

Abstract
The 21-member connexin gene family exhibits distinct tissue expression patterns that can cause a diverse array of over 30 inherited connexin-linked diseases ranging from deafness to skin defects and blindness. Intriguingly, germline mutations can cause disease in one tissue while other tissues that abundantly express the mutant connexin remain disease free highlighting the importance of the cellular context of mutant expression. Modelling connexin pathologies in genetically-modified mice and tissue-relevant cells has informed extensively on no less than a dozen gain- and loss-of function mechanisms that underpin disease. This review focuses on how a deeper molecular understanding of the over 930 mutations in 11 connexin encoding genes is foundational for creating a framework for therapeutic interventions.
Keywords: connexins, gap junctions, inherited disease, mutations
Connexins are complex and multifunctional
Connexins (see Glossary) are a family of 21 tetraspanning integral membrane proteins that, after synthesis in the endoplasmic reticulum (ER), hexamerize to form structures termed “connexons” or “hemichannels” (Figure 1A). Upon their delivery to the cell surface, connexons pair with connexons from a contacting cell to form gap junction channels (Figure 1B). The term “gap junction” refers to the collection of densely packed connexin channels, like seen in the myocardium (Figure 1C), that allow the direct intercellular exchange of ions and other members of the metabolome that are smaller than ~1000 Da [1]. Gap junctional intercellular communication (GJIC) includes the exchange of important signaling molecules like IP3, calcium, cyclic AMP, ATP, glucose and short peptides like glutathione [2]. Overall, channels formed of different connexin family members have diverse permeability characteristics (i.e., permselectivity) to small molecules of variable charge and shape [3]. The molecular determinants governing permselectivity have been enriched by the solving of the crystal structure for Cx26 at 3.5 Å and by obtaining CryoEM near atomic resolution (1.9 Å) structures of native Cx46/50 channels isolated from the lens [4,5] (Figure 1D). Cells often express two or more connexin isoforms that can intermix leading to an immense array of intercellular channel compositions (e.g., homomeric, heteromeric, homotypic, heterotypic) that further serve to selectively regulate the cellular exchange of ions and metabolome constituents. It is notable that connexins are dynamic in most cell types with relatively short half-lives of 1–5 hours allowing for yet another means of attenuating GJIC [6].
Figure 1:

Connexin localization and channel assembly. A) Diagrammatic view of an undocked connexin oligomer referred to as a hemichannel given its capacity to open in an undocked state. B) Schematic representation of a prototypical gap junction channel denoting two docked hexamers of connexins (connexons). Arrows in A and B represent a channel pore that allows the regulated passage of small molecules of less than 1 kD. C) Connexins are found in every organ system as exemplified by the magnified area of the heart depicting contacting cardiomyocytes within the myocardium where Cx43 is found abundantly at sites of intercalated discs (brown staining, e.g., at arrows). Bar = 100 μm. D) A three-dimensional 1.9 Å resolution reconstruction of a native lens Cx46/50 gap junction channel within a dual lipid nanodisc as revealed by CryoEM (Image obtained with permission from Flores et al., 2020, Nature Communications 11:4331) [4]. Images in A, B and C were partially generated using Biorender (https://biorender.com/).
Cells may also harbor regulatable connexin hemichannels that can selectively open to exchange small signaling molecules between the cytoplasm and extracellular milieu as recently revealed by the high resolution CryoEM structure of human Cx31.3 [7]. As an example, connexin hemichannels appear to play important regulatory roles in the bone, opening in response to shear stress and releasing prostaglandin PGE2 that can regulate bone formation [8]. Other non-canonical roles for connexins involve their extensive interactome that can form protein scaffolds to engage in crosstalk with constituent signaling pathways that regulate, not only gap junction formation/stability, but also the status of tight junctions, adherens junctions and desmosomes [9–12]. In addition, many connexins are subject to an orchestrated series of phosphorylation/dephosphorylation events that regulate their life cycle and gating properties [13,14]. These and other non-canonical connexin activities including potential roles for alternate start codons that produce truncated species and connexin engagement in nuclear and mitochondria function have been discussed elsewhere [15–19]. This review focuses on human germline mutations and variants found within the coding regions of connexin genes (Table 1) and the cellular mechanisms underlying a plethora of diseases affecting multiple organ systems. Together with the ability to interrogate and mechanistically subclassify disease-linked mutants and the near atomic knowledge of connexin channel structure, it is now possible to envision, design and deploy smart 21st century therapeutics (Box 1).
Table 1.
Connexin genes linked to inherited diseasesa
| Gene | Protein | Mutations/variants reported |
|---|---|---|
| GJE1 | C×23 | - |
| GJB7 | C×25 | - |
| GJB2 | C×26 | 175 |
| GJB6 | C×30 | 6 |
| GJC3 | C×30.2/31.3 | - |
| GJB4 | C×30.3 | 8 |
| GJB3 | C×31 | 35 |
| GJB5 | C×31.1 | - |
| GJD3 | C×31.9 | - |
| GJB1 | C×32 | 431 |
| GJD2 | C×36 | - |
| GJA4 | C×37 | - |
| GJA5 | C×40 | 10 |
| GJD4 | C×40.1 | - |
| GJA1 | C×43 | 98 |
| GJC1 | C×45 | 2 |
| GJA3 | C×46 | 45 |
| GJC2 | C×47 | 51 |
| GJA8 | C×50 | 70 |
| GJA9 | C×59 | - |
| GJA10 | C×62 | - |
Red color denotes the gene, protein and approximate number of mutations/variants linked to inherited disease
Box 1: Longitudinal epidemiological studies and therapeutic considerations.
With the growing awareness and knowledge related to the scope of connexin gene mutations/variants found within society, it is now possible to predict that new connexin mutation linkages to disease will be discovered particularly related to aging. Building from earlier platforms like the Genome Aggregation Database (https://www.nature.com/immersive/d42859-020-00002-x/index.html), this approach is largely being made possible by ongoing large-scale genome sequencing projects (https://allofus.nih.gov/; https://www.astrazeneca.com/) designed to generate unprecedented databases. Here, the genomes of millions of participants are being sequenced and archived along with longitudinal medical data, family histories and lifestyle indicators. This cohort will undoubtedly include a large subset of participants that harbor connexin gene mutations. As an example, Cx26 has multifaceted linkages to cancer onset and progression [94], and Cx26 is known to be expressed in a wide range of tissues and organs such as the esophagus, liver, bladder and breast. Given the frequency of GJB2 (Cx26) mutations that lead to SNHL and skin diseases, it will be possible to determine the relative risk of these subjects developing cancer or other diseases as properly functioning Cx26 may indeed be necessary for life-long health. The question will still remain as to whether any newly identified pathologies or existing connexin-linked diseases can be mitigated or cured. Successful therapeutic designs may hinge on the knowledge of how a specific mutant is affecting connexin function. When considering the subclass of connexin mutants that are considered trafficking defective mutants, therapies that improve ER function and protein folding may show benefit. For example, treatment of HeLa cells engineered to express the trafficking defective Cx50 D47A mutant with the molecular chaperone 4-phenlybutyrate improved its delivery to the cell surface and gap junction formation, but disappointedly failed to improve opacity in a Cx50 D47A mouse model of human congenital cataracts [95]. In cases where the pathology is linked to leaky hemichannels, FDA-approved mefloquine has shown some promise to suppress hyperactive hemichannels found in a Cx26 G45E mutant mouse model of KID syndrome [60,91]. Supporting this approach, Kuang and colleagues developed a novel monoclonal antibody that acts as an antagonist to block Cx30 A88V hyperactive hemichannels [61]. In a mouse model of Clouston syndrome, abEC1.1 antibody treatment largely mitigated or reversed the skin disease phenotype [61], highlighting a strategy which may have considerable utility for other Cx26-linked pathologies of the skin [96]. In yet another scenario, a mutant allele-specific siRNA was successfully used to repair skin damage induced by a heterozygous Cx26 D50N mutation in patient-derived keratinocytes and in a human-mouse skin graft model of KID syndrome [92].
