

Abstract

A scalable four-step synthesis of molnupiravir from cytidine is described herein. The attractiveness of this approach is its fully chemical nature involving inexpensive reagents and more environmentally friendly solvents such as water, isopropanol, acetonitrile, and acetone. Isolation and purification procedures are improved in comparison to our earlier study as all intermediates can be isolated via recrystallization. The key steps in the synthesis, namely, ester formation, hydroxyamination, and deprotection were carried out on a multigram scale to afford molnupiravir in 36–41% yield with an average purity of 98 wt % by qNMR and 99 area% by HPLC.
Keywords: COVID-19, antivirals, molnupiravir, EIDD-2801, MK-4482
Introduction
Molnupiravir (also known as EIDD-2801 and MK-4482) is a promising drug candidate for treating COVID-19. Merck licensed the compound from Ridgeback Biotherapeutics in 2020, and clinical results with outpatients advanced molnupiravir to Phase 3 clinical trials.1 Molnupiravir offers complementary advantages over remdesivir such as oral bioavailability and structural simplicity, thus reducing the manufacturing complexity.2,3 The original synthesis of molnupiravir developed by Painter utilizes uridine (1) and proceeds in five steps with a low overall yield (17% overall; two steps have assumed yields) with the aid of an acetonide-protecting group strategy (Scheme 1).4
Scheme 1. Discovery Route to Molnupiravir.

As the above synthesis had a high step count accompanied by a low yield, an alternative synthesis of molnupiravir was necessary. In our original synthetic route, we found that Novozyme 435, a common lipase enzyme, could selectively esterify the primary hydroxy groups of cytosine derivatives without the need for acetonide protection, thus reducing the overall step count to two (Route I, Scheme 2).5a,5 The order of this two-step sequence can be varied either by carrying out the hydroxyamination of cytidine (7) first followed by regioselective acylation or vice versa. Also, Kappe and co-workers in the year 2020 reported a high-yield synthesis of molnupiravir from uridine.6 In a recent preprint disclosure, Merck demonstrated a similar enzymatic strategy for molnupiravir using ribose 9 and uracil proceeding in 69% yield over three steps.7 Although the enzymatic routes are indeed attractive because of the low step count, we felt that demonstrating a scalable, nonenzymatic reaction sequence would still be valuable to ensure maximum global access to this important drug candidate.
Scheme 2. Development Routes toward Molnupiravir.
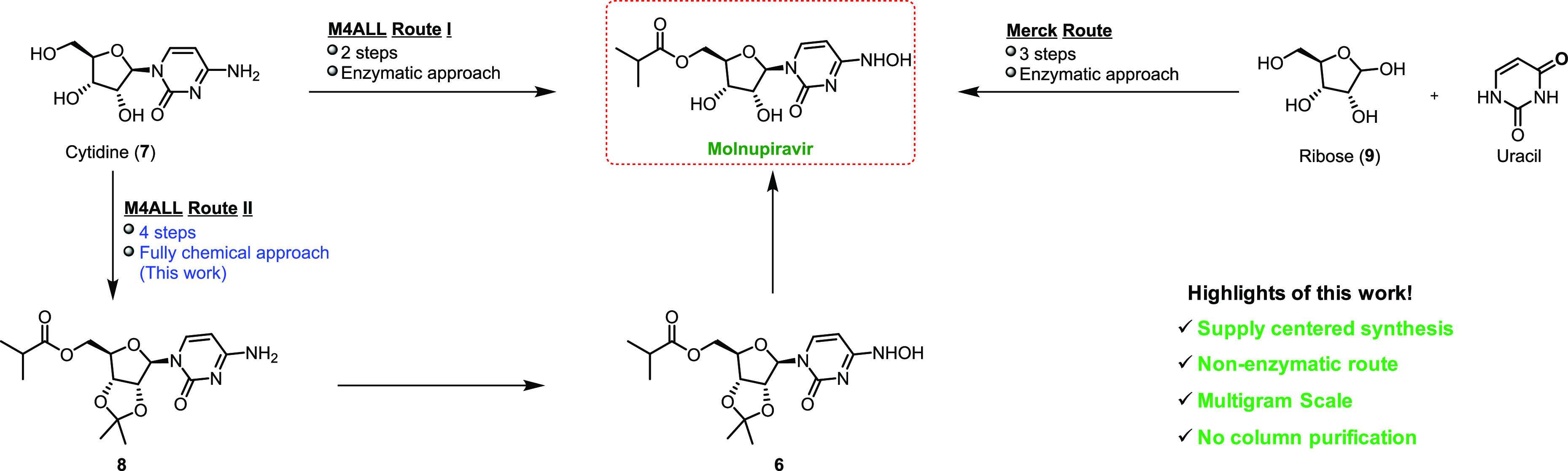
Our preliminary results on a fully chemical route to molnupiravir (Route II, Scheme 2) were disclosed recently.5b The details of our acetonide approach to molnupiravir are shown in Scheme 3. The synthesis began with protection of 7 as its acetonide 10 in 94% yield using 2,2-dimethoxypropane in conjunction with acetone and sulfuric acid. Chemoselective esterification was then accomplished using isobutyric anhydride and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) in acetonitrile to afford the ester 8 in 78% isolated yield. Hydroxyamination gave the penultimate intermediate 6 in excellent yield (96%), and upon deprotection with formic acid, molnupiravir was isolated in 42% overall yield in four steps. Alternatively, we can reduce one step in this sequence by using a solution of water/IPA (60/40), whereby molnupiravir is obtained in a single step from 8 in an overall 39% yield (Step 3′).
