

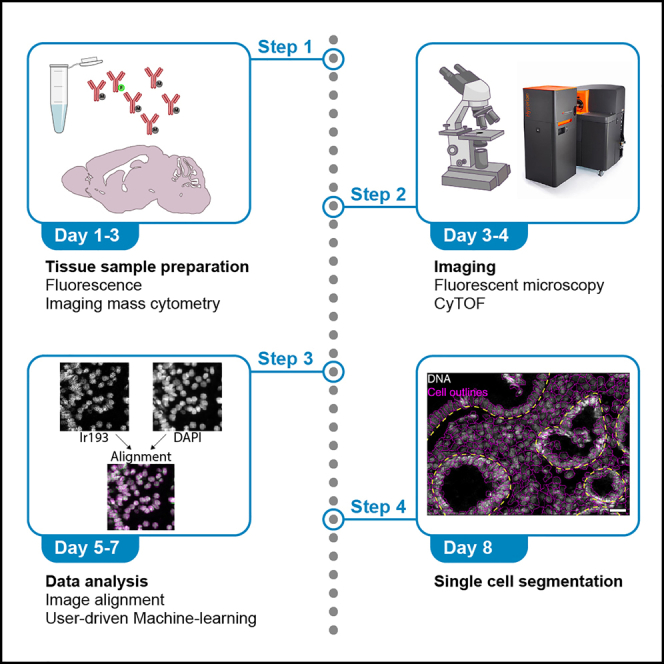
Summary
Exploring tissue heterogeneity on a single-cell level by imaging mass cytometry (IMC) remains challenging because of its limiting resolution. We previously demonstrated that combining higher resolution fluorescence with IMC data in the analysis pipeline resulted in high-quality single-cell segmentation. Here, we provide a step-by-step workflow of this MATISSE pipeline, including instructions regarding the staining procedure, and the analysis route to generate single-cell data.
For complete details on the use and execution of this protocol, please refer to Baars et al., 2021.
Subject areas: Bioinformatics, Cell Biology, Single Cell, Flow Cytometry/Mass Cytometry, Microscopy, Antibody, Biotechnology and bioengineering
Graphical abstract
Highlights
-
•
High-plex tissue staining combining isotope-labeled antibodies with DNA intercalator
-
•
Imaging mass cytometry (IMC) and fluorescent microscopy in a single workflow
-
•
Combined data processing pipeline of IMC and fluorescent images
-
•
The MATISSE pipeline generates high-quality single-cell segmentation of IMC data
Exploring tissue heterogeneity on a single-cell level by imaging mass cytometry (IMC) remains challenging because of its limiting resolution. We previously demonstrated that combining higher resolution fluorescence with IMC data in the analysis pipeline resulted in high-quality single-cell segmentation. Here, we provide a step-by-step workflow of this MATISSE pipeline, including instructions regarding the staining procedure, and the analysis route to generate single-cell data.
Before you begin
Tissue heterogeneity has become an important focus of research to gain mechanistic insights in disease and therapy response by multiparametric exploration of tissue landscapes. Studying cells in their native tissue environment is essential for this purpose, for instance by using imaging mass cytometry (IMC). To date, single-cell segmentation of IMC data remains challenging due to the limiting resolution of 1 μM pixel size. We have previously demonstrated that integrating higher resolution fluorescent data in the segmentation pipeline significantly improved both the quality and quantity of detected single cells in the tissue. This pipeline, MATISSE (iMaging mAss cytometry mIcroscopy Single-cell SegmEntation), contains both wet-lab and computational steps to combine high-resolution fluorescence microscopy images of nuclei with multiplex IMC imaging data for the purpose of single-cell data generation (Baars et al., 2021). All steps of the pipeline make use of standard lab reagents and immunohistochemical staining procedures, as well as open-access image analysis tools and scripts, which allows easy implementation by scientific communities and research laboratories. The protocol below describes the specific steps for combining IMC and DAPI staining on five human colorectal biopsies, and the subsequent image analysis pipeline (illustrated in Figure 1). This protocol can also be used for combining IMC staining with other fluorescent staining data, and for analysis of tissues other than colorectal biopsies, including human skin, small intestine, liver, small-cell lung cancer, and melanoma tissue.
Figure 1.

MATISSE workflow
Execution of successful experiments will require experience in imaging approaches, as well as image analysis. Therefore, before attempting the MATISSE workflow, we recommend users should be familiar and comfortable with running CellProfiler & Ilastik (Sommer et al., 2011; Carpenter et al., 2006). In addition, intermediate understanding of R language is required (Gillespie and Lovelace, 2016).
Folder naming and structure
Many components in this pipeline are automated and therefore depend heavily on well-defined filenames and folder structures. For instance, to conveniently associate the fluorescent and IMC images of the same tissue region automatically, one should use a unique sample identifier (i.e., sample ID and subregion within-sample) in the filenames used to store the fluorescent and IMC data. Before data acquisition, it is advised to decide on what regions to acquire data on, and what names to give these regions. Figure 2 shows an example of acquired tissue regions using the MATISSE pipeline and the used names. We propose the following (or similar) naming pattern: [Slide ID]_[Patient/Tumor ID]_[Tile Region].
CRITICAL: Make sure to use an identical file name for the DAPI tile-region scan and IMC region of interest (ROI) that need to be registered as indicated in Figure 2.
Note: Always use a numeric format for naming regions. Try to avoid the use of special characters, like slashes, dots, commas, or spaces in file names. Use underscores strictly to separate naming sections as shown above.
Figure 2.