Breadth, severity, and mode of inheriting connexin-linked diseases
Probably one of the most underappreciated aspects of inheritable connexin-linked diseases is their extensive prevalence in society. Many distinct connexin-linked diseases have been documented in accordance with the Online Mendelian Inheritance in Man repository [20,21]. The most recent assessment has revealed over 930 unique mutations/variants found across 11 connexin genes (Table 1) that are linked to 30+ clinically distinct diseases. Exact tabulation of unique human diseases linked to connexins is challenging owing to the scarcity of some genotype/phenotype linkages and incidences of overlapping phenotypes as well as clinical diseases that appear identical yet are caused by different connexin genes. Many connexin gene variants are also insufficiently characterized to firmly establish their role in being causative of disease. While skin diseases represent the largest subset of connexin-linked diseases (>7 in total), congenital sensorineural hearing loss (SNHL) is by far the most common as GJB2 (Cx26)-linked SNHL occurs in ~1/2000 children within many world populations [22,23]. Not surprisingly commercial DNA sequencing companies routinely screen for two variants of the GJB2 gene due to their high frequency in society (e.g., https://www.23andme.com/en-ca/test-info/), and it is now standard operating procedure to sequence the GJB2 and GJB6 (Cx30) genes when children present with hearing loss. Where connexin mutant-linked diseases will clinically present has been a research mystery in many cases owing to the ubiquitous distribution of some commonly found connexins like Cx43.
Connexin-linked inherited pathologies can be fatal or lead to severe morbidities that drastically compromise life-long health. One form of hypomyelinating disease, known as Pelizaeus-Merzbacher-like disease, is caused by mutations in GJC2 (Cx47) which severely affects the brain and spinal cord, dramatically shortening life expectancy [24]. Likewise, keratitis-ichthyosis-deafness (KID) syndrome is caused by rare GJB2 gene mutations that lead to severe skin disease, profound hearing loss, often blindness, and premature childhood death in patients harboring A88V or G45E mutants [25]. That said, the vast majority of connexin-linked diseases are not typically life-shortening but rather can impose severe morbidities as seen in SNHL [22]. In other cases, like congenital cataracts linked to GJA3 (Cx46) and GJA8 (Cx50) gene mutations, corrective surgery is a viable option for some patients [26]. Since connexin-linked diseases can be both debilitating and severe, research to identify disease mechanisms, effective treatments and possibly even cures is critical.
Except for a couple of digenic scenarios related to deafness, connexin-linked diseases are typically monogenic in nature involving a single connexin encoding gene mutation that is transmitted via autosomal dominant, autosomal recessive and X-linked dominant modes of inheritance. Autosomal recessive connexin gene mutations are the most common owing largely to links between GJB2 mutations and SNHL [22]. Affected individuals rarely have disease or compromised organ function outside the cochlea raising queries as to why other tissues enriched in Cx26 like the esophagus, liver, bladder and female breast apparently remain disease-free. Conversely, with the possible exclusion of the genes encoding Cx46 and Cx50 where disease is confined to the lens, individuals that harbor autosomal dominant connexin-linked diseases often have syndromic disease involving two or more distinct organ systems that are compromised. In this group, autosomal dominant GJA1 and GJB2 gene mutations have been extensively studied due to their role in multiple organ dysfunction and a collection of skin diseases, respectively [1,27]. The final X-linked category of inheritance is populated solely by the GJB1 (Cx32) gene where mutations are causal of the demyelinating disease known as Charcot Marie Tooth Disease X1 (CMTX1) [28]. As a collective, inheritable connexin-linked diseases are highly prevalent in society with over 3% of the population acting as carriers; a number that can be much higher in parts of the world where GJB2 mutations linked to SNHL are endemic [22].
Genotype to phenotype to disease
Sensory organs
GJB2 (Cx26)-linked SNHL is often caused by damage or loss of hair cells in the sensory cells of the organ of Corti which is unexpected given that hair cells are not connected to adjacent cells via gap junctions nor do they express connexins [29]. However, large Cx26 and Cx30 containing gap junctions are found abundantly throughout the supporting cell network of the cochlear epithelium linking SNHL to the necessity for these gap junctions, and likely hemichannels as well, to be intact and fully functional [29–31]. This suggests that Cx26-linked hair cell loss is in fact a downstream consequence of defective or altered Cx26 function in non-sensory cells during development or early in life [32]. The complexity underpinning the pathophysiology of how Cx26 dysregulation and connexins in general are linked to congenital SNHL has been reviewed recently elsewhere in considerable detail [33–36]. Of all the GJB2 mutations, the c.35delG mutation is the most prevalent variant in Western Europe and the Americas while c.235delC is the most widespread mutation in East Asia [22,37]. Truncated Cx26 encoded by either of these mutations is insufficient for channel formation and is functionally equivalent to a Cx26 knockout. Together with c.167delT, R143W, W24X and V37I, these six mutations account for the most common forms of congenital SNHL found worldwide [22,37], even though the current tabulation of GJB2 variants is at least 175. The prevalence of GJB2 allele variants in carriers implies an evolutionary heterozygous advantage that may be related to thickening of the epidermis found in some individuals [38].
Both GJA3 (Cx46) and GJA8 (Cx50) gene mutations have been causally linked to ~22% of all congenital nonsyndromic cataracts [26]. These connexins play central roles in water, signaling molecules and ion exchange in the avascular lens containing enucleated fiber cells [39]. Cx50 and Cx46 appear to be highly engaged in functional hemichannel activity which may alone, or in partnership with gap junction channels, be critically important for lens homeostasis and maintenance of its transparency [40–42]. The absence of cellular exchange of glucose, reduced glutathione, calcium or even unknown metabolites may all play a critical role in the loss of lens opacities. Currently, there are ~45 GJA3 and ~70 GJA8 unique mutations linked to congenital cataracts in humans, the vast majority of which are subject to autosomal dominant inheritance (https://cat-map.wustl.edu/home/cat-map-variant-file/) [43]. The bulk of these mutations are single amino acid substitutions found prior to amino acid 225 suggesting the first half of these connexin encoding genes are rich in mutational hotspots. Mechanistically, cataract-linked mutations in GJA3 and GJA8 have been shown to both drive the production of leaky or pathological hemichannels leading to cellular apoptosis or the inhibition of hemichannel function that may disrupt lens nutrient homeostasis [39].