Scheme 3. Our Initial Acetonide Approach to Molnupiravir (M4ALL Route II).

While this three-step approach appears attractive, challenges foreseen to control the impurity profile of molnupiravir due to telescoping the entire operation made it less favorable for further development. Also, there is some safety and environmental concerns related to hydroxylamine. It was reported that hydroxylamine may explode on heating.8 It is also an irritant to respiratory tract, eyes, skin, and other mucous membranes making it a possible mutagen.9 However, hydroxylamine and its derivatives are more safely handled in the form of salts. The products from Steps 1 and 3 were isolated by crystallization; however, column chromatography was used to isolate the products of Step 2 and molnupiravir.5b This prompted us to explore further optimization to avoid some of these drawbacks.
We now report herein an optimized, operationally simpler process featuring chromatography-free isolation. The scalability of the entire process has been demonstrated by synthesizing multigram quantities of molnupiravir.
Results and Discussion
Impurity Profiling in Four-Step Synthesis of Molnupiravir
Prior to further developmental work, we identified the impurities in each step of our route either by direct isolation from the reaction mixtures or by synthesis of authentic materials for comparison with reaction in-process control data (Figure 1). Step 1 is very clean and no impurities were observed either during the reaction or after the isolation of the product, whereas the key impurities generated in Step 2 were the diacylated side compound 11 and the N-acylated compound 12. For Step 3, the key impurities in the crude reaction mixture were the starting material amine 8, molnupiravir, and the acetonide-deprotected compound 13 of the starting material. For Step 4, the key impurities were 6, 8, EIDD-1931, 13, and uridine (1).
Figure 1.

Side products in Step 2, Step 3, and Step 4.
Step 1: Acetonide Protection—Impurity Profile and Reaction Optimization
The acetonide protection of the vicinal diol of cytidine was performed using 2,2-dimethoxypropane (5 equiv) in acetone mediated by sulfuric acid (2.3 equiv) to afford 10, which crystallized from the reaction mixture as its sulfate salt and was isolated in 94% yield with 95 wt % purity based on quantitative nuclear magnetic resonance (qNMR) spectroscopy. Purity by high-performance liquid chromatography (HPLC) was 100%.
Step 2: Esterification—Impurity Profile and Reaction Optimization
A time profile for the acylation of 10 was recorded to determine the formation of product and impurities in this reaction (see Figure S1, Supporting Information). When the reaction was performed using isobutyric anhydride (1.1 equiv) and triethylamine (2.5 equiv) as the base in the presence of catalytic N,N-dimethylamino pyridine (DMAP, 20 mol %) in acetonitrile as a solvent at room temperature for 18 h, it was observed that within first 30 min, the reaction proceeded to more than 85% conversion and also that conversion quickly tapered off over the course of additional reaction time. When the reaction was stirred for additional 17 h of reaction period, only a marginal increase in product formation was observed along with the two side products, namely, over-acylated compound 11 and the N-acylated compound 12 (Figure 1) in 6.3 and 0.2 area % by HPLC (LCAP), respectively.
We were curious to know if 11 could be transformed to the desired product 6 of the subsequent step by transamination with hydroxylamine since this would eliminate the need for purging while providing a yield boost for the subsequent step. To probe this possibility, 11 was prepared from O-monoacylated 8 by reaction with iso-butyryl chloride (Scheme 4). Isolated 11 was treated with 3.2 equiv of hydroxylamine sulfate in 70% IPA-H2O at 78 °C for 24 h. This reaction led to the formation of at least seven compounds by HPLC, of which the major constituents were 6 and molnupiravir in LCAP 21 and 31%, respectively. Although we were disappointed that this reaction did not lead to a higher level of the desired compound 6, it did provide some useful insights for Step 3. In particular, this reaction profile matches that seen for the hydroxyamination step (Step 3), which suggests that the source of impurities in that reaction stems from side reactions arising from 11 (Scheme 4). This experiment clearly demonstrates that minimization of side product 11 in Step 2 is critical.