Schematic overview of DAPI and IMC tissue region selection
Blue: tissue selection for DAPI tile-region scanning. Red: tissue selection for IMC ablation. Please note that the tissue region selection for DAPI scanning is always bigger than the IMC region selection. We propose the following naming pattern for DAPI and IMC images: [Slide ID]_[Patient/Tumor ID]_[Tile Region].
Installation of software
Timing: 45 min
-
1.
Download Fiji, Ilastik, CellProfiler, Python, R, and Rstudio before starting the MATISSE pipeline. Links for downloads are included in the key resources table.
Installation of Fiji plugins
In order to correctly project Z-stacks using the provided scripts with the MATISSE pipeline, a modified EDF plugin is required. Additionally, several Fiji plugins must be installed.
-
2.
Download the custom EDF plugin from the VercoulenLab GitHub repository (https://github.com/VercoulenLab/MATISSEStarProtocol).
-
3.
Unpack the EDF.zip file to the fiji.app/plugins folder on your computer.
-
4.
Install Microscope Image Stitching tool (MIST) Flow and ImageScience plugins via Fiji>Help>Update>Manage Update Sites. Click on the required plugins, close the window and click on apply changes. Restart Fiji to complete the installation.
-
5.
Install Scale Invariant Feature Transform (SIFT), Landmark correspondence, Multi-Scale Oriented Patches (MOPS), FeatureJ, and MorphoLibJ plugins via the links provided in the key resources table.
Download scripts used in the MATISSE pipeline
-
6.
Download the MATISSE pipeline scripts from the VercoulenLab GitHub repository (https://github.com/VercoulenLab/MATISSEStarProtocol).
Installation of IMC tools
-
7.
Install Python 3.8.
-
8.
Install IMC tools by typing “python -m pip install imctools==2.1.4” in a terminal.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Alpha-Smooth Muscle Actin Monoclonal Antibody, Clone 1A4 | Thermo Fisher Scientific | Cat# 14-9760-82; RRID: AB_2572996 |
| Recombinant Anti-CD14 Antibody, Clone EPR3653 | Abcam | Cat# ab214438; RRID: AB_2889158 |
| Recombinant Anti-CD16 Antibody, Clone EPR16784 | Abcam | Cat# ab215977; RRID: AB_2877105 |
| Anti-CD20-161Dy Monoclonal Antibody, Clone H1 | Fluidigm | Cat# 3161029D; RRID: AB_2811016 |
| Anti-CD3 Antibody Polyclonal Antibody | Agilent | Cat# A0452; RRID: AB_2335677 |
| Recombinant Anti-CD4 Antibody, Clone EPR6855 | Abcam | Cat# ab181724; RRID: AB_264377 |
| Anti-CD45 Monoclonal Antibody, Clone D9M8I | Cell Signaling Technology | Cat# 13917; RRID: AB_2750898 |
| Anti-CD45RO Monoclonal Antibody, Clone UCHL1 | Cell Signaling Technology | Cat# 55618; RRID: AB_2799491 |
| Anti-CD68-159Tb Monoclonal Antibody, Clone KP1 | Fluidigm | Cat# 3159035D; RRID: AB_2810859 |
| Anti-CD8a-162Dy Monoclonal Antibody, Clone C8/144B | Fluidigm | Cat# 3162034D; RRID: AB_2811053 |
| Anti-E-Cadherin Monoclonal Antibody, Clone 24E10 | Cell Signaling Technology | Cat# 3195; RRID: AB_2291471 |
| Recombinant Anti-FOXP3 Monoclonal Antibody, Clone 236A/E7 | Abcam | Cat# ab96048; RRID: AB_10861686 |
| Anti-Histone H3 Monoclonal Antibody, Clone D1H2 | Cell Signaling Technology | Cat# 4499; RRID: AB_10544537 |
| Anti-IL-17 Polyclonal Antibody | R&D Systems | Cat# AF-317-NA; RRID: AB_354463 |
| Anti-Ki-67 Monoclonal Antibody, Clone B56 | BD Biosciences | Cat# 556003; RRID: AB_396287 |
| Recombinant Anti-Lamin B1 Antibody, Clone EPR8985(B) | Abcam | Cat# ab220797; RRID: AB_2889226 |
| Anti-Pan-Keratin Monoclonal Antibody, Clone C11 | Cell Signaling Technology | Cat# 4545; RRID: AB_490860 |
| Anti-TCRγδ Monoclonal Antibody, Clone H-41 | Santa Cruz | Cat# sc-100289; RRID: AB_11300061 |
| Human TruStain FcXTM (Fc Receptor Blocking Solution) Antibody | BioLegend | Cat# 422302; RRID: AB_2818986 |
| Biological samples | ||
| Human colorectal biopsies | This study | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| DAPI (4′,6-Diamindino-2′-Phenyindole, Dihydrochloride) | Sigma-Aldrich | Cat# D9542; CAS: 28718-90-3 |
| Cell-ID Intercalator-Ir | Fluidigm | Cat# 201192B |
| Xylene | Klinipath | Cat# 4055-9005; Cas 1330-20-7 |
| Ethanol absolute ≥99% | Klinipath | Cat# 4099-9005; Cas 64-17-5 |
| Ethylenediaminetetraacetic acid disodium salt dihydrate (EDTA) | Sigma-Aldrich | Cat# 27285; Cas 6381-92-6 |
| 2-Amino-2-methyl-1,3-propanediol (Tris base) | Sigma-Aldrich | Cat# 10708976001; Cas 77-86-1 |
| Bovine Serum Albumin (BSA) | Sigma-Aldrich | Cat# A9647; Cas 9048-46-8 |
| Sodium Chloride (NaCl) | Sigma-Aldrich | Cat# 31434; Cas 7647-14-5 |
| Tween 20 | Sigma-Aldrich | Cat# P9416; Cas. 9005-64-5 |
| Hydrochloric acid fuming 37% | Sigma-Aldrich | Cat# 100317; Cas 7647-01-0 |
| Sodium hydroxide | Sigma-Aldrich | Cat# 106498; Cas 1310-73-2 |
| Maxpar X8 141Pr Labeling Kit – 4 Rxn | Fluidigm | Cat# 202241A |
| Maxpar X8 142Nd Labeling Kit – 4 Rxn | Fluidigm | Cat# 201142A |
| Maxpar X8 143Nd Labeling Kit – 4 Rxn | Fluidigm | Cat# 201143A |
| Maxpar X8 144Nd Labeling Kit – 4 Rxn | Fluidigm | Cat# 201144A |
| Maxpar X8 146Nd Labeling Kit – 4 Rxn | Fluidigm | Cat# 201146A |
| Maxpar X8 148Nd Labeling Kit – 4 Rxn | Fluidigm | Cat# 201148A |
| Maxpar X8 152Sm Labeling Kit – 4 Rxn | Fluidigm | Cat# 201152A |
| Maxpar X8 156Gd Labeling Kit – 4 Rxn | Fluidigm | Cat# 201155A |
| Maxpar X8 159Tb Labeling Kit – 4 Rxn | Fluidigm | Cat# 201159A |
| Maxpar X8 161Dy Labeling Kit – 4 Rxn | Fluidigm | Cat# 201161A |
| Maxpar X8 162Dy Labeling Kit – 4 Rxn | Fluidigm | Cat# 201162A |
| Maxpar X8 167Er Labeling Kit – 4 Rxn | Fluidigm | Cat# 201167A |
| Maxpar X8 170Er Labeling Kit – 4 Rxn | Fluidigm | Cat# 201170A |
| Maxpar X8 174Yb Labeling Kit – 4 Rxn | Fluidigm | Cat# 201174A |
| Maxpar X8 176Yb Labeling Kit – 4 Rxn | Fluidigm | Cat# 201176A |
| PAP pen for immunostaining (5 mm tip) | Sigma-Aldrich | Cat# Z377821 |
| Critical commercial assays | ||
| Maxpar X8 Labeling kit | Fluidigm | Cat# 201300 |
| Deposited data | ||
| Raw and Processed MATTISE Data | Baars et al., 2021 | https://zenodo.org/record/4727873 |
| Software and algorithms | ||
| Fiji (version 2.0.0-rc-69/1.52p) | Schindelin et al., 2012 | https:/fiji.sc/ |
| Microscope Image Stitching tool (MIST), Fiji Plugin | Chalfoun et al., 2017 | https://github.com/usnistgov/MIST |
| Scale Invariant Feature Transform (SIFT) Flow, Fiji Plugin | (Liu et al., 2008) | https://github.com/OpenGenus/SIFT-Scale-Invariant-Feature-Transform |
| Landmark correspondence, Fiji Plugin | Stephan Saalfeld | https://imagej.net/Landmark_Correspondences |
| Multi-Scale Oriented Patches (MOPS), Fiji Plugin | (Brown et al., 2005) | https://github.com/axtimwalde/mpicbg/ |
| FeatureJ (version 1.6.2), Fiji Plugin | (Meijering, 2002) | https://github.com/imagescience/FeatureJ/ |
| MorphoLibJ (version 1.4.3), Fiji Plugin | (Legland et al., 2016) | https://github.com/ijpb/MorphoLibJ/ |
| ImageScience (version 3.1.0), Fiji Plugin | Erik Meijering | https://github.com/ImageScience/ImageScience |
| Modified Extended Depth of Field (EDF), Fiji Plugin | Forster et al., 2004 | Original: https://github.com/Biomedical-Imaging-Group/EDF-Extended-Depth-of-Field Modified: https://github.com/VercoulenLab/MATISSEStarProtocol |
| Imctools (version 2.1.7) | Bodenmiller lab | https://github.com/BodenmillerGroup/imctools |
| Ilastik (version 1.3.3) | (Sommer et al., 2011) | https://www.ilastik.org |
| CellProfiler (version 3.1.9) | Carpenter et al., 2006 | https://cellprofiler.org |
| Python (version 3.8.7) | Python Software Foundation | https://www.python.org/ |
| R (version 4.0.0) | R Core Team | https://www.r-project.org/ |
| Rstudio (version 1.4) | Rstudio Team | https://cran.r-project.org/bin/windows/base/old/ |
| Zen 2.6 Blue Edition (version 2.6.76.00000) | Carl Zeiss Microscopy | https:/www.zeiss.com |
| Fluoresence_StackGeneration2.ijm | This paper | https://github.com/VercoulenLab/MATISSEStarProtocol |
| Fluoresence_ExtendedDepthOfField.ijm | This paper | https://github.com/VercoulenLab/MATISSEStarProtocol |
| Fluoresence_TileRegionStitchName_ToSubfolder.ijm | This paper | https://github.com/VercoulenLab/MATISSEStarProtocol |
| Mcd_to_ometiff.py | This paper | https://github.com/VercoulenLab/MATISSEStarProtocol |
| OME-tiff-channelnames_imctools2.ijm | This paper | https://github.com/VercoulenLab/MATISSEStarProtocol |
| OME-renamer.ijm | This paper | https://github.com/VercoulenLab/MATISSEStarProtocol |