Demyelinating diseases:
CMTX1 caused by mutations in GJB1 (Cx32) is the second most prevalent (~1:50,000) connexin related genetic disease which is more common and severe in males [44]. The vast majority of the over 430 GJB1 (Cx32) gene mutations are either missense or frameshift in nature and are found throughout the entire length of the gene (http://hihg.med.miami.edu/code/http/cmt/public_html/index.html#/). Functional studies on CMTX1-causing mutations performed in a variety of reference cell types indicate that they mostly cause loss of Cx32 channel function [45]. This is particularly intriguing as Cx32 is not only found at paranodes but is also known to form unique intracellular (i.e., reflexive) gap junction channels at Schmidt-Lanterman incisures of Schwann cells [46]. Here, Cx32 channels are thought to allow for the transfer of small metabolites and potassium ions across the Schwann cell myelin sheath. However, dye diffusion along this radial pathway was not slower in Cx32-null mice [47] raising questions as to whether the pathogenesis of CMT1X is fully linked to the role of Cx32 at the Schmidt-Lanterman incisures. In keeping with this notion, in a recent report where the CMTX1-causing R220X truncated mutant was interrogated, the authors postulated that Schwann cell myelination of axons required intracellular calcium regulated opening of Cx32 hemichannels and ATP release [48]. Since Cx32 is also found in oligodendrocytes of the central nervous system (CNS), patients harbouring GJB1 mutations may present with episodic CNS dysfunction [49], but this appears to be uncommon suggesting that other co-expressed connexins act to protect the CNS from dysfunction. Beyond the nervous system, we are not aware of any evidence that CMTX1 patients present with any disease in the liver or pancreas where Cx32 is highly expressed [50]. How cells in these Cx32-rich organs developmentally adapt to escape clinically detectable disease remains an enigma especially given the lack of compensatory connexin expression to fulfil the role of defective Cx32 channels.
Oligodendrocytes of the CNS abundantly express Cx47 and nearly 40 autosomal recessive GJC2 (Cx47) mutations have been linked to Pelizeaus-Merzbacher-like disease (PMLD1), a severe dysmyelinating disease that manifests early in life [24]. A few additional cases of hereditary spastic paraparesis have been reported in patients with GJC2 mutations and autosomal dominant GJC2 mutations have been linked to lymphedema [51]. Although not yet extensively studied, evidence points to Cx47 causing PMLD1 via a loss of channel and/or hemichannel function [52], but other gain-of-function mechanisms have not been ruled-out.
Skin diseases
The epidermis of the skin expresses upwards of eight connexins that are spatially and temporally regulated as basal keratinocytes proliferate, differentiate and migrate during human epidermis renewal every 2–4 weeks [53]. Mutations in five connexin genes (GJB2, GJB3, GJB4, GJB6 and GJA1) have been linked to more than seven skin diseases that present with both distinct and overlapping clinical symptoms [21,23,54]. These skin diseases are typically subject to autosomal dominant inheritance and many are syndromic in nature affecting hearing or other organ functions. Remarkably, erythrokeratodermia variabilis et progressiva (EKVP) can be attributed to single missense mutations in GJB3, GJB4 or GJA1 [55] raising questions as to how mutant connexin isoforms with unique expression patterns within the epidermis cause the same clinical disease. In order to inform on how mutations in five connexin genes lead to skin disease, several genetically-modified mouse models have been engineered to mimic the various human diseases with GJB2 being the most frequently studied [56]. Collectively, while all these genetically-modified mice did confirm and inform on important functional roles for connexins in the epidermis, in most cases they did not completely mimic the human disease. This can at least partially be attributed to the fact that human skin is several layers thicker than mouse skin and connexin expression (particularly during development), localization, response to wounding and functional roles in the thin skin of mice are not identical to human [57]. One prevailing theme that did emerge to account for a host of incurable connexin skin pathologies was the ability of Cx26, Cx30 and Cx43 mutants to form pathological pores or leaky hemichannels [58–61].
Multiorgan defects
Given that Cx43 is expressed in >50% of all human cell types, it is not surprising that mutations in GJA1 (Cx43) cause the multiorgan developmental disorder known as oculodentodigital dysplasia (ODDD) and related disorders [62,63]. Of the mutations/variants that have been tabulated from the published literature, all but four were attributed to autosomal dominant inheritance (Figure 2A). Interestingly, while GJA1 gene mutations commonly cause ODDD, and to a much lesser frequency EKVP, variants that require further confirmation have been associated with no less than four additional diseases [21,59] (Figure 2A). Nevertheless, this spectrum of GJA1-linked diseases raises the question as to how compromised Cx43 cellular function could have selective and differential impact on only some Cx43-rich tissues and not others (Figure 2B). In cardiomyocytes of the heart, for example, gap junctions present at the intercalated discs that allow beat synchrony express primarily Cx43 but only a miniscule fraction of patients harboring GJA1 mutations exhibit cardiac disease. Given that cardiomyocytes infrequently co-express other connexin isoforms, maybe there remains sufficient Cx43 function bestowed by the wild type Cx43 allele to avoid disease. Here again genetically-modified mice that model ODDD have advanced our understanding of the wholistic role Cx43 plays in maintaining life-long health [64–66]. Collectively, all of these mice harbored relatively mild cardiac pathologies not typically seen in ODDD patients reflecting some innate limitation in modelling ODDD in mice. Nevertheless, they provided evidence to suggest that ODDD patients may in fact have subclinical diseases related to hearing, bladder function, and even lactation that could present later in life for some patients [67–69], but this concept will need to be tracked longitudinally (Box 1). Going forward these and other mouse models of connexin pathologies should help to determine whether a newly discovered connexin gene variant or polymorphism is linked to disease. For instance, polymorphisms of GJA4 (Cx37) have been associated with polycystic ovarian syndrome in a south Indian population [70], and a trafficking defective Cx37 G316S variant was recently identified in patients with primary ovarian insufficiency further suggesting a possible link between Cx37 and ovarian disease [71].