Scheme 4. Synthesis of Diacylated Side Product 11 and Its Subsequent Hydroxyamination Products.

A systematic optimization of key variables in Step 2 was conducted to maximize the formation of the monoacylated product 8 and minimize the diacylation side reaction leading to compound 11. The results of this optimization are provided in Table 1. The optimization was performed using the base (2.1 equiv) in the presence of catalytic DMAP (20 mol %) in acetonitrile as a solvent over the period of 8 h reaction time. However, after screening some of the bases as shown in Table 1 (entries 1–5), it was observed that the reaction performed well with DBU as the base and gives a good yield of the desired product within 3 h of reaction time (entries 5–11). Furthermore, the temperature and reaction concentration were studied (entries 5–11) using DBU as the base and it was observed that the most impactful result from this parameter screening is the role of the reaction concentration on the conversion of the starting material 10 to the product 8. Increasing the amount of the solvent from 2.5 to 10 V (entries 5–8) increased the LCAP of 8 from 89 to 94% and reduced the overall contribution from the impurities 10, 11, and 12 from 11 to 4.3%. Also, reaction temperature has a major effect on the formation of byproduct 11 and it was observed that decreasing the reaction temperature from 19 to 0 °C decreases the formation of byproduct 11 from 6.2 to 2.2% (entry 5 vs entry 9). Because the formation of byproduct 11 (major byproduct) requires two consecutive second-order reactions, here we reasoned that decreasing the reaction temperature would lead to a dramatic decrease in the rate of the second acylation reaction.
Table 1. Optimization of Regioselective Acylation of 10.

| entrya | base | temp (°C) | solvent (V) | LCAPb,c |
|||
|---|---|---|---|---|---|---|---|
| 8 | 10 | 11 | 12 | ||||
| 1 | Et3N | 19 | 10 | 59 | 0 | 17 | 0.5 |
| 2 | Bu3N | 19 | 10 | 52.6 | 0 | 21.6 | 1.1 |
| 3 | DIPEA | 19 | 10 | 60.4 | 0 | 17.5 | 0 |
| 4 | lutidine | 19 | 10 | 34.5 | 0 | 30.2 | 6.5 |
| 5 | DBU | 19 | 10 | 93.5 | 0.2 | 6.1 | 0.1 |
| 6 | DBU | 19 | 2.5 | 89.3 | 7.1 | 3.6 | 0.3 |
| 7 | DBU | 19 | 5 | 89.4 | 5.3 | 4.3 | 0.3 |
| 8 | DBU | 19 | 7 | 92.5 | 0.4 | 6.1 | 0.1 |
| 9 | DBU | 0 | 10 | 94.7 | 2.2 | 2.1 | 0.6 |
| 10 | DBU | 40 | 10 | 88.7 | 6 | 4.6 | 0.4 |
| 11 | DBU | 56 | 10 | 85.5 | 8.5 | 5.4 | 0.6 |
Reaction conditions: isobutyric anhydride (1.1 equiv), base (2.1 equiv), DMAP (20 mol %), and CH3CN.
Reaction time: 8 h for entries 1–4 and 3 h for entries 5–11.
LCAP at 260 nm.
After optimization of the key reaction variables for the regioselective acylation of 10, we found the optimal conditions for the formation of 8 in a high yield to be isobutyric anhydride (1.1 equiv) and DBU (2.1 equiv) in CH3CN (10 V) at 0 °C for a reaction time of 3 h (entry 9). Although DBU was found to be the optimal base for this reaction, its removal from the reaction mixture is challenging. In our first study, chromatographic separation was used to isolate the pure compound, but this will be costly on scale. Given the large pKa difference between DBU (∼12.5) and the cytidine amine functionality (∼4.2), we reasoned that a mild acidic workup could potentially remove DBU from the organic layer without extraction of the product 8. The reaction mixture was concentrated to remove acetonitrile and then dissolved in dichloromethane. Several aqueous acid washes were performed for DBU removal, including 10% ammonium chloride, 10% acetic acid, 10% sulfuric acid, and 85% H3PO4. It was observed that using 10% acetic acid resulted in the maximum recovery of the ester 8 and extraction of DBU into the aqueous layer. The acid wash was followed with a saturated sodium bicarbonate wash to remove the residual acetic acid. The product was then isolated by concentrating the organic layer to dryness. The purity of the final product was found to be 84 wt % by qNMR in chloroform-d, and LCAP was 94.7% for 8, 2.1% for 11, and 0.6% for 12.
