

Abstract
Anti-tubulin agents, such as paclitaxel, have been used extensively for treatment of several types of cancer, including ovarian, lung, breast, and pancreatic cancers. Despite their wide use in cancer treatment, however, patient response is highly variable and drug resistance remains a major clinical issue. Protein kinase RNA-activated (PKR) plays a critical role in immune response to viral infection. We identified PKR as a phospho-protein in response to anti-tubulin agents and this phosphorylation occurs independent of its own kinase activity. PKR is phosphorylated by cyclin-dependent kinase 1 (CDK1) during anti-tubulin treatment and unperturbed mitosis and that PKR regulates mitotic progression in a phosphorylation-dependent manner. Furthermore, inactivation of PKR confers resistance to paclitaxel in ovarian and breast cancer cells in vitro and in vivo. PKR expression levels and activity are decreased in chemotherapeutic recurrent ovarian cancer patients. Mechanistically, our findings suggest that PKR controls paclitaxel chemosensitivity through repressing Bcl2 expression. Pharmacological inhibition of Bcl2 with FDA-approved agent venetoclax overcomes paclitaxel resistance in preclinical animal models of ovarian cancer. Our results suggest that PKR is a critical determinant of paclitaxel cytotoxicity and that PKR-Bcl2 axis as a potential therapeutic target for the treatment of recurrent drug-resistant ovarian tumors.
Keywords: PKR, phosphorylation, paclitaxel, chemosensitivity, Bcl2
Introduction
Anti-tubulin drugs, such as paclitaxel (Taxol) and docetaxel, are widely used for various malignancies, including ovarian, breast, non-small cell lung, and pancreatic cancers. Despite their wide use in cancer treatment, however, patient responses are unpredictable, and drug resistance (both inherent and acquired) is common and remains a major challenge in clinic (1,2).
Protein kinase R (PKR), also known as eukaryotic translation initiation factor 2-alpha kinase 2 (EIF2AK2), was originally identified as an interferon inducible double-stranded RNA (dsRNA) activated kinase (3–5). Upon infection, host PKR is activated through binding to viral RNAs and conformational change (4). PKR activation (auto-phosphorylated) leads to the phosphorylation and inactivation of a subunit of eukaryotic initiation factor 2 (EIF2S1/EIF2-α), which results in the suppression of global protein translation and subsequent the induction of apoptosis and inflammation (3,4). Besides dsRNAs, PKR can be activated by cellular proteins and RNAs in response to various cellular stress such as metabolic stress (6,7), oxidative stress (8), calcium depletion (9), endoplasmic reticulum stress (7,10), and genotoxic drugs (11,12).
In addition to its well-established role in the innate immune response (3,4,13). PKR is also involved in cell growth and tumorigenesis (3,14,15). Early studies suggest PKR as a tumor suppressor (16,17). Consistent with these observations, PKR is suppressed or inactivated in head and neck squamous cell carcinoma (18), hepatocellular carcinoma (19), lung (20), colon tumors (21), and leukemia (22). Furthermore, increased PKR expression positively correlates with improved prognosis in some of these cancers. A recent study reported that PKR antagonizes HER2+ breast tumorigenesis and improves the efficacy of Trastuzumab therapy (23). PKR has also been implicated in the anti-tumor activity of DNA-damaging agents such as doxorubicin, etoposide, and 5-FU (11,12,24,25). However, other studies suggest a tumor-promoting role of PKR in several types of cancer including breast (26), colon (27), melanoma (27), acute myeloid leukemia (28,29), and lung adenocarcinoma (30).
Upon activation, PKR functions in signal transduction and transcriptional control through the nuclear factor κB (NF-κB), the signal transducer and activator of transcription protein 1/3 (STAT1/3), p53, activating transcription factors (ATFs), phosphatase and tensin homolog (PTEN), mitogen-activated protein kinase (MAPK), and the toll-like receptors (TLKs) (3,4). Accumulated evidence highlights the importance of PKR as a molecular target for chemotherapeutics and its potential as a biomarker in human malignancies. Despite the known function of PKR in response to various stresses, whether anti-tubulin drugs deliver the stress signal to PKR signaling and the role of PKR in anti-tubulin chemotherapeutics are unclear.
Using a Phos-tag approach, we identified PKR as a protein that becomes phosphorylated in response to anti-tubulin drug treatment and show that the phosphorylation occurs independent of its own kinase activity. We also demonstrate that alterations of PKR expression determine paclitaxel chemosensitivity, and that PKR regulates paclitaxel response by repressing B-cell lymphoma 2 (Bcl2) in vitro and in vivo. Since FDA-approved agents for Bcl2 inhibition are available, our results have clinical implications to improve treatment outcomes in patients undergoing anti-tubulin therapy.
Materials and methods
CRISPR/Cas9-mediated PKR knockout
An EGFP sequence was inserted into the pX330A_D10A-1×2 all-in-one vector (Addgene 58772) to establish the CRISPR vector expressing EGFP (31). To construct the all-in-one CRISPR/Cas9n plasmid targeting PKR, the sense and anti-sense oligonucleotides were synthesized, annealed, and Golden Gate assembled into the pX330A_D10A-1×2-EGFP and pX330S-2 vectors as described previously (31). The guide RNA sequences are as follows: PKR#1A-forward: GACCTCCACATGATAGGAGGT; PKR#1B-forward: GTACTACTCCCTGCTTCTGA; PKR#2A-forward: GAGTGTGCATCGGGGGTGCAT; PKR#2B-forward: GTCTGGGCAATTCTATTGATA. The resulting pX330A_D10A-1×2-EGFP-PKR#1 or -PKR#2 construct was transiently transfected into cells and GFP-positive clones were selected 48 h post-transfection by flow cytometry-based cell sorting. Single-cell clones were expanded and PKR deletion was screened by Western blotting analysis and genomic sequencing.
Cell culture and transfection
HEK293T, HeLa, SKOV3, TOV21G, OVCAR3, OVCAR8, and MCF7 cell lines were purchased from American Type Culture Collection (ATCC). The cell lines were authenticated at ATCC and were used at low (<30) passages. SKOV3 cells were maintained in McCoy’s 5A media, TOV21G cells were cultured in 1:1 mixture of MCDB 105 media and Medium 199, and MCF7 cells were maintained in MEM media with 0.01 mg/ml human recombinant insulin. Other cell lines were maintained in DMEM media and all cell lines were supplemented with 10% FBS and 1% penicillin/streptomycin. The isogenic cell lines (SKOV3 and SKOV3-TR) were obtained from Dr. Michael Seiden (32). Attractene (Qiagen) was used for transient overexpression following the manufacturer’s instructions. Nocodazole (100 ng/ml for 16–20 h) and Taxol (0.1 μM for 16 h) from LC laboratories were used to arrest cells in mitosis phase unless otherwise indicated. PKR inhibitor imidazole-oxindole C16 was from Sigma. Bcl2-specific inhibitor venetoclax/ABT199 were from AbbVie and Genentech (animal use) and LC Laboratory (for in vitro use). The use of other kinase inhibitors has been described (33,34).
Expression constructs
The original PKR cDNA was purchased from Harvard Medical School (HsCD0034–1151). To make the lentiviral PKR expression constructs, the above full-length PKR cDNA was cloned into the pSIN4-Flag-IRES-puro vector (31). Point mutations were generated by the QuikChange Site Directed PCR Mutagenesis Kit (Agilent) and verified by Sanger sequencing. The pCDH-puro-Bcl2 construct was purchased from Addgene (46971) and the cDNA was cloned into the pSIN4-Flag-IRES-neo vector (35). The pSIN4-Flag-IRES-neo vector was made by replacing the puromycin-coding sequence of the pSIN4-Flag-IRES-puro vector with a neomycin-coding sequence. The GFP-Cyclin B-R42A and pcDNA3-CDK1-AF (718) plasmids were from Jonathon Pines (Addgene 61849 and 39872).
Establishment of cell lines
Stable overexpression and re-expression of PKR (wild type, 3A:S83A/S456A/S542A mutant, kinase-dead: K296R mutant) in PKR-KO cells were achieved by lentivirus-mediated infection and selection. Ectopic expression of Bcl2 was also achieved by a lentivirus-mediated approach. The transduced cells were then selected with 800 μg/ml of neomycin (at 48 hours post-infection) to establish cell lines stably expressing exogenous proteins. HeLa-RFP and HeLa-PKR-KO-RFP cell lines were achieved by lentivirus-mediated infection of pHIV-H2BmRFP vector (Addgene 18982). The transduced and RFP-positive cells were selected by flow cytometry-based sorting. SKOV3-PKR-KO cells expressing luciferase were obtained by infection of pLenti PGK V5-LUC-Puro (W543–1) vector (Addgene 19360) and followed by puromycin selection.
Antibodies
The anti-PKR monoclonal antibody from Santa Cruz Biotechnology (sc-6282, 1:1000) and Cell Signaling Technology (12297, 1:2000) was used throughout the study. The anti-p-T446 PKR antibody was from Abcam (ab32036, 1:1000). Anti-CDK1 antibody (9116, 1:1000) was from Cell Signaling Technology. Anti-cyclin B1 (sc-245, 1:2000), anti-CDC27 (sc-9972, 1:1000), anti-GFP (sc-9996, 1:2000), and anti-β-actin (sc-47778, 1:2000) antibodies were obtained from Santa Cruz Biotechnology. Anti-Flag antibody (F1804, 1:2000) was from Sigma-Aldrich. Rabbit polyclonal phospho-specific antibodies against PKR S83, S456, and S542 were generated and purified by AbMart, Inc. The peptides used for immunizing rabbits were EKKAV-pS-PLLLT (S83), TLRYM-pS-PEQIS (S456), and TVWKK-pS-PEKNE (S542). The corresponding non-phosphorylated peptides were also synthesized and used for antibody purification and blocking assays. Other antibodies used in this study were provided in the supplemental table.