| OME_ExtractChannel2.ijm | This paper | https://github.com/VercoulenLab/MATISSEStarProtocol |
| Fluoresence_Registration_FileList.ijm | This paper | https://github.com/VercoulenLab/MATISSEStarProtocol |
| RandomImageCrop2.ijm | This paper | https://github.com/VercoulenLab/MATISSEStarProtocol |
| OME_ExtractmachineLearningStack2.ijm | This paper | https://github.com/VercoulenLab/MATISSEStarProtocol |
| RandomImageCrop_ApplyToOthers2.ijm | This paper | https://github.com/VercoulenLab/MATISSEStarProtocol |
| ManualAnnotation_Training | Baars et al., 2021 | https://github.com/VercoulenLab/MATISSE-Pipeline |
| FilterAdditionStack.ijm | Baars et al., 2021 | https://github.com/VercoulenLab/MATISSE-Pipeline |
| Ilastik_Apply_Training.sh | This paper | https://github.com/VercoulenLab/MATISSEStarProtocol |
| Training_model.sh | This paper | https://github.com/VercoulenLab/MATISSEStarProtocol |
| CP-Pipeline.cppipe | Baars et al., 2021 | https://github.com/VercoulenLab/MATISSE-Pipeline |
| ExtractSingleCellDataParallel.r | Baars et al., 2021 | https://github.com/VercoulenLab/MATISSE-Pipeline |
| Other | ||
| Surgipath X-tra adhesive pre-cleaned Micro Slides | Leica Biosystems | Cat# 3008203 |
| EasyDip Slide Staining Rack | Simport Scientific | Cat# M905-12DGY |
| EasyDip Slide Staining System | Simport Scientific | Cat# M906 |
| Z1 imager | Zeiss | Cat# 423900-9901-000 |
| Objective EC “Plan-Neofluar” 5×/0.16 M27 | Zeiss | Part# 420330-9901-000 |
| Objective EC “Plan-Neofluar” 20×/0.50 M27 | Zeiss | Part# 420350-9900-000 |
| Filterset 49 | Zeiss | Part# 488049-9901-000 |
| Filterset 45 | Zeiss | Part# 000000-1114-459 |
| Zeiss Axiocam 503 Mono Camera System | Zeiss | Part# 426559-0000-000 |
| Hyperion Imaging System | Fluidigm | Part# 108001 |
| Eppendorf Centrifuge | Eppendorf | Cat# 5430R |
| Waterbath | Adamas Instrumenten | Cat# WB85 |
| pH measurement system | Radiometer | Cat# PHM220 |
| Tissue Heating plate | Marshall Scientific | Cat# 12621-092 |
| Shaking plate | Kika Labtechniek | Cat# KS250basic |
| Milli-Q System | Merck | Cat# ZIQP0D000 |
| Dell Precision T5820 – Xeon W-2133, Linux 16.04 | Dell | Cat# xctopt5820corexemea |
Materials and equipment
Microscope
Slides were imaged on a Zeiss Z1 imager using a 5× and 20× dry objective (Zeiss, EC Plan-NEOFLUAR 5×/ 420330-9901; Zeiss, EC Plan-NEOFLUAR 20×/0.5 420350-9900) with mercury lamp as light source, in combination with a 45 and 49 filter set. The system was equipped with an Axiocam 503 mono camera system and ZEN pro 2.6 software with Z-stack, timelapse, and Tiles & Position module licences.
Importantly, the MATISSE pipeline works independent of the used microscope. In this protocol, all imaging steps are illustrated for one microscope option. Other microscope setups with tile-scanning option, z-stack, anchor points, tiff export, 20× dry objective, and 14+ bit detector are also compatible with this pipeline.
Imaging mass cytometer
Slides were ablated on a Hyperion Imaging System.
Computer hardware
System memory (RAM): required 16 GB, recommended: 64 GB; Processors: required 8 Core 16 threads, recommended: 12 Core 24 threads.
Note: The full MATISSE pipeline was designed for Ubuntu 18 LTS and Windows 7 & 10 systems. The majority of the steps in the MATISSE pipeline are also compatible with MacOS, although small adjustments of the scripts might be required.
Tissue sections
Four μm thick tissue sections were used for this protocol. Slides were baked for 120 min at 60°C on a heating plate to ensure all water evaporated, and to ensure the tissue was properly attached to the glass slide.
Antibodies
Validated antibodies were manually labeled with isotopes using the MaxparX8 labeling protocol (https://www.fluidigm.com/binaries/content/documents/fluidigm/resources/maxpar-antibody-labeling-user-guide/maxpar-antibody-labeling-user-guide/fluidigm%3Afile). All labeled antibodies should be titrated and validated before starting the MATISSE pipeline.
Preparation of buffers
10× TBS
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris base | 500 mM | 60.6 g |
| Sodium Chloride | 1500 mM | 87.6 g |
| MQ | n/a | ∼950 mL |
| 1 M HCl | n/a | ∼50 mL |
| Total | n/a | 1 L |
Store at 18°C–22°C for a maximum of 6 months.
Note: First dissolve Tris base and Sodium Chloride in 900 mL Milli-Q (MQ). Adjust the pH to 7.4 with 1 M HCl. Then adjust the volume of the buffer to 1 L using MQ.
1× TBST
| Reagent | Final concentration | Amount |
|---|---|---|
| 10× TBS buffer | 50 mM Tris base, 150 mM NaCl | 100 mL |
| Tween 20 | n/a | 1000 μL |
| MQ | n/a | 900 mL |
| Total | n/a | 1 L |
Store at 18°C–22°C for a maximum of 6 months.
10× Tris-EDTA
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris Base | 10 mM | 1.21 g |
| EDTA | 1 mM | 0.392 g |
| MQ | n/a | ∼950 mL |
| 1 M NaOH | n/a | ∼50 mL |
| Total | n/a | 1 L |
Store at 18°C–22°C for a maximum of 6 months.
Note: First dissolve Tris Base and EDTA in 950 mL MQ. Adjust pH to 9.5 with 1 M NaOH. Then adjust the volume of the buffer to 1 L using MQ.
1× Tris-EDTA
| Reagent | Final concentration | Amount |
|---|---|---|
| 10× Tris-EDTA buffer | 10 mM Tris base, 1 mM EDTA | 100 mL |
| MQ | n/a | 900 mL |
| Total | n/a | 1 L |
Store at 18°C–22°C for a maximum of 6 months.
Preparation of BSA stocks
0.5% BSA/TBST
| Reagent | Final concentration | Amount |
|---|---|---|
| BSA | 0.5% | 0.5 g |
| TBST | n/a | 100 mL |
| Total | n/a | 100 mL |
Store at -20°C for a maximum of 6 months. Avoid freeze thaw cycles.
3% BSA/TBST
| Reagent | Final concentration | Amount |
|---|---|---|
| BSA | 3% | 3 g |
| TBST | n/a | 100 mL |
| Total | n/a | 100 mL |
Store at −20°C for a maximum of 6 months. Avoid freeze thaw cycles.
Preparation of blocking and staining solutions
Prepare blocking solution, antibody cocktail, and DNA-intercalator mix shortly before using. Decide on the volume based on the size of the tissue. Use 50 μL for small tissue biopsies (∼10 × 10 mm) and 100 μL for regular sized tissues (∼20 × 20 mm). Consider using a volume up to 300 μL when staining a tissue microarray (∼35 × 20 mm). Here, examples are given for preparation of blocking and staining solutions for 5 regular-sized tissues (500 μL).
Blocking solution
| Reagent | Final concentration | Amount |
|---|---|---|
| Human TruStain FcXTM | 1:100 | 5 μL |
| 3% BSA in TBST | n/a | 495 μL |
| Total | n/a | 500 μL |
Store at 4°C for a maximum of 24 h.
DNA-intercalator
| Reagent | Final concentration | Amount |
|---|---|---|
| Ir-intercalator | 1.25 μM | 1.2 μL |
| DAPI | 20 ng/mL | 0.5 μL |
| 1× TBST | n/a | 498.25 μL |
| Total | n/a | 500 μL |
Store at 4°C for a maximum of 24 h.
Step-by-step method details
Tissue staining
In this step, tissue sections are prepared, stained with isotope-labeled antibodies, as well as metal-based (Iridium) and fluorescent (DAPI) DNA intercalators.
Day 1
-
1.
Place FFPE tissue sections in a slide rack.
-
2.Dewax tissue sections in xylene for 2 × 10 min in containers in the fume hood.
-
a.In the meanwhile, heat up a water bath to 96°C.
-
a.
-
3.Rehydrate tissue sections in descending grades of ethanol in containers in the fume hood:
-
a.100% ethanol, 10 min
-
b.95% ethanol, 5 min
-
c.80% ethanol, 5 min
-
d.70% ethanol, 5 min
-
e.MQ, 3 min
-
a.
-
4.Place the rack with tissue sections in a container with TBST for 10 min in order to wash the slides.
-
a.In the meantime: fill #number∗50 mL Falcon tubes with Tris-EDTA antigen retrieval buffer (∼35 mL per tube) and preheat in the water bath to 96°C.
-
a.
-
5.
Insert the tissue sections into the 50 mL tube with preheated Tris-EDTA buffer and incubate at 96°C for 30 min in the water bath.
Note: Two tissue sections fit back-to-back in a 50 mL tube.
-
6.
Take the tubes containing Tris-EDTA buffer and the tissue sections from the water bath and cool down on a lab bench for approximately 5–10 min.
-
7.
Place the tissue sections back in a rack and wash with TBST for 10 min in a container.
-
8.
Dry the back of the glass slide with a paper tissue, and the glass surrounding the tissue with an aspirator pump.
-
9.
Circle the tissue with hydrophobic peroxidase-antiperoxidase (PAP) pen. Keep a 1–2 mm distance between tissue and PAP barrier.
Note: Make sure the PAP barrier is properly closed so no fluid can leak or spill over to other tissues on the glass slide (Figure 3A).
Figure 3.