A comparison of HPLC data for an in-process control versus the organic layer post 10% acetic acid and bicarbonate workup shows that the relative ratios of cytosine-containing materials were relatively unaffected by the extraction conditions. A trace amount of DBU was detected (0.5 area%) in the final product 8 (Figure S2, Supporting Information).
Step 3: Hydroxyamination—Impurity Profile and Reaction Optimization
In our preliminary study, the transamination of 8 using hydroxylamine sulfate resulted in 96% yield (94 wt % purity) of 6. Unreacted hydroxylamine sulfate was removed by filtration after dissolution of 6 in a minimal amount of acetonitrile. However, in our previous study,5b the starting material for this reaction had been purified by column chromatography. Conducting the same transamination reaction with ester 8 (84 wt % purity) that was obtained using our optimized acetic acid workup gives a mixture of the product 6, molnupiravir, and some unreacted starting material (Scheme 5). A typical distribution of products at the end of 20 h of the reaction period with an internal reaction temperature of 73 °C is 86% product 6, 6% of starting material 8, 3% molnupiravir, and ∼1% of the acetonide-deprotected ester 13 by HPLC. After isolation, this translated to 89% average-adjusted isolated yield with 73 wt % purity from duplicate runs before removal of unreacted hydroxylamine sulfate.
Scheme 5. Hydroxyamination and Side Products Formed.

Qualitative solubility studies were carried out using different solvents (Figure S3, Supporting Information) for purification of the product 6 (Table 2) and two methods were identified. The first method (Method A) involves recrystallization of crude 6 (73 wt % purity) from 2.5 V of isopropyl acetate. The mass recovery using this method is 66% and the purity is 98 wt % by qNMR in acetone-d6 and more 6 was present in the mother liquor from Method A. In the second purification method (Method B), recrystallization of the crude 6 (73% purity) from 2.5 V of acetonitrile provided 64% adjusted isolated yield of 6 with a purity of 96 wt % by qNMR. Additional product 6 (second crop) from Method B was isolated by concentrating the mother liquor and recrystallizing the remaining solid in 2.5 V of acetonitrile. This secondary isolation affords an additional 11% mass yield of material with a purity of 90 wt % by qNMR.
Table 2. Recrystallization of Crude Product 6 in Different Solvents.
| entry | solvent | solvent (V) | % mass recoverya | wt % purityb |
|---|---|---|---|---|
| 1 | chlorobenzene | 10 | 53 | 98 |
| 2 | CPME | 10 | 23 | 97 |
| 3 | anisole | 10 | 30 | 69 |
| 4 | toluene/chlorobenzene (1:1) | 5 | 61 | 96 |
| 5 | toluene | 10 | 84 | 82 |
| 6 | i-PrOAc | 2.5 | 66 | 98 |
| 7 | CH3CN | 2.5 | 64 | 96 |
Adjusted to purity.
By qNMR.
The two methods described above afford the Step 3 product 6 of >96% purity. More notably, the amount of 13 was reduced postpurification by either of these methods from 1.7 area% to 0.1 area% (see the HPLC trace in Figure S4, Supporting Information, a peak at a retention time of 4.998 min). The key impurities in 6 obtained by either of these methods were molnupiravir and 8 (2.6 area% and 0.3 area% by Method A and 2.1 area% and 0.5 area% by Method B). Method A was preferred for scale up as it provided 6 with a higher purity.
Step 4: Acetonide Deprotection—Impurity Profile and Optimization
With access to a relatively pure 6 from Step 3, screening of conditions for deprotection of the acetonide group in 6 was next performed (see Figure S5, Supporting Information). Most protic acids such as sulfuric acid, phosphoric acid, hydrochloric acid, or Lewis acids such as zirconium tetrachloride provided molnupiravir in low to modest yields (<50%) (as estimated by HPLC area% of the crude reaction mixtures) along with a variety of side products. For example, increased formation of 8 and 13 under some reaction conditions (TFA in EtOAc, TFA in DCM or HCl in DCM, and H2SO4 in DCM) possibly indicates a self-oxidation reduction, previously reported with aryl hydroxylamines.10 Of all the acids screened, only neat formic acid and trifluoroacetic acid provide conversions to molnupiravir in >90% yield. Finally, formic acid was selected for further development as it gave consistent results over different scales during the development of molnupiravir.