Phos-tag and Western blot analysis
Phos-tag was obtained from Wako Pure Chemical Industries, Ltd. (catalog no. 304–93521) and used at a concentration of 10 μM (with 100 μM MnCl2) in 8% SDS-acrylamide gels. After separation, the gels were equilibrated in 1X transfer buffer (25 mM Tris, 192 mM glycine, 10% methanol) containing 10 mM EDTA two times, each for 10 min. The gels were then soaked in transfer buffer (without EDTA) for another 10 min and followed by transferring onto PVDF membranes (Millipore). Western blotting, immunoprecipitation, and lambda phosphatase treatment assays were done as described (36).
Immunofluorescence staining, confocal microscopy, and live-cell imaging
Cells were fixed with 100% cold methanol at −20°C for 10 min, and then permeabilized with 1% Triton X-100 in PBS for 15 min at room temperature. Nonspecific epitopes were blocked with 4% BSA in PBS for 1 h and cells were then incubated with the primary antibodies (at 1:50 dilutions for both p-S83 PKR and p-S456 PKR) in 4% BSA/PBS solution for overnight at 4°C (37). Texas Red (GE Healthcare) and/or Alexa Fluor 594-conjugated (Molecular Probes) anti-rabbit/mouse IgG were incubated with the cells for 60 min with 4% BSA in PBS at room temperature. After washing the cells three times (10 min each wash) with PBS, the stained cells were mounted with ProLong Gold antifade reagent with DAPI (Thermo Fisher) and visualized with an upright, inverted, Axiovert 200 M Zeiss fluorescence microscope (Carl Zeiss). The Slidebook software (version 4.2, Intelligent Imaging Innovations) was used for analyzing and processing all immunofluorescence images. For peptide blocking assays, the phospho-PKR antibodies were diluted at 1:500 by an excess of phosphorylated peptides (2 μg/mL for 1 h at room temperature) in PBS. The phospho-peptide-containing antibody was then used for staining in parallel with staining using antibodies with no peptide or corresponding non-phospho-peptide (33).
Live cell imaging was performed as we previously described (38). Briefly, cells were seeded in Nunc™ 96-Well Optical-Bottom (165305, Thermo Fisher) at 1,000 cells per well in a volume of 100 μl. After an overnight incubation, cells were washed with PBS and incubated with Fluorescent-free medium (A1896701, Thermo Fisher). Imaging was performed using a Cellomics Arrayscan VTI Fluorescent Microscope Imager (Thermo Fisher) with a 20X objective and collected every 5 min for 24 h. Image sequences were viewed using NIH ImageJ software (http://rsbweb.nih.gov/ij/) and cell behavior was analyzed manually.
Assessment of apoptosis
Caspase-Glo 3/7 assay:
Cells were plated in white-walled 96-well plates (NUNC, 136101, Thermo Fisher) for 24 hours and treated with Taxol, venetocalx, C16 or the combination for an additional 24 hours. Equal volume of Caspase-Glo 3/7 reagent was subsequently added into each well and incubated for 30 minutes at room temperature in the dark. Apoptosis was measured by Caspase-Glo 3/7 Assay Kit (Promega, G8090) according to the manufacturer’s instructions and quantified on a lumiometer. Background luminescence (cell culture medium without cells and Caspase-Glo 3/7 Reagent) was subtracted from all measurements. Experiments were performed for at least three biological times.
Annexin V/PI staining:
The Dead Cell Apoptosis Kit with Annexin V Alexa Fluor™ 488 and propidium iodide (Invitrogen, V13241) was used for examination of apoptosis. Cells were cultured in 6-well plates and treated with drugs at the indicated concentrations for 24 hours. Cell suspension preparation and staining were done according to the procedures described in the kit’s manual. Stained cells were analyzed by flow cytometry with fluorescence emission at 530 nm and 575 nm. Both early (Annexin V positive/PI negative) and late (Annexin V positive/PI positive) apoptotic proportions for each population were included. Experiments were performed at least three times.
Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) assay:
TUNEL assays were performed to detect cell death on tumor samples by In Situ Cell Death Detection Kit, Fluorescein (Roche), according to the manufacturer’s instructions. The slides were mounted by DAPI (Thermo Fisher) and then evaluated by Axiovert 200 M Zeiss fluorescence microscope (Carl Zeiss).
Cleaved caspase-3 immunohistochemistry (IHC) staining:
IHC staining with cleaved caspase 3 antibody (1:100 dilutions, Cell Signaling Technology) was also used to detect apoptosis on fixed tumors by using a VECTASTAIN Elite ABC-Peroxidase Kits following the manufacturer’s instructions (PK-6101, VECTOR LAB INC).
Clonogenic assay
2,000 cells were seeded in six-well plates and incubated overnight. The cells were then treated with the indicated concentrations of paclitaxel for 24 hours. The drug was removed and surviving cells were left to form colonies. After 14 days of incubation, colonies were fixed with 4% paraformaldehyde for 15 minutes at room temperature and then washed with PBS. The fixed cells were stained with 0.5% crystal violet for thirty minutes, rinsed with tap water, and air-dried overnight. Quantification was achieved by ImageJ software.
Cell proliferation and cell migration assays
For cell proliferation assays, cells (SKOV3, MCF7, and TOV21G: 100,000 per well) were seeded in a six-well plate in triplicate. Cells were counted by the automated cell counter (Invitrogen). In vitro analysis of migration (SKOV3 and MCF7 cells: 1×106 cells and TOV21G: 5×105 cells) was assessed using Transwell system (Corning), according to the manufacturer’s instructions. The migratory cells were stained with 0.1% crystal violet and counted manually. Data are collected from at least three independent experiments.
Human phospho-kinase antibody array
The relative levels of phosphorylation of 43 kinases were measured using the Human Phospho-Kinase Array Kit (ARY003B, R&D Systems) according to the manufacturer’s instructions. Briefly, SKOV3 cells and SKOV3-KO cells were treated with 100 nM Taxol for 24 hours and then lysed in immunoprecipitation lysis buffer (Pierce). The arrays were then blocked with blocking buffer provided by the Kit and incubated with 500 μg of cell lysate/proteins overnight at 4°C. After washing, the arrays were incubated with a horseradish peroxidase (HRP)-conjugated secondary antibody and developed by enhanced luminol-based chemiluminescent reagent (Pierce).
IHC staining and scoring
The tissue array consists of matched recurrent/resistant and primary/naive from 14 ovarian cancer patients (39). Slides deparaffinization, antigen retrieving, and blocking were performed as we have described (40). The arrays were stained with anti-phospho- PKR T446 antibody (ab32036, 1:200 dilutions, Abcam) by using a VECTASTAIN Elite ABC-Peroxidase Kits following the manufacturer’s instructions (PK-6101, VECTOR LAB INC). Mouse tumors from SKOV3 xenografts (control and PKR knockout) were used to validate the antibody. Cleaved caspase 3 IHC staining (1:100 dilutions) was also used to detect cell death in tumors. Cell nuclei were stained with hematoxylin. Ventana iScan HT (Roche) was used for slide scanning with a 20X objective lens. Scoring and statistical analysis were done as we previously described (40).
Animal models
In the subcutaneous tumor model, SKOV3, SKOV3-PKR-KO, and SKOV3-PKR-OE cells were mixed with an equal volume of Matrigel (363201002, R&D Systems) and injected subcutaneously (5X106 cells/injection) on both flanks into female athymic nu/nu mice of 5–6 weeks old. Drug treatment started as soon as the tumor became palpable. Mice were treated with PBS or paclitaxel (Hospira) intraperitoneally daily for two weeks with 2 days drug off at the end of each week. Tumor growth was measured by an electronic caliper and the tumor volume (V) was calculated by the formula: V = 0.5 × length × width2. At the end of experiments, all tumors were excised, weighed, and stored for subsequent histological and biochemistry analysis.
In the intraperitoneal tumor model, SKOV3-PKR-KO-Luciferase cells (1X107) were injected intraperitoneally into 5–6-weeks old female immunodeficient nude mice. Drug treatment was initiated when luminescence signal reached >2×107 photons/sec/cm2/sr. Mice were randomly grouped and were treated with vehicle control/PBS, paclitaxel (12 mg/kg/d) (Hospira, IP injection), venetoclax (100 mg/kg/d) (AbbVie/Genentech, oral gavage), or the combination. Venetoclax was dissolved in 60% Phosal 50 PG (368315, Lipoid), 30% poly-ethylene glycol (P3265, Sigma-Aldrich), and 10% ethanol. Prior to imaging, animals were injected with D-luciferin through IP (150 mg/kg, Sigma-Aldrich) in 150 μl PBS. Tumor burden was measured weekly using the IVIS Spectrum In Vivo Imaging System (Perkin Elmer). All quantitative fluorescence measurements and analyses were performed using the Living Image software (V4.5.1, Perkin Elmer). The animals were housed in pathogen-free facilities. All animal experiments were approved by the University of Nebraska Medical Center Institutional Animal Care and Use Committee.