Antibody incubation
(A) Correct and incorrect example of surrounded tissue by PAP. The incorrect example shows spillover of fluid between different tissue samples on the same glass slide. Additionally, leakage of fluid can also happen when tissues are correctly surrounded by PAP. This can be prevented by decreasing the buffer volume.
(B) Humidified staining tray containing tissue sections incubating with blocking buffer.
-
10.
Put the slides in a staining tray (with lid) and pipette 100 μL blocking solution on the tissue. Incubate 60 min at 18°C–22°C.
Note: Wet paper towels with demi water and add to the staining tray to ensure a humidified environment (Figure 3B).
-
11.
Carefully remove the blocking solution from the tissue using an aspirator pump (Methods video S1).
-
12.
Add 100 μL antibody cocktail (Table 1) to the tissues and incubate for 16–20 h at 4°C in the humidified staining tray with a closed lid.
Table 1.
Antibody mix IMC
| Reagent | Final concentration | Amount |
|---|---|---|
| Anti-alpha-SMA-141Pr | 1:300 (=1.7 μg/mL) | 1.7 μL |
| Anti-CD14-144Nd | 1:200 (=2.5 μg/mL) | 2.5 μL |
| Anti-CD16-146Nd | 1:200 (=2.5 μg/mL) | 2.5 μL |
| Anti-CD20-161Dy | 1:300 (=1.7 μg/mL) | 1.7 μL |
| Anti-CD3-170Er | 1:100 (=5 μg/mL) | 5.0 μL |
| Anti-CD4-156Gd | 1:100 (=5 μg/mL) | 5.0 μL |
| Anti-CD45-143Nd | 1:100 (=5 μg/mL) | 5.0 μL |
| Anti-CD45RO-152Sm | 1:100 (=5 μg/mL) | 5.0 μL |
| Anti-CD68-159Tb | 1:600 (=0.8 μg/mL) | 0.8 μL |
| Anti-CD8a-162Dy | 1:200 (=2.5 μg/mL) | 2.5 μL |
| Anti-E-Cadherin-142Nd | 1:150 (=3.3 μg/mL) | 3.3 μL |
| Anti-FOXP3-155Gd | 1:50 (=10 μg/mL) | 10.0 μL |
| Anti-Histon H3-176Yb | 1:600 (=0.8 μg/mL) | 0.8 μL |
| Anti-IL-17-167Er | 1:100 (=5 μg/mL) | 5.0 μL |
| Anti-Ki-67-168Er | 1:200 (=2.5 μg/mL) | 2.5 μL |
| Anti-Lamin B1-113ln;115ln | 1:100 (=5 μg/mL) | 5.0 μL |
| Anti-Pankeratin-148Nd | 1:200 (=2.5 μg/mL) | 2.5 μL |
| Anti-TCRγδ -174Yb | 1:50 (=10 μg/mL) | 10.0 μL |
| 0.5% BSA/TBST | n/a | 429.2 μL |
| Total | n/a | 500 μL |
Store at 4°C for a maximum of 24 h.
Day 2
-
13.
Carefully remove the staining solution from the slide using an aspirator pump.
-
14.
Place the slides in a rack and wash 3 × 5 min in TBST in a container on a shaking plate at 50–100 RPM.
-
15.
Dry the back of the glass slide, and the glass surrounding the tissue with paper towels and place the tissue sections in the staining tray.
-
16.
Pipette 100 μL of DNA intercalator mix on the tissue. Incubate for 60 min at 18°C–22°C.
-
17.
Carefully remove the DNA intercalator mix from the slide using an aspirator pump.
-
18.
Place the tissue sections in a rack and wash in MQ for 3 min in a container.
-
19.
Remove the slides from the rack, and carefully remove excess liquid with a paper towel and aspirator pump.
-
20.
Air dry the slides for at least 20 min at 18°C–22°C.
Pause point: Stained tissue sections can be stored at 4°C in the dark until time of DAPI imaging. It is recommended to not store the stained tissue sections longer than 6 weeks before DAPI imaging due to potential fading of the DAPI signal over time.
Fluorescent imaging
In this step, tile-region scans are obtained of the DAPI staining using a fluorescent microscope. Focus points and Z-stacks are used to ensure that all parts of the scanned tissue are captured in focus. Depending on the size of the scan, obtaining 2 tile-regions of 5 different tissue biopsies (located on 5 different glass slides) takes approximately 6 h (4 mm2 per tissue, 20× scan) to 10 h (25 mm2 per tissue, 20× scan) including settings.
Note: Decide on which tissue regions you want to acquire data on. After IMC ablation, fluorescent images can no longer be obtained. Always make a DAPI tile-region scan bigger in size than the region of interest for ablation by IMC as indicated in Figure 2.
CRUCIAL: Use a dry objective for imaging since the tissue sections are not mounted with a cover slip.
-
21.
Start the fluorescent microscope and Zen 2.6 Blue edition software.
-
22.Select the correct settings as illustrated in Figure 4.
-
a.In the acquisition tab, enable “Z-stack” and “Tiles”. Enable “Autosave” in order to automatically save all (raw) files to a specific location.
-
b.Select the channels tab and select DAPI.
-
c.Make sure binning is set to 1 × 1, and detector bit-depth is set to maximum value.
-
d.In the Multidimensional Acquisition window (Figure 4), select the Tiles tab. Now select the options drop-down and set the “tile overlap” at 10 percent and the “travel in tile regions” on Meander.
-
a.
Note: Other “travel in tile regions” methods can be used but will not be recognized by default in the scripts included in the MATISSE pipeline.
-
23.
Set the focus and exposure time at 5× magnification. In general, the DAPI exposure time is between 5 and 250 ms.
Note: Do not use auto exposure during tile-region scanning. All images should be acquired using identical exposure settings.
-
24.Calibrate the X/Y position of your glass slide.
-
a.From the tiles menu, select advanced setup.
-
b.Locate the frosty end (top left for upright microscope systems) of the glass slide using binocular vision.
-
c.In the sample carrier drop-down menu, select slide 25 × 75 mm.
-
d.Click on calibrate in the sample carrier drop-down menu. Move the lighting path from binocular to the camera. In the step 1/3 window, click next (located at the left bottom of the window). In the step 2/3 window choose “set Zero” and click on next. In the step 3/3 window choose “set current X/Y” and click finish. The stage is now calibrated to the inserted microscopy slide which allows easy orientation and mapping of the tissue (Figure 5).
-
a.
-
25.Make a preview DAPI scan at 5× magnification.
-
a.In the tile region set-up (located at the bottom of the screen), select setup by “Contour” (Figure 5). Select the rectangle tool.
-
b.Draw a region on the screen of the region of interest for preview scanning (Figure 5). Alternatively, make a preview of the full slide.
-
c.Set the focus for the camera using the “live” mode.
-
d.Go to the “Preview Scan” tab and hit the “Start Preview Scan” button. Now the microscope will start to generate the slide preview.
-
a.
-
26.Make a detailed 20× DAPI scan.
-
a.Switch the microscope objective to a dry 20× objective
-
b.Set the exposure time for DAPI at 20× (∼5–50 ms).
-
c.Draw the region of interest for 20× scanning on the preview scan (Figure 5) by using the rectangle tool in the “Contour” tab.
-
d.In the Tiles tab, go to the “Tile regions” drop-down menu and deselect the Tile region that represents the generated preview scan (Figure 6A).
-
e.Select the newly created tile region and rename to the desired region name.CRITICAL: Naming your files correctly and consistently is extremely important Please carefully read the Folder naming and structure paragraph of this protocol before naming your regions and files for data acquisition.
-
f.In the Multidimensional Acquisition window, select the Z-Stack tab. In the Z-Stack tab, select the “Center” option. Additionally, set the number of slices to 9–12 and the interval to 1.5 μm (Figure 6B). Making Z-stacks ensures that the entire tile-scan is in focus.
-
g.Add focus points to the tile region to correct for focus drift. Distribute focus points evenly over the tile region (Figure 6C).
-
h.In the tiles tab, select “focus surface (verify)” in the drop-down menu. Add focus points throughout the panorama by clicking the “+” in the local (per tile region) box.
-
i.Click on “verify tile regions/positions” in the “local (per tile region)” box. A window will appear listing all focus points. Select the first focus point and click “move to current point”. Turn on ‘live’ mode. Adjust the focus of the current focus point and click on “Set Z & Move to Next” (Figure 6D). Do so for all focus points. Close the window when all points are verified.Note: A 20× scan of 2000 × 2000 μm takes approximately 10 minutes.
-
a.
-
27.
When additional regions need to be scanned on the same glass slide, repeat steps 25 and 26.
-
28.
In the Acquisition tab, click on “Start Experiment”.
-
29.
Save the czi file. Before naming the file, please carefully read the Folder naming and structure paragraph of this protocol.
-
30.Image export
-
a.Click on the processing tab in the left upper corner. Select the option “Batch”
-
b.Select “Image export” in the “Batch method” drop-down window.
-
c.Select export parameters as illustrated in Figure 7. Importantly, export all files in tiff format without compression and generate a .xml file.
-
d.Add all .czi files containing the tile region images to the file que in the “Batch processing” window via the “+ Add” button.
-
e.Choose the saving location. Deselect “Use input Folder as output folder”, click on “…”, and navigate to the folder in which the images should be stored.
-
f.Click on Apply in the processing tab.
-
a.
-
31.
Store the exported folder with DAPI .tiff files locally on your computer in a folder called “FLUOR_data”.
Note: Every .czi file results in export of one folder with DAPI .tiff files with the same name as the czi file (i.e. S1_T20 for the example illustrated in the Folder naming and structure paragraph of this protocol). This exported folder can contain DAPI .tiff files from multiple tile regions (i.e. S1_T20_R1 and S1_T20_R2).
Figure 4.