A range of recrystallization conditions was evaluated to purify molnupiravir (Table 3). Using 10 volumes of 1:1 EtOAc/acetonitrile, the condition used for purification via the enzymatic route,5d an 82% mass recovery of molnupiravir was obtained in three crops with 97 wt % purity (HPLC area % purity of the product was 97.8% and area% purity of known impurities 6 and 8 was 0.4 and 0.3%, respectively). Improved purity could be obtained by crystallization from five volumes of 1:1 n-BuOH/water with 99.7 wt % purity (the HPLC area % purity of the product was 99.7% and area% purity of the known impurity 8 was 0.2%, entry 4) or two volumes of water with 98.5 wt % purity (the HPLC area% purity of the product was 98.6% and area% purity of known impurities 6 and 8 were 0.3 and 0.8%, respectively, entry 3) although with reduced mass recovery. With two volumes of n-BuOH/water (1:1), precipitation of the product was observed with a lower purity. Based on the balance of purity of molnupiravir and its recovery, water was selected as the final crystallization solvent.
Table 3. Recrystallization of Molnupiravir from Various Solvents.
| entry | solvent | solvent (V) | % mass recoverya | wt % purityb |
|---|---|---|---|---|
| 1 | IPA | 10 | 91 | 89 |
| 2 | IPA–EtOAc (1:1) | 10 | 18 | |
| 3 | H2O | 2 | 69 | 98.5 |
| 4 | n-BuOH–H2O (1:1) | 5 | 59 | 99.7 |
| 5 | CH3CN | 10 | 95 | 95.7 |
| 6 | EtOAc–CH3CN (1:1) | 10 | 82c | 97 |
Adjusted to purity.
By qNMR.
Combined from three crops.
Scalability
A preliminary evaluation of the scalability of the entire sequence was conducted at 100 g scale. Acetonide protection proceeded smoothly to afford the product of Step 1 in an excellent yield and purity (>95%) on a 100 g scale (Table 4). The optimized Step 2 conditions resulted in >90% isolated yield with good purity on a 150 g scale. At a 130 g scale, the hydroxyamination (Step 3) showed modest yields of the product (70–76%) postrecrystallization with isopropyl acetate with good purity (∼95%) The final deprotection on a 80 g scale afforded molnupiravir in 59–61% yield with 98.5% wt % purity, which represents an overall yield of 36–41% over the four steps. Consistent results were observed in repetition batches on varying scales for each step as can be seen below in Table 4. The filtrate from the final recyrstallization contained additional molnupiravir (14%). The impurity profile of the molnupiravir obtained from this route indicates the presence of 6 (0.3 area%) and 8 (0.8 area%) as the two main impurities (Figure S6, Supporting Information).
Table 4. Scale Up of the Four-Step Sequence to Molnupiravir.

| step | scale (g) | LCAP of producta | wt % purityb | yield (%)c |
|---|---|---|---|---|
| 1 | 100 | 99.1 | 95.0 | 94 |
| 300 | 99.2 | 95.5 | 92 | |
| 2 | 150 | 98.6 | 89.0 | 92 |
| 140 | 94.0 | 85.8 | 95 | |
| 3 | 130 | 97.3 | 94.0 | 70 |
| 120 | 98.7 | 95.0 | 76 | |
| 4 | 80 | 98.6 | 98.5 | 59 |
| 100 | 99.6 | 97.6 | 61 |
LCAP at 260 nm.
By qNMR.
Yield adjusted to purity.
Conclusions
In summary, improvements and reaction details for our four-step route to molnupiravir from highly available cytidine are disclosed. Compound 10 from Step 1 is isolated directly from the reaction by filtration and washing, whereas compound 8 from Step 2 is obtained directly by washing the organic layer with 10% acetic acid. The pure compound 6 from Step 3 is obtained by recrystallization of the crude from isopropyl acetate, and molnupiravir from Step 4 is obtained by recrystallization from water. We have demonstrated comparable overall yields to our previous study and we have also been able to substitute column chromatographic purification with simple purification procedures that can be performed at a large scale.
Experimental Section
General
For all compounds, 1H and 13C NMR spectra were recorded using a Bruker Avance III 600 MHz spectrometer. Chemical shifts were measured relative to the residual solvent resonance for 1H and 13C NMR (CDCl3 = 7.26 ppm for 1H and 77.0 ppm for 13C, DMSO-d6 = 2.50 ppm for 1H and 39.5 ppm for 13C, CD3OD = 3.31 ppm for 1H and 49.0 ppm for 13C, and D2O = 4.79 ppm for 1H). Coupling constants J are reported in hertz (Hz). The following abbreviations were used to denote signal multiplicity: s, singlet; d, doublet; t, triplet; q, quartet; p, pentet; dd, doublet of doublet; ddd, doublet of doublet of doublet; dt, double of triplet; ddt, doublet of doublet of triplet; m, multiplet; and br, broad. Reactions were monitored by HPLC using the methods indicated. Quantitative NMR measurements were done using either mesitylene or 1,3,5-trimethoxybenzene as the internal standard. Glassware was oven-dried at 120 °C, assembled while hot, and cooled to an ambient temperature under an inert atmosphere. Unless otherwise noted, reactions involving air-sensitive reagents and/or requiring anhydrous conditions were performed under a nitrogen atmosphere.