Statistical analysis
Statistical significance was evaluated using a two-tailed, unpaired Student’s t-test. The generalized linear mixed effects model with generalized logit link for multinomial distributed data was used to model the IHC data. The odds of having medium score or strong score instead of weak score were compared between the parental samples and resistant samples. The Wilcoxon rank sum test was used to compare the IHC staining data between groups. A P value of <0.05 was considered as indicating statistical significance.
Results
CDK1 phosphorylates PKR during anti-tubulin treatment
To understand the regulation and role of PKR in mitosis and anti-tubulin agent-induced cytotoxicity, we treated various cancer cells with Taxol or nocodazole (arrest cells in mitosis) and investigated the response of PKR using a Phos-tag method (41). We found that PKR protein was upshifted during Taxol or nocodazole treatment (Fig. 1A, B).
Figure 1. CDK1/cyclin B1 kinase complex phosphorylates PKR during mitotic arrest.

(A, B) HeLa (cervical cancer cell), U2OS (osteosarcoma cell), RKO (colorectal cancer cell), TOV21G (ovarian cancer), and SKOV3 (ovarian cancer) cells were treated with DMSO, nocodazole (100 ng/mL for 16 h; Noco), or Taxol (100 nM for 16 h) and total cell lysates were probed with the indicated antibodies on regular or Phos-tag SDS-polyacrylamide gels. Increased cyclin B1 levels or shifted CDC27 marks cells in mitosis.
(C) HeLa cells were treated with Taxol as indicated and total cell lysates were further treated with (+) or without (−) λ phosphatase (ppase). Increased cyclin B1 levels marks cells in mitosis.
(D) PKR activity (autophosphorylation at T446) is not altered during anti-tubulin treatment. HeLa cells were treated as in A.
(E) PKR kinase activity is not required for its mobility shift. HeLa cells expressing PKR-wild type (WT), PKR-2TA (T446A/T451A), or PKR-KD (kinase dead) were subjected to Taxol treatment and the mobility shift of PKR was examined regular and Phos-tag SDS polyacrylamide gels.
(F) HeLa cells were treated with Taxol together with or without various kinase inhibitors as we previously described (33,34,38). Inhibitors were added (with MG132 to prevent cyclin B1 from degradation and cells from exiting from mitosis) 1.5 h before harvesting the cells. Total cell lysates were electrophoresed on regular and Phos-tag SDS-polyacrylamide gels and probed with the indicated antibodies.
(G) HeLa cells expressing Flag-PKR or Flag-PKR-3A (S83A/S456A/S542A) were treated with Taxol for 24 h and exogenous PKR was immunoprecipitated by Flag antibodies. Samples were probed with phospho-specific antibodies for PKR.
(H) CDK1/Cyclin B1 induced PKR phosphorylation. HEK293T cells were transfected with expression constructs as indicated. GFP-Cyc B1-CA: GFP-Cyclin B1-R42A (a nondegradable/constitutive active mutant). Flag-CDK1-CA: Flag-CDK1 T14A/Y15A (nonphosphorylatable/constitutive active CDK1).
(I) HeLa cells expressing Flag-PKR were treated as indicated. Immunoprecipitated Flag-PKR and total cell lysates were subjected to Western blotting with the indicated antibodies. Increased p-Aurora marks cells in mitosis.
(J) Mitotic phosphorylation does not affect PKR activity (autophosphorylation at T446). HEK293T cells were transfected with expression constructs as indicated and total cell lysates were analyzed by Western blotting.
(K, L) HeLa parental or PKR-knockout (KO) cells were arrested at mitosis by Taxol and stained with antibodies against S83 PKR (K) and S456 PKR (L). Nuclei were stained with DAPI. PKR-KO HeLa cells were used to validate the specificity of the phospho-antibodies.
(M, N) Immunofluorescence (IF) staining of p-PKR S83 (M) and p-PKR S456 (N) in freely cycling HeLa cells. White and yellow arrows (in K-N) mark the metaphase and interphase cells, respectively.
Lambda phosphatase treatment largely converted all up-shifted bands to fast-migrating bands, confirming that the mobility shift of PKR during mitotic arrest is caused by phosphorylation (Fig. 1C). In response to viral infection and other stresses, PKR is activated by autophosphorylation at T446 and T451 (3,42). However, phosphorylation at T446 is not changed during nocodazole or Taxol treatment (Fig. 1D). Furthermore, the kinase-dead (KD) or T446A/T451A mutant PKR was shifted as efficiently as wild type PKR during Taxol treatment (Fig. 1E). These observations suggest that PKR kinase activity is not required for PKR phosphorylation in response to Taxol, and that PKR is phosphorylated on sites other than the autophosphorylation sites. We next used various kinase inhibitors to identify the candidate kinase for PKR phosphorylation. Inhibition of Aurora-A, -B, -C (with VX680), JNK1/2 (with SP600125), MEK-ERK kinases (with U0126), p38 (with SB203580), mTOR (with rapamycin), Akt (MK2206), or Plk1 (with BI2536) failed to alter phosphorylation of PKR during Taxol treatment (Fig. 1F). However, treatment with RO3306 (CDK1 inhibitor) or Purvalanol A (CDK1/2/5 inhibitor) significantly inhibited the mobility shift/phosphorylation of PKR (Fig. 1F). These data suggest that CDK1, a well-known master mitotic kinase, is a relevant kinase for PKR phosphorylation induced during Taxol or nocodazole treatment.
Through database-based analysis (https://www.phosphosite.org), we identified S83, S456, and S542 as potential novel phosphorylation sites in PKR. Interestingly, all sites are within proline-directed consensus sequences, which are in agreement with CDK1 (CDKs) phosphorylation sites (43). We generated phospho-specific antibodies against S83, S456, and S542 and examined whether PKR phosphorylation at these sites occurs in cells. Taxol treatment significantly increased the phosphorylation of S83 and S456 of transfected PKR (Fig. 1G). Phosphorylation at S542 was not detected due to poor quality of the antibody. The signal was abolished by mutating serines to alanines (Fig. 1G), suggesting that these phospho-antibodies specifically recognize phosphorylated PKR during Taxol treatment. Constitutive activation of CDK1 (a T14A/Y15F mutant) or Cyclin B1 (R42A mutant; a binding partner and activator of CDK1) stimulated PKR phosphorylation on S83 and S456 sites (Fig. 1H). Using kinase inhibitors, we further demonstrated that phosphorylation of PKR is CDK1 kinase-dependent (Fig. 1I). In contrast, CDK1-mediated phosphorylation did not affect PKR autophosphorylation at T446 (Fig. 1J).
We further performed immunofluorescence staining using the described antibodies in Taxol-treated cells. The phospho-S83 and phospho-S456 signals were significantly higher in Taxol-arrested prometaphase cells (white arrows, condensed chromosomes) when compared with interphase cells (yellow arrows) (Fig. 1K, L). Further verifying antibody specificity, the staining signals from phospho-S83 and phospho-S456 were abolished in CRISPR-Cas9-mediated PKR-knockout (KO) cells (Fig. 1K, L). The phospho-signal was also detected in mitotic cells in an asynchronous culture (Fig. 1M, N, white arrows), suggesting that phosphorylation of PKR occurs during normal mitosis.
PKR is required for precise mitosis
To explore PKR’s possible role in regulating mitotic processes, we generated PKR-KO HeLa-RFP-H2B cells and performed fluorescent live-cell imaging. PKR-KO cells have a marked shortened mitotic length (from nuclear-envelope break down (NEBD) to telophase) when compared with parental cells (Fig. 2A, B). The difference is largely due to accelerated anaphase onset (Fig. 2A, C). Re-expression of wild type PKR in PKR-KO cells rescued this phenotype, however addback of the non-phosphorylatable mutant PKR (PKR-3A) failed to do so, supporting that PKR phosphorylation is required for proper mitotic progression (Fig. 2B, C).
Figure 2. Phosphorylation of PKR is required for precise mitosis in cancer cells.

(A) PKR deletion shortens mitotic length (from NEBD to anaphase onset) in HeLa cells. RFP-HeLa and RFP-HeLa-PKR-KO (knockout) cells were seeded in 96-well plate and incubated overnight. Live-cell imaging was performed using a Fluorescent Microscope Imager with a 20X objective and collected every 5 min for 24 h. Representative cells are shown.
(B, C) Various cell lines were subjected to live-cell imaging and mitotic length was quantified. NEBD: nuclear envelope breakdown; Ana: anaphase onset. Data in B, C were collected from 100 mitotic cells for each group (mean ± SD of three independent experiments). **: p<0.01; ***: p<0.001 (unpaired Student’s t test).
(D, E) Knockdown of PKR resulted in massive mitotic defects in HeLa and SKOV3 cells. Representative photos of normal mitosis (control) and mitotic abnormalities (PKR-KO) in HeLa cells were shown in D. Cells were stained with β-tubulin, γ-tubulin antibodies, and DAPI to visualize microtubules (green), centrosomes (red), and chromosomes (blue), respectively. Data in E were collected from 150 mitotic cells for each group (mean ± SD of three independent experiments).
(F) Establishment of cell lines expressing exogenous PKR-WT, PKR-3A, or PKR-KD (kinase dead) in HeLa PKR-KO cells.
(G-J) Qualification of cells with mitotic defects in cell lines established in F. Data were collected from 150 mitotic cells for each group (mean ± SD of three independent experiments). ***: p<0.001 (unpaired Student’s t test).