Figure 2:
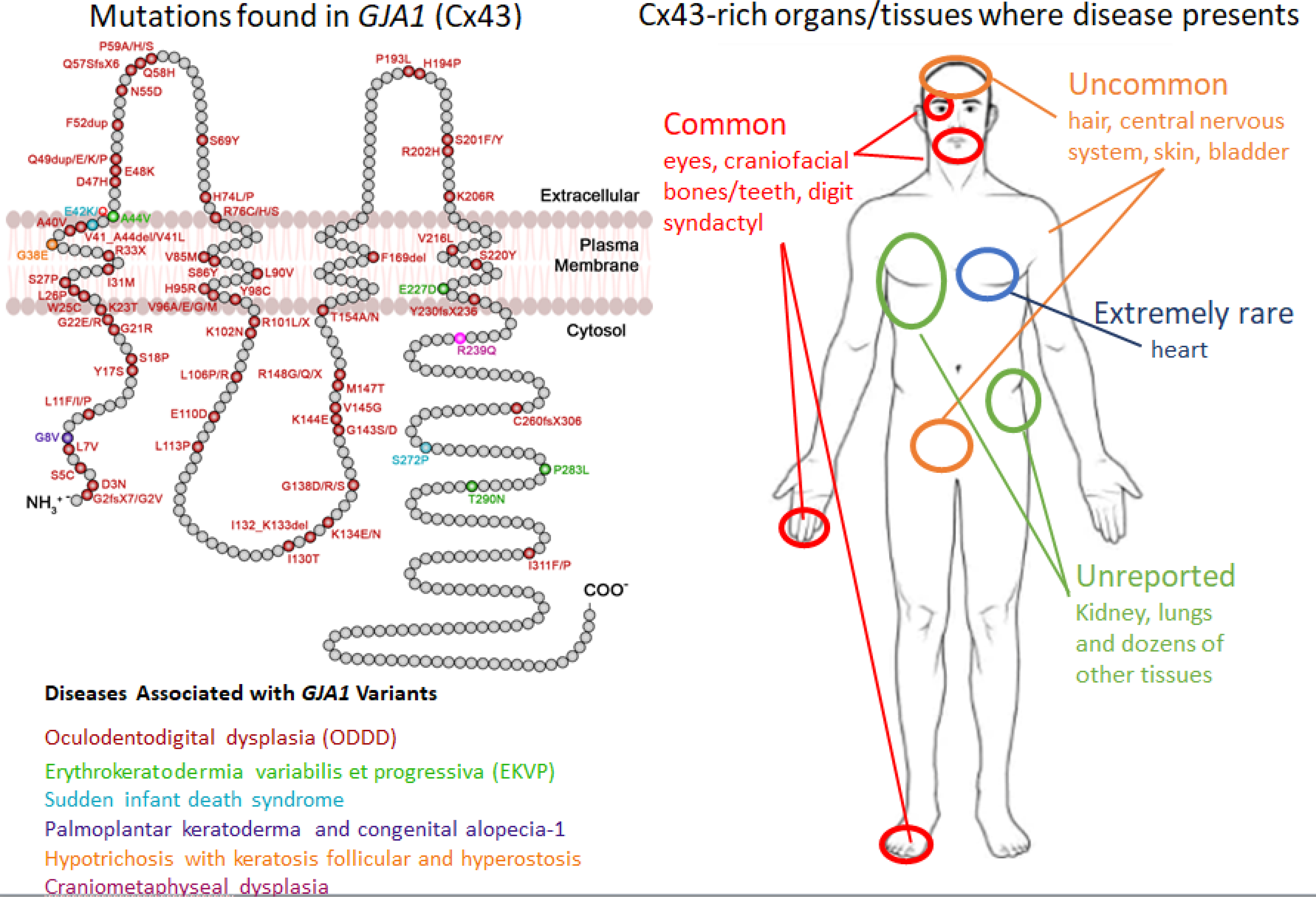
Schematic map of GJA1 gene mutations/variants, the reported inherited disease and the tissues/organs affected. The polypeptide model of Cx43 denotes the position and amino acid change of gene variants/mutations representing six inherited diseases. Given the frequency, ODDD is by far the most documented disease linked to GJA1 mutations. EKVP, while rare, has significant linkages to GJA1 mutations, but the remaining denoted diseases and potentially others are less well established and may need further confirmation. The human silhouette denotes the approximate location of Cx43-rich tissues/organs that commonly, uncommonly, rarely or essentially never present with abnormalities or disease in patients harboring GJA1 gene mutations.
Atrial Fibrillation and Conduction
Since Cx40 is highly expressed in the cardiac atrium, evidence continues to mount that upwards of ten germline autosomal dominant GJA5 (Cx40) mutations represent an independent risk factor for a small subset of idiopathic atrial fibrillation (AF) [72–74]. Functional studies have revealed that many, but not all, of the Cx40 mutants exhibit loss of GJIC or gain of hemichannel function pointing to mechanisms that may inform on how Cx40 channel dysfunction alters cardiac rhythm [75,76]. This notion that GJA5 mutations are associated with AF was further supported when a Cx40 A96S mutant mouse was generated and found to have prolonged episodes of induced AF [77]. Most recently a heterozygous GJC1 (Cx45) M235L variant that failed to traffic to the cell surface has been identified that co-segregated with AF across three generations [78]. Further linking GJC1 mutations to disease, a patient harboring a Cx45 R75H mutant resulted in a rare defective form of inherited cardiac conduction and craniofacial abnormalities similar to that seen in ODDD [79].
Cellular mechanisms underpinning disease
A deep understanding of the breadth and impact of congenital connexin pathologies remains incomplete even though their combined frequencies suggest they represent one of the largest cohorts of inherited diseases found in humans. The community continues to unravel the reason why several human organs that harbor abundant levels of connexin mutants remain free of clinically detectable disease while other organs are severely compromised. While redundancies in connexin subtype expression patterns and residual mutant channel function provide some explanation for this physiological outcome, this knowledge is insufficient to explain how connexin gene mutations lead to either non-syndromic or syndromic disease. The answer may rest in part on knowing how each connexin gene mutation affects the mutant connexin life cycle and function. At present there are no less than 12 mechanisms that reflect both loss- and gain- of-function outcomes (see Figure 3 for a schematic representation).
Figure 3:

Mechanisms underpinning connexin linked diseases. Many connexin-linked mutants have been interrogated in mouse models of disease, human tissues, tissue-relevant cells and reference models, and loss- or gain-of-function mechanisms have now been discerned that can be mapped to the life cycle of connexins. A total of eight loss-of-function (a-h) and four gain-of-function mechanisms (i-l) are denoted in yellow and reflect mutants that: a.) fail to fold correctly and pass ER quality control mechanisms; b.) escape the ER but accumulate in the Golgi apparatus; c.) traffic to the cell surface but lack normal hemichannel function; d.) lack the capacity to dock to form a complete gap junction channel; e.) assemble into gap junction channels that have reduced function or abnormal gating properties; f.) assemble into gap junction channels that are functionally inactive; g.) acquire a prolonged or shortened half-life; h.) exhibit a defective relationship with the interactome; i.) acquire an increased ability to co-oligomerize with co-expressed wild type connexins; j.) establish constitutively active or leaky hemichannels; k.) exhibit functionally dysregulated connexin hemichannels when mixed with co-expressed wild type connexins; l.) established pathological heterotypic gap junction channels with co-expressed wild type connexins with attenuated function. Part of this image was generated using Biorender (https://biorender.com/).