Cytidine 7 was purchased from Chem-Impex. All other reagents and solvents were purchased from Aldrich Chemical Company, Fisher Scientific, Alfa Aesar, Acros Organics, Oakwood, or TCI. Liquid reagents were purified by distillation when necessary. Unless otherwise noted, solid reagents were used without further purification.
Acronyms: DBU (1,8-diazabicylo[5.4.0]undec-7-ene), MTBE (methyl tert-butyl ether), CPME (cyclopentyl methyl ether), DMAP (N,N-dinethylaminopyridine), LCAP (Area % by HPLC), qNMR (Quantitative NMR in the presence of an internal standard), DCM (methylene chloride), and IPA (isopropanol).
4-Amino-1-((3aR,4R,6R,6aR)-6-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)pyrimidin-2(1H)-one (1) Sulfuric Acid Salt
To the mechanically stirred 2000 mL three-neck round-bottom flask, cytidine 1 (100 g, 0.41 mol, 1 equiv) and anhydrous acetone (1300 mL) were added followed by 2,2-dimethoxypropane (251.9 mL, 2.055 mol, 5 equiv) under a nitrogen atmosphere. Neat sulfuric acid (50.7 mL, 0.94 mol, 2.3 equiv) was added to the above suspension and left for stirring for 15 h. The insoluble residue was filtered, and the solid precipitate was washed multiple times with acetone (1000 mL) followed by MTBE (400 mL). The solid was left for drying under vacuum for a day to obtain 155 g (94% corrected yield, 95 wt % purity by NMR in DMSO-d6) of compound 10 as an off-white solid.
Data matched with those previously reported.5b
((3aR,4R,6R,6aR)-6-(4-Amino-2-oxopyrimidin-1(2H)-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl isobutyrate (8)
To the mechanically stirred 2000 mL three-neck round-bottom flask, compound 10 (150 g, 95 wt % purity, 0.37 mol, 1 equiv) and dry acetonitrile (1500 mL) followed by DMAP (9.13 g, 0.075 mol, 0.2 equiv) and DBU (117.4 mL, 0.78 mol, 2.1 equiv) were added at room temperature. The reaction mixture was stirred for 10 min and isobutyric anhydride (65.11 mL, 0.39 mol, 1.05 equiv) was added dropwise at 0 °C in two equal portions at half hour intervals each and the reaction mixture was maintained for 2 h at the same temperature. The reaction mixture was then directly concentrated under a reduced pressure to afford a waxy solid. The resultant material was redissolved in dichloromethane (600 mL) and washed with 10% acetic acid (1000 mL) once. To the organic layer in a 2000 mL three-neck round-bottom flask was then added a clear solution of saturated sodium bicarbonate (1000 mL) dropwise with stirring until effervescence ceased. The layers were then separated, and the dichloromethane layer was dried over anhydrous sodium sulfate and concentrated under a reduced pressure to obtain 136.5 g (91.9% corrected yield, 89.3 wt % purity by NMR in DMSO-d6) of compound 8 as a white foamy solid.
1H NMR (600 MHz, CD3OD)
δ 7.90 (s, 1H), 7.63 (d, J = 7.4 Hz, 1H), 5.86 (d, J = 7.4 Hz, 1H), 5.71 (d, J = 6.9 Hz, 1H), 5.03 (dd, J = 4.7, 1.6 Hz, 1H), 4.84 (dd, J = 6.3, 3.4 Hz, 1H), 4.31 (m, 1H), 2.55 (septet, J = 7 Hz, 1H), 1.53 (s, 3H), 1.34 (s, 3H), 1.13 (ddd, J = 3.2, 1.8, 1.3 Hz, 6H) ppm.
13C NMR (151 MHz, CD3OD)
δ 178.49, 168.31, 158.14, 145.15, 115.34, 97.21, 96.25, 87.04, 86.71, 83.36, 65.86, 35.35, 27.74, 25.75, 19.75, 19.60, 19.52 ppm.
Data matched with those previously reported.5b
((3aR,4R,6R,6aR)-6-(4-(Hydroxyamino)-2-oxopyrimidin-1(2H)-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl isobutyrate (6)
To the mechanically stirred 2000 mL three-neck round-bottom flask, compound 8 (130 g, 89.3 wt % purity, 0.33 mol, 1 equiv) and hydroxylamine sulfate (172.7 g, 1.05 mol, 3.2 equiv) followed by 70% IPA (1300 mL) were added and the resultant solution was heated to an internal temperature of 72–73 °C for 19 h at which time HPLC showed the formation of a product in addition to the starting material and molnupiravir. At this juncture, the two layers were separated. The top layer (IPA) was concentrated to afford a thick residue. To this, acetonitrile (200 mL) was added as a cosolvent and concentrated to dryness. To the crude product, isopropyl acetate (375 mL) was added and heated to 80–85 °C for 30 min while stirring. It was then cooled to room temperature under slow stirring for 12 h and the solid which formed was filtered and washed with isopropyl acetate (150 mL) and dried under a vacuum pressure to obtain 84.8 g (70.1% corrected yield, 97% purity by HPLC, 94 wt % purity by NMR in acetone-d6) of compound 6 as a white powdery solid.