Deletion of PKR in HeLa (cervical cancer) and SKOV3 (ovarian cancer) cells resulted in a significant higher percentage of cells with mitotic defects including spindle disorganization (including multipolar spindles, β-tubulin staining), supernumerary centrosomes (γ-tubulin staining), and chromosome lagging/misalignment/segregation (DAPI staining) (Fig. 2D, E). These phenotypes were largely rescued by re-expressing wild type PKR in PKR-KO cells (Fig. 2F–J). In contrast, the phospho-deficient mutant (PKR-3A) or the kinase dead form (PKR-KD) failed to restore these defects (Fig. 2G–J), suggesting that both mitotic phosphorylation and kinase activity of PKR are essential for precise mitosis.
PKR levels determine paclitaxel chemosensitivity in cancer cells
To examine the role of PKR in cancer cell survival and Taxol-induced cytotoxicity, we knocked out PKR in SKOV3, TOV21G (ovarian cancer), and MCF7 (breast cancer) cells (with two independent gRNAs for each cell line) in addition to HeLa cells (Fig. 3A). Deletion of PKR did not induce significant apoptosis under normal cell culture conditions. In contrast, significant changes in PARP cleavage, a canonical indicator of apoptosis, were observed when cells were treated with Taxol. Cells with PKR deletion were resistant to Taxol treatment as compared to control cells in HeLa, MCF7, SKOV3, and TOV21G lines (Fig. 3A). Caspase3/7 activity measurements and annexin-V staining (additional assays for apoptosis) confirmed PARP cleavage data (Fig. 3B).
Figure 3. PKR regulates Taxol chemosensitivity in cancer cells.

(A) CRISPR/cas9n-mediated deletion of PKR promotes Taxol resistance. Two independent guide RNAs (KO-1 and KO-2) were used in each cell line. Cells were treated with Taxol (100 nM) for 24 h and total cell lysates were probed with the indicated antibodies. Cleaved (Cl)-PARP serves as an apoptotic marker.
(B) Cells were incubated with Taxol (100 nM) for 24 h and the caspase 3/7 activities were measured by Caspase-Glo 3/7 Assay (Promega). Cell death was also determined by flow cytometry-based Annexin-V/PI staining in SKOV3 cells. **: p<0.01, ***: p<0.001 (unpaired Student’s t-test).
(C-E) MCF7, SKOV3, and OVCAR8 cells were treated with DMSO (control), PKR inhibitor C16 (1 μM), Taxol (100 nM) or the combination. Apoptosis was determined by Western blot analysis using cleaved (cl)-PARP.
(F, G) MCF7, SKOV3, OVCAR8, and TOV21G cells were treated with DMSO (control), PKR inhibitor C16 (1 μM), Taxol (T, 100 nM) or the combination. Apoptosis was determined by Caspase-Glo 3/7 Assay (Promega) and flow cytometry-based Annexin-V/PI staining. **: p<0.01; ***: p<0.001 (unpaired Student’s t-test).
(H) Western blots confirm establishment of PKR-overexpressing cell lines.
(I, J) Enhanced expression of PKR promotes cell death in response to Taxol treatment. TOV21G, SKOV3 or MCF7 (control and PKR-OE) cells were treated with Taxol (100 nM) for 24 h. Apoptosis was determined by the levels of cleaved PARP (I) or caspase3/7 activities (J).
(K) PKR regulates survival in response to Taxol treatment. Clonogenic assays were performed in SKOV3 and MCF7 (control, PKR-KO, PKR-OE) cells. OE: overexpression. Data in panels B, F, G, J, and K are expressed as mean ± SEM from three independent experiments. *: p<0.05, **: p<0.01, ***: p<0.001 (unpaired Student’s t-test).
We also examined the cellular response to the PKR selective inhibitors C16 and 2-AP (44–47). No significant effects on survival and apoptosis of MCF7, SKOV3, and OVCAR8 (another ovarian cancer cell line) cells were observed when C16 was used at 1 μM (Fig. 3C–G). However, combined treatment of C16 and Taxol greatly decreased cytotoxicity compared with Taxol treatment alone, suggesting that inhibition of PKR induces resistance to Taxol treatment (Fig. 3C–G).
We next established TOV21G, SKOV3, and MCF7 cells expressing vector (control) or PKR (Fig. 3H). Ectopic expression of PKR in these cells did not significantly affect cell proliferation or apoptosis. However, consistent with the PKR knockout cell phenotype (Fig. 3C–G), ectopic expression of PKR significantly increased Taxol cytotoxicity in SKOV3, TOV21G, and MCF7 cells (Fig. 3I, J). These observations indicate that PKR regulates Taxol chemosensitivity in ovarian and breast cancer cells.
We next performed clonogenic assays to examine the effect of PKR in cell survival in response to Taxol treatment. As shown in Figure 3K, deletion of PKR significantly promoted cell survival after Taxol treatment compared with control cells. In agreement, overexpression of PKR reduced survival in SKOV3 and MCF7 cells in response to Taxol (Fig. 3K; S1A, B). Together, these data suggest that PKR is a critical regulator of Taxol chemosensitivity in cancer cells.
PKR regulates paclitaxel chemosensitivity in ovarian cancer in vivo
We further explored the biological significance of PKR in cancer cell growth. Deletion of PKR significantly reduced cell proliferation rates (Fig. 4A–C) and migratory ability (Fig. 4D–I) of MCF7, TOV21G, and SKOV3 cells. Importantly, SKOV3-PKR-KO cells formed significantly smaller tumors compared to control cells in immunodeficient mice (Fig. 4J, K). While Taxol treatment inhibited the tumor growth of SKOV3 parental cells, it did not further decrease tumor sizes in SKOV3-PKR-KO cells (Fig. 4J, K). These data suggest that deletion of PKR inhibits tumor growth but may also promote Taxol resistance. Ectopic expression of PKR failed to alter the tumor growth in SKOV3 cells without drug treatment, however, in line with loss-of-function phenotypes, PKR overexpression greatly sensitized SKOV3 cells to Taxol treatment (Fig. 4L, M). Elevated levels of apoptosis (revealed by cleaved caspase 3) were detected in Taxol treated SKOV3-overexpression tumors (Fig. 4N). Therefore, loss-of-function and gain-of-function assays suggest that PKR controls Taxol chemosensitivity in vivo.
Figure 4. PKR promotes tumorigenesis and paclitaxel sensitivity in vivo.
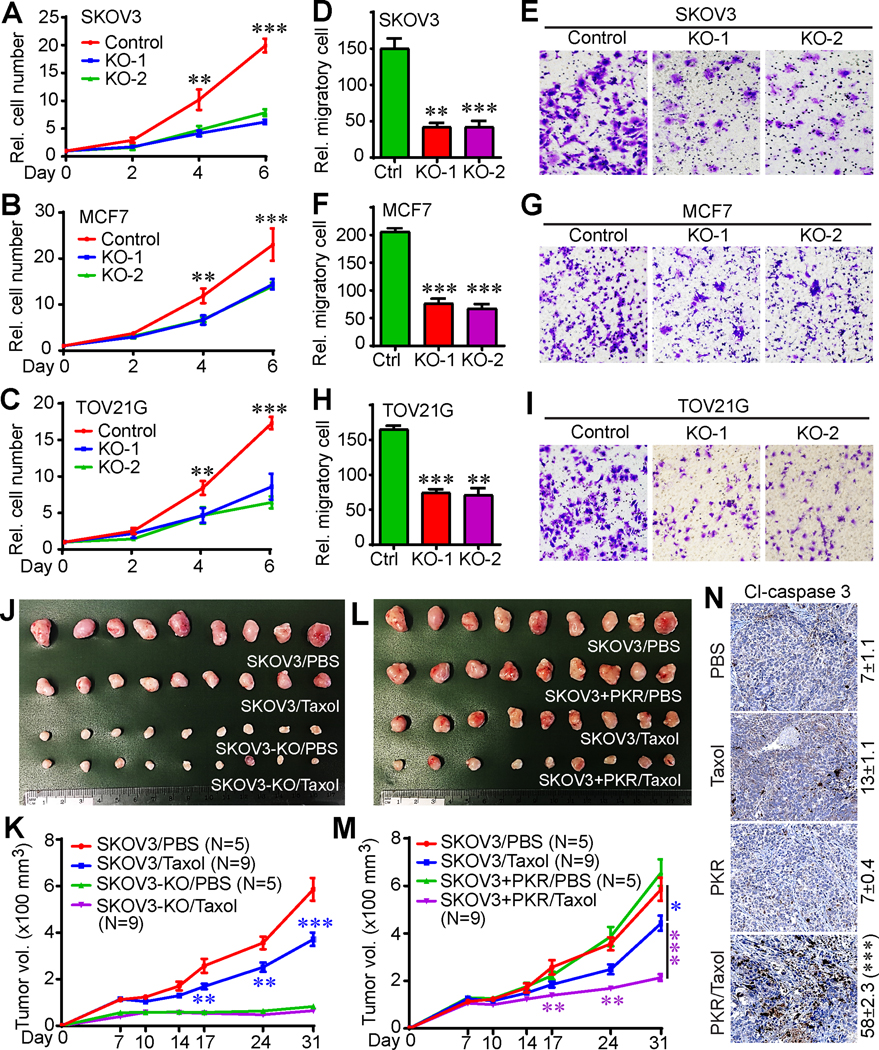
(A-C) Cell proliferation curves in SKOV3 (A), MCF7 (B), and TOV21G (C) cell lines.
(D-I) Cell migration (Transwell assay) in parental and PKR-KO SKOV3, MCF7, and TOV21G cells. Representative fields were shown in E (SKOV3), G (MCF7), and I (TOV21G). Data in panels A-D, F, and H are expressed as mean ± SEM from three independent experiments. **: p<0.01, ***: p<0.001 (unpaired Student’s t-test).