Loss-of-function
First, in the loss-of-function category, there is a substantial subclass of connexin gene mutations linked to connexin pathologies that lead to mutants that are severely truncated and rapidly degraded or mutants that fail to fold correctly and pass quality control mechanisms housed within the endoplasmic reticulum (ER)(Figure 3, left side) [78]. Some of these mutants may cause ER stress, the unfolded protein response or even cell death as exemplified by the Cx31 G45E mutant associated with EKVP [80] or the Cx50 D47A mutant linked to cataracts [81]. Second, trafficking defective mutants may escape the quality control mechanisms of the ER and accumulate in the Golgi apparatus as seen for several CMT1X-linked Cx32 mutants [45]. These mutants were deemed possible candidates for gene replacement strategies, but they were found to sequester co-expressed wild type Cx32 preventing its delivery to the cell surface [45]. Third, mutants might traffic to the cell surface but lack normal hemichannel function [82,83]. Fourth, mutants, especially when the mutation occurs within one of the two extracellular loops, may lack the capacity to dock with a connexin from a contacting cell and form a complete gap junction channel [84]. Fifth, mutants might form gap junction channels that have reduced function or abnormal gating properties [85]. Sixth, mutants may proceed to assemble a prototypical gap junction, but the resulting channels are functionally inactive [86,87]. Seventh, amino acid substitutions may occur in connexin motifs known to alter its normally short half-life [88] potentially resulting in pathologies related to a reduction or surplus of GJIC. Finally, mutants can lose the ability to bind their native interactome which is certainly the case for frameshift mutants where the C-terminal is deleted as we know that most of the interactome binds to this connexin domain [9]. Of note, mutants are not confined to exhibiting only one of these defective properties as any pathological mutant may exhibit two or more of these characteristics [89].
Gain-of-function
Connexins are highly restrictive as to which connexin subtypes they will oligomerize when co-expressed in the same cell. However, connexin gene mutations can dramatically change these restrictions allowing for mutant connexins to interact and attenuate the function of a wide range of unmutated connexin isoforms [90] (Figure 3, right side). Another somewhat common gain-of-function effect can occur when mutant connexins form constitutively active or leaky hemichannels that appear at the cell surface [54,59–61,91]. These aberrant pores can drive cells into apoptosis or destroy the integrity of the cytosol as critical members of the metabolome escape from the cell. Mixed oligomers of mutant and wild type connexins can form hemichannels that are functional but exhibit abnormal gating properties or distinctly different permeability characteristics. Identifying and characterizing heteromeric hemichannels at the cell surface is a challenge but has been approached by co-expressing Cx26 mutants with wild type Cx43 to study potential mechanisms underpinning palmoplantar keratoderma [90]. Still in other situations, mutant containing connexons may gain the capacity to dock with connexons of different types and form pathological heterotypic gap junction channels that have attenuated function owing to the mutant trans-dominantly inhibiting the functional capability of wild type connexins [54]. As in the case of loss-of-function mechanisms, a single mutant may exhibit more than a single gain-of-function mechanism and even exhibit both gain- and loss-of-function characteristics. It is also important to note that some mechanistic changes may be rare (e.g., a change in a connexin half-life) while others may be far more common (e.g., leaky hemichannels).
Concluding Remarks
At present there are no prescribed treatments for inherited connexin-linked diseases beyond what can be achieved through surgical interventions for syndactyly, camptodactyly, congenital cataracts and, in some cases, hearing loss. Now that considerable progress has been made towards the mechanistic understanding of a dozen categories of disease-linked connexin mutants, new therapeutics can be envisioned, designed and tested. One can imagine scenarios where the high-resolution knowledge of connexin channels could be harnessed to screen for a class of drugs that could preferentially upregulate wild type connexin channel function to alleviate the detrimental effects of mutant connexin expression (See Outstanding Questions). Conversely, gene targeting strategies to specifically reduce the expression of mutant alleles have already shown some efficacy as has deployment of antibodies to block leaky hemichannels in models of skin disease [61,92]. These strategies establish a proof-of-concept and show considerable promise that effective treatments targeting the root causes of connexin-linked diseases are indeed possible. Nevertheless, for all proposed treatment strategies, it is important to recognize that unmutated connexins must continue to function well in both affected and surrounding tissues to avoid treatment-induced side effects. Going forward, new insights into the breath of inherited connexin-linked diseases are likely to emerge through knowledge obtained from ongoing large-scale gene sequencing and longitudinal epidemiological studies that will report on disease onset and progression. With larger numbers of aged subjects, previously unidentified connexin pathologies are likely to become evident. Revelations from these studies will further drive efforts to find new therapeutics as the community strives to mitigate the severity of connexin pathologies [93].
Outstanding questions.
Why do germline mutations in a specific connexin gene cause disease symptoms in one tissue but not in another where the connexin is predominately expressed? Are the effects of these mutations subclinical, benign, or compensated for by other co-expressed connexins or unknown genes?
Can the current knowledge of the connexin channel structure and the mechanisms associated with gain-and loss-of-function mutations be exploited for designing new treatment technologies?
What role does the connexin interactome play in diseases linked to connexin isoforms? At present, other than Cx43, the interactome of the 10-remaining disease-linked connexin isoforms is far less well understood.
Given the widespread expression of some connexins and the regional expression of others, will genome editing techniques or other smart therapeutics be curative for some connexin-based diseases?
Will new genome sequencing databases and epidemiological association studies allow us to link connexin gene mutations to diseases associated with aging such as some types of cancer, Parkinson’s disease, cardiac diseases, diabetes or Alzheimer’s disease?
How many additional diseases will be definitively linked to connexin gene mutations? Will connexin gene variants associated with sudden infant death syndrome, female infertility and other extremely rare situations prove to be causal of disease?
Given that disease severity caused by specific connexin gene mutations appears to vary in different populations/ethnic groups, what other genes play a role in disease penetrance?
Highlights.
Mutations in no less than 11 connexin encoding genes cause at least 30 human diseases with a combined frequency that place the connexin gene family near the top in worldwide prevalence of inherited diseases.
Germline connexin gene mutations can cause disease in one tissue while other tissues that abundantly express the mutant connexin remain disease-free indicating specialized roles for connexins in different cell types.
Connexin gene mutations can lead to precise organ defects (e.g., sensorineural hearing loss, congenital cataracts) or be syndromic causing disease in multiple organs (e.g., oculodentodigital dysplasia).
Disease-linked connexin mutants appear to affect both canonical and non-canonical roles that can lead to cell dysfunction or death.
Connexin mutations cause disease via at least a dozen gain- and loss-of function mechanisms.
Acknowledgements
The authors would like to thank Meng Xi Sun for drafting some of the artwork shown in Figure 2 and both Meng Xi Sun and Ryan Yee who engaged in extensive literature searches that helped establish the framework for this review. We would also like to thank Drs. Donglin Bai and Joell Solan for providing a critical read and valuable comments. Given reference limitations and the requirement to focus on articles published within the past 5 years, the authors apologize for the many primary and seminal articles that were not cited. Work related to gap junctions in the authors’ labs was funded by the Canadian Institutes of Health Research (Grants 148630, 148584) to DWL and the US National Institutes of Health (GM55632) to PDL.