Tile-region scanning settings Zeiss Z1 imager
Figure 5.

Generating tile-region scan
Tile settings used on the Zeiss Z1 imager for scanning tile-regions. (1) Starting position of the cursor after X/Y position calibration. (2) Selection of a region on the glass slide for preview scanning. (3) Selection of a region of interest for 20× scanning on the preview scan.
Figure 6.

Setting focus points
(A and B) Settings used on the Zeiss Z1 imager for (A) adding focus points to tile scans, and (B) setting Z-stacks.
(C) Representative image of a selected region of interest for 20× scanning projected on the preview scan. The yellow circles indicate the set focus points.
(D) Pop-up window for verification of tile regions/positions.
Figure 7.

Export settings Zeiss Z1 imager
Stitching DAPI panorama
In this step, the exported tile-regions are stitched using Fiji scripts. First, single-image .tiff exported from the Zen software are concatenated into multi-image stacks to recreate the original Z-stack. Then, all stacks are converted to single in-focus images by the extended depth of field (EDF) principle. Finally, all EDF-projected images are stitched using the MIST plugin to create a full seamless panorama of the original tile-region.
The scripts expect a specific folder structure. The images from the different processing steps are stored in the following folders: “Stacks”, “EDF”, and “Stitch”. In total, the processing of 10 DAPI tile regions (2 tile regions of 5 tissue biopsies) consisting of 60 tiles takes approximately 8 h on a recommended workstation.
Note: Make sure to have the exported “_info.xml” file present next to the exported .tiff files (this file should be exported by the Zen software in the same folder as the .tiff files)
Note: When running Fiji steps, do not interfere with your computer.
Folder structure:
/FLUOR_data
/S1_T20
/+Stacks
/+EDF
/+Stitch
-
32.Concatenate single-image .tiff files to multi-image stacks.
-
a.Open script Fluorescence_StackGeneration2.ijm in Fiji (by dragging it into Fiji) and press run.
-
b.In the pop-up window, select the folder with all exported .tiff files from the panorama (S1_T20 folder in example) and click open.
-
c.The script will now process and store all acquired image stacks.
-
d.The script is finished when the log screen prints “DONE”. A “Stack” folder is now created containing stacked .tiff files. Troubleshooting 1
-
a.
-
33.Convert stacks to single-plane images.
-
a.Open script Fluorescence_ExtendedDepthOfField.ijm in Fiji (by dragging it into Fiji) and press run.
-
b.In the pop-up window, select the FLUOR_data folder and click open.
-
c.Running the script takes a while (approximately 30 min for 60 tile regions using npar = 3 settings. It will take a maximum of 15 min when using npar = 6 settings). The script is finished when the pop-up window “Processing completed!” appears. An “EDF” folder is now created with .tiff images.
-
a.
-
34.Create a seamless panorama by stitching all EDF-projected stacks.
-
a.Open script Fluorescence_TileRegionStitchName_ToSubfolder.ijm in Fiji (by dragging it into Fiji) and press run.
-
b.In the pop-up window, select the folder that contains your exported .tiff files (S1_T20 folder in example) and click open.
-
c.The script is finished when the log screen prints “DONE”. A “Stitch” folder is now created. This folder contains your stitched panorama.
-
a.
Note: Consider deleting the raw microcopy files when all tile-regions are correctly stitched in order to save storing space on your computer.
IMC ablation and export
In this step, IMC is used to ablate a tissue region from the prepared tissue section. It takes approximately 1 h for machine warm-up, and tuning. Ablating 2 regions (500 × 500 μm) of 5 tissue sections (located on different glass slides) takes approximately 8.5 h.
-
35.Ablate tissue regions from the stained tissue sections using the Hyperion according to the standard Fluidigm IMC protocol.
-
a.Name the regions (ROI) according to your naming scheme.
-
a.
-
36.
Save the mcd file.
Note: The ablated tissue area must be part of the prepared DAPI panorama (Figure 2).
-
37.Export the ablated regions of interest from .mcd files using Python and imctools.
-
a.Open Anaconda and launch Spyder.
-
b.Run the mcd_to_ometiff.py script.
-
i.Indicate the path of the input folder in line 79.
-
ii.Indicate the path of the output folder in line 80.
-
i.
-
c.ome.tiff files are now stored in the indicated output folder.
-
a.
Alternatives: Run the script in step 37 in PyCharm.
-
38.Extract IMC channel names.
-
a.Open an ome.tiff file in Fiji and run script OME-tiff-channelnames_imctools2.ijm. This script will give you an overview of the different IMC channel names present in the file. This list is required during downstream analysis.
-
a.
-
39.Rename the ome.tiff files according to the given ROI names during IMC acquisition.
-
a.Open script OME-renamer.ijm in Fiji and press run.
-
b.In the pop-up window, select the folder with extracted ome.tiff files from step 37 and click open.
-
c.The ome.tiff files exported in step 37 are now renamed according to the given ROI names during IMC acquisition.
-
d.Store the renamed stacked 32-bit .tiff files locally on your computer in a folder called “OMETIF”. Place the OMETIF folder in a folder called “IMC_data”.
-
a.
Image registration
In this step, the DAPI panorama is aligned with the Ir193 IMC images. First of all, the single Ir193 .tiff files are extracted from the combined 32-bit .tiff stacks that were created in step 39. Then, Fiji is used to align the DAPI panorama with the Ir193 IMC images using the SIFT and Landmark correspondence plugins. The “Ir193” and “Registration” folders are generated while running the scripts. The “FileList.csv file” is created manually while following the image registration steps. Image registration of 10 images takes approximately 3 h.
Folder structure:
/FLUOR_data
/S1_T20
/Stacks
/EDF
/+Registration
/+Keypoints
/+Overlay
/+FileList.csv file
/Stitch
/IMC_data
/OMETIF
/+Ir193
-
40.Determine the correct brightness and contrast for the DNA-Ir193 IMC channel.
-
a.Open the combined 32-bit .ome.tiff IMC images in Fiji. Scroll through the stack and identify the Ir193 channel (see step 38). Write down the number of this channel.
-
b.Open the Brightness/Contrast window using Image >> Adjust >> Brightness/Contrast. Set the minimum display brightness to 0. Drag the “maximum” down until the signal is saturated (can be checked by clicking LUT >> HiLo, red indicates saturated signal). Some pixels should show saturation, but not all. Identify the maximum intensity threshold in the Brightness/Contrast window and write down this threshold.
-
a.
CRUCIAL: Setting the correct brightness/contrast is crucial for further analyses. Examples of incorrect and correct settings are illustrated in Figure 8. Use identical brightness/contrast settings for multiple regions obtained during the same staining procedure and IMC run.
-
41.Extract DNA-Ir193 images from the stacked 32-bit .ome.tiff IMC files.
-
a.Open the OME_ExtractChannel2.ijm script in Fiji (by dragging it into Fiji).
-
i.Indicate the channel number of the Ir193 channel in line 2.
-
ii.Indicate the correct max intensity threshold for the Ir193 channel in line 4.
-
i.
-
b.Run the script.
-
c.In the first pop-up window, select the OMETIF folder.
-
d.In the second pop-up window, select the IMC_data folder.
-
e.The script is finished when the pop-up window “done” appears. An “Ir193” folder is now created in the IMC_data folder. This folder contains the single Ir193 .tiff files.
-
a.
-
42.Create a table in a comma-separated file (.csv file) where the DAPI and Ir193 images of the same regions are linked as illustrated in Figure 9A. The content of the file should be as follows:
-
a.Column 1: [Path to FLUOR Stitch folder]
-
b.Column 2: [Path to IMC Ir193 folder]
-
c.Column 3: [Name of the region = output name]
-
a.
Note: A header for the columns can be included in the first row of the .csv file (for instance: FLUOR, IMC, Name as indicated in Figure 9A).
-
43.
Save the file as “FileList.csv” in the FLUOR_data folder.
-
44.Register the DAPI and IMC images:
-
a.Open the script: Fluorescence_Registration_FileList.ijm in Fiji (by dragging it into Fiji) and click on run.Note: Adapt the scale parameter in the script (lines 18–20) to 1/pixel size for the used microscope settings. Default parameters are suggested for the Zeiss Z1 imager for a 20× objective.
-
b.Select the FLUOR_data folder. The script will start running now. This takes approximately 1–3 min per image.
-
c.The script is finished when the pop-up window “done” appears. The script will generate a “Registration” folder, as well as “Keypoints” and “Overlay” folders.
-
i.In the Registration folder, the cropped and registered DAPI images are saved.
-
ii.In the Overlay folder, a registered overlay image is saved.
-
i.
-
a.
-
45.
Check the overlay images located in the Overlay folder. If the DAPI images and Ir193 IMC images are properly overlayed, the image shows an overlay of blue (DAPI) and red (Ir193) colors, together creating magenta color. An example is illustrated in Figure 9B. Troubleshooting 2
Figure 8.

Brightness and contrast settings for IMC images
Examples are shown of correct and incorrect brightness and contrast settings of Ir193 IMC images. The original Ir193 images are shown, as well as images with HiLo display settings (blue: low-end saturated pixels; red: high-end saturated pixels). The brightness/contrast window indicates the used threshold for each image. Scale bar indicates 200 μm.
Figure 9.