(J, K) Deletion of PKR inhibited tumor growth, but promoted paclitaxel resistance in animals. Cells were injected subcutaneously into female athymic mice on both flanks. Paclitaxel (12 mg/kg) was injected through intraperitoneal every other day for 2 weeks. The initial treatment was done at day 7 post cell injection. Tumors were excised and photographed at the endpoint (J).
(L, M) Enhanced expression of PKR improved paclitaxel sensitivity in vivo. A lower dose of paclitaxel (8 mg/kg) was used in this experiment. The control groups (SKOV3/PBS) in panels J and K are identical to control groups in panels L and M. Data in panels K and M are expressed as mean ± SEM from all tumors of each group. *: p<0.05, **: p<0.01, ***: p<0.001 (unpaired Student’s t-test).
(N) Immunohistochemistry (IHC) staining of cleaved caspase 3 to determine cell death in tumor samples in panel L. Cl-caspase 3-positive cells were quantified from three random fields in each tumor. ***: p<0.001 (unpaired Student’s t-test).
PKR phosphorylation is essential for cancer cell growth and Taxol chemosensitivity
To determine if phosphorylation of PKR is involved in cell growth and Taxol chemosensitivity, we re-expressed wild type PKR or non-phosphorylatable mutant PKR (PKR-3A) in SKOV3 and MCF7 PKR-KO cells (Fig. 5A, D). Importantly, re-expression of wild type PKR completely rescued cell proliferation and migration in PKR-KO cells (Fig. 5B, C, E–G). However, expression of the non-phosphorylatable mutant PKR failed to restore or only partially restored these characteristics (Fig. 5B, C, E–G), suggesting that mitotic phosphorylation is required for PKR activity in SKOV3 and MCF7 cells. In addition, addback of PKR completely restored the cell death effect in PKR-KO cells induced by Taxol (Fig. 5H–J). However, re-expression of PKR-3A failed to do so (Fig. 5H–J), suggesting that CDK1-meditated phosphorylation is required for PKR-driven chemosensitivity in response to Taxol. Together, these studies elucidate a previously undefined regulatory mechanism of PKR in response to anti-tubulin chemotherapeutics and identify PKR as a kinase regulating Taxol cytotoxicity in cancer cells.
Figure 5. Mitotic phosphorylation of PKR is required for cancer cell growth and paclitaxel chemosensitivity.

(A) Establishment of cell lines with exogenous PKR-WT or PKR-3A (S83A/S456A/S542A) in SKOV3-PKR-KO cells.
(B, C) Cell proliferation curves and cell migration assays (Transwell assay) with cell lines established in A.
(D) Establishment of cell lines with exogenous PKR-WT or PKR-3A (S83A/S456A/S542A) in MCF-7-PKR-KO cells.
(E-G) Cell proliferation curves and cell migration assays (Transwell assay) with cell lines established in D. Representative images of cell migration were shown in G.
(H) Cell lines from A and D were treated with DMSO or Taxol (100 nM) for 24 h. Total cell lysates were probed with cleaved-PARP to determine apoptosis.
(I, J) Cell lines from A and D were treated with Taxol (100 nM) for 24 h and the caspase 3/7 activities in these cells were analyzed by Caspase-Glo 3/7 Assay. Data in panels B, C, E, F, I, and J are expressed as mean ± SEM from three biological replicates. *: p<0.05, **: p<0.01, ***: p<0.001 (unpaired Student’s t-test).
PKR regulates the apoptotic pathway in ovarian cancer cells
To define the signaling pathway downstream of PKR, we compared kinase activity using a phospho-kinase protein array (R&D Systems, consists of 43 phospho-kinases) in control and PKR-KO TOV21G cells (Fig. S2A). We found p-c-Jun levels was elevated in TOV21G-KO cells compared with control cells. In addition, the levels of p-p27 T198, p-LCK, p-p53 (S46 and S392), and p-RSK S380 were significantly decreased upon PKR deletion (Fig. S2A, B). We further validated these findings in SKOV3 and TOV21G cells. Taxol treatment greatly induced p53 and its phosphorylation at S392, in contrast, this induction was largely blocked in PKR-KO cells (Fig. S2C, D). Phosphorylation of RSK was confirmed (reduced) in both TOV21G and SKOV3 PKR-KO cells (Fig. S2E). We were unable to confirm the increased p-c-Jun in PKR-KO cells. Akt-PI3K-PTEN, JAK-STAT3, β-catenin, NF-kB, MAPK (ERK and p38), and Hippo-YAP signaling activities were not significantly altered upon PKR deletion (Fig. S2E, F).
The Bcl2 family proteins are key regulators of apoptosis. Several reports demonstrated that inactivation of Bcl2, B-cell lymphoma-extra large (Bcl-xL) and myeloid cell leukemia 1 (Mcl1) (by phosphorylation and/or protein destruction) is essential to potentiate cell death in response to anti-tubulin agents (48–52). To further study the mechanism of PKR-induced apoptosis in response to Taxol in cancer cells, the expression levels of pro- and anti-apoptotic proteins were examined in control and PKR-KO cells. Notably, PKR deletion significantly upregulates the anti-apoptotic protein Bcl2 and downregulates the pro-apoptotic protein Bcl2-associated X protein (Bax) (Fig. 6A). Bcl2 levels were also increased in MCF7 cell upon PKR deletion (Fig. 6B).
Figure 6. PKR regulates paclitaxel sensitivity through Bcl2.

(A) Expression of apoptosis regulators in SKOV3 parental and SKOV3-PKR-KO cell lines. Cells were treated with Taxol (100 nM) for 24 h and total cell lysates were probed with the indicated antibodies.
(B) Bcl2 expression is increased in MCF7-PKR-KO cells compared with control cells.
(C-H) Bcl2 inhibition reverses Taxol resistance in SKOV3-PKR-KO, TOV21G-PKR-KO, and MCF7-PKR-KO cells. Cells were treated with DMSO (control), Bcl2 inhibitor venetoclax (Ven, 10 μM), Taxol (100 nM) or the combination for 24 h. Apoptosis was determined by cleaved-PARP on Western blots (C). Cell apoptosis was also determined by measuring the caspase3/7 activity (D-F) and flow cytometry-based Annexin-V positivity (G, H).
(I-K) Establishment of cell lines with enhanced expression of both PKR and Bcl2. TOV21G, SKOV3, and MCF7 cells with PKR-overexpression (TOV21G+PKR, SKOV3+PKR, and MCF7+PKR) were established first and were then used for ectopic expression of Bcl2.
(L, M) Enhanced expression of Bcl2 confers resistance to Taxol. Cell lines established in H-J were treated with Taxol (100 nM) for 24 h and apoptosis was determined by cleaved-PARP expression (L) and caspase 3/7 activities (M).
(N) Overexpression of Bcl2 results in mitotic defects SKOV3 cells. Data were collected from 150 mitotic cells for each group (mean ± SD of three independent experiments). *: p<0.05; **: p<0.01 (unpaired Student’s t test).
(O) Expression levels of PKR and Bcl2 in parental and Taxol-resistant (TR) SKOV3 cells.
(P, Q) SKOV3-TR cells were treated with Taxol (250 nM) for 24 h or 48 h and apoptosis was determined by cleaved-PARP expression (P) and caspase 3/7 activities (Q).
(R) Phospho-PKR (autophosphorylation at T446, Abcam) levels is decreased in recurrent ovarian tumors. IHC staining with phospho-PKR on ovarian cancer tissue microarrays consisting of matched naïve and resistant tumors (39). Staining and scoring were done as we described (40). +: low expression; ++: moderate expression; +++: strong expression. P=0.0028 (Wilcoxon rank sum test). Data in panels D-H, M, and Q are expressed as mean ± SEM from three independent experiments. ***: p<0.001 (unpaired Student’s t-test).
Inhibition of Bcl2 overcomes Taxol resistance in vitro
Bcl2 proteins are the targets for chemotherapeutics and Bcl2 selective inhibitor venetoclax (ABT199) is FDA-approved for the treatment of certain types of leukemia. While PKR-KO cells are largely resistant to Taxol treatment, addition of venetoclax significantly enhanced Taxol-induced cell death/apoptosis in SKOV3, TOV21G, and MCF7 PKR-KO cells (Fig. 6C–H). Venetoclax treatment alone did not cause significant cell death in these cells (Fig. 6C–H). Consistently, while ectopic expression of PKR enhanced Taxol-induced cell death (Figs. 3, 4), these cells became resistant to Taxol treatment when Bcl2 was overexpressed (Fig. 6I–M). Interestingly, in line with the mitotic defects in PKR-KO, overexpression of Bcl2 in SKOV3 cells resulted in similar mitotic dysregulation (Fig. 6N). Furthermore, inhibition of Bcl2 restored the PKR-KO cells to the control level of mitotic defects (Fig. 6N). These findings suggest that PKR regulates Taxol chemosensitivity through Bcl2.
Importantly, PKR protein levels and PKR activity (revealed by autophosphorylation at T446) were reduced in previously established Taxol-resistant (TR) SKOV3 cells when compared to the corresponding parental cells (PT) (32) (Fig. 6O). Consistently, Bcl2 expression was greatly increased in SKOV3-TR cells (Fig. 6O). Again, Bcl2 inhibition re-sensitized SKOV3-TR cells to Taxol treatment (Fig. 6P, Q). These observations are consistent with the notion that reduced PKR activity contributes resistance to Taxol treatment by upregulating Bcl2, and that inhibition of Bcl2 overcomes Taxol resistance in ovarian cancer cells.