Glossary
- Atrial Fibrillation
uncoordinated, usually rapid heartbeat that occurs when the two atria do not properly synchronize electrical signals.
- Charcot-Marie-Tooth Disease X1 (CMTX1)
caused by mutations in the GJB1 (Cx32) gene found on the X chromosome that results in demyelination and damage to peripheral nerves.
- Congenital Cataract
clouding of the eye lens that is present at birth.
- Connexin genes
there are 21 human connexin genes that are named starting with GJ (denoting gap junction) followed by a subfamily designation letter according to their evolutionary similarities denoted as “A, B, C, D, E” and a number designation according to their order of discovery. Thus, the most prevalent connexin gene expressed in the human body is denoted as “GJA1”.
- Connexin
tetraspanning integral membrane protein that is abbreviated with a “Cx” prefix followed by its approximate predicted molecular mass in kilodaltons. Thus, the most prevalent connexin found in the human body is termed “Cx43”.
- Connexon
an assembly intermediate of 6 connexin proteins formed in the endoplasmic reticulum/Golgi apparatus that exists prior to docking with an apposing connexon from an adjacent cell to form a complete gap junction channel.
- Gap Junction
collection of densely packed cell-to-cell channels that allow the direct intercellular exchange of ions and other members of the metabolome that are smaller than ~1000 Da.
- Hemichannel
a connexon at the cell surface that has the capacity to gate open in some physiological and pathological situations
- Heteromeric
the 6 connexins within the connexon are not the same.
- Heterotypic
gap junction channel formed from at least 2 different connexins.
- Homomeric
all 6 connexins within the connexon are the same.
- Homotypic
gap junction channel formed of 12 connexins that are the same.
- Congenital Sensorineural Hearing Loss
caused by developmental defects in the structures of the inner ear or auditory nerve.
- Oculodentodigital dysplasia (ODDD)
a multi-organ, mostly autosomal dominant, developmental disease that displays a wide variety of symptoms including facial, eye, digit, nose, mouth, teeth and neurological abnormalities.
Footnotes
Declaration of Interests
DWL has no competing interest. PDL is a non-paid member of the scientific advisory board of FirstString Research and has been granted stock options (currently no value) for his service.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- 1.Leybaert L et al. (2017) Connexins in Cardiovascular and Neurovascular Health and Disease: Pharmacological Implications. Pharmacol. Rev. 69, 396–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harris AL (2018) Electrical coupling and its channels. J. Gen. Physiol. 150, 1606–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koval M et al. (2014) Mix and match: investigating heteromeric and heterotypic gap junction channels in model systems and native tissues. FEBS Lett. 588, 1193–1204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Flores JA et al. (2020) Connexin-46/50 in a dynamic lipid environment resolved by CryoEM at 1.9 A. Nat. Commun. 11, 4331. 10.1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Myers JB et al. (2018) Structure of native lens connexin 46/50 intercellular channels by cryo-EM. Nature 564, 372–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Laird DW (2006) Life cycle of connexins in health and disease. Biochem. J. 394, 527–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee HJ et al. (2020) Cryo-EM structure of human Cx31.3/GJC3 connexin hemichannel. Sci. Adv. 6, eaba4996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li G et al. (2021) Osteocytic Connexin43 Channels Regulate Bone-Muscle Crosstalk. Cells 10. 237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leithe E et al. (2018) The connexin 43 C-terminus: A tail of many tales. Biochim. Biophys. Acta 1860, 48–64 [DOI] [PubMed] [Google Scholar]
- 10.Solan JL et al. (2019) Cx43 phosphorylation-mediated effects on ERK and Akt protect against ischemia reperfusion injury and alter the stability of the stress-inducible protein NDRG1. J. Biol. Chem. 294, 11762–11771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu W et al. (2020) Gap junction-mediated cell-to-cell communication in oral development and oral diseases: a concise review of research progress. Int. J. Oral. Sci. 12, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Green KJ et al. (2019) Desmosomes: Essential contributors to an integrated intercellular junction network. F1000Res 8. F1000 Faculty Rev-2150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Solan JL and Lampe PD (2018) Spatio-temporal regulation of connexin43 phosphorylation and gap junction dynamics. Biochim. Biophys. Acta. 1860, 83–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Solan JL and Lampe PD (2020) Src Regulation of Cx43 Phosphorylation and Gap Junction Turnover. Biomolecules 10. E1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xiao S et al. (2020) Auxiliary trafficking subunit GJA1–20k protects connexin-43 from degradation and limits ventricular arrhythmias. J. Clin. Invest. 130, 4858–4870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zeitz MJ et al. (2019) Dynamic UTR Usage Regulates Alternative Translation to Modulate Gap Junction Formation during Stress and Aging. Cell Rep. 27, 2737–2747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Epifantseva I and Shaw RM (2018) Intracellular trafficking pathways of Cx43 gap junction channels. Biochim. Biophys. Acta 1860, 40–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boengler K and Schulz R (2017) Connexin 43 and Mitochondria in Cardiovascular Health and Disease. Adv. Exp. Med. Biol. 982, 227–246 [DOI] [PubMed] [Google Scholar]
- 19.Kotini M et al. (2018) Gap junction protein Connexin-43 is a direct transcriptional regulator of N-cadherin in vivo. Nat. Commun. 9, 3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kelly JJ et al. (2015) Mechanisms linking connexin mutations to human diseases. Cell Tissue Res. 360, 701–721 [DOI] [PubMed] [Google Scholar]
- 21.Srinivas M et al. (2018) Human diseases associated with connexin mutations. Biochim. Biophys. Acta 1860, 192–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chan DK and Chang KW (2014) GJB2-associated hearing loss: systematic review of worldwide prevalence, genotype, and auditory phenotype. Laryngoscope 124, E34–53 [DOI] [PubMed] [Google Scholar]
- 23.Laird DW et al. (2017) SnapShot: Connexins and Disease. Cell 170, 1260–1260 e1261 [DOI] [PubMed] [Google Scholar]
- 24.Abrams CK (2019) Diseases of connexins expressed in myelinating glia. Neurosci. Lett. 695, 91–99 [DOI] [PubMed] [Google Scholar]