DAPI and Ir193 image registration
(A) Example of the table (.csv file) for image matching and subsequent registration.
(B) Overlay (right) of DAPI (left image) and Ir193 (center image) images after registration. Blue: DAPI; Red: Ir193. Scale bar indicates 200 μm.
Select subset of data for training and validation
In this step, data is extracted for training purposes. We suggest using 10% surface area of every obtained image for training purposes. In order to keep the 10% selection of every image as unbiased as possible, random crop regions are obtained from the DAPI images and IMC channels in this step. One 10% crop region will be used for training purposes, the other 10% crop region will be used for validation of the training in the next step. The “MachineLearning”, “Training”, and “Validation” folders are generated while running the scripts, as well as the “SubsetROICoords.txt” file. Selecting data subsets from 10 images takes approximately 2 h.
Folder structure:
/FLUOR_data
/S1_T20
/Stacks
/EDF
/Registration
/Keypoints
/Overlay
/FileList.csv file
/Stitch
/+Training
/+Validation
/+SubsetROICoords.txt
/IMC_data
/OMETIF
/Ir193
/+MachineLearning
/+Training
/+Validation
-
46.Extract training and validation data from the DAPI images.
-
a.Open an aligned DAPI image in Fiji.
-
b.Open the script: RandomImageCrop2.ijm in Fiji (by dragging it into Fiji) and press run.Note: Adapt the scale parameter in the script (lines 7–9) to 1/pixel size for the used microscope settings. Default parameters are suggested for the Zeiss Z1 imager for a 20× objective.
-
c.In the pop-up window, select the size of the crops you would like to make in micrometers. The default setting is 158 × 158 μm (10% of a 500 × 500 μm image). Click “OK”.
-
d.Two proposed crop regions are shown on the DAPI image (Figure 10A). A pop-up window will appear with the question whether you are satisfied with the suggested regions. If yes, click “OK”.
-
e.If you are not satisfied with the suggested regions (for instance because they are located outside the tissue area), deselect “satisfied” and click “OK”. New crop regions will be suggested.
-
f.In the end, two non-overlapping regions per image will be generated. One image will be stored in a folder called “Training”. The other image will be stored in a folder called “Validation”.
-
a.
-
47.Select IMC channels:
-
a.Open the stacked 32-bit .tiff IMC images (located in the OMETIF folder) in Fiji. Scroll through the stack and identify + note down the numbers of the channels you would like to include for downstream analyses (use the table generated in step 38). In the example illustrated in this protocol, all markers included in the antibody cocktail (Table 1) were included for downstream analyses.Note: The order of the chosen channels is important to write down, since this information is not visible when opening the .tiff stack in Ilastik.
-
b.Determine the correct maximum intensity thresholds for the chosen channels as explained in step 40. These thresholds should work for all images inside the dataset.
-
c.Open the script: OME_ExtractMachineLearningStack2.ijm in Fiji (by dragging it into Fiji).
-
i.Indicate in line 1 the numbers of the channels you would like to include for membrane training.
-
ii.Indicate in line 2 the maximum intensity thresholds per channel.
-
i.
-
d.Run the script.
-
i.In the first pop-up window, select the OMETIF folder with 32-bit ome.tiff IMC files.
-
ii.In the second pop-up window, select the IMC_data folder.
-
i.
-
e.The script is finished when the pop-up window “done” appears. A “MachineLearning” folder is now created in the IMC_data folder. This folder contains the multichannel IMC images of the indicated channels.
-
a.
-
48.Extract training and validation data of the IMC images.
-
a.Open script RandomImageCrop_ApplyToOthers2.ijm in Fiji and press run.
-
b.In the pop-up window, select the “MachineLearning” folder that contains the stacked IMC images and click open.
-
c.In the second pop-up window, select the “SubsetROICoords.txt” file generated with the script in step 46 and click open.
-
d.The stacked IMC images are now cropped with identical coordinates as the fluorescent images as illustrated in Figure 10B and stored in the created “Training” and “Validation” folders within the MachineLearning folder.
-
a.
Figure 10.

Generation of training and validation subsets
Training and validation subsets are generated from the original fluorescent and IMC data.
(A) Two random crops consisting of 10% of the total tissue area are suggested within the original DAPI image. Scale bar indicates 50 μm.
(B) IMC image stacks are cropped with identical coordinates as defined in the DAPI images in (A). Scale bars indicate 25 μm.
DAPI probability maps
In this step, probability maps are generated based on the DAPI images. First, manual nucleus annotations are generated in Fiji. Next, a data enrichment step is performed in Fiji using the FeatureJ, MorphoLibJ plugins. Finally, these manual annotations and the enriched images are used for training machine learning in Ilastik software. A validation step is included in order to verify whether the training is sufficient. The “Annotations”, “Morph-Feat”, and “ProbabilityMaps” folders are generated while running the scripts. Generating DAPI probability maps for 10 158 × 158 μm training images takes approximately 7 h.
Folder structure:
/FLUOR_data
/S1_T20
/Stacks
/EDF
/Registration
/Keypoints
/Overlay
/FileList.csv file
/Stitch
/Training
/+Annotations
/+Mask
/+Overlay
/+Morph-Feat
/Validation
/+Morph-Feat
/SubsetROICoords.txt
/+Morph-Feat
-
49.Generating nucleus annotations data:
-
a.Open a single DAPI .tiff image from the Training folder in Fiji and then open the MannualAnnotation_Training.ijm script Fiji (by dragging it into Fiji) and click on run.
-
b.Choose “Nuclei” in the pop-up screen.
-
c.Zoom in and annotate nuclei using the brush tool (or another selection tool). Make sure to color the entire surface area of a nucleus, up to the nuclear border. Press B to store the drawn annotation. Click on “oke” when finished. Figure 11 shows annotation examples.Note: Annotate nuclei with distinguishable nuclear borders. Hence, only annotate the nuclei you are confident about.Note: It is recommended that different people annotate nuclei for training in order to obtain reliable training data.
-
d.Now annotate background pixels by selecting “Background” in the pop-up screen.
-
e.Select the areas in the images without nuclei using the brush tool (or another selection tool). Also draw closely around annotated nuclei. Press B to store the drawn annotation. Click on “OK” when finished with making annotations. Figure 11 shows examples of annotated background pixels.
-
f.Click on “save” when you are finished with all annotations. This will store your annotations in the “Annotations” folder.
-
a.
-
50.Data enrichment:
-
a.Open the FilterAdditionStack.ijm script in Fiji and press run. This script will result in data enrichment which will be used in the next step to generate DAPI prediction maps. In total, this script needs to be run 3 times: (1) for the training dataset, (2) the validation dataset, and (3) the actual dataset. Generated image features and 2D segmentation images are stored in the “Morph-Feat” folders.
-
i.Run 1: In the pop-up window, select the Training folder.
-
ii.Run 2: In the pop-up window, select the Validation folder.
-
iii.Run 3: In the pop-up window, select the Stitch folder.
-
i.
-
a.
Note: From this point onwards, the steps in the MATISSE pipeline do not depend on folder structure anymore.
-
51.Generate DAPI prediction maps on training images:
-
a.Start the Ilastik software and select the pixel classification workflow.
-
b.In the Input Data tab, add the images from the Morph-Feat folder inside the training folder.
-
c.Select all the added files and right click. Click on “Edit shared properties” and change storage from “relative link” to “copy into project file”. Click on OK.
-
d.In the Feature Selection tab, click on “Select Features”. Select all features with sigma 1. Click on OK.
-
e.Proceed to the Training tab and add 1 label to the already present labels, 3 in total.
-
f.In the “Current View” box, select the first image from your input data. Then right click on “labels” and click on import. Navigate to your annotations folder >> Mask >> and select the annotated image with identical name as selected in the current view. Click OK in the pop-up window. The manual annotations (generated in step 49) of this image will now appear on the screen.Note: Step 51f only works if users have generated annotations of all categories in all images. Otherwise, annotations will be put in the wrong category.
-
g.Repeat step 51f for every image present in the project.
-
h.Click live update (This takes ∼5 min for 10 training images. This step takes longer depending on the number of images and annotations included in the project).
-
i.Closely investigate the prediction maps. You can do this by scrolling down in the left window and selectively visualizing the prediction for label 1, 2, and 3 by clicking on the eye. Confirm that nuclei and background are properly identified (i.e., nuclear borders are properly defined, nuclei are not merged, and nuclei are not split into multiple events). In case nuclei and background are not properly identified, add additional annotations to the training set by repeating steps 49–51.
-
j.Turn off live update.
-
a.
-
52.Generation of DAPI probability maps of validation data:
-
a.In the Prediction Export tab, select Probabilities in the Source drop down menu. Then click on the ‘Choose Export Image Settings…’ button.
-
i.Change export format to multipage tiff.
-
ii.Change “Convert to Data Type” to unsigned 32-bit.
-
iii.In the output file info box, specify the saving directory and add a naming pattern.
-
iv.Click on OK.CRITICAL: In order to use the CellProfiler pipeline for cell segmentation during downstream analysis, make sure that the names of the exported DAPI probability maps (step 52 + 53) and IMC probability maps (step 55 + 56) are identical.
-
i.
-
b.In the Batch Processing tab, click on ‘Select Raw Data Files...’. Select the images within the Morph-Feat folder of the validation data. Then click on “Process all files”.
-
c.Closely investigate the probability maps and confirm that nuclei and background are properly identified. Troubleshooting 3
-
a.
-
53.Generation of DAPI probability maps of full dataset:
-
a.If nuclei and background are properly identified, proceed with generating DAPI probability maps of the full dataset by repeating step 52a and 52b. Select the images within the Morph-Feat folder of the Stitch folder. Specify the saving directory and naming pattern.
-
a.
Figure 11.