To determine the clinical relevance of PKR in chemoresistance, we performed immunohistochemistry (IHC) staining to analyze the kinase activity levels (p-T446) of PKR in paired, recurrent post-chemotherapy (paclitaxel and carboplatin) high-grade serous ovarian cancer and their primary untreated tumors (39). Bcl2 antibody is not suitable for IHC staining. Of interest, p-T446 levels of PKR were significantly lower in recurrent tumors compared to primary tumors (Fig. 6R). These data suggest the potential association between PKR inactivation and Taxol chemoresistance.
Pharmacological inhibition of Bcl2 overcomes Taxol resistance in vivo
We further validate our hypothesis in preclinical animal models. SKOV3-PKR-KO-Luciferase cells were injected intraperitoneally into immunodeficient mice and tumor growth was assessed following Taxol, venetoclax, or combination treatment (Fig. 7A). Venetoclax treatment alone did not suppress tumor growth and the response to Taxol treatment was heterogeneous (Fig. 7B, C). Strikingly, combined therapy of venetoclax and Taxol almost completely suppressed tumor growth (Fig. 7B–D). Moreover, no obvious side effects were detected in combined therapy-treated animals (no body weight loss) (Fig. 7E). Consistent with cell culture models, massive apoptosis was detected in venetoclax and Taxol treated animals (Fig. 7F–H). These observations indicate that combination of venetoclax with Taxol overcomes Taxol resistance in ovarian cancer.
Figure 7. Bcl2 inhibition reverses paclitaxel resistance in vivo.
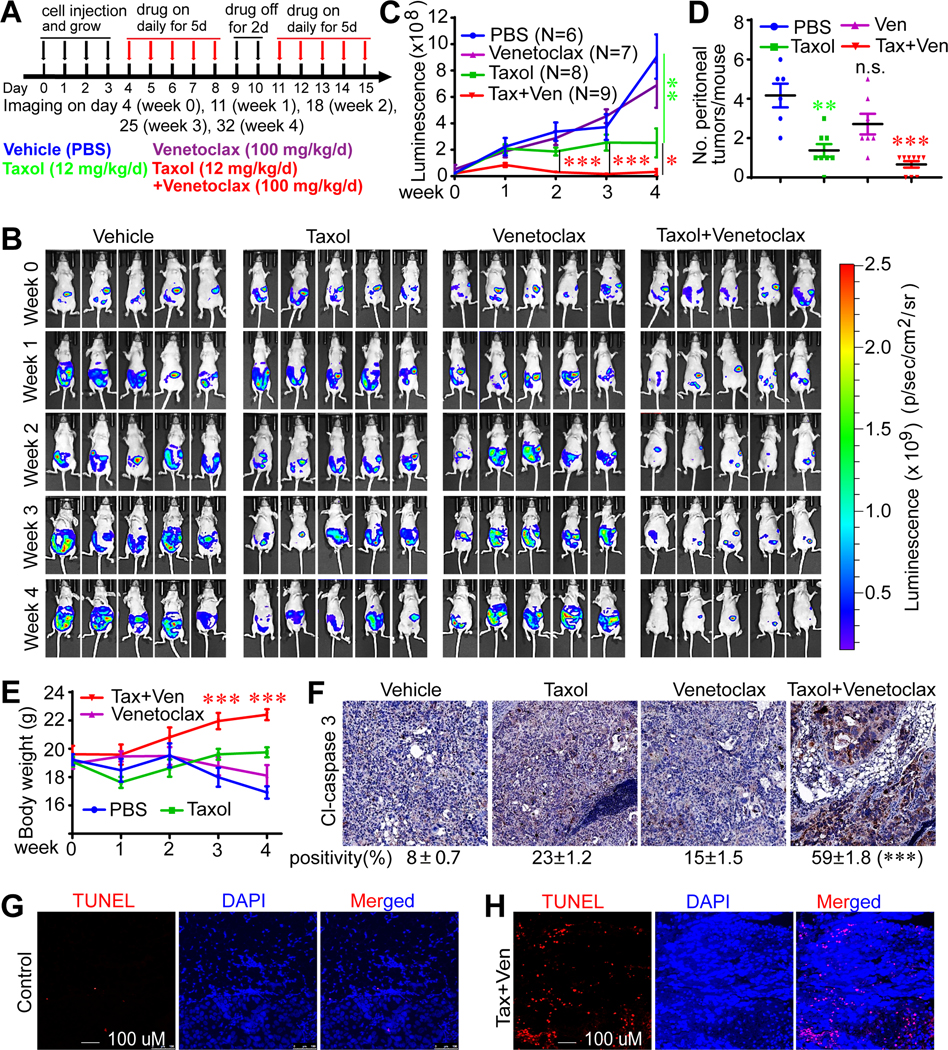
(A) A schematic diagram for drug treatment. Ten million SKOV3-PKR-KO-Luciferase cells were injected intraperitoneally into 5–6-week old female immunodeficient (athymic nude) mice and were randomly grouped 4 days post injection. Mice were treated with PBS (control), paclitaxel intraperitoneally, venetoclax orally or the combination.
(B) The luminescence images of the intraperitoneal tumor xenografts from four different treatment groups at the indicated time points.
(C) Quantification of the luminescence signals from B.
(D) Number of peritoneal tumors was examined at the end of the experiments. Data in panels C and D are expressed as mean ± SEM from all tumors of each group. *: p<0.05, **: p<0.01, ***: p<0.001 (unpaired Student’s t-test). N.S.: not significant.
(E) Body weight of animals in B. ***: p<0.001 (Taxol+Venetoclax group vs PBS control group, unpaired Student’s t-test).
(F-H) Combined treatment of Taxol and Bcl2 inhibition induced apoptosis determined by cleaved caspase 3 IHC staining (F) and TUNEL staining (G, H). Cl-caspase 3-positive cells were quantified from three random fields in each tumor (F). ***: p<0.001 (unpaired Student’s t-test).
Discussion
The mitotic window is considered the most vulnerable period of the cell cycle and is the target of anti-cancer drugs including Taxol (1,53). Intact mitotic checkpoints are essential for Taxol chemosensitivity, therefore mitotic regulators are often linked to Taxol response. PKR has recently been shown to be activated (phosphorylation of T446 and T451) during mitosis and depletion of PKR led to delay in mitotic progression and defects in cytokinesis by regulating multiple mitotic factors (54). In line with this report, we identified PKR as a mitotic-phosphoprotein and observed that PKR deletion causes massive mitotic defects (Figs. 1, 2). However, in contrast to this study, we did not detect PKR activation (autophosphorylation at pT446) in response to anti-tubulin-induced mitotic arrest (Fig. 1). Instead, we identified CDK1-mediated novel phosphorylation sites (S83, S456, and S542) on PKR, independent of its own kinase activity and T446 phosphorylation (Fig. 1). We further demonstrated that mitotic phosphorylation of PKR is essential for its role in mitotic progression and Taxol chemosensitivity in various cancer cell lines (Figs. 2, 3, and 5). Thus, our findings reveal a new layer of regulation of PKR and suggest that inactivation of PKR contributes to Taxol resistance through regulating the mitotic machinery.
Tp53 status influences mitotic checkpoint function, cell cycle progression, senescence, and cell death in response to anti-tubulin drugs. The link between PKR and p53 has been described. For example, PKR associates with and directly phosphorylates p53 at the S392 residue (55). Consistent with this finding, we showed that Taxol-induced p53 S392 phosphorylation was largely blocked in PKR knockout cells (Fig. S2). PKR is also a p53 target gene (56) and PKR is required for p53 induction by Taxol (Fig. S2C, D), suggesting a positive feedback loop between two proteins. These previous and our initial observations prompted us to delete PKR in various cancer cell lines with different p53 status. MCF7 and TOV21G cells are p53 intact (wild type) and SKOV3 cells are p53 null, thus, our findings suggest that PKR-mediated mitotic progression and Taxol chemosensitivity is unlikely to be p53-dependent. Bcl2 has been shown to be phosphorylated during mitosis and this phosphorylation is essential for Taxol-induced cell death (48,49). Our data suggest that enhanced expression of Bcl2 resulted in mitotic defects and promoted Taxol resistance in SKOV3 cells (Fig. 6L–N). Furthermore, Bcl2 inhibition in PKR knockout cells restored these cells to normal mitosis and subsequently re-sensitized cells to Taxol treatment (Fig. 6C–H, N). Altogether, these observations suggest that Bcl2 is likely downstream PKR linking mitotic defects to Taxol chemosensitivity. Future studies are required to determine the precise mechanisms through which the PKR-Bcl2 axis connects the mitotic machinery and Taxol cytotoxicity.
Our study showed that PKR negatively regulates Bcl2 expression in ovarian cancer cells, which is consistent with previous reports (57,58). PKR activation leads to the phosphorylation and inactivation of EIF2S1/EIF2-α, which results in the suppression of protein synthesis (3). Therefore, it is expected that PKR inactivation might increase Bcl2 expression through increased protein translation. However, in our case, we did not detect significant decrease of p-eIF2a upon PKR deletion (Fig. S2F), suggesting that protein translation is unlikely contributing to the increased Bcl2 expression in PKR-KO cells. In addition to its role in regulating protein translation, PKR plays a role in signal transduction and transcriptional control through the NF-kB, STAT1/3, p53, ATF, PTEN, MAPKs (JNK1/2 and p38), and the toll-like receptors (TLKs) (3). It will be interesting to investigate whether any of these signaling molecules is involved in regulating Bcl2 expression.
