Inducible deletion of epidermal Dicer and Drosha reveals multiple functions for miRNAs in postnatal skin. Development 2012;139(8):1405–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urman NM, Mirza A, Atwood SX, Whitson RJ, Sarin KY, Tang JY, et al. Tumor-Derived Suppressor of Fused Mutations Reveal Hedgehog Pathway Interactions. PloS one 2016;11(12):e0168031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal VP, Ortonne N, Schedl A. SOX9 expression is a general marker of basal cell carcinoma and adnexal-related neoplasms. Journal of cutaneous pathology 2008;35(4):373–9. [DOI] [PubMed] [Google Scholar]
- Waikel RL, Wang XJ, Roop DR. Targeted expression of c-Myc in the epidermis alters normal proliferation, differentiation and UV-B induced apoptosis. Oncogene 1999;18(34):4870–8. [DOI] [PubMed] [Google Scholar]
- Wang C, Wu H, Evron T, Vardy E, Han GW, Huang XP, et al. Structural basis for Smoothened receptor modulation and chemoresistance to anticancer drugs. Nature communications 2014;5:4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter M, Moser MA, Meunier D, Fischer C, Machat G, Mattes K, et al. Divergent roles of HDAC1 and HDAC2 in the regulation of epidermal development and tumorigenesis. The EMBO journal 2013;32(24):3176–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Ping XL, Lee PK, Wu XL, Yao YJ, Zhang MJ, et al. Role of PTCH and p53 genes in early-onset basal cell carcinoma. The American journal of pathology 2001;158(2):381–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that the main data supporting the findings of this study are available within the article and its Supplemental Information files. All correspondence and material requests should be addressed to Sarah E. Millar. This study includes no data deposited in external repositories.