1H NMR (600 MHz, CD3OD)
δ 6.85 (d, J = 8.2 Hz, 1H), 5.69 (d, J = 2.2 Hz, 1H), 5.57 (d, J = 8.2 Hz, 1H), 4.97–4.99 (dd, J = 6.4, 2.2 Hz, 1H), 4.79–4.81 (dd, J = 6.3, 4.8 Hz, 1H), 4.26 (d, J = 5.3 Hz, 2H), 4.21 (q, J = 4.9 Hz, 1H), 2.60 (septet, J = 7 Hz, 1H), 1.53 (s, 3H), 1.34 (s, 3H), 1.15–1.17 (dd, J = 7, 1.8 Hz, 6H) ppm.
13C NMR (151 MHz, CD3OD)
δ 178.61, 151.42, 146.49, 134.21, 115.73, 99.73, 94.53, 85.62, 85.58, 82.87, 65.54, 35.36, 30.97, 27.79, 25.82, 19.61, 19.58 ppm.
Data matched with those previously reported.5b
((2R,3S,4R,5R)-3,4-Dihydroxy-5-(4-(hydroxyamino)-2-oxopyrimidin-1(2H)-yl)tetrahydrofuran-2-yl)methyl isobutyrate (Molnupiravir)
To the mechanically stirred 2000 mL three-neck round-bottom flask, compound 6 (80 g, 94 wt % purity, 0.20 mol) followed by formic acid (1300 mL) was added. The resultant solution was stirred at room temperature for 7 h. The solvent was removed under a reduced pressure and fresh EtOH (500 mL) was added. The resultant solution was again concentrated under vacuum to afford a waxy solid as a crude product. For recrystallization from water, to the crude (70 g), water (140 mL) was charged and heated to 60–65 °C for 10 min. The reaction mixture was cooled to 25–30 °C and slowly stirred for 12 h. The solid was filtered and the wet solid was washed with MTBE (210 mL) and dried under vacuum to get 40.0 g (58.7% corrected yield, 98.5 wt % purity by NMR in methanol-d4 and 98.3% purity by HPLC at 260 nm) of pure molnupiravir as an off-white solid.
1H NMR (600 MHz, CD3OD)
δ 6.91 (d, J = 8.2 Hz, 1H), 5.82 (d, J = 4.8 Hz, 1H), 5.61 (d, J = 8.2 Hz, 1H), 4.29 (d, J = 3.6 Hz, 2H), 4.14 (t, J = 4.9 Hz, 1H), 4.08 (p, J = 4.9 Hz, 2H), 2.62 (septet, J = 7.0 Hz, 1H), 1.19 (d, J = 7.0 Hz, 6H) ppm.
13C NMR (151 MHz, CD3OD)
δ 178.6, 151.81, 146.44, 132.04, 99.84, 90.74, 82.88, 74.67, 71.80, 65.23, 35.45, 27.49, 19.65, 19.61 ppm.
Data matched with those previously reported.5b
Acknowledgments
This work was supported, in whole or in part, by the Bill & Melinda Gates Foundation [OPP1176590]. Under the grant conditions of the Foundation, a Creative Commons Attribution 4.0 Generic License has already been assigned to the Author Accepted Manuscript version that might arise from this submission. We would like to express gratitude to Trevor Laird and John Dillon for insightful discussions and suggestions. We also thank Silpa Sundaram (BMGF) and Dr. Susan Hershenson (BMGF) for fostering an ecosystem where rapid decisions on project direction can be made. Authors are grateful to Ryan Nelson and G. Michael Laidlaw for their inputs in editing the manuscript.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.oprd.1c00219.