PKR exerts its function through variable signaling pathways likely in a cell type-dependent manner (3,4). Surprisingly, most of the known signaling downstream PKR including the major substrate eIF2α are not altered upon PKR inactivation in ovarian cancer cells (SKOV3 and TOV21G, Fig. S2F). In contrast, the RSK activity but not its upstream ERK1/2 kinases is greatly reduced in PKR knockout cells (Fig. S2E). Given that PKR positively regulates RSK activity and that RSK promotes cell survival in most conditions, it is unlikely that RSK mediates PKR’s role in Taxol chemosensitivity. However, these findings do possibly favor a model in which PKR controls cell/tumor growth through RSK.
Our findings also suggest that activation of PKR may be beneficial to enhance the Taxol-induced cytotoxicity in ovarian cancer cells (Fig. 4) and compounds that phenocopy PKR activation could be explored (59). However, targeting PKR requires special attention as PKR activation may be oncogenic in certain context (4). The identification of Bcl2 as a downstream mediator for PKR-driven Taxol chemosensitivity provides an alternative and probably better approach to overcome Taxol resistance. The Bcl2-specific inhibitor venetoclax (ABT199) is FDA-approved to treat multiple hematological malignancies including acute myeloid leukemia, chronic lymphocytic leukemia, and small lymphocytic leukemia (60). Venetoclax (in combination with other agents) is also being studied in the treatment of solid tumors including breast cancer and castration resistant prostate cancer (https://www.clinicaltrails.gov). Our study demonstrates that combining venetoclax with paclitaxel suppressed Taxol-resistant (SKOV3) tumor growth (Fig. 7). SKOV3 (and TOV21G) are not high-grade serous ovarian cancer (HGSOC) lines (61), the most prevalent subtype of ovarian cancer, therefore, it is worth using HGSOC cells to further validate our model. Nevertheless, since venetoclax is an FDA-approved agent, our findings revealed a new option for the treatment of recurrent ovarian cancer patients and could be rapidly tested in the clinic.
Supplementary Material
Acknowledgments
We are very grateful to Dr. Michael Seiden for the SKOV3/SKOV3-Taxol resistant cell lines (32). All fluorescence images were acquired by Zeiss LSM 710 or LSM 800 confocal microscopes at the Advanced Microscopy Core at the University of Nebraska Medical Center. The core is supported in part by grant P30 GM106397 from the National Institutes of Health (NIH). Research in the Dong laboratory is supported by Fred & Pamela Buffett Cancer Center Support Grant (P30 CA036727), grants P30 GM106397 and R01 GM109066 from the NIH. We also thank Dr. Joyce Solheim for critical reading and comments on the manuscript.
Footnotes
Competing interest: The authors declare no potential conflicts of interest.
References
- (1).Dominguez-Brauer C, Thu KL, Mason JM, Blaser H, Bray MR, Mak TW. Targeting Mitosis in Cancer: Emerging Strategies. Mol Cell 2015;60: 524–536. [DOI] [PubMed] [Google Scholar]
- (2).Gascoigne KE, Taylor SS. How do anti-mitotic drugs kill cancer cells? J Cell Sci 2009;122: 2579–2585. [DOI] [PubMed] [Google Scholar]
- (3).Garcia-Ortega MB, Lopez GJ, Jimenez G, Garcia-Garcia JA, Conde V, Boulaiz H, et al. Clinical and therapeutic potential of protein kinase PKR in cancer and metabolism. Expert Rev Mol Med 2017;19: e9. [DOI] [PubMed] [Google Scholar]
- (4).Lee YS, Kunkeaw N, Lee YS. Protein kinase R and its cellular regulators in cancer: An active player or a surveillant? Wiley Interdiscip Rev RNA 2019: e1558. [DOI] [PubMed] [Google Scholar]
- (5).Meurs E, Chong K, Galabru J, Thomas NS, Kerr IM, Williams BR, et al. Molecular cloning and characterization of the human double-stranded RNA-activated protein kinase induced by interferon. Cell 1990;62: 379–390. [DOI] [PubMed] [Google Scholar]
- (6).Carvalho BM, Oliveira AG, Ueno M, Araujo TG, Guadagnini D, Carvalho-Filho MA, et al. Modulation of double-stranded RNA-activated protein kinase in insulin sensitive tissues of obese humans. Obesity (Silver Spring) 2013;21: 2452–2457. [DOI] [PubMed] [Google Scholar]
- (7).Nakamura T, Furuhashi M, Li P, Cao H, Tuncman G, Sonenberg N, et al. Double-stranded RNA-dependent protein kinase links pathogen sensing with stress and metabolic homeostasis. Cell 2010;140: 338–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Pyo CW, Lee SH, Choi SY. Oxidative stress induces PKR-dependent apoptosis via IFN-gamma activation signaling in Jurkat T cells. Biochem Biophys Res Commun 2008;377: 1001–1006. [DOI] [PubMed] [Google Scholar]
- (9).Srivastava SP, Davies MV, Kaufman RJ. Calcium depletion from the endoplasmic reticulum activates the double-stranded RNA-dependent protein kinase (PKR) to inhibit protein synthesis. J Biol Chem 1995;270: 16619–16624. [DOI] [PubMed] [Google Scholar]
- (10).Lee ES, Yoon CH, Kim YS, Bae YS. The double-strand RNA-dependent protein kinase PKR plays a significant role in a sustained ER stress-induced apoptosis. FEBS Lett 2007;581: 4325–4332. [DOI] [PubMed] [Google Scholar]
- (11).Garcia MA, Carrasco E, Aguilera M, Alvarez P, Rivas C, Campos JM, et al. The chemotherapeutic drug 5-fluorouracil promotes PKR-mediated apoptosis in a p53-independent manner in colon and breast cancer cells. PLoS One 2011;6: e23887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Peidis P, Papadakis AI, Muaddi H, Richard S, Koromilas AE. Doxorubicin bypasses the cytoprotective effects of eIF2alpha phosphorylation and promotes PKR-mediated cell death. Cell Death Differ 2011;18: 145–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Dzananovic E, McKenna SA, Patel TR. Viral proteins targeting host protein kinase R to evade an innate immune response: a mini review. Biotechnol Genet Eng Rev 2018;34: 33–59. [DOI] [PubMed] [Google Scholar]
- (14).Garcia MA, Gil J, Ventoso I, Guerra S, Domingo E, Rivas C, et al. Impact of protein kinase PKR in cell biology: from antiviral to antiproliferative action. Microbiol Mol Biol Rev 2006;70: 1032–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Marchal JA, Lopez GJ, Peran M, Comino A, Delgado JR, Garcia-Garcia JA, et al. The impact of PKR activation: from neurodegeneration to cancer. FASEB J 2014;28: 1965–1974. [DOI] [PubMed] [Google Scholar]
- (16).Meurs EF, Galabru J, Barber GN, Katze MG, Hovanessian AG. Tumor suppressor function of the interferon-induced double-stranded RNA-activated protein kinase. Proc Natl Acad Sci U S A 1993;90: 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Mundschau LJ, Faller DV. Oncogenic ras induces an inhibitor of double-stranded RNA-dependent eukaryotic initiation factor 2 alpha-kinase activation. J Biol Chem 1992;267: 23092–23098. [PubMed] [Google Scholar]
- (18).Haines GK, Panos RJ, Bak PM, Brown T, Zielinski M, Leyland J, et al. Interferon-responsive protein kinase (p68) and proliferating cell nuclear antigen are inversely distributed in head and neck squamous cell carcinoma. Tumour Biol 1998;19: 52–59. [DOI] [PubMed] [Google Scholar]
- (19).Shimada A, Shiota G, Miyata H, Kamahora T, Kawasaki H, Shiraki K, et al. Aberrant expression of double-stranded RNA-dependent protein kinase in hepatocytes of chronic hepatitis and differentiated hepatocellular carcinoma. Cancer Res 1998;58: 4434–4438. [PubMed] [Google Scholar]
- (20).He Y, Correa AM, Raso MG, Hofstetter WL, Fang B, Behrens C, et al. The role of PKR/eIF2alpha signaling pathway in prognosis of non-small cell lung cancer. PLoS One 2011;6: e24855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Schmidt S, Gay D, Uthe FW, Denk S, Paauwe M, Matthes N, et al. A MYC-GCN2-eIF2alpha negative feedback loop limits protein synthesis to prevent MYC-dependent apoptosis in colorectal cancer. Nat Cell Biol 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Beretta L, Gabbay M, Berger R, Hanash SM, Sonenberg N. Expression of the protein kinase PKR in modulated by IRF-1 and is reduced in 5q- associated leukemias. Oncogene 1996;12: 1593–1596. [PubMed] [Google Scholar]
- (23).Darini C, Ghaddar N, Chabot C, Assaker G, Sabri S, Wang S, et al. An integrated stress response via PKR suppresses HER2+ cancers and improves trastuzumab therapy. Nat Commun 2019;10: 2139–019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Li X, Wu Z, An X, Mei Q, Bai M, Hanski L, et al. Blockade of the LRP16-PKR-NF-kappaB signaling axis sensitizes colorectal carcinoma cells to DNA-damaging cytotoxic therapy. Elife 2017;6: 10.7554/eLife.27301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Ruvolo VR, Kurinna SM, Karanjeet KB, Schuster TF, Martelli AM, McCubrey JA, et al. PKR regulates B56(alpha)-mediated BCL2 phosphatase activity in acute lymphoblastic leukemia-derived REH cells. J Biol Chem 2008;283: 35474–35485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Haines GK, Cajulis R, Hayden R, Duda R, Talamonti M, Radosevich JA. Expression of the double-stranded RNA-dependent protein kinase (p68) in human breast tissues. Tumour Biol 1996;17: 5–12. [DOI] [PubMed] [Google Scholar]