- 25.Lilly E et al. (2019) More than keratitis, ichthyosis, and deafness: Multisystem effects of lethal GJB2 mutations. J. Am. Acad. Dermatol. 80, 617–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shiels A and Hejtmancik JF (2019) Biology of Inherited Cataracts and Opportunities for Treatment. Annu. Rev. Vis. Sci. 5, 123–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Delmar M et al. (2018) Connexins and Disease. Cold Spring Harb Perspect Biol. 10. a029348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cisterna BA et al. (2019) Role of Connexin-Based Gap Junction Channels in Communication of Myelin Sheath in Schwann Cells. Front Cell Neurosci. 13, 69. 10.3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mammano F (2019) Inner Ear Connexin Channels: Roles in Development and Maintenance of Cochlear Function. Cold Spring Harb Perspect. Med. 9. a033233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ceriani F et al. (2016) Critical role of ATP-induced ATP release for Ca2+ signaling in non-sensory cell networks of the developing cochlea. Proc. Natl. Acad. Sci. U S A 113, E7194–E7201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhu Y et al. (2013) Active cochlear amplification is dependent on supporting cell gap junctions. Nat. Commun. 4, 1786. 10.1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnson SL et al. (2017) Connexin-mediated signalling in non-sensory cells is crucial for the development of sensory inner hair cells in the mouse cochlea. J. Neurosci. 37, 258–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu X et al. (2019) Structure and Function of Cochlear Gap Junctions and Implications for the Translation of Cochlear Gene Therapies. Front Cell Neurosci. 13, 529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Verselis VK (2019) Connexin hemichannels and cochlear function. Neurosci Lett 695, 40–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao HB (2017) Hypothesis of K(+)-Recycling Defect Is Not a Primary Deafness Mechanism for Cx26 (GJB2) Deficiency. Front Mol. Neurosci. 10, 162. 10.3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mammano F and Bortolozzi M (2018) Ca(2+) signaling, apoptosis and autophagy in the developing cochlea: Milestones to hearing acquisition. Cell Calcium 70, 117–126 [DOI] [PubMed] [Google Scholar]
- 37.Azadegan-Dehkordi F et al. (2019) Update of spectrum c.35delG and c.−23+1G>A mutations on the GJB2 gene in individuals with autosomal recessive nonsyndromic hearing loss. Ann. Hum. Genet. 83, 1–10 [DOI] [PubMed] [Google Scholar]
- 38.Guastalla P et al. (2009) Detection of epidermal thickening in GJB2 carriers with epidermal US. Radiology 251, 280–286 [DOI] [PubMed] [Google Scholar]
- 39.Berthoud VM and Ngezahayo A (2017) Focus on lens connexins. BMC Cell Biol. 18, 6. 10.1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brink PR et al. (2020) Lens Connexin Channels Show Differential Permeability to Signaling Molecules. Int. J. Mol. Sci. 21. E6943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shi W et al. (2018) Connexin hemichannels mediate glutathione transport and protect lens fiber cells from oxidative stress. J. Cell Sci. 131. jcs212506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hu Z et al. (2018) Cataract-associated connexin 46 mutation alters its interaction with calmodulin and function of hemichannels. J. Biol. Chem. 293, 2573–2585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shiels A et al. (2010) Cat-Map: putting cataract on the map. Mol. Vis. 16, 2007–2015. 216 [PMC free article] [PubMed] [Google Scholar]
- 44.Panosyan FB et al. (2017) Cross-sectional analysis of a large cohort with X-linked Charcot-Marie-Tooth disease (CMTX1). Neurology 89, 927–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kyriakoudi S et al. (2017) Golgi-retained Cx32 mutants interfere with gene addition therapy for CMT1X. Hum. Mol. Genet. 26, 1622–1633 [DOI] [PubMed] [Google Scholar]
- 46.Bortolozzi M (2018) What’s the Function of Connexin 32 in the Peripheral Nervous System? Front. Mol. Neurosci. 11, 227. 10.3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Balice-Gordon RJ et al. (1998) Functional gap junctions in the schwann cell myelin sheath. J. Cell Biol. 142, 1095–1104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carrer A et al. (2018) Cx32 hemichannel opening by cytosolic Ca2+ is inhibited by the R220X mutation that causes Charcot-Marie-Tooth disease. Hum. Mol. Genet. 27, 80–94 [DOI] [PubMed] [Google Scholar]
- 49.Tian D et al. (2021) Systematic review of CMTX1 patients with episodic neurological dysfunction. Ann. Clin. Transl. Neurol. 8, 213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tachikawa M et al. (2020) Targeted Proteomics-Based Quantitative Protein Atlas of Pannexin and Connexin Subtypes in Mouse and Human Tissues and Cancer Cell Lines. J. Pharm. Sci. 109, 1161–1168 [DOI] [PubMed] [Google Scholar]
- 51.Ferrell RE et al. (2010) GJC2 missense mutations cause human lymphedema. Am. J. Hum. Genet. 86, 943–948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Castorena-Gonzalez JA et al. (2018) Mechanisms of Connexin-Related Lymphedema. Circ. Res. 123, 964–985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Churko JM and Laird DW (2013) Gap junction remodeling in skin repair following wounding and disease. Physiology 28, 190–198 [DOI] [PubMed] [Google Scholar]
- 54.Lilly E et al. (2016) Connexin channels in congenital skin disorders. Semin. Cell Dev. Biol. 50, 4–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ishida-Yamamoto A (2016) Erythrokeratodermia variabilis et progressiva. J. Dermatol. 43, 280–285 [DOI] [PubMed] [Google Scholar]
- 56.Dobrowolski R and Willecke K (2009) Connexin-caused genetic diseases and corresponding mouse models. Antioxid. Redox Signal. 11, 283–295 [DOI] [PubMed] [Google Scholar]
- 57.Garcia IE et al. (2016) From Hyperactive Connexin26 Hemichannels to Impairments in Epidermal Calcium Gradient and Permeability Barrier in the Keratitis-Ichthyosis-Deafness Syndrome. J. Invest. Dermatol. 136, 574–583 [DOI] [PubMed] [Google Scholar]
- 58.Srinivas M et al. (2019) Connexin43 mutations linked to skin disease have augmented hemichannel activity. Sci. Rep. 9, 19. 10.1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cocozzelli AG and White TW (2019) Connexin 43 Mutations Lead to Increased Hemichannel Functionality in Skin Disease. Int. J. Mol. Sci. 20. E6186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mese G et al. (2011) The Cx26-G45E mutation displays increased hemichannel activity in a mouse model of the lethal form of keratitis-ichthyosis-deafness syndrome. Mol. Biol. Cell 22, 4776–4786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kuang Y et al. (2020) A potent antagonist antibody targeting connexin hemichannels alleviates Clouston syndrome symptoms in mutant mice. EBioMedicine 57, 102825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Paznekas WA et al. (2009) GJA1 mutations, variants, and connexin 43 dysfunction as it relates to the oculodentodigital dysplasia phenotype. Hum. Mutat. 30, 724–733 [DOI] [PubMed] [Google Scholar]
- 63.Kumar V et al. (2020) Oculodentodigital Dysplasia: A Case Report and Major Review of the Eye and Ocular Adnexa Features of 295 Reported Cases. Case Rep. Ophthalmol. Med. 2020, 6535974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Flenniken AM et al. (2005) A Gja1 missense mutation in a mouse model of oculodentodigital dysplasia. Development 132, 4375–4386 [DOI] [PubMed] [Google Scholar]
- 65.Kalcheva N et al. (2007) Gap junction remodeling and cardiac arrhythmogenesis in a murine model of oculodentodigital dysplasia. Proc. Natl. Acad. Sci. U S A 104, 20512–20516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dobrowolski R et al. (2008) The conditional connexin43G138R mouse mutant represents a new model of hereditary oculodentodigital dysplasia in humans. Hum. Mol. Genet. 17, 539–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Abitbol JM et al. (2018) Mice harbouring an oculodentodigital dysplasia-linked Cx43 G60S mutation have severe hearing loss. J. Cell Sci. 131. jcs214635. [DOI] [PubMed] [Google Scholar]
- 68.Huang T et al. (2014) Myogenic bladder defects in mouse models of human oculodentodigital dysplasia. Biochem. J. 457, 441–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stewart MK et al. (2013) The severity of mammary gland developmental defects is linked to the overall functional status of Cx43 as revealed by genetically modified mice. Biochem. J. 449, 401–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Guruvaiah P et al. (2016) Analysis of Connexin37 gene C1019T polymorphism and PCOS susceptibility in South Indian population: case-control study. Eur. J. Obstet. Gynecol. Reprod. Biol. 196, 17–20 [DOI] [PubMed] [Google Scholar]
- 71.Bachelot A et al. (2018) A common African variant of human connexin 37 is associated with Caucasian primary ovarian insufficiency and has a deleterious effect in vitro. Int. J. Mol. Med. 41, 640–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Noureldin M et al. (2018) Functional Characterization of Novel Atrial Fibrillation-Linked GJA5 (Cx40) Mutants. Int. J. Mol. Sci. 19. E977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sun Y et al. (2013) Novel germline GJA5/connexin40 mutations associated with lone atrial fibrillation impair gap junctional intercellular communication. Hum. Mutat. 34, 603–609 [DOI] [PubMed] [Google Scholar]
- 74.Gollob MH et al. (2006) Somatic mutations in the connexin 40 gene (GJA5) in atrial fibrillation. N. Engl. J. Med. 354, 2677–2688 [DOI] [PubMed] [Google Scholar]
- 75.Sun Y et al. (2014) Atrial fibrillation-linked germline GJA5/connexin40 mutants showed an increased hemichannel function. PLoS One 9, e95125. 10.1371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bai D (2014) Atrial fibrillation-linked GJA5/connexin40 mutants impaired gap junctions via different mechanisms. FEBS Lett. 588, 1238–1243 [DOI] [PubMed] [Google Scholar]
- 77.Lubkemeier I et al. (2013) The Connexin40A96S mutation from a patient with atrial fibrillation causes decreased atrial conduction velocities and sustained episodes of induced atrial fibrillation in mice. J. Mol. Cell Cardiol. 65, 19–32 [DOI] [PubMed] [Google Scholar]
- 78.Li RG et al. (2021) Connexin45 (GJC1) loss-of-function mutation contributes to familial atrial fibrillation and conduction disease. Heart Rhythm. 18, 684–693 [DOI] [PubMed] [Google Scholar]
- 79.Seki A et al. (2017) Progressive Atrial Conduction Defects Associated With Bone Malformation Caused by a Connexin-45 Mutation. J. Am. Coll. Cardiol. 70, 358–370 [DOI] [PubMed] [Google Scholar]
- 80.Easton JA et al. (2019) A rare missense mutation in GJB3 (Cx31G45E) is associated with a unique cellular phenotype resulting in necrotic cell death. Exp. Dermatol. 28, 1106–1113 [DOI] [PubMed] [Google Scholar]
- 81.Berthoud VM et al. (2016) The Cataract-linked Mutant Connexin50D47A Causes Endoplasmic Reticulum Stress in Mouse Lenses. J. Biol. Chem. 291, 17569–17578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Taki T et al. (2018) Roles of aberrant hemichannel activities due to mutant connexin26 in the pathogenesis of KID syndrome. Sci. Rep. 8, 12824. 10.1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Garcia IE et al. (2018) The syndromic deafness mutation G12R impairs fast and slow gating in Cx26 hemichannels. J. Gen. Physiol. 150, 697–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bai D (2016) Structural analysis of key gap junction domains--Lessons from genome data and disease-linked mutants. Semin. Cell Dev. Biol. 50, 74–82 [DOI] [PubMed] [Google Scholar]
- 85.Sanchez HA et al. (2014) Altered inhibition of Cx26 hemichannels by pH and Zn2+ in the A40V mutation associated with keratitis-ichthyosis-deafness syndrome. J. Biol. Chem. 289, 21519–21532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shibayama J et al. (2005) Functional characterization of connexin43 mutations found in patients with oculodentodigital dysplasia. Circ. Res. 96, e83–91 [DOI] [PubMed] [Google Scholar]
- 87.Chen L et al. (2017) The connexin 46 mutant (V44M) impairs gap junction function causing congenital cataract. J. Genet. 96, 969–976 [DOI] [PubMed] [Google Scholar]
- 88.Thevenin AF et al. (2017) Phosphorylation regulates connexin43/ZO-1 binding and release, an important step in gap junction turnover. Mol. Biol. Cell 28, 3595–3608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Abrams CK et al. (2018) Alterations at Arg(76) of human connexin 46, a residue associated with cataract formation, cause loss of gap junction formation but preserve hemichannel function. Am. J. Physiol. Cell Physiol. 315, C623–C635 [DOI] [PubMed] [Google Scholar]
- 90.Shuja Z et al. (2016) Connexin26 Mutations Causing Palmoplantar Keratoderma and Deafness Interact with Connexin43, Modifying Gap Junction and Hemichannel Properties. J. Invest. Dermatol. 136, 225–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Levit NA et al. (2015) Aberrant connexin26 hemichannels underlying keratitis-ichthyosis-deafness syndrome are potently inhibited by mefloquine. J. Invest. Dermatol. 135, 1033–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lee MY et al. (2019) Allele-Specific Sirna Corrects Aberrant Cellular Phenotype in Keratitis-Ichthyosis-Deafness Syndrome Keratinocytes. J. Invest. Dermatol. 140,1035–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Acosta ML et al. (2021) Connexin therapeutics: blocking connexin hemichannel pores is distinct from blocking pannexin channels or gap junctions. Neural Regen. Res. 16, 482–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Aasen T et al. (2017) Gap junctions and cancer: communicating for 50 years. Nat. Rev. Cancer 17, 74. 10.1038 [DOI] [PubMed] [Google Scholar]
- 95.Jara O et al. (2018) Chemical chaperone treatment improves levels and distributions of connexins in Cx50D47A mouse lenses. Exp. Eye Res. 175, 192–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Xu L et al. (2017) Design and Characterization of a Human Monoclonal Antibody that Modulates Mutant Connexin 26 Hemichannels Implicated in Deafness and Skin Disorders. Front. Mol. Neurosci. 10, 298. 10.3389 [DOI] [PMC free article] [PubMed] [Google Scholar]