Reaction optimization, experimental details, copies of 1H and 13C NMR spectra of all new compounds, and HPLC method (PDF)
Author Contributions
∥ V.G. and A.L.K. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Recent news articles see; a Kropff A. “Coronavirus” https://www.wtsp.com/article/news/health/coronavirus/antiviral-drug-molnupiravir-showing-promise-in-trials-against-coronavirus/67-87f24932-8244-439a-a664-ba6aa21aac01, Published Mar 26, 2021.; b Merck media “News release”. https://www.merck.com/news/merck-and-ridgeback-biotherapeutics-provide-update-on-progress-of-clinical-development-program-for-molnupiravir-an-investigational-oral-therapeutic-for-the-treatment-of-mild-to-moderate-covid-19/
- Sheahan T. P.; Sims A. C.; Zhou S.; Graham R. L.; Pruijssers A. J.; Agostini M. L.; Leist S. R.; Schäfer A.; Dinnon K. H. III; Stevens L. J.; Chappell J. D.; Lu X.; Hughes T. M.; George A. S.; Hill C. S.; Montgomery S. A.; Brown A. J.; Bluemling G. R.; Natchus M. G.; Saindane M.; Kolykhalov A. A.; Painter G.; Harcourt J.; Tamin A.; Thornburg N. J.; Swanstrom R.; Denison M. R.; Baric R. S. An orally bioavailable broad-spectrum antiviral inhibits SARS-CoV-2 in human airway epithelial cell cultures and multiple coronaviruses in mice. Sci. Transl. Med. 2020, 12, 541. 10.1126/scitranslmed.abb5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox R. M.; Wolf J. D.; Plemper R. K. Therapeutically Administered Ribonucleoside Analogue MK-4482/EIDD-2801 Blocks SARS-CoV-2 Transmission in Ferrets. Nat. Microbiol. 2021, 6, 11–18. 10.1038/s41564-020-00835-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Painter G. R.; Perryman D.; Bluemling G. R.. 4’-Halogen Containing Nucleotide and Nucleoside Therapeutic Compositions and Uses Related Thereto, G.R.WO2019173602 September 12, 2019.; b Painter G.; Bluemling G.; Natchus M.; Guthrie D.. N4-Hydroxycytidine and Derivatives and Anti-Viral Uses Related Thereto, WO2019113462, June 13, 2019.
- a Vasudevan N.; Ahlqvist G. P.; McGeough C. P.; Paymode D. J.; Cardoso F. S. P.; Lucas T.; Dietz J.-P.; Opatz T.; Jamison T. F.; Gupton F. B.; Snead D. R. A Concise Route to MK-4482 (EIDD-2801) from Cytidine. Chem. Commun. 2020, 56, 13363–13364. 10.1039/D0CC05944G. [DOI] [PubMed] [Google Scholar]; b Gopalsamuthiram V.; Williams C.; Noble J.; Jamison T. F.; Gupton B. F.; Snead D. R. A Concise Route to MK-4482 (EIDD-2801) from Cytidine: Part 2. Synlett 2021, 32, 326–328. 10.1055/a-1275-2848. [DOI] [Google Scholar]; c Ahlqvist G. P.; McGeough C. P.; Senanayake C.; Armstrong J. D.; Yadaw A.; Roy S.; Ahmad S.; Snead D. R.; Jamison T. F. Progress Toward a Large-Scale Synthesis of Molnupiravir (MK-4482, EIDD-2801) from Cytidine. ACS Omega 2021, 6, 10396–10402. 10.1021/acsomega.1c00772. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Paymode D. J.; Vasudevan N.; Ahmad S.; Kadam A. L.; Cardoso F. S. P.; Burns J. M.; Cook D. W.; Stringham R. W.; Snead D. R. Toward a Practical, Two-Step Process for Molnupiravir: Direct Hydroxyamination of Cytidine Followed by Selective Esterification. Org. Process Res. Dev. 2021, 25, 1822–1830. 10.1021/acs.oprd.1c00033. [DOI] [Google Scholar]
- Steiner A.; Znidar D.; Ötvös S. B.; Snead D. R.; Dallinger D.; Kappe C. O. A High-Yielding Synthesis of EIDD-2801 from Uridine. Eur. J. Org. Chem. 2020, 2020, 6736–6739. 10.1002/ejoc.202001340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benkovics T.; McIntosh J.; Silverman S.; Kong J.; Maligres P.; Itoh T.; Yang H.; Huffman M.; Verma D.; Pan W.; Ho H.-I.; Vroom J.; Knight A.; Hurtak J.; Morris W.; Strotman N.; Murphy G.; Maloney K.; Fier P.. Evolving to an Ideal Synthesis of Molnupiravir, an Investigational Treatment for COVID-19. ChemRxiv. 2020, 10.26434/chemrxiv.13472373.v1. [DOI] [Google Scholar]
- Japan Science and Technology Agency Failure Knowledge Database Archived 2007-12-20 at the Wayback Machine.
- MSDS Sigma-Aldrich.
- Yang C.-H.; Lin Y.-C. The Self Oxidation Reduction of N-Arylhydroxylamines. J. Chin. Chem. Soc. 1987, 34, 19–24. 10.1002/jccs.198700004. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.