Manual annotations
Nuclei (red) and background (green) are manually annotated in the training images. Scale bar indicates 20 μm.
IMC probability maps
In this step, IMC probability maps for cell membranes and nuclei are generated based on the IMC images in Ilastik software. Generating IMC probability maps for 10 158 × 158 μm training images takes approximately 8 h.
-
54.Generate IMC probability maps:
-
a.Start the Ilastik software and select the pixel classification workflow.
-
b.In the Input Data tab, add the images in the IMC MachineLearning training folder to the project.
-
c.Select all the added files and right click. Click on “Edit shared properties” and change storage from “relative link” to “copy into project file”. Click on OK.
-
d.In the Feature Selection tab, click on “Select Features”. Select all features with omega 0.3 to 1.6 and click on OK.
-
e.Proceed to the Training tab and add the labels you would like to train for. For instance: (1) background, (2) immune cell membrane, (3) immune cell nucleus.Note: Write down the included labels and their names in their respective order! You need this for further steps.Note: It is recommended that different people annotate images for training in order to obtain reliable segmentation data.
-
f.In the current view box, select an image of interest. In the left window, go to “Raw Input” and browse through the different IMC channels of this image using the number indicated in the box. Each number corresponds to an IMC channel included in the machine learning image stack, starting at 0. In the example presented in Figure 12, Iridium (nuclei), CD45, CD3, and CD8 were chosen for training.
-
g.Identify a cell with a clear membrane signal (example in Figure 12). Then scroll through the different channels and make sure a nucleus is present for the chosen cell.
-
h.Start making pixel annotations of membranes and nuclei. A few things to keep in mind:
-
i.Use multiple channels to annotate the membrane of a cell. For instance, start making membrane annotations based on the CD45 signal. When finished, scroll through the IMC channels to see whether this cell is also CD3+ or CD8+. If there are white/light grey pixels in these channels that are not annotated yet, but belong to this specific cell, add pixel annotations based on this signal.
-
ii.Only annotate pixels that give a clear signal. Hence, annotate white and light grey pixels, but not black pixels! This means that sometimes, membrane annotations do not completely surround the nucleus annotation (Figure 12).
-
iii.You can annotate membranes of a cell without annotating the nucleus.
-
iv.It is possible to annotate multiple cell types based on different markers.
-
i.
-
i.Turn on live update and closely investigate probabilities. You can do this by scrolling down in the left window and visualizing the prediction for the different labels by clicking on the eye. Confirm that all classes are properly identified. Troubleshooting 4
-
j.Turn off live update.
-
a.
-
55.Generation of IMC probability maps of the validation data:
-
a.In the Batch Processing tab, add the images in the IMC MachineLearning validation folder to the project.
-
b.Export the probability maps as described in step 52.
-
a.
-
56.Generation of IMC probability maps of the full dataset:
-
a.If nuclei, membranes, and background are properly identified (Figure 12B), proceed with generating IMC probability maps of the full dataset by repeating step 52a and b. Select the images within the MachineLearning folder. The prediction export of 10 images takes approximately 15 min.
-
a.
Figure 12.

IMC annotations and probability maps
IMC images are used for machine learning to create membrane and nucleus probability maps.
(A) different channels are used for annotating membranes (red) and nuclei (blue). Scale bars indicate 10 μm (top rows), 25 μm (bottom row).
(B) Exported probability maps of the example shown in (A). Scale bar indicates 25 μm.
Single-cell segmentation
In this step, single-cell segmentation is performed in CellProfiler software using the DAPI and IMC probability maps generated with Ilastik.
-
57.
Start the CellProfiler software and import the CP-Pipeline.cppipe via file > import > Pipeline from file. The CellProfiler panel is divided into three sections as shown in Figure 13.
-
58.Configuring image input for CellProfiler:
-
a.Select the Images module. A box will appear in the module settings panel prompting for files to be placed into the box. Drag-and-drop the folders containing IMC probability maps and DAPI probability maps.
-
a.
-
59.Configure metadata:
-
a.Select the Metadata module and click on “yes” to “Extract metadata?”.
-
b.In the drop-down menu “Metadata extraction method”, select “Extract from image file headers”.
-
c.Change “Extract metadata from” to “Images matching a rule”.
-
d.Select the rule criteria which looks like “File”, “Does”, “Contain”, and write the name “IMC” in the empty box. Click “+” to add another rule and write “DAPI” in the empty box.
-
e.Click on the “update metadata” button below the horizontal divider.
-
a.
-
60.Configure naming patterns:
-
a.Select the NamesAndTypes module. All image types to be used by CellProfiler have to be identified based on their filename.
-
b.Assign a name to “image matching rules”.
-
c.Select the rule criteria “File”, “Does”, “End with”. Define criteria that are specifically true for all IMC and DAPI probability map tiff files and write the name in the box “Probability”. You can add extra rules and name of the images by clicking “+”.
-
i.IMC prob → “IMC_prob_membrane”, “IMC_prob_nucleus”.
-
ii.FLUOR prob → “FLUOR_prob_membrane”, “FLUOR_prob_nucleus”
-
i.
-
d.Name to assign these images: Enter a suitable descriptive name to identify the image for later use in the pipeline; downstream modules will then refer to the image by this name for processing. For example, “IMC_prob_membrane” can be used to indicate that all images associated with this type represent the membrane channel from the probability maps generated with the IMC data.
-
e.Press the “Update” button below the horizontal divider to index and display a table containing all identified images. Each row displays an unique image set, and the image types are listed in columns.
-
a.
-
61.
Select the module “IdentifyingPrimaryObject”. This will be used to identify nuclei within the DAPI probability maps nuclear category.
Optional: We have used the Otsu method for threshold strategy which automatically finds thresholds by dividing image pixels into two categories (foreground and background) by minimizing the variance within each class. The threshold method and value can be changed if required. Troubleshooting 5
Note: By selecting “intensity” in this module in the CellProfiler pipeline as the de-clumping method, a line will be drawn between the areas of two nuclei whose boundaries intercept. These nuclei will then be separated and identified as distinct nuclei.
-
62.
Choose ‘IdentifyingSecondaryObject’. This module will propagate outwards from the identified nuclei to create new objects that cover both the nuclei and edges. IdentifySecondaryObject modules require a FLUOR image plus nuclear primary objects which have already been identified.
Note: At this stage the pipeline has identified all nuclei in the probability maps and translated that information into a segmentation map that represents all identified nuclei as illustrated in Figure 14, and Figure 15.
Figure 13.

Screenshots CellProfiler interface
The CellProfiler pipeline panel is divided into three sections: the "Modules Image Input" in which to specify information about the images to be processed, the "Modules Analysis" which are executed sequentially to process the images, collect the measurements, and write the output. The "Module Settings Panel" provides the customizable settings for each selected module in the Pipeline panel.
Figure 14.

Identification of primary and secondary objects
(A and B) (A) Identification of primary object and (B) Identification of secondary objects of a particular tissue region in CellProfiler. Image axes are pixels. Image resolution is 0.65 μm/pixel.
Figure 15.

Single-cell segmentation map
Display of regions of interest (ROI) showing an overlay of DAPI (white), and the predicted cell outlines for nuclear segmentation (magenta). Scale bar indicates 200 μm (left) and 25 μm (right).
-
63.
This pipeline will save the segmentation maps and filenames in the “object folder” (folder naming specified by the user in the CellProfiler pipeline) based on IMC input names with suffixes for e.g., IMC_primary_object or IMC_secondary_object.
-
64.
Run the pipeline in test mode by pressing the button “Start test Mode” to check whether each module is working correctly. Repeat this on another image set to make sure set parameters are adequate.
-
65.
Start the analysis by pressing the “Analyze Images” button. The output from the CellProfiler pipeline is a comma-separated value (.csv) file summarizing the key values extracted during the image-processing step along with the images.
Single-cell data generation
Single-cell data is generated by extracting pixel intensities from unscaled 32-bit images for all channels for all cells represented in the segmentation maps.
-
66.MATISSE single-sell data generation:
-
a.Open script ExtractSingleCellDataParallel.r in RStudio.
-
b.Input the paths of the folder “Object” where all image files are stored.
-
c.Make sure all input images can be recognized using the associated filename patterns mentioned in step 63.
-
d.After completion of the script, single-cell data is stored in the file "SingleCellData-MATISSE.Rda".
-
a.
Expected outcomes
This protocol allows improved single-cell segmentation using combined IMC and fluorescence, and enables accurate prediction of tissue microenvironment in different tissue types. The final outcome of this protocol is the generation of single-cell data for all channels represented in segmentation maps as illustrated in Figure 16.
Figure 16.

Extraction of single-cell data
Matrix showing single-cell data generation in RStudio (right) from the CellProfiler segmentation map (left). Scale bar indicates 200 μm.
Limitations
The quality of single-cell segmentation is influenced by the quality of the tissue and the IMC staining. It is therefore recommended to exclude badly mounted and damaged tissues from the experiment. Additionally, it is recommended to ensure all antibodies in the IMC panel are tested and titrated. Importantly, the choices that are made for the IMC panel will have an effect on the outcome of the single-cell segmentation. Hence, a membrane staining needs to be included for all cell types of interest, to allow membrane-based segmentation. Users are responsible for making sure the segmentation is adequate.
As nucleus recognition heavily depends on manual annotations, it is essential that the user annotation quality is high (annotations need to be made by experienced users). Furthermore, tissue cell density is an important limitation of cell segmentation. Tissues with extremely high cell densities (such as tonsil) are difficult to annotate, since nuclei overlap in tissue sections, and can therefore result in nonoptimal segmentation maps. The MATISSE protocol is computationally time intensive in terms of training, and generating output data for individual nuclei and cells present in all imaged tissue regions.
Troubleshooting
Problem 1
Fiji scripts are not running and/or are producing errors while running (step 32).
Potential solution
In general, most issues with Fiji scripts arise when not strictly following the folder structure as explained in the Folder naming and structure paragraph of this protocol. Make sure all files and folders are in the expected locations and select the correct folders while running the scripts as indicated in the protocol.
Problem 2
Image alignment is not working properly (step 45), thereby generating an alignment image that does not show proper overlay of DAPI and Ir193 images as illustrated in Figure 17. Alternatively, no aligned image is generated in the output. Often the cause is that the IMC image is covering substantially less surface area in comparison to the stitched DAPI image, thereby increasing the risk of false keypoint identification.
Figure 17.

Troubleshooting 2, incorrect image alignment
Example of incorrect and correct image alignment of DAPI (blue) and Ir193 (red) images. Overlay of these colors is shown in magenta. Scale bar indicates 100 μm (top) or 25 μm (lower panels).
Potential solution
First trace which images have problems. Then open both Ir193 and stitched DAPI images of the troublesome tissue region. Crop the stitched DAPI image to the approximate region where the IMC data was acquired. Store as a separate file. Perform the alignment again using the script, after adding the paths of this image pair to the filelist. This pre-cropping makes the finding of SIFT keypoints more focused and therefore more likely to result in valid matches.
Problem 3
DAPI probability maps of the validation data set show incorrectly identified nuclei (step 52, Figure 18).
Figure 18.

Troubleshooting 3, incorrect DAPI probability maps
Yellow arrows show examples of incorrect nucleus identification in DAPI probability maps. In the original DAPI image it is clear that no nuclei are present in these locations. Red arrows show examples of merged nuclear surface prediction and absence of edge detection in the DAPI probability maps. Scale bar indicates 25 μm.
Potential solution
Repeat steps 49–52 and include additional annotations of nuclei and background on the training dataset and add these to the trained Ilastik project, then retrain. Specifically add annotations for cells that are incorrectly identified (pointy cells, areas with high cell density etc.). If the prediction maps in the training dataset look fine but the validation training dataset is still not optimal, consider reselection of training data to ensure it is representative for the entire dataset.
Problem 4
IMC probability maps show incorrectly identified membranes and nuclei (step 54).
Potential solution
Add more annotations to the training images until membranes, nuclei and background are correctly predicted. Try to specifically include new annotations for the regions/pixels that are not properly identified in the probability maps. Training is sufficient when individual cells are identified (no merged cells, every cell has a predicted nucleus and membrane etc.).
Problem 5
The most likely sources of errors while running the CellProfiler pipeline (step 61) are the thresholding to binary and the primary object (nuclei) identification parameters.
Potential solution
The intensity threshold affects the decision of whether each pixel will be considered foreground or background. A higher threshold value will result in only the brightest regions being identified, whereas a lower threshold value will include dim regions. So, it is highly recommended to check whether the automatically calculated threshold was unusually high or low by trying a different thresholding method (Otsu, manual, robust background, or minimum cross entropy).
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Yvonne Vercoulen (Y.Vercoulen@umcutrecht.nl).
Materials availability
This study did not generate new unique reagents.
Acknowledgments
We would like to thank UCyTOF.nl for CyTOF facility support, the CMM ICT support team for ICT infrastructure and tailored solutions, and UBEC for HPC support. Furthermore, we would like to thank Livio Kleij for imaging support, Domenico Castigliego for providing tissue sections, and Jeanette H.W. Leusen (UMCU, and TigaTx B.V.) for constructive meetings and funding co-acquisition. Finally, we thank Pascalle Hemelop for help with the supporting video in this protocol. This work has been supported by grants from cancergenomicscenter.nl NWO (Dutch Research Council) Gravitation 024.001.028, Stichting WKZ Fonds Grant ‘TREAT-PID’, and Health Holland Grant number TKI2018 ‘TumMyTOF’ (to Y.V., J.H.W.L., and TigaTx B.V.). Y.V. is a Center for Unusual Collaborations (CuCo) Fellow, consortium Structures of Strength.
Author contributions
Methodology, M.J.D.B., D.K., N.S. Software, M.J.D.B., N.S. Validation, D.K., S.D., M.A. Writing – original draft, D.K., N.S. Writing – Review & Editing, M.J.D.B., S.D., M.A., Y.V. Supervision Y.V. Funding acquisition, Y.V.
Declaration of interests
Y.V. receives funding through a Public-private partnership grant (TKI-Health Holland) with TigaTx B.V.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2021.101034.
Data and code availability
All generated image analysis scripts used in the MATISSE pipeline are publicly available in the VercoulenLab GitHub repository (https://github.com/VercoulenLab/MATISSEStarProtocol). All raw and processed data of the MATISSE pipeline are publicly available via https://zenodo.org/record/4727873. Importantly, the publicly available MATISSE data does not follow the naming patterns as described in this protocol.
References
- Baars M.J.D., Sinha N., Amini M., Pieterman-Bos A., van Dam S., Ganpat M.M.P., Laclé M.M., Oldenburg B., Vercoulen Y. MATISSE: a method for improved single cell segmentation in imaging mass cytometry. BMC Biol. 2021;19:99. doi: 10.1186/s12915-021-01043-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown M., Szeliski R., Winder S. IEEE; 2005. Multi-image matching using multi-scale oriented Patches; pp. 510–517. (2005 IEEE Computer Society Conference on Computer Vision and Pattern Recognition (CVPR’05)). [DOI] [Google Scholar]
- Carpenter A.E., Jones T.R., Lamprecht M.R., Clarke C., Kang I.H., Friman O., Guertin D.A., Chang J.H., Lindquist R.A., Moffat J., et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006;7:R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalfoun J., Majurski M., Blattner T., Bhadrijaju K., Keyrouz W., Bajcsy P., Brady M. MIST: accurate and scalable microscopy image stitching tool with stage modeling and error minimization. Sci. Rep. 2017;7:4988. doi: 10.1038/s41598-017-04567-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster B., van de Ville D., Berent J., Sage D., Unser M. Complex wavelets for extended depth-of-field: a new method for the fusion of multichannel microscopy images. Microsc. Res. Tech. 2004;65:33–42. doi: 10.1002/jemt.20092. [DOI] [PubMed] [Google Scholar]
- Gillespie C., Lovelace R. O’Reilly Media, Inc; 2016. Efficient R Programming. [Google Scholar]
- Legland D., Arganda-Carreras I., Andrey P. MorphoLibJ: integrated library and plugins for mathematical morphology with ImageJ. Bioinformatics. 2016;32:3532–3534. doi: 10.1093/bioinformatics/btw413. [DOI] [PubMed] [Google Scholar]
- Liu C., Yuen J., Torralba A. SIFT flow: dense correspondence across scenes and its applications. IEEE Trans Pattern Anal Mach Intell. 2011;33:978–994. doi: 10.1109/TPAMI.2010.147. [DOI] [PubMed] [Google Scholar]
- Meijering E. 2002. FeatureJ: An ImageJ Plugin Suite for Image Feature Extraction. http://imagescience.org/meijering/software/featurej/ [Google Scholar]
- Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer C., Straehle C., Koethe U., Hamprecht F.A. Eighth IEEE international symposium on biomedical imaging (ISBI) proceedings. 2011. Ilastik: interactive leraning and segmentation toolkit; pp. 230–233. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All generated image analysis scripts used in the MATISSE pipeline are publicly available in the VercoulenLab GitHub repository (https://github.com/VercoulenLab/MATISSEStarProtocol). All raw and processed data of the MATISSE pipeline are publicly available via https://zenodo.org/record/4727873. Importantly, the publicly available MATISSE data does not follow the naming patterns as described in this protocol.