- (27).Kim SH, Gunnery S, Choe JK, Mathews MB. Neoplastic progression in melanoma and colon cancer is associated with increased expression and activity of the interferon-inducible protein kinase, PKR. Oncogene 2002;21: 8741–8748. [DOI] [PubMed] [Google Scholar]
- (28).Basu S, Panayiotidis P, Hart SM, He LZ, Man A, Hoffbrand AV, et al. Role of double-stranded RNA-activated protein kinase in human hematological malignancies. Cancer Res 1997;57: 943–947. [PubMed] [Google Scholar]
- (29).Blalock WL, Grimaldi C, Fala F, Follo M, Horn S, Basecke J, et al. PKR activity is required for acute leukemic cell maintenance and growth: a role for PKR-mediated phosphatase activity to regulate GSK-3 phosphorylation. J Cell Physiol 2009;221: 232–241. [DOI] [PubMed] [Google Scholar]
- (30).Roh MS, Kwak JY, Kim SJ, Lee HW, Kwon HC, Hwang TH, et al. Expression of double-stranded RNA-activated protein kinase in small-size peripheral adenocarcinoma of the lung. Pathol Int 2005;55: 688–693. [DOI] [PubMed] [Google Scholar]
- (31).Stauffer S, Zeng Y, Zhou J, Chen X, Chen Y, Dong J. CDK1-mediated mitotic phosphorylation of PBK is involved in cytokinesis and inhibits its oncogenic activity. Cell Signal 2017;39: 74–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Lamendola DE, Duan Z, Yusuf RZ, Seiden MV. Molecular description of evolving paclitaxel resistance in the SKOV-3 human ovarian carcinoma cell line. Cancer Res 2003;63: 2200–2205. [PubMed] [Google Scholar]
- (33).Yang S, Zhang L, Liu M, Chong R, Ding SJ, Chen Y, et al. CDK1 phosphorylation of YAP promotes mitotic defects and cell motility and is essential for neoplastic transformation. Cancer Res 2013;73: 6722–6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Zhou J, Zeng Y, Cui L, Chen X, Stauffer S, Wang Z, et al. Zyxin promotes colon cancer tumorigenesis in a mitotic phosphorylation-dependent manner and through CDK8-mediated YAP activation. Proc Natl Acad Sci U S A 2018;115: E6760–E6769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Wang Z, Chen X, Zhong MZ, Yang S, Zhou J, Klinkebiel DL, et al. Cyclin-dependent kinase 1-mediated phosphorylation of YES links mitotic arrest and apoptosis during antitubulin chemotherapy. Cell Signal 2018;52: 137–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Xiao L, Chen Y, Ji M, Dong J. KIBRA regulates Hippo signaling activity via interactions with large tumor suppressor kinases. J Biol Chem 2011;286: 7788–7796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Zhang L, Iyer J, Chowdhury A, Ji M, Xiao L, Yang S, et al. KIBRA regulates aurora kinase activity and is required for precise chromosome alignment during mitosis. J Biol Chem 2012;287: 34069–34077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Stauffer S, Zeng Y, Santos M, Zhou J, Chen Y, Dong J. Cyclin-dependent kinase 1-mediated AMPK phosphorylation regulates chromosome alignment and mitotic progression. J Cell Sci 2019;132: 10.1242/jcs.236000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Yu Y, Gaillard S, Phillip JM, Huang TC, Pinto SM, Tessarollo NG, et al. Inhibition of Spleen Tyrosine Kinase Potentiates Paclitaxel-Induced Cytotoxicity in Ovarian Cancer Cells by Stabilizing Microtubules. Cancer Cell 2015;28: 82–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Zhang L, Yang S, Chen X, Stauffer S, Yu F, Lele SM, et al. The hippo pathway effector YAP regulates motility, invasion, and castration-resistant growth of prostate cancer cells. Mol Cell Biol 2015;35: 1350–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Chen X, Stauffer S, Chen Y, Dong J. Ajuba Phosphorylation by CDK1 Promotes Cell Proliferation and Tumorigenesis. J Biol Chem 2016;291: 14761–14772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Romano PR, Garcia-Barrio MT, Zhang X, Wang Q, Taylor DR, Zhang F, et al. Autophosphorylation in the activation loop is required for full kinase activity in vivo of human and yeast eukaryotic initiation factor 2alpha kinases PKR and GCN2. Mol Cell Biol 1998;18: 2282–2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Nigg EA. Cellular substrates of p34(cdc2) and its companion cyclin-dependent kinases. Trends Cell Biol 1993;3: 296–301. [DOI] [PubMed] [Google Scholar]
- (44).Tronel C, Page G, Bodard S, Chalon S, Antier D. The specific PKR inhibitor C16 prevents apoptosis and IL-1beta production in an acute excitotoxic rat model with a neuroinflammatory component. Neurochem Int 2014;64: 73–83. [DOI] [PubMed] [Google Scholar]
- (45).Xiao J, Tan Y, Li Y, Luo Y. The Specific Protein Kinase R (PKR) Inhibitor C16 Protects Neonatal Hypoxia-Ischemia Brain Damages by Inhibiting Neuroinflammation in a Neonatal Rat Model. Med Sci Monit 2016;22: 5074–5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Barage S, Kulkarni A, Pal JK, Joshi M. Unravelling the structural interactions between PKR kinase domain and its small molecule inhibitors using computational approaches. J Mol Graph Model 2017;75: 322–329. [DOI] [PubMed] [Google Scholar]
- (47).Wang Y, Men M, Xie B, Shan J, Wang C, Liu J, et al. Inhibition of PKR protects against H2O2-induced injury on neonatal cardiac myocytes by attenuating apoptosis and inflammation. Sci Rep 2016;6: 38753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Barille-Nion S, Bah N, Vequaud E, Juin P. Regulation of cancer cell survival by BCL2 family members upon prolonged mitotic arrest: opportunities for anticancer therapy. Anticancer Res 2012;32: 4225–4233. [PubMed] [Google Scholar]
- (49).Eichhorn JM, Sakurikar N, Alford SE, Chu R, Chambers TC. Critical role of anti-apoptotic Bcl-2 protein phosphorylation in mitotic death. Cell Death Dis 2013;4: e834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Harley ME, Allan LA, Sanderson HS, Clarke PR. Phosphorylation of Mcl-1 by CDK1-cyclin B1 initiates its Cdc20-dependent destruction during mitotic arrest. EMBO J 2010;29: 2407–2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Terrano DT, Upreti M, Chambers TC. Cyclin-dependent kinase 1-mediated Bcl-xL/Bcl-2 phosphorylation acts as a functional link coupling mitotic arrest and apoptosis. Mol Cell Biol 2010;30: 640–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Wertz IE, Kusam S, Lam C, Okamoto T, Sandoval W, Anderson DJ, et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature 2011;471: 110–114. [DOI] [PubMed] [Google Scholar]
- (53).Henriques AC, Ribeiro D, Pedrosa J, Sarmento B, Silva PMA, Bousbaa H. Mitosis inhibitors in anticancer therapy: When blocking the exit becomes a solution. Cancer Lett 2019;440–441: 64–81. [DOI] [PubMed] [Google Scholar]
- (54).Kim Y, Lee JH, Park JE, Cho J, Yi H, Kim VN. PKR is activated by cellular dsRNAs during mitosis and acts as a mitotic regulator. Genes Dev 2014;28: 1310–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Cuddihy AR, Wong AH, Tam NW, Li S, Koromilas AE. The double-stranded RNA activated protein kinase PKR physically associates with the tumor suppressor p53 protein and phosphorylates human p53 on serine 392 in vitro. Oncogene 1999;18: 2690–2702. [DOI] [PubMed] [Google Scholar]
- (56).Yoon CH, Lee ES, Lim DS, Bae YS. PKR, a p53 target gene, plays a crucial role in the tumor-suppressor function of p53. Proc Natl Acad Sci U S A 2009;106: 7852–7857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Hu CW, Yin GF, Wang XR, Ren BW, Zhang WG, Bai QL, et al. IL-24 Induces Apoptosis via Upregulation of RNA-Activated Protein Kinase and Enhances Temozolomide-Induced Apoptosis in Glioma Cells. Oncol Res 2014;22: 159–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Tang Y, Wang Z, Yang J, Zheng W, Chen D, Wu G, et al. Polycystin-1 inhibits eIF2alpha phosphorylation and cell apoptosis through a PKR-eIF2alpha pathway. Sci Rep 2017;7: 11493–017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Natarajan A, Fan YH, Chen H, Guo Y, Iyasere J, Harbinski F, et al. 3,3-Diaryl-1,3-Dihydroindol-2-Ones as Antiproliferatives Mediated by Translation Initiation Inhibition. J Med Chem 2004;47: 1882–1885. [DOI] [PubMed] [Google Scholar]
- (60).Cang S, Iragavarapu C, Savooji J, Song Y, Liu D. ABT-199 (venetoclax) and BCL-2 inhibitors in clinical development. J Hematol Oncol 2015;8: 129–015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Domcke S, Sinha R, Levine DA, Sander C, Schultz N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat Commun 2013;4: